

Abstract
This study outlines the methodology to model hydrophobic deep eutectic solvent (HDES) interactions to obtain computational results that accurately represent experimental results of furfural removal from water. Computational prediction with high accuracy of HDES behavior could elucidate hydrogen bond interaction in HDES. COSMOtherm modeling and experimental evaluation demonstrated that both decanoic and dodecanoic acid-based HDES can remove furfural from water even at very low concentrations of 0.1 mol %. The modeling methodology considered salts as independent cations, which were paired with the hydrogen bond donor (HBD) species. These resulted in computational predictions of liquid–liquid equilibrium (LLE) between tetra n-alkyl ammonium bromide salt-based HDES with >95% accuracy of experimental results. The COSMOtherm modeling methodology strengthens the understanding of HDES by considering intermolecular forces that affect electron density (σ) of the HDES components to determine the LLE of the HDES-aqueous system. This results in a deep eutectic phase that has a positive sigma potential (potentials, μ(σ), up to 0.1 kcal/mol Å2) at charge densities associated with hydrogen bonding (±0.0084 e/Å2). Though n-alkyl ammonium salts ranging from tetramethyl- to tetraoctylammonium bromide were considered in the computational model, only pentyl- and longer alkyl chains displayed hydrophobic behavior with less than 1% salt loss to the aqueous phase. However, there was still significant water uptake in the eutectic phase (final phase composition containing greater than 60 mol and 12% by mass) for the hydrophobic DES.
1. Introduction
Deep eutectic solvents (DES) are a relatively new class of solvents that can optimize a solvent’s physical properties by complex interactions through multiple solvent components. DES are characterized by the mixing of two or more compounds that form a single phase with a lower melting point than any of the individual components. These components can be metal chlorides, weak acids and bases, or functionalized aromatic species. Generally, DES components are hydrogen bond acceptors (HBA) and hydrogen bond donors (HBD). Choline chloride (HBA) and urea (HBD) at 1:2 molar ratio was first shown to have a deep eutectic interaction by Abbot et al.1 The DES formed has a melting point of 12 °C, which is significantly lower than the melting points of choline chloride (300 °C) and urea (133 °C). Since then, additional DES have been shown to form using HBA of quaternary phosphonium and ammonium salts along with HBD of nearly any compound with an acidic hydrogen2−4 Unfortunately, most of these examples of DES are hydrophilic. Applications for these DES have been limited to organic systems and gas absorption from dehydrated gas streams.2,5−7 Water can be added to decrease DES viscosity, though processes that involve significant amounts of water can disrupt the DES phase significantly and change the properties of the solvents, which includes absorption parameters, polarity, density, and viscosity.8−10
The physical properties of the DES can be tuned and enhanced by altering HBA and HBD. This includes changing solubility and miscibility in water drastically compared to the individual DES components. Van Osch et al. demonstrated that utilizing quaternary ammonium salts (HBA) with long alkyl chains paired with long-chain acids (C8–C18, HBD) could be used to promote hydrophobicity in the DES, even though the HBA itself is soluble in water.11 This new category of DES, therefore referred to as hydrophobic deep eutectic solvents (HDES), allows for further application to extraction processes from an aqueous medium. One such application includes the recovery of valuable platform organic chemicals from aqueous solutions (whether from processed biomass, wastewater, or post thermochemical process liquid).12,13 Furfural is one such platform chemical that can be easily made through the hydrolysis of sugars in a water medium.14−16 Usually, furfural concentration in wastewater or process water can be found as high as 1000 mg/L with optimal conditions.17 This concentration varies with the operating parameters as well as the feedstock used. Furfural often repolymerizes into larger molecules at higher concentration and hydrolysis temperatures.18 Therefore, the concentration of furfural is often maintained low. Therefore, several stages of purification are required at the downstream processing. Generally, fluid extraction processes (such as liquid–liquid equilibrium extraction or supercritical extraction) are desirable for furfural purification since thermal methods can degrade the furfural or cause polymerization of the furan monomers.19,20
HDES could be a potential solvent for furfural removal from water. However, with overwhelmed possibilities of DES components (HBA and HBD), experimental methodologies are limited in their capacity to process the enormous possible combinations of components at varying ratios. Dietz et al. tackled a small subset of HDES to remove furfural from water using experimental methodologies and found that furfural has a 4:1 distribution ratio between water and HDES.21 This work is promising but only covers a small amount of solvents when considering all the possible combinations of HBA and HBD that can form HDES. Computational methods are therefore required to screen and process DES. However, modeling DES solvents and the eutectic interactions are not straightforward, as electrostatic interactions along with partial hydrogen bonding and solvation shells result in complex bonding networks. Depending on the type of HBA and HBD being modeled, methods such as the nonrandom two-liquid model (NRTL) can have errors up to 20% when predicting the solubility of salts in phosphonium-based DES.22 Multiple studies have even found that considering eutectic interactions for DES compounds made less accurate predictions than considering ideal solutions when using perturbed chain statistical associating fluid theory (PC-SAFT)-based models.23,24 These methods need adjustments to account for the hydrogen bonding that occurs in the DES. Studies that have explicitly incorporated corrections for hydrogen bonding from experimental data have achieved SAFT predictions closer to real values.25 However, additional corrections may be needed since, with HBA and HBD interactions, the electron density is shifted around the molecules, which can induce various dipoles that affect even nonhydrogen-based interactions.26 Therefore, molecular simulations and probabilistic models are required to evaluate DES solvent properties at higher accuracies. Density functional theory (DFT) is one such method for modeling the interactions between molecules by first determining electron distribution. COSMOthermTR is a software suite from Dassault Systèmes (previously Cosmologic.de) that utilizes DFT calculations to determine electrostatic interactions in liquids.27 This modeling methodology specifically focuses on electrostatic interactions as well as hydrogen bond affinities, which is more relevant for DES, as it represents an ionic bond in the halide salt as well as the eutectic hydrogen bond that is formed. Previous studies have used COSMOtherm to predict the solubility of hexane and benzene in hydrophilic quaternary ammonium salt-based DES.28,29 Overall, the predictions have been relatively close with root-mean-square deviations averaging 5%; however, the modeling methodologies are not always transferrable among solvents. A successful method for reline (choline chloride/urea 1:2 molar ratio) that considers its unique hydrogen bonding states is likely to be unsuccessful for HDES as tetraoctylammonium bromide and decanoic acid do not have the same bonding and intermolecular behaviors.
One of the limitations of the existing COSMO models, and other DFT models that evaluate electrostatic interactions of DES, is that intermolecular forces that redistribute electron density rapidly drive up the complexity and computational effort. To overcome the limitation, several studies assumed that electron densities of solutions, noted with sigma (σ, unit of e/Å2), are linear combinations of the component species.27,30,31 However, the charge densities of these solutions are not exactly linear combinations.32 The potential associated with the electron density distribution, noted as μ(σ) and referred to as sigma potential (kcal/mol Å2), can be used to elucidate intermolecular bonding. Negative sigma potential values show energetically favorable interactions, and potentials at high- and low-charge densities can be used to predict if a molecule will participate in hydrogen bonding. There are significant deviations in the area associated with hydrogen bond donation in DES, which will likely to increase COSMO model accuracy. Therefore, this study will evaluate the modeling of HDES by methods previously used on ionic liquid modeling (i.e., separate modeling of anionic and cationic species in the ammonium salt). These methods are expected to be more accurate (less than 5% error compared to experiments) for predicting furfural distribution in DES–water systems. This model will be validated by rigorous experiments that consider the effect of alkyl chain length on the ammonium salt and the size of the carboxylic acid.
2. Results and Discussion
2.1. Validation of COSMO Model Consistency
The modeling methodology used for this study was tested against previously verified models such as the similar COSMO-based model detailed by Hizaddin et al., which examined denitrification of fuels using hydrophilic DES.33 This modeling methodology predicted lower solubilities and selectivity of nitrogen-containing compounds such as pyrrole in a DES made of N4444Br and n-hexanoic acid (1:2 molar ratio). This model predicted a selectivity of pyrrole over diesel of 123 and a maximum capacity (determined at infinite dilution) of pyrrole of 6.1, whereas Hizaddin et al. reported selectivity of 207 and 24.6, respectively.33 In general, this difference is caused by the newer model predicting a lower degree of hydrogen bonding interaction between the solute species and the DES. This is a result of the newer model recalculating the electron distribution of the DES components after they have been mixed together, whereas Hizaddin et al. assumed a linear combination of sigma profiles.33 The result of these differences is that Hizaddin’s model has electron density potentials of N4444Br and n-hexanoic acid DES that do not go below −0.5 kcal/(mol Å2) at positive charge densities, whereas this model predicts larger values of −0.8 kcal/(mol Å2) at the same conditions. It should be noted that Hizaddin et al. also report errors lower than 5% between model and experimental data sets.33 This shows that models are not necessarily transferrable between solutions and solvents; their model was appropriate for their conditions, while the model used here is more appropriate for evaluating HDES.
Perturbed chain statistical associating fluid theory (PC-SAFT)-based models are also commonly used for modeling the electrostatic interactions found in DES. Dietz et al. developed a model for predicting CO2 solubility in HDES.34 Compared to the PC-SAFT model, the described COSMO methodology predicts a higher solubility of CO2 in a [N4444]+-based DES (mole fraction of CO2 at 0.20 compared to 0.14 at 1 MPa and 25 °C in N4444Cl and decanoic acid at a 1:2 molar ratio). This difference in prediction is consistent with other PC-SAFT models that also predicted a lower solubility of furans in ammonium and nonammonium-based HDES than those measured by experimental methods.24 Dietz et al. have reported, using a similar PC-SAFT model, that N8888Br/decnaoic acid (1:2 molar ratio) HDES has a high solubility of 5-hydroxymethyl furfural (HMF), whereas this model predicted full miscibility between the HDES and HMF.24 Both of these PC-SAFT models do not consider hydrogen bonding effects explicitly that occur between both the HDES components as well as the HDES and the target solute, but rather use a generic association term between components that includes all intermolecular interactions.24,34
Conformers of all quaternary ammonium cations were generated using COSMOconfx. Conformers and their respective energy compared to the ideal geometry are shown in Table S1. Conformers are considered as significant, when they have energies less than 4 kcal/mol higher than the ideal geometry.35 This is in agreement with more robust experimental studies that have investigated conformer energies of smaller quaternary ammonium salts.35 The addition of these additional conformers is significant, as nonideal geometries may actually have lower energy when interacting with additional solvents or solutes. DES have been shown to exist in multiple conformer states even without additional species being added to the solution.36 The addition of these conformers does introduce additional complexity to the calculations but should not be ignored when screening such a wide range of solvent compositions.
2.2. Charge Density Distribution of Individual Species and DES
Electron density distribution (distribution of σ(e/Å2)) is the basis for the equilibria calculations performed in COSMOtherm, with hydrophobic and hydrophilic species have drastically different profiles.32 Electron density distribution is commonly referred to as the sigma profile or σ-profile. For example, water has a dispersed profile over both positive and negative charge densities (±0.015 e/Å2), while hexane has a sharp peak at a neutral charge density that quickly falls off and does not extend beyond ±0.005 e/Å2..37 The total and maximum values of these electron density distribution profiles are dependent on the size of the molecule, so hexane has a much higher peak than water (nearly 40 Å2 compared to less than 5 Å2). Water’s sigma profile extends into the zones associated with hydrogen bond donation while hexane does not. Similar evaluations can be used to qualitatively compare different molecules before quantitatively comparing them using the calculations performed by the COSMO software.
Figure 1 shows the sigma profile of the various quaternary cations along with the sigma distributions of the DES and their respective components. The major sigma profile peak for all species occurs between −0.005 and −0.002 e/Å2 in the nonpolar region, with longer alkyl chains leading to higher peaks (as expected since there is more surface area on the longer molecules). These tightly grouped distributions show that the ammonium cation has a fairly consistent charge around the molecule. This can be mostly attributed to the longer alkyl chains, which have the significant surface area and only slightly positive charges (causing the peak to occur in the slightly negative electron density range of −0.005 to −0.002 e/Å2). All of the cations, when corrected for having a positive charge, correctly lack significant electron density at charges associated with hydrogen bond acceptance (electron density greater than 0.0084 e/Å2). In the DES itself, the halogen is what will be participating in the hydrogen bond interaction with the donor species (decanoic and dodecanoic acid).38
Figure 1.

Sigma potential profiles of tetraalkyl ammonium cations, decanoic acid, and dodecanoic acid with respect to charge density.
The sigma potentials elucidate additional information about the hydrogen bond accepting and donating capabilities of the HDES investigated and their individual components. These potentials, commonly referred to as sigma potentials or μ(σ), show how a molecule would favorably or unfavorably interact in a charged environment. These potentials are generally divided into two regions, the central region that shows nonpolar interactions (charge density < ±0.0084 e/Å2), and the outer regions that show hydrogen bond acceptance or donation capabilities (charge density > ±0.0084 e/Å2). Figures 2 and 3 show these profiles along with their molar and mass-based averages. All profiles can be found in the Supporitng Information.
Figure 2.

Sigma potential of N4444Br and decanoic acid HDES with respect to charge density. All other profiles are found in the Supporitng Information.
Figure 3.

Sigma potential of N4444Br and dodecanoic acid HDES with respect to charge density. All other profiles are found in the Supporting Information.
The x-axis for all figures is the charge density and spans both the hydrogen bonding and nonpolar interacting regions, while the y-axis refers to the sigma potential the molecule experiences when exposed to an environment with that charge. Each subfigure shows a different ammonium salt (from N4444Br to N8888Br) with its respective sigma potential profile, along with the potentials of its associated HDES. A key distinction between sigma profiles and sigma potential profiles is that charge density in the former refers to the molecule’s charge density, and charge density in the latter refer to the charge density of a surface not on the molecule. Thus, high electron density on a sigma profile would be reflected by a peak in the positive electron density region and mostly likely a favorable, negative potential in the negative electron density region of the sigma potential profile. Each HDES has three potentials: the potential calculated by COSMO (which was used for determining LLE), the potential determined by percent mass contribution, and the potential determined by percent mole contribution. The mass- and mole-based potentials, which were not used in any calculations, are shown to compare to methods used in previous studies that have examined the sigma potentials of DES.27,30,31 As expected from the sigma profiles, the sigma potentials of the ammonium salts (with the halogen now included) predict that these species can accept hydrogen bonds but cannot donate the hydrogens that they do not have. All N####Br species have negative sigma potentials below −0.0084 e/Å2 and positive sigma potentials above 0.0084 e/Å2. Lengthening the alkyl chain has little effect on the sigma potential of the HBA at negative charge densities, with values ranging from −0.809 to −0.798 kcal/mol Å2 for N4444Br and N8888Br, respectively, at −0.3 e/Å2. At positive charge densities, lengthening the alkyl chain increases the potential, with values ranging from 0.36 to 0.428 kcal/mol Å2 for N4444Br and N8888Br, respectively, at 0.3 e/Å2.
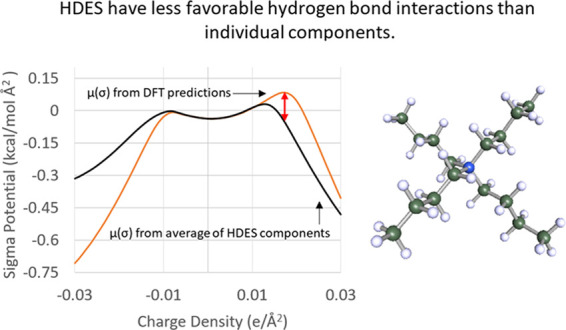
The HDES themselves have a positive potential after the hydrogen-bond-donating threshold (charge density = 0.0084 e/Å2) and only display donor affinity after charge densities of 0.022 e/Å2. This difference between the predicted molar average and actual sigma potentials is significant, as generally molar averages are considered acceptable prediction methods for sigma values.30,31,39 The largest difference between the molar average and model values occurs in the hydrogen bond donor region at 0.022 e/Å2, where the linear combination value is close to zero, and the model value is higher at around 0.1 kcal/mol Å2. This positive potential indicates that for there to be favorable hydrogen bonding interactions the bonding species must have an electron density greater than 0.022 e/Å2. In general, this would mean that the model predicts less extensive hydrogen bonding on species that are not strongly electronegative. For evaluating the performance of the HDES in removing furfural, this prediction of energetically unfavorable hydrogen bonds would also suggest that van der Waals interactions will be the primary mechanism for furfural removal.
Variations in the ammonium salt and the carboxylic acid had minimal effect on the sigma potential of the HDES phase. For all ammonium salts, the related HDES formed by either decanoic or dodecanoic acid have sigma potentials within a range of 0.006 kcal/mol Å2 (such as N4444Br when paired with decanoic acid compared to N4444Br paired with dodecanoic acid). Figures 4 and 5 show the sigma potentials of the decanoic and dodecanoic acid-based DES, respectively, with each DES’s potential being relative to N4444Br. Each figure shows two clear inflection points around the hydrogen bonding thresholds at approximately ±0.008e/Å2. Between these values (e/Å2 < ±0.008), the largest ammonium salts have the most negative potentials, but even so there is only a difference of 0.004 kcal/mol Å2. At charge densities greater than the hydrogen bonding threshold (e/Å2 > ±0.008), the larger the ammonium salt the more positive (and thus unfavorable) the sigma potential. This is significant as it shows that simply increasing the salt’s size, in this case increasing the alkyl chain length, will not make the HDES more available to hydrogen bonding (the main mechanism for the formation of the eutectic phase). The potential difference between the species is approximately 3% for each step in alkyl chain length (such as pentyl- > hexyl) in the hydrogen bonding regions (e/Å2 > ±0.008).
Figure 4.
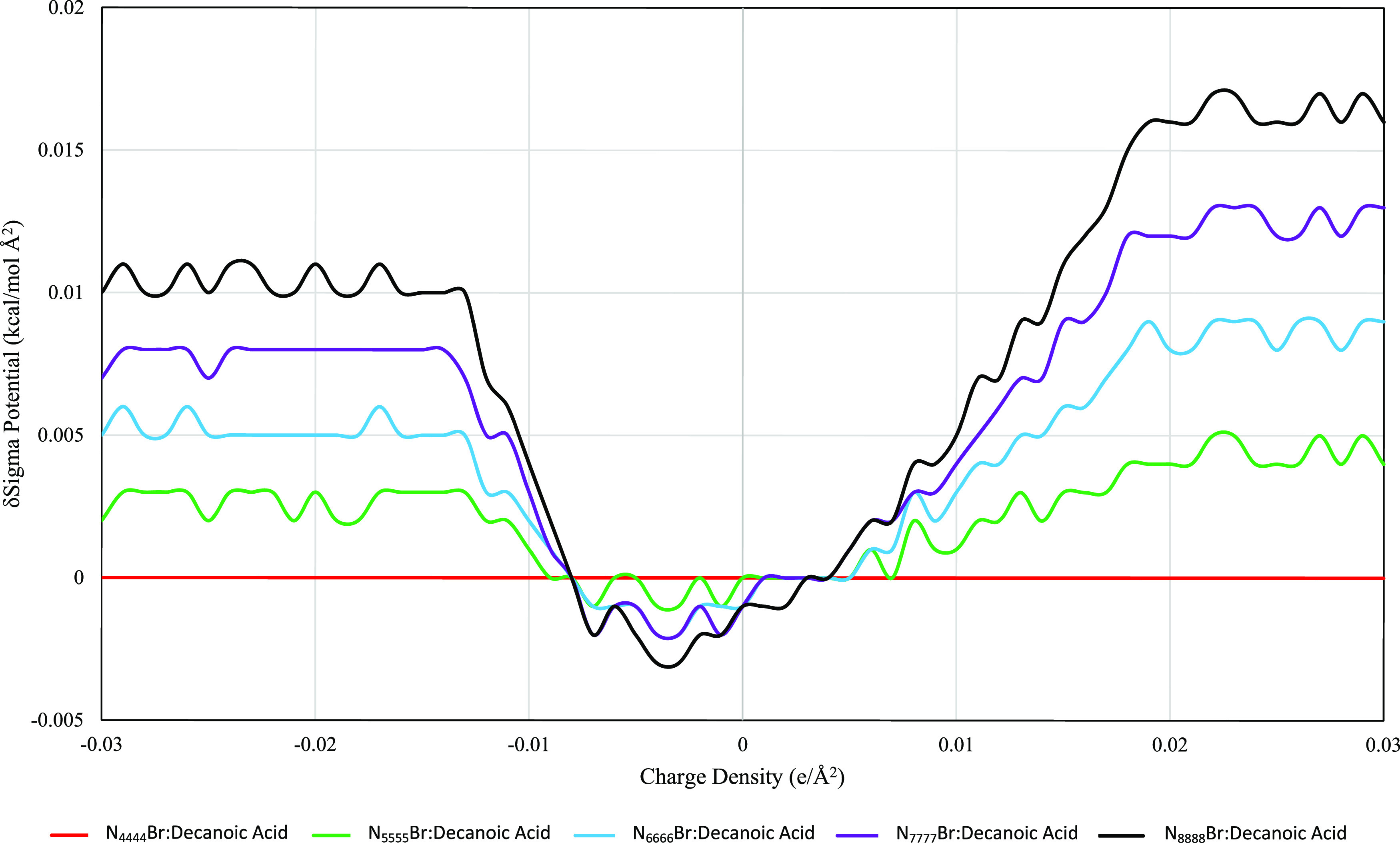
Stacked δSigma potential of N####Br decanoic acid HDES with respect to charge density; δSigma potential refers to relative potential compared to N4444Br decanoic DES.
Figure 5.
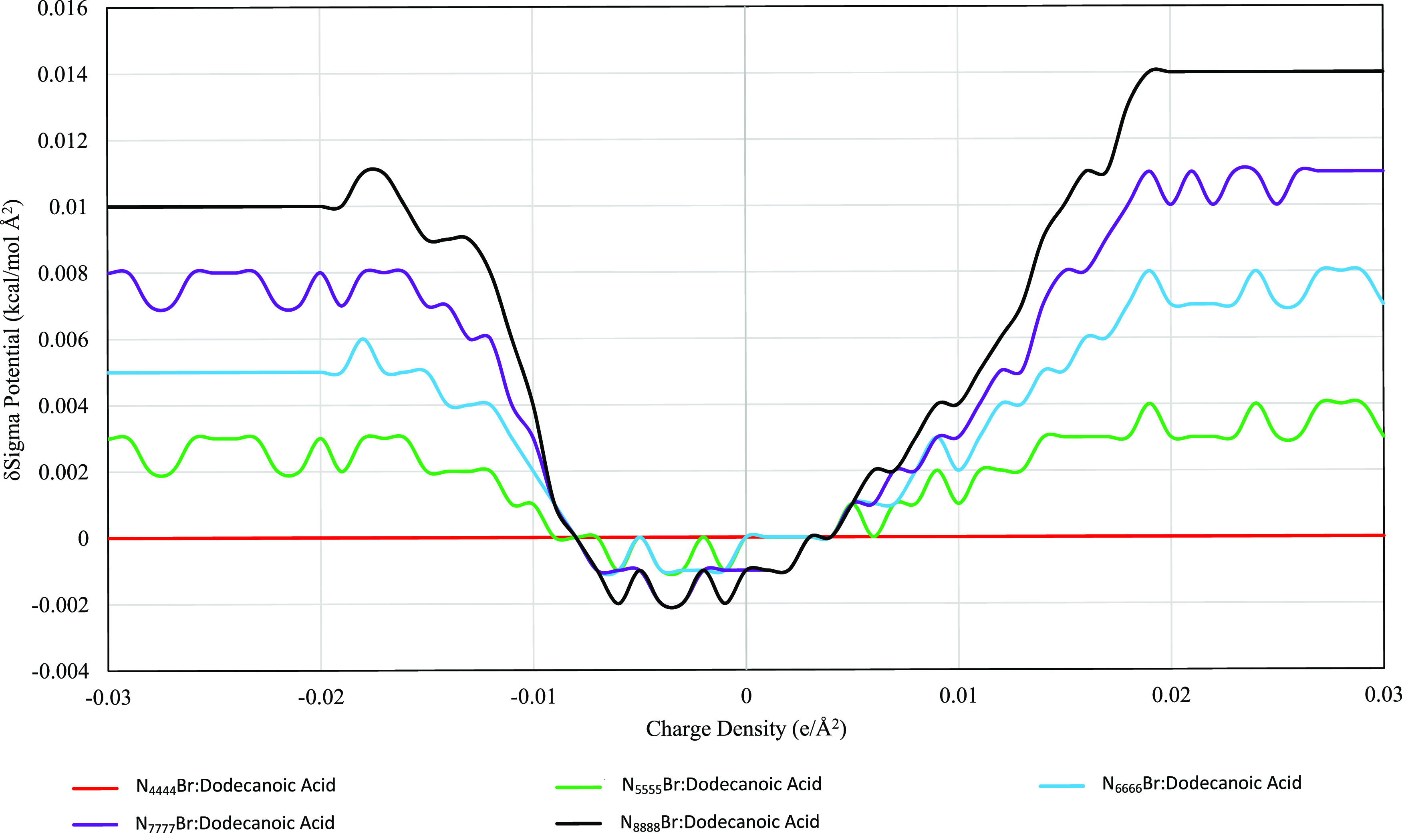
Stacked δSigma potential of N####Br dodecanoic acid HDES with respect to charge density; δSigma potential refers to relative potential compared to N4444Br dodecanoic HDES.
2.3. Furfural Extraction from Water and Salt Loss
LLE were modeled for systems of HDES made from decanoic and dodecanoic acid using N4444Br to N8888Br, at a 1:3 molar ratio. These were mixed with low concentration furfural solutions. Table 1 shows how each system was evaluated, and Table 2 shows the finial equilibrium reached in each system. In all systems, there is a two-phase distribution. Generally, phase 1 refers to the HDES-rich phase (mostly nonpolar), and phase 2 refers to the water-rich phase (mostly polar). N4444 and N5555 systems, when paired with either acid, do not keep their eutectic ratios as more of the ammonium salt moves into the aqueous phase than the carboxylic acid does. N4444Br and decanoic acid are at a 1:11.2 ratio and N5555Br and decanoic acid are at a 1:7.4 ratio in their respective nonaqueous phases. In dodecanoic acid, these ratios are 1:11.1 and 1:7.6 for N4444Br and N5555Br, respectively. This change in the ratio is because the ammonium salt has a stronger interaction with water than with the HBD. This causes most of the ammonium salt to move into the water phase (more than 70% for N4444Br and more than 55% for N5555Br when paired with either acid). Due to this significant mixing with water, DES with ammonium salts with chains shorter than five carbons can be considered hydrophilic, and those with longer chains can be considered hydrophobic (though there is still significant mixing of the two phases, a clear aqueous and nonaqueous phase).
Table 1. Computational and Experimental LLE Combinations of HBA and HBD for Furfural Removal from Water by HDES.
| aqueous phase | HDES phase | computationally evaluated | experimentally evaluated | ||
|---|---|---|---|---|---|
| water | furfural | decanoic acid | N4444Br | √ | |
| N5555Br | √ | ||||
| N6666Br | √ | √ | |||
| N7777Br | √ | ||||
| N8888Br | √ | √ | |||
| dodecanoic acid | N4444Br | √ | |||
| N5555Br | √ | ||||
| N6666Br | √ | √ | |||
| N7777Br | √ | ||||
| N8888Br | √ | √ | |||
Table 2. Equilibrium Phase Distribution of Furfural Solutions With N#### Bromide and Decanoic Acid/Dodecanoic Acid HDES at 40 °C and 1 atoma.
| DES component |
phase 1 species (mMol) |
phase 2 species (mMol) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| HBD | HBA | HBD | HBA | water | furfural | HBD | HBA | water | furfural |
| decanoic acid | N4444Br | 597.1 | 53.1 | 453 | 5.7 | 2.9 | 146.9 | 9547 | 4.3 |
| decanoic acid | N5555Br | 599.9 | 81.6 | 1698.8 | 8.4 | 0.1 | 118.4 | 8301.2 | 1.6 |
| decanoic acid | N6666Br | 599.9 | 199.9 | 1670 ± 80b | 8.5 ± 0.1b | 0.1 | 0.1 | 8330 ± 80b | 1.5 ± 0.1b |
| decanoic acid | N7777Br | 599.9 | 199.9 | 1481.7 | 8.4 | 0.1 | 0.1 | 8518.3 | 1.6 |
| decanoic acid | N8888Br | 599.9 | 191.4 | 1230 ± 60b | 7.9 ± 0.1b | 0.1 | 8.6 | 8770 ± 60b | 2.1 ± 0.1b |
| dodecanoic acid | N4444Br | 599.4 | 53.9 | 346.7 | 5.4 | 0.6 | 146.1 | 9653.3 | 4.6 |
| dodecanoic acid | N5555Br | 599.6 | 78.7 | 488.6 | 5.4 | 0.4 | 121.3 | 9511.4 | 4.6 |
| dodecanoic acid | N6666Br | 599.9 | 199.9 | 1380 ± 30b | 8.4 ± 0.1b | 0.1 | 0.1 | 8620 ± 30b | 1.6 ± 0.1b |
| dodecanoic acid | N7777Br | 599.9 | 199.9 | 1251.6 | 8.3 | 0.1 | 0.1 | 8748.4 | 1.7 |
| dodecanoic acid | N8888Br | 600 | 188.5 | 1030 ± 40b | 7.6 ± 0.1b | 0 | 11.5 | 8970 ± 40b | 2.4 ± 0.1b |
Initial conditions used 0.6 moles HBD, 0.2 moles HBA, 10 moles water, and 0.01 moles furfural.
Values with standard deviations shown are experimentally determined values.
Though there is little HBA loss in the longer chained HDES, there is a large amount of water uptake into the HDES phase. This additional water uptake may be beneficial for furfural extraction though, as N6666 removes more total furfural than N8888 (85% compared to 80%, respectively). Another explanation can be found in Figures 4 and 5, which show that increasing the ammonium salt’s alkyl chain length decreases the energetic favorability of hydrogen bonding. Phase distributions were confirmed using experimental methods previously described. The calculations were confirmed within ±5% relative error of the results presented in Table 2. Experimental values are shown in Table 2 with standard deviations, and computation values are shown without standard deviations. These results indicate a clear cutoff for HDES that can be used with aqueous systems with minimal solvent loss. This is consistent with previous experimental research, which has shown that DES with ammonium salts with shorter alkyl chain than N5555 are water miscible.11,40,41 For N4444Br with decanoic acid, there is a loss of salt leaching of 0. 28 g/g water at equilibrium. This is comparable to measurements taken by van Osch et al. using similar solvents (0.35 g of N4444Cl lost per gram of water).11 Longer alkyl chains drastically reduce the amount of salt leaching, with less than 1 mol % of N6666Br and N7777Br leaching into the aqueous phase. N8888Br has slightly higher leaching, with 4.3 and 5.8% loss with decanoic and dodecanoic acid, respectively. This increase, despite having the longest alkyl chains of any of the HBA, is most likely due to weakened interactions between the HBA and HBD in the HDES. Of the ammonium salts that do not dissolve into water, N6666Br extracts the most furfural (85%), though it also takes up the most water (67 mol %/13.7 mass % water with decanoic acid and 63 mol %/ 12.7 mass % water with dodecanoic acid). This is far more water than is predicted by PC-SAFT models, which predict less than 4 mass % of water in N8888Br/decanoic acid HDES.24 For N8888Br/decanoic acid, specifically the modeled and experimental results match previous research that measured a 4:1 ratio of distribution of furfural between the DES and aqueous phases even with less acid, suggesting that the salt is primarily responsible for furfural removal.21 Increasing the hydrophobicity of the acid lowers the amount of water in the HDES phase, but has no significant impact on the amount of furfural removed. This was true for both HDES that did and did not keep their ratios of 1:3 HBA/HBD in the HDES phase.
3. Conclusions
Hydrophobic deep eutectic solvents were modeled using COSMOtherm software suite. This process included density functional theory (DFT)-based modeling of the electron distribution of the quaternary ammonium salts and evaluation of the various geometric conformers that may appear. The separate consideration of the bromide anion and ammonium cation resulted in an accurate prediction for the HDES’s ability to remove furfural from water. This step resulted in a sigma potential distribution that was dissimilar from the linear combination of the sigma potentials of the component species, specifically in the hydrogen bond-donating region. The HDES did not display donor affinity until after charge densities of 0.022 e/Å2, well beyond the normal threshold for hydrogen bonding of 0.0084. Shorter alkyl chains on the ammonium salts generally reduced the ability to remove furfural form water, as there was significant salt leaching into the aqueous phase. Future research into hydrophobic deep eutectic solvents should consider N5555Br as the smallest ammonium salt that does not have significant salt loss in water.
4. Materials and Methods
4.1. Modeling Process of N-Quaternary Ammonium Cations
COSMOtherm’s TURBOMOLEX module was used to create COSMO profiles for HDES components. These COSMO profiles are the energetically favorable configuration of the components, whose initial electron distributions are calculated using the tri-zeta valence polarizable (TZVP) parameters. The Supporting Information shows the two such configurations of tetra octyl ammonium ion before and after minimizing energy by optimizing geometry that results from this molecular modeling process (Figure S1). Using the COSMO parameter (meaning each atom as an ideal conductor) selection along with the TZVP parameter set the modeled molecules are considered as ideal conductor where electron density can move across the molecules’ surface. This is preferable for the HDES components, as capturing the correct electronic nature of the molecules is vital for correctly macroscopic behavior of the model. This is particularly important when there is such a significant polarity difference between phases, such as liquid–liquid extractions (LLE) between hydrophobic (dielectric constant below 15) and aqueous phases (dielectric constant above 75).
N-Quaternary ammonium cations ranging from tetramethylammonium, [N1111]+, to tetraoctylammonium, [N8888]+, were modeled using the methods described above. The central nitrogen was considered with an SP3 hybridization and the overall molecular charge was considered +1 for all species (as they were modeled as cations). This independent modeling of cations and anion is meant to reflect experimental observations of eutectic solvent arrangement in real solutions.42 Additional conformers were generated using COSMO’s confX18 module. A basis set of TZVP was also used for conformer generation. This module determines the various geometric confers that exist by performing the same calculations in TURBOMOLEX but with intermediate steps of adjusting the dihedral bonding angles. The relative energy of each conformer is then compared to the original molecule. Similar molecules are then grouped together, so that even if hundreds of geometric variations exist, the whole set can be approximated by a few (<10) unique geometric configurations. The conformers used in this study can be found in the Supporting Information.
4.2. COSMOtherm Liquid–Liquid Extraction (LLE) of Furfural from Water
COSMOtherm’s LLE property calculation option was used to determine the equilibrium state of various HDES when mixed with water and furfural. All calculations for equilibria were performed at 40 °C and 1 bar, which matched with the experimental conditions. Only N4444Br–N8888Br were considered as the lower alkyl chain lengths than N4444Br are fully miscible in water and do not form a separate phase readily.43 For each case, 0.6 moles of acid (either decanoic or dodecanoic) and 0.2 moles of HDES (0.2 moles of [N####]+ along with 0.2 moles of [Br]−) were considered Phase 1, and 10 moles of water with 0.01 moles of furfural were considered Phase 2. This low concentration of furfural was selected to mimic experimental measurements of furfural in treated wastewaters.17 The COSMO profiles of all acids, water, and furfural were all extracted from the COSMO TVZP molecular library. Both acids were considered with five conformers, while furfural had two conformers. Extended options of 500 000 iterations (though most converged in less than 1000 and all converged before 100 000) and a desired convergence threshold of 1×10–6 was used for all LLE calculations (convergence refers to a relative change in concentrations between calculation iterations).
4.3. Experimental Determination of Liquid–Liquid Extraction (LLE) of Furfural from Water
Phase equilibria between HDES and furfural solutions were determined by the mixing of the two individual phases into a centrifuge tube that was kept at 40 °C in a water bath at atmospheric pressure. HDES were prepped from N4444Br, N6666Br, and N8888Br and decanoic and dodecanoic acid combinations at a 1:3 molar ratio, as shown in Table 1. This ratio of 1:3 for salt/acid was selected to minimize salt leach into the aqueous phase.11 Additionally, previous works have shown that hydrophobic DES have strong melting point depressions even outside the ideal eutectic ratio.44 Osch et al. have also noted in previous works that increasing the amount of carboxylic acid in HDES can extend HDES life when regenerating after extraction has been finished.45 All chemicals were purchased from Fisher Scientific (Hampton, NH). The samples were allowed to mix for 4 h before analysis by ultraviolet–visible (UV–vis). Furfural concentration in the aqueous phase was determined by UV–vis using a Hach DR6000 UV–vis spectrometer (Loveland, CO). The water capacity of the HDES was determined through the addition of 100 μL of water into a known mass of HDES, also at 40 °C. The HDES was considered saturated when there was a visible clear aqueous phase, which would appear below the HDES phase. The mixture was allowed 30 min between water additions to come to equilibrium, though most solutions seemed to visually separate after saturation within a minute. All experimental samples were kept in a desiccator and covered in plastic film to prevent the uptake of atmospheric moisture. All experimental conditions were completed in triplicates. Table 1 shows all solvent combinations evaluated using computational modeling and experimental methods. All combinations are at a 1:3 molar ratio between HBA and HBD.
Acknowledgments
This work was funded through the Petroleum Research Fund by the American Chemical Society (PRF # 60342-DNI9) and by the National Science Foundation under Grant No. 1856058.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02665.
Conformer energy and configurations for all tetraalkyl ammonium cations, additional plots of sigma potential of hydrophobic deep eutectic solvents (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Abbott A. P.; Capper G.; Davies D.; Rasheed R. K.; Tambyrajah V. Novel Solvent Properties of Choline Chloride/Urea Mixtures. Chem. Commun. 2003, 70–71. 10.1039/b210714g. [DOI] [PubMed] [Google Scholar]
- Abbott A. P.; Capper G.; Davies D. L.; McKenzie K. J.; Obi S. U. Solubility of Metal Oxides in Deep Eutectic Solvents Based on Choline Chloride. J. Chem. Eng. Data 2006, 51, 1280–1282. 10.1021/je060038c. [DOI] [Google Scholar]
- Shahbaz K.; Baroutian S.; Mjalli F. S.; Hashim M. A.; AlNashef I. M. Densities of Ammonium and Phosphonium Based Deep Eutectic Solvents: Prediction Using Artificial Intelligence and Group Contribution Techniques. Thermochim. Acta 2012, 527, 59–66. 10.1016/j.tca.2011.10.010. [DOI] [Google Scholar]
- Ribeiro B. D.; Florindo C.; Iff L. C.; Coelho M. A. Z.; Marrucho I. M. Menthol-Based Eutectic Mixtures: Hydrophobic Low Viscosity Solvents. ACS Sustainable Chem. Eng. 2015, 3, 2469–2477. 10.1021/acssuschemeng.5b00532. [DOI] [Google Scholar]
- Hizaddin H. F.; Hadj-Kali M. K.; Ramalingam A.; Ali Hashim M. Extractive Denitrogenation of Diesel Fuel Using Ammonium- and Phosphonium-Based Deep Eutectic Solvents. J. Chem. Thermodyn. 2016, 95, 164–173. 10.1016/j.jct.2015.12.009. [DOI] [Google Scholar]
- Oliveira F. S.; Pereiro A. B.; Rebelo L. P. N.; Marrucho I. M. Deep Eutectic Solvents as Extraction Media for Azeotropic Mixtures. Green Chem. 2013, 15, 1326–1330. 10.1039/c3gc37030e. [DOI] [Google Scholar]
- Rodriguez N. R.; Requejo P. F.; Kroon M. C. Aliphatic–Aromatic Separation Using Deep Eutectic Solvents as Extracting Agents. Ind. Eng. Chem. Res. 2015, 54, 11404–11412. 10.1021/acs.iecr.5b02611. [DOI] [Google Scholar]
- Hammond O. S.; Bowron D. T.; Edler K. J. The Effect of Water upon Deep Eutectic Solvent Nanostructure: An Unusual Transition from Ionic Mixture to Aqueous Solution. Angew. Chem., Int. Ed. 2017, 56, 9782–9785. 10.1002/anie.201702486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A.; Pandey S. Solvatochromic Probe Behavior within Choline Chloride-Based Deep Eutectic Solvents: Effect of Temperature and Water. J. Phys. Chem. B 2014, 118, 14652–14661. 10.1021/jp510420h. [DOI] [PubMed] [Google Scholar]
- Shah D.; Mjalli F. S. Effect of Water on the Thermo-Physical Properties of Reline: An Experimental and Molecular Simulation Based Approach. Phys. Chem. Chem. Phys. 2014, 16, 23900–23907. 10.1039/C4CP02600D. [DOI] [PubMed] [Google Scholar]
- van Osch D. J. G. P.; Zubeir L. F.; van den Bruinhorst A.; Rocha M. A. A.; Kroon M. C. Hydrophobic Deep Eutectic Solvents as Water-Immiscible Extractants. Green Chem. 2015, 17, 4518–4521. 10.1039/C5GC01451D. [DOI] [Google Scholar]
- Dai Y.; Rozema E.; Verpoorte R.; Choi Y. H. Application of Natural Deep Eutectic Solvents to the Extraction of Anthocyanins from Catharanthus Roseus with High Extractability and Stability Replacing Conventional Organic Solvents. J. Chromatogr. A 2016, 1434, 50–56. 10.1016/j.chroma.2016.01.037. [DOI] [PubMed] [Google Scholar]
- Tang X.; Zuo M.; Li Z.; Liu H.; Xiong C.; Zeng X.; Sun Y.; Hu L.; Liu S.; Lei T.; Lin L. Green Processing of Lignocellulosic Biomass and Its Derivatives in Deep Eutectic Solvents. ChemSusChem 2017, 10, 2696–2706. 10.1002/cssc.201700457. [DOI] [PubMed] [Google Scholar]
- Yemiş O.; Mazza G. Acid-Catalyzed Conversion of Xylose, Xylan and Straw into Furfural by Microwave-Assisted Reaction. Bioresour. Technol. 2011, 102, 7371–7378. 10.1016/j.biortech.2011.04.050. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Kumar R.; Wyman C. E. Enhanced Yields of Furfural and Other Products by Simultaneous Solvent Extraction during Thermochemical Treatment of Cellulosic Biomass. RSC Adv. 2013, 3, 9809–9819. 10.1039/c3ra41857j. [DOI] [Google Scholar]
- Lynam J. G.; Coronella C. J.; Yan W.; Reza M. T.; Vasquez V. R. Acetic Acid and Lithium Chloride Effects on Hydrothermal Carbonization of Lignocellulosic Biomass. Bioresour. Technol. 2011, 102, 6192–6199. 10.1016/j.biortech.2011.02.035. [DOI] [PubMed] [Google Scholar]
- Reza M. T.; Rottler E.; Herklotz L.; Wirth B. Hydrothermal Carbonization (HTC) of Wheat Straw: Influence of Feedwater PH Prepared by Acetic Acid and Potassium Hydroxide. Bioresour. Technol. 2015, 182, 336–344. 10.1016/j.biortech.2015.02.024. [DOI] [PubMed] [Google Scholar]
- McKillip W. J.Chemistry of Furan Polymers. In Adhesives from Renewable Resources, ACS Symposium Series; American Chemical Society, 1989; Vol. 385, pp 408–423. [Google Scholar]
- Hillyer J. C.; Nicewander D. A.. Inhibiting Polymerization of Furfural. U.S. Patent US2,440,4421948.
- Sangarunlert W.; Piumsomboon P.; Ngamprasertsith S. Furfural Production by Acid Hydrolysis and Supercritical Carbon Dioxide Extraction from Rice Husk. Korean J. Chem. Eng. 2007, 24, 936–941. 10.1007/s11814-007-0101-z. [DOI] [Google Scholar]
- Dietz C. H. J. T.; Gallucci F.; van Sint Annaland M.; Held C.; Kroon M. C. 110th Anniversary: Distribution Coefficients of Furfural and 5-Hydroxymethylfurfural in Hydrophobic Deep Eutectic Solvent + Water Systems: Experiments and Perturbed-Chain Statistical Associating Fluid Theory Predictions. Ind. Eng. Chem. Res. 2019, 58, 4240–4247. 10.1021/acs.iecr.8b06234. [DOI] [Google Scholar]
- Ghareh Bagh F. S.; Hadj-Kali M. K. O.; Mjalli F. S.; Hashim M. A.; AlNashef I. M. Solubility of Sodium Chloride in Phosphonium-Based Deep Eutectic Solvents. J. Mol. Liq. 2014, 199, 344–351. 10.1016/j.molliq.2014.09.025. [DOI] [Google Scholar]
- Martins M. A. R.; Pinho S. P.; Coutinho J. A. P. Insights into the Nature of Eutectic and Deep Eutectic Mixtures. J. Solution Chem. 2019, 48, 962–982. 10.1007/s10953-018-0793-1. [DOI] [Google Scholar]
- Dietz C. H. J. T.; Erve A.; Kroon M. C.; van Sint Annaland M.; Gallucci F.; Held C. Thermodynamic Properties of Hydrophobic Deep Eutectic Solvents and Solubility of Water and HMF in Them: Measurements and PC-SAFT Modeling. Fluid Phase Equilib. 2019, 489, 75–82. 10.1016/j.fluid.2019.02.010. [DOI] [Google Scholar]
- Crespo E. A.; Silva L. P.; Lloret J. O.; Carvalho P. J.; Vega L. F.; Llovell F.; Coutinho J. A. P. A Methodology to Parameterize SAFT-Type Equations of State for Solid Precursors of Deep Eutectic Solvents: The Example of Cholinium Chloride. Phys. Chem. Chem. Phys. 2019, 21, 15046–15061. 10.1039/C9CP02548K. [DOI] [PubMed] [Google Scholar]
- Pandey A.; Rai R.; Pal M.; Pandey S. How Polar Are Choline Chloride-Based Deep Eutectic Solvents?. Phys. Chem. Chem. Phys. 2013, 16, 1559–1568. 10.1039/C3CP53456A. [DOI] [PubMed] [Google Scholar]
- Eckert F.; Klamt A. Fast Solvent Screening via Quantum Chemistry: COSMO-RS Approach. AIChE J. 2002, 48, 369–385. 10.1002/aic.690480220. [DOI] [Google Scholar]
- Rodriguez N. R.; Gerlach T.; Scheepers D.; Kroon M. C.; Smirnova I. Experimental Determination of the LLE Data of Systems Consisting of {hexane+benzene+deep Eutectic Solvent} and Prediction Using the Conductor-like Screening Model for Real Solvents. J. Chem. Thermodyn. 2017, 104, 128–137. 10.1016/j.jct.2016.09.021. [DOI] [Google Scholar]
- Salleh Z.; Wazeer I.; Mulyono S.; El-blidi L.; Hashim M. A.; Hadj-Kali M. K. Efficient Removal of Benzene from Cyclohexane-Benzene Mixtures Using Deep Eutectic Solvents – COSMO-RS Screening and Experimental Validation. J. Chem. Thermodyn. 2017, 104, 33–44. 10.1016/j.jct.2016.09.002. [DOI] [Google Scholar]
- Han J.; Dai C.; Yu G.; Lei Z. Parameterization of COSMO-RS Model for Ionic Liquids. Green Energy Environ. 2018, 3, 247–265. 10.1016/j.gee.2018.01.001. [DOI] [Google Scholar]
- Islam M. R.; Chen C.-C. COSMO-SAC Sigma Profile Generation with Conceptual Segment Concept. Ind. Eng. Chem. Res. 2015, 54, 4441–4454. 10.1021/ie503829b. [DOI] [Google Scholar]
- Klamt A.; Eckert F. COSMO-RS: A Novel and Efficient Method for the a Priori Prediction of Thermophysical Data of Liquids. Fluid Phase Equilib. 2000, 172, 43–72. 10.1016/S0378-3812(00)00357-5. [DOI] [Google Scholar]
- Hizaddin H. F.; Ramalingam A.; Hashim M. A.; Hadj-Kali M. K. O. Evaluating the FiPerformance of Deep Eutectic Solvents for Use in Extractive Denitrification of Liquid Fuels by the Conductor-like Screening Model for Real Solvents. J. Chem. Eng. Data 2014, 59, 3470–3487. 10.1021/je5004302. [DOI] [Google Scholar]
- Dietz C. H. J. T.; van Osch D. J. G. P.; Kroon M. C.; Sadowski G.; van Sint Annaland M.; Gallucci F.; Zubeir L. F.; Held C. PC-SAFT Modeling of CO2 Solubilities in Hydrophobic Deep Eutectic Solvents. Fluid Phase Equilib. 2017, 448, 94–98. 10.1016/j.fluid.2017.03.028. [DOI] [Google Scholar]
- Luzhkov V. B.; Österberg F.; Acharya P.; Chattopadhyaya J.; Åqvist J. Computational and NMR Study of Quaternary Ammonium Ion Conformations in Solution. Phys. Chem. Chem. Phys. 2002, 4, 4640–4647. 10.1039/B203526J. [DOI] [Google Scholar]
- Kaur S.; Malik A.; Kashyap H. K. Anatomy of Microscopic Structure of Ethaline Deep Eutectic Solvent Decoded through Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 8291–8299. 10.1021/acs.jpcb.9b06624. [DOI] [PubMed] [Google Scholar]
- Mullins E.; Oldland R.; Liu Y. A.; Wang S.; Sandler S. I.; Chen C.-C.; Zwolak M.; Seavey K. C. Sigma-Profile Database for Using COSMO-Based Thermodynamic Methods. Ind. Eng. Chem. Res. 2006, 45, 4389–4415. 10.1021/ie060370h. [DOI] [Google Scholar]
- Hammond O. S.; Bowron D. T.; Edler K. J. Liquid Structure of the Choline Chloride-Urea Deep Eutectic Solvent (Reline) from Neutron Diffraction and Atomistic Modelling. Green Chem. 2016, 18, 2736–2744. 10.1039/C5GC02914G. [DOI] [Google Scholar]
- Mehler C.; Klamt A.; Peukert W. Use of COSMO-RS for the Prediction of Adsorption Equilibria. AIChE J. 2002, 48, 1093–1099. 10.1002/aic.690480518. [DOI] [Google Scholar]
- Florindo C.; Branco L. C.; Marrucho I. M. Development of Hydrophobic Deep Eutectic Solvents for Extraction of Pesticides from Aqueous Environments. Fluid Phase Equilib. 2017, 448, 135–142. 10.1016/j.fluid.2017.04.002. [DOI] [Google Scholar]
- Cao J.; Yang M.; Cao F.; Wang J.; Su E. Tailor-Made Hydrophobic Deep Eutectic Solvents for Cleaner Extraction of Polyprenyl Acetates from Ginkgo Biloba Leaves. J. Cleaner Prod. 2017, 152, 399–405. 10.1016/j.jclepro.2017.03.140. [DOI] [Google Scholar]
- Ashworth C. R.; Matthews R. P.; Welton T.; Hunt P. A. Doubly Ionic Hydrogen Bond Interactions within the Choline Chloride–Urea Deep Eutectic Solvent. Phys. Chem. Chem. Phys. 2016, 18, 18145–18160. 10.1039/C6CP02815B. [DOI] [PubMed] [Google Scholar]
- Pontes P. V. A.; Crespo E. A.; Martins M. A. R.; Silva L. P.; Neves C. M. S. S.; Maximo G. J.; Hubinger M. D.; Batista E. A. C.; Pinho S. P.; Coutinho J. A. P.; Sadowski G.; Held C. Measurement and PC-SAFT Modeling of Solid-Liquid Equilibrium of Deep Eutectic Solvents of Quaternary Ammonium Chlorides and Carboxylic Acids. Fluid Phase Equilib. 2017, 448, 69–80. 10.1016/j.fluid.2017.04.007. [DOI] [Google Scholar]
- Makoś P.; Przyjazny A.; Boczkaj G. Hydrophobic Deep Eutectic Solvents as “Green” Extraction Media for Polycyclic Aromatic Hydrocarbons in Aqueous Samples. J. Chromatogr. A 2018, 1570, 28–37. 10.1016/j.chroma.2018.07.070. [DOI] [PubMed] [Google Scholar]
- van Osch D. J. G. P.; Parmentier D.; Dietz C. H. J. T.; van den Bruinhorst A.; Tuinier R.; Kroon M. C. Removal of Alkali and Transition Metal Ions from Water with Hydrophobic Deep Eutectic Solvents. Chem. Commun. 2016, 52, 11987–11990. 10.1039/C6CC06105B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.