

Abstract
Chronic hepatitis C virus (HCV) infection is one of the major etiological factors for hepatocellular carcinoma (HCC). The treatment options for HCC are limited for lack of a convenient animal model for study with HCV infection and liver pathogenesis. We aimed at developing a patient-derived xenograft (PDX) tumor in mouse from HCV associated HCC patient and evaluated therapeutic potential. After resection of the primary tumor from patient liver, excess viable tumor was implanted into highly immunodeficient mice. A mouse xenograft tumor line was developed and the tumor was successfully passaged for at least three rounds in immunodeficient mice. The patient’s primary tumor and the mouse xenografts were histologically similar. Genetic profiling by short tandem repeat analysis verified that the HCC-PDX model was derived from the HCC clinical specimen. HCV RNA present in the patient liver specimen was undetectable after passage as xenograft tumors in mouse. Human albumin, α−1-antitrypsin, glypican 3, α-SMA, and collagen type 1A2 markers were detected in human original tumor tissues and xenograft tumors. Both the patient primary tumor and the xenograft tumors had a significantly higher level of c-Kit mRNA. Treatment of HCC-PDX xenograft tumor bearing mice with c-Kit inhibitor, imatinib, significantly reduced tumor growth, and phospho-Akt and cyclin D1 expression, as compared to untreated control tumors.
Conclusion:
Our results demonstrated an establishment of HCV associated HCC PDX model as a powerful tool for evaluating candidate drugs. Information on molecular changes in cancer associated specific gene expression will facilitate for efficient targeted therapy and treatment strategy.
Keywords: hepatitis C virus, hepatocellular carcinoma, patient derived xenograft (PDX) model, c-Kit, imatinib
Introduction
The incidence and mortality of liver cancer are increasing at an alarming rate in the USA (1, 2). In contrast to the stable and declining incidence rates for other major cancers, liver cancer shows an increase, annualized at 3.6% for men and 2.9% for women, and a rising death rate for both sexes. Minimal advances in agents targeting specific molecular features of liver cancer have been achieved over the last 10 years. Since the development of sorafenib in 2007, there have been several phase III clinical trials of other inhibitors that have failed to show superiority (3, 4). FDA approved regorafenib- the congener of sorafenib, and nivolumab -a programmed cell death 1(PD1) inhibitor antibody, for second line treatment in patients who developed resistance or were intolerant to sorafenib. Median overall survival benefit of these drugs is short (~3 months), and only a subset of sorafenib resistant patients (~11%) respond to regorafenib. A recent Phase II clinical trial reported that nivolumab produced a response rate of ~14% and a 13.8-month survival rate of 74% (3). Surgical resection and orthotropic liver transplantation are potentially curative options for treating HCC, although the use of these approaches is limited to one-third of patients, and donor organ availability is low. Moreover, the 5-year recurrence of HCC after surgical resection approaches ~70%. (5). There have been minimal advances in the medical treatment of HCC, and this is related to the complex tumor genesis mechanisms, including mutations and tumor heterogeneity. Another important factor hindering the advancement of medical therapies is the absence of a good preclinical model that will translate to successful phase III clinical trials. Thus, there is an overarching challenge for developing alternative more efficacious therapies for HCC.
Silent liver disease progression from chronic HCV infection is an important factor in the etiology of cirrhosis and HCC (6–10). Although, a major global research effort illuminated many aspects of HCV life cycle in facilitating direct acting antiviral (DAA) development, uncertainties remain for clinical efficacy of antiviral therapy among patients with chronic HCV infection and decompensated cirrhosis (11). Even those cured from viremia remain at significantly elevated risk for HCC, as are unresolved HCV patients (12–15). Independent groups of investigators have observed that a sustained virological response (SVR) after DAA treatment is not associated with a reduced incidence of HCC (16, 17).
Liver carcinogenesis requires initiation of neoplastic clones, and progression to clinically diagnosed malignancy (18). Cancer stem cells are a small sub-population of the total cancer cell population, and stem cells tend to be resistant to chemotherapeutic agents. c-Kit is a receptor for stem cell factor (SCF) and is highly expressed in spheres generated from HCV associated transformed human hepatocytes (THH) (10). SCF is involved in proliferation and differentiation of numerous embryonic, adult hematopoietic, neural, and primordial stem cells (19). The uncontrolled activity of c-Kit contributes to the human tumor formation. c-Kit-SCF axis signaling activates PI3K/AKT, Src kinase, and JAK/STAT pathways. SCF plays a key role in hepatic regeneration after injury. Since we do not have a small animal model to study with HCV infection and associated HCC, we developed a patient-derived xenograft (PDX) model using surgically resected liver cancer tissues and successfully implanted, and passaged in immunodeficient mice. c-Kit expression was higher in HCC patients and HCC-PDX passaged tumors. Treatment of HCC-PDX mouse model with imatinib significantly inhibited tumor growth.
Experimental Procedures
ESTABLISHMENT OF HCC PDX TUMOR MOUSE MODEL
Tumors selected for establishment of PDX mouse model were from HCV-associated HCC patients who had tumor resections. Excess resected HCC tissues were used from 4 patients after separating portioned required for clinical frozen sections and histopathological analyses in the pathology laboratory at the Saint Louis University Hospital. Tissues were collected under the protocol (#27805) approved by the Saint Louis University Institutional Review Board. Patient consents were obtained and viable tumors were collected from de-identified resected liver samples. Thin slices of tumor tissue were placed in culture medium immediately after resection and kept on ice for implantation. The time from collection to implantation in highly immunodeficient NOD/SCID gamma (NSG) mice was less than 4 hours.
HCC liver specimens were cut into small pieces (1–3 mm3) and directly implanted into NSG mice. Mice were anesthetized using isoflurane (1–3%), skin aseptically prepared, and a small dorsal midline incision (<10 mm) was made at the level of the flank. Tumor tissues were placed in bilateral subcutaneous pockets and the incision was closed. Lidocaine was infiltrated at the wound edges to control postoperative pain. When tumor volume reached >600 mm3, the mouse was humanely sacrificed (CO2 asphyxiation) and the tumors were cryopreserved or explanted for passage in another NSG mouse using the same protocol.
SHORT TANDEM REPEAT (STR) ANALYSIS
DNA was isolated from the original patient tumor and PDX tumors passaged (P0, P1 and P2) in mice. Target DNA (10 ng) was amplified by multiplex polymerase chain reaction for STR analysis using GenePrint24 System (Promega Corporation, WI). STR analysis was performed on different chromosomes at 24 loci. The following autosomal STR loci were analyzed: CSF1PO, FGA, TH01, TPOX, vWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11, D10S1248, D22S1045, D2S441, D1S1656, D12S391, D2S1338, D19S433, DYS391, Penta D and Penta E plus Amelogenin for gender determination. Following amplification, size- and color-based separation and detection of STR loci was performed using capillary electrophoresis. The data were analyzed by GeneMapper Software (ThermoFisher Scientific).
XENOGRAFT TUMOR GROWTH AND IMATINIB TREATMENT
Transformed human hepatocytes (THH) were injected subcutaneously (106 cells/site) into the flank of nude mice for generation in tumor. In a separate set of experiment, HCC-PDX P0 tumors were implanted into NSG mice. When tumors were palpable, tumor bearing mice were randomly divided into two groups for control (regular drinking water) or c-Kit inhibitor (imatinib) treatment given through drinking water (600 mg/L) as described earlier (20). The imatinib-supplemented water was replaced every three days. Tumor growth was examined weekly until the end of each experiment. Tumor volume in control and imatinib treated mice were compared using a two-tailed T-test as previously described (21). All statistical analyses were two sided and values <0.05 was considered significant.
RNA ISOLATION AND QUANTITATIVE REVERSE TRANSCRIPTION-PCR (qRT-PCR)
RNA was isolated from the original patient tumor (HCC8) and PDX tumors using a TRIZOL reagent (Life Technologies) and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized using random hexamers and a Superscript III reverse transcriptase kit (Invitrogen, CA). Real-time PCR was performed for quantitation of gene expression using TaqMan Universal PCR master mix and 6-carboxyfluorescein (FAM)-MGB probes for albumin (assay ID: Hs000609411_m1), glypican 3 (assay ID: Hs01018936_m1), smooth muscle actin alpha 2- human (assay ID: Hs00426835_g1) and mouse (assay ID: Mm00725412_s1), Collagen, type I, α 2 human (assay ID:Hs00164099_m1) and mouse (assay ID: Mm00483888_m1) as per manufacture’s protocol (Thermo Fisher Scientific). 18S rRNA (assay ID: Hs03928985_g1) was used as an endogenous control. Gene expression analysis for c-Kit (FP: 5’ GAGTTGGCCCTAGACTTAGAAG 3’; RP: 5’ TCTTTGTGATCCGACCATGAG 3’) and α−1 antitrypsin (FP: 5’ GGCCATACCCATGTCTATCC 3’; RP: 5’ TTCACCACTTTTCCCATGAA 3’) was carried out by SYBR green based detection system (Thermo Fisher Scientific). 18S rRNA (FR: 5’ GTCATAAGCTTGCGTTGATT 3’ and RP: 5’ TAGTCAAGTTCGACCGTCTT 3’) was used as endogenous control. The relative gene expression was analyzed by using the 2−ddCT formula (ddCT = dCT of the sample − dCT of the untreated control). Each sample was analyzed in triplicate.
Results and Discussion
DEVELOPMENT AND CHARACTERIZATION OF HCV ASSOCIATED HCC-PDX TUMORS
HCC tumors obtained after surgery were subcutaneously implanted into NSG mice as described in the Materials and Methods section. The patient information is summarized in the Supplementary Table 1. Four patient (all males, ages ranged from 63–68) tumors were used as primary tumors. Xenograft tumor was developed after 6–7 weeks from patient #8 (named HCC8) and was successfully passaged in mice for three generations. Only one primary patient tumor out of 4 (25%) was successfully engrafted and passaged to the next mouse. Tumors take rate at P0, P1 and P2 passages were 30%, 100% and 100%, respectively. Cryopreserved tumors were also used from first and second generation xenografts, and successfully engrafted in a subsequent generation of mice. Tumor samples were evaluated for the presence of HCV genome. HCV RNA was detected in the initial tumor from patient, however, tumors in subsequently passaged through mice were negative for HCV genome, suggesting that human tumor environment in mouse does not support HCV replication in transplanted liver tissue. Tumor tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin for evaluation by light microscopy. Histological analysis showed that the PDX tumor at all passages retained the histological features of the most poorly differentiated component of the original carcinoma (Fig. 1, panel A). An unusual feature of this donor patient’s tumor was that it occurred in a non-cirrhotic liver. The chronic hepatitis had mild activity (grade 2) by histology, which may explain why the liver had not progressed to cirrhosis. Another interesting feature of this tumor is that it is classified as a biphenotypic “combined hepatocellular-cholangiocarcinoma” (cHCC-CCA) (22), and the poorly differentiated non-hepatocellular component represented in the tumor xenograft has histologic and biochemical features intermediate between HCC and CCA. We measured for RNA expression of α−1antitrypsin, human albumin, and glypican-3 in original specimen, and PDX tumor samples. Similar levels of α−1 antitrypsin and human albumin were observed in all the tumor samples (Fig. 1, panel B). Although the passaged xenograft tumors had the same “intermediate cell” appearance, all the PDX tumors expressed classic hepatocellular antigen glypican-3. These tumors are poorly differentiated and appeared to be more aggressive. We also tested the α−1 antitrypsin and albumin in non-tumor adjacent tissue (NTT) from the same patient. The expression level of α−1 antitrypsin and albumin were comparable between HCC and NTT (data not shown). We also examined the expression of human and mouse α-smooth muscle actin, and collagen type 1A2 in these tumor samples. Biochemical analysis indicated that tumor specimens from patient and PDX display the presence of human fibrosis markers (Fig. 1, panel B). However, tumor grown in mice exhibited mouse fibrosis marker as previously observed (23), suggesting species specific tumor associated fibroblast migration in the respective tumor regions.
Figure 1: Characterization of HCV associated HCC-PDX tumor.
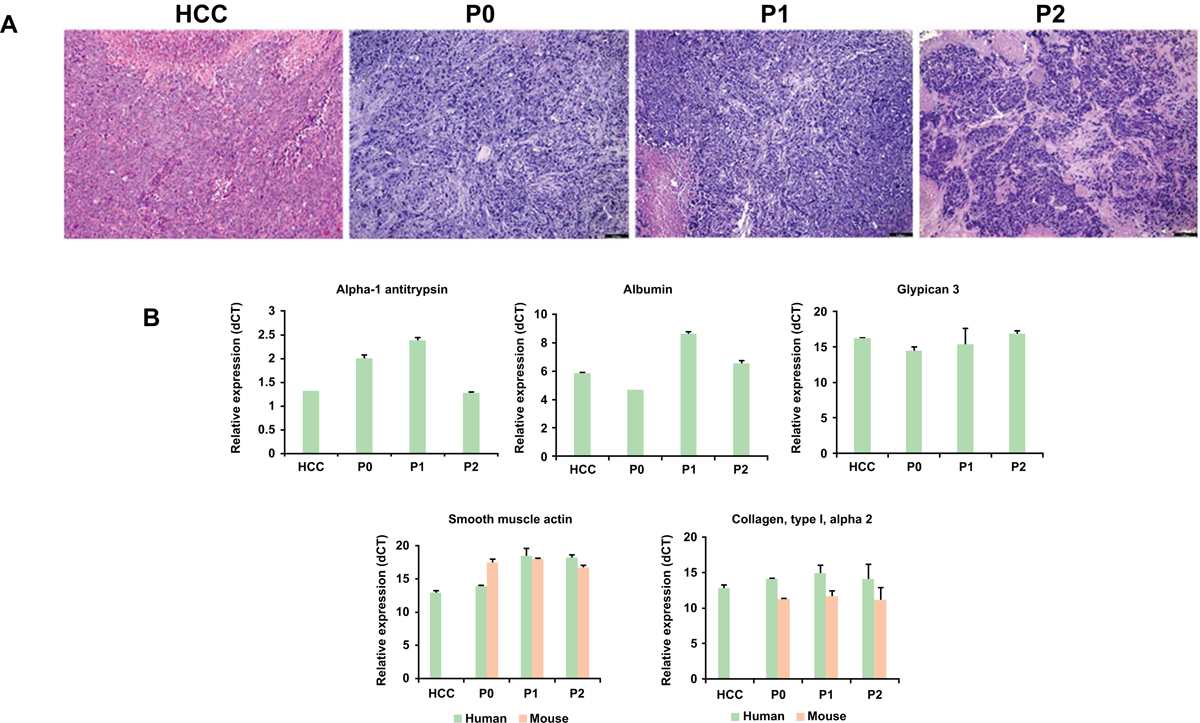
Panel A: Comparative histology of original patient tumor (HCC8) and tumors from PDXs at different passages (P0–P2). Representative micrographs of hematoxylin & eosin staining are shown. PDX tumors retain the histopathologic characteristics of the original samples throughout the passages in mice. Magnification 10X. Panel B: Relative mRNA expression (dCT) of α−1 antitrypsin, albumin, glypican-3, human and mouse α-smooth muscle actin, and collagen type 1A2 in tumors from HCC patient and PDX were performed by qRT-PCR. 18S rRNA gene was used as an internal control. Small bar indicates standard error.
STR PROFILE OF PDX TUMORS
Next, we performed microsatellite STR identity analyses to establish that the PDX model tumor is derived from the human HCC primary tumor. For this, we examined autosomal STR loci: CSF1PO, FGA, TH01, TPOX, vWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11, D10S1248, D22S1045, D2S441, D1S1656, D12S391, D2S1338, D19S433, Penta D and Penta E by co-amplification in each sample. Amelogenin for gender determination was also examined. In addition, the male-specific DYS391 locus was included to identify null Y allele results for Amelogenin. The STR profiles of PDX tumors and the HCC8 primary patient tumor were similar and compatible (Fig. 2 and Table 1). The PDX passage 2 (P2) xenograft tumor has become homozygotic at several loci that were heterozygous in the primary tumor, such as Penta E, D16S539, D2S1338, Penta D, D21S11, DYS391, D12S391, and FGA.
Figure 2: Authentication of HCC-PDX samples by short Tandem Repeats (STR) profiling.
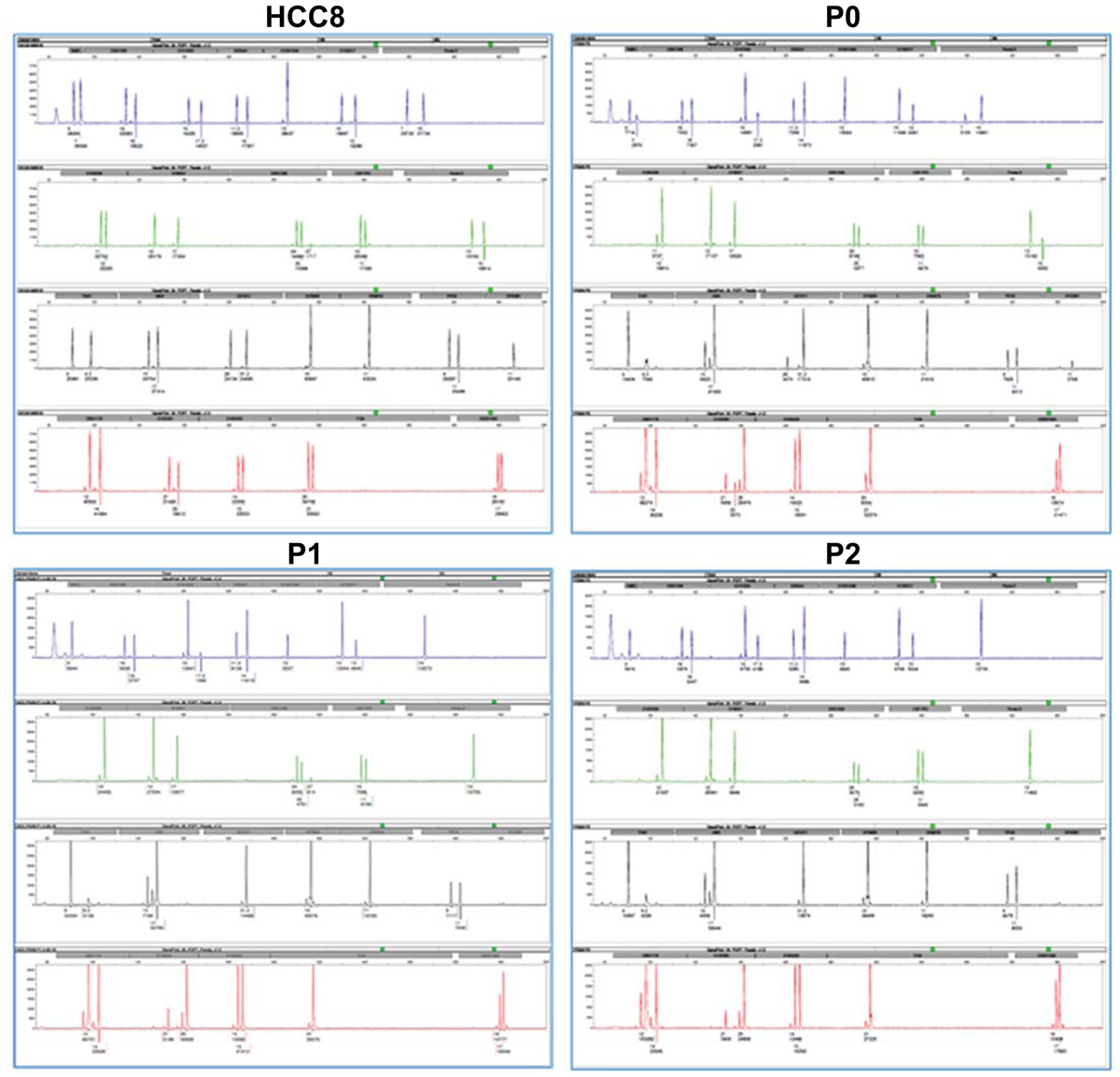
STR profiles of original tumor (HCC8) and PDX tumor at P0, P1 and P2 passages are shown.
Table 1:
STR profiles of original tumor (HCC8) and PDX tumors at P0, P1 and P2
| STR locus | Chromosomallocation | HCC8 | P0 | P1 | P2 |
|---|---|---|---|---|---|
| Amelogenin | Xp22.1–22.3, Y | X, Y | X, Y | X | X |
| D3S1358 | 3p21.31 | 16, 18 | 16, 18 | 16, 18 | 16, 18 |
| D1S1656 | 1q42 | 15, 17.3 | 15, 17.3 | 15, 17.3 | 15, 17.3 |
| D2S441 | 2p14 | 11.3, 14 | 11.3, 14 | 11.3, 14 | 11.3, 14 |
| D10S1248 | 10q26.3 | 13 | 13 | 13 | 13 |
| D13S317 | 13q31.1 | 10, 13 | 10, 13 | 10, 13 | 10, 13 |
| PentaE | 15q26.2 | 7, 10 | 7, 10 | 10 | 10 |
| D16S539 | 16q24.1 | 11, 12 | 11, 12 | 12 | 12 |
| D18S51 | 18q21.33 | 12, 17 | 12, 17 | 12, 17 | 12, 17 |
| D2S1338 | 2q35 | 24, 25, 27 | 24, 25, 27 | 24, 25, 27 | 24, 25 |
| CSF1PO | 5q33.1 | 10, 11 | 10, 11 | 10, 11 | 10, 11 |
| PentaD | 21q22.3 | 13, 15 | 13, 15 | 13 | 13 |
| TH01 | 11p15.5 | 6, 9.3 | 6, 9.3 | 6, 9.3 | 6, 9.3 |
| vWA | 12p13.31 | 15, 17 | 15, 17 | 15, 17 | 15, 17 |
| D21S11 | 21q21.1 | 28, 31.2 | 28, 31.2 | 31.2 | 31.2 |
| D7S820 | 7q21.11 | 10 | 10 | 10 | 10 |
| D5S818 | 5q23.2 | 11 | 11 | 11 | 11 |
| TPOX | 2p25.3 | 9, 11 | 9, 11 | 9, 11 | 9, 11 |
| DYS391 | Y | 11 | 11 | -- | -- |
| D8S1179 | 8q24.13 | 12, 14 | 12, 14 | 12, 14 | 12, 14 |
| D12S391 | 12p12 | 21, 23 | 21, 23, 25 | 21, 25 | 21, 25 |
| D19S433 | 19q12 | 14, 15 | 14, 15 | 14, 15 | 14, 15 |
| FGA | 4q28 | 20, 21 | 20, 21 | 21 | 21 |
| D22SA045 | 22q12.3 | 16, 17 | 16, 17 | 16, 17 | 16, 17 |
INHIBITION OF HCC GROWTH BY IMATINIB
We examined the expression of c-Kit in tumor and adjacent non-tumor tissue of the same patient HCC8 tumor, and a high level of c-Kit expression was noted (Fig. 3, panel A). We also observed higher level of c-Kit expression in HCC-PDX tumors. The protooncogene c-Kit is expressed in a wide variety of human cancers. We have previously observed that c-Kit inhibitor reduces sphere forming cell growth generated on low-binding plate from HCV associated transformed human hepatocytes (THH) (10). We also observed a higher expression status of c-Kit in HCC liver specimens from other etiology (data not shown). We next examined the efficacy of c-Kit inhibitor in our newly developed HCC-PDX and THH xenograft mouse models. We have previously shown a high level of c-Kit expression in THH (10). THH were implanted into nude mice and when tumors were palpable, mice were randomly divided in two groups. The treatment group of mice were given the c-Kit inhibitor imatinib in their drinking water at doses selected based on the literature for other gastrointestinal tumors (20). The control group of mice received unmodified drinking water. HCC tumor volumes were reduced in c-Kit inhibitor imatinib treated animals (Fig. 3, panel B) indicating the in vivo efficacy of imatinib in a HCC xenograft mouse model. We subsequently tested imatinib in our newly developed HCV associated HCC-PDX model. Ten NSG mice in each group were used and animals were treated with imatinib as described above. Tumor volumes in HCV associated HCC-PDX were significantly reduced following imatinib treatment as compared to that of control group (Fig. 3, panels C and D). We next examined the representative downstream molecules of c-Kit signaling (Fig. 4, panel A) in HCC-PDX tumors following imatinib treatment. Lysates from control and imatinib treated PDX tumors were analyzed by Western blot analysis for phospho-Akt and cyclin D1 expression using specific antibodies. A significant reduction of phospho-Akt (Fig. 4, panel B) and cyclin D1 (Fig. 4, panel C) expression was observed in imatinib treated PDX tumors as compared to that of control, suggesting that imatinib treatment disrupts the c-Kit signaling in HCV associated HCC-PDX tumors.
Figure 3: Therapeutic potential of c-Kit inhibitor in HCV-HCC PDX tumor.
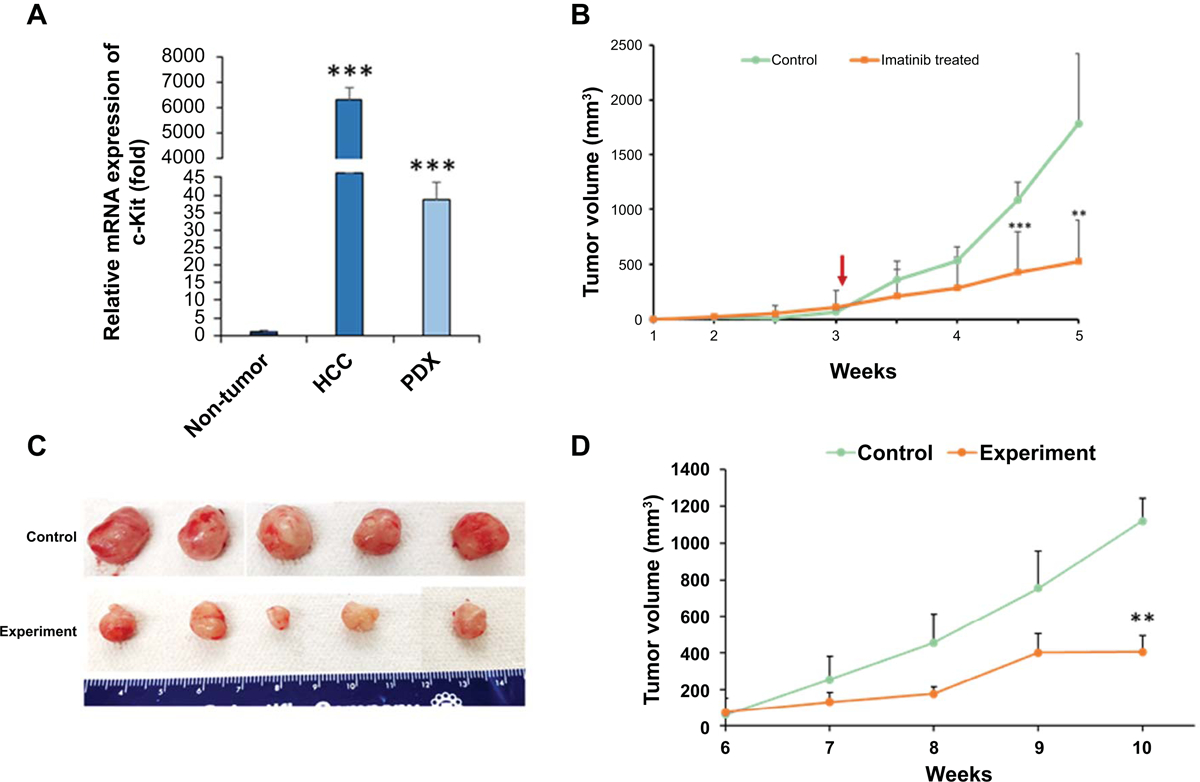
Panel A: Relative mRNA expression (fold) of c-Kit in original tumor and PDX samples as compared to adjacent non-tumor tissue analyzed by qRT-PCR. 18S gene was used as an internal control. Small bar indicates standard error (***p<0.001). Panel B Transformed human hepatocytes (1 ×106) were injected subcutaneously into the flank of nude mice. After palpable tumor development (>75mm3), mice were randomly divided into negative control and experimental groups. Experimental group received c-Kit inhibitor imatinib in drinking water. Tumors were measured using a slide caliper and tumor volumes were calculated. Arrow indicates starting point of imatinib. Small bar indicates standard error (**, p<0.01; *** p<0.001). Panel C: Cryopreserved PDX tumor (P0) was reconstituted and pieces of tumors were implanted subcutaneously into the flank of NSG mice. After palpable tumor development (6 weeks after implantation), mice were randomly divided into two groups. The imatinib treatment in drinking water was started. Mice were humanely sacrificed at 4 weeks after treatment. Representative images of tumors from control and experimental groups are shown. Panel D: Tumors were measured using a slide caliper and tumor volumes were calculated. Small bar indicates standard error (**, p<0.01).
Figure 4: Imatinib treatment reduces AKT and cyclin D1 expression in HCV-HCC PDX tumors.

Panel A: A schematic representation of c-Kit downstream molecules. Panel B: Lysates from control PDX tumors and imatinib-treated tumors were subjected to western blot analysis for phospho-Akt (Thr308) expression using specific antibody. The blot was reprobed for total AKT and actin (for comparison of protein load) using specific antibodies. Densitometric scanning results are presented as bar diagram. Panel C: Lysates from control PDX tumors and imatinib-treated tumors were subjected to western blot analysis for cyclin D1 expression using specific antibody. The blot was reprobed with antibody to actin for comparison of protein load. Densitometric scanning results are presented as bar diagram. *p < 0.05.
In this study, we established HCV associated human liver cancer xenograft tumor in mice which provides a clinically relevant small animal model. Tumors could be successfully passaged and retained the original characteristics. Our HCC-PDX model showed remarkable similarities in the phenotype and molecular details of the primary human tumor. Human albumin and α-antitrypsin markers were present in patient and passaged xenograft tumors. The model was authenticated by STR analysis and the tumors could also be successfully passaged from cryopreserved samples. We demonstrated that the serial propagation of xenograft tumors in mice did not significantly change the biological characteristics. This xenograft model served as a useful model for in vivo testing of potential chemotherapeutic agents against HCC.
We showed previously that spheres from HCV associated THH display higher c-Kit expression (10). All of our chronically HCV infected HCC patients had a higher c-Kit expression status. In a separate study, c-Kit mRNA and protein expression were also detected in ~27% HCV associated HCC from a Taiwanese cohort (24). We also observed high a c-Kit expression level in PDX xenograft tumors. Gain-of-function from mutations results in constitutive c-Kit activation and play a pathogenic role in several cancers, including ~95% gastrointestinal stromal tumors (GIST) (25, 26), leading to uncontrolled proliferation and resistance to apoptosis. Imatinib significantly reduces GIST tumor growth and is a FDA approved drug for GIST, especially in metastasis with minimum side effects (27). We also showed that transformed hepatocytes and HCC-PDX tumors respond well following imatinib treatment. Lin et al. (2008) reported earlier from their Phase II study that imatinib as a monotherapy had little efficacy in HCC (28). However, only six out of 15 patients had histologic confirmation of HCC and none of the samples were tested for c-Kit expression. HCC is a heterogeneous tumor with varying molecular heterogeneity. Since treatment for HCC is very limited and none of the current regimen has given >50% efficacy, molecular testing and targeted treatment will likely be most helpful. Therefore, HCC-PDX mouse model will be important for evaluation of new or additional targeted chemotherapeutic agents for effective treatment regimen.
Supplementary Material
Acknowledgement:
We thank Adrian Di Bisceglie for helpful suggestions, and Saint Louis Pathology DNA laboratory for STR analysis.
Financial support: This work was supported by research grants R21CA188472 and R01 DK081817 from the National Institutes of Health, Saint Louis University Liver Center Seed Grant, and a grant from Lottie Caroline Hardy Charitable Trust.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J Carcinog 2017;16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu Z, Li Q, et al. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J Exp Clin Cancer Res 2019;38:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marisi G, Cucchetti A, Ulivi P, Canale M, Cabibbo G, Solaini L, Foschi FG, et al. Ten years of sorafenib in hepatocellular carcinoma: Are there any predictive and/or prognostic markers? World J Gastroenterol 2018;24:4152–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villanueva A Hepatocellular Carcinoma. N Engl J Med 2019;380:1450–1462. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee A, Meyer K, Mazumdar B, Ray RB, Ray R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J Virol 2010;84:5936–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bose SK, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Hepatitis C virus induces epithelial-mesenchymal transition in primary human hepatocytes. J Virol 2012;86:13621–13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 2013;13:123–135. [DOI] [PubMed] [Google Scholar]
- 9.Rusyn I, Lemon SM. Mechanisms of HCV-induced liver cancer: what did we learn from in vitro and animal studies? Cancer Lett 2014;345:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwon YC, Bose SK, Steele R, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Promotion of Cancer Stem-Like Cell Properties in Hepatitis C Virus-Infected Hepatocytes. J Virol 2015;89:11549–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Meer AJ, Berenguer M. Reversion of disease manifestations after HCV eradication. J Hepatol 2016;65:S95–S108. [DOI] [PubMed] [Google Scholar]
- 12.van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, Duarte-Rojo A, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012;308:2584–2593. [DOI] [PubMed] [Google Scholar]
- 13.Aleman S, Rahbin N, Weiland O, Davidsdottir L, Hedenstierna M, Rose N, Verbaan H, et al. A risk for hepatocellular carcinoma persists long-term after sustained virologic response in patients with hepatitis C-associated liver cirrhosis. Clin Infect Dis 2013;57:230–236. [DOI] [PubMed] [Google Scholar]
- 14.Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J Hepatol 2014;61:S79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamdane N, Juhling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, Bandiera S, et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019;156:2313–2329 e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reig M, Marino Z, Perello C, Inarrairaegui M, Ribeiro A, Lens S, Diaz A, et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol 2016;65:719–726. [DOI] [PubMed] [Google Scholar]
- 17.Conti F, Buonfiglioli F, Scuteri A, Crespi C, Bolondi L, Caraceni P, Foschi FG, et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J Hepatol 2016;65:727–733. [DOI] [PubMed] [Google Scholar]
- 18.Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res 2015;4:256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broudy VC. Stem cell factor and hematopoiesis. Blood 1997;90:1345–1364. [PubMed] [Google Scholar]
- 20.Vitiello GA, Medina BD, Zeng S, Bowler TG, Zhang JQ, Loo JK, Param NJ, et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin Cancer Res 2018;24:972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patra T, Meyer K, Ray RB, Ray R. Hepatitis C Virus Mediated Inhibition of miR-181c Activates ATM Signaling and Promotes Hepatocyte Growth. Hepatology 2019; doi: 10.1002/hep.30893. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciarra A, Park YN, Sempoux C. Updates in the diagnosis of combined hepatocellular-cholangiocarcinoma. Hum Pathol 2020; pii: S0046–8177(19)30206–0. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki R, Devhare P, Ray RB, Ray R. Hepatitis C virus-induced tumor-initiating cancer stem-like cells activate stromal fibroblasts in a xenograft tumor model. Hepatology 2017;66:1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung CY, Yeh KT, Hsu NC, Chang JH, Lin JT, Horng HC, Chang CS. Expression of c-kit protooncogene in human hepatocellular carcinoma. Cancer Lett 2005;217:231–236. [DOI] [PubMed] [Google Scholar]
- 25.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61:8118–8121. [PubMed] [Google Scholar]
- 26.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580. [DOI] [PubMed] [Google Scholar]
- 27.Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med 2012;63:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin AY, Fisher GA, So S, Tang C, Levitt L. Phase II study of imatinib in unresectable hepatocellular carcinoma. Am J Clin Oncol 2008;31:84–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.