

Abstract
Background and Aims
Vacuolar H+‐ATP complex (V‐ATPase) is a multisubunit protein complex required for acidification of intracellular compartments. At least five different factors are known to be essential for its assembly in the endoplasmic reticulum (ER). Genetic defects in four of these V‐ATPase assembly factors show overlapping clinical features, including steatotic liver disease and mild hypercholesterolemia. An exception is the assembly factor vacuolar ATPase assembly integral membrane protein (VMA21), whose X‐linked mutations lead to autophagic myopathy.
Approach and Results
Here, we report pathogenic variants in VMA21 in male patients with abnormal protein glycosylation that result in mild cholestasis, chronic elevation of aminotransferases, elevation of (low‐density lipoprotein) cholesterol and steatosis in hepatocytes. We also show that the VMA21 variants lead to V‐ATPase misassembly and dysfunction. As a consequence, lysosomal acidification and degradation of phagocytosed materials are impaired, causing lipid droplet (LD) accumulation in autolysosomes. Moreover, VMA21 deficiency triggers ER stress and sequestration of unesterified cholesterol in lysosomes, thereby activating the sterol response element‐binding protein–mediated cholesterol synthesis pathways.
Conclusions
Together, our data suggest that impaired lipophagy, ER stress, and increased cholesterol synthesis lead to LD accumulation and hepatic steatosis. V‐ATPase assembly defects are thus a form of hereditary liver disease with implications for the pathogenesis of nonalcoholic fatty liver disease.
Abbreviations
- ALT
alanine aminotransferase
- apoCIII
apolipoprotein CIII
- ATP6AP1 and ATP6AP2
ATPase H+ transporting accessory protein 1 and 2
- AST
aspartate aminotransferase
- CDG
congenital disorders of glycosylation
- CTSB
cathepsin B
- ER
endoplasmic reticulum
- LDL
low‐density lipoprotein
- LD
lipid droplet
- MALDI‐TOF
matrix‐assisted laser/desorption ionization time‐of‐flight
- NPC
Niemann–Pick type C disease
- PERK
protein kinase R–like endoplasmic reticulum kinase
- SREBP1
sterol response element‐binding protein‐1
- TIEF
isoelectric focusing of serum transferrin
- V‐ATPase
vacuolar H+‐APT complex
The vacuolar H+‐ATP complexes (V‐ATPases) are large multisubunit protein complexes that are responsible for the pH homeostasis of cytoplasmic organelles.( 1 ) They are organized into two domains: a membrane‐integral V0 domain responsible for proton translocation and a cytosolic V1 domain, which carries out adenosine triphosphate hydrolysis.( 1 ) For the biogenesis of the V‐ATPases, studies in Saccharomyces cerevisiae have shown that V0 and V1 domains are assembled separately in the endoplasmic reticulum (ER) and cytosol, respectively. The assembly of the V0 domain was shown to depend on a set of ER‐resident chaperones( 2 , 3 ): Vma12p, Vma21p, Voa1p, Pkr1p, and Vma22p.( 3 , 4 , 5 , 6 , 7 ) In the first step of the assembly, Vma21p comes into contact with subunit c′, and this interaction promotes the assembly of the proteolipid subunits into a ring.( 3 , 4 ) In a second parallel step, a complex of Vma12p and Vma22p interacts transiently with subunit a of the V0 domain and mediates its assembly with the proteolipid ring.( 2 ) Once the V0 is fully assembled, Vma21p escorts the V0 domain to the cis‐Golgi, where it will bind the V1 sector to form the functional V‐ATPase.( 3 ) Owing to its ER retention motif KKXX, Vma21p is transported back to the ER to assist in additional rounds of V0 assembly.( 3 ) Vma12p, Vma21p, and Vma22p correspond to transmembrane protein 199 (TMEM199), vacuolar ATPase assembly integral membrane protein (VMA21), and coiled‐coil domain containing protein 115 (CCDC115), respectively, in mammals. Voa1p has been replaced by ATPase H+ Transporting Accessory Protein 1 and 2 (ATP6AP1 and ATP6AP2),( 8 , 9 ) whereas Pkr1p does not have a mammalian ortholog. Whether the assembly of the mammalian V0 is mechanistically similar to yeast V0 assembly is currently unclear.( 10 )
Lessons on the phenotypic consequences of V‐ATPase assembly dysfunction can be learned from human genetic diseases. Patients with mutations in four of the assembly factor genes, ATP6AP1, ATP6AP2, TMEM199, and CCDC115, present with a heterogeneous spectrum of disease symptoms.( 8 , 11 , 12 , 13 ) Interestingly, the organ that is affected in all patients is the liver. Hepatic features include steatosis in hepatocytes and lipids and variable hepatic injury ranging from chronic elevations of liver aminotransferases to end‐stage liver disease necessitating liver transplantation. All patients were also found to have abnormal serum protein N‐glycosylation and O‐glycosylation.( 8 , 11 , 12 , 13 ) However, the cause of hepatic dysfunction is unknown.
In contrast to the phenotypes of these four V‐ATPase assembly defects, pathogenic variants in VMA21 have been associated with X‐linked myopathy with excessive autophagy (XMEA; MIM 310440) characterized by progressive vacuolisation and atrophy of skeletal muscle.( 14 , 15 , 16 , 17 , 18 ) XMEA can have a milder course with onset after the age of 5 years with slow progression( 14 , 17 , 19 ) and a prenatal/neonatal presentation with a more severe phenotype of congenital autophagic vacuolar myopathy associated with death in infancy.
Here, we present VMA21 deficiency as autophagic hepatopathy with steatohepatitis and chronic hypertransaminasemia. Mechanistically, it is caused by defective lipophagy and can be readily diagnosed by screening for abnormal glycosylation in patient sera. Our results expand the clinical spectrum of VMA21 deficiency from solely a muscle disease to an autophagic hepatopathy and indicate V‐ATPase assembly as a general mechanism that can be further studied to understand defective lipophagy in more common fatty liver disease.
Patients and Methods
Patient Materials
Blood and fibroblasts of affected individuals were obtained for diagnostics of inborn errors of metabolism and used together with parental DNA after written informed consent from parents and treating physicians, in accordance with the 1975 Declaration of Helsinki.
Glycosylation Studies
Screening for congenital disorders of glycosylation (CDG) was carried out as described.( 12 ) Plasma N‐glycan profiling was performed by matrix‐assisted laser/desorption ionization time‐of‐flight (MALDI‐TOF) mass spectrometry of permethylated glycans( 20 ) using 10 μL of plasma. High‐resolution mass spectrometry of intact transferrin was performed on a 6540 nanochip quadruple time‐of‐flight (QTOF, Agilent) according to published protocols.( 21 )
Cell Culture
Primary fibroblasts from patients and controls were grown from a skin biopsy and cultured in Dulbecco’s modified Eagle’s medium (DMEM; high glucose 4,500 mg/L, Thermo Fisher) supplemented with 10% fetal bovine serum, 1% l‐glutamine, and 1% penicillin/streptomycin at 37°C under 5% carbon dioxide. Research on patients’ cells was prospectively reviewed and approved by the Ethical Committees of the University Hospital of Nijmegen.
BODIPY, Cathepsin B, Filipin, and LysoSensor Staining
For BODIPY and filipin labeling, fibroblasts or Huh7 cells were fixed for 10 minutes in 4% paraformaldehyde and incubated with BODIPY 493/503 (2.5 μg∙mL−1, Molecular Probes) and Hoechst (0.5 μg∙mL−1) diluted in phosphate‐buffered saline (PBS)‐Triton 0.1% for 2 hours RT or with filipin (0.05 mg/mL, F9765, Sigma) in PBS for 1 hour RT.
For LysoSensor and cathepsin B (CTSB) labeling, fibroblasts were grown on MatTek glass bottom dishes and incubated for 20 minutes at 37°C with Magic Red Cathepsin assay (ImmunoChemistry Technologies, LLC) or 5 minutes at 37°C with LysoSensor Green DND‐189 (50 nM, Molecular Probes) diluted in DMEM. The probe‐containing medium was replaced with fresh medium, and the cells were observed live using a confocal microscope.
To study the effect of proton pump inhibition, fibroblasts grown on glass coverslips were incubated for 1 hour in Bafilomycin A1 (Baf A1) 100 nM (Calbiochem) diluted in prewarmed complete DMEM.
Results
Unexplained Hepatopathy With Abnormal Plasma Protein Glycosylation
Index patient 1 (P1) is a 26‐year old man whose newborn period and infancy were uneventful. During routine blood tests at the age of 3 years, he was found to have elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST). At the age of 6 years, chronic hepatitis had developed with elevated aminotransferases and high cholesterol (321 mg/dL). Wilson disease (WD) was suspected because of low serum ceruloplasmin and copper and slightly elevated urinary copper excretion (Supporting Table S1). Treatment with zinc was initiated, resulting in reduced levels of cholesterol. After 2 years, treatment was stopped because of the occurrence of pallor, anemia, and severe neutropenia, and cholesterol levels increased again. Because of continuing liver abnormalities, a liver biopsy was performed at the age of 12 years, which revealed a normal liver architecture without features of inflammation or fibrosis. However, microvesicular steatosis was found in 10%‐20% of hepatocytes (Supporting Fig. S1E), hepatocytes were irregularly shaped, and periodic acid–Schiff staining showed positive areas of glycogen (Supporting Fig. S1A‐D). Ultrastructural investigations in hepatocytes revealed the presence of lipid droplets (LDs) inside autolysosomes or lysosome‐like structures, abundant glycogen, autophagic vacuoles, and multivesicular structures similar to Niemann–Pick disease type C (NPC)( 22 ) (Fig. 1B,D). In the majority of the cells, the Golgi appeared dilated and occasionally fragmented, with accumulation of electron‐dense material within both cisterns and vesicles (Fig. 1A,B). Kupffer cells contained numerous lysosomes filled with electron‐dense material, an expanded Golgi, and large multivesicular bodies (MVB) in the cytoplasm (Fig. 1C,D). Because WD and other causes for the unexplained hepatitis, such as hemochromatosis, viral hepatitis, autoimmune hepatitis, and alpha1‐antitrypsin deficiency, were excluded, biochemical diagnostics for metabolic causes of liver abnormalities was initiated. Routine metabolic tests were normal, including amino acids, organic acids, and bile acids. However, screening for CDG as part of the routine metabolic investigations was abnormal. Analysis of N‐glycosylation by isofocusing of plasma transferring revealed a Golgi glycosylation disorder with increased disialotransferrin and trisialotransferrin (Fig. 3A). In addition, analysis of mucin O‐glycosylation by isofocusing of apolipoprotein CIII (apoCIII) was altered, showing reduced Golgi sialic acid incorporation by increased apoCIII‐1 and decreased apoCIII‐2 isoforms (Fig. 3A).
FIG. 1.

Ultrastructure of hepatocytes and Kupffer cells of a liver biopsy of CDG‐P1. (A‐B) Hepatocytes showing dilated Golgi (g), autolysosomes (a), multivesicular bodies (MVB), lipolysosomes (LP) with incorporated bubbles containing fibro‐granular material in the bile canalicular zone (bc) and lipid droplets (LD). (C,D) Kupffer cells with lysosomes (L) or multivesicular bodies (MVB) containing dense deposits.
FIG. 3.
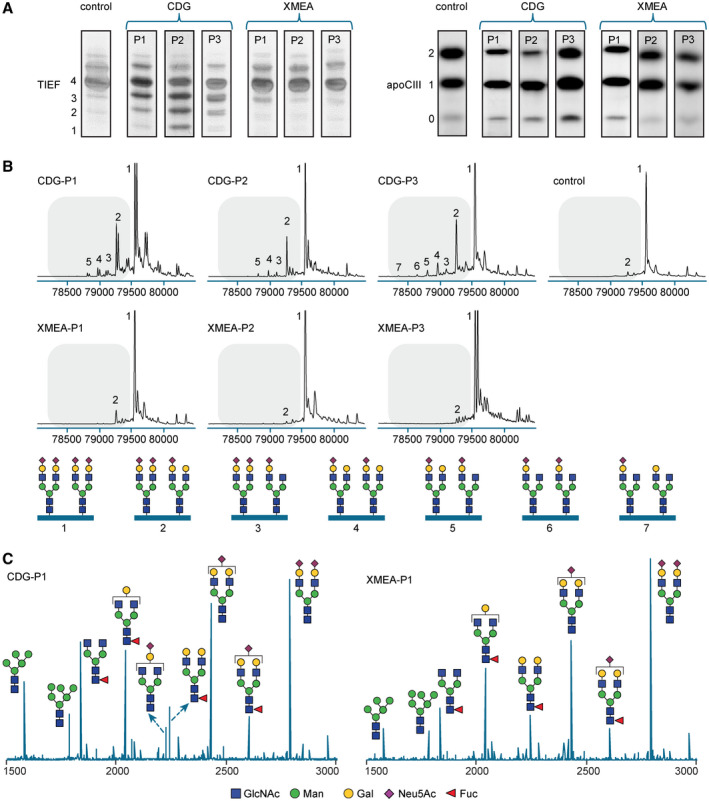
CDG screening and glycosylation analysis. (A) CDG screening results of patients with CDG and XMEA and a control. TIEF (left) was used for assaying N‐glycosylation and apoCIII isofocusing (right) was used for assaying mucin type O‐glycosylation. The accompanying numbers represent the total number of sialic acids in the different protein isoforms. (B) Nanochip‐C8‐QTOF mass spectra of transferrin isolated from patients with CDG‐P1/2/3 and XMEA‐P1/2/3 and a healthy control. Peak 1 (79,556 amu) is the intact transferrin protein with two attached complete glycans. Any subsequent loss of sialic acid and/or galactose was calculated based on the mass difference with the main peak (e.g., loss of one sialic acid [purple diamond, peak 2]). Transferrin glycoforms of the individual peaks are indicated below the profiles. Relative levels are presented in Supporting Table S5. Double peaks for CDG‐P1 and XMEA‐P3 indicate the presence of a polymorphism in the transferrin protein. (C) Representative MALDI‐TOF spectra of plasma‐derived N‐glycans of CDG‐P1 and XMEA‐P1. Spectra of all patients and a control are shown in Supporting Fig. S4. N‐Acetylglucosamine (GlcNAc), mannose (Man), galactose (Gal), N‐Acetylneuraminic acid (Neu5Ac), fucose (Fuc).
Identification of Variants in X‐Linked VMA21 Causative for a CDG
To identify genetic variants underlying the hepatopathy with abnormal glycosylation, whole exome sequencing was performed on P1. Genetic variants were ordered using a described prioritization scheme,( 23 ) by which VMA21 was identified as candidate gene. VMA21 is located on ChrXq28 and comprises three exons. The mutation c.188A>G, which is present in heterozygous form in the mother and absent in the father, results in a substitution of asparagine at position 63 for glycine (p.Asn63Gly; Fig. 2A‐C, Supporting Tables S2 and S3). p.Asn63Gly is localized in the luminal loop region of the protein just before the second predicted transmembrane region.
FIG. 2.

Identification of missense mutations in VMA21. (A) Pedigree and segregation status of VMA21 variants in families F1, F2, and F3. Partial chromatograms show X‐linked segregation for all patients. On the left side, family F1 shows the segregation of the missense mutation (c.188A>G; F1‐II‐2). In the middle, the pedigree for family F2 shows the segregation of the pathogenic mutation in the 5’ untranslated region (UTR) of VMA21 (c.‐10C>T) identified in patient P2 (F2‐II‐1). On the right side, the pedigree for family F3 shows the segregation of the missense mutation (c.52A>G) to patient P3 (F3‐II‐3). (B) Exon structure of human VMA21 (in yellow) and the domain structure of the encoded protein (in white and gray boxes), the numbers delimiting the different domains refer to amino acid positions. The blue dotted lines indicate the positions of the missense variants within the families, both at the nucleotide and the protein level (blue boxes). (C) The sequence alignment shows the conservation of the affected amino acids of VMA21 in Homo sapiens (H. sapiens), Mus musculus (M. musculus), Xenopus laevis (X. laevis), Saccharomyces cerevisiae (S. cerevisiae), and Drosophila melanogaster (D. melanogaster). The ER retrieval motif KKXX is present only in S. cerevisiae.
To identify additional patients with VMA21 variants, we selected a subgroup of undiagnosed male patients with a combination of liver symptoms and positive CDG screening results indicative of a Golgi glycosylation disorder. Sanger sequencing of VMA21 in this patient cohort resulted in the identification of genetic variants in VMA21 in two patients (P2 and P3). P2 showed a hemizygous variant in the 5′ untranslated region of VMA21 (c.‐10C>T), which was detected as heterozygous in the mother and was absent in the father, in agreement with X‐linked recessive inheritance (Fig. 2A). The c.‐10C>T mutation results in a new ATG initiation codon, 11 bp upstream of the first exon, that could lead to a frameshift and a premature stop codon at position c.26‐28 in the protein. P3 showed a hemizygous variant resulting in a predicted missense substitution of arginine for glycine (p.Arg18Gly). Analysis of complementary DNA showed the formation of a predominant alternative transcript generated by disruption of the 5′ splice donor site and use of a cryptic splice site, which resulted in the inclusion of 93 base pairs of intronic sequence. On the protein level, the new splice variant results in a premature stop codon after the arginine to glycine substitution (p.Arg18Gly*; Supporting Fig. S2A,B).
With respect to the hepatic symptoms, CDG‐P2 and CDG‐P3 showed mild signs of cholestasis that improved over time, chronic elevation of aminotransferases (ALT and AST), and elevated gamma‐glutamyltransferase (GGT) and alkaline phosphatase (Supporting Table S1). Synthetic liver function was normal in all patients, and no evidence for other causes of hepatitis, such as autoimmune hepatitis, hemochromatosis, and viral hepatitis, could be found. As in CDG‐P1, elevated total cholesterol with increased low‐density lipoprotein (LDL) cholesterol was observed repeatedly for CDG‐P3, which was accompanied by elevated secretion of apolipoprotein B (Supporting Table S4). Cholesterol levels were normal for CDG‐P2. Additional clinical signs and symptoms of patients P2 and P3 as well as all hepatic laboratory values are reported in the supporting case reports and Supporting Table S1.
With respect to glycosylation of plasma proteins, CDG‐P2 and CDG‐P3 showed glycosylation abnormalities that were highly similar to CDG‐P1. Isoelectric focusing of serum transferrin (TIEF) of all three cases showed an accumulation of undersialylated isoforms, whereas apoCIII‐IEF showed undersialylation with a slight decrease of apoCIII‐2 and increase of apoCIII‐1 isoforms (Fig. 3A). To obtain more insight into the glycosylation abnormalities, we performed high‐resolution QTOF mass spectrometry analysis of hepatocyte‐derived plasma transferrin and MALDI‐TOF analysis of total plasma‐derived N‐glycans. All three patients with CDG showed transferrin isoforms with an increase of truncated glycans lacking sialic acid and/or galactose (peaks 2‐7, Fig. 3B, Supporting Fig. S3, and Supporting Table S5). Similar abnormalities were seen in the total plasma‐derived N‐glycans of the 3 patients with CDG with a marked accumulation of a truncated glycan lacking both galactose and sialic acid at m/z 2,227 (Fig. 3C, Supporting Fig. S4). Comparison with the other V‐ATPase assembly defects ATP6AP1‐CDG, ATP6AP2‐CDG, CCDC115‐CDG, and TMEM199‐CDG reveals highly similar abnormalities of a combined N‐glycosylation and O‐glycosylation disorder and the presence of truncated glycans lacking galactose and sialic acid. Together, these results indicate VMA21 variants as the underlying cause of a CDG with hepatopathy with increased aminotransferases and LDL cholesterol as more common/subclinical features.
Comparison of VMA21‐CDG and VMA21‐XMEA Variants
Previously, VMA21 deficiency has only been associated with XMEA.( 14 ) Nevertheless, increased aminotransferases have been reported as incidental findings,( 16 , 24 ) and, recently, a described mutation has been reported in a patient with hepatic failure of unknown cause.( 25 ) Therefore, we studied liver parameters of a patient with XMEA (XMEA‐P1), which has been reported with a vacuolar myopathy because of a p.Gly91Ala missense substitution in VMA21.( 16 ) We found normal aminotransferases (ALT 54 [ref. 10‐70 U/L], AST 35 [ref. 15‐45 U/L]), increased GGT (181 [ref. 15‐115 U/L]), high cholesterol (5.8 [ref. <6 mmol/L]) with increased LDL cholesterol (4.1 [ref. <3.0 mmol/L]) and low normal ceruloplasmin (206 [ref. 160‐450 mg/L]. Creatine kinase (CK) was elevated as reported (593 [ref. 40‐280 U/L]; Supporting Table S1). Together with the finding of hypercholesterolemia and increased aminotransferases in the 3 patients with CDG, this indicates that the liver could be more frequently involved in VMA21 deficiency and that this clinical phenotype is more similar to the one of the other V‐ATPase assembly defects.( 11 , 12 , 13 ) In contrast, TIEF and apoCIII‐IEF results for 3 patients with XMEA,( 14 ) XMEA‐P1, XMEA‐P2 (c.*6A>G), and XMEA‐P3 (c.164‐7T>G) were normal. Similarly, more detailed studies on protein glycosylation by high‐resolution QTOF mass spectrometry analysis of transferrin and MALDI‐TOF analysis of total plasma‐derived N‐glycans (Fig. 3B,C, Supporting Figs. S3 and S4 and Supporting Table S5) were normal for the three patients with XMEA.
VMA21 Variants are Hypomorphic and Reduce Protein Expression
To functionally validate the VMA21 variants, we employed CDG fibroblasts and compared them with fibroblasts from a healthy control and fibroblasts from XMEA‐P1. Strongly reduced VMA21 protein expression was found for CDG‐P2 (c.10C>T) and CDG‐P3 (p.Arg18Gly), whereas CDG‐P1 (p.Asp63Gly) and XMEA‐P1 showed a milder reduction (Fig. 4A). Interestingly, analysis of mRNA expression by real‐time quantitative PCR (qPCR) showed strongly reduced levels for all patients with CDG, suggesting mRNA instability as a cause for reduced protein levels (Fig. 4B). In contrast, although the c.272C>G (XMEA) mutation was reported to abolish a predicted splice enhancer site, it only slightly reduced mRNA expression in fibroblasts (Fig. 4B).
FIG. 4.

Characterization of the VMA21 variants. (A) Western blot showing the endogenous level of VMA21 in control and patient fibroblasts using anti‐VMA21 antibodies. Results were normalized to the loading control β‐actin. The results were calculated on three independent experiments. (B) VMA21 mRNA quantification by qPCR in control and patient fibroblasts. The results were calculated on three independent experiments. (C) Western blot showing the steady‐state levels of the V1 subunits in control and patient fibroblasts using anti‐ATP6V1B1/2 and anti‐ATP6V1D1 antibodies. Results were normalized to the loading control β‐actin. The results were calculated on five and four independent experiments, respectively. (D) Western blot showing the steady‐state levels of the V0 subunits in control and patient fibroblasts using anti‐ATP6V0D1 and anti‐ATP6V0C antibodies. Results were normalized to the loading control β‐actin. The results were calculated on five and three independent experiments, respectively. All data are mean ± SEM; statistical significance was determined by ordinary one‐way ANOVA followed by a Bonferroni multiple comparisons test. Levels of significance: ns, not significant, *P < 0.05, **P < 0.01.
VMA21 Variants Impair V‐ATPase Assembly
To study the impact of VMA21 variants on the assembly of the V‐ATPase V0 and V1 domains, western blot analysis was performed. As expected, steady‐state levels of V1 subunits ATP6V1D1 and ATP6V1B1/2 in total cell extracts remained unaffected in control, CDG, and XMEA fibroblasts, confirming that the assembly of the V1 domain, which occurs independently in the cytosol,( 14 ) is not impaired by mutations in the ER assembly factor VMA21 (Fig. 4C). By contrast, expression of the V0 subunits ATP6V0D1 and ATP6V0C was reduced in fibroblasts from patients with CDG and patients with XMEA, indicating impaired V0 assembly in the ER, thus leading to an overall defect in V‐ATPase assembly (Fig. 4D).
Although any reduction in expression levels is sufficient to explain reduced assembly, we tested by overexpression whether the mutated proteins could have decreased interactions with V‐ATPase components. On transient transfection of Myc‐tagged VMA21R18G, VMA21D63G, and VMA21G91A in HEK293T cells, the levels of the three overexpressed proteins were comparable with VMA21WT (Supporting Fig. S5A,B). However, they showed reduced interaction with the assembly factor ATP6AP2 and V0 subunit ATP6V0C (Supporting Fig. S5C,D). This suggests that in addition to the lower expression levels, the missense mutations might also impair protein interactions of VMA21, interfering with proper assembly of the V‐ATPase.
VMA21 Variants Cause Reduced Proton Pump Function
The function of VMA21 is best understood in yeast cells.( 2 ) VMA21 and yeast Vma21p share 30% similarity, although VMA21 lacks the C‐terminal dilysine motif necessary for ER retrieval( 23 ) (Fig. 2B,C). To study the effect of the identified variants on yeast V‐ATPase function, we used an established yeast assay for proton pump activity that is based on the dependence of pump activity for survival and growth in the presence of elevated divalent cations, such as zinc( 2 , 26 , 27 , 28 ) (Fig. 5A). It has been shown that strains lacking a functional Vma21p are unable to grow in these nonpermissive conditions.( 29 ) Therefore, the human sequences carrying the appropriate amino acid changes were introduced by homologous recombination into the yeast genomic locus. Although the expression of VMA21WT rescued the yeast growth under elevated zinc conditions (Fig. 5A), CDG and XMEA variants impaired this rescue (Fig. 5A).
FIG. 5.

VMA21 variants cause reduced proton pump function. (A) V‐ATPase‐dependent growth tests on permissive (pH 5) or nonpermissive (4 or 5 mM ZnCl2) conditions of strains expressing VMA21WT, VMA21R18G, VMA21D63G, and VMA21G91A compared with Vma21p and ΔVma21p. (B) LysoSensor Green DND‐189 staining of patients and control fibroblasts. Results were calculated on two independent experiments with >30 cells/genotype. Scale bars: 20 µm. (C) LysoTracker staining of patient and control fibroblasts. Results were calculated on two independent experiments with >10 cells/genotype. Scale bars: 20 µm. (D) Magic Red staining for cathepsin B activity of control and patient fibroblasts, in untreated conditions or on 1 hour of bafilomycin A1 (Baf A1) 100 nM treatment. Results were calculated on 3 independent experiments with >30 cells/genotype. Scale bars: 15 µm. All data are mean ± SEM; statistical significance was determined by ordinary one‐way ANOVA followed by a Bonferroni multiple comparisons test. Levels of significance: ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To study proton pump dysfunction in patient fibroblasts, we used LysoSensor, a dye able to emit fluorescence only inside acidic cellular compartments and whose fluorescence intensity has an inverse correlation with the pH value,( 30 ) and LysoTracker, a dye able to label acidic compartments. Live fibroblasts from both CDG and XMEA‐P1 showed a strong reduction in number and intensity of both LysoSensor (Fig. 5B, Supporting Fig. S6A) and LysoTracker‐positive punctae compared with control fibroblasts (Fig. 5C, Supporting Fig. S6B). Taken together, these results indicate that human VMA21 variants reduce V‐ATPase function.
VMA21 Deficiency Leads to Autophagic Defects
To understand if impaired organellar acidification results in decreased lysosomal degradation, we tested lysosomal CTSB activity( 31 , 32 , 33 ) using a cresyl violet fluorophore fused to a peptide sequence that can be cleaved by CTSB. By live cell imaging, we observed a strong reduction in CTSB activity for all CDG and XMEA‐P1 cells (Fig. 5D, Supporting Fig. S6C). Use of V‐ATPase inhibitor bafilomycin A1 as positive control showed a similar reduction (Fig. 5D, Supporting Fig. S6C).
We next tested whether reduced lysosomal acidification and degradation also affected lysosomal morphology. Immunofluorescence showed a strongly increased overall intensity of the lysosomal marker lysosomal‐associated membrane glycoprotein 1 (LAMP1) in VMA21‐deficient fibroblasts (Fig. 6A, Supporting Fig. S7A). LAMP1‐positive vesicles also appeared to be enlarged, suggesting impairment in the turnover of these organelles (Fig. 6A, Supporting Fig. S7A). This was supported by the increased levels of LAMP1 in western blotting of fibroblasts from patients with CDG and XMEA‐P1 (Fig. 6B, Supporting Fig. S7B).
FIG. 6.

VMA21 deficiency leads to autophagic defects. (A) Immunofluorescence analyses of control and patient fibroblasts showing LAMP1‐positive vesicles. Scale bars: 20 µm. Lower panels are magnifications of the insets demarked in upper panels. Scale bars: 10 µm. (B,C) Western blot of LAMP1, p62, and LC3 in control and patient fibroblasts. Results were normalized to the loading control β‐actin. The results were calculated on three, two, and two independent experiments, respectively. (D) LAMP1 and LC3 staining in control and patient fibroblasts. Scale bars: 20 µm. Lower panels are magnifications of the insets demarked in upper panels. Scale bars: 20 µm. The analysis of the LAMP1/LC3 colocalization was performed by using the JACoP plug‐in (ImageJ) and quantified by using Pearson’s colocalization coefficient. The colocalization between LAMP1 and LC3 was calculated on 3 independent experiments with 15 cells/genotype. All data are mean ± SEM; statistical significance was determined by ordinary one‐way ANOVA followed by a Bonferroni multiple comparisons test. Levels of significance: ns, not significant, *P < 0.05, **P < 0.01.
With regard to the autophagy pathway, we found an accumulation of the autophagosome‐associated lipidated form of microtubule‐associated protein 1 light chain 3 (LC3‐II) for the patients with CDG and XMEA‐P1 (Fig. 6C, Supporting Fig. S7C). Furthermore, we found an accumulation of the autophagosomal substrate p62, suggesting decreased autophagic degradation because of a block in autophagic flux (Fig. 6B, Supporting Fig. S7B).
LAMP1 punctae showed an extensive colocalization with light chain 3 (LC3) in both control and patients fibroblasts, indicating that autophagosomes were able to fuse with ATP–deficient lysosomes (Fig. 6D, Supporting Figs. S7D and S8G). Similar to studies in Drosophila fat bodies,( 34 ) the accumulation of nonfunctional lysosomes led to a block in autophagic flux and the formation of giant autolysosomes (Fig. 6A, Supporting Figs. S7A and S8B). Importantly, small interfering RNA–mediated knockdown of VMA21 in human Huh7 hepatocytes was able to recapitulate the LAMP1, p62, and LC3 accumulation, confirming the hypomorphic effects of the VMA21 variants on the lysosomal‐autophagic pathway (Supporting Fig. S8A‐D).
VMA21 Variants Lead to ER Stress and Lipid Abnormalities in Patient Fibroblasts
Because of the steatotic liver phenotypes and accumulation of intracellular LDs observed in patient hepatocytes, we next tested whether impaired lysosomal degradation could be the cause of LD accumulation. LDs are cytoplasmic organelles that have a core of triglycerides and cholesterol surrounded by a single layer of phospholipids.( 35 ) LDs are formed in the ER, where triglycerides are synthesized from fatty acids.( 36 ) LD degradation can either occur through cytoplasmic lipases( 37 ) or in the context of autophagy (lipophagy).( 38 )
The number and size of LDs was assessed by BODIPY staining in patient and control fibroblasts. CDG and XMEA cells showed a clear accumulation of LDs compared with the control fibroblasts (Fig. 7A, Supporting Figs. S8F and S9A). Importantly, treatment with bafilomycin A1 increased the LD number in control fibroblasts (Fig. 7A, Supporting Figs. S8F and S9A), confirming that lysosomal acidification is key to LD turnover. By contrast, bafilomycin A1 treatment in patient fibroblasts did not further decrease the LD number, suggesting that in these cells an additional suppression of acidification‐dependent LD turnover may not be possible. Interestingly, the increase in LDs was also accompanied by an increase of total triglycerides in patient cells (Fig. 7C, Supporting Fig. S9C).
FIG. 7.

VMA21 mutations cause impaired lipid metabolism. (A) BODIPY staining of LDs in control and patient fibroblasts. The staining was performed in untreated samples or on 1 hour 100 nM bafilomycin A1. The results were calculated on 3 independent experiments with >10 cells/genotype. Scale bars: 15 µm. (B) Immunolabeling of unesterified cholesterol by filipin staining in control and patient fibroblasts. The intensity of the staining was calculated on three independent experiments with >10 cells/genotype. Scale bars: 20 µm. (C) Total triglyceride (TAG) levels assayed enzymatically in control and patient fibroblasts. The results were calculated on three independent experiments. All data are mean ± SEM; statistical significance was determined by ordinary one‐way ANOVA followed by a Bonferroni multiple comparisons test. Levels of significance: ns, not significant, *P < 0.05, ***P < 0.001. ****P < 0.0001.
In the lysosomal storage disease NPC, cholesterol accumulates inside the lysosomes because of defective extraction through the NPC1 and NPC2 proteins.( 39 ) As lysosomal cholesterol extraction occurs in a pH‐dependent manner,( 40 ) we tested whether the hypercholesterolemia observed in the patients with CDG could also be linked to decreased lysosomal acidification. Therefore, we labeled unesterified cholesterol with the filipin dye. As expected, the staining revealed an accumulation of cholesterol in vesicular structures of CDG fibroblasts compared with control cells, whereas the XMEA fibroblasts showed only a small tendency to accumulate cholesterol (Fig. 7B, Supporting Figs. S8E and S9B). Similar to the LDs, pretreatment of the cells with the autophagy inhibitor bafilomycin A1 led to accumulation of cholesterol in both control and XMEA fibroblasts but not in CDG cells (Fig. 7B, Supporting Figs. S8E and S9B).
When cholesterol is sequestered in lysosomes, the cholesterol content in the ER may be lower, which in turn can trigger lipogenic pathways by cleavage of the sterol response element‐binding protein‐1 (SREBP1).( 39 , 41 ) To test whether such de novo lipogenic pathways could be responsible for the observed hypercholesterolemia in the VMA21 patients, SREBP1 activation and cleavage was analyzed by western blotting. This revealed an increased amount of mature SREBP1 in CDG but not in control and XMEA fibroblasts (Fig. 8A, Supporting Fig. S10A). Taken together, this suggests that VMA21 deficiency leads to LD and cholesterol accumulation inside lysosomal‐like structures, contributing to elevated plasma (LDL) cholesterol by increasing de novo SREBP1‐mediated lipogenesis.
FIG. 8.

Characterization of the lipid abnormalities in patient fibroblasts. (A) Western blot on control and patient fibroblasts showing the full‐length (precursor) and cleaved form (mature) of SREBP1. Results were normalized to the loading control α‐tubulin. The results are representative of three independent experiments. (B) Western blot of total PERK (Tot PERK) and phosphorylated (Thr980) PERK (pPERK) in control and patient fibroblasts. Results were normalized to the loading control β‐actin. The results were calculated in three independent experiments. (C) ATF4 and CHOP mRNA quantification in control and patient fibroblasts. The results are representative of three independent experiments. (D) Immunofluorescence analyses of control and patient fibroblasts showing GRP94 staining in patient and control fibroblasts. The results are representative of three independent experiments. Scale bars: 15 µm. (E) GRP94 mRNA quantification in control and patient fibroblasts. The results were calculated on three independent experiments. All data are mean ± SEM, statistical significance was determined by ordinary one‐way ANOVA followed by a Bonferroni multiple comparisons test. Levels of significance: ns, not significant, *P < 0.05, **P < 0.01, ****P < 0.0001.
CDG Mutations Cause Protein Kinase R–Like ER Kinase–Mediated ER Stress
As defective V0 assembly has also been associated with ER stress, which is related to LD formation in hepatocytes,( 9 , 42 ) we studied ER stress in CDG cells. We tested all three unfolded protein response (UPR) stress pathways (Supporting Fig. S11A‐E) but only found a strong activation of the protein kinase R–like ER kinase (PERK) branch in the CDG but not in control or XMEA fibroblasts. This was demonstrated by the autophosphorylation of PERK on Thr980 (Fig. 8B, Supporting Fig. S10B). PERK activation led to the up‐regulation of several UPR target genes, including the activating transcription factor 4 (ATF4), CCAAT/enhancer binding protein homologous protein (CHOP), and glucose‐regulated protein 94 (GRP94), as demonstrated by qPCR and immunostaining (Fig. 8C‐E, Supporting Fig. S10C‐E). Together, these results suggest that ER stress because of V‐ATPase misassembly may contribute to the steatotic phenotype in the patients.
Discussion
Here, we identified deficiency of V‐ATPase assembly factor VMA21 leading to a heterogeneous clinical spectrum with hepatic features of chronic hypertransaminasemia and mild hyperlipidemia with increased LDL cholesterol. All individuals displayed abnormal glycosylation of hepatocyte‐derived proteins, suggesting that the combination of liver symptoms and CDG should guide clinicians toward genetic testing of VMA21 and other V‐ATPase assembly factors.
Our functional studies demonstrated that both VMA21‐CDG and VMA21‐XMEA variants are hypomorphic mutations lowering mRNA and protein levels. As a consequence of VMA21 reduction, there was an impairment of V‐ATPase assembly, which was marked by reduced V0 subunit expression and reduced interaction with V0 subunit themselves and the ER assembly factor ATP6AP2, that led to reduced V‐ATPase activity. This in turn caused reduced lysosomal acidification and protease activation as well as the inability to execute the final steps of the (auto)lysosomal degradation pathway. In fibroblasts, the impaired lysosomal acidification led to defective lipophagy, consistent with the presence of enlarged LD‐containing autolysosomes in the hepatocytes of the liver biopsy of CDG‐P1.
The observed cholesterol accumulation in patient fibroblasts may have been caused by reduced lysosomal acidification. Interestingly, it has been demonstrated that the activity of lysosomal protease CTSB in the lysosomal storage disorder NPC can be impaired by both altered lysosomal pH and the accumulation of lipid material inside lysosomes.( 43 ) In analogy, the accumulated cholesterol in VMA21‐deficient cells may impair lysosomal function. As we additionally observed an increased cleavage of SREBP1, it can be hypothesized that the failure to properly extract cholesterol from the lysosomes leads to an overall decline of cellular cholesterol, which in turn triggers SREBP1 cleavage and de novo production of cholesterol.( 40 ) As newly synthesized cholesterol can directly be secreted into the serum together with lipoprotein particles, the hypercholesterolemia observed in the patients may be a result of SREBP1 activation and de novo cholesterol synthesis. Interestingly, the PERK branch of the UPR, which was shown to be activated in the CDG cells, is also able to activate SREBP1‐mediated de novo lipogenesis,( 42 , 47 ) suggesting that the ER stress responses might further contribute to cholesterol accumulation. This could also be the case for the LD accumulation, as increased LD formation has been linked to ER stress in many instances, particularly in hepatocytes.( 46 )
What causes the ER stress in VMA21‐deficient cells is unclear. Lack of ATP6AP2, another ER assembly factor, has been associated with ER stress,( 9 ) suggesting that it may be directly related to the defective V0 assembly.( 23 , 47 ) However, in plants, it has also been shown that VMA21 interacts with and helps ER‐derived vesicles protein 41‐46 (Erv41p‐Erv46p),( 42 ) an early secretory‐localized complex involved in the protein loading into coat protein complex II vesicles for transport to the Golgi. Lack of Vma21p in ∆Vma21p strains resulted in a complete absence of the vesicle specific SNAP receptor Bos1p in the COPII vesicles,( 42 ) suggesting an impaired ER‐to‐Golgi transport also in fibroblasts from patients with CDG. Likewise, because of the involvement of Vma21p in retrograde transport back to the ER, disturbance of ER and Golgi homeostasis might also stem from defects in this trafficking pathway.
Although the phenotypes of patients with XMEA and patients with CDG are different,( 14 , 15 ) we found mild differences in patient fibroblasts. Both CDG and XMEA variants showed reduced protein expression, V‐ATPase misassembly, and dysfunction, whereas some differences were seen with regard to ER stress and cholesterol impairment. Given that liver involvement has already been described in patients with XMEA, ranging from increased liver aminotransferases to fatal hepatic failure,( 16 , 24 , 25 ) whereas CK elevation could be observed in one of the patients with CDG (P2), it seems clear that more patients are needed to fully appreciate the phenotypic spectrum of VMA21 deficiency. The elevated LDL cholesterol levels in XMEA‐P1 suggests further mechanistic overlap between the CDG and XMEA variants. Nevertheless, glycosylation abnormalities were not detected in patients with XMEA, and the CDG variants had a stronger impact on VMA21 expression and PERK phosphorylation than the XMEA mutation tested. Therefore, it could be that stronger reduction of VMA21 correlates with a stronger effect on ER stress and glycosylation and that these processes seem to be more sensitive to VMA21 protein dosage compared with the lysosomal acidification. Tissue‐specific expression of modulator genes, alternative transcripts of VMA21, or differential expression levels of V‐ATPase assembly factors need to be investigated to further explain the tissue‐specific clinical differences between the patient groups.
VMA21 deficiency is the last of five known human V‐ATPase assembly factors associated with hepatic steatosis.( 8 , 11 , 12 , 13 ) In some cases, this new disease class progresses to severe fatty liver disease. Our findings of defective lipophagy as a mechanism underlying LD accumulation are therefore of broader relevance to understand the pathophysiology of hepatic steatosis in the other V‐ATPase assembly defects but also bear significant relevance to understand the pathogenesis of nonalcoholic fatty liver disease (NAFLD).( 32 ) As LD accumulation is a hallmark of NAFLD and autophagy has been implicated in disease pathology and as being a treatment target for NAFLD, understanding the mechanisms underlying defective lipophagy is important.
In summary, we identify three individuals with a CDG featured by autophagic defects and ER stress caused by different variants in VMA21. Our data suggest X‐linked VMA21 deficiency as a cause of mild chronic liver disease. It is thus advised that patients with signs of steatohepatitis and chronically elevated aminotransferases are tested for lipid profiles and plasma protein glycosylation, followed by genetic testing of the V‐ATPase assembly factors. A better understanding of how ER stress and autophagic defects contribute to liver disease in these rare diseases may aid the development of new therapeutic strategies for NAFLD. Therefore, our work underscores the notion that insights from rare genetic diseases can hold important lessons for common diseases and indicate treatment targets for both.
Author Contributions
M.C.S. designed (with the help of G. L. M.), performed, and analyzed the experiments in patient fibroblasts, HEK293T, and Huh7 cells. L.A.G., M.R., and T.H.S. performed yeast studies and analyzed and interpreted the data. A.A. and S.T. performed and analyzed genetic and biochemical studies in plasma and fibroblasts. K.R. and M.H. performed glycosylation screening. E.C. performed and interpreted ultrastructural studies. L.E.L., W.A.R.O., J.W.J., and A.G.H. performed and analyzed the blood lipids and lipoproteins on patients’ sera. J.C.J., C.F., L.H., P.P., E.M., and P.S. performed clinical evaluation. C.G., R.R., and J.A.V. performed and supervised genetic studies. M.S. and D.J.L. conceived and supervised the project and analyzed the data. M.S., M.C.S. and D.J.L. wrote the paper. All authors contributed to parts of the paper on their specific expertise and critically reviewed the paper.
Supporting information
Supplementary Material
Acknowledgment
We thank Hannu Kalimo, Berge Minassian, and Alessandra Ruggieri for providing plasma samples from the 3 patients with XMEA. We are grateful to Paul Saftig for providing antibodies. We thank Anne‐Marije Pennekamp, Han Levels, and Aldo Grefhorst for their analyses of fast protein liquid chromatography and Selectra lipid profiles of the involved patients.
This work was financially supported by the Radboudumc (grant to D.L., R.R., and J.V.), grants from the Dutch Organisation for Scientific Research (ZONMW‐NWO; Medium Investment Grant 40‐00506‐98‐9001 and VIDI Grant 91713359 to D.J.L.), the National Institutes of Health Grant GM 38006 (to T.H.S.), the ATIP‐Avenir program, and the Fondation Bettencourt‐Schueller (Liliane Bettencourt Chair of Developmental Biology) as well as state funding by the Agence Nationale de la Recherche (ANR) under the “Investissements d’avenir” program (ANR‐10‐IAHU‐01) and the NEPHROFLY (ANR‐14‐ACHN‐0013) grant (to M.S.). A.G.H. is supported by the Gilead Research scholarship grant, the Amsterdam UMC Fellowship grant, and a Health~Holland TKI‐PPP grant. J.W.J. is supported by grants from The Netherlands Organization for Scientific Research (VICI grant 016.176.640 to J.W.J.) and European Foundation for the Study of Diabetes (award supported by EFSD/Novo Nordisk).
In addition, financial support was obtained from EUROGLYCAN‐omics (ERARE18‐117) under the frame of E‐Rare‐3, the ERA‐Net for Research on Rare Diseases.
Potential conflict of interest: Nothing to report.
Contributor Information
Alessandra Rugierri, Email: alessandra.ruggieri@istituto-besta.it.
Matias Simons, Email: matias.simons@med.uni-heidelberg.de.
Dirk J. Lefeber, Email: Dirk.Lefeber@radboudumc.nl.
References
Author names in bold designate shared co‐first authorship.
- 1. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007;8:917‐929. http://www.nature.com/articles/nrm2272 [DOI] [PubMed] [Google Scholar]
- 2. Graham LA, Flannery AR, Stevens TH. Structure and assembly of the yeast V‐ATPase. J Bioenerg Biomembr 2003;35:301‐312. [DOI] [PubMed] [Google Scholar]
- 3. Malkus P, Graham LA, Stevens TH, Schekman R. Role of Vma21p in assembly and transport of the yeast vacuolar ATPase. Mol Biol Cell 2004;15:5075‐5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davis‐Kaplan SR, Compton MA, Flannery AR, Ward DM, Kaplan J, Stevens TH, et al. PKR1 encodes an assembly factor for the yeast V‐type ATPase. J Biol Chem 2006;281:32025‐32035. [DOI] [PubMed] [Google Scholar]
- 5. Hill KJ, Stevens TH. Vma22p is a novel endoplasmic reticulum‐associated protein required for assembly of the yeast vacuolar H(+)‐ATPase complex. J Biol Chem 1995;270:22329‐22336. [DOI] [PubMed] [Google Scholar]
- 6. Hirata R, Umemoto N, Ho MN, Ohya Y, Stevens TH, Anraku Y. VMA12 is essential for assembly of the vacuolar H(+)‐ATPase subunits onto the vacuolar membrane in Saccharomyces cerevisiae . J Biol Chem 1993;268:961‐967. [PubMed] [Google Scholar]
- 7. Ryan M, Graham LA, Stevens TH. Voa1p functions in V‐ATPase assembly in the yeast endoplasmic reticulum. Mol Biol Cell 2008;19:5131‐5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jansen EJR, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun 2016;27:11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guida MC, Hermle T, Graham LA, Hauser V, Ryan M, Stevens TH, et al. ATP6AP2 functions as a V‐ATPase assembly factor in the endoplasmic reticulum. Mol Biol Cell 2018;29:2156‐2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miles AL, Burr SP, Grice GL, Nathan JA. The vacuolar‐ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1α prolyl hydroxylation by regulating cellular iron levels. eLife 2017;15:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jansen JC, Timal S, van Scherpenzeel M, Michelakakis H, Vicogne D, Ashikov A, et al. TMEM199 deficiency is a disorder of golgi homeostasis characterized by elevated aminotransferases, alkaline phosphatase, and cholesterol and abnormal glycosylation. Am J Hum Genet 2016;98:322‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jansen JC, Cirak S, van Scherpenzeel M, Timal S, Reunert J, Rust S, et al. CCDC115 deficiency causes a disorder of golgi homeostasis with abnormal protein glycosylation. Am J Hum Genet 2016;98:310‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rujano MA, Cannata Serio M, Panasyuk G, Péanne R, Reunert J, Rymen D, et al. Mutations in the X‐linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J Exp Med 2017;214:3707‐3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol (Berl) 2013;125:439‐457. [DOI] [PubMed] [Google Scholar]
- 15. Crockett CD, Ruggieri A, Gujrati M, Zallek CM, Ramachandran N, Minassian BA, et al. Late adult‐onset of X‐linked myopathy with excessive autophagy. Muscle Nerve 2014;50:138‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saraste A, Koskenvuo JW, Airaksinen J, Ramachandran N, Munteanu I, Udd B, et al. No cardiomyopathy in X‐linked myopathy with excessive autophagy. Neuromuscul Disord 2015;25:485‐487. [DOI] [PubMed] [Google Scholar]
- 17. Munteanu I, Ramachandran N, Ruggieri A, Awaya T, Nishino I, Minassian BA. Congenital autophagic vacuolar myopathy is allelic to X‐linked myopathy with excessive autophagy. Neurology 2015;84:1714‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munteanu I, Kalimo H, Saraste A, Nishino I, Minassian BA. Cardiac autophagic vacuolation in severe X‐linked myopathy with excessive autophagy. Neuromuscul Disord 2017;27:185‐187. [DOI] [PubMed] [Google Scholar]
- 19. Mercier S, Magot A, Caillon F, Isidor B, David A, Ferrer X, et al. Muscle magnetic resonance imaging abnormalities in X‐linked myopathy with excessive autophagy. Muscle Nerve 2015;52:673‐680. [DOI] [PubMed] [Google Scholar]
- 20. Morelle W, Michalski J‐C. Analysis of protein glycosylation by mass spectrometry. Nat Protoc 2007;2:1585‐1602. [DOI] [PubMed] [Google Scholar]
- 21. van Scherpenzeel M, Steenbergen G, Morava E, Wevers RA, Lefeber DJ. High‐resolution mass spectrometry glycoprofiling of intact transferrin for diagnosis and subtype identification in the congenital disorders of glycosylation. Transl Res J Lab Clin Med 2015;166:639‐649.e1. [DOI] [PubMed] [Google Scholar]
- 22. Pacheco CD, Lieberman AP. The pathogenesis of Niemann‐Pick type C disease: a role for autophagy? Expert Rev Mol Med 2008;10:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hill KJ, Stevens TH. Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di‐lysine motif and is required for the assembly of the vacuolar H(+)‐ATPase complex. Mol Biol Cell 1994;5:1039‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruggieri A, Ramachandran N, Wang P, Haan E, Kneebone C, Manavis J, et al. Non‐coding VMA21 deletions cause X‐linked myopathy with excessive autophagy. Neuromuscul Disord 2015;25:207‐211. [DOI] [PubMed] [Google Scholar]
- 25. Ackerley CA, Cooper MA, Munoz DG, Minassian BA. Fatal hepatic failure and pontine and extrapontine myelinolysis in XMEA. Neurology 2016;87:1417‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klionsky DJ, Herman PK, Emr SD. The fungal vacuole: composition, function, and biogenesis. Microbiol Rev 1990;54:266‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eide DJ, Clark S, Nair TM, Gehl M, Gribskov M, Guerinot ML, et al. Characterization of the yeast ionome: a genome‐wide analysis of nutrient mineral and trace element homeostasis in Saccharomyces cerevisiae . Genome Biol 2005;6:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kane PM. The where, when, and how of organelle acidification by the yeast vacuolar H+‐ATPase. Microbiol Mol Biol Rev 2006;70:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ho MN, Hill KJ, Lindorfer MA, Stevens TH. Isolation of vacuolar membrane H(+)‐ATPase‐deficient yeast mutants; the VMA5 and VMA4 genes are essential for assembly and activity of the vacuolar H(+)‐ATPase. J Biol Chem 1993;268:221‐227. [PubMed] [Google Scholar]
- 30. Zhou J, Tan S‐H, Nicolas V, Bauvy C, Yang N‐D, Zhang J, et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome‐lysosome fusion. Cell Res 2013;23:508‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sloane BF. Cathepsin B and cystatins: evidence for a role in cancer progression. Semin Cancer Biol 1990;1:137‐152. [PubMed] [Google Scholar]
- 32. Mao Y, Yu F, Wang J, Guo C, Fan X. Autophagy: a new target for nonalcoholic fatty liver disease therapy. Hepatic Med Evid Res 2016;8:27‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest 2010;120:3421‐3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome‐lysosome fusion is independent of V‐ATPase‐mediated acidification. Nat Commun 2015;11:7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leung‐Toung R, Li W, Tam TF, Karimian K. Thiol‐dependent enzymes and their inhibitors: a review. Curr Med Chem 2002;9:979‐1002. [DOI] [PubMed] [Google Scholar]
- 36. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 2019;20:137‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yen C‐LE, Stone SJ, Koliwad S, Harris C, Farese RV. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 2008;49:2283‐2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 2011;121:2102‐2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vance JE, Karten B. Niemann‐Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res 2014;55:1609‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deffieu MS, Pfeffer SR. Niemann‐Pick type C 1 function requires lumenal domain residues that mediate cholesterol‐dependent NPC2 binding. Proc Natl Acad Sci U S A 2011;108:18932‐18936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Welsh LM, Tong AHY, Boone C, Jensen ON, Otte S. Genetic and molecular interactions of the Erv41p‐Erv46p complex involved in transport between the endoplasmic reticulum and Golgi complex. J Cell Sci 2006;119(Pt 22):4730‐4740. [DOI] [PubMed] [Google Scholar]
- 43. Elrick MJ, Lieberman AP. Autophagic dysfunction in a lysosomal storage disorder due to impaired proteolysis. Autophagy. 2013;9:234‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bobrovnikova‐Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, et al. PERK‐dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci U S A 2008;105:16314‐16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 1997;89:331‐340. https://linkinghub.elsevier.com/retrieve/pii/S0092867400802135 [DOI] [PubMed] [Google Scholar]
- 46. Rutkowski DT. Liver function and dysfunction—a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J 2019;286:356‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Esmail S, Kartner N, Yao Y, Kim JW, Reithmeier RAF, Manolson MF. N‐linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H+ ‐ATPase (V‐ATPase) biosynthesis. J Cell Biochem 2018;119:861‐875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material