

Abstract
Previous studies have reported robust inflammatory cell infiltration, synthesis of IgG, B-cell clonal expansion, deposition of immune complexes and complement within cerebral cavernous malformation (CCM) lesions. B-cell depletion has also been shown to reduce the maturation of CCM in murine models. We hypothesize that antigen(s) within the lesional milieu perpetuate the pathogenetic immune responses in CCMs. This study aims to identify those putative antigen(s) using monoclonal antibodies (mAbs) derived from plasma cells found in surgically removed human CCM lesions. We produced human mAbs from laser capture micro-dissected plasma cells from four CCM patients, and also germline-reverted versions. CCM mAbs were assayed using immunofluorescence on central nervous system (CNS) tissues and immunocytochemistry on human primary cell lines. Antigen characterization was performed using a combination of confocal microscopy, immunoprecipitation and mass spectrometry. Affinity was determined by enzyme-linked immunosorbent assay, and specificity by multi-color confocal microscopy and quantitative co-localization. CCM mAbs bound CNS tissue, especially endothelial cells and astrocytes. Non-muscle myosin heavy chain IIA (NMMHCIIA), vimentin and tubulin are three cytoskeleton proteins that were commonly targeted. Selection of cytoskeleton proteins by plasma cells was supported by a high frequency of immunoglobulin variable region somatic hypermutations, high affinity and selectivity of mAbs in their affinity matured forms, and profoundly reduced affinity and selectivity in the germline reverted forms. Antibodies produced by plasma cells in CCM lesions commonly target cytoplasmic and cytoskeletal autoantigens including NMMHCIIA, vimentin and tubulin that are abundant in endothelial cells and astrocytes. Binding to, and selection on autoantigen(s) in the lesional milieu likely perpetuates the pathogenetic immune response in CCMs. Blocking this in situ autoimmune response may yield a novel treatment for CCM.
Keywords: Cerebral cavernous malformation, Recombinant monoclonal antibody, Autoimmunity, Cytoskeleton proteins
1. Introduction
Cerebral cavernous malformations (CCMs) are common cerebrovascular malformations predisposing patients to a lifetime risk of seizures, symptomatic hemorrhage and focal neurological deficits [1]. Two forms of CCM disease exist: sporadic and familial. The sporadic form typically manifests a solitary lesion that is clustered with a developmental venous anomaly. The familial form harbors multifocal lesions throughout the brain and has been associated with autosomal dominant germline mutations in one of three CCM genes (CCM1/KRIT1, CCM2/OSM or CCM3/PDCD10). No histological differences exist between sporadic and familial CCM lesions. The lesions consist of multiple caverns lined by a layer of endothelium with defective inter-cellular junctions and basal lamina, and scant pericytes [2, 3], leading to a leaky blood-brain barrier [3, 4]. Lesions exhibit hallmarks of blood leakage and gliotic reaction. It has been suggested that these features result in a milieu suited to antigenic challenge and a local adaptive immune response, which in turn contributes to lesion proliferation and clinical manifestations [5, 6].
Animal models with Ccm gene loss have significantly contributed to the understanding of the disease pathobiology. The same inflammatory cell infiltrates were documented in murine chronic models as human [7]. Our recent transcriptomic analysis indicated that an immune response was a prominent feature in chronic murine CCM models [8]. Moreover, we have shown that B-cell depletion in murine models impedes the development and maturation of CCMs [9]. Our previous studies revealed that robust immune cell infiltration, as well as membrane attack complex co-localization with IgG, associated with the CCM phenotype [10]. Both familial and sporadic cases, regardless of history of recent clinically overt hemorrhage and/or recent growth, show prominent antibody producing cells (B-cells and plasma cells) [11]. Furthermore, cDNA analyses from laser-capture-microdissection isolated plasma cells was consistent with oligoclonal IgG expansion in CCMs [10]. These data indicate an in situ antigen driven lymphocyte activation and the involvement of these cells and their progeny antibodies in CCM lesion inflammation.
The putative antigens driving in situ adaptive immunity in CCM lesional tissue have not been identified. We hypothesize that antigens in tissue adjacent to the defective CCM lesional endothelium contribute to the immune response in situ. The purpose of our study was to use monoclonal antibodies (mAbs) derived from sequences of immunoglobulin transcripts from plasma cells within human CCM lesions to identify targeted antigens.
2. Materials and Methods
2.1. Patient samples
Surgically removed CCM specimens were collected with informed consent under a protocol approved by the University of Chicago Institutional Review Board. These CCM specimens were collected and fresh frozen in optimal cutting temperature (OCT, ThermoFisher Scientific, Waltham, MA). Presence of CCM disease was confirmed by surgical pathology diagnosis at the University of Chicago Medicine by a neuropathologist.
2.2. Laser capture microdissection and CCM lesion-associated immunoglobulin cloning
OCT embedded CCM tissues were cryosectioned and mounted on membrane slides (Leica Microsystems, Wetzler, Germany), fixed in acetone, washed in phosphate buffered saline (PBS), blocked by DAKO dual endogenous enzyme block (Agilent Technologies, Santa Clara, CA), stained by anti-CD138 antibody (ThermoFisher Scientific), developed and visualized using Envision Detection Systems peroxidase/DAB (Agilent Technologies). Individual CD138 positive cells were isolated by using an LMD 6500 laser capture microdissection system (Leica Microsystems) and collected in tubes containing 10 μl of SuperScript IV first strand buffer (ThermoFisher Scientific), and 20 U RNasin ribonuclease inhibitors (Promega, Madison, WI, USA). Cell lysis was carried out with 20 μl SuperScript IV first strand buffer containing 1% IGEPAL CA-630 (MilliporeSigma, Burlington, MA, USA), and RNA was reverse transcribed by using a SuperScript IV Synthesis System kit (ThermoFisher Scientific).
Cloning of immunoglobulin variable region genes was performed as previously described [12]. Briefly, variable gene segments of the immunoglobulin heavy (γ) and light (κ/λ) chains were separately amplified from the cDNA of the same single cell and run through agarose gel electrophoresis to confirm amplification of a product length consistent with IgG variable regions. Cloned light chain PCR products were inserted into expression vectors containing Igκ or Igλ sequences as previously described. The Igγ chain expression vector (FJ475055), into which the heavy chain variable region sequence was inserted, was modified from the original plasmid (as described by Kinloch et al. [13]) by inserting a nucleotide sequence (GACTACAAAGACGATGACGACAAG) immediately 5’ to the stop codon, yielding a C-terminal FLAG tag on each translated heavy chain. The heavy chain variable region sequences from four CCM lesion-derived antibody variable region pairs previously generated [10] were also subcloned into the new FLAG-heavy chain vector. All respective variable region pairings were expressed as CCM lesion derived recombinant human FLAG tag expressing monoclonal antibodies (“CCM-rhFLAG-mAb”).
2.3. Immunoglobulin gene sequence analysis and germline-reverted antibody
Sequence analysis of cloned Ig heavy chain and light chain genes was performed using the international ImMunoGeneTics information system (IMGT) V-QUEST freeware (http://www.imgt.org/IMGT_vquest). Ig variable region gene segment usage, CDR3 length and V gene segment somatic hypermutation was first determined. Replacement and silent mutation frequencies in framework regions (FWRs) and complementarity-determining regions (CDRs) were calculated for each region based on the absolute number of nucleotides in all analyzed sequences. The germline sequences of the antibodies prior to somatic hypermutation were predicted using V-QUEST module of IMGT based on the closest matching germline V-D-J (heavy chain) and V-J (light chain) sequences. The variable regions of the predicted germline sequence were synthesized (GenScript, Piscataway, NJ) and subcloned into respective expression heavy and light chain expression vectors. Insertion of variable region sequences was verified by sequencing as previously described [12].
2.4. Cell Culture
HEK293A cells (ThermoFisher Scientific) were maintained in DMEM (ThermoFisher Scientific), 10% fetal bovine serum (FBS) (ThermoFisher Scientific), 0.1 mM MEM non-essential amino acids (ThermoFisher Scientific) and subcultured every 3–5 days. Cells were used below 30 passages. Primary human brain microvascular endothelial cells (HBMEC) isolated from human brain tissue (ScienCell Research Laboratories, Carlsbad, CA) were cultured in endothelial cell growth medium (ECM, ScienCell) containing 5% FBS, 1% endothelial cell growth supplement and 100 units/ml penicillin/streptomycin in poly-L-lysine coated culture vessels (Corning Life Science, Tewksbury, MA). HBMEC cells were subcultured every 3 to 4 days and used between passages 5 and 10. Primary human cortical astrocytes isolated from fetal cerebral cortex (ScienCell) were cultured in astrocyte medium (ScienCell) containing 2% FBS, 1% astrocyte growth supplement and 100 units/ml penicillin/streptomycin in poly-L-lysine coated culture vessels and subcultured every 3 and 4 days and used between passages 5 and 10. All cells were cultured in a 37°C, 5% CO2 incubator.
2.5. Transfection and production of recombinant human FLAG-tagged IgG monoclonal antibody
Antibody heavy and light chain pairings (in respective expression vectors) were cotransfected into HEK-293A cells and purified from supernatants using protein A agarose (ThermoFisher Scientific) as CCM-rhFLAG-mAbs using the method previously described [12].
2.6. Immunofluorescent staining
Resected CCM lesions were embedded in OCT (ThermoFisher Scientific), sectioned at 5 μm, and mounted on positively charged slides (ThermoFisher Scientific). Tissue sections were fixed in acetone at −20°C for 15 minutes, washed with PBS, and permeabilized with 0.5% Nonidet P40 in PBS. Following blocking in 10% normal donkey serum (NDS, Jackson ImmunoResearch, West Grove, PA) diluted in PBS, tissue sections were incubated with CCM-rhFLAG-mAbs, sheep anti-human CD31 antibody (AF806, R&D Systems, Minneapolis, MN) and mouse anti-glial fibrillary acidic protein (GFAP) antibody (clone GA5, Cell Signaling, Danvers, MA) diluted in PBS/5% NDS. Slides were washed with PBS, and incubated with a rat anti-FLAG antibody (clone L5, BioLegend, San Diego, CA, USA). Following 1% NDS in PBS wash, slides were then incubated with Alexa Flour 488-conjugated donkey anti-rat antibody (Jackson ImmunoResearch) to detect the anti-FLAG antibody. Alexa Flour 594-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch) was added to detect anti-GFAP antibody, and Alexa Flour 647-conjugated donkey anti-sheep antibody (Jackson ImmunoResearch) to detect anti-CD31 antibody. Counter staining for double stranded DNA was performed with Hoechst 33342 (ThermoFisher Scientific). Slides were then washed with PBS, and subsequently water, and mounted using Prolong Gold anti-fade mounting medium (ThermoFisher Scientific) and covered with coverslips.
HBMEC and human astrocyte staining was performed using the method previously developed [14]. Briefly, cells cultured on poly-L-lysine coated microscope slides (Tekdon, Myakka City, FL) were fixed in −20°C methanol for 5 minutes, blocked in 10% NDS in PBS, and incubated with CCM-rhFLAG-mAb, and mouse anti-vimentin (clone V9, Agilent Technologies), mouse anti-myosin9 (clone 5D9D2, Proteintech, Rosement, IL), or mouse anti-α tubulin (clone DM1A, Cell Signaling Technology, Denver, MA), and (where indicated) rabbit anti-ENO1 (PA-29660, ThermoFisher Scientific) for 1 hour at room temperature. Normal human IgG isotype control antibody (31154. ThermoFisher Scientific) was used as a negative control. After washing, samples were incubated with 5% NDS in PBS containing Alexa Flour 488-conjugated donkey anti-human antibody, Alexa Flour 594-conjugated donkey anti-mouse antibody, Alexa Flour 647-conjugated donkey anti-rabbit antibody (all Jackson ImmunoResearch), and Hoechst 33342 (ThermoFisher Scientific). Slides were then washed and mounted in Prolong Gold antifade reagent and sealed as before.
2.7. Fluorescent microscopy and image processing
Images were acquired using an SP8 laser scanning confocal microscope (Leica Microsystems) driven by Leica Application Suite X software. Human tissue sections were imaged with 20x and 40x oil immersion objectives (numerical aperture (NA)=0.7 and 1.25, respectively). Image files were exported as grey scale TIF images, assigned false colors and converted to RGB format using Image J (National Institutes of Health, Bethesda, MD). HBMEC and astrocyte samples were imaged by using a 63X oil immersion objective (NA=1.4). Raw image files were exported as TIF files, and processed in Image J. For manual mean pixel intensity (MPI) measurements of whole cells (a surrogate measure of the CCM-rhFLAG-mAbs cellular reactivity), the ENO1 channel was used for guiding manual/hand segmentation of whole cell areas. Cell membranes were defined using the polygon function in Image J. MPIs of individual cells (5 fields of view (FOV)) for the channel acquired for the CCM-rhFLAG-mAbs stain were quantified using the measure function in Image J.
CytoSkaler [13] was used for automated segmentation of individual cells and to quantify pixel intensity co-variance coefficients between different raw image channels representing respective stains. Briefly, the CytoSkaler machine learning algorithm uses the ENO1, nuclear, and cytoskeletal raw image files to segment individual whole cell areas, their respective nuclear and cytoplasmic zones, and subcellular areas abundant for the stained cytoskeletal protein in each whole cell. Following whole cell and subcellular compartment segmentation, for each cell within a FOV, co-variance coefficients between indicated channels (i.e. CCM-rhFLAG-mAb and anti-NMMHCIIA/anti-vimentin/anti-tubulin) were calculated between the pixel intensity values for all pixels within that given cell.
2.8. Co-immunoprecipitation and mass spectrometry
HBMECs and astrocytes cultured in T75 flasks were washed twice with PBS. Cells were lysed with 1 ml of cold IP lysis buffer (ThermoFisher Scientific) at 4°C. Cell lysate was collected and centrifuged at 14000 g at 4°C for 15 minutes. Supernatant was precleared with protein G agarose beads (ThermoFisher Scientific) at 4°C for 30 minutes and incubated with 60 μg CCM mAb and protein G agarose beads overnight at 4°C. Human IgG isotype control antibody was used as control. Following centrifugation at 1000 g at 4°C for 1 minute, beads were washed in ice-cold immunoprecipitation wash buffer (25mM Tris, 150mM NaCl; pH 7.2), resuspended in Lane Marker reducing sample buffer (ThermoFisher Scientific), and heated at 95˚C for 10 minutes. Samples were subjected to 4–20% Tris-Glycine SDS-PAGE gel electrophoresis (Bio-Rad Laboratories). and silver staining (ThermoFisher Scientific) to visualize co-immunoprecipitated protein. For mass spectrometry, samples were run for 5 minutes and the full height gel band for each lane was excised. Excised gel slabs were washed with 100 mM ammonium bicarbonate in acetonitrile and reduced with 10 mM dithiothreitol at 50°C for 30 minutes. Cysteine residues were alkylated with 100 mM iodoacetamide in the dark for 30 minutes at room temperature. Excised gel bands were washed in 100 mM ammonium bicarbonate in acetonitrile and incubated with 600 ng trypsin overnight at 37°C. Following solubilization, tryptic digested peptides were lyophilized then reconstituted in 5% acetonitrile and 0.1% formic acid in water and injected onto a trap column (150um ID × 3 cm, in-house packed with ReproSil C18, 3 μm) coupled with an analytical column (75 um ID × 10.5cm, PicoChip column packed with ReproSil C18, 3 μm) (New Objectives, Woburn, MA). Samples were separated using a linear gradient of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) over 120 minutes using a Dionex UltiMate 3000 Rapid Separation nanoLC system (ThermoFisher Scientific). Mass spectrometry data were obtained on an Orbitrap Elite Mass spectrometer (ThermoFisher Scientific). Peptides were searched using Mascot v2.5.1 (Matrix Science, Boston, MA) against the uniport Swiss-Prot Human database (2019). Scaffold v.4.8.4 (Proteome Software, Portland, OR) was used to visualize proteomic data. Peptide identification was accepted at >99% probability, and protein identifications were accepted at >99% probability to achieve a false discovery rate of <1%. At least two different peptides were required to define of a protein candidate. Proteins were considered to be specifically enriched by CCM-rhFLAG-mAb when there was both >10-fold total spectrum count and >2-fold total spectrum count relative to total spectrum count of peptides identified in control samples.
2.9. Enzyme linked immunosorbent assay (ELISA)
For ELISA, 10 μg/ml recombinant human vimentin (C-terminally His tagged and expressed in Rosetta Red E.coli as described previously [15]) recombinant human non-muscle myosin heavy chain IIA (NMMHCIIA, N-terminally FLAG tagged and expressed in Sf-9 cells as described previously[16]), and purified porcine brain tubulin (Cytoskeleton, Denver, CO) diluted in PBS were used to coat 96 well Costar ELISA plates (Corning Life Sciences) overnight at 4°C. Plates were then washed with ultrapure water and incubated with blocking buffer (PBS/3% BSA) for 1 h. Plates were incubated with CCM-rhFLAG-mAbs at three fold dilutions (30 to 0.041 μg/ml in blocking buffer) at room temperature for 2 h, and washed three times with 0.01% Tween-20 in PBS. Bound CCM-rhFLAG-mAb was detected using HRP-conjugated anti-human IgG antibody (109–035-098, Jackson ImmunoResearch) diluted in blocking buffer at room temperature for 1 h, and washed three times with 0.01% Tween-20 in PBS. Plates were incubated with Super AquaBlue ELISA substrate (ThermoFisher Scientific) at room temperature for 30 minutes, and absorbance was measured at 405 nm by using an 800 TS microplate reader (Agilent Technologies). Antibody concentration and corresponding OD405 values were used to calculate antibody binding affinity to individual antigens by using the nonlinear regression module of GraphPad Prism Version 7.0e (GraphPad Software, San Diego, CA).
2.10. Statistical analysis
Data analysis was performed using GraphPad 7.0e. Data displayed in graphs represent mean ± SEM, Statistical significance was assessed by 2 tailed Student’s t test. For all analyses, P values less than 0.05 were considered significant.
3. Results
3.1. Characterization of CCM in situ-expressed immunoglobulins
Plasma cells were identified by CD138 staining of CCM fresh frozen tissue, laser micro-dissected and used for subsequent RT-PCR and antibody variable region cloning. From more than 200 individual plasma cells collected, successful cloning of both heavy and light chain antibody variable regions occurred for six plasma cells. These six antibodies, in combination with 4 antibodies identified in our previous study, were used for subsequent analyses. These 10 antibodies included five from patient 1, three from patient 2, one from patient 3 and one from patient 4 (Supplementary Table 1).
To determine immunoglobulin heavy and light chain characteristics, sequences of the CCM lesion derived antibody variable region pairs were analyzed using IMGT/V-QUEST[17]. The length of the VH complementary determining region 3 (VH CDR3) ranged from 14 to 23 amino acids (Mean±SD = 16.9 ± 3.1), while the VL CDR3 ranged from 11 to 14 amino acids (Mean±SD = 11.4 ± 1.0) (Supplementary Table 2). The number of somatic mutations identified in the immunoglobulin heavy and light chain coding sequences were estimated to be 25±7 and 13±6, respectively. The frequency of replacement mutations within the heavy and light chain variable regions are shown in Supplementary Fig. 1.
3.2. CCM lesion associated mAbs bind to brain tissues
To determine the histological distribution and cellular origins of antigens reactive with antibodies generated from resident in situ plasma cells in CCM lesions, FLAG tagged recombinant antibodies were produced, “CCM-rhFLAG-mAbs”. We first determined if these CCM-rhFLAG-mAbs could detect antigens in CCM lesions. Surgically resected CCM lesions were cryosectioned and stained with CCM-rhFLAG-mAbs. Six out of 10 CCM-rhFLAG-mAbs (H1B, H1E, H2A, H2C, H3A and H4A) showed significant reactivity with variant staining intensity, and this staining pattern was reproduced in different CCM lesional tissues (Supplementary Fig. 2). Antibody H1B yielded the strongest staining as approximated by a more intense fluorescence. CCM-rhFLAG-mAbs primarily stained structures surrounding dilated microvessels (caverns) within CCM lesions and between caverns (Supplementary Figure. 3). Co-staining of CCM-rhFLAG-mAbs with the endothelial cell marker CD31 and astrocyte marker GFAP showed that the more reactive CCM-rhFLAG-mAbs bounded to antigens in endothelial cells and astrocytes, although additional cells and structures also harbored reactive antigen (Fig. 1).
Fig. 1. CCM-rhFLAG-mAbs co-localize with CD31 and GFAP positive cells in resected CCM tissue.
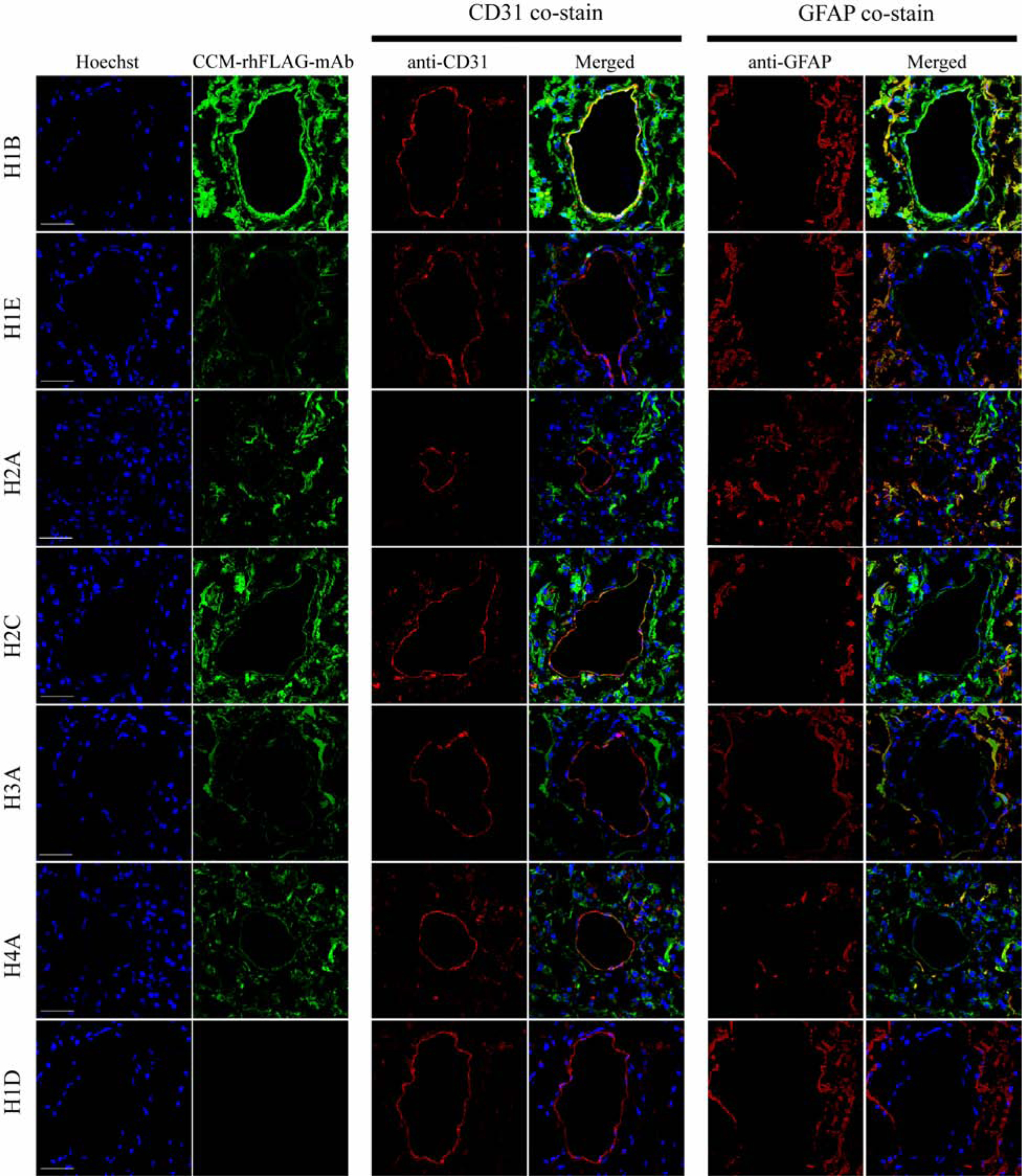
Scanning confocal microscopy showing that six CCM-rhFLAG-mAb bound structures (green, second column from left, top six rows) are often expressed within CD31 (red, middle two columns) positive endothelium surrounding unstained caverns. CCM-rhFLAG-mAbs bound structures are also abundantly expressed within GFAP positive (red, right two columns) astrocytes cells. Unlike the other six CCM-rhFLAG-mAbs, H1D CCM-rhFLAG-mAb (bottom row) does not show detectable staining signal. DNA (blue, first column from left) is stained with Hoechst 33342. Scale bars: 50 μm.
Since the CCM-rhFLAG-mAbs stained endothelial cells and astrocytes in CCM lesions, we next tested if these antibodies could stain similar cells in normal brain tissue, to gain insight into whether driving antigens are readily available prior to disease onset. Autopsied cerebellar tissues were sectioned and stained with CCM-rhFLAG-mAbs, and co-stained for endothelial cells and astrocytes (Fig. 2). The staining was generally weaker for autopsied normal brain tissue (Supplementary Fig. 4), suggesting that although there are local antigens present in the absence of disease, these antigens become more abundant during in situ inflammation. H1B, and H2C antibodies bound to both endothelial cells and astrocytes while H1E, H2A, H3A, H4A antibodies bound to astrocytes and other cells in normal brain.
Fig. 2. CCM-rhFLAG-mAbs co-localize with CD31 and GFAP positive cells in normal brain tissue.
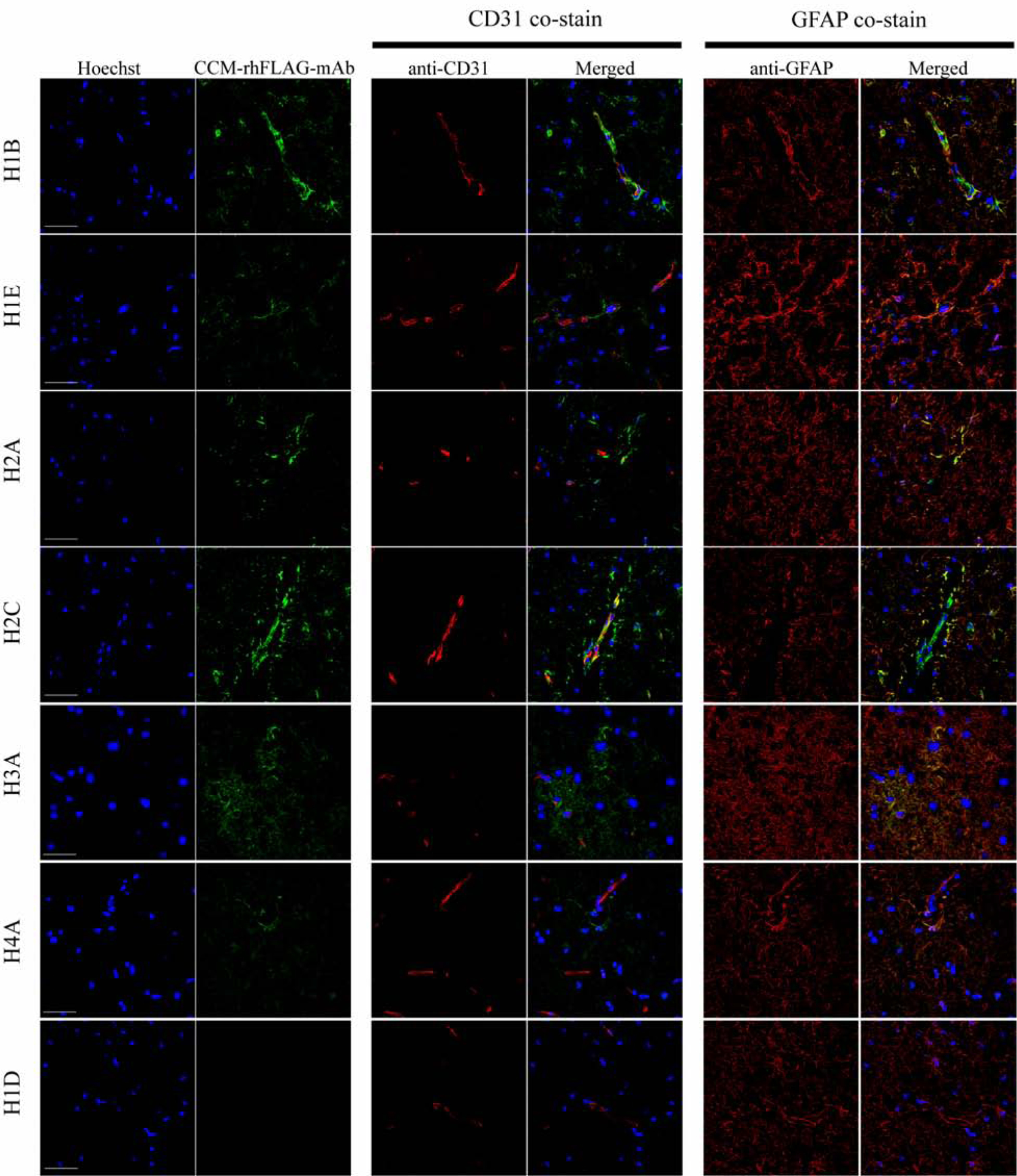
Scanning confocal microscopy showing six CCM-rhFLAG-mAb (green, second column from left, top six rows) bound structures within CD31 (red, middle two columns) positive endothelial cell of vessels. The CCM-rhFLAG-mAb (green) bound structures are also found in GFAP (red, right two columns) positive astrocytes adjacent to vessels. Unlike the other six mAbs, H1D CCM-rhFLAG-mAb (bottom row) does not show detectable staining signal. DNA (blue, first column from left) is stained with Hoechst 33342. Scale bars: 50 μm. 2:
Taken together, these studies suggest that antibodies generated by in situ plasma cells are reactive with readily available locally produced antigens particularly abundant in endothelial cells and astrocytes, which are not dependent on a diseased state, but become more available during inflammation.
3.3. CCM lesion-derived antibodies bind to cultured endothelial cells and astrocytes
We then sought to identify target antigens by using readily available culturable cells representative of cells found in situ. Six of the most reactive CCM-rhFLAG-mAbs were tested for reactivity with brain microvascular endothelial cells and astrocytes. Indirect immunofluorescence showed that the CCM-rhFLAG-mAbs reactive to brain tissues also bound cultured primary HBMEC and astrocytes (Fig. 3A). These antibodies primarily stained structures within the cytoplasm. While most of the reactive antibodies exhibited diffuse staining patterns, H1B bound preferentially to filamentous structures. Similar to that observed for CCM lesion staining, H1B reactivity also appeared to yield the highest cellular reactivity. This was confirmed by measuring whole cell MPI (a surrogate for antibody reactivity) for different CCM-rhFLAG-mAbs (Fig. 3B). These data support endothelial cells and astrocytes harboring the targets of in situ produced antibodies and the cells discussed above being relevant and readily available reagents for downstream antigen characterization.
Fig. 3. CCM-rhFLAG-mAbs stain cultured HBMECs and astrocytes.
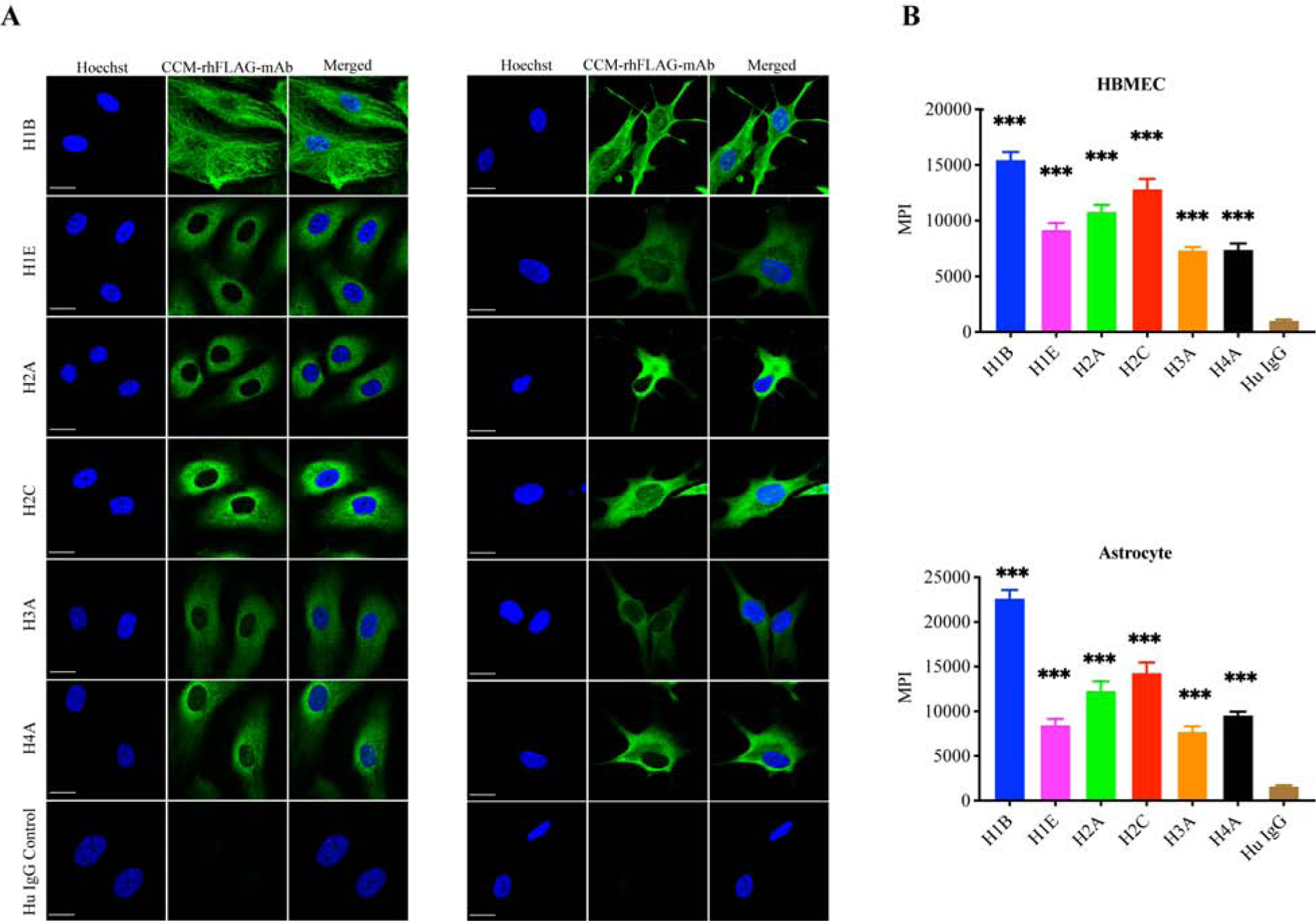
(A)Scanning confocal microscopy showing cytoplasmic staining with six CCM-rhFLAG-mAbs (green) using cultured HBMECs (left three columns) and astrocytes (right three columns). The human IgG isotype control (bottom row) has negligible reactivity. DNA (blue) is stained with Hoechst 33342. Scale bar: 20 μm. (B) Reactivity of CCM-rhFLAG-mAbs with HBMEC (top panel) and astrocytes (bottom panel) measured as whole cell area (n=10) mean pixel intensities (MPIs). Data are represented as mean±SEM.***p<0.001.
3.4. CCM lesion-derived antibodies bind to cytoskeletal components
CCM-rhFLAG-mAbs (H1B and H2C) were used to immunoprecipitate their binding partners by using HBMEC and astrocyte lysates. Human IgG isotype antibody was used as a negative control. CCM-rhFLAG-mAbs binding partners were identified by mass spectrometry (Supplementary Table 3). Out of the candidate antigens identified by mass spectrometry, NMMHIIA and vimentin were the proteins most abundantly identified in the precipitates formed between H1B and H2C and HBMEC and astrocyte lysates. In addition, different tubulin α and β chain isoforms were also commonly precipitated with CCM lesion derived antibodies.
We next co-stained HBMECs and astrocytes with CCM-rhFLAG-mAbs and commercially available antibodies raised against candidate antigens NMMHIIA, vimentin, and tubulin (Fig. 4A–B) to garner support for direct binding to antigens identified by mass spectrometry. Both H1B and H2C CCM-rhFLAG-mAbs partially colocalized with NMMHIIA, vimentin, and tubulin in both HBMECs and astrocytes. H1B preferentially co-localized with vimentin, and H2C preferentially co-localized with NMMHIIA in both cell types.
Fig. 4. CCM-rhFLAG-mAbs co-localize with cytoskeletal components.
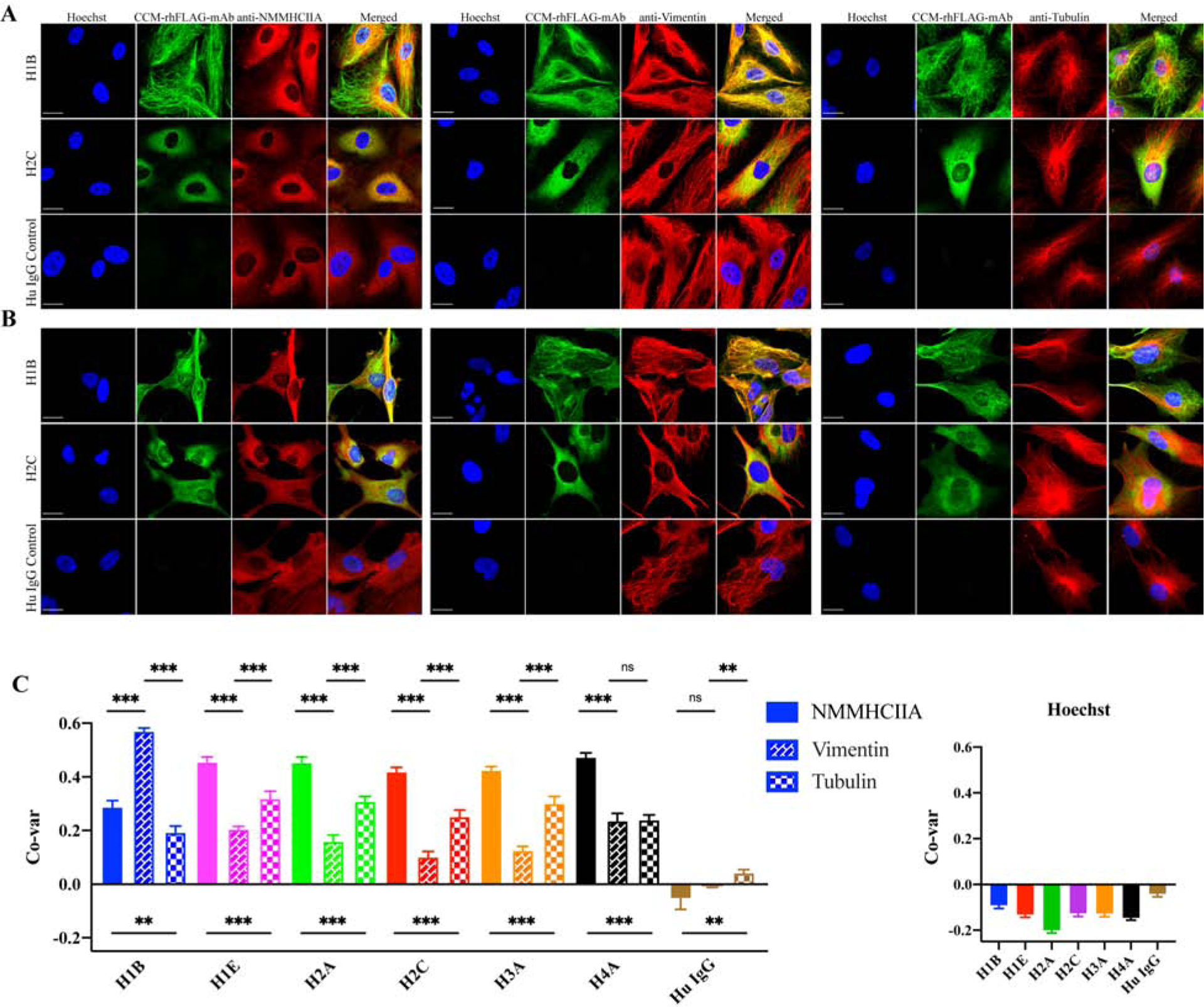
CCM-rhFLAG-mAbs (H1B, H2C) co-staining with anti-NMMHCIIA, anti-vimentin and anti-α tubulin antibodies in HBMECs (A) and astrocytes (B). (C) Co-variance (Co-var) correlation analyses of pixel intensities between signals generated by biding of six reactive CCM-rhFLAG-mAbs and either anti-NMMHCIIA, anti-vimentin, anti-α tubulin antibodies (Left panel) or Hoechst 33342 (DNA reactive) in HBMECs. Scale bars: 20 μm. Data are represented as mean±SEM (n>40). *p<0.05, ***p<0.001.
All six reactive CCM-rhFLAG-mAbs were next quantitatively assayed for preferential binding to candidate antigens when offered a mixture of different cellular antigens, by once more co-staining with anti-NMMHCIIA, anti-vimentin or anti-tubulin using HBMECs. This time, CytoSkaler software was used to perform automated whole cell segmentation, and subsequent co-variance correlation analysis between pixel intensities for the total pixels within the segmented cellular areas. Cytokskaler analyses showed that five of the six CCM-rhFLAG-mAbs (H1E, H2A, H2C, H3A and H4A) had higher co-variance with NMMHCIIA (with mean co-variance correlation coefficients of 0.45, 0.45, 0.42, 0.42 and 0.47, respectively, p<0.001), indicating that these antibodies are more selective for NMMHCIIA, or co-distributed structures than for vimentin or tubulin. In contrast, H1B was more selective for vimentin (mean co-variance correlation = 0.57, p<0.001) (Fig. 4C).
We next determined the direct binding affinity of CCM-rhFLAG-mAbs to NMMHIIA, vimentin, and tubulin by performing ELISAs. H1B showed strong binding affinity to purified NMMHIIA (dissociation constant (Kd)=1.21 × 10−8 M), vimentin (Kd=1.80 × 10−8 M), and tubulin (Kd=7.34 × 10−9 M) proteins (Fig. 5, Supplementary Table 4). The H2A, H2C, and H3A antibodies had lower affinities for these proteins. H1E and H4A also had weak to moderate interactions (Kd between 10−6 and 10−7 M) with these same proteins.
Fig. 5. CCM-rhFLAG-mAbs bind to purified NMMHCIIA, vimentin, and tubulin proteins.
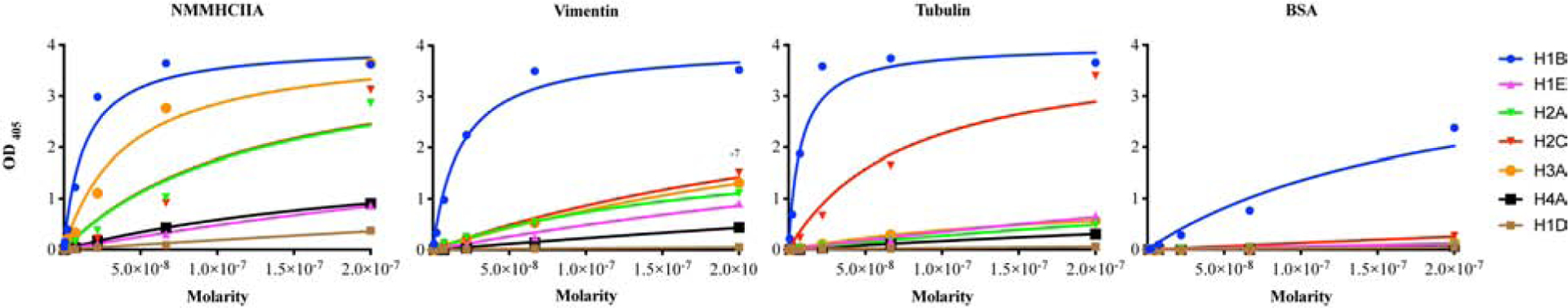
Binding of six reactive CCM-rhFLAG-mAbs (H1B, H1E, H2A, H2C, H3A, H4A) and a non-reactive mAb (H1D) to purified NMMHCIIA, vimentin, tubulin, and bovine serum albumin proteins as determined by ELISA.
Taken together, the results from co-immunoprecipitation, immunofluorescent staining, and ELISAs support cytoskeletal associated proteins NMMHIIA, vimentin, and tubulin as target of antibodies synthesized within the CCM lesion milieu.
3.5. Reduced reactivity in germline reverted CCM lesion-associated antibodies
In silico analyses of variable region sequences demonstrated that CCM lesion-associated antibodies have extensive somatic hypermutations. This suggests that these in situ generated antibodies, produced by local parent plasma cells have undergone affinity maturation, following selection on epitopes on antigens such as those characterized above. To test this, germline reverted antibodies (H1Brev and H2Crev) were generated, in which the heavy and light chains were reverted to their (H1B and H2C) predicted germline sequences. The mAbs represented the B-cell receptors (BCRs) that were likely to have been expressed when those plasma cells were in their ancestral/naïve B-cell cell state. Their abilities to bind to the candidate antigens characterized above were compared to mature CCM-rhFLAG-mAbs. ELISAs showed that H1Brev had lower binding to purified NMMHIIA (Kd=1.75 × 10−7 M), vimentin (Kd=1.18 × 10−7 M), and tubulin (Kd=2.01 × 10−7 M) relative to H1B antibody. In addition, H2Crev exhibited 2-fold lower affinity to NMMHIIA, 12-fold lower to vimentin and 50-fold lower tubulin reactivity compared to H2C antibody (Fig. 6A, Supplementary Table 4). Immunofluorescent staining studies also demonstrated that both H1Brev and H2Crev antibodies had weaker HBMEC staining (Fig. 6B–C). Co-variance analyses showed that H1B had higher selectivity for vimentin than H1Brev, while H2C has higher selectivity to NMMHCIIA than H2Crev (Fig. 6D). The increase in specificity (as measured by co-localization) with affinity maturation is consistent with selection on these putative cytoskeletal antigens, or other antigens that share common epitopes. Increase in both affinity and specificity for the candidate antigens identified herein gives credence to a new model of immunopathology in which, during CCM, in situ derived antigens are driving local lymphocytic activation and inflammation.
Fig. 6. Reduced antigen binding of germline reverted (rev) rhFLAG-mAbs.
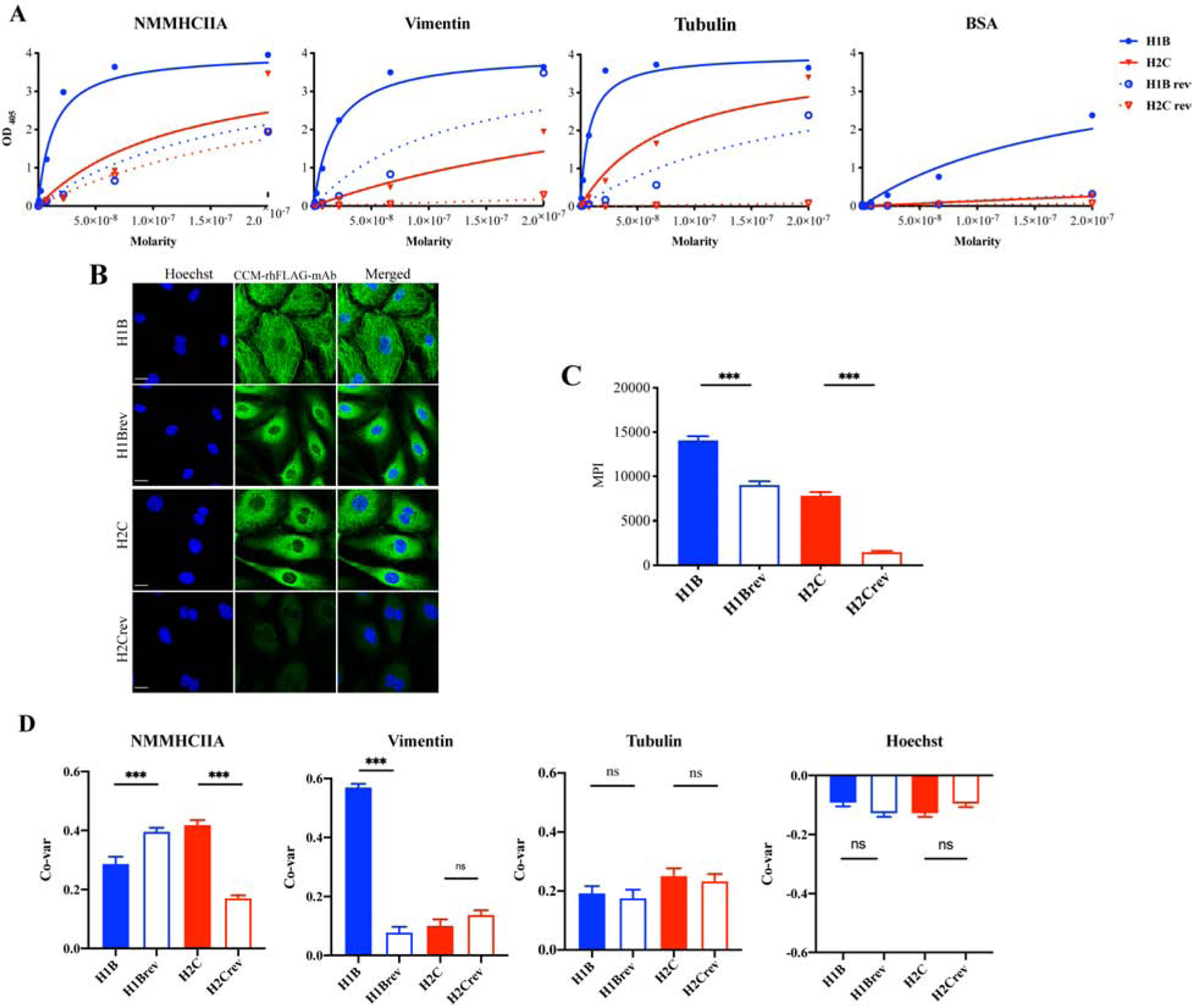
(A) Binding of CCM rhFLAG-mAbs and their germline reverted antibodies (H1B, H1Brev, H2C, and H2Crev) to purified NMMHCIIA, vimentin and tubulin as determined by ELISA. (B) Indirect Immunofluorescence staining of HBMECs with CCM rhFLAG-mAbs and their germline reverted antibodies, (C) Reactivity of CCM rhFLAG-mAbs and their germline reverted antibodies to HBMECs was measured as whole cell area mean pixel intensities (MPIs) (n=10 cells). (D) Co-variance (Co-var) correlation analyses of pixel intensities of rhFLAG-mAbs and anti-NMMHCIIA, anti-vimentin, anti-α tubulin antibodies, and DNA (stained by Hoechst 33342) in HBMECs (n>40). Scale bar: 20 μm. Data are represented as mean±SEM. *p<0.05, ***p<0.001.
4. Discussion
We previously reported multiple lines of evidence suggestive of the pathogenic in situ adaptive immunity in CCM lesions including an abundance of B-cells, in situ somatic hypermutation and adjacent IgG-containing immune complex formation [10]. In this study, we produced recombinant IgG antibodies (CCM-rhFLAG-mAbs) with variable regions corresponding to those produced by plasma cells within CCM lesional tissues, to determine their binding targets. We determined that the CCM lesion associated antibodies primarily react with cytoplasmic autoantigens, especially cytoskeleton proteins. We postulate that targeting of these antigens participates in CCM disease pathogenesis.
The immunofluorescent results herein showed that six of the ten CCM-rhFLAG-mAbs that we evaluated have high affinity with CCM lesional tissues, notably structures adjacent to caverns. These CCM-rhFLAG-mAbs often bind both endothelial and astrocytes cells within diseased and normal brain tissue. Our data suggest that antigens that are capable of supporting local lymphocytic activation and/or immune complex formation are present in the lesional milieu. We also showed CCM-rhFLAG-mAbs binding to cytoskeletal and cytoplasmic antigens in cultured primary HBMECs and astrocytes. Thus, autoreactive antigens presenting from injured/activated endothelial cells and astrocytes in CCM lesions could be targeted by locally produced plasma cell derived antibodies. Importantly, targets of in situ plasma cell derived antibodies may not be limited to these cell types; they may also react with other neurovascular unit components such as neurons, pericytes and other cell types.
The mass spectrometry results herein did not identify antigens specifically expressed by endothelium or astrocytes, suggesting that CCM-rhFLAG-mAbs may target various cell types within CCM lesional tissue. Notably, the most abundant recognized targets in both cell types were cytoskeleton proteins with the most common including NMMHCIIA, vimentin, and tubulin. Despite the high antibody sequence variability, the reactive CCM-rhFLAG-mAbs all recognized these three cytoskeletal components with variable affinity, suggesting the existence of common epitopes on these antigens. Importantly, five of the six reactive CCM-rhFLAG-mAbs were more selective toward NMMHCIIA, suggesting that NMMHCIIA is the more potent antigen driving B-cell selection. The cross-reactivity of CCM-rhFLAG-mAbs for different antigens is not surprising, as this phenomenon has been demonstrated for antibodies derived from multiple autoimmune diseases [14, 18, 19]. Autoantibody selection in CCM disease may be due to selection on different antigens, sharing common epitopes, during the evolution of the B-cells that ultimately yield the plasma cells in the CCM lesion.
NMMHCIIA, vimentin and tubulin can be expressed by various cell types in human brain [20–22]. Previous proteomic analyses showed that genetic mutation of any of the CCM genes altered expression of cytoskeletal proteins in the endothelial cells, especially NMMHCIIA [23]. NMMHCIIA is responsible for the formation of intercellular contractility and tension through interactions with actin cytoskeleton, the function of NMMHCIIA depends mainly on the Rho GTPase family [20]. A loss of any CCM gene has been associated with increased RhoA activity, which leads to vascular hyperpermeability [24]. In addition, vimentin is a marker for mesenchymal cells, and a disruption of the CCM1/KRIT1 gene has been shown to induce endothelial-mesenchymal transition (EndMT), which contributes to development of vascular malformations [25]. A dysregulation of EndMT has also been observed in atherosclerotic lesions, and has been suggested to be linked to increased vimentin levels [26]. Finally, tubulins are involved in cell movement, intracellular tracking and mitosis [21]. CCM1/KRIT1 has been shown to co-localize within the same complex than β–tubulin [27]. A dysregulation of tubulin may contribute to increased cellular permeability through KRIT1. Cytoskeletal components have been shown to be targets of multiple autoimmune diseases, such as rheumatoid arthritis [28], systemic lupus erythematosus [14], and inflammatory bowel disease [29], suggesting similarity between CCM and the in situ inflammatory response in these other more canonical autoimmune diseases.
Our previous study showed mature B- and T-cells aggregated within CCM lesions, where they are undergoing in situ clonal expansion and somatic hypermutation [10]. Such tertiary lymphoid structures (TLS) are similar to those seen in other canonical autoimmune diseases [30–32]. Importantly, TLS have been associated with poor prognosis of disease [33] and malignancy of development [34]. The germline reverted mAbs generally had reduced reactivity to the candidate antigens identified herein, consistent with antigen driven affinity maturation on these putative antigens from low affinity B-cell receptor precursors. Interestingly, the germline reverted mAbs retained relatively high affinity for NMMHCIIA, indicating that V-D-J rearrangement in the absence of somatic hypermutation is sufficient for strong reactivity with NMMHCIIA, and that naïve B-cells with reactivity for this antigen are escaping central tolerance, and ultimately able to select on antigen that becomes accessible during CCM formation. In addition, co-variance analyses showed that H1B had higher selectivity for vimentin than H1Brev, while H2C had higher selectivity to NMMHCIIA. These results demonstrate that CCM lesion derived antibodies have increased binding affinity to cytoskeletal components following affinity maturation, and that they can have different antigenic preferences in a cellular milieu.
How the intracellular antigens get exposed to extracellular environments in order to activate surface BCRs is an ongoing area of study. The first hypothesis proposes that apoptosis play an important role as a source of cytoplasmic self-antigens. During the process of programmed cell death, apoptotic blebs appear on the cell surface and microparticles are released from the cell [35]. Notably, the CCM3/PDCD10 encodes as a protein upregulated during apoptosis, indicating CCM proteins may indeed affect endothelial death [36]. Our recent study showed that loss of KRIT1 expression promotes intestinal epithelial cell apoptosis following TNF treatment [37]. The second hypothesis is the exposure of antigens by stimulation. Vimentin can be secreted following stimulation with inflammatory cytokines [38]. Importantly, an increase in TNF-α plasma levels have previously been associated with a more severe clinical course in CCM patients [39]. Similarly, NMMHCIIA can be expressed on the surface of endothelial cells following stimulation [40]. Other routes of antigen release include NETosis and METosis, which have been observed in other autoimmune diseases [41, 42]. Another hypothesis for reactivity with cellular antigens is that CCM lesion derived antibodies may have been selected on non-human tissue antigens with shared epitopes. This phenomenon has been reported for anti-streptococcal antibodies, which demonstrate cross-reactivity with myosin, vimentin and tubulin in rheumatic carditis and Sydenham’s chorea [43, 44]. Future studies with epitope mapping should address the question of cross-reactivity of mAbs across various antigens.
Results herein do not directly address the pathogenicity of the defined immune response in CCM lesions. Previous studies in murine models recapitulating the human disease has demonstrated that B-cell infiltrates develop in more mature multicavernous hemorrhagic CCM lesions, rather than in perimordial lesions characterized by capillary ectasia without bleeding [7]. Similarly, inflammatory transcriptome differentiates mature from acute CCM lesions in murine models [8]. Importantly, our previous study showed that B cell-depletion effectively blunts lesion development in chronic CCM disease model [9]. This result suggests that CCM disease-associated B-cells, and/or the antibodies derived from them could participate in lesion development. Our previous research had also demonstrated complement activation in association with the inflammatory niches in CCM lesions [10]. The functional role of in situ produced antibodies to cytoskeletal proteins is an exciting avenue for future study.
We cannot confirm that mAbs do not react with other antigenic triggers than those identified herein. And our study did not assess mAb binding to tissues outside the brain and CCM lesions, although it is unclear if such binding would directly impact pathogenicity of this disease, where lesions arise primarily in the central nervous system parenchymal milieu. Another major limitation of our study was the small antibody sample size. We used laser capture microdissection to obtain plasma cells from CCM lesions, and subsequently generated mAbs to represent the BCRs of cells selected in situ, and the soluble antibodies that are produced by local plasma cells. However, the rate of success of PCR amplification of both heavy and light chains was relatively low. Therefore, our study was restricted to a small number of specimens and mAbs preventing us from more powerfully ascertaining the frequency of plasma cells that generate antibodies with reactivity for the antigens characterized herein. Despite these limitations, the antibodies characterized in this study have given us the first insight into the antigens commonly targeted by locally produced antibodies, and have opened a new avenue of research into the immunopathogenesis of CCM.
5. Conclusions
This is the first study to characterize the antigens targeted by resident plasma cells in CCMs. Moreover, we have demonstrated an affinity maturation from low level BCR precursors for these candidate antigens. Taken together, our results suggest that CCM is similar to other chronic inflammatory conditions with evidence of in situ adaptive responses to cytoskeletal proteins and anecdotal evidence of B-cell depletion therapy success. These data open the door to studying a novel previously unappreciated aspect of CCM, namely in situ tertiary lymphoid organogenesis, and more specifically its potential immunopathogenic significance and range of therapeutic targets therein.
Supplementary Material
Highlights.
Antibodies produced by plasma cells in CCM lesions are primary bound to endothelial cells and astrocytes.
CCM-related autoantibodies target mainly cytoskeletal autoantigens including NMMHCIIA, vimentin and tubulin.
An autoimmune reaction within lesions suggests novel mechanisms related to physiopathogenesis of CCMs.
Characterizing the relationships between the autoimmune reaction and clinical outcomes may open novel treatments for CCM disease.
Acknowledgements
We thank Dr. Xiong Liu (NIH) for generously providing recombinant NMMHCIIA. Proteomics services were performed by the Northwestern Proteomics Core Facility supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569.
Funding
This research was partially supported by a grant from the Safadi Program of Excellence in Clinical and Translational Neuroscience; The National Institutes of Health (P01 NS092521) grants, the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR000430), the William and Judith Davis Fund in Neurovascular Surgery Research at the University of Chicago to IAA; National Natural Science Foundation of China (81771276), Key Project of Natural Science Foundation of Heilongjiang Province of China (ZD2016015), and Foundation for High-Level Returned Overseas talents of Ministry of Human Resources and Social Security of China to CS and DZ; and from the Sigrid Juselius Foundation, Maud Kuistila Foundation and Emil Aaltonen Foundation to JK.
Abbreviations
- BCR
B cell receptor
- CCM
Cerebral cavernous malformation
- CDR
Complementary determining region
- CNS
Central nervous system
- EndMT
Endothelial-mesenchymal transition
- FOV
Fields of view
- FWR
Framework region
- GFAP
Glial fibrillary acidic protein
- HBMEC
Human brain microvascular endothelial cell
- IMGT
ImMunoGeneTics
- KRIT1
Krev interaction trapped 1
- mAb
Monoclonal antibody
- MPI
Mean pixel intensity
- NA
Numerical aperture
- NDS
Normal donkey serum
- NMMHCIIA
Non-muscle myosin heavy chain IIa
- OCT
Optimal cutting temperature
- OSM
Osmosensing scaffold for MEKK3
- PDCD10
Programmed cell death 10
- ROCK
Rho-associated protein kinase
- TLS
Tertiary lymphoid structure
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
Authors declare no conflict of interest.
Research data
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
- [1].Awad IA, Polster SP. Cavernous angiomas: deconstructing a neurosurgical disease. J Neurosurg. 131 (2019) 1–13, 10.3171/2019.3.JNS181724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry. 71 (2001) 188–92, 10.1136/jnnp.71.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tanriover G, Sozen B, Seker A, Kilic T, Gunel M, Demir N. Ultrastructural analysis of vascular features in cerebral cavernous malformations. Clin Neurol Neurosurg. 115 (2013) 438–44, 10.1016/j.clineuro.2012.06.023. [DOI] [PubMed] [Google Scholar]
- [4].Tu J, Stoodley MA, Morgan MK, Storer KP. Ultrastructural characteristics of hemorrhagic, nonhemorrhagic, and recurrent cavernous malformations. J Neurosurg. 103 (2005) 903–9, 10.3171/jns.2005.103.5.0903. [DOI] [PubMed] [Google Scholar]
- [5].Shenkar R, Shi C, Check IJ, Lipton HL, Awad IA. Concepts and hypotheses: inflammatory hypothesis in the pathogenesis of cerebral cavernous malformations. Neurosurgery. 61 (2007) 693–702; discussion −3, 10.1227/01.NEU.0000298897.38979.07. [DOI] [PubMed] [Google Scholar]
- [6].Shi C, Shenkar R, Batjer HH, Check IJ, Awad IA. Oligoclonal immune response in cerebral cavernous malformations. Laboratory investigation. J Neurosurg. 107 (2007) 1023–6, 10.3171/JNS-07/11/1023. [DOI] [PubMed] [Google Scholar]
- [7].Zeineddine HA, Girard R, Saadat L, Shen L, Lightle R, Moore T et al. Phenotypic characterization of murine models of cerebral cavernous malformations. Lab Invest. 99 (2019) 319–30, 10.1038/s41374-018-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koskimaki J, Zhang D, Li Y, Saadat L, Moore T, Lightle R et al. Transcriptome clarifies mechanisms of lesion genesis versus progression in models of Ccm3 cerebral cavernous malformations. Acta Neuropathol Commun. 7 (2019) 132, 10.1186/s40478-019-0789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi C, Shenkar R, Zeineddine HA, Girard R, Fam MD, Austin C et al. B-cell depletion reduces the maturation of cerebral cavernous malformations in murine models. J Neuroimmune Pharmacol. 11 (2016) 369–77, 10.1007/s11481-016-9670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shi C, Shenkar R, Kinloch A, Henderson SG, Shaaya M, Chong AS et al. Immune complex formation and in situ B-cell clonal expansion in human cerebral cavernous malformations. J Neuroimmunol. 272 (2014) 67–75, 10.1016/j.jneuroim.2014.04.016. [DOI] [PubMed] [Google Scholar]
- [11].Shi C, Shenkar R, Du H, Duckworth E, Raja H, Batjer HH et al. Immune response in human cerebral cavernous malformations. Stroke. 40 (2009) 1659–65, 10.1161/STROKEAHA.108.538769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 4 (2009) 372–84, 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kinloch AJ, Mohsin A, Asano Y, Henry C, Mor Vaknin N, Legendre L et al. A novel image analysis program, “CytoSkaler”, demonstrates that anti-vimentin antibody affinity maturation in lupus tubulointerstitial nephritis also results in more selective antigen targeting 2019 ACR/ARP Annual Meeting paper and manuscript in preparation.
- [14].Kinloch AJ, Chang A, Ko K, Henry Dunand CJ, Henderson S, Maienschein-Cline M et al. Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol. 66 (2014) 3359–70, 10.1002/art.38888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kinloch AJ, Kaiser Y, Wolfgeher D, Ai J, Eklund A, Clark MR et al. In situ humoral immunity to vimentin in HLA-DRB1*03(+) patients with pulmonary sarcoidosis. Front Immunol. 9 (2018) 1516, 10.3389/fimmu.2018.01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu X, Shu S, Korn ED. Polymerization pathway of mammalian nonmuscle myosin 2s. Proc Natl Acad Sci U S A. 115 (2018) E7101–E8, 10.1073/pnas.1808800115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lefranc MP, Ehrenmann F, Kossida S, Giudicelli V, Duroux P. Use of IMGT((R)) databases and tools for antibody engineering and humanization. Methods Mol Biol. 1827 (2018) 35–69, 10.1007/978-1-4939-8648-4_3. [DOI] [PubMed] [Google Scholar]
- [18].Steen J, Forsstrom B, Sahlstrom P, Odowd V, Israelsson L, Krishnamurthy A et al. Recognition of amino acid motifs, rather than specific proteins, by human plasma cell-derived monoclonal antibodies to posttranslationally modified proteins in rheumatoid arthritis. Arthritis Rheumatol. 71 (2019) 196–209, 10.1002/art.40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Connolly AM, Pestronk A. Anti-tubulin autoantibodies in acquired demyelinating polyneuropathies. J Infect Dis. 176 Suppl 2 (1997) S157–9, 10.1086/513801. [DOI] [PubMed] [Google Scholar]
- [20].Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of nonmuscle myosin IIA in human disease. Gene. 664 (2018) 152–67, 10.1016/j.gene.2018.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Breuss MW, Leca I, Gstrein T, Hansen AH, Keays DA. Tubulins and brain development - The origins of functional specification. Mol Cell Neurosci. 84 (2017) 58–67, 10.1016/j.mcn.2017.03.002. [DOI] [PubMed] [Google Scholar]
- [22].Dahl D, Strocchi P, Bignami A. Vimentin in the central nervous system. A study of the mesenchymal-type intermediate filament-protein in Wallerian degeneration and in postnatal rat development by two-dimensional gel electrophoresis. Differentiation. 22 (1982) 185–90, 10.1111/j.1432-0436.1982.tb01248.x. [DOI] [PubMed] [Google Scholar]
- [23].Baxter SS, Dibble CF, Byrd WC, Carlson J, Mack CR, Saldarriaga I et al. Role of cytoskeletal proteins in cerebral cavernous malformation signaling pathways: a proteomic analysis. Mol Biosyst. 10 (2014) 1881–9, 10.1039/c3mb70199a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 207 (2010) 881–96, 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 498 (2013) 492–6, 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- [26].Danielsson F, Peterson MK, Caldeira Araujo H, Lautenschlager F, Gad AKB. Vimentin diversity in health and disease. Cells. 7 (2018) 10.3390/cells7100147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gunel M, Laurans MS, Shin D, DiLuna ML, Voorhees J, Choate K et al. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A. 99 (2002) 10677–82, 10.1073/pnas.122354499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shrivastav M, Mittal B, Aggarwal A, Misra R. Autoantibodies against cytoskeletal proteins in rheumatoid arthritis. Clin Rheumatol. 21 (2002) 505–10, 10.1007/s100670200124. [DOI] [PubMed] [Google Scholar]
- [29].Zauli D, Crespi C, Dall’Amore P, Bianchi FB, Pisi E. Antibodies to the cytoskeleton components and other autoantibodies in inflammatory bowel disease. Digestion. 32 (1985) 140–4, 10.1159/000199232. [DOI] [PubMed] [Google Scholar]
- [30].Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. 186 (2011) 1849–60, 10.4049/jimmunol.1001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 116 (2006) 3183–94, 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW et al. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain. 134 (2011) 534–41, 10.1093/brain/awq350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hsieh C, Chang A, Brandt D, Guttikonda R, Utset TO, Clark MR. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res (Hoboken). 63 (2011) 865–74, 10.1002/acr.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol. 9 (2018) 1952, 10.3389/fimmu.2018.01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. 125 (2015) 2194–202, 10.1172/JCI78084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M. Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke. 40 (2009) 1474–81, 10.1161/STROKEAHA.108.527135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Y, Li Y, Zou J, Polster SP, Lightle R, Moore T et al. The cerebral cavernous malformation disease causing gene KRIT1 participates in intestinal epithelial barrier maintenance and regulation. FASEB J. 33 (2019) 2132–43, 10.1096/fj.201800343R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 5 (2003) 59–63, 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- [39].Girard R, Zeineddine HA, Fam MD, Mayampurath A, Cao Y, Shi C et al. Plasma biomarkers of inflammation reflect seizures and hemorrhagic activity of cerebral cavernous malformations. Transl Stroke Res. 9 (2018) 34–43, 10.1007/s12975-017-0561-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun Y, Qi Y, Liu C, Gao W, Chen P, Fu L et al. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J Virol. 88 (2014) 237–48, 10.1128/JVI.02141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Berthelot JM, Le Goff B, Neel A, Maugars Y, Hamidou M. NETosis: At the crossroads of rheumatoid arthritis, lupus, and vasculitis. Joint Bone Spine. 84 (2017) 255–62, 10.1016/j.jbspin.2016.05.013. [DOI] [PubMed] [Google Scholar]
- [42].Doster RS, Rogers LM, Gaddy JA, Aronoff DM. Macrophage extracellular traps: a scoping review. J Innate Immun. 10 (2018) 3–13, 10.1159/000480373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. 106 (2000) 217–24, 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kirvan CA, Cox CJ, Swedo SE, Cunningham MW. Tubulin is a neuronal target of autoantibodies in Sydenham’s chorea. J Immunol. 178 (2007) 7412–21, 10.4049/jimmunol.178.11.7412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.