

Abstract
Axons are essential for nervous system function and axonal pathology is a common hallmark of many neurodegenerative diseases. Over a century and a half after the original description of Wallerian axon degeneration, advances over the past five years have heralded the emergence of a comprehensive, mechanistic model of an endogenous axon degenerative process that can be activated by both injury and disease. Axonal integrity is maintained by the opposing actions of the survival factors NMNAT2 and STMN2 and pro-degenerative molecules DLK and SARM1. The balance between axon survival and self-destruction is intimately tied to axonal NAD+ metabolism. These mechanistic insights may enable axon-protective therapies for a variety of human neurodegenerative diseases including peripheral neuropathy, traumatic brain injury and potentially ALS and Parkinson’s.
Wallerian Degeneration: 1850–2012
When an axon is physically separated from its cell body, the portion distal to the injury site undergoes a stereotypical fragmentation known as Wallerian degeneration. This field of research has its origin in experiments performed in the mid-19th century with the observation of the degeneration of the nerve fibers innervating the frog tongue after transection[1]. The stereotypic fragmentation of the nerve was originally believed to be due to a passive wasting of the nerve after its disconnection from its source of nutrients in the neuronal soma.
The first evidence that axon degeneration is an active rather than passive process came in 1989 with the serendipitous discovery of a peculiar mouse strain with drastically slowed Wallerian degeneration, which came to be known as Wallerian degeneration slow (WldS)[2]. In addition to nerve transection, other axonal insults also result in Wallerian-like axon degeneration[3]. Importantly, the WldS mouse provides protection from some of these insults, like the chemotherapy drug vincristine, but also in disease models such as progressive motor neuronopathy, suggesting the existence of a common program of axon degeneration that is activated in a variety of injury and disease conditions[4,5].
WldS encodes an unnatural fusion protein that includes the entire NMNAT1 protein, a nicotinamide mononucleotide adenylyl transferase that converts nicotinamide mononucleotide (NMN) into nicotinamide adenine dinucleotide (NAD+)[6]. The axon protection afforded by WldS is explained by the axonal localization and enzymatic activity of overexpressed NMNAT1 protein[7–9], implicating the NAD+ metabolic pathway in axonal health and destruction. Although treatment with various NAD+ precursors or NAD+ itself can result in axon protection and the enzymatic activity of WldS or NMNAT1 is required for protection, their expression does not result in higher NAD+ levels within the axon, as was expected from an NAD+-synthesizing enzyme[8,10,11].
A molecular understanding for the axoprotection afforded by misexpression of WldS/NMNAT1 awaited another seminal discovery, the identification of SARM1 as an essential pro-degenerative molecule[12]. Loss of SARM1 in fruit flies or mice provides potent cell-autonomous axon protection comparable to WldS expression, demonstrating that SARM1 is required for axon degeneration[12,13] (Fig. 1). SARM1 has N-terminal Armadillo-like repeats, two sterile alpha motifs (SAMs), and a Toll/interleukin-1 receptor (TIR) domain (Fig. 2). TIR domains are the signature signaling domain in innate immune signaling, so prior work focused on SARM1’s potential role in innate immunity[14].
Fig. 1.
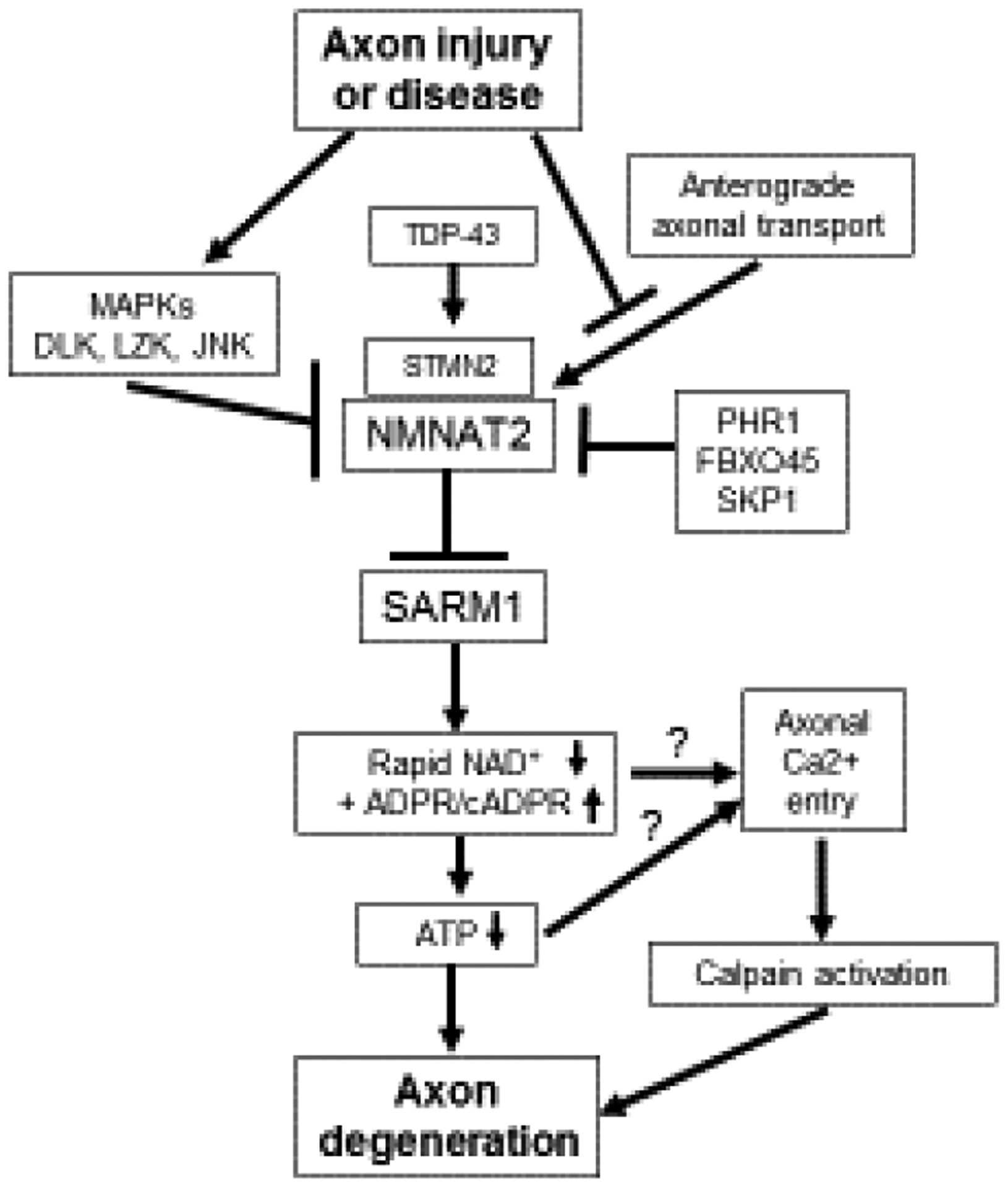
A schematic describing the endogenous Wallerian degeneration pathway.
Fig. 2.
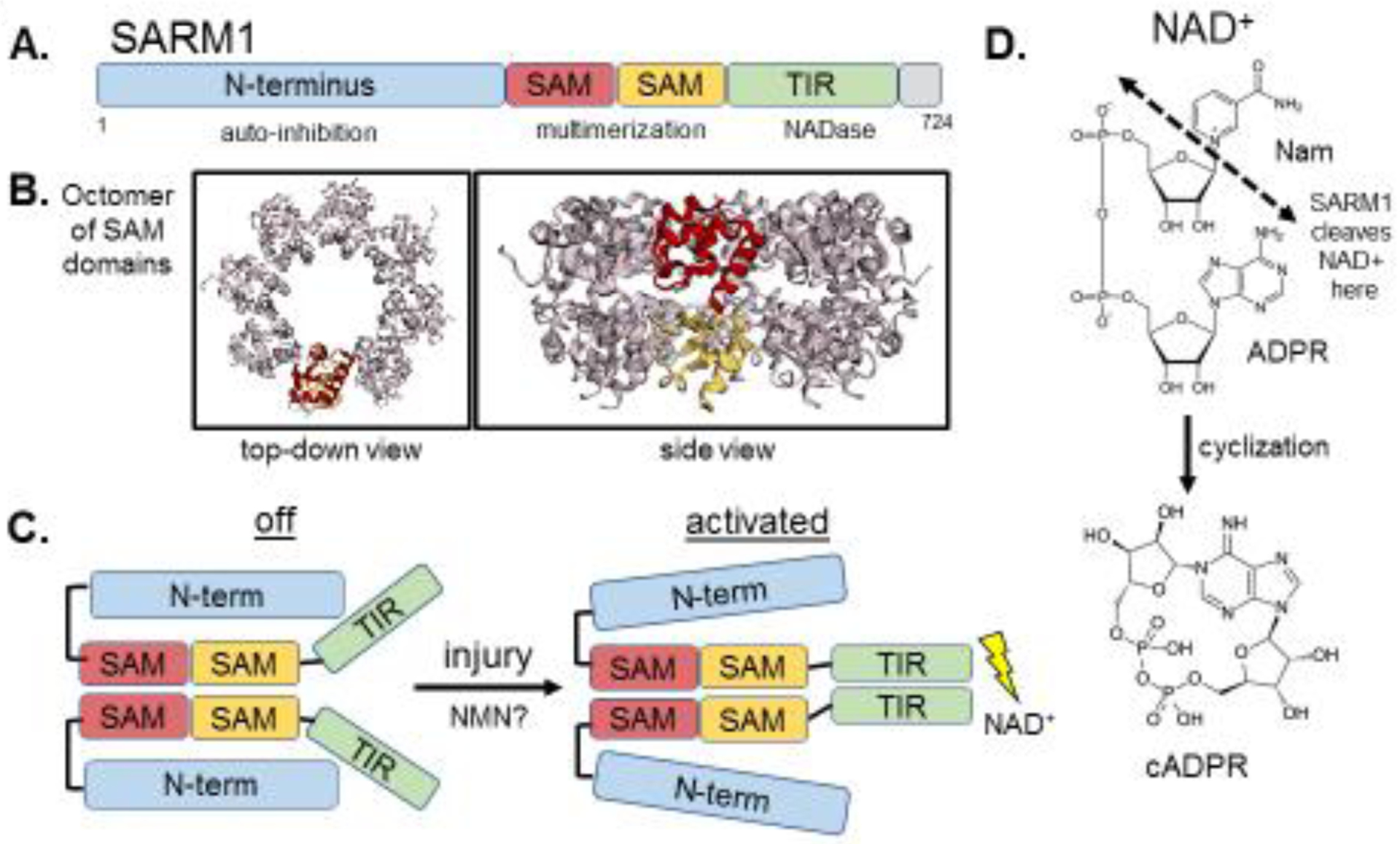
A. SARM1 contains an auto-inhibitory N-terminus, two tandem SAM domains and a C-terminal TIR domain. B. The crystal structure of SARM1’s SAM domains, which form an octomer (Image of PDB ID:6QWV[26], created with EzMol[76]). The side-view shows that the tandem SAM domains form two stacking rings in the octomer. C. A schematic of SARM1 at rest, where the N-terminus interacts with the TIR, preventing its dimerization, and after injury-induced activation, where the N-terminus-TIR interaction is disrupted and the TIR’s multimerize, leading to enzymatic degradation of NAD+. D. SARM1 cleaves NAD+ into Nam and ADPR, and can also cyclize ADPR into cyclic ADPR.
SARM1 is the founding member of a novel class of NAD-consuming enzymes
Our understanding of how SARM1 triggers axon degeneration emerged from detailed structure/function studies that came to the surprising conclusion that SARM1 has a novel enzymatic function that is essential for axon death. In healthy axons, SARM1 has an auto-inhibitory N-terminal region that restrains the degenerative capability of the two SAM domains and a C-terminal TIR domain[13] (Fig. 2). SARM1’s SAM domains mediate its multimerization and mimicking this function with forced dimerization of the TIR domain alone is sufficient to cause axon destruction and neuronal death, as well as a rapid decline in the levels of axonal NAD+[13,15], a phenomenon previously described during axon injury[10]. Importantly, a rapid loss of NAD+ is sufficient to drive axon degeneration[15], suggesting that activated SARM1’s ability to destroy axons is intimately linked to its regulation of NAD+ (Fig. 2). However, TIR domains function as scaffolding proteins in innate immunity signaling pathways[16], and so this known function provided no obvious clues to the mechanism by which SARM1 mediates NAD+ destruction.
After fruitless attempts to identify known NAD+-consuming enzymes that might cooperate with SARM1, the SARM1 TIR domain was demonstrated to have intrinsic NADase activity, cleaving NAD+ and producing nicotinamide, ADPR and cyclic ADPR[17] (Fig. 2). This enzymatic activity relies on a catalytic glutamate residue in the TIR domain and is essential for SARM1 to mediate axon degeneration[17] (Fig. 1). Interestingly, SARM1 is the major producer of neuronal cADPR, which also serves as a biomarker of SARM1 activity[18]. The identification of SARM1 as an enzyme redefined our understanding of the TIR domain, leading to the discovery of enzymatic TIR domains from multiple proteins in species as evolutionarily ancient as bacteria and archaea[19]. Remarkably, SARM1-triggered axon loss is the paradigm for an ancient cell death mechanism that is conserved across biological kingdoms. TIR-containing proteins in plants that mediate leaf cell death in response to pathogen recognition use the same highly conserved catalytic glutamate as SARM1’s TIR domain to degrade NAD+ and promote cell death[20,21].
While SARM1’s TIR domain promotes pathological rapid depletion of NAD+ and axon self-destruction after injury, SARM1’s TIR domain or other TIR-containing proteins could drive subtler cell signaling through NADase activity. In Drosophila and C. elegans SARM1 regulates signaling pathways[22,23], and yet the TIR enzyme function is conserved in invertebrates[17,24]. It will be important to investigate whether such signaling utilizes this NADase function. Indeed, such a non-degenerative signaling role may occur on the proximal axon side after injury in mammals, where SARM1 transmits a transcriptional immune response[25]. The potential for such a signaling role is supported by the recent finding that there is a low level of basal SARM1 NADase activity in uninjured neurons[18].
The structure of SARM1 has begun to be revealed during the past year. SARM1’s isolated SAM domains form an octameric ring that is necessary for its function[21,26] (Fig. 2). Crystal structures of SARM1’s TIR domain as well as related TIR domains from other organisms revealed a likely binding pocket for NAD+ containing the catalytic glutamate and described an interaction between the catalytic pocket and the BB loop of the TIR domain, a conserved region that participates in injury-induced SARM1 activation via an interaction with the auto-inhibitory N-terminus[21,27]. The precise interactions between SARM1’s N-terminus and TIR domain at rest and after injury-induced activation (Fig. 2) remain to be determined and will likely require structural characterization of the full-length protein.
The survival factor NMNAT2 restrains pro-degenerative SARM1 activity
Expression of WldS/NMNAT1 results in axon protection, but until recently it was unknown how its gain-of-function mechanism was connected to the function of the endogenous axon pro-degenerative protein SARM1. Over the past decade, NMNAT2 has emerged as the endogenous NMNAT enzyme present in healthy axons where it functions as an axon survival factor to restrain SARM1 from its destructive activity (Fig. 1).
NMNAT2 is a labile protein in axons and after injury it is rapidly degraded prior to axon fragmentation[28] (Fig. 1). Remarkably, loss of NMNAT2 from axons is sufficient to induce axon degeneration that is entirely dependent on SARM1. Nmnat2−/− mice are embryonic lethal but they can be rescued by expression of wlds/NMNAT1, a more stable version of NMNAT2, or by genetic loss of sarm1[28–30].
The regulation of NMNAT2 protein levels is a major determinant of axon health. Axonal NMNAT2 can be palmitoylated, a post-translational modification affecting both its trafficking and stability[31]. The non-palmitoylated axonal NMNAT2 is regulated by an atypical E3 ligase complex consisting of PHR1, FBXO45 and SKP1[32] (Fig. 1). Reduction of these genes results in increased levels of NMNAT2 and potent axon protection[33–37]. Palmitoylated axonal NMNAT2 is differentially regulated by a neuronal JNK stress kinase pathway activated by the MAP3K DLK and its paralog leucine-zipper kinase/LZK[32,38,39] (Fig. 1). MAPK signaling promotes the degradation of palmitoylated NMNAT2 and genetic or pharmacologic inhibition of MAPK signaling results in increased NMNAT2 levels and axon protection[32,40]. Axonal stressors that activate DLK also decrease NMNAT2 protein levels, sensitizing axons to SARM1-dependent degeneration[41] (Fig. 1). Interestingly, even a partial decrease in NMNAT2 can lead to peripheral nerve axonopathy in aged mice[42].
The mechanism of SARM1 activation remains an open question. Loss of NMNAT2 is a common effect of many injuries that lead to SARM1 activation, including axotomy, treatment with chemotherapy drugs vincristine and bortezomib, and mitochondrial toxins[28,41,43,44]. Since loss of NMNAT2 results in SARM1 activation and the ability of NMNATs to protect axons is dependent on their enzymatic activity, it was proposed that the NMNAT2 substrate NMN can promote axon degeneration, potentially via SARM1 activation[45,46] (Fig. 3). Increased NMN levels occur in injured axons as NMNAT2 is lost prior to degeneration and inhibition of the NMN-generating enzyme NAMPT provides axonal protection[45].
Fig. 3.
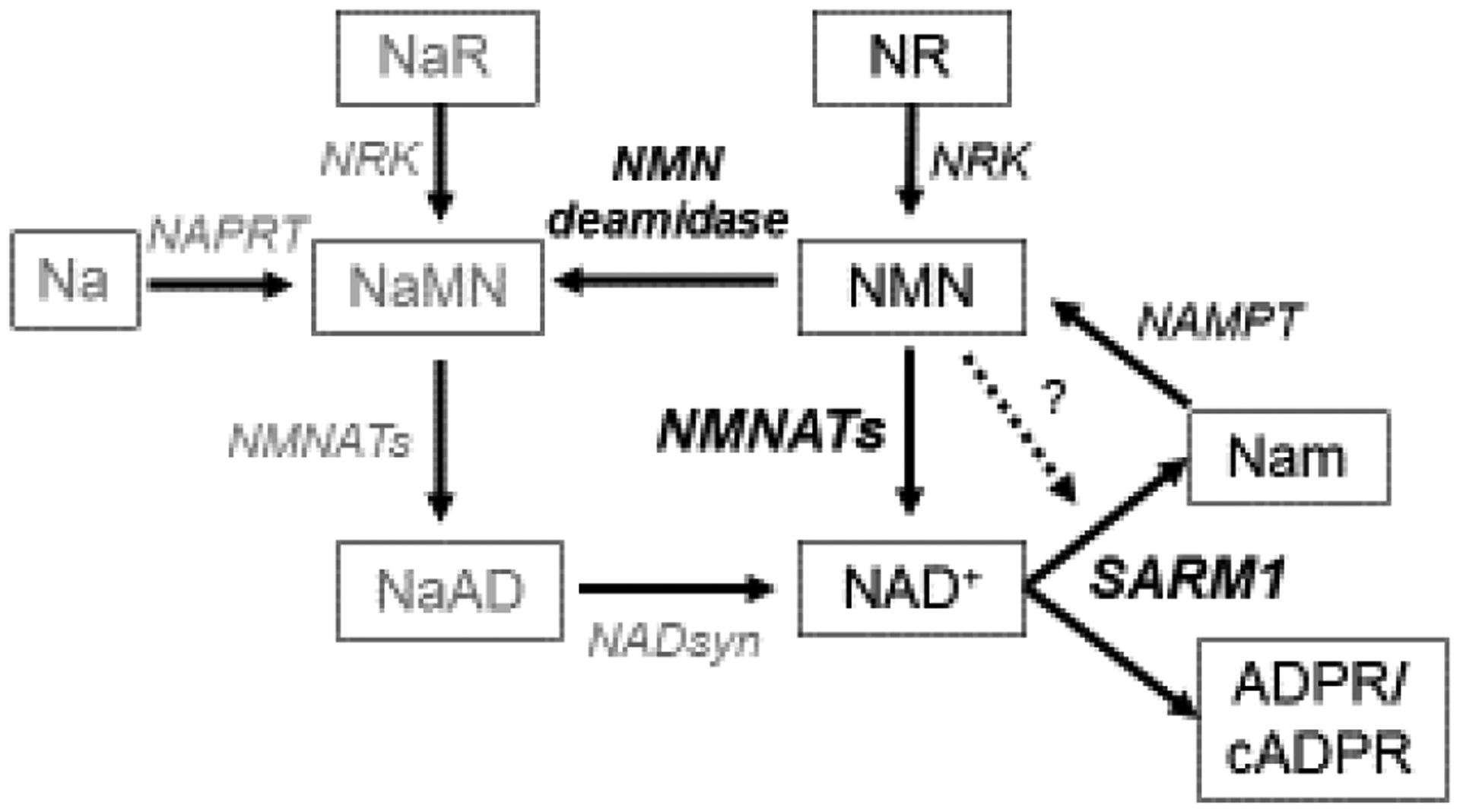
The NAD+ metabolic pathway. In the axon, NMN is turned into NAD+ by NMNAT2. When activated, SARM1 degrades NAD+ into nicotinamide (Nam) and ADPR or cyclic ADPR (cADPR). NMN may activate SARM1 (indicated by dashed line). Nam can be converted back to NMN by NAMPT. Another source of NMN is NR phosphorylated by NRK. The bacterial enzyme NMN deamidase can convert NMN to its deamidated counterpart, nicotinic acid mononucleotide (NaMN). Nicotinic acid riboside (NaR) and nicotinic acid (Na) can be alternate sources of NaMN (via NRK or NAPRT, respectively). NaMN can be also be made into NAD+ via conversion to NaAD by NMNAT2 and then by NADsyn into NAD+.
A potential pro-degenerative role for NMN is bolstered by experiments showing potent axon protection after injury or genetic loss of Nmnat2 from expression of the bacterial NMN-consuming enzyme NMN deamidase[45,47,48] (Fig. 3). However, manipulations of the NAD+ pathway that result in high NMN don’t necessarily promote axon degeneration[49], and can even be compatible with robust axon protection (nicotinamide riboside (NR) treatment, or NAMPT expression)[47]. Limiting NMN accumulation while treating with the NAD+ precursor nicotinic acid riboside (NaR) can provide lasting axon protection from vincristine, suggesting that perhaps both lowering NMN and raising NAD+ are important for axon protection[49] (Fig. 3). Surprisingly, NMNAT1 and NMN deamidase both functionally protect axons by keeping SARM1 from being activated to degrade NAD+ after injury, but not via an obvious common effect on NMN or NAD+ levels[47] (Fig. 1).
NMN may promote axon degeneration by activating SARM1 (Fig. 3). Indeed, NMN stimulates SARM1-dependent Ca2+ entry into injured axons[46]. A recent study offered the most direct evidence yet that NMN can activate SARM1. A chemically-modified and cell-permeant version of NMN, CZ-48, induced SARM1 NADase activity and cADPR production in HEK cells, potentially through increasing multimerization of SARM1’s TIR domains[50]. CZ-48 or NMN itself could directly activate purified SARM1’s NADase activity[50]. These results await further investigation in neurons. Since CZ-48 is a close analog of NMN, it could also inhibit NMNAT2, which would indirectly alter SARM1 activation. If NMN can stimulate SARM1’s axon self-destruction behavior, it will be of great importance to determine its mechanism of activation and how axons can survive or even maintain protection in the presence of elevated levels of NMN[47].
The Wallerian axon degeneration pathway is activated in many neurodegenerative conditions and can be therapeutically inhibited
While Wallerian degeneration is a specific pathological response to injury, the SARM1 pathway mediating Wallerian degeneration also promotes axon loss in various neurodegenerative conditions. The WldS mouse provides functional improvement in models of glaucoma, ischemia, Parkinson’s and Charcot-Marie-Tooth neuropathy[51]. These phenotypes likely reflect inhibition of SARM1 by WldS. Importantly, loss of SARM1 is much more potent than expression of WldS in rescuing Nmnat2−/− mice[52], implying that in mouse models of neurodegeneration that have been helped by WldS expression, the effect of SARM1 inhibition will be stronger.
Indeed, recent studies demonstrate that the absence of SARM1 is profoundly neuroprotective in a number of models of neurodegeneration including peripheral neuropathy. Peripheral neuropathy is the most common neurodegenerative disease, and involves a dying-back axon loss without cell body death, and so is a prime candidate to be mediated by SARM1. A series of studies demonstrate that SARM1 is essential for the development of chemotherapy-induced peripheral neuropathy in response to mechanistically distinct chemotherapeutics[43,53,54]. Loss of SARM1 is also protective in models of diabetic neuropathy[55]. These initial results suggest that SARM1-mediated axon destruction is a common mechanism in a variety of peripheral neuropathies and that its inhibition is a promising therapeutic strategy.
SARM1 also plays a role in central nervous system axon degeneration. After traumatic brain injury (TBI), mice lacking SARM1 have preserved neurological function and improved long-term axon integrity[56–59]. The axon degeneration pathway is also implicated in adult-onset neurodegeneration conditions such as amyotrophic lateral sclerosis (ALS). Activation of the pro-degenerative DLK MAP3K pathway is observed in a number of neurodegenerative mouse models and in Alzheimer’s and ALS patient samples[60], and activated DLK can sensitize axons to SARM1-destruction via lowering levels of the survival factors NMNAT2 and STMN2/SCG10, another labile axonal protein[41,61] (Fig. 1). Excitingly, direct links have recently emerged between two neurodegenerative diseases and the Wallerian degeneration pathway. Loss of STMN2 protein is a key downstream factor in TDP-43-associated ALS[62,63] and Parkinson’s disease[64] (Fig. 1), suggesting that the Wallerian degeneration pathway may be worth pursuing as a therapeutic approach in these neurodegenerative conditions. Thus far, mouse models of TDP-43- and SOD1-ALS have seen some or no benefit, respectively, from loss of SARM1[65,66], but the human data on STMN2 encourages further investigation.
The first mutations in endogenous Wallerian degeneration genes causing human disease were described in 2019. Missense mutations in NMNAT2 were found in two siblings with childhood onset polyneuropathy and accompanying erythromelalgia[67] and two stillborn siblings with fetal akinesia deformation sequence[68]. These NMNAT2 mutations range from partial to complete loss-of-function, with correlated severity of disease phenotypes[67,68]. The discovery of these mutations underscores the relevance of the Wallerian degeneration pathway to human disease and implies that even mild loss-of-function alleles in NMNAT2 or mutations in other genes in the pathway like SARM1 may directly cause or sensitize people to various neuropathies or neurodegenerative conditions.
Finally, if the Wallerian degeneration pathway is playing a pro-degenerative role in these various neurodegenerative conditions, it will be of great therapeutic interest to inhibit it. Manipulations that result in increased NMNAT2 should be axon protective and efforts are ongoing to create pharmacological DLK inhibitors or to find small molecules that alter NMNAT2 levels[32,69,70]. The discovery of enzymatic NADase activity in SARM1’s TIR domain also makes it an attractive candidate for the development of small molecule inhibitors. Recent insights into the SARM1 catalytic mechanism may facilitate the development of such an inhibitor[24]. In addition, AAV-mediated delivery of a potent dominant-negative SARM1 transgene provides profound in vivo axon protection[71], raising hopes for multiple lines of therapeutic modalities targeting the Wallerian degeneration pathway.
Conclusions
A comprehensive mechanistic understanding of the axon degeneration pathway emerged over the past five years. Essential axon survival factors like NMNAT2 and STMN2 are continuously supplied to axons and when this supply is lost or their levels are otherwise reduced, the pro-degenerative protein SARM1 is activated to destroy NAD+ and cause irreversible axon fragmentation (Fig. 1). The levels of these axon survival factors are tuned by key regulators, including an E3 ligase complex consisting of PHR1, FBXO45 and SKP1, a neuronal stress kinase pathway featuring dual MAP3Ks DLK and LZK, and now a central neurodegenerative disease protein, TDP-43 (Fig. 1). This mechanistic understanding is leading to the development of therapeutic targets for neurodegenerative disease.
Despite this rapid progress, several important questions remain. How are the axonal levels of the survival factors NMNAT2 and STMN2 regulated in vivo? How do DLK and the MAPK pathway promote NMNAT2 degradation? How does disruption of mitochondrial function result in lowered NMNAT2 and STMN2 and will other mitochondrial diseases result in SARM1 activation[44,72]? How is NMNAT2 palmitoylation regulated and what is the function of the different pools of palmitoylated vs. non-palmitoylated NMNAT2 in axons?
The details of SARM1 activation are also not yet fully understood. If NMN can activate SARM1, how does this occur? How do NMNAT1 and NMN deamidase keep SARM1 from being activated by injury? Are there other triggers for SARM1 activation? Is SARM1 multimerization regulated in a similar manner to other large signaling complexes like the inflammasome[73]? How does loss of NAD+ lead to axon fragmentation and are there meaningful steps downstream of SARM1, such as the yet unknown function of the Drosophila gene Axed[74]? Is axonal ATP loss in the axon a direct consequence of NAD+ decline and, if so, how does the resulting local energy deficit promote axon fragmentation[39,75]? Surprisingly, although there is a substantial increase in cADPR in injured axons that precedes morphological fragmentation, manipulations that raise or lower cADPR levels in axons do not change the time-course of degeneration, demonstrating that cADPR is likely not a pro-degenerative byproduct of SARM1’s cleavage of NAD+[18]. However, a potential pro-degenerative role for nicotinamide and ADPR, SARM1’s other byproducts, remains to be investigated.
There is great promise of a meaningful role for the SARM1 axon degeneration pathway in human disease. A major question in the field is which diseases show activation of the pathway and which approach will be the most fruitful for therapeutic intervention to promote axonal health and nervous system function. The identification of cADPR as a gene-sensitive biomarker of SARM1 activity in nerves and plasma neurofilament light chain (NfL) as a biomarker of axon fragmentation provides the first molecular assays of SARM1-dependent axon degeneration in vivo[18]. These biomarkers should aid in determining the status of SARM1 activation and the efficacy of therapies designed to inhibit the pathway. With the current pace of progress and the recent elucidation of the axon degeneration pathway, we can expect answers to many of these questions in the near future.
Highlights.
A mechanistic understanding of the endogenous axon degeneration pathway has emerged over the past few years.
Axonal health is maintained by a balance of axon survival factors NMNAT2 and STMN2 and pro-degenerative molecules DLK and SARM1.
The pro-degenerative protein SARM1 is the founding member of the TIR-domain family of NAD+-consuming enzymes and this enzymatic activity of SARM1 is essential for axon degeneration.
The endogenous axon degeneration pathway is activated in many neurodegenerative conditions and the pathway can be therapeutically targeted in several ways that are likely to be beneficial to treating human diseases.
Funding:
This work was supported by National Institutes of Health grants R01CA219866 and RO1NS087632.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and reHview of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement:
A. DiAntonio and Washington University are inventors on patents related to this work. A. DiAntonio is a cofounder of Disarm Therapeutics and a member of its scientific advisory board. The authors declare no additional competing financial interests.
References
- 1.Waller A: Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres. Philosophical Transactions of the Royal Society of London 1850, 140:423–429. [Google Scholar]
- 2.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S: Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci 1989, 1:27–33. [DOI] [PubMed] [Google Scholar]
- 3.Schlaepfer WW: Vincristine-induced axonal alterations in rat peripheral nerve. J Neuropathol Exp Neurol 1971, 30:488–505. [DOI] [PubMed] [Google Scholar]
- 4.Wang M-S, Wu Y, Culver DG, Glass JD: The Gene for Slow Wallerian Degeneration (Wlds) Is Also Protective against Vincristine Neuropathy. Neurobiol Dis 2001, 8:155–161. [DOI] [PubMed] [Google Scholar]
- 5.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC: Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol 2003, 13:669–673. [DOI] [PubMed] [Google Scholar]
- 6.Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH: An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A 1998, 95:9985–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araki T, Sasaki Y, Milbrandt J: Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305:1010–1013. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki Y, Vohra BPS, Lund FE, Milbrandt J: Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci 2009, 29:5525–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP: Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci 2010, 30:13291–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z: A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 2005, 170:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki Y, Araki T, Milbrandt J: Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 2006, 26:8484–8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. : dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J: Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci 2013, 33:13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Neill LAJ, Bowie AG: The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007, 7:353–364. [DOI] [PubMed] [Google Scholar]
- 15.**.Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J: SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015, 348:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that NAD+ degradation occurs downstream of SARM1 activation and is sufficient to promote axon degeneration.
- 16.Narayanan KB, Park HH: Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis 2015, 20:196–209. [DOI] [PubMed] [Google Scholar]
- 17.**.Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J: The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93:1334–1343.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study described a novel NADase activity in SARM1’s TIR domain that is necessary for its role in promoting axon degeneration.
- 18.Sasaki Y, Engber TM, Hughes RO, Figley MD, Wu T, Bosanac T, Devraj R, Milbrandt J, Krauss R, DiAntonio A: cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp Neurol 2020, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, Milbrandt J: TIR Domain Proteins Are an Ancient Family of NAD+-Consuming Enzymes. Curr Biol 2018, 28:421–430.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan L, Essuman K, Anderson RG, Sasaki Y, Monteiro F, Chung E-H, Nishimura EO, DiAntonio A, Milbrandt J, Dangl JL, et al. : TIR domains of plant immune receptors are NAD+-cleaving enzymes that promote cell death. Science 2019, 365:799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.**.Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, Qi T, Gilley J, Lai J-S, Rank MX, et al. : NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 2019, 365:793–799. [DOI] [PubMed] [Google Scholar]; This study described crystal structures of SARM1’s TIR domain and SAM domains and, with Wan et al., demonstrated that TIR domain NADase activity mediates an ancient degenerative mechanism conserved from plants to animals.
- 22.Chuang C-F, Bargmann CI: A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev 2005, 19:270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaughlin CN, Nechipurenko IV, Liu N, Broihier HT: A Toll receptor-FoxO pathway represses Pavarotti/MKLP1 to promote microtubule dynamics in motoneurons. J Cell Biol 2016, 214:459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loring HS, Icso JD, Nemmara VV, Thompson PR: Initial Kinetic Characterization of Sterile Alpha and Toll/Interleukin Receptor Motif-Containing Protein 1. Biochemistry 2020, doi: 10.1021/acs.biochem.9b01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.*.Wang Q, Zhang S, Liu T, Wang H, Liu K, Wang Q, Zeng W: Sarm1/Myd88–5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep 2018, 23:716–724. [DOI] [PubMed] [Google Scholar]; This study demonstrated a non-degenerative role for SARM1 signaling to regulate a transcriptional immune response on the proximal side of the axon after injury.
- 26.*.Sporny M, Guez-Haddad J, Lebendiker M, Ulisse V, Volf A, Mim C, Isupov MN, Opatowsky Y: Structural Evidence for an Octameric Ring Arrangement of SARM1. J Mol Biol 2019, 431:3591–3605. [DOI] [PubMed] [Google Scholar]; This study provided the first structural evidence that SARM1’s SAM domains form a closed octomeric ring.
- 27.Summers DW, Gibson DA, DiAntonio A, Milbrandt J: SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc Natl Acad Sci U S A 2016, 113:E6271–E6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilley J, Coleman MP: Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 2010, 8:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilley J, Adalbert R, Yu G, Coleman MP: Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci 2013, 33:13410–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.**.Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP: Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep 2015, 10:1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided strong genetic evidence that loss of NMNAT2 leads to SARM1-dependent axon destruction.
- 31.Milde S, Gilley J, Coleman MP: Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol 2013, 11:e1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Summers DW, Milbrandt J, DiAntonio A: Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc Natl Acad Sci U S A 2018, 115:E8746–E8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA: The Highwire Ubiquitin Ligase Promotes Axonal Degeneration by Tuning Levels of Nmnat Protein. PLoS Biol 2012, 10:e1001440–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babetto E, Beirowski B, Russler EV, Milbrandt J, DiAntonio A: The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep 2013, 3:1422–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brace EJ, Wu C, Valakh V, DiAntonio A: SkpA restrains synaptic terminal growth during development and promotes axonal degeneration following injury. J Neurosci 2014, 34:8398–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamagishi Y, Tessier-Lavigne M: An Atypical SCF-like Ubiquitin Ligase Complex Promotes Wallerian Degeneration through Regulation of Axonal Nmnat2. Cell Rep 2016, 17:774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desbois M, Crawley O, Evans PR, Baker ST, Masuho I, Yasuda R, Grill B: PAM forms an atypical SCF ubiquitin ligase complex that ubiquitinates and degrades NMNAT2. J Biol Chem 2018, 293:13897–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A: A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci 2009, 12:387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M: Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 2015, 160:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A: MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 2017, 6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.*.Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A: DLK Activation Synergizes with Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Mol Neurobiol 2019, doi: 10.1007/s12035-019-01796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that activated DLK can promote loss of the axon survival factors NMNAT2 and STMN2 which sensitizes axons to SARM1-dependent axon degeneration.
- 42.*.Gilley J, Mayer PR, Yu G, Coleman MP: Low levels of NMNAT2 compromise axon development and survival. Hum Mol Genet 2019, 28:448–458. [DOI] [PubMed] [Google Scholar]; This study showed that even moderate reduction in levels of NMNAT2 can lead to deficits in axon survival and peripheral neuropathy in aged mice.
- 43.Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, DiAntonio A: Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 2019, 4(17):e129920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, Lucci C, Sanchez-Martinez A, Whitworth AJ, Conforti L, et al. : Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis 2020, 134:104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.**.Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, et al. : A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ 2015, 22:731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to demonstrate that increased NMN levels after injury can promote axon degeneration.
- 46.Loreto A, Di Stefano M, Gering M, Conforti L: Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca(2+) Influx but Only Modestly Influenced by Mitochondria. Cell Rep 2015, 13:2539–2552. [DOI] [PubMed] [Google Scholar]
- 47.*.Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J: NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 2016, 5:1010. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided evidence that the axoprotective effect of WldS/NMNAT1 is to inhibit SARM1 from consuming NAD+ after injury.
- 48.Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, Webster J, Donaldson LF, Gering M, Raffaelli N, et al. : NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr Biol 2017, 27:784–794. [DOI] [PubMed] [Google Scholar]
- 49.Liu H-W, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C, Goodman RH: Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proc Natl Acad Sci U S A 2018, 27:201809392–201809396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.*.Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, Li T, Li SL, Lu JG, Zhang L, et al. : A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 2019, 15:452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that NMN can directly activate SARM1’s NADase activity in vitro, possibly by promoting the multimerization of SARM1’s TIR domains.
- 51.Conforti L, Gilley J, Coleman MP: Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 2014, 15:394–409. [DOI] [PubMed] [Google Scholar]
- 52.Gilley J, Ribchester RR, Coleman MP: Sarm1 Deletion, but Not WldS, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. CellReports 2017, 21:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A: Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139:3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turkiew E, Falconer D, Reed N, Höke A: Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J Peripher Nerv Syst 2017, 22:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng Y, Liu J, Luan Y, Liu Z, Lai H, Zhong W, Yang Y, Yu H, Feng N, Wang H, et al. : Sarm1 Gene Deficiency Attenuates Diabetic Peripheral Neuropathy in Mice. Diabetes 2019, 68:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown RH Jr: Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain 2016, 139:1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ziogas NK, Koliatsos VE: Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms with High-Resolution Anatomical Methods. J Neurosci 2018, 38:4031–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marion CM, McDaniel DP, Armstrong RC: Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp Neurol 2019, 321:113040. [DOI] [PubMed] [Google Scholar]
- 59.Maynard ME, Redell JB, Zhao J, Hood KN, Vita SM, Kobori N, Dash PK: Sarm1 loss reduces axonal damage and improves cognitive outcome after repetitive mild closed head injury. Exp Neurol 2020, 327:113207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Sengupta Ghosh A, Jiang Z, Lee S-H, et al. : Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci Transl Med 2017, 9:eaag0394. [DOI] [PubMed] [Google Scholar]
- 61.Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A: SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A 2012, 109:E3696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.*.Melamed Z ‘ev, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. : Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 2019, 22:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, along with Klim et al., showed that loss of the axon survival factor STMN2 is a key downstream event in TDP-43-ALS.
- 63.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. : ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 2019, 22:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Zhang Y, Wang M, Song W-M, Shen Q, McKenzie A, Choi I, Zhou X, Pan P-Y, Yue Z, et al. : The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat Commun 2019, 10:5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters OM, Lewis EA, Osterloh JM, Weiss A, Salameh JS, Metterville J, Brown RH Jr, Freeman MR: Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 2018, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White MA, Lin Z, Kim E, Henstridge CM, Pena Altamira E, Hunt CK, Burchill E, Callaghan I, Loreto A, Brown-Wright H, et al. : Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathologica Communications 2019, 7:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.*.Huppke P, Wegener E, Gilley J, Angeletti C, Kurth I, Drenth JPH, Stadelmann C, Barrantes-Freer A, Brück W, Thiele H, et al. : Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp Neurol 2019, 320:112958. [DOI] [PubMed] [Google Scholar]; This study, along with Lukacs et al., first showed that mutations in NMNAT2 can lead to human disease.
- 68.Lukacs M, Gilley J, Zhu Y, Orsomando G, Angeletti C, Liu J, Yang X, Park J, Hopkin RJ, Coleman MP, et al. : Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Exp Neurol 2019, 320:112961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siu M, Sengupta Ghosh A, Lewcock JW: Dual Leucine Zipper Kinase Inhibitors for the Treatment of Neurodegeneration. J Med Chem 2018, 61:8078–8087. [DOI] [PubMed] [Google Scholar]
- 70.Ali YO, Bradley G, Lu H-C: Screening with an NMNAT2-MSD platform identifies small molecules that modulate NMNAT2 levels in cortical neurons. Sci Rep 2017, 7:43846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.*.Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, Park J, DiAntonio A, Milbrandt J: Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med 2019, 216:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed the first evidence that a therapy targeting SARM1 can protect axons after injury in vivo.
- 72.Summers DW, DiAntonio A, Milbrandt J: Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci 2014, 34:9338–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nanson JD, Kobe B, Ve T: Death, TIR, and RHIM: Self- assembling domains involved in innate immunity and cell- death signaling. J Leukoc Biol 2019, 105:363–375. [DOI] [PubMed] [Google Scholar]
- 74.Neukomm LJ, Burdett TC, Seeds AM, Hampel S, Coutinho-Budd JC, Farley JE, Wong J, Karadeniz YB, Osterloh JM, Sheehan AE, et al. : Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron 2017, 95:78–91.e5. [DOI] [PubMed] [Google Scholar]
- 75.Schlaepfer WW: Effects of energy deprivation on Wallerian degeneration in isolated segments of rat peripheral nerve. Brain Res 1974, 78:71–81. [DOI] [PubMed] [Google Scholar]
- 76.Reynolds CR, Islam SA, Sternberg MJE: EzMol: A Web Server Wizard for the Rapid Visualization and Image Production of Protein and Nucleic Acid Structures. J Mol Biol 2018, 430:2244–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]