

Abstract
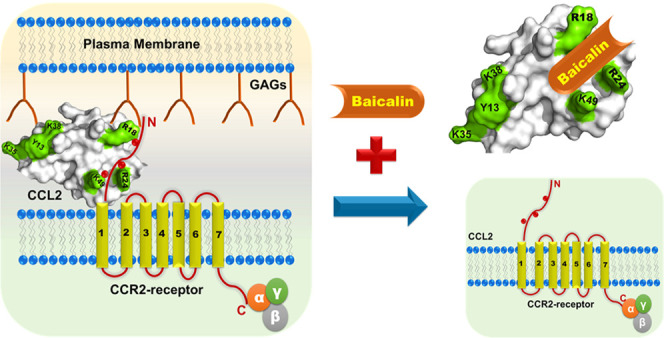
An integrated and controlled migration of leukocytes is necessary for the legitimate functioning and maintenance of the immune system. Chemokines and their receptors play a decisive role in regulating the leukocyte migration to the site of inflammation, a phenomena often referred to as chemotaxis. Chemokines and their receptors have become significant targets for therapeutic intervention considering their potential to regulate the immune system. Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a preeminent member of CC chemokine family that facilitates crucial roles by orchestrating the recruitment of monocytes into inflamed tissues. Baicalin (BA), a major bioactive flavonoid, has been reported to attenuate chemokine-regulated leukocyte trafficking. However, no molecular details pertaining to its direct binding to chemokine(s)/receptor(s) are available till date. In the current study, using an array of monomers/dimers of human and murine CCL2 orthologs (hCCL2/mCCL2), we have shown that BA binds to the CCL2 protein specifically with nanomolar affinity (Kd = 270 ± 20 nM). NMR-based studies established that BA binds CCL2 in a specific pocket involving the N-terminal, β1- and β3-sheets. Docking studies suggested that the residues T16, N17, R18, I20, R24, K49, E50, I51, and C52 are majorly involved in complex formation through a combination of H-bonds and hydrophobic interactions. As the residues R18, R24, and K49 of hCCL2 are crucial determinants of monocyte trafficking through receptor/glycosaminoglycans (GAG) binding in CCL2 human/murine orthologs, we propose that baicalin engaging these residues in complex formation will result in attenuation of CCL2 binding to the receptor/GAGs, thus inhibiting the chemokine-regulated leukocyte trafficking.
1. Introduction
Inflammation is a pivotal biological response of the immune system, elicited due to various injurious stimuli and toxic compounds such as damaged cells and pathogens.1,2 During acute inflammatory conditions, several molecular and cellular interactions adroitly abate the impending infection. This mitigation process is regulated by various immune mediators such as cytokines, interferons (IFNs), chemokines, colony-stimulating factors (CSFs), and tumor necrosis factors (TNFs).3 Chemokines are a specific subclass of cytokines that contribute to the restoration and resolution of tissue homeostasis. Chemokines are the small, secreted chemotactic proteins that direct the migration of immune cells to the foci of inflammation and function as intracellular messengers.4 Based on the chromosomal location, the position of conserved cysteine residues, and their specific cell targets, these chemotactic proteins have been classified into four families: CXC, CX3C, CC, and C.5 Chemokines interact with G-protein-coupled receptors (GPCRs) embedded in the cell membranes of leukocytes and glycosaminoglycans (GAGs) present on the endothelial cell surface during leukocyte migration.4,6 As the chemokine–receptor/GAG interaction is a fundamental pivot that regulates chemokine-mediated leukocyte trafficking, their involvement in several inflammatory and infectious conditions is well documented.7 For instance, an enhanced level of peripheral blood during the hepatitis C virus (HCV) infection is explicitly arbitrated by chemokines and their receptors.8 Moreover, these interacting partners play critical roles in tumor progression,9 autoimmune diseases,10 lung infection,11 and several neurodegenerative conditions.12
The monocyte chemoattractant protein (MCP) family is a small subfamily of CC chemokines that mediate immune responses in various inflammatory processes.13,14 The family constitutes four small, secreted, and structurally related proteins known as MCP-1, -2, -3, and -4.15 Among these four members, MCP-1, also termed as CCL2, is the most studied member of the MCP family and is a potent agonist for monocytes, memory T cells, basophils, and dendritic cells.16 Earlier, NMR studies have unveiled that the monomeric structure of CCL2 comprises an N-terminal loop and four antiparallel β-strands, followed by a C-terminal helix (Figure 1A).17 In solution, CCL2 forms a symmetric dimer topology where both the monomeric units are placed antiparallel and interact through intermonomer β-strand contacts, thus forming a canonical CC chemokine dimer (Figure 1B).18−20 A recent study on murine CCL2 suggested that it also exhibits similar dimeric topology to that of human orthologs with some minor structural differences at the C-terminal end.21 CCL2 has been reported to be associated with various deleterious inflammatory ailments, including atherosclerosis,22 tumor neovascularity,23 tuberculosis,24 inflammatory bowel diseases,25 and multiple sclerosis.26 In these clinical manifestations, it is apparent that to exacerbate the inflammation, CCL2 induces the activation of leukocytes, specifically macrophages/monocytes, in the tissues by interacting with its GPCR receptor CCR2.
Figure 1.
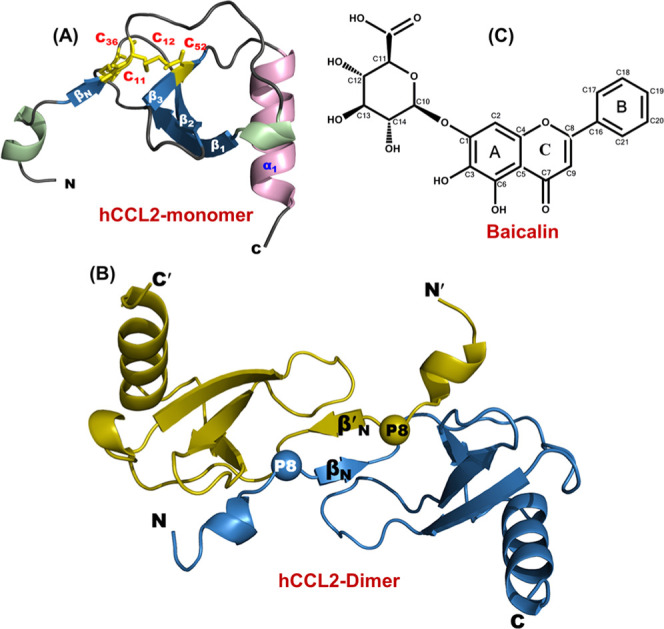
Structural characteristics of CCL2 protein and baicalin (BA): (A) Three-dimensional structure of human CCL2 protein’s monomeric subunit depicting all of the structural elements (PDB ID: 1dok). The structure comprises a long N-terminal, three antiparallel β-strands, and a C-terminal α-helix. The disulfide bonds are highlighted in yellow (stick representation). (B) Three-dimensional structure of the human CCL2 dimer (PDB ID: 1dok). The proline residues involved in the arm-exchange process are represented by spheres. (C) Chemical structure of flavonoid baicalin (BA).
Since most of the chemokines tend to oligomerize at higher concentrations, an ardent effort has been inflicted to depict the stoichiometry of chemokine–receptor interactions.27−29 It has been widely accepted that most of the CC chemokines interact with their cognate receptors in their monomer conformations.30−32 In line with this, the monomeric variant of human CCL2 (P8A*) preferably interacts and activates its receptor CCR2 to that of its dimeric conformation.30 Chemokine–receptor interactions arbitrate a significant immune response under various inflammatory conditions and prevent the damage of the host. Several monoclonal antibodies and receptor antagonists have been identified to regulate the interactions on the CCL2–CCR2 axis.33−35 In the same context, Wang et al. have reported the disruption of chemokine–receptor interactions using the flavonoid baicalin (BA) with an array of CXC/CC chemokines, which resulted in reduced chemotactic activity.36 Baicalin (7-glucuronic acid, 5,6-dihydroxyflavone), the flavone glycoside extracted from Scutellaria baicalensis Georgi, has been used as a potent anti-inflammatory agent in Asian traditional medicine (Figure 1C).36 Further, it has also been reported that baicalin dramatically hinders the superantigen-mediated generation of various cytokines and chemokines from human PMBC cells and inhibits the T-cell proliferation induced by the staphylococcal superantigens.37 In addition to this, baicalin also exhibits diverse therapeutic properties such as anti-allergic, antitumor, antioxidant, antibronchitis, and antinephritis and anticardiovascular actions.38−40
Although baicalin has been reported as a potent anti-inflammatory agent and it regulates the chemotactic activity of chemokines, no molecular/structural details are available regarding its interaction with chemokine oligomers (monomer/dimers). The current study is designed to elucidate the binding features of baicalin to the chemokine CCL2. As CCL2 exhibits the monomer–dimer equilibrium, the interaction of baicalin was investigated using both CCL2 dimers and monomers. To obtain a comprehensive knowledge about the binding, CCL2 orthologs from both human and murine species were studied against baicalin. Fluorescence spectroscopy and multidimensional solution NMR spectroscopy techniques and molecular docking tools were applied to unravel the molecular interactions. Our results unveiled that baicalin binds specifically to CCL2 monomers and dimers with similar affinities (Kd in range of ∼270 ± 20 nM). Further, the extensive overlap of the chemokine binding pocket observed in this study with that of the GAG/receptor binding sites substantiates the earlier cell-based reports of baicalin interference in chemokine–receptor interactions and thus the attenuated chemotactic activity of chemokines in its presence.
2. Results
2.1. Assessing the Oligomeric State of the P8A Mutant in CCL2 Orthologs
The oligomeric state is crucial for activation and regulation of proteins. The principles of the oligomerization process remain unclear especially when these oligomers are formed through a domain swap or arm-exchange mechanism. The frequent existence of proline residues at the dimeric interface has been extensively reported.41 It has been suggested that these residues impose restraints on the conformation of the protein and assist in oligomerization through the “arm-exchange” process.41 Earlier studies have reported that mutation of proline into alanine (P8A) in the wild-type (WT) hCCL2 resulted in an obligate monomer that potentially interacts and activates its receptor (CCR2) (Figure 1B).30 Sequence analysis of CCL2 chemokines from primate and rodent families has suggested that the proline residue at the 8th position is highly conserved among all of the members (Figures S1 and 2A). These observations are consistent with the previous studies, where the conserved profile of the proline residue at the 8th position has been reported to exert an essential impact on the efficiency of the dimerization.41 To generate the monomeric variants of the CCL2 orthologs, we have constructed the P8A mutant proteins using the human/murine CCL2-WT genes (Figure S2A–F and Table S2). The mutant proteins along with the wild-type constructs of both human and murine species were expressed and purified using a series of chromatographic techniques to obtain the pure proteins (Figures 2B, S3A–E, and Table S2).
Figure 2.
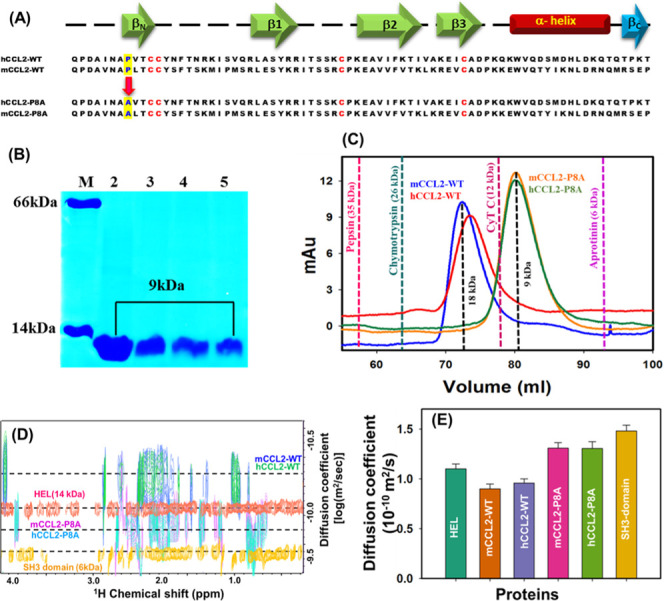
Comparative biophysical characterization of monomeric and dimeric CCL2 orthologs: (A) Sequence alignment of murine and human CCL2-WT and monomeric orthologs. The proline-to-alanine mutation is highlighted in blue, and marked with an arrow. The conserved cysteine residues in proteins are highlighted in red. The secondary structure elements are shown on the top of the sequences, and the presence of C-terminal βc-strand present specifically in mCCL2 is highlighted in dark cyan. (B) Pure protein profiles of CCL2 orthologs: Lane M, marker bovine serum albumin (BSA) (66 kDa) and hen egg lysozyme (HEL) (14kDa); Lane 2, pure mCCL2-WT protein (9 kDa); Lane 3, pure murine mCCL2-P8A protein (9 kDa); Lane 4, pure hCCL2-WT protein (9 kDa); and Lane 5, pure hCCL2-P8A protein (9 kDa). (C) Size exclusion chromatography (SEC) profile for mCCL2-WT (blue), mCCL2-P8A (orange), hCCL2-WT (red), and hCCL2-P8A (green) proteins. The standard molecular weight (MW) proteins are depicted by dotted lines at their respective elution maxima, which are describing their MW. (D) Two-dimensional diffusion-ordered spectroscopy (2D-DOSY) profiles for mCCL2-WT (blue), hCCL2-WT (green), mCCL2-P8A (purple), and hCCL2-P8A (cyan) proteins. The standard reference proteins hen egg lysozyme (HEL) and chicken SH3 protein domain are shown in orange and dark yellow, respectively. (E) DOSY-diffusion plot between the molecular weight and diffusion coefficients. The histograms of mCCL2-WT and hCCL2-WT are highlighted in orange and light blue, whereas mCCL2-P8A and hCCL2-P8A are highlighted in pink and green, respectively. Standard proteins are highlighted in teal (HEL) and dark yellow (SH3 domain), respectively.
To unravel the impact of the proline mutation on oligomerization of the mCCL2-P8A mutant, size exclusion chromatography (SEC) and proton-based 2D-DOSY measurements were carried out on the wild-type and P8A mutant proteins. The elution profiles of both mutant proteins were compared with standard proteins along with their respective WT proteins (mCCL2 and hCCL2). In contrast to the wild-type proteins, which exist in dimeric conformations (∼18 kDa), both P8A mutant proteins were eluted corresponding to the molecular weight of ∼9 kDa, thus suggesting the monomeric nature of the P8A mutants in both the orthologs (Figure 2C).21 The observed dimeric state of the wild-type CCL2 ortholog proteins and the monomeric conformations of the P8A mutants are in line with the earlier published reports.30,42
Further, to substantiate the oligomerization results obtained from the SEC, NMR-based translational diffusion measurements were performed. The diffusion profiles of CCL2 oligomeric variants and the standard proteins are presented in Figure 2D. Diffusion coefficient (D) values of 1.309 × 10–10 and 1.305 × 10–10 m2 s–1 were obtained for mCCL2-P8A and hCCL2-P8A and 0.9 × 10–10 and 0.96 × 10–10 m2 s–1 for mCCL2-WT and hCCL2-WT, respectively. The obtained D values are consistent with the previously reported D values of other CC monomeric and dimeric chemokines (Figure 2E).43,44 The monomeric nature of the P8A mutants and the dimeric nature of the wild-type proteins were also independently assessed by comparing the diffusion coefficients of these proteins with standard proteins HEL and SH3 domain (Figure 2E). All of these results comprehensively establish the dimeric conformation of the wild-type proteins and the monomeric conformation of the P8A mutants of both CCL2 orthologs at the chosen experimental conditions.
2.2. Secondary and Tertiary Structural Features of CCL2-P8A
It is essential to characterize the secondary and tertiary structural features of CCL2-P8A, as structural changes are associated with various aspects of functional capabilities. Far-UV circular dichroism (CD) (190–250 nm) offers an imperative means to monitor the changes that occur in the secondary structure of the protein.45,46 To assess the secondary structural features, the CD profiles of all four CCL2 orthologs have been compared (Figure 3A). Although the wild-type spectra and P8A spectra of both the orthologs overlaid well, certain differences in the spectral features between the wild-type and monomeric CCL2 were observed. Henceforth, to evaluate the secondary structural characteristics, the CD spectra of all of the four conformers are subjected to quantitative measurements using Dichroweb-K2D software (Table S3). These results unveiled that all four CCL2 orthologs exhibit a similar percentage of secondary structural content, thus indicating that the mutation of P8A and the formation of monomer do not influence the CCL2 secondary structural features in murine, which is also in line with its human counterpart.30
Figure 3.
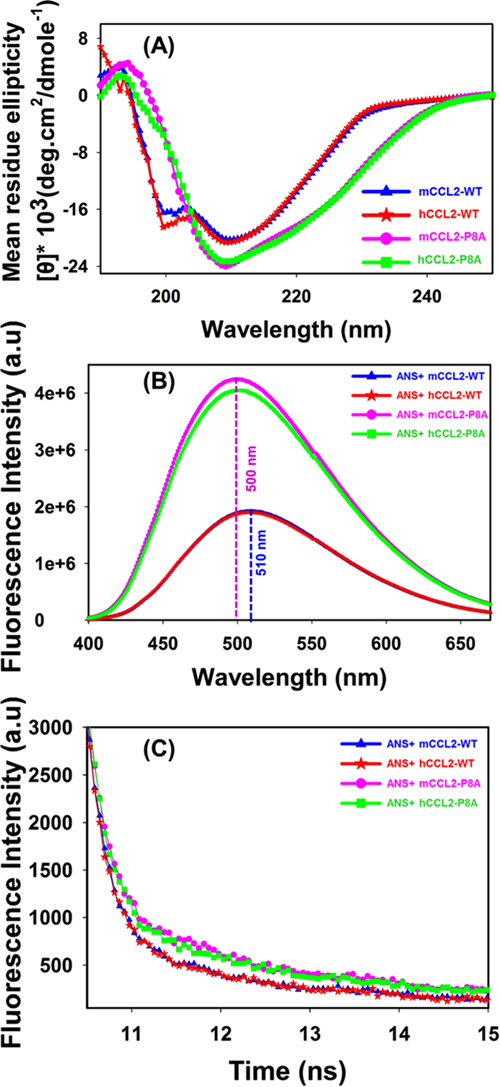
Structural characterization of CCL2 ortholog monomers and dimers: (A) Overlay of the far-UV CD profiles of WT (human, red; murine, blue) and monomeric (human, green; murine, pink) proteins of CCL2 orthologs. (B) Overlay of ANS fluorescence spectra of WT (human, red; murine, blue) and monomeric (human, green; murine, pink) proteins of CCL2 orthologs. (C) Overlay of ANS fluorescence decay profiles of WT (human, red; murine, blue) and monomeric (human, green; murine, pink) proteins of CCL2 orthologs.
To elucidate the tertiary structural characteristics of CCL2 orthologs, intrinsic tryptophan- and ANS-based fluorescence measurements were performed. For intrinsic fluorescence experiments, tryptophan (W59) was used as a fluorophore, and the fluorescence emission profiles of all proteins were compared. No noticeable spectral shift/intensity was observed for the measured CCL2 proteins, thus signifying no measurable/significant changes in the Trp environment (Figure S4). Further, to dissect the differential tertiary structural/surface characteristics of CCL2 constructs, ANS emission profiles of four proteins were analyzed. In comparison to WT proteins, a noticeable change in the ANS fluorescence profile was observed for both monomers, although both the monomers exhibited similar spectral profiles (Figure 3B). The binding of ANS to CCL2-WT proteins has resulted in a sharp decrease of ∼2.2 times in the fluorescence intensity as compared to their monomeric counterparts. It is also noteworthy that, along with significant changes in the ANS intensity, a spectral shift of ∼10 ± 2 nm in the emission spectra was also observed for both WT proteins as compared to monomeric proteins. Such differential spectral profiles of ANS binding to CCL2 monomer proteins suggest that these monomeric proteins possess a more exposed hydrophobic surface than that of the WT proteins.
The differential nature of the fluorophore environment (Trp and ANS) in the monomers and dimers of CCL2 orthologs was also assessed in their excited states using the fluorescence lifetime decays. Tryptophan lifetime decay profiles were observed to be similar for both monomers and dimers, suggesting the similar electronic environment of the excited states of CCL2 proteins irrespective of the oligomerization characteristics (Figure S5). These lifetime results are in line with the steady-state tryptophan results discussed in the above paragraph. In contrast to Trp behavior, distinguishable fluorescence decay profiles of ANS were observed between WT and monomeric CCL2 orthologs (Figure 3C). In general, the ANS fluorescence decay is uniexponential with a lifetime of ∼0.25 ns. However, when it binds to proteins, it follows triexponential decay.47−49 The triexponential decay represents two different types of ANS binding to the protein molecules (τ2, τ3), along with the decay of free ANS (τ1). The two decays corresponding to the ANS–protein complex include a shorter decay (τ2), where ANS binds to the surface of proteins, and a longer decay (τ3), representing the binding of ANS specifically to the protein hydrophobic core. In the case of CCL2 proteins, we have noticed that the lifetime values and the amplitudes of different lifetimes varied significantly between the wild-type and monomeric proteins, although they are similar between the orthologs (Table 1). The increase in lifetime/amplitude of the longer decay (τ3) in monomers as compared to their wild-type/dimeric counterparts clearly evidences the enhanced binding of ANS at their hydrophobic core. This can be attributed to the loss of dimeric contacts and exposure of the hydrophobic surface in monomeric CCL2 proteins, as also observed in steady-state ANS experiments (Figure 3B).
Table 1. Fluorescence Lifetime Values of Free ANS and ANS Complexed to CCL2 Wild-Type Proteins and Monomeric Proteins from Murine and Human Speciesa.
| ANS | τ1 (ns) | τ2 (ns) | τ3 (ns) | average lifetime (ns) | χ2 |
|---|---|---|---|---|---|
| ANS | 0.25 (100) | 0.25 | 0.99 | ||
| mCCL2-WT | 0.23 (37) | 2.5 (38) | 9.2 (25) | 3.3 | 1.1 |
| hCCL2-WT | 0.23 (38) | 2.6 (37) | 9.3 (25) | 3.3 | 1.2 |
| mCCL2-P8A | 0.25 (46) | 2.3 (26) | 11.5 (28) | 3.9 | 1.1 |
| hCCL2-P8A | 0.25 (45) | 2.2 (27) | 11.2 (28) | 3.8 | 1.3 |
The values of the relative amplitude for each lifetime are presented in the parenthesis.
2.3. Residuewise Comparative Analysis of Dimeric and Monomeric mCCL2 Proteins
The biophysical studies using CD and fluorescence suggested that the monomeric and the wild-type/dimeric proteins have similar secondary structures; however, their intrinsic tertiary structures exhibited differences due to the loss of quaternary interaction. Using multidimensional NMR spectroscopy, the authors have established that the monomeric hCCL2 (P8A) has similar secondary structural features to those of the dimeric CCL2 protein, although some significant differences in the position of the resonances were observed in the 1H–15N heteronuclear single quantum coherence (HSQC) spectra of the monomer.30 The differences/chemical shift perturbations (CSP) were attributed to the local structural changes due to the loss of quaternary interactions at the dimer interface.
As no such information is available for the mCCL2-P8A protein, first we assessed its concentration-dependent oligomerization property. To rule out the oligomerization/aggregation at higher protein concentrations, HSQC spectra were recorded at 50 and 500 μM concentrations for mCCL2-P8A (Figure S6). At both concentrations, HSQC yielded identical spectra with a single set of well-dispersed NH cross-peaks, indicating the existence of mCCL2-P8A as an exclusive monomer in the measured experimental conditions. Further, to assess such local changes in the monomeric mCCL2 (P8A) protein, we have compared the 1H–15N chemical shifts of mCCL2-P8A with those of the dimeric mCCL2 protein (∼800 μM). Overlay of the HSQC spectra for both proteins is shown in Figure 4A. It is worth noting that wild-type mCCL2 forms a dimer at high concentrations (∼800 μM) and equilibrium of monomers and dimers at low concentrations (∼100 μM), thus resulting in more number of resonances at the low concentrations.21 The HSQC spectral overlay suggested that ∼70% NH cross-peaks were present at their respective positions, and certain resonances were specifically shifted. To identify the resonances with substantial shifts between the monomeric and dimeric mCCL2 proteins, a chemical shift perturbation (CSP) map has been plotted using 40 unambiguous resonances (Figure 4B). CSP results suggested that residues A7, L9, T10, C12, S14, T16, I20, R30, and A53 have shown significant perturbations. Among these residues, A7, L9, T10, C12, S14, T16, and I20 are present in the N-terminal portion and constitute the dimer interface. Certain extent of the chemical shift perturbation for the residues A7, L9, and T10 in the N-terminal can be attributed to the mutational effect of the neighboring residue (P8A). Other perturbed residues such as R30 and A53 can be due to relay of perturbations. For example, CCL2 comprises a disulfide bond between the residues C12 and C52, which can potentially relay the perturbations to A53. All of the identified perturbed residues are represented as spheres on the monomeric structure of mCCL2 and on the surface of the dimeric CCL2 (Figure 4B inset, C). Mapping of these residues on the dimeric CCL2 structures clearly depicts that the perturbations are majorly present at the dimer interface, and the observed changes in the chemical shifts are due to change in the local environment of these residues due to loss of dimeric contacts. These observations are consistent with the fact that the loss of interface contacts might have perturbed the chemical environment of affected residues. These results indeed are in line with the hCCL2 results and also corroborated with the other CC chemokines.30,43 Hence, these results establish that the substantial changes observed in the chemical shift values of the monomer are exclusively due to the loss of dimer interface and the mCCL2-P8A mutant exists exclusively in the monomeric conformation even at high concentrations.
Figure 4.
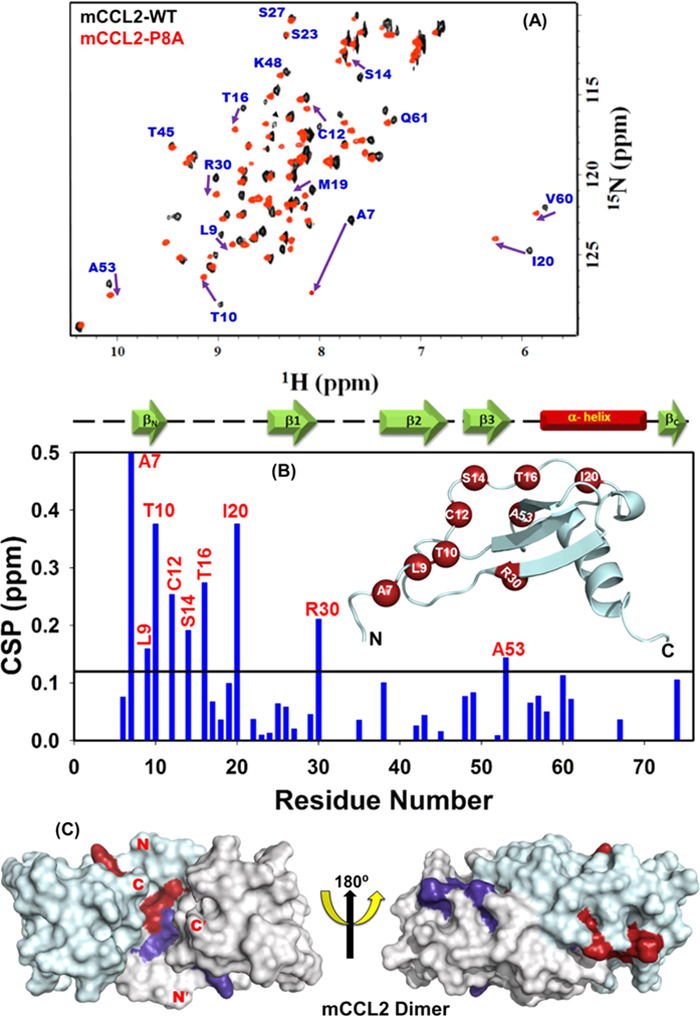
NMR characterization of mCCL2-P8A: (A) Overlay of 1H–15N HSQC spectra of mCCL2-WT (black) and mCCL2-P8A monomer (red). The arrows are representing the residues showing significant chemical shift perturbation from the WT protein. (B) Chemical shift perturbation (CSP) map depicting the change in the chemical shift values for the residues of the mCCL2-P8A protein in comparison to mCCL2-WT. The solid black line represents the cutoff CSP value. The secondary structure elements are shown on the top of the CSP map. The inset represents the monomeric structure of the mCCL2 protein; the perturbed residues are shown as dark red spheres. (C) Surface structure of the mCCL2 dimer representing the perturbed residues in dark red and dark purple on each monomeric subunit, respectively.
2.4. Measuring the Binding Affinity of Baicalin to CCL2 Orthologs
A priori knowledge of baicalin-regulated chemotactic activity of proinflammatory chemokines instilled us to explore the plausibility and mechanism of direct interaction of BA with the CCL2 chemokine. Further, measurements of these interactions using orthologous proteins also provide information about the varied/conserved nature of the binding surfaces under evolutionary perspective, as chemokines are one of the known families of immune proteins that are rapidly evolving.50−53 As previously reported, the hCCL2-P8A monomer activates its receptor with significant efficiency compared to its dimeric counterpart;30 it is also essential to evaluate the binding preference of baicalin toward both the monomeric and dimeric conformations. Considering these perspectives, we have chosen both human and murine CCL2-WT and monomeric proteins to investigate the binding interaction with BA using fluorescence quenching experiments. All of the four CCL2 proteins were titrated with the increasing concentration of BA flavonoid, as described in the experimental section. It was observed that the intrinsic fluorescence intensity of CCL2 orthologs decreased significantly upon increasing the BA concentration, suggesting a prominent interaction between BA and CCL2 orthologs (Figure 5A–D). It is worth noting that, along with intensity changes, a bathochromic shift of ∼8 nm for wild-type proteins and ∼11 nm for monomers was observed in the emission spectra of CCL2 proteins upon binding to BA. The red shift in fluorescence emission strongly supports the substantial involvement of hydrogen bonding interactions between CCL2 and BA.54
Figure 5.

Fluorescence quenching profiles of CCL2 orthologs: (A–D) Fluorescence quenching profiles of mCCL2-WT, mCCL2-P8A, hCCL2-WT, and hCCL2-P8A, respectively. (E–H) Stem–Volmer plots of mCCL2-WT, mCCL2-P8A, hCCL2-WT, and hCCL2-P8A, respectively. (I–L) Double-log plots of mCCL2-WT, mCCL2-P8A, hCCL2-WT, and hCCL2-P8A, respectively. In figure (A–D), the inset A–F represents Apo protein (without BA), protein–BA (1:0.1), protein–BA (1:0.2), protein–BA (1:0.4), protein–BA (1:0.5), protein–BA (1:0.7), and protein–BA (1: 1).
The BA-mediated fluorescence quenching was assessed using the Stern–Volmer equation. The quenching constant (Kq) value for all CCL2 orthologs was determined by analyzing the linear regression plot of F0/F versus [Q] (Figure 5E–H). The value of Kq for CCL2 wild-type proteins was found to be 5.1 × 1012 L mol–1 s–1 for mCCL2-WT, 5.2 × 1012 L mol–1 s–1 for hCCL2-WT, 6.2 × 1012 L mol–1 s–1 for mCCL2-P8A, and 6.1 × 1012 L mol–1 s–1 for hCCL2-P8A (Table 2). In general, the quenching process follows different mechanisms such as dynamic quenching, static quenching, or both. These quenching processes can be distinguished by their dependence on the quenching constant Ksv.55 As previously reported, for dynamic quenching, the value of the maximum dynamic quenching constant (Kq) can be 2.0 × 1010 L mol–1 s–1.55,56 Considering that the quenching constants obtained here upon interaction with BA are far higher than those of the maximum limit of dynamic quenching, we believe that the quenching process in the CCL2–BA interaction is static quenching. Further, the binding constant and binding sites were calculated by fitting the data to a double-logarithmic equation (Figure 5I–L). The estimated dissociation constant (Kd) values for CCL2 wild-type and monomeric proteins were found to be in the range of 270 ± 20 nM, and the number of binding sites per monomer is around 1, suggesting that one monomer of CCL2 binds to one baicalin molecule (Table 2). The obtained Kd values suggest that both the CCL2 orthologs in their wild-type and the monomeric conformations bind to BA with similar binding affinity. Further, the observed binding constant of 270 ± 20 nM establishes that BA interacts with the CCL2 orthologs very tightly.
Table 2. Binding Parameters for the Interaction of CCL2 Orthologs with Baicalin (BA)a.
| name of protein | quenching constant (Kq) M–1 s–1 | dissociation constant (Kd) [nM] | number of binding site | R2 |
|---|---|---|---|---|
| mCCL2-WT | 5.1 × 1012 | 290 ± 20 | 1.2 ± 0.1 | 0.99 |
| hCCL2-WT | 5.2 × 1012 | 250 ± 20 | 1.1 ± 0.1 | 0.98 |
| mP8A | 6.2 × 1012 | 270 ± 20 | 1.1 ± 0.2 | 0.99 |
| hP8A | 6.1 × 1012 | 260 ± 20 | 1.2 ± 0.1 | 0.98 |
For all CCL2 proteins, the concentrations were obtained as a monomeric unit.
2.5. Deciphering the Baicalin Binding Sites on the CCL2 Protein Using NMR Spectroscopy
The fluorescence experiments have suggested that the oligomeric and orthologous nature of CCL2 does not impose any significant effect on the binding affinity for baicalin. Hence, to unveil the residue-level insights for baicalin binding using NMR spectroscopy, the mCCL2-WT protein was used at 100 μM concentration, where one can simultaneously monitor the dimeric and monomeric resonances. Such protein equilibrium under a slow exchange regime allows measuring the binding behavior of both the species (monomer and dimer). The binding of BA to the mCCL2-WT protein was determined by saturating the mCCL2-WT protein with BA in a ratio of 1:5 (mCCL2-WT/BA). It was observed that a subset of NH resonances was significantly perturbed for both the monomeric and dimeric conformations of mCCL2 (Figure 6A,B). Henceforth, to gain more insights into the binding pattern between mCCL2 and BA, the chemical shift perturbation (CSP) approach was used. CSP is an exquisitely sensitive method to describe the binding interactions and is routinely used for studying protein–ligand interactions.57,58 The sequence-specific CSP map was plotted for all of the residues of the mCCL2 dimer and 17 unambiguously identified residues of monomeric conformation at a molar ratio of 1:5 (mCCL2-WT/BA) (Figure 6C) using the mCCL2-WT protein (∼100 μM) as a reference. The obtained CSP profile suggests that only a specific group of residues is significantly perturbed in both the monomeric and dimeric conformations, suggesting a similar binding surface of interaction in both the conformers (Figure 6C). Moreover, these similar perturbations and no significant effect on the dimeric–-monomeric equilibrium of mCCL2 upon addition of BA, as observed from the peak intensity ratios of the dimer and monomer (data not shown), also indicate that the binding site is not at the dimer interface. The perturbed residues include L9, T10, C12, S17, K18, and I20 from the N-terminal; S23 and R24 from the 310 helix; L25, E26, and S27 from the β1-sheet; T45 and K46 from the 3rd loop; R49 and V51 from the β3-sheet; and L67 from the C-terminal α-helix. All of the residues exhibiting perturbation have been marked on one subunit of the molecular structure of the mCCL2 dimeric protein (Figure 6D). These observations establish that BA binds to the mCCL2-WT protein at the N-terminal by involving the β1- and β3-sheets.
Figure 6.
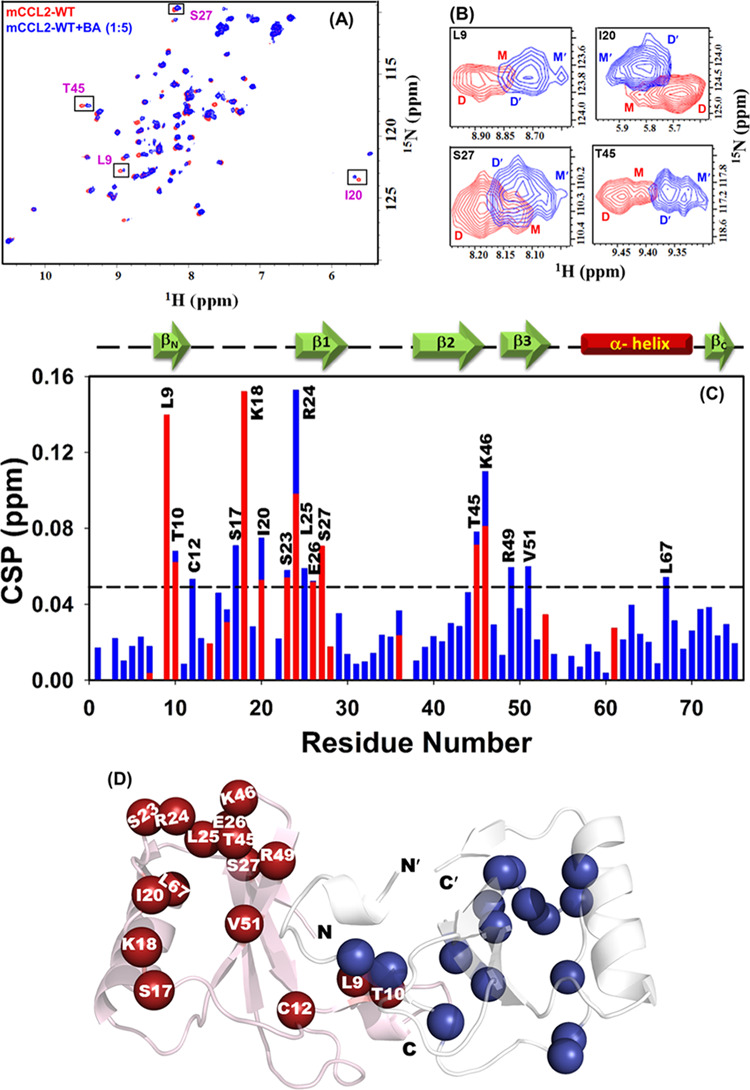
NMR elucidation of BA binding to the mCCL2-WT protein: (A) 1H–15N HSQC spectral overlay for mCCL2-WT/apo (red) and BA–mCCL2-WT complex (blue). Representative resonances exhibiting dimer–monomer equilibrium are boxed. (B) Zoom-in section of all of the boxed residues shown in figure (A). (C) Chemical shift perturbation (CSP) plot of mCCL2-WT and BA interactions. The dotted horizontal black line represents the cutoff value for perturbed residues. The secondary structure elements are shown on the top of the CSP plot. (D) Residues showing significant chemical shift perturbation are shown on the mCCL2 dimeric structure as spheres. The residues are marked only in one subunit for clarity.
2.6. Analyzing the Binding Interaction of BA with CCL2 Proteins Using Molecular Docking
Taking the NMR-based studies in the background, the molecular docking was executed to unravel the atomic-level interaction between BA and CCL2 monomers. The docking study was performed using the AutoDock 4.2 tool by incorporating the perturbed residues obtained from NMR-based CSP studies. It was observed that BA binds specifically in a pocket that is away from the dimer interface (Figure 7A,B). The binding energy for the BA and mCCL2 monomer interaction was observed to be ∼−6.5 kcal mol–1. The binding of BA to mCCL2/hCCL2 was mediated by various types of interactions including hydrophobic, hydrogen, and electrostatic interactions. For the BA–mCCL2 complex, the primary residues observed to be involved in the formation of hydrophobic interactions were I20 and V51. For example, Cδ of I20 was interacting with the C3 and C4 moieties of BA, respectively, whereas Cβ of V51 was interacting with the C3 moiety of BA. Further, residues T16, S17, K18, R24, and R49 were involved in the formation of hydrogen bond contacts. The essential interactions include (T16) Hγ–O1 (BA), (S17) NH–O1 (BA), (K18) HZ3–O8 (BA), (R24) NH2–O4 (BA), and (R49) NH2–O5 (BA). Apart from these hydrophobic and H-bond interactions, an electrostatic interaction between the O group of M19 residues and the O9H group of BA was also observed. Some of these crucial interactions involved in BA–mCCL2 interactions have been depicted in the molecular structure (Figure 7C,D), and all of the possible contacts have been summarized in Table 3. Considering the similarity in the human and murine CCL2 protein sequences and the CCL2–BA complex dissociation constants observed in the earlier section, docking studies were also performed for human CCL2 monomeric protein using the same surface grid/NMR-based CSP map of mCCL2 (Figure 7E,F). The binding energy for BA and hCCL2 monomer interactions was observed to be ∼−6.3 kcal mol–1. The crucial residues observed to be involved in the formation of hydrophobic interactions were I20 and I51. For example, the Cγ1 group of I20 was interacting with the C9 moiety of BA, while the Cγ2 group of I51 was interacting with the C16 moiety of BA. Additionally, residues T16, N17, R18, R24, K49, E50, and C52 were involved in the formation of hydrogen bond interactions. The crucial interactions include (T16) Hγ1–O4 (BA), (N17) Oδ1–O11 (BA), (R18) NH–O11 (BA), (R24) NH1–O8 (BA), (K49) Hζ2–O6H (BA), (E50) OA–C16 (BA), and (C52) NH–O19 (BA). Some of these essential interactions involved in BA–hCCL2 interactions have been depicted in the molecular structure (Figure 7G,H), and all of the possible contacts have been summarized in Table 3. The collective fluorescence, NMR, and docking-based studies establish that BA interacts specifically with CCL2 protein orthologs in both their monomeric and dimeric conformations.
Figure 7.
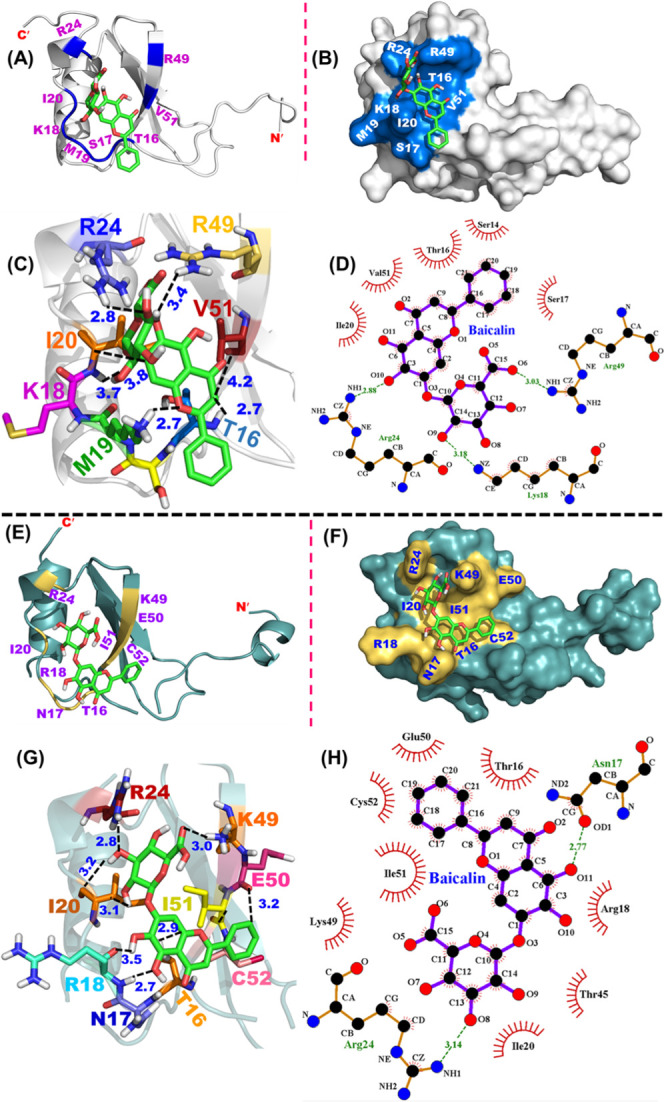
Docking of BA on mCCL2-WT and hCCL2-WT proteins: (A) Docking of BA onto the mCCL2 monomer. The BA binding pocket is highlighted in marine blue, and the interacting residues are shown in purple. (B) Representation of the BA binding pocket onto the surface structure of the mCCL2 monomer. (C) BA–mCCL2 monomer depicting various interactions. Black dotted lines depict the interactions. Numbers in Å represent the spatial proximity of the interactions. (D) LigPlot showing the representative hydrogen bonding contacts and hydrophobic interactions in the BA–mCCL2 monomer complex. Dotted green lines indicate hydrogen bonding interactions, while the hydrophobic interactions are indicated by an arc along with spokes directing toward the ligand binding position. (E) Docking of BA onto the hCCL2 monomer. The BA binding pocket is highlighted in pale yellow, and the interacting residues are shown in purple. (F) Representation of the BA binding pocket onto the surface structure of the hCCL2 monomer. (G) BA–hCCL2 monomer depicting various interactions. Black dotted lines depict the interactions. Numbers in Å represent the spatial proximity of the interactions. (H) LigPlot showing the representative hydrogen bonding contacts and hydrophobic interactions in the BA–hCCL2 monomer complex. Dotted green lines indicate hydrogen bonding interactions, while the hydrophobic interactions are indicated by an arc along with spokes directing toward the ligand binding position.
Table 3. Summary of All possible Contacts Obtained from Docking and LigPlot for the BA–mCCL2 and BA–hCCL2 Complexesa.
| mCCL2 | baicalin | type of interaction & distance | hCCL2 | baicalin | type of interaction & distance |
|---|---|---|---|---|---|
| T16 (Hγ) | O1 | H-bond (2.7 Å) | T16 (Cβ) | C8 | HPI (3.3 Å) |
| T16 (Cβ) | C8 | HPI (3.2 Å) | T16 (Oγ1) | C7 | HPI (3.1 Å) |
| T16 (γO) | C5 | HPI (3.6 Å) | T16 (Hγ1) | O4 | H-bond (2.9 Å) |
| T16 (Cβ) | C4 | HPI (3.8 Å) | N17 (Oδ1) | O11 | H-bond (2.1 Å) |
| S17 (Hγ) | C18 | HPI (3.0 Å) | N17 (2Hδ2) | O11H | H-bond (3.4 Å) |
| S17 (NH) | O1 | H-bond (3.0 Å) | N17 (Oδ1) | O2 | H-bond (2.5 Å) |
| S17 (γOH) | C19 | HPI (3.2 Å) | N17 (NH) | O11H | H-bond (2.7 Å) |
| S17 (NH) | C18 | HPI (3.4 Å) | R18 (Cβ) | C10 | HPI (3.5 Å) |
| K18 (HZ3) | O8 | H-bond (3.5 Å) | R18 (NH) | O11 | H-bond (3.2 Å) |
| K18 (NZ) | O9 | H-bond (3.2 Å) | R18 (NH) | O11H | H-bond (3.3 Å) |
| K18 (HZ2) | O9 | H-bond (2.6 Å) | R24 (NH1) | O8 | H-bond (3.1 Å) |
| K18 (NZ) | O9H | H-bond (3.0 Å) | R24 (Cδ) | C8 | HPI (3.1 Å) |
| K18 (Cδ) | C2 | HPI (3.3 Å) | R24 (NH1) | O8 | H-bond (3.1 Å) |
| M19 (O) | O9H | ES (6.6 Å) | R24 (2HH1) | O8H | H-bond (2.8 Å) |
| I20 (Cδ) | C4 | HPI (3.3 Å) | I20 (Cγ1) | C9 | HPI (3.1 Å) |
| I20 (Cδ) | C3 | HPI (3.2 Å) | I20 (Cγ1) | C10 | HPI (3.2 Å) |
| R24 (NH2) | O4 | H-bond (3.1 Å) | K49 (Nζ) | C5 | HPI (2.9 Å) |
| R24 (1HH2) | O3 | H-bond (3.2 Å) | K49 (Hζ3) | C15 | HPI (2.2 Å) |
| R24 (NH1) | O5 | H-bond (2.9 Å) | K49 (Hζ3) | O6H | H-bond (3.0 Å) |
| R24 (CZ) | C15 | HPI (4.0 Å) | K49 (Hζ2) | O6H | H-bond (3.3 Å) |
| R24 (1HH1) | O10 | H-bond (2.1 Å) | E50 (OA) | C16 | H-bond (3.2 Å) |
| R49 (NH2) | O6 | H-bond (3.1 Å) | I51 (Cγ2) | O1 | H-bond (3.0 Å) |
| R49 (NH2) | O5 | H-bond (2.6 Å) | I51 (Cγ2) | C17 | HPI (3.3 Å) |
| R49 (2HH1) | O5 | H-bond (2.2 Å) | I51 (Cγ2) | C1 | HPI (3.3 Å) |
| R49 (NH1) | O5 | H-bond (2.2 Å) | C52 (NH) | C19 | HPI (2.5 Å) |
| V51 (Cγ1) | C4 | HPI (2.9 Å) | C52 (NH) | C20 | HPI (2.4 Å) |
| V51 (Cβ) | C3 | HPI (3.9 Å) | C52 (NH) | C21 | HPI (2.9 Å) |
HPI, H-bond, and ES depict the hydrophobic interaction, hydrogen bond, and electrostatic interaction, respectively.
3. Discussion
3.1. Dissecting the Biophysical Characteristics of the mCCL2-P8A Variant
Chemokine functioning involves synergistic processes, including chemokine oligomerization and binding to the G-protein-coupled receptors (GPCRs) and GAGs.59 Oligomerization of chemokines is also regulated by GPCRs and GAGs, as GAGs induce oligomerization of chemokines60 and receptors disrupt the oligomers as the chemokine monomers are reported to be high-affinity ligands to them.30,61 On similar lines, the monomeric variant of the hCCL2 chemokine (P8A) has been reported to confer a higher binding affinity for CCR2 than that of its dimeric WT conformation.30 The proline residue (P8) in CCL2 inflicts some restraints on its conformation, thus assisting the process of oligomerization through the arm-exchange mechanism.41 Proline-mediated oligomerization through the arm-exchange process has also been reported for bleomycin resistance protein (BRP)41 and bovine ribonuclease A (RNaseA).41,62 In line with these studies, mutation of P8A in mCCL2 disrupted the dimer interface significantly, thus resulting in a monomeric conformation, as evidenced by 2D-DOSY results (Figure 2D). The obtained diffusion coefficients (D’s) are consistent with previously reported D values for other monomeric and dimeric CC and CXC chemokines.43,44 For example, the reported D value (0.813 × 1010 m2 s–1) for the CCL27 protein is less than that of the CCL2 dimer (0.9 × 1010 m2 s–1) and P8A monomer (1.3 × 1010 m2 s–1), suggesting a higher-order oligomerization, specifically the tetramer formation for CCL27.43
The obtained P8A variants of human and murine CCL2 orthologs have exhibited similar secondary structural features to those of their dimer counterparts (Figure 3A). However, loss of quaternary interactions at the dimer interface resulted in significant differences in the hydrophobic surfaces of the monomers and dimers, as identified from ANS fluorescence experiments (Figure 3B). The monomers exposed more hydrophobic patches as compared to the dimeric proteins. The differences in the tertiary/quaternary structure at the dimer interface of CCL2-P8A monomers were further supported by NMR HSQC-based CSP experiments, where a specific set of peaks at the dimer interface residues was significantly perturbed (Figure 4B). Furthermore, these P8A monomers remained as monomers even at a high concentration of ∼0.5 mM (Figure S6). Coherent with this study, a report on the enzyme dimethylarginine dimethylaminohydrolase (DDAH) that exists in a monomer–dimer equilibrium (Kd = 500 nM) suggested that DDAH gets converted into a stable monomeric conformation by substituting crucial interface residues.63 Further, biochemical and NMR experiments suggested that the monomeric DDAH retains 95% catalytic activity and exists as a monomer even at 1 mM concentration. These observations strongly establish the candidature of P8A proteins as monomeric variants of h/mCCL2 to elucidate their structure–function relationships and to study the molecular interactions with their interacting partners to dissect the molecular details of the leukocyte migration phenomenon.
3.2. Molecular Insights into Baicalin-Mediated Attenuation of Chemokine-Based Leukocyte Trafficking and Its Implications as an Alternate Therapeutic Molecule
Owing to the phenomenal involvement in various immune-related diseases, G-protein-coupled receptors, glycosaminoglycans, and chemokines became astounding therapeutic targets during the last two decades. Indeed, to target pathological conditions, identification and development of small molecules that specifically target the GPCR–chemokine axis have become the foremost approach of pharmaceutical industries. Approximately 30% of FDA-approved small inhibitors have been identified to target and block the GPCR–chemokine axis.64 Since it is widely accepted that the binding of chemokine–GAG interactions also regulates the functioning of chemokines during injurious conditions, few researchers have investigated the inhibition of the GAG–chemokine axis along with the receptor–chemokine axis as an alternative strategy.65 On this line, various small chemokine binding proteins (CKBPs) from many parasites and viruses,66 immunomodulatory proteins such as evasins,67,68 chemokine mimetics such as mutated and truncated chemokines,69 aptamers,70 and other small-molecule compounds binding to chemokines/receptors71,72 have been identified as potential blockers/inhibitors of the GPCR/GAG–chemokine axis. Alternatively, the natural plant products such as flavonoids have been used to treat various injurious ailments, and due to their low toxic nature and safe consumption, they have drawn remarkable attention in new alternative medicine. Baicalin (BA) flavonoid has been described to exhibit several pharmacological activities, comprising anti-inflammatory, antiviral, antioxidative, and antiproliferative activities.36,73−75 BA has also been affirmed to target several immunomodulatory proteins, such as chemokines, and attenuate their ability to induce cell recruitment by interfering with the chemokine–receptor interaction.36 A study on HIV-1 infection revealed that BA exhibits substantial anti-inflammatory effect by interfering with the binding of HIV-1 Env domains with chemokine coreceptors, as it blocked the HIV-1 entry of target cells at the initial stage.76
In an effort to elucidate the nature of the molecular interactions of baicalin chemokines, we have chosen CCL2 chemokine orthologs in both dimeric and monomeric forms. The results have delineated that the BA binding site comprises the N-terminal end along with β1- and β3-sheets of CCL2. The binding is mediated by both hydrogen bonds and hydrophobic interactions. Indeed, the N-terminal of CCL2 is the primary binding surface for its receptor CCR2 interaction. According to the most accepted two-site model of receptor–chemokine interactions for CC chemokines, residues from the N-terminal and second and third β-sheets predominantly interact with the cognate receptor.77 For CCL2, the major receptor binding residues include Y13, R18, R24, K35, K38, and K49.30 Comparative analysis of the receptor binding surface of CCL2 with the BA binding pocket has suggested that these two surfaces are extensively overlapped (Figure 8A,B and Table 4). The patch of the basic residues (R18, K19, R24, and K49) is crucial for receptor interaction as D25 and D27 residues of the DYDY motif of the CCR2 receptor through electrostatic interactions/H-bonds, which were acidic in nature.78 Interestingly, a close look into the GAG binding surface of mCCL2 has suggested that K18, R24, and K49 residues are the overlapping residues for receptor, GAG, and BA binding on the CCL2 protein (Figure 8C). Hence, it is evident that the observed binding surface for BA shows a noticeable concurrency with the essential binding residues of the receptor and GAG binding domains on CCL2. These observations essentially provide the rationale that BA may potentially target both the chemokine–receptor and chemokine–GAG axes, thereby abrogating the CCL2-mediated leukocyte tariffing as evidenced by reported in vitro studies (Figure 8D).36
Figure 8.

Comparative analysis of baicalin binding surface with the receptor and GAG binding surfaces: (A) Surface representation of the hCCL2 monomer depicting the crucial BA binding residues (purple and pink). The overlapping residues among the receptor, GAG, and BA binding on the hCCL2 surface are highlighted in pink. (B) Surface representation of the hCCL2 monomer depicting the crucial receptor-binding residues (red). (C) Surface representation of the hCCL2 monomer depicting the crucial GAG binding residues (green). (D) Schematic showing the baicalin-induced dissociation of the receptor/GAG–CCL2 complex.
Table 4. Comparative Analysis of the Binding Energy, Kd Values, and Interacting Residues for BA–mCCL2 and BA–hCCL2 Complexes.
| ligand | protein | binding energy | Kd [nM] | interacting residues |
|---|---|---|---|---|
| baicalin | mCCL2 (monomer) | –6.54 | 270 ± 20 | T16, S17, K18, M19, I20, R24, R49, V51 |
| baicalin | hCCL2 (monomer) | –6.39 | 260 ± 20 | T16, N17, R18, I20, R24, K49, E50, I51, C52 |
Indeed, several molecules have been designed to target either the chemokine–receptor or chemokine–GAG axis individually or both simultaneously. For example, vaccinia virus protein VV-35 kDa interacts with the CCL2 chemokine specifically with the receptor binding surface with a stoichiometry of 1:1.79 The crucial binding residues observed for the CCL2–VV-35 kDa complex were Y13, R18, K19, R24, K38, and K49. It was also reported that the binding affinity of the VV-35 kDa protein for the P8A monomeric variant is similar to that of wild-type dimeric CCL2. The observed interaction of baicalin with CCL2 wild-type and P8A monomers with a similar affinity and 1:1 stoichiometry of the BA–CCL2 monomer as depicted by fluorescence quenching experiments are in line with the binding studies reported on CCL2 with CKBPs (Table 4). In line with these results, the crystal structure of the dimeric quorum sensing protein TraR complex with the bound ligand N-(3-oxo-octanoyl)-l-homoserine lactone is also reported for a binding stoichiometry of 1:1 of ligand–TraR-monomer.80 NMR-based intensity measurements suggested no significant change (data not shown) in the dimer–monomer ratio of NH resonances in the absence and presence of baicalin, thus establishing that baicalin binding on CCL2 does not alter the oligomerization characteristics of the CCL2 protein, and it binds far from the dimer interface. Such a behavior was also observed on a dimeric corticotropin-releasing factor receptor type 1 (CRF1R), which belongs to class B of GPCRs. Interaction of various agonists and antagonist ligands with the CRF1R protein does not alter the monomer–dimer ratio/equilibrium of the protein even at a high concentration of ligand, suggesting that the ligand binding and dimerization are two independent events for CRF1R.81 The independent nature of ligand binding and dimerization was also reported for the Gp96 (GRP94) glycoprotein, as the authors reported that peptide/ligand/monomer stoichiometry was similar for the dimer and the higher-order homo-oligomeric conformation.82 Further, it was observed that the interaction of baicalin with monomers and dimers of CCL2 was specific, as evidenced by the perturbation of a subset of NH resonances (Figure 6C), and the binding interactions are majorly governed by hydrogen bonding and hydrophobic interactions (Tables 3 and 4). Similar to BA–CCL2 interactions, binding of chemokine binding protein vCKBP-2 with CCL2 also evidenced that the interactions are essentially mediated through H-bonding and hydrophobic interactions, and the vCCI binding surface on CCL2 is largely overlapped with the CCR2 binding surface.83
The CCL2 binding mirror-image aptamer (NOX-E36, also termed as Spiegelmer) consisting of l-ribonucleotides covers the entire GAG binding domain of CCL2, thus obstructing both GAG and receptor binding surfaces and abrogating the chemotactic activity of CCL2.70 The l-aptamer binds to R18, K19, R24, H66, and K49 through hydrogen bonds. Similar to the human counterpart, a murine CCL2-specific Spiegelmer (mNOX-E36) was also reported to be active in various animal models in obstructing CCL2–GAG interactions.70,84 Several of the residues observed for l-aptamers and CKBPs on the CCL2 surface are very similar to the residues observed for baicalin in the present study. On similar lines, a set of various small CCL5 binders and chimeric molecules has been screened on CCL5 proteins. The small non-carbohydrate/binder molecules interacted with CCL5 at the GAG binding surface at 30s and 40s loops of the CCL5 chemokine with the Kd values in the range of ∼20–60 μM. As the 40s loop is also the major site for receptor interaction, these molecules were found to constrain the binding interaction of CCL5 to its receptors CCR1 and CCR5.85 Likewise, recently, a molecular study identified the binding pocket for using naphthalene derivatives on CXCL3, which is in the close vicinity of the GAG binding domain.49 Similar to these compounds, CCL2–BA interactions observed in this study suggest that baicalin can potentially interfere with both the receptor and GAG interactions of the CCL2 chemokine. As BA has also been recognized as a potent anti-inflammatory agent providing inherent therapeutic benefits for the treatment of chronic and acute inflammatory reactions,40,86 considering the experimental outcomes and the potential benefits of BA together, it can be tested as a promising chemokine antagonist for therapeutic intervention in animal studies to strengthen its role further as a immunomodulatory agent/nutraceutical agent.
4. Concluding Remarks
In summary, the study suggests that the mCCL2-P8A variant exclusively exists in a monomeric conformation. The P8A monomers possess similar secondary structure contents to those of the dimers, whereas the hydrophobic surfaces were altered due to loss of dimerization contacts. Fluorescence studies have established that BA flavonoid interacts with CCL2 orthologs with a substantial nanomolar affinity irrespective of their oligomeric/orthologous behaviors. Further, the NMR and docking studies established that the BA binding surface on CCL2 comprises N-terminal, β1 and β3-sheets, and both these molecules are complexed through an array of hydrophobic and hydrogen bonding interactions. As the observed binding surface for BA on CCL2 extensively overlaps with the receptor/GAG binding surfaces, it can be postulated that BA binding to CCL2 disrupts/weakens its cognate receptor/GAG interactions, thus attenuating the chemotactic activity in its presence as reported earlier using in vitro studies. The study also points the fact of exploring baicalin and other potential flavonoids as nutraceutical immunomodulatory agents for regulating inflammatory conditions.
5. Materials and Methods
5.1. Site-Directed Mutagenesis, Expression, and Purification of the Monomeric Variant of Murine and Human CCL2
The monomeric variants of murine CCL2 and human CCL2 were generated using standard the QuickChange site-directed mutagenesis (SDM) method. Note that, hereafter, monomers of murine and human CCL2 are termed as mCCL2-P8A and hCCL-P8A, respectively, while the wild-type (dimers) are represented as mCCL2-WT and hCCL2-WT, respectively. Briefly, for both monomeric mutants (P8A), PCR-based gene amplification was performed using mCCL2-WT and hCCL2-WT genes as a template along with appropriate forward and reverse primers (Table S1) and inserted into the pET expression vector.21 The PCR products were checked in 0.8% DNA agarose gel, and the desired mutants were confirmed by Sanger DNA sequencing. Both monomeric mutant proteins and the wild-type constructs of human and murine CCL2 proteins were expressed and purified as described elsewhere.21
5.2. Size Exclusion Chromatography (SEC)
Size exclusion chromatography (SEC) was carried out using a 120 mL (16/60) volume of the Superdex 75 AKTA prime FPLC column system. All four CCL2 proteins (monomers and wild-type dimers of human and murine CCL2 orthologs) of ∼2 mg mL–1 concentrations were directly injected into the pre-equilibrated column in 50 mM sodium phosphate and 100 mM NaCl with 1% glycerol (pH 6.0) buffer. All of the SEC experiments were carried out at a flow rate of 1 mL min–1, and the elution profile of the proteins was observed at 280 nm wavelength. The oligomeric states/molecular weights of the proteins were assessed by comparing the elution profiles with those of standard proteins (pepsin, chymotrypsin, aprotinin, and cytochrome C). The standard proteins were loaded onto the same FPLC column under identical buffer conditions.
5.3. Optical Spectroscopy
For all optical spectroscopy experiments, the protein samples of 50 μM (20 mM Tris and 50 mM NaCl, at pH 7) concentration were used.
5.3.1. Circular Dichroism (CD) Spectroscopy
All far-UV CD measurements were performed on a Peltier controlled Jasco J-1500 CD spectrophotometer at 25 °C using a 1 mm path length quartz cuvette. For CCL2 orthologs (WT and monomers), the CD spectra were recorded from 190 to 250 nm wavelength as described elsewhere.87 The quantitative measurement of the secondary structural elements of CCL2 orthologs was carried out using DICROWEB-K2D software with the default parameters (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml).88
5.3.2. Steady-State Fluorescence Spectroscopy
All of the steady-state fluorescence experiments were acquired at 25 °C temperature on a Fluorolog spectrophotometer equipped with a xenon lamp source. The spectra were acquired at room temperature using a 4 mm path length quartz cuvette. The spectral bandwidths of excitation and emission slits were kept constant at 5 nm. The emission profiles of intrinsic tryptophan fluorescence of CCL2 orthologs were monitored by exciting them at 295 nm wavelength. To monitor the binding of ANS to CCL2 ortholog proteins, ANS was excited at a wavelength of 380 nm, and the emission spectra were acquired from 400 to 650 nm wavelength. For ANS binding experiments, a ratio of 1:5 (ANS/protein) was used for all samples.
5.3.3. Fluorescence Lifetime Spectroscopy (FLS)
Fluorescence lifetime experiments were recorded with a Horiba Jobin Yvon FluoroCube system for all of the four CCL2 ortholog proteins. The tryptophan-based lifetime decay profiles were obtained using an excitation wavelength of 295 nm and emission wavelength of 340 nm. Similarly for ANS-based studies, the 380 nm wavelength was used for excitation and 470 nm wavelength was used to collect the emission. The lifetime profiles were analyzed using the multiexponential decay functions.89
5.3.4. Baicalin Quenching Measurements
For baicalin quenching measurements, all of the fluorescence titrations were recorded under identical buffer conditions and at a fixed concentration of protein (50 μM) as described in the steady-state fluorescence spectroscopy section. For BA, a stock solution of 20 mM was prepared by dissolving it into dimethyl sulfoxide (DMSO). The titrand (BA) was added in incremental steps of 5 μM ranging from 5 to 100 μM. For titrations, the protein sample was excited at 295 nm, and the emission profile was monitored from 300 to 450 nm. To achieve thermodynamic equilibrium, the protein–BA complex was preincubated for ∼15 min at room temperature. All of the fluorescence experiments were recorded in triplicate to validate the binding data. For analyzing the quenching data, the following Stern–Volmer equation was used.56
| 1 |
where F0 and F are the intensities of fluorescence without and with the presence of quencher, respectively; [Q] is the concentration of quencher; and Ksv is the Stern–Volmer quenching constant and can be described as Ksv = Kqτ0.55,56Kq is the rate constant for quenching, and τ0 is the lifetime of the fluorophore compound without quencher and equals 10–8 s.55 The relationship between the fluorescence quenching intensity and the concentration of BA can be described by the following binding constant formula.
| 2 |
where Ka is a binding constant and n is the number of binding sites.
5.4. Nuclear Magnetic Resonance Spectroscopy
5.4.1. NMR Sample Preparation
All of the NMR experiments were recorded on a Bruker 500 MHz spectrometer equipped with a TXI probe at 298 K. For all NMR-based experiments (except for 2D translational diffusion spectroscopy), 15N-labeled mCCL2-WT and mCCL2-P8A protein samples were prepared in 50 mM sodium phosphate and 50 mM sodium chloride buffer (pH 6.0) in 10% D2O. For 2D-DOSY, unlabeled protein samples were prepared and dissolved in 100% D2O solvent. DOSY experiments were recorded on a Bruker 800 MHz spectrometer as described elsewhere.89,90 For mCCL2-P8A, 1H–15N HSQC spectra were recorded in the concentration range of 50–500 μM.
5.4.2. Baicalin Titrations
For titration experiments, HSQC spectra were recorded at a fixed concentration of 100 μM for both proteins and an increasing concentration of BA in the ratio (P/L) of 1:0.5, 1:1, 1:2, and 1:5. All HSQC spectra were recorded with sweep widths (SWs) of 12 and 26 ppm for 1H and 15N dimensions, respectively. For each HSQC spectrum, 128 scans and 128 complex increments were used and the offset values of 1H and 15N were set to 4.7 and 120 ppm, respectively. TOPSPIN 3.2 software was used to process all HSQC spectra. The residue-specific chemical shift perturbations (CSPs) for the mCCL2-WT–BA complex were evaluated using the following equation.
| 3 |
where ΔδH and ΔδN are the change in the chemical shift values of 1H and 15N, respectively.
5.4.3. Two-Dimensional Diffusion-Ordered Spectroscopy (2D-DOSY) Experiments
For all four proteins, a concentration of ∼500 μM was used to acquire the 2D-DOSY experiments. The translational diffusion coefficients of all proteins were estimated by nonlinear least-squares fitting of intensity data as described elsewhere.44 To assess the molecular weight, the two standard proteins hen egg lysozyme (HEL, MW ∼ 14.3 kDa) and the chicken SH3 domain (SH3, MW ∼ 7.2 kDa) were used as a reference.
5.5. Molecular Docking
The molecular docking approach was used to unravel the atomic-level interactions between the protein and BA complex. The AutoDock 4.2 tool was used to dock BA (PubChem SID: 329831336) onto the monomeric subunits of human and murine CCL2 proteins using the CS-Rosetta structural model for mCCL2-WT21 and reported NMR structure of hCCL2-WT (PDB ID: 1dok).17 For ligand (BA) (IUPAC name: (2S, 3S, 4S, 5R, 6S)-6-(5,6-dihydroxy-4-oxo-2-phenylchromen-7-yl) oxy-3, 4, 5-trihydroxyoxane-2-carboxylic acid) (Figure 1C), the sdf file format was used, which was extracted from the PubChem database. The docking programme was performed using a hybrid genetic algorithm, Lamarckian genetic algorithm (LGA), which utilizes a parameter-based free-energy scoring function for estimating binding energy.91,92 Kollman (6) and Gasteiger charges were computed, added, and allocated to all atoms on monomeric CCL2. Based on CSP results, the grid box was defined according to the identified region of the mCCL2/hCCL2 protein and centered at 60.41, 2.64, and 14.25. The dimensions of the grid for docking were selected as 74 × 82 × 84, with 0.37 Å spacing. The LigPlot+ graphical plots were constructed using LigPlot+ software, and the PDB-format file was used as a primary input to the program.93 PyMol software was used to analyze the docking results obtained from AutoDock.
Acknowledgments
K.M.P. acknowledges the receipt of Grants CRG/2018/001329 and SERB-SB/YS/LS-380/2013 from SERB-DST, and DBT-IYBA fellowship BT/07/IYBA/2013-19. N.J. acknowledges the receipt of JRF & SRF fellowship from UGC. The authors acknowledge the support of biophysical and 500 MHz NMR instrumentation facilities at the Institute Instrumentation Centre, IIT-Roorkee, and 800 MHz NMR facility at CMBR, Lucknow.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03428.
Details of the primer sequences for the site-directed mutagenesis (SDM) (Table S1); information of total protein yield (Table S2); description of the percentage of secondary structural elements of CCL2 orthologs (Table S3); CCL2 multiple sequence alignment profile (Figure S1); SDM, protein expression, and purification profiles of CCL2 orthologs (Figures S2 and S3); intrinsic tryptophan fluorescence and lifetime decay profiles of CCL2 orthologs (Figures S4 and S5); and concentration-dependent 15N HSQC profile of P8A (Figure S6) (PDF)
Author Contributions
K.M.P. designed the project. N.J. and D.K. performed the experiments. N.J. and K.M.P. analyzed the data. N.J. and K.M.P. wrote the manuscript. All of the authors reviewed and accepted the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell 2010, 140, 771–776. 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Chen L.; Deng H.; Cui H.; Fang J.; Zuo Z.; Deng J.; Li Y.; Wang X.; Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204. 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulkhaleq L.; Assi M.; Abdullah R.; Zamri-Saad M.; Taufiq-Yap Y.; Hezmee M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627. 10.14202/vetworld.2018.627-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poluri K. Chemokines: the holy messengers of leukocyte trafficking. Austin J. Biotechnol. Bioeng. 2014, 1, 1–3. [Google Scholar]
- Schall T. J.; Bacon K. B. Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 1994, 6, 865–873. 10.1016/0952-7915(94)90006-X. [DOI] [PubMed] [Google Scholar]
- Gulati K.; Poluri K. M. Mechanistic and therapeutic overview of glycosaminoglycans: the unsung heroes of biomolecular signaling. Glycoconjugate J. 2016, 33, 1–17. 10.1007/s10719-015-9642-2. [DOI] [PubMed] [Google Scholar]
- Speyer C. L.; Ward P. A. Role of endothelial chemokines and their receptors during inflammation. J. Invest. Surg. 2011, 24, 18–27. 10.3109/08941939.2010.521232. [DOI] [PubMed] [Google Scholar]
- Zeremski M.; Petrovic L.; Talal A. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J. Viral Hepatitis 2007, 14, 675–687. 10.1111/j.1365-2893.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- Vandercappellen J.; Van Damme J.; Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- Choi J.; Selmi C.; Leung P. S.; Kenny T. P.; Roskams T.; Gershwin M. E. Chemokine and chemokine receptors in autoimmunity: the case of primary biliary cholangitis. Expert Rev. Clin. Immunol. 2016, 12, 661–672. 10.1586/1744666X.2016.1147956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’AMBROSIO D.; Mariani M.; Panina-Bordignon P.; Sinigaglia F. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, 1266–1275. 10.1164/ajrccm.164.7.2103011. [DOI] [PubMed] [Google Scholar]
- Savarin-Vuaillat C.; Ransohoff R. M. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce?. Neurotherapeutics 2007, 4, 590–601. 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulkower K.; Brosnan C. F.; Aquino D. A.; Cammer W.; Kulshrestha S.; Guida M. P.; Rapoport D. A.; Berman J. W. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J. Immunol. 1993, 150, 2525–2533. [PubMed] [Google Scholar]
- Glabinski A. R.; Balasingam V.; Tani M.; Kunkel S. L.; Strieter R. M.; Yong V. W.; Ransohoff R. M. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 1996, 156, 4363–4368. [PubMed] [Google Scholar]
- Van Coillie E.; Van Damme J.; Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. 1999, 10, 61–86. 10.1016/S1359-6101(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Charo I. F.; Ransohoff R. M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Handel T. M.; Domaille P. J. Heteronuclear (1H, 13C, 15N) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry 1996, 35, 6569–6584. 10.1021/bi9602270. [DOI] [PubMed] [Google Scholar]
- Lubkowski J.; Bujacz G.; Boqué L.; Alexander W. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 1997, 4, 64–69. 10.1038/nsb0197-64. [DOI] [PubMed] [Google Scholar]
- Wang X.; Watson C.; Sharp J. S.; Handel T. M.; Prestegard J. H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. 10.1016/j.str.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyakov A.; Obmolova G.; Gilliland G. L. Structural insights into chemokine CCL17 recognition by antibody M116. Biochem. Biophys. Rep. 2018, 13, 27–31. 10.1016/j.bbrep.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N.; Nagar N.; Gulati K.; Gangele K.; Mishra A.; Kumar D.; Poluri K. M. Dissecting the differential structural and dynamics features of CCL2 chemokine orthologs. Int. J. Biol. Macromol. 2020, 156, 239–251. 10.1016/j.ijbiomac.2020.04.067. [DOI] [PubMed] [Google Scholar]
- Chen Y.-L.; Chang Y.-J.; Jiang M. J. Monocyte chemotactic protein-1 gene and protein expression in atherogenesis of hypercholesterolemic rabbits. Atherosclerosis 1999, 143, 115–123. 10.1016/S0021-9150(98)00285-8. [DOI] [PubMed] [Google Scholar]
- Wang J.; Ou Z.; Hou Y.; Luo J.; Shen Z.; Ding J.; Shao Z. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene 2006, 25, 7201–7211. 10.1038/sj.onc.1209703. [DOI] [PubMed] [Google Scholar]
- Flores-Villanueva P. O.; Ruiz-Morales J. A.; Song C.-H.; Flores L. M.; Jo E.-K.; Montaño M.; Barnes P. F.; Selman M.; Granados J. A functional promoter polymorphism in monocyte chemoattractant protein–1 is associated with increased susceptibility to pulmonary tuberculosis. J. Exp. Med. 2005, 202, 1649–1658. 10.1084/jem.20050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoettl T.; Hausmann M.; Herlyn M.; Gunckel M.; Dirmeier A.; Falk W.; Herfarth H.; Schoelmerich J.; Rogler G. Monocyte chemoattractant protein-1 (MCP-1) inhibits the intestinal-like differentiation of monocytes. Clin. Exp. Immunol. 2006, 145, 190–199. 10.1111/j.1365-2249.2006.03113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanuma N.; Sakuma H.; Sasaki A.; Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006, 112, 195–204. 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- Lowman H. B.; Fairbrother W. J.; Slagle P. H.; Kabakoff R.; Hebert C. A.; Liu J.; Shire S. Monomeric variants of IL-8: Effects of side chain substitutions and solution conditions upon dimer formation. Protein Sci. 1997, 6, 598–608. 10.1002/pro.5560060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong S. R.; Hebert C. A.; Lowman H. B.; Liu J.; Shire S.; Deforge L. E.; Gillece-Castro B. L.; Mcdowell R. IL-8 single-chain homodimers and heterodimers: Interactions with the chemokine receptors CXCR1, CXCR2, and DARC. Protein Sci. 1997, 6, 609–617. 10.1002/pro.5560060310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Rollins B. J. A dominant negative inhibitor indicates that monocyte chemoattractant protein 1 functions as a dimer. Mol. Cell. Biol. 1995, 15, 4851–4855. 10.1128/MCB.15.9.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paavola C. D.; Hemmerich S.; Grunberger D.; Polsky I.; Bloom A.; Freedman R.; Mulkins M.; Bhakta S.; McCarley D.; Wiesent L. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 1998, 273, 33157–33165. 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- Laurence J. S.; Blanpain C.; Burgner J. W.; Parmentier M.; LiWang P. J. CC chemokine MIP-1β can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 2000, 39, 3401–3409. 10.1021/bi9923196. [DOI] [PubMed] [Google Scholar]
- Tan J. H.; Canals M.; Ludeman J. P.; Wedderburn J.; Boston C.; Butler S. J.; Carrick A. M.; Parody T. R.; Taleski D.; Christopoulos A. Design and receptor interactions of obligate dimeric mutant of chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 2012, 287, 14692–14702. 10.1074/jbc.M111.334201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard O. Z.; Ben-Baruch A.; Oppenheim J. J. Chemokines: progress toward identifying molecular targets for therapeutic agents. Trends Biotechnol. 1996, 14, 46–51. 10.1016/0167-7799(96)80920-6. [DOI] [PubMed] [Google Scholar]
- Van Acker F.; Voss H.; Timmerman H. Chemokines: structure, receptors and functions. A new target for inflammation and asthma therapy?. Mediators Inflamm. 1996, 5, 393–416. 10.1155/S0962935196000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekido N.; Mukaida N.; Harada A.; Nakanishi I.; Watanabe Y.; Matsushima K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993, 365, 654–657. 10.1038/365654a0. [DOI] [PubMed] [Google Scholar]
- Li B. Q.; Fu T.; Gong W.-H.; Dunlop N.; Kung H.-f.; Yan Y.; Kang J.; Wang J. M. The flavonoid baicalin exhibits anti-inflammatory activity by binding to chemokines. Immunopharmacology 2000, 49, 295–306. 10.1016/S0162-3109(00)00244-7. [DOI] [PubMed] [Google Scholar]
- Krakauer T.; Li B. Q.; Young H. A. The flavonoid baicalin inhibits superantigen-induced inflammatory cytokines and chemokines. FEBS Lett. 2001, 500, 52–55. 10.1016/S0014-5793(01)02584-4. [DOI] [PubMed] [Google Scholar]
- Kubo M.; Matsuda H.; Tanaka M.; Kimura Y.; Okuda H.; Higashino M.; Tani T.; Namba K.; Arichi S. Studies on Scutellariae radix. VII. Anti-arthritic and anti-inflammatory actions of methanolic extract and flavonoid components from Scutellariae radix. Chem. Pharm. Bull. 1984, 32, 2724–2729. 10.1248/cpb.32.2724. [DOI] [PubMed] [Google Scholar]
- Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat. Rev. 2009, 35, 57–68. 10.1016/j.ctrv.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Gao Z.; Huang K.; Yang X.; Xu H. Free radical scavenging and antioxidant activities of flavonoids extracted from the radix of Scutellaria baicalensis Georgi. Biochim. Biophys. Acta, Gen. Subj. 1999, 1472, 643–650. 10.1016/S0304-4165(99)00152-X. [DOI] [PubMed] [Google Scholar]
- Bergdoll M.; Remy M.-H.; Cagnon C.; Masson J.-M.; Dumas P. Proline-dependent oligomerization with arm exchange. Structure 1997, 5, 391–401. 10.1016/S0969-2126(97)00196-2. [DOI] [PubMed] [Google Scholar]
- Paolini J. F.; Willard D.; Consler T.; Luther M.; Krangel M. S. The chemokines IL-8, monocyte chemoattractant protein-1, and I-309 are monomers at physiologically relevant concentrations. J. Immunol. 1994, 153, 2704–2717. [PubMed] [Google Scholar]
- Jansma A. L.; Kirkpatrick J. P.; Hsu A. R.; Handel T. M.; Nietlispach D. NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J. Biol. Chem. 2010, 285, 14424–14437. 10.1074/jbc.M109.091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph P. R. B.; Poluri K. M.; Gangavarapu P.; Rajagopalan L.; Raghuwanshi S.; Richardson R. M.; Garofalo R. P.; Rajarathnam K. Proline substitution of dimer interface β-strand residues as a strategy for the design of functional monomeric proteins. Biophys. J. 2013, 105, 1491–1501. 10.1016/j.bpj.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venyaminov S. Y.; Yang J. T.. Determination of Protein Secondary Structure. In Circular Dichroism and the Conformational Analysis of Biomolecules; Springer, 1996; pp 69–107. [Google Scholar]
- Sreerama N.; Venyaminov S. Y.; Woody R. W. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 2000, 287, 243–251. 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- Collini M.; D’Alfonso L.; Baldini G. New insight on β-lactoglobulin binding sites by 1-anilinonaphthalene-8-sulfonate fluorescence decay. Protein Sci. 2000, 9, 1968–1974. 10.1110/ps.9.10.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Winter S.; Löber G. Use of fluorescence decay times of 8-ANS-protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophys. Chem. 1996, 60, 79–88. 10.1016/0301-4622(96)00009-9. [DOI] [PubMed] [Google Scholar]
- Gulati K.; Gangele K.; Kumar D.; Poluri K. M. An inter-switch between hydrophobic and charged amino acids generated druggable small molecule binding pocket in chemokine paralog CXCL3. Arch. Biochem. Biophys. 2019, 662, 121–128. 10.1016/j.abb.2018.12.007. [DOI] [PubMed] [Google Scholar]
- Gulati K.; Jamsandekar M.; Poluri K. M. Mechanistic insights into molecular evolution of species-specific differential glycosaminoglycan binding surfaces in growth-related oncogene chemokines. R. Soc. Open Sci. 2017, 4, 171059 10.1098/rsos.171059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangele K.; Jamsandekar M.; Mishra A.; Poluri K. M. Unraveling the evolutionary origin of ELR motif using fish CXC chemokine CXCL8. Fish Shellfish Immunol. 2019, 93, 17–27. 10.1016/j.fsi.2019.07.034. [DOI] [PubMed] [Google Scholar]
- Zlotnik A.; Yoshie O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries M. E.; Kelvin A. A.; Xu L.; Ran L.; Robinson J.; Kelvin D. J. Defining the origins and evolution of the chemokine/chemokine receptor system. J. Immunol. 2006, 176, 401–415. 10.4049/jimmunol.176.1.401. [DOI] [PubMed] [Google Scholar]
- Pletnev S.; Pletneva N. V.; Souslova E. A.; Chudakov D. M.; Lukyanov S.; Wlodawer A.; Dauter Z.; Pletnev V. Structural basis for bathochromic shift of fluorescence in far-red fluorescent proteins eqFP650 and eqFP670. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 1088–1097. 10.1107/S0907444912020598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X.; Dan-Dan D.; Zhi-Juan C.; Qiong X.; Liang J.-Y.; Jian-Zhong L. Interaction between serum albumin and four flavones by fluorescence spectroscopy and molecular docking. Chin. J. Anal. Chem. 2010, 38, 483–487. 10.1016/S1872-2040(09)60034-7. [DOI] [Google Scholar]
- Xiao J. B.; Chen J. W.; Cao H.; Ren F. L.; Yang C. S.; Chen Y.; Xu M. Study of the interaction between baicalin and bovine serum albumin by multi-spectroscopic method. J. Photochem. Photobiol., A 2007, 191, 222–227. 10.1016/j.jphotochem.2007.04.027. [DOI] [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Li Y.; Kang C. Solution NMR spectroscopy in target-based drug discovery. Molecules 2017, 22, 1399. 10.3390/molecules22091399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanga C.; Handel T. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 2011, 317, 590–601. 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogewerf A. J.; Kuschert G. S.; Proudfoot A. E.; Borlat F.; Clark-Lewis I.; Power C. A.; Wells T. N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sharp J. S.; Handel T. M.; Prestegard J. H.. Chemokine Oligomerization in Cell Signaling and Migration. In Progress in Molecular Biology and Translational Science; Elsevier, 2013; Vol. 117, pp 531–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato A.; Cafaro V.; D’Alessio G. Ribonuclease A can be transformed into a dimeric ribonuclease with antitumor activity. J. Biol. Chem. 1994, 269, 17394–17396. [PubMed] [Google Scholar]
- Plevin M. J.; Magalhães B. S.; Harris R.; Sankar A.; Perkins S. J.; Driscoll P. C. Characterization and manipulation of the Pseudomonas aeruginosa dimethylarginine dimethylaminohydrolase monomer–dimer equilibrium. J. Mol. Biol. 2004, 341, 171–184. 10.1016/j.jmb.2004.05.057. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. The druggable genome. Nat. Rev. Drug Discovery 2002, 1, 727–730. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Crijns H.; Vanheule V.; Proost P. Targeting Chemokine—Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 2020, 11. 10.3389/fimmu.2020.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.; Bryant N. A.; Alcami A.. Soluble Chemokine Binding Proteins Encoded by Viruses. In Chemokines in Viral Infections; Plenum Publishers, 2002. [Google Scholar]
- Denisov S. S.; Ippel J. H.; Heinzmann A. C.; Koenen R. R.; Ortega-Gomez A.; Soehnlein O.; Hackeng T. M.; Dijkgraaf I. Tick saliva protein Evasin-3 modulates chemotaxis by disrupting CXCL8 interactions with glycosaminoglycans and CXCR2. J. Biol. Chem. 2019, 294, 12370–12379. 10.1074/jbc.RA119.008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J. M.; Losberger C.; Déruaz M.; Power C. A.; Proudfoot A. E.; Shaw J. P. Structural basis of chemokine sequestration by a tick chemokine binding protein: the crystal structure of the complex between Evasin-1 and CCL3. PLoS One 2009, 4, e8514 10.1371/journal.pone.0008514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton J.; Neil S.; Wintle J.; Clark-Lewis I.; Moore H.; Lam C.; Auer M.; Hub E.; Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 1997, 91, 385–395. 10.1016/S0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- Oberthür D.; Achenbach J.; Gabdulkhakov A.; Buchner K.; Maasch C.; Falke S.; Rehders D.; Klussmann S.; Betzel C. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat. Commun. 2015, 6, 7923 10.1038/ncomms7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz B. J.; Grovit-Ferbas K.; Sharron M. P.; Mao S.-H.; Goetz M. B.; Daar E. S.; Doms R. W.; O’Brien W. A. A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J. Exp. Med. 1997, 186, 1395–1400. 10.1084/jem.186.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselgesser J.; Ng H. P.; Liang M.; Zheng W.; May K.; Bauman J. G.; Monahan S.; Islam I.; Wei G. P.; Ghannam A. Identification and characterization of small molecule functional antagonists of the CCR1 chemokine receptor. J. Biol. Chem. 1998, 273, 15687–15692. 10.1074/jbc.273.25.15687. [DOI] [PubMed] [Google Scholar]
- Lee Y.-C.; Chuah A. M.; Yamaguchi T.; Takamura H.; Matoba T. Antioxidant activity of traditional Chinese medicinal herbs. Food Sci. Technol. Res. 2008, 14, 205–210. 10.3136/fstr.14.205. [DOI] [Google Scholar]
- Huang W.-H.; Lee A.-R.; Yang C.-H. Antioxidative and anti-inflammatory activities of polyhydroxyflavonoids of Scutellaria baicalensis GEORGI. Biosci., Biotechnol., Biochem. 2006, 70, 2371–2380. 10.1271/bbb.50698. [DOI] [PubMed] [Google Scholar]
- Li B.; Fu T.; Yan Y.; Baylor N. W.; Ruscetti F. W.; Kung H. Inhibition of HIV infection by baicalin--a flavonoid compound purified from Chinese herbal medicine. Cell. Mol. Biol. Res. 1993, 39, 119–124. [PubMed] [Google Scholar]
- Li B. Q.; Fu T.; Dongyan Y.; Mikovits J. A.; Ruscetti F. W.; Wang J. M. Flavonoid baicalin inhibits HIV-1 infection at the level of viral entry. Biochem. Biophys. Res. Commun. 2000, 276, 534–538. 10.1006/bbrc.2000.3485. [DOI] [PubMed] [Google Scholar]
- Tan J. H.; Ludeman J. P.; Wedderburn J.; Canals M.; Hall P.; Butler S. J.; Taleski D.; Christopoulos A.; Hickey M. J.; Payne R. J. Tyrosine sulfation of chemokine receptor CCR2 enhances interactions with both monomeric and dimeric forms of the chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 2013, 288, 10024–10034. 10.1074/jbc.M112.447359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerich S.; Paavola C.; Bloom A.; Bhakta S.; Freedman R.; Grunberger D.; Krstenansky J.; Lee S.; McCarley D.; Mulkins M. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry 1999, 38, 13013–13025. 10.1021/bi991029m. [DOI] [PubMed] [Google Scholar]
- Seet B. T.; Singh R.; Paavola C.; Lau E. K.; Handel T. M.; McFadden G. Molecular determinants for CC-chemokine recognition by a poxvirus CC-chemokine inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9008–9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gargioli C.; Muraglia E.; Cortese R.; De Francesco R.; Neddermann P.; Di Marco S. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J. 2002, 21, 4393–4401. 10.1093/emboj/cdf459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraetke O.; Wiesner B.; Eichhorst J.; Furkert J.; Bienert M.; Beyermann M. Dimerization of corticotropin-releasing factor receptor type 1 is not coupled to ligand binding. J. Recept. Signal Transduction 2005, 25, 251–276. 10.1080/10799890500468838. [DOI] [PubMed] [Google Scholar]
- Thorne M. E.; McQuade K. L. Heat-induced oligomerization of gp96 occurs via a site distinct from substrate binding and is regulated by ATP. Biochem. Biophys. Res. Commun. 2004, 323, 1163–1171. 10.1016/j.bbrc.2004.08.215. [DOI] [PubMed] [Google Scholar]
- Beck C. G.; Studer C.; Zuber J.-F.; Demange B. J.; Manning U.; Urfer R. The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 2001, 276, 43270–43276. 10.1074/jbc.M106305200. [DOI] [PubMed] [Google Scholar]
- Ninichuk V.; Clauss S.; Kulkarni O.; Schmid H.; Segerer S.; Radomska E.; Eulberg D.; Buchner K.; Selve N.; Klussmann S. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36–3′ PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol. 2008, 172, 628–637. 10.2353/ajpath.2008.070601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot A. E. Glycosaminoglycan analogs as a novel anti-inflammatory strategy. Front. Immunol. 2012, 3, 293. 10.3389/fimmu.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinda B.; Dinda S.; DasSharma S.; Banik R.; Chakraborty A.; Dinda M. Therapeutic potentials of baicalin and its aglycone, baicalein against inflammatory disorders. Eur. J. Med. Chem. 2017, 131, 68–80. 10.1016/j.ejmech.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Gulati K.; Gangele K.; Agarwal N.; Jamsandekar M.; Kumar D.; Poluri K. M. Molecular cloning and biophysical characterization of CXCL3 chemokine. Int. J. Biol. Macromol. 2018, 107, 575–584. 10.1016/j.ijbiomac.2017.09.032. [DOI] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Kumar D.; Poluri K. M. Unraveling the differential structural stability and dynamics features of T7 endolysin partially folded conformations. Biochim. Biophys. Acta, Gen. Subj. 2018, 1862, 924–935. 10.1016/j.bbagen.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Kumar D.; Poluri K. M. Elucidating the pH-dependent structural transition of T7 bacteriophage endolysin. Biochemistry 2016, 55, 4614–4625. 10.1021/acs.biochem.6b00240. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. . [DOI] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.