

Abstract
Purpose:
Head and neck cancers (HNSCC) are routinely treated with radiotherapy; however normal tissue toxicity remains a concern. Therefore, it is important to validate treatment modalities combining molecularly targeted agents with radiotherapy to improve the therapeutic ratio. The aim of this study was to assess the ability of the PARP inhibitor niraparib (MK-4827) alone, or in combination with cell cycle checkpoint abrogating drugs targeting Chk1 (MK-8776) or Wee1 (MK-1775), to radiosensitize HNSCCs in the context of HPV status.
Materials and Methods:
Accepted Manuscript PARP1, PARP2, Chk1 or Wee1 shRNA constructs were analyzed from an in vivo shRNA screen of HNSCC xenografts comparing radiosensitization differences between HPV(+) and HPV(−) tumors. Radiosensitization by niraparib alone or in combination with MK-8776 or MK-1775 was assessed by clonogenic survival in HPV(−) and HPV(+) cells; and the role of p16 in determining response was explored. Relative expressions of DNA repair genes were compared by PCR array in HPV(+) and HPV(−) cells, and following siRNA-mediated knockdown of TRIP12 in HPV(−) cells.
Results:
In vivo shRNA screening showed a modest preferential radiosensitization by Wee1 and PARP2 in HPV(−) and Chk1 in HPV(+) tumor models. Niraparib alone enhanced the radiosensitivity of all HNSCC cell lines tested. However, HPV(−) cells were sensitized to a greater degree, as suggested by the shRNA screen. When combined with MK-8776 or MK-1775, radiosensitization was further enhanced in an HPV dependent manner with HPV(+) cells enhanced by MK-8776 and HPV(−) cells enhanced by MK-1775. A PCR array for DNA repair genes showed PARP and HR proteins BRCA1 and RAD51 were much lower in HPV(+) cells than in HPV(−). Similarly, directly knocking down p16-dependent TRIP12 decreased expression of these same genes. Overexpressing p16 decreased TRIP12 expression and increased radiosensitivity in HPV(−) HN5. However, while PARP inhibition led to significant radiosensitization in the control, it led to no further significant radiosensitization in p16 overexpressing cells. Forced p16 expression in HPV(−) HN5 increased accumulation in G1 and subG1 and limited progression to S phase, thus reducing effectiveness of PARP inhibition.
Conclusions:
Niraparib effectively radiosensitizes HNSCCs with a greater benefit seen in HPV(−). HPV status also plays a role in response to MK-8776 or MK-1775 when combined with niraparib due to differences in DNA repair mechanisms. This study suggests that using cell cycle abrogators in combination with PARP inhibitors may be a beneficial treatment option in HNSCC, but also emphasizes the importance of HPV status when considering effective treatment strategies.
Keywords: HPV, PARP, Chk1, Wee1, HNSCC
Introduction
Human papilloma virus (HPV) infection leads to various types of squamous cell carcinomas including those of the head and neck (HNSCC). Radiation alone or in combination with cytotoxic chemotherapy forms the backbone of therapy for this disease, leading to high rates of survival even in locally advanced disease. However, this treatment can have dramatic toxicity leading to many clinical trials which are focused on enhancing the therapeutic ratio in HPV positive (HPV(+)) HNSCC. In fact, recent data from a phase 1 trial in HNSCC combining radiation, cetuximab and PARP inhibition have been published showing acceptable toxicity and good outcomes (Karam et al. 2018). Though, further randomized data is needed to potentially incorporate this combination in clinic.
In addition to potentially improving outcomes in HPV(+) HNSCC, studying the mechanisms involved in the response of HPV-related malignancies to radiation-based therapy may reveal new targets that can be utilized for treating other non-HPV derived malignancies (HPV(−)). It is known that HPV(+) HNSCCs are generally more sensitive to radiation both in pre-clinical models and in patients (Ang et al. 2010; Ang and Sturgis 2012; Wang, Zhang, et al. 2017). Exploring the reasons behind this difference in response may reveal druggable targets that could be used to improve the radiosensitivity of more radioresistant cancers.
Poly(ADP-Ribose) polymerase (PARP) inhibitors enhance radiosensitivity by interfering with the repair of sublethal single strand DNA breaks (SSB) by trapping PARP and thus blocking DNA replication leading to lethal double strand DNA breaks (DSB) (Godon et al. 2008; Chalmers et al. 2010; Rouleau et al. 2010; Murai et al. 2012; Bridges et al. 2014; Guster et al. 2014). DSBs are preferentially repaired by homologous recombination (HR) in the S and G2 phase of the cell cycle. When HR is not possible, DSBs are repaired by non-homologous end joining (NHEJ) which is more error prone and can lead to genomic instability and eventual cell death. Cells with defective homologous DNA repair pathways have been shown to have an enhanced response to PARP inhibition (Farmer et al. 2005; McCabe et al. 2005; McCabe et al. 2006). Therefore, we reasoned that targeting the HR pathway with a Chk1 or Wee1 inhibitor in combination with PARP inhibition would lead to an improved response to radiation.
In a previous collaborative project with Dr. Raymond E. Meyn, we tested several DNA repair inhibitors to determine their efficacy as radiosensitizers and explored the mechanisms of their actions. From this work we found that MK-4827, a PARP inhibitor, MK-1775, a Wee1 kinase inhibitor, and MK-8776, a Chk1 kinase inhibitor, are potent radioenhancers in various tumor types including lung, breast and prostate (Bridges et al. 2011; Wang et al. 2012; Bridges et al. 2014; Bridges et al. 2016). Here we present an analysis of in vivo shRNA screening data for these specific DNA-damage repair proteins showing a modest preferential radiosensitization of HPV(−) tumors by Wee1 inhibition and of HPV(+) tumors by Chk1 inhibition. We then expand upon Dr. Meyn’s previous work with combinations of each of these inhibitors in HNSCC, in the context of HPV status.
Materials and methods
Cell lines
HEK-293T (293T), FaDu, UPCI:SCC154 (SCC154) and UPCI:SCC152 (SCC152) cell lines were obtained from the American Type Culture Collection (ATCC). HN5 and UMSCC47 cell lines were obtained from Dr. Jeffery Myers, UT MD Anderson Cancer Center, Houston, TX, USA. FaDu, UPCI:SCC154 and UPCI:SCC152 cells were maintained in MEM (Gibco) supplemented with 10% heat inactivated Fetal Bovine Serum (Sigma), 1% MEM NEAA (Gibco), Sodium Pyruvate (Gibco) and Penicillin Streptomycin Solution (Corning). UMSCC47 cells were maintained in DMEM (Gibco) supplemented with 10% heat inactivated Fetal Bovine Serum (Sigma), 2% MEM Vitamin Solution (Gibco), 1% MEM NEAA (Gibco), Sodium Pyruvate (Gibco) and Penicillin Streptomycin Solution (Corning). 293T and HN5 cells were maintained in DMEM/F-12 50/50 (Corning) supplemented with 10% heat inactivated Fetal Bovine Serum (Sigma) and 1% Penicillin Streptomycin Solution (Corning). All cell lines used were authenticated by STR fingerprinting by the Characterized Cell Line Core Facility at UT MD Anderson.
In vivo shRNA screening
Our group and others have adopted and optimized a classical shRNA screen to function in vivo in the context of an intact host stroma and tumor microenvironment, as described by Carugo and colleagues (Carugo et al. 2016). Briefly, in this study, HNSCC cells were transduced with a pooled library of barcoded shRNAs (10 shRNAs per gene). Cells were transduced with a multiplicity of infection (MOI) of <1 to express a single shRNA per cell, which was validated for each cell-derived xenograft model prior to the experiment. Infected cells were then briefly selected with puromycin (1–2 days), implanted subQ into the flank of nude mice and tumors were allowed to establish. Once the tumors had reached approximately 100 mm3, each was irradiated at 2 Gy/treatment to a total dose needed to achieve 20% tumor volume reduction over two weeks (typically 6–10 Gy total dose). Approximately two weeks following completion of radiation, the resultant tumor tissue was harvested, genomic DNA isolated and shRNA abundance determined by deep sequencing. The representation of shRNAs in tumor tissue was compared to the reference control cells collected immediately prior to injection. The data shown represent percentiles of median fold change of the abundance of the shRNA constructs for each given gene compared to reference.
Inhibitors
Niraparib (MK-4827), MK-1775 and MK-8776 (APExBIO Technology LLC, Houston, Texas).
MTS assay
In a 96 well plate, 2000–4000 cells were plated/well. Cells were treated with MK-4827 with doses ranging from 0–20μM for 24h with 6 replicates/dose. MTS assay was performed using CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega) according to manufacturer’s instruction.
Clonogenic survival assay
Clonogenicity was tested following inhibitor treatment and irradiation using an X-RAD 320 biological irradiator (Precision X-Ray) as previously described (Bridges et al. 2011). Briefly, exponentially growing cells were treated with inhibitor (1μM MK-4827,200nM MK-8776 and/or 200nM MK-1775) for 1h then irradiated. 24h later cells were trypsinized and replated in triplicate into 6-well dishes and allowed to grow for 10–21 days for colony formation. Colonies were then stained with 0.25% crystal violet in methanol and colonies with more than 50 cells were counted. The percent plating efficiency was determined by dividing the number of colonies by the number of cells plated and survival curves were generated by extrapolation from radiation surviving fractions using alpha/beta analysis with GraphPad Prism (v 7.0) (Graphpad). Each experiment was plated in triplicate and repeated at least three independent times. Error bars represent standard error.
siRNA transfection
HN5 cells were transfected with 100nM of TRIP12 siRNA (Dharmacon, onTarget plus) or mock transfected by electroporating in 100 microliters transfection reagent T (Lonza, VCA-1002) using program T-001 on the Amaxa Nucleofector II. After electroporation, cells were washed and then plated in antibiotic free media containing 1% serum overnight. Normal growth media was added the following day for 24h hours followed by RNA extraction and subsequent PCR array analysis.
PCR array
Extracted RNA was prepared using RNeasy Kit (Qiagen). We used iScript Reverse Transcription Supermix (Bio-Rad) to convert total RNA into cDNA. cDNA was labeled using iTaq Universal SYBR green supermix (Bio-Rad). 8ng cDNA was loaded per well into a PrimePCR array plate for DNA damage signaling pathway (Bio-Rad, SAB Target List) H96 customized to add TRIP12. We ran RT-PCR using BioRad CFX Connect and compared samples using BioRad CFX Manager.
Over-expression of p16
Stable over-expression of p16 or rfp pLOC Turbo Lentiviral vector (Precision LentiORF, Dharmacon) were co-transfected with lentiviral plasmids DR.8 and VSVG (Addgene) in HEK-293T for 48h using Fugene (Promega) transfection reagent. Media plus lentivirus was then filtered through a 0.45 micron PES syringe filter and added to cells. 5 μg/ml polybrene was added and cells were transduced for 6h. Transduction procedure was repeated for 2 consecutive days. 3 days after transduction, stable expressing cells were selected with 10 μg/ml blasticidin.
Western blot analysis
Following treatment, cells were lysed with extraction buffer containing 20mM Hepes, pH 7.9, 400mM NaCl, 0.1mM EDTA pH 8.0, 0.1mM EGTA pH 7.0 and XPert protease and phosphatase inhibitors were added at 1:100 dilution (GenDepot) then sonicated. Equal amounts of protein were loaded into 4–15% gradient polyacrylamide gels (Bio-Rad) then transferred to PVDF membranes for 10min at 25V using Trans-Blot Turbo (Bio-Rad). Membranes were incubated in 5% dry milk for 1h then incubated in primary antibody p16 1:1000 (BD Biosciences), Trip12 1:10,000 (Abcam) or Actin 1:2000 (Cell Signaling Technologies) overnight at 4°C. Immunoblots were detected using horseradish peroxidase-conjugated secondary antibodies (GE) and ECL2 chemiluminescent substrate (Pierce).
Cell cycle analysis
Cells were trypsinized and collected at 4 or 24h after irradiation and washed with PBS. They were then fixed in 70% ethanol at 4°C for 1h followed by 2 PBS washes. Cells were then stained with APO-DIRECT PI/RNase Staining Buffer (BD Biosciences) for 30 min. and analyzed using the BD Accuri C6 flow cytometer and software (BD Biosciences).
Statistics
In vitro data was analyzed using GraphPad Prism (v 7.0) (Graphpad). Individual values on clonogenic assay were analyzed using Two-Way ANOVA analysis with post-hoc comparisons between values. Analysis of in vivo shRNA screening data was performed using R software. All p-values shown are two-sided.
Results and discussion
Analysis of in vivo shRNA screening data reveals a modest HPV specificity for radiosensitization
We had previously performed in vivo shRNA screening of tumors generated from five different HNSCC cell lines (2 HPV(+) and 3 HPV(−)) generally using the method described by Carrugo and colleagues (Carugo et al. 2016). We first examined the magnitude of radiosensitization seen in the in vivo screen following expression of the shRNA constructs for i) PARP1 or PARP2 (both targeted by niraparib), ii) Chk1 or iii) Wee1. We then compared the magnitude of this effect between the HPV(+) and HPV(−) tumor models (Figure 1A). There was a modest preferential radiosensitization by Wee1 and PARP2 seen on the screen in HPV(−) models, and a modest preferential radiosensitization of HPV(+) tumor models by Chk1 inhibition. No difference was seen in the in vivo screen in radiosensitization induced by PARP1 inhibition between HPV(+) and HPV(−) tumor models.
Figure 1: PARPi radiosensitizes HNSCC cells with little cytotoxicity as a single agent.
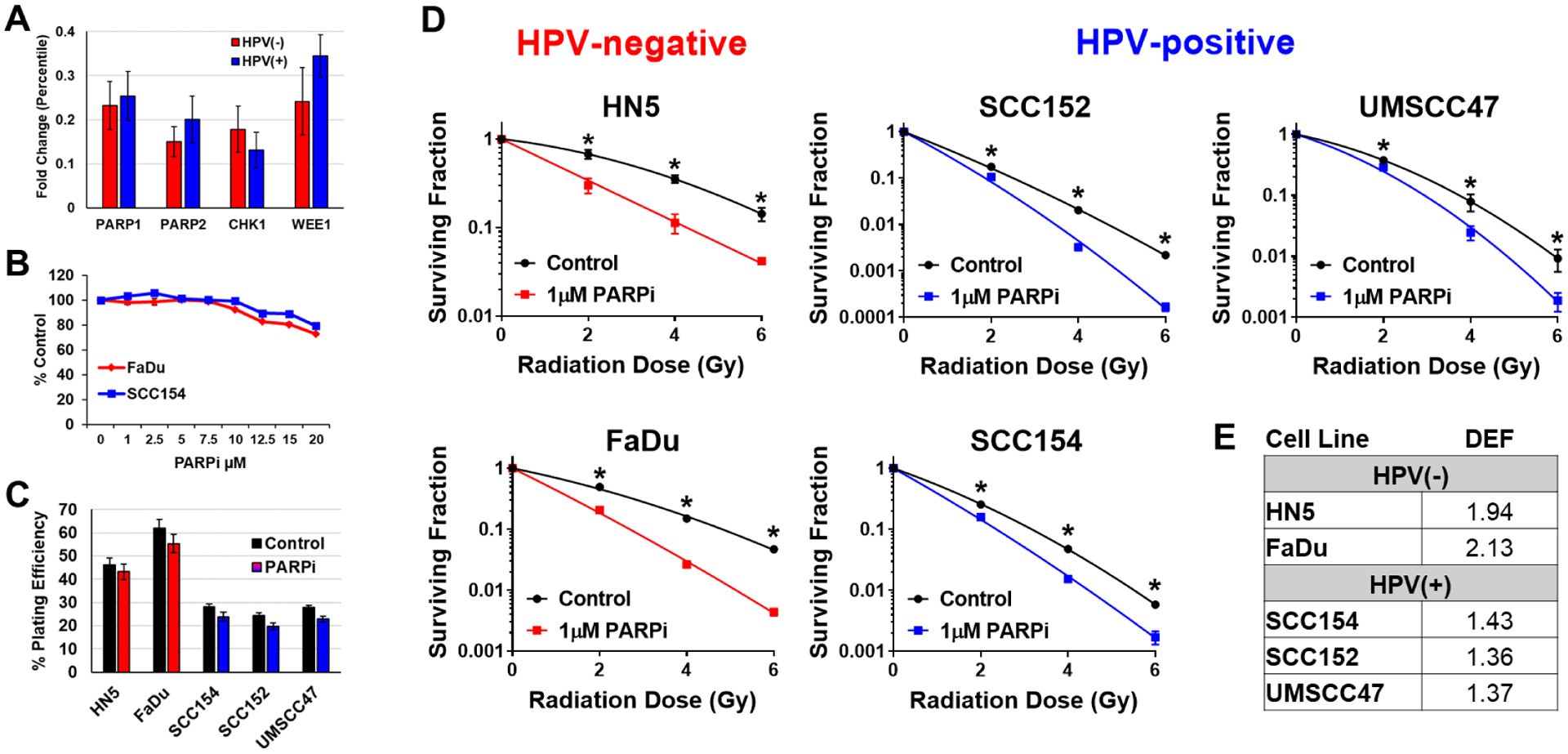
(A) In vivo shRNA screening of tumors generated from five different HNSCC cell lines (2 HPV(+) and 3 HPV(−)) comparing the difference in magnitude of radiosensitization between HPV(−) and HPV(+) tumor models following the expression of the shRNA constructs for PARP1, PARP2, Chk1 or Wee1. (B) MTS assay at 24h after treatment with niraparib (PARPi) at doses of 0–20μM with 6 replicates/dose. (C) Plating efficiencies in HPV(−) (red) and HPV(+) (blue) cell lines taken from CSAs shown in D. (D) Cells were treated with 1μM niraparib (PARPi) for 1h then irradiated with 0–6Gy and replated 24h later for colony formation. (E) Radiation dose enhancement factors (DEF) calculated at surviving fraction of 0.5 in HPV(−) and HPV(+) cell lines following treatment with niraparib (PARPi) and radiation.
Niraparib radiosensitizes human HNSCC cells
Previously we showed that PARP inhibition by niraparib radiosensitized lung and breast cancer cell lines in a p53-independent manner while not radiosensitizing normal lung fibroblasts (Bridges et al. 2014). Moreover, our screen suggested a modest radiosensitization by PARP inhibition in general as well as, in the case of PARP2 inhibition, a possible preferential radiosensitization of HPV(−) models. Therefore, we assessed whether HPV status could play a role in determining radioenhancement from PARP inhibition in HNSCC. Three HPV(+) and two HPV(−) HNSCC cell lines were used in this study and their HPV status had previously been established. Radiosensitization was assessed using clonogenic survival assays with 1μM niraparib 1h prior to and 24h postradiation. Cytotoxicity was minimal in all cell lines tested independent of HPV status up to a 20μM concentration (Figure 1B) and had a minimal effect on plating efficiency at 1μM (Figure 1C). Optimal in vitro sequencing of niraparib and radiation time course had previously been established by measuring PARP enzymatic activity using a chemiluminescent assay (Bridges et al. 2014).
Clonogenic survival assay curves for HPV(−) cell lines HN5 and FaDu, and HPV(+) cell lines UMSCC47, UPCI:SCC152 and UPCI:SCC154 were generated (Figure 1D). The radiation dose enhancement factors (DEF) were determined by calculating the ratio of radiation doses needed to achieve a 50% surviving fraction and are presented in Figure 1E. Despite their differential HPV status, all five HNSCC cell lines were radiosensitized by 1μM niraparib with significant differences in SF2, SF4 and SF6, but interestingly, similar to the in vivo screening data, HPV(−) cell lines were enhanced to a greater degree (Figure 1D–E).
Exogenous expression of HPV surrogate marker p16INK4a in HPV(−) cells limits niraparib radioenhancement by blocking G1/S transition
p16 is known to be overexpressed in HPV(+) cancers and is a surrogate marker of HPV status (El-Naggar and Westra 2012; Stephen et al. 2013). We previously established that p16 regulates ubiquitin-dependent homologous DNA repair by down-regulating a key E3 ubiquitin ligase TRIP12, which is needed for efficient BRCA1 dependent DNA repair. TRIP12 was found to have higher basal level expression in HPV(−) than HPV(+) cell lines, and when TRIP12 was knocked down directly with siRNA in HPV(−) cells, BRCA1 foci formation was reduced leading to a more radiosensitive phenotype (Wang, Zhang, et al. 2017). Since PARP inhibitors have been shown to be more effective as single agents in tumors deficient in HR (Farmer et al. 2005; McCabe et al. 2005; McCabe et al. 2006), we had previously expected that HPV(+) cell lines would also respond better to PARP inhibition due to their reduced BRCA1 expression. However, based on our in vivo screening data and in vitro validation data we wished to explore this question in more detail.
In comparing HPV(+) UMSCC47 with HPV(−) HN5 by PCR array, we found that PARP as well as HR proteins BRCA1 and RAD51 were much lower in UMSCC47 (Figure 2A). We also show that directly knocking down p16-dependent TRIP12 lowers expression of these same HR proteins suggesting that these proteins are regulated by TRIP12 (Figure 2B). If HR expression levels were the sole predictor for response to PARP inhibition, we would expect to see preferential cytotoxicity in HR reduced HPV(+) cells as well as preferential radioenhancement due to the inefficient repair of PARP inhibitor induced DNA lesions in the G2/M checkpoint.
Figure 2: Exogenous expression of p16 in HPV(−) cells down-regulates HR proteins through TRIP12 and limits niraparib radioenhancement by blocking G1/S transition.
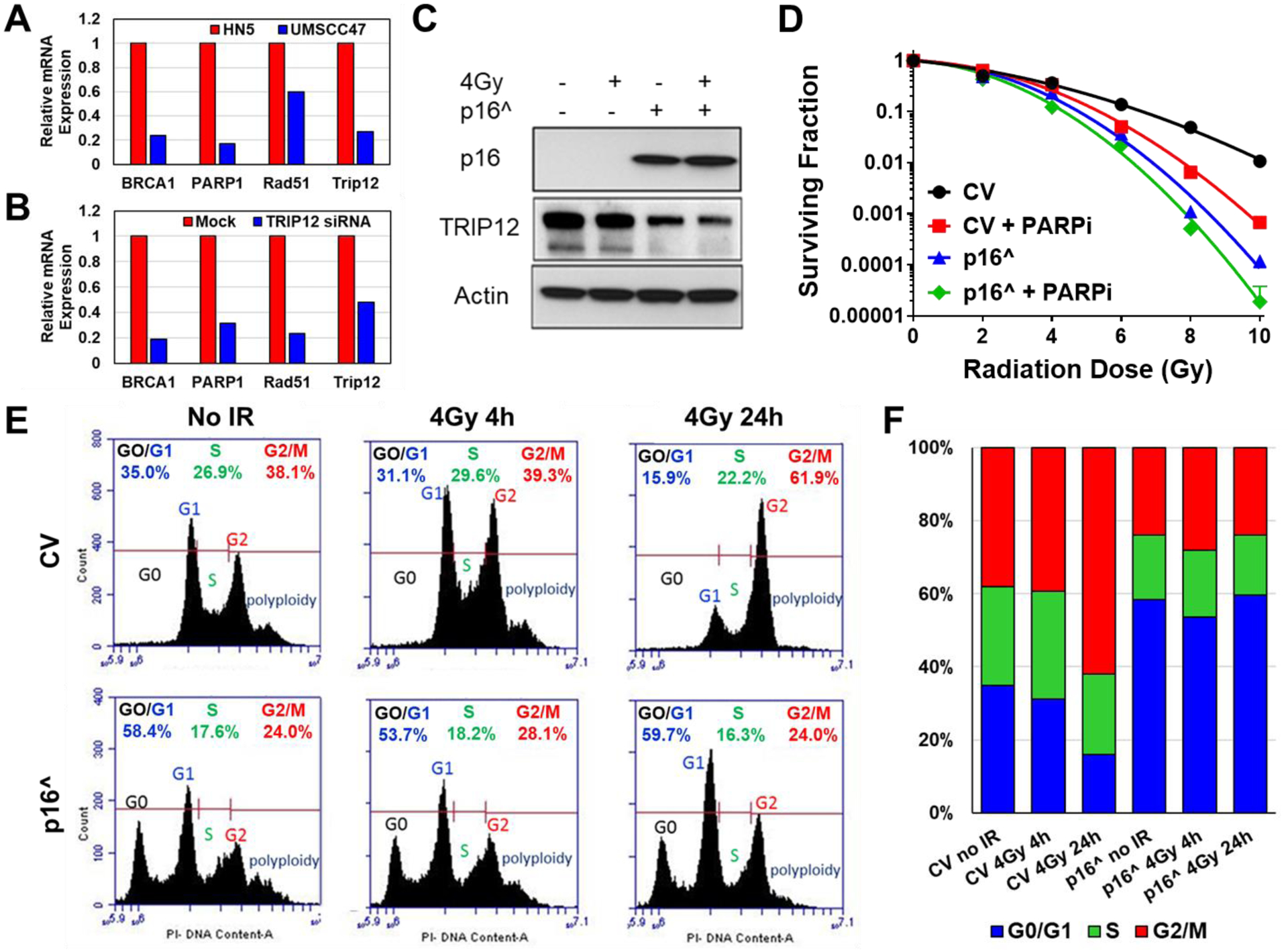
(A) PCR array comparing relative mRNA expressions in HPV(+) UMSCC47 (blue) normalized to HPV(−) HN5 (red). (B) PCR array comparing relative mRNA expressions in HPV(−) HN5 following siRNA knockdown of TRIP12 (blue) normalized to HN5 mock transfected (Mock). (C) Western blot following p16 overexpression (p16^) in HN5 cells. (D) p16 was overexpressed in HN5 cells then treated with 1μM niraparib (PARPi) for 1h followed by irradiation with 0–10Gy and replated 24h later for colony formation. (E) Cell cycle histograms following p16 overexpression and IR in HN5 cells. (F) Cell cycle distribution from E.
However, this was not the case as illustrated in Figure 1. This demonstrates that mere expression levels of HR proteins cannot accurately predict response to the combination of radiation and PARP inhibition, but that HR mutations may be required for synthetic lethality.
Further examining the relationship between PARP inhibition, HPV status and p16, we forced expression of p16 in the HPV(−) HN5 cell line. Predictably, overexpression of p16 led to decreased expression of TRIP12 (Figure 2C) and increased radiosensitivity (Figure 2D). However, while niraparib led to significant radiosensitization in the control vector, the combination of p16 overexpression with PARP inhibition led to no further significant radiosensitization (Figure 2D).
To examine the mechanism of p16 effect on PARP radioenhancement, we performed cell cycle analysis on control vector (CV) and p16 expressing cells (p16^) after 4Gy radiation (Figure 2E–F). Cell cycle data showed that HN5 control vector cells complete progression through S phase to G2/M in 24h. However, after forced p16 expression, cells accumulate in the G1 and subG1 phases and cannot progress to S phase as efficiently. Since forced expression of p16 in HPV(−) cells induces senescence through the Rb/E2F pathway and stalls cells in G0/G1, this cell cycle distribution shift would explain the decreased effectiveness of PARP inhibition. Because as we demonstrated before, PARP inhibition converts SSB to DSB during DNA replication in the S phase of the cell cycle (Bridges et al. 2014). However, by means of viral protein E7, HPV(+) cells can inactivate Rb (Munger et al. 2004; Liu et al. 2018) allowing cell cycle progression into S phase even in the presence of p16 (Demers et al. 1994; Hickman et al. 1994; Slebos et al. 1994) and can therefore still give a moderate response to PARP inhibition.
Inhibition of Wee1 or Chk1 leads to further radiosensitization in HNSCC
We next tested whether we could further enhance radiosensitivity by targeting the HR pathway in combination with PARP inhibition using checkpoint abrogators MK-8776 and MK-1775. Chk1 inhibitors have been shown previously to radioenhance p53-defective HNSCC, lung and breast cell lines (Bridges et al. 2016) as well as HPV(+) HNSCCs (Busch et al. 2013; Busch et al. 2017) without radioenhancing normal lung fibroblasts (Bridges et al. 2016). Chk1 is a cell cycle checkpoint protein kinase involved in cell cycle arrest in S phase, G2/M and in mitosis. When combined with niraparib, the Chk1 inhibitor MK-8776 further enhanced radiosensitivity in the HPV(+) UPCI:SCC154 cell line, but had no additional benefit in the HPV(−) FaDu cell line (Figure 3A–C).
Figure 3: Inhibition of Wee1 or Chk1 leads to further radiosensitization in HNSCC.
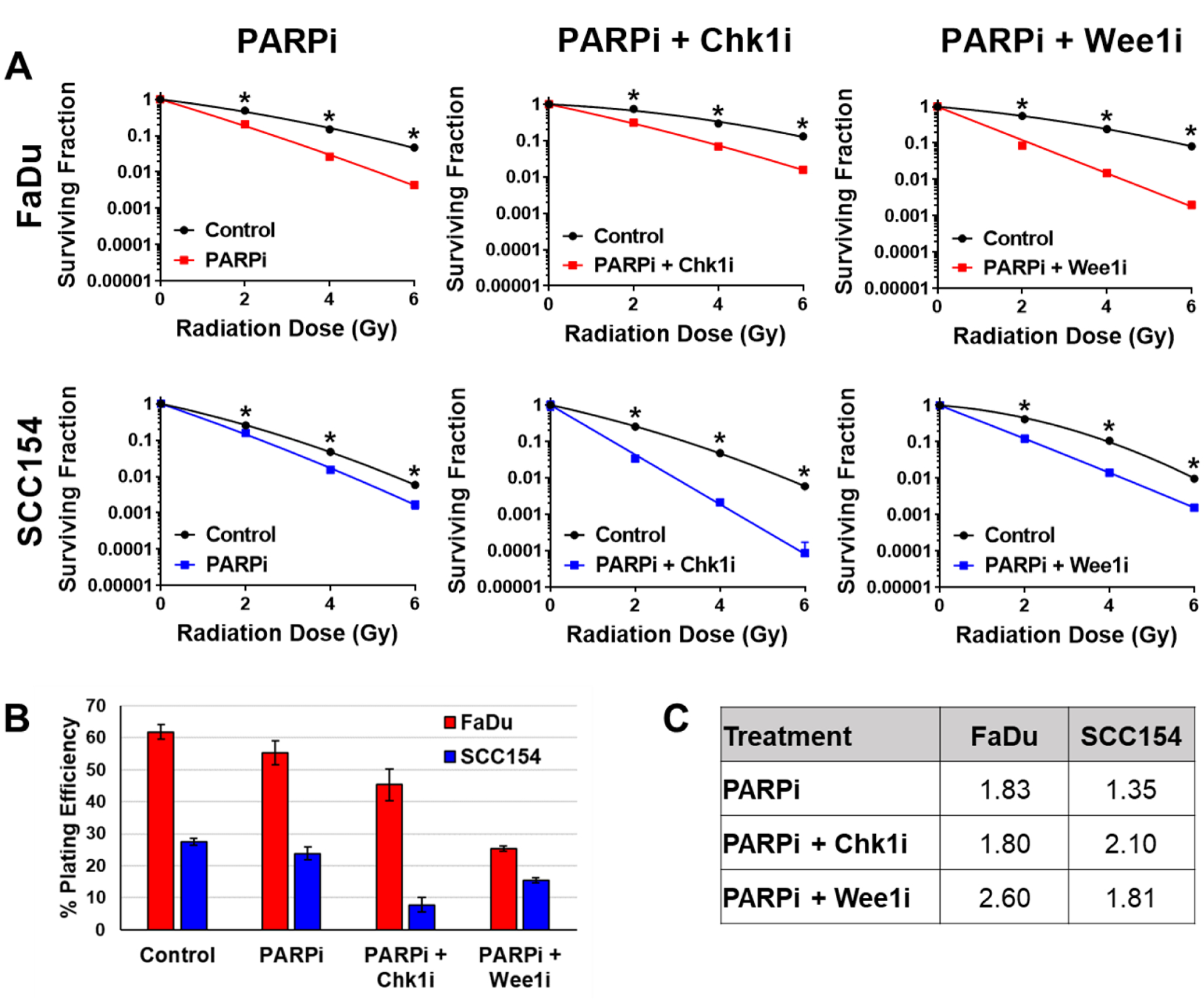
(A) Cells were treated with 1μM niraparib (PARPi) alone or in combination with 200nM MK-8776 (Chk1i) or MK-1775 (Wee1i) for 1h then irradiated with 0–6Gy and replated 24h later for colony formation. (B) Plating efficiencies in HPV(−) FaDu (red) and HPV(+) SCC154 (blue) cell lines taken from CSAs shown in A. (C) Radiation dose enhancement factors (DEF) calculated at surviving fraction of 0.1 in HPV(−) FaDu and HPV(+) SCC154 cell lines.
Alternatively, MK-1775 is a Wee1 inhibitor that we have shown enhances radiosensitivity in lung, breast and prostate cancer cell lines but not in normal lung fibroblasts by significantly abrogating the G2/M checkpoint, leading to premature entry into mitosis and subsequent mitotic cell death (Bridges et al. 2011). The combination of the Wee1 inhibitor MK-1775 with niraparib had little additional radiosensitization in the HPV(+) UPCI:SCC154 cell line, but led to dramatically increased radioenhancement in the HPV(−) FaDu cells (Figure 3A–C). This differential response of Chk1 versus Wee1 inhibition was unexpected since Chk1 is immediately upstream of Wee1 in the G2/M DDR checkpoint. However, this could be explained by inherent differences in the DDR options available in HPV(+) versus HPV(−) cell lines.
HPV(+) cells express p16 which downregulates TRIP12 leading to reduced BRCA1 foci formation and reduced HR capacity (Wang, Zhang, et al. 2017). HPV(+) cells have also been shown to have higher levels of 53BP1 foci following radiation than HPV(−) cells indicative of a greater reliance on NHEJ (Wang, Wang, et al. 2017; Wang, Zhang, et al. 2017). However, in HPV(+) cells p53 is ubiquitinated and degraded due to E6 viral oncogene expression (Munger et al. 2004; Liu et al. 2018) which disrupts G1 arrest thus limiting their ability to perform NHEJ during G1 arrest (Kessis et al. 1993; Havre et al. 1995). Therefore, HPV(+) cells have limited DNA repair options and rely more heavily on S phase checkpoints or performing NHEJ outside of G1 than other non-HPV derived cancers. In addition to its role in DNA damage response in G2/M, Chk1 is also involved in maintenance of genome integrity through replication fork stabilization and plays a critical role in the intra-S-phase checkpoint (Bartek et al. 2004). With so few DDR options, interfering with S phase checkpoints by inhibition of Chk1 and PARP could lead to catastrophic genomic instability following radiation induced DNA damage in HPV(+) cancers.
On the other hand, HPV(−) cells have higher TRIP12 expression and therefore a more efficient HR pathway, and likely rely heavily on HR instead in the G2 phase of cell cycle. Wee1 inhibitor MK-1775 was previously shown to more effectively abrogate G2/M than the Chk1 inhibitor MK-8776 (Bridges et al. 2016). These cells therefore are more likely to respond to Wee1 inhibition which abrogates the G2 checkpoint causing the cells to progress into mitosis with unrepaired DNA damage leading to mitotic death.
In conclusion, we have shown that the PARP inhibitor, niraparib, effectively radiosensitizes HNSCCs with a greater benefit seen in HPV(−) cell lines. Furthermore, the addition of molecularly targeted cell cycle checkpoint abrogators targeting Chk1 or Wee1 can increase the radiosensitizing benefits seen with niraparib in HNSCC cells in an HPV dependent fashion. While HPV(−) cells showed no additional benefit from adding the Chk1 inhibitor, they had a dramatically improved response to the addition of the Wee1 inhibitor. Conversely, the addition of Wee1 inhibition had a modest improvement in radiosensitivity in HPV(+) cells, but inhibition of Chk1 had a much stronger enhancing effect. This study suggests that using cell cycle abrogators in combination with PARP inhibitors may be a beneficial treatment option in HNSCC and warrants a more in-depth investigation with an emphasis on the role of HPV status for optimizing treatment strategies.
Acknowledgements
This work was supported by the National Cancer Institute under Grant R01 CA168485 (HS); the National Institute of Dental and Craniofacial Research under Grants R01 DE028105 (HS), R01 DE028061 (CP and HS), U01DE025181 (JM and MF); and the Cancer Research Institute of Texas (CPRIT) by RP150293. The Characterized Cell Line Core Facility was supported by the Cancer Center Support Grant P30 NIH CA016672.
Footnotes
Disclosure of interest
The authors report no conflict of interest.
References
- Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C et al. 2010. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 363(1):24–35. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang KK, Sturgis EM. 2012. Human papillomavirus as a marker of the natural history and response to therapy of head and neck squamous cell carcinoma. Seminars in radiation oncology. 22(2):128–142. eng. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. 2004. Checking on DNA damage in S phase. Nature reviews Molecular cell biology. 5(10):792–804. eng. [DOI] [PubMed] [Google Scholar]
- Bridges KA, Chen X, Liu H, Rock C, Buchholz TA, Shumway SD, Skinner HD, Meyn RE. 2016. MK-8776, a novel chk1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Oncotarget. 7(44):71660–71672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. 2011. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 17(17):5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges KA, Toniatti C, Buser CA, Liu H, Buchholz TA, Meyn RE. 2014. Niraparib (MK-4827), a novel poly(ADP-Ribose) polymerase inhibitor, radiosensitizes human lung and breast cancer cells. Oncotarget. 5(13):5076–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch CJ, Kriegs M, Laban S, Tribius S, Knecht R, Petersen C, Dikomey E, Rieckmann T. 2013. HPV-positive HNSCC cell lines but not primary human fibroblasts are radiosensitized by the inhibition of Chk1. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 108(3):495–499. eng. [DOI] [PubMed] [Google Scholar]
- Busch CJ, Kroger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C, Munscher A, Rothkamm K, Rieckmann T. 2017. G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 122(2):260–266. eng. [DOI] [PubMed] [Google Scholar]
- Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D, Cicalese A, Shah PK, Viale A, Pettazzoni PF et al. 2016. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell reports. 16(1):133–147. eng. [DOI] [PubMed] [Google Scholar]
- Chalmers AJ, Lakshman M, Chan N, Bristow RG. 2010. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Seminars in radiation oncology. 20(4):274–281. eng. [DOI] [PubMed] [Google Scholar]
- Demers GW, Foster SA, Halbert CL, Galloway DA. 1994. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proceedings of the National Academy of Sciences of the United States of America. 91(10):4382–4386. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Naggar AK, Westra WH. 2012. p16 expression as a surrogate marker for HPV-related oropharyngeal carcinoma: a guide for interpretative relevance and consistency. Head & neck. 34(4):459–461. eng. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434(7035):917–921. eng. [DOI] [PubMed] [Google Scholar]
- Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V. 2008. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic acids research. 36(13):4454–4464. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guster JD, Weissleder SV, Busch CJ, Kriegs M, Petersen C, Knecht R, Dikomey E, Rieckmann T. 2014. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 113(3):345–351. eng. [DOI] [PubMed] [Google Scholar]
- Havre PA, Yuan J, Hedrick L, Cho KR, Glazer PM. 1995. p53 inactivation by HPV16 E6 results in increased mutagenesis in human cells. Cancer research. 55(19):4420–4424. eng. [PubMed] [Google Scholar]
- Hickman ES, Picksley SM, Vousden KH. 1994. Cells expressing HPV16 E7 continue cell cycle progression following DNA damage induced p53 activation. Oncogene. 9(8):2177–2181. eng. [PubMed] [Google Scholar]
- Karam SD, Reddy K, Blatchford PJ, Waxweiler T, DeLouize AM, Oweida A, Somerset H, Marshall C, Young C, Davies KD et al. 2018. Final Report of a Phase I Trial of Olaparib with Cetuximab and Radiation for Heavy Smoker Patients with Locally Advanced Head and Neck Cancer. Clin Cancer Res. 24(20):4949–4959. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessis TD, Slebos RJ, Nelson WG, Kastan MB, Plunkett BS, Han SM, Lorincz AT, Hedrick L, Cho KR. 1993. Human papillomavirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 90(9):3988–3992. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Mann D, Sinha UK, Kokot NC. 2018. The molecular mechanisms of increased radiosensitivity of HPV-positive oropharyngeal squamous cell carcinoma (OPSCC): an extensive review. Journal of otolaryngology - head & neck surgery = Le Journal d’oto-rhino-laryngologie et de chirurgie cervico-faciale. 47(1):59. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. 2005. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer biology & therapy. 4(9):934–936. eng. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ et al. 2006. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer research. 66(16):8109–8115. eng. [DOI] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. 2004. Mechanisms of human papillomavirus-induced oncogenesis. Journal of virology. 78(21):11451–11460. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. 2012. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 72(21):5588–5599. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. 2010. PARP inhibition: PARP1 and beyond. Nature reviews Cancer. 10(4):293–301. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebos RJ, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB, Cho KR. 1994. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proceedings of the National Academy of Sciences of the United States of America. 91(12):5320–5324. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen JK, Divine G, Chen KM, Chitale D, Havard S, Worsham MJ. 2013. Significance of p16 in Site-specific HPV Positive and HPV Negative Head and Neck Squamous Cell Carcinoma. Cancer and clinical oncology. 2(1):51–61. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mason KA, Ang KK, Buchholz T, Valdecanas D, Mathur A, Buser-Doepner C, Toniatti C, Milas L. 2012. MK-4827, a PARP-1/−2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Investigational new drugs. 30(6):2113–2120. eng. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang X, Li Y, Han S, Zhu J, Wang X, Molkentine DP, Blanchard P, Yang Y, Zhang R et al. 2017. Human papillomavirus status and the relative biological effectiveness of proton radiotherapy in head and neck cancer cells. Head & neck. 39(4):708–715. eng. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang P, Molkentine DP, Chen C, Molkentine JM, Piao H, Raju U, Zhang J, Valdecanas DR, Tailor RC et al. 2017. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene. 36(6):820–828. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]