

Abstract
Healthy peripheral nerves encounter, with increased frequency, numerous chemical, biological, and biomechanical forces. Over time and with increasing age, these forces collectively contribute to the pathophysiology of a spectrum of traumatic, metabolic, and/or immune-mediated peripheral nerve disorders. The blood-nerve barrier (BNB) serves as a critical first-line defense against chemical and biologic insults while biomechanical forces are continuously buffered by a dense array of longitudinally orientated epineural collagen fibers exhibiting high-tensile strength. As emphasized throughout this Experimental Neurology Special Issue, the BNB is best characterized as a functionally dynamic multicellular vascular unit comprised of not only highly specialized endoneurial endothelial cells, but also associated perineurial cells, pericytes, Schwann cells, basement membrane, and invested axons. The composition of the BNB, while anatomically distinct, is not functionally dissimilar to that of the well characterized neurovascular unit of the central nervous system. While the BNB lacks a glial limitans and an astrocytic endfoot layer, the primary function of both vascular units is to establish, maintain, and protect an optimal endoneurial (PNS) or interstitial (CNS) fluid microenvironment that is vital for proper neuronal function. Altered endoneurial homeostasis as a secondary consequence of BNB dysregulation is considered an early pathological event in the course of a variety of traumatic, immune-mediated, or metabolically acquired peripheral neuropathies. In this review, emerging experimental advancements targeting the endoneurial microvasculature for the therapeutic management of immune-mediated inflammatory peripheral neuropathies, including the AIDP variant of Guillain-Barré syndrome, are discussed.
Keywords: Blood Nerve Barrier, Endoneurial Endothelial Cells, Perineurial Cells, Pericytes, Basement Membrane, Therapeutics, Leukocytes, Chronic Pain, Nanotherapy
INTRODUCTION
The anatomical composition and physiologic uniqueness of the blood-nerve barrier (BNB) has been the subject of many excellent review articles, (Kanda, 2013; Reinhold and Rittner, 2017; Richner et al., 2018) including those within this Experimental Neurology Special Issue. In contrast to the neurovascular unit of the blood-brain barrier (BBB), the BNB neurovascular unit is associated with two relatively distinct anatomical sites (i) a concentric multilayered restrictive physical barrier consisting of perineurial epithelioid myofibroblasts concentrated within the innermost layers of the perineurium and (ii) an endoneurial microvascular functional unit consisting of pericytes and non-fenestrated tight-junction forming endothelial cells each enveloped by a continuous basement membrane. Despite being anatomically distinct from the BBB, previous experimental studies have demonstrated that the BNB exhibits molecular and biophysical mechanisms that exquisitely serve to maintain peripheral nerve endoneurial homeostasis (reviewed in (Greathouse et al., 2016; Ubogu, 2013)). Early permeability studies demonstrate qualitatively similar properties of the BNB to that of the BBB and include (i) restricted passage of IgG antibodies and of albumin and (ii) selective transport of insulin, transferrin, and nerve growth factor (Olsson, 1966; Poduslo et al., 1994). Recent studies have raised awareness that reciprocal crosstalk between peripheral neurons and adjacent endoneurial blood vessels occurs not only during fetal and postnatal development but throughout adulthood and during neural repair (Wild et al., 2017). These findings support the emerging concept of a functional neurovascular unit operating within the peripheral nervous system and must be taken into account when considering novel therapeutic approaches for the management of the diverse array of peripheral neuropathies.
Whereas the perineurium presents a formidable restrictive barrier to systemic therapeutic strategies, the endoneurial microvasculature is the penultimate interface between the arteriole blood supply and that of the endoneurial microenvironment (Kanda, 2013). By limiting recruitment and paracellular passage of activated monocytes/leukocytes, emerging therapeutic strategies targeting the endoneurial microvasculature tight-junctions are considered promising and potentially effective adjuncts to current standard of care treatment of inflammatory disorders (Getter et al., 2019; Langert and Brey, 2018; Upadhyay, 2014). This is particularly relevant when considering immune-mediated inflammatory neuropathies (Shimizu and Kanda, 2015).
Transendothelial migration (paracellular trafficking) of leukocytes into peripheral nerves is recognized as an early pathologic hallmark of acquired inflammatory demyelinating nerve disorders (Greathouse et al., 2016; Kieseier et al., 2018; Maiuolo et al., 2019; Ubogu, 2015; Zhang et al., 2019). Often acting in concert with cellular adaptive immunity, many inflammatory peripheral neuropathies (eg., Guillain-Barré Syndrome and it’s clinical subtypes; chronic inflammatory demyelinating polyradiculoneuropathy; multifocal motor neuropathy; MGUS neuropathy) also involve pathologic humoral adaptive immune responses (Lawlor et al., 2002; Querol et al., 2017; Sarkey et al., 2007; Schafflick et al., 2017; Zhang et al., 2019). Exactly how pathogenic antibodies gain access to the endoneurium remains a matter of debate. An intact BNB would be expected to restrict access of circulating immunoglobulins. However, it well documented that endothelial cells of the BBB are not entirely impermeable to macromolecules, including IgG antibodies. In contrast to utilizing clathrin-dependent receptor-mediated transcellular transport (Villasenor et al., 2019), recent studies suggest that IgG immunoglobulins cross central endothelial barriers using a mechanism involving nonspecific fluid-phase transcytosis while transport of IgG across peripheral endothelial barriers is dominated by a mechanism involving caveolae-dependent receptor-mediated (FcRn) transcellular transport (Ruano-Salguero and Lee, 2020). Although it remains unclear whether endoneurial endothelial cells are capable of IgG transcytosis, a recent characterization study of the human BNB transcriptome reported the presence of an Fc IgG receptor and transporter transcript (Palladino et al., 2017).
Clinical and preclinical studies further suggest that the innate immune response in the form of macrophage lineage cell endoneurial infiltrates also plays a pivotal role at eliciting nerve injury in inflammatory nerve disorders. Although seemingly distinct, innate and adaptive immune responses are not mutually independent, but rather are functionally linked in part by binding of immune complexes to cellular Fc-gamma receptors (FcγRs) expressed on infiltrating macrophages/monocytes (Hogarth, 2002; Nimmerjahn and Ravetch, 2008; Takai, 2002). Given that both innate and adaptive immune responses play key roles in the pathogenesis of inflammatory nerve disorders, strategies reviewed below that selectively target and disrupt paracellular trafficking or transcytosis into peripheral nerves offers new and promising options for improved therapeutic management of affected patients (Figure).
Figure:
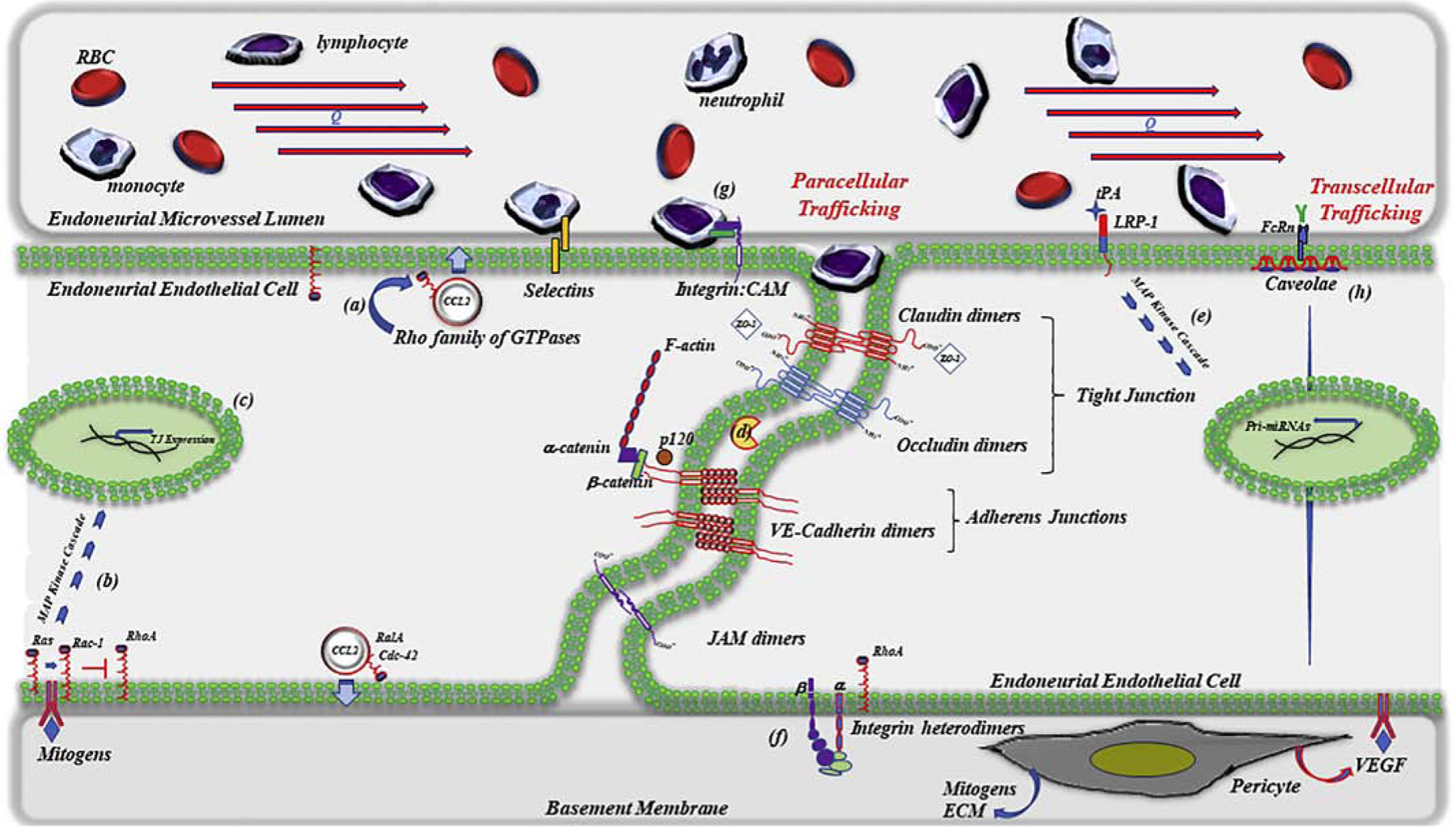
Illustration of a serpentine junctional interface between two endoneurial endothelial cells emphasizing multiple putative intracellular and extracellular therapeutic molecular targets currently under intense investigation for the management of inflammatory peripheral neuropathies/neuropathic pain. Strategies include preclinical evaluation of inhibitors/activators that are designed to either limit paracellular trafficking of leukocytes across perineurial/endothelial barriers or, in the case of neuropathic pain, transiently open restrictive perineurial/endothelial barriers. Shown is a partial selection of targets of interest which includes (a) the Rho family of small monomeric GTPases affecting chemokine release (b & c) mitogen-stimulated GTPase-dependent upregulation of tight junction protein expression (d) matrix metalloproteinases / TIMP-1 (e) tPA-LRP-1 dependent expression of claudin-specific miRNAs (f) ECM-integrin facilitated GTPase-mediated localization and stabilization of VE-cadherins (g) integrin-CAM signaling and (h) caveolae-dependent transcellular trafficking. Therapeutic strategies currently approved for the clinical management of inflammatory neuropathies include non-specific immune modulating corticosteroids, intravenous immunoglobulins, or plasmapheresis. Clinical strategies used for the management of neuropathic pain, while pharmacologically numerous, do not currently target the BNB.
Alternatively, whereas strategies designed to restrict transcytosis of harmful autoantibodies or paracellular trafficking of autoreactive leukocytes may prove beneficial to patients with inflammatory disorders, the management of other neurologic disorders need to consider strategies that are designed to open, rather than restrict, endothelial barriers (Figure). Delivery of certain classes of hydrophilic analgesics (e.g. opioid peptides) for the management of intractable peripheral nerve pain, for example, can be particularly problematic in patients with an intact BNB. To address this challenge, several distinct strategies designed to transiently open the BNB are currently under intense experimental investigation (for a recent review, see (Reinhold and Rittner, 2017)). Early studies showed that hypertonic solutions could transiently breach the BBB (Rapoport, 2000). Approximately a decade later, perineural injection of hypertonic saline was shown to transiently open the BNB, not by mechanical disruption but by a receptor-mediated mechanism leading to reduced expression of tight-junction proteins (Hackel et al., 2012a; Hackel et al., 2012b). Experimental studies specifically targeting tight-junction proteins using siRNA (Hackel et al., 2012a), peptidomimetics (Staat et al., 2015), or miRNAs (Yang et al., 2016) have also proved effective at transiently opening the BNB without eliciting detectable peripheral nerve damage. Exploitation of transcytosis represents an attractive alternative by which to deliver hydrophilic therapeutic agents/therapeutic antibodies across an intact BNB. This, however, remains currently speculative, as additional studies demonstrating functional relevance of transcytosis at the BNB are currently lacking. Despite pre-clinical advancements, there appears to be a paucity in clinical trials that are designed to evaluate the blood-nerve barrier as a targetable strategy for the management of neuropathic pain. Out of 2908 currently registered (ClinicalTrials.gov) clinical trials that address chronic pain, and 1055 trials that specifically address neuropathic pain, none appear to be exploring strategies that transiently breach the BNB. Similarly, none of the 964 clinical trials registered with the European Union Clinical Trials Register that address chronic pain, and 253 trials that specifically address neuropathic pain, utilize this potential strategy. There is, however, a single non-interventional observational clinical trial registered with the German Clinical Trials Register (DRKS00017731) that is investigating the role for tight junction proteins, including claudin-5, as potential biomarkers for inflammatory and non-inflammatory polyneuropathies. Alternatively, emerging pre-clinical advancements in bioengineering strategies are showing promise as a way to facilitate local, controlled targeted delivery of therapeutics to peripheral nerves as a function of underlying BNB integrity, as recently reviewed (Langert and Brey, 2018).
ENDONEURIAL ENDOTHELIAL CELLS
Within a given species, endoneurial endothelial cells of the peripheral nerve microvasculature are uniquely distinct from endothelial cells that establish the microvasculature of the epineurium and perineurium. Endothelial cells of the precapillary arterioles that form the vasa nervosum, which courses lengthwise along the external surface of the epineurium, exhibit numerous fenestrations while lacking tight- or adherens-junction proteins (Ubogu, 2013). As these vessels branch and penetrate the concentric layers of the perineurium they emerge as capillaries within the endoneurium. The endothelial cells of these endoneurial capillaries are quite different from those of the precapillary arterioles. Endoneurial endothelial cells exhibit properties uniquely characteristic of a functional blood barrier, having lost their fenestrations and now are more prominently connected to adjacent endothelial cells by expressing a continuous array of tight- and adherens-junction proteins. Early ultrastructural studies of endoneurial microvessels within biopsy specimens from patients with immune-mediated neuropathies, however, exhibit fenestrations or intercellular gaps along with a loss of tight-junctions (Kanda et al., 2004; Lach et al., 1993; Meier et al., 1984). Breakdown of the BNB is considered a critical event in the pathophysiology of inflammatory demyelinating polyradiculoneuropathies, allowing pathogenic autoantibodies access to the normally restricted endoneurium (Abe et al., 2012; Maiuolo et al., 2019; Mathey et al., 2015; Shimizu and Kanda, 2015). However, despite circumstantial evidence from biopsy and serological studies showing antibody deposition on the outer surface of Schwann cells and compact myelin in some affected patients (Dalakas and Engel, 1980; Hays et al., 1988), there remains much contention surrounding the pathogenicity of myelin-directed autoantibodies (Liu et al., 2018). By comparison, ultrastructural studies of nodal and paranodal regions of affected nerves show abnormalities in Schwann cell microvilli and paranodal glial loops (Cifuentes-Diaz et al., 2011), with between 10–30 percent of CIDP patients exhibiting serum IgG antibodies binding to either nodal or paranodal regions (Devaux et al., 2012). Regardless of causality, the most effective strategies currently approved for the clinical management of GBS patients are limited and restricted to the use of nonselective immune-modulating therapies (intravenous immunoglobulin or plasma exchange).
Although the pathologic mechanism responsible for immune-mediated disruption of endoneurial endothelial tight-junction integrity remains unclear, the use of cultured primary or immortalized endoneurial endothelial cell lines has implicated several endogenous mediators (humoral, inflammatory cytokines, VEGF) as potential therapeutic targets (Gironi et al., 2010; Kanda et al., 2003; Reddy et al., 2013; Shimizu et al., 2011). Given that breakdown of the BNB plays a key role in the progression of peripheral nerve injury and ensuing long-term neurologic deficits, elucidating the molecular mechanisms that regulate the BNB integrity is anticipated to facilitate the development of novel therapeutic strategies for the management of immune-mediated inflammatory neuropathies.
As an established first-line treatment for most inflammatory neuropathies (Muley et al., 2008), it is interesting to note that corticosteroids have been shown to promote marked expression of the tight-junction proteins in cultured endothelial cells (Felinski et al., 2008; Firestone and Kapadia, 2014; Kashiwamura et al., 2011). Not all cultured endothelial cells are, however, phenotypically alike. Endothelial cells from different vascular beds within a given species exhibit organ-specific structural and functional heterogeneity (Feng et al., 2007; Yano et al., 2007). Early morphometric studies suggest that minor interspecies differences may even exist between endoneurial microvessels (Bell and Weddell, 1984a, b). A recent molecular study characterized the transcriptome from human endoneurial microvessels as consisting of 12881 unique transcripts expressed by endoneurial endothelial cells inclusive of an impressive array of transporters (509), chemokine and chemokine receptors, as well as over 100 junctional complex proteins (22 tight junction/JAMs; 45 adherens junction/associated; 52 cell-junction/adaptor proteins) (Palladino et al., 2017). Primary endoneurial endothelial cells isolated from different species by different laboratories (Abe et al., 2012; Argall et al., 1994; Kanda et al., 1997; Kanda et al., 2000; Kanda et al., 2003; Langert et al., 2013b; Sano et al., 2007; Ubogu, 2013; Yosef and Ubogu, 2013; Yosef et al., 2010) appear to have, however, remarkably similar cellular and molecular characteristics (Table 1). One notable distinction, however, is with observed transendothelial electrical resistance (TEER), a widely accepted quantitative measure of tight-junction integrity. Depending on the experimental conditions employed, cultures of human primary endoneurial endothelial cells exhibit a 7–10 fold increase in TEER (Ubogu, 2013) compared with cultured human brain primary capillary endothelial cells (Davidson et al., 2009) or with human or rat primary endoneurial endothelial cells (Abe et al., 2012; Langert et al., 2013b). While these findings suggest the possibility of an interspecies difference, it is well established that peripheral nerve pericytes maintain BNB function by releasing mitogens (Oishi et al., 2019; Ubogu, 2013), including basic fibroblast growth factor (bFGF), which has been shown to enhance the expression of endothelial tight-junction proteins (Shimizu et al., 2010). Moderate concentrations (0.5–5 ng/ml) of bFGF-2 were reported to enhance BBB tight-junction protein expression ex vivo in organotypic cortical slice cultures (Bendfeldt et al., 2007). A recent in vitro study further demonstrated that bFGF-induces upregulation of tight-junction proteins in cultured human brain microvascular endothelial cells (Wang et al., 2016). Culturing human primary endoneurial endothelial cells in the presence of recombinant human bFGF may, thus, account for some of the differences in observed TEER values between these two species. Collectively, pre-clinical findings support a role for certain mitogens at preserving or restoring BNB integrity. Reported limitations of targeted mitogen therapy in peripheral neuropathies/chronic pain, however, warrants due caution with regards to any localized therapeutic application of these agents.
Table 1.
Cellular and Molecular Characteristics of Cultured Primary Endoneurial Endothelial Cells
| Species | Cellular Characteristics | Molecular Characteristics |
|---|---|---|
| Rat |
|
|
| Cow |
|
|
| Human |
|
|
TARGETING INTRACELLULAR MEDIATORS OF BNB INTEGRITY
G-Proteins Modulators
Monomeric GTP binding proteins (G-proteins) belonging to the Rho family of GTPases are actively engaged at regulating transendothelial paracellular trafficking of leukocytes across endoneurial endothelial microvessel interfaces (Infante and Ridley, 2013; Sarkey et al., 2007; Wang et al., 2016). As a result, interest in isoprenylated G-proteins as potentially novel therapeutic targets for the management of immune-mediated neuropathies is growing (Langert et al., 2017; Langert et al., 2014; Langert et al., 2013a; Sarkey et al., 2007). Belonging to the Ras superfamily of monomeric G-proteins, the Rho family consists of over 20 members which have been categorized into eight distinct subfamilies. Among these subfamilies, Rho, Rac1, and Cdc42 are best studied and largely regulate cytoskeletal dynamics affecting a diverse array of intracellular processes including cell polarity, intracellular vesicle trafficking, endocytosis, cell cycle progression, cell contractility, and the formation of stress fibers or focal adhesions (Burridge and Wennerberg, 2004; Mackay and Hall, 1998).
The participation of Rho family of G-proteins in the regulation of leukocyte paracellular trafficking across endoneurial endothelial barriers first emerged from experimental studies using statins, well-tolerated inhibitors of the cholesterol biosynthetic pathway (Adamson and Greenwood, 2003; Greenwood and Mason, 2007). By limiting the endogenous production of bioactive sesquiterpene (farnesyl) and diterpene (geranylgeranyl) isoprenoids, statins prevent post-translational prenylation of G-proteins, including members of the Rho subfamily. Although the biochemical consequences of post-translational prenylation remains to be fully elucidated, one biophysical attribute is to enhance the hydrophobicity of nascent G-proteins thereby facilitating intracellular membrane localization and subsequent activation of these key biomolecular switches (Stubbs and Von Zee, 2012). Early observational studies provided the first functional evidence that a short-term high-dose course of statins markedly restricts transendothelial trafficking of autoreactive leukocytes into peripheral nerves and safely attenuates the development and progression of experimental autoimmune neuritis (EAN), a well-established animal model of AIDP/GBS (Sarkey et al., 2007). Follow-up mechanistic studies demonstrated that statins limit transendothelial migration of autoreactive leukocytes into peripheral nerves by inhibiting TNF-α mediated Cdc42-faciliated secretion of endoneurial endothelial cell-expressed CCL2, a chemokine implicated in GBS (Langert et al., 2014; Langert et al., 2013a). These reported findings agree well with previous studies documenting CCL2 chemokines, in addition to CCL5, CXCL8, and CXCL10 chemokines, as well-known mediators of leukocyte migration across endothelial barriers (Oppenheim et al., 1991; Subileau et al., 2009). While rare, but serious, adverse side effects associated with chronic systemic statin use currently prohibit their therapeutic application for the management of inflammatory peripheral neuropathies (Aiman et al., 2014; Golomb and Evans, 2008), selective localized targeting of endoneurial endothelial cell chemokine release remains a potentially effective therapeutic strategy by which to limit the recruitment and/or migration of autoreactive leukocytes across cytokine-activated endoneurial microvessels (Figure 1). Given that CC-chemokines are also known to promote inflammation-driven angiogenesis, specific inhibition of CC-chemokine signaling are being currently explored as a novel therapeutic strategy for the management of angiogenesis associated pathological diseases (Ridiandries et al., 2016; Ridiandries et al., 2017). Experimental studies have further identified various chemokines as key mediators of communication between neurons and neighboring non-neuronal cells (Ramesh et al., 2013). In addition to limiting leukocyte paracellular recruitment across the BNB, therapeutic approaches that further disrupt chemokine-mediated communication between neurons and infiltrating or resident non-neuronal cells are being evaluated for the management of chronic inflammatory pain (Akgun et al., 2015; Montague and Malcangio, 2017; Ubogu, 2011; White et al., 2007). Despite showing promise in early preclinical trials, the systemic use of chemokine antagonists as a therapeutic strategy for the management of chronic inflammatory disorders, including pain, has yet to be realized in phase clinical trials (Horuk, 2009a, b; Yekkirala et al., 2017).
An alternative mechanism by which statins may limit leukocyte trafficking across endothelial barriers is by direct enhancement of endothelial cell tight-junction formation. By suppressing endogenous isoprenylation of Rap1A (a member of the Ras oncogene family of small G-proteins), statins indirectly stimulate mitogen activated protein (MAP) kinase ERK5 in cultured human cardiac microvascular endothelial cells (Wilkinson et al., 2018). ERK5 is one of four major mitogen activated kinases (Buschbeck and Ullrich, 2005) and is critically needed to preserve vascular integrity in adult mice. Growth factor-stimulated activation of MAP kinases, including ERK5, typically results in nuclear translocation of this kinase and subsequent transcriptional regulation (Kondoh et al., 2006). However, statin-mediated activation of ERK5 in endothelial cells uniquely promotes translocation of this MAP kinase to the plasma membrane. Whether ERK5 is expressed by endoneurial endothelial cells and, if so, exactly how ERK5 enhances endothelial barrier integrity at this subcellular domain remains to be determined but may involve facilitating localized accumulation of zonula occludens-1 (Tornavaca et al., 2015) scaffolding proteins to subcortical F-actin tight-junction domains. The effect of statins on BNB integrity are not without precedence, as similar statin-mediated increases in tight-junction formation have been observed with cultured primary endothelial cells from human pulmonary artery (Chen et al., 2014) or from rat brain (Morofuji et al., 2010). Although it remains to be determined whether G-proteins play a key role in regulating BNB tight-junction formation, mitogen (bFGF)-mediated upregulation of tight-junction protein expression in human brain microvascular endothelial cells occurs, in part, through a mechanism that involves Rac1-dependent inhibition of RhoA (Wang et al., 2016). Localized application of inhibitors of G-protein signaling (Langert et al., 2017) represent an attractive experimental alternative to the therapeutic management of not only immune-mediated but also metabolic peripheral neuropathies (Shah and Singh, 2006). While the development and clinical application of inhibitors that target small GTPases is not exactly novel (Prieto-Dominguez et al., 2019), additional studies are warranted before translational value of this strategy can be fully realized for the management of acquired peripheral nerve disorders.
Claudin Modulators
In contrast to strategies designed to protect the endoneurial microenvironment by enhancing BNB integrity, there are some neurologic disorders such as chronic pain that may be best managed clinically by transient relaxation of BNB integrity. This is particularly necessary when considering the targeted delivery of hydrophilic therapeutics (e.g., opioid receptor peptidomimetics, voltage-activated sodium channel blockers) which, unlike hydrophobic compounds, do not readily diffuse across an intact BNB. Targeted disruption of the BNB is neither novel nor particularly difficult to experimentally achieve. Early studies showed that peripherally administered hypertonic saline elicits a transient breach of the BNB thereby enhancing antinociceptive properties of applied opioid peptides (Rittner et al., 2009; Stein, 2013). Mechanistic studies revealed that hypertonic saline relaxes the BNB integrity through a matrix metalloperoteinase-9 (MMP-9) facilitated, LDL receptor-related protein-1 (LRP-1) stimulated, ERK1/2-dependent down-regulation of claudin-1, a tetraspanin barrier-forming tight-junction protein expressed within the perineurium (Hackel et al., 2012b). Alternatively, upregulation of MMPs following nerve injury may alter BNB integrity through degradation of various endothelial tight junction proteins (Chernov et al., 2015; Qin et al., 2016; Wang et al., 2018; Yu et al., 2016). In contrast, tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) is similarly upregulated in response to peripheral nerve injury (Chernov et al., 2015; Kim et al., 2012; Remacle et al., 2018) which may serve to temporally and spatially regulate MMP-induced changes in BNB integrity. Given the pro-algesic properties of multiple MMPs, local inhibition of these extracellular zinc proteases with exogenous TIMP therapy has been reported in preclinical studies to exhibit potent analgesic beneficial properties (Kawasaki et al., 2008). However, while MMPs have been the focus for inhibitor design for many decades, MMP inhibitors have failed in various clinical trials owing to a lack of isoform specificity thus necessitating further development (Arkadash et al., 2017).
An analogous approach by which to down-regulation of perineurial claudin-1 expression includes the use of the LRP-1 agonist tissue plasminogen activator (tPA), which upregulates in an ERK1/2-dependent manner the expression of transcriptional repressors microRNA-29b or microRNA-183 (Yang et al., 2016). Advancements using specific siRNAs to target perineurial expression of tight-junction proteins, including claudin-1, also shows experimental promise (Hackel et al., 2012a; Rittner et al., 2012). Alternatively, perineurial application of a claudin-1 peptidomimetics (C1C2), which binds to the first extracellular loop of claudin-1, has been shown to selectively and safely relax the BNB integrity allowing access of hydrophilic analgesics to axonal nociceptors (Sauer et al., 2014; Staat et al., 2015).
In contrast to localized expression of claudin-1 at the perineurial barrier, claudin-5 is a tetraspanin tight-junction protein that is largely localized to junctional contacts between endoneurial endothelial cells and is considered a key regulator of both peripheral and central paracellular permeability (Lux et al., 2020; Wang et al., 2018). Most recently, regulated expression of claudin-5 has been proposed as a novel therapeutic strategy by which to better manage a variety of disparate CNS neurologic disorders including Alzheimer’s disease, multiple sclerosis, depression and schizophrenia (Greene et al., 2019). Previous studies, however, have raised concerns regarding the functional role of claudin-5 at regulating BNB permeability, and thus its role in BNB permeability remains controversial. Immunohistochemical studies of sural nerve biopsy specimens from patients with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP, n=10) showed a ~35% reduction in claudin-5 expression within endoneurial microvessels compared with disease-control nerves from patients with Churg-Strauss syndrome (n=6), hereditary neuropathy (n=6), or with nutritional (B1-deficient, n=4) neuropathy (Kanda et al., 2004). As a ubiquitously expressed tight-junction protein, reduced expression of endoneurial endothelial claudin-5 might be expected to enhance BNB permeability. However, the loss of claudin-5 immunoreactivity did not correlate with endoneurial/sub-perineurial edema in sural nerves from CIDP patients. Another tetraspanin protein implicated in the maintenance of BNB integrity is occludin. Levels of occludin immunoreactivity in endoneurial microvessels did not change appreciably in inflammatory neuropathies (Kanda et al., 2004). These findings would suggest that, unlike CNS expressed claudin-5, molecular strategies designed to selectively alter endoneurial endothelial claudin-5 / occludin expression may not be as effective at altering the BNB integrity.
TARGETING EXTRACELLULAR MEDIATORS OF BNB INTEGRITY
Pericytes, Basement Membranes, and Integrin Modulators
Molecular details responsible for cell-surface tethering, rolling, adhesion and paracellular/transcellular diapedesis of activated leukocytes out of circulating blood, across endothelial barriers, and into the perivascular space are well documented and have been the subject of many excellent reviews (Ley et al., 2007; Liu et al., 2004; Mamdouh et al., 2009; Muller, 2011, 2014). Less clear, however, are details on how transmigrating leukocytes subsequently traverse basement membranes, which physically encase endoneurial endothelial cells and associated abluminal pericytes and functionally serve as a potentially rate-limiting protective barrier to the endoneurium.
Pericytes are perivascular mural cells that extend along nearly every capillary in the human body and are embedded in the vascular basement membrane of microvasculature (Armulik et al., 2011a; Armulik et al., 2011b; Zhao and Chappell, 2019). They are particularly enriched in endoneurial microvessels and are now recognized as key cellular contributors of BNB integrity (Shimizu and Kanda, 2015; Shimizu et al., 2008). Recent studies support the existence of pericytes subtypes (ensheathing, mesh, and thin-strand) that correspond to their physical location on any given microvascular network (Zhao and Chappell, 2019). The influence of the local neurovascular microenvironment together with phenotypic subtypes may very well contribute to the unique phenotype of peripheral nerve pericytes. In addition to serving as a local source of essential neurotrophic factors (NGF, BDNF, GDNF), peripheral nerve pericytes are largely responsible for establishing and maintaining the biochemical composition of endothelial basement membranes through their synthesis and release of several prominent extracellular matrix constituents including collagen type IV, fibronectin, vitronectin, and TIMP-1 (Richner et al., 2018; Shimizu et al., 2010; Zhao and Chappell, 2019). An additional major functional component of basement membranes produced by pericytes include glycoproteins belonging to the laminin family. Three distinct laminin chains (cell binding domain α, β, γ) combine to form up to twelve different heterotrimeric isoforms. Of these isoforms, only laminin 411 (α4β1γ1 or laminin 8) and laminin 511 (α5β1γ1 or laminin 10) are found in endothelial basement membranes (Frieser et al., 1997; Sixt et al., 2001). Post capillary venule endothelial cell expression of laminin 511 appears variable while expression of laminin 411 is ubiquitous. Venules lacking laminin 511 are permissive to leukocyte diapedesis, while in vitro studies show that laminin 511 selectively inhibits transmigration of autoreactive T cells (Wu et al., 2009). These findings suggest that differential expression of laminin isoforms may play a role in directly regulating leukocyte transendothelial trafficking (Fujikawa et al., 2017; Sixt et al., 2001).
The mechanism by which laminins are able to enhance BNB integrity and limit leukocyte diapedesis was recently elucidated and involves stabilization of endothelial cell adherens junctions (Song et al., 2017). Paracellular transmigration of leukocytes across endoneurial endothelial cell-cell interfaces is governed by junctional localization of both tight junction and adherens junctions (cadherens) proteins. Localization of VE-cadherin to junctional compartments appears to contribute to the regulation of leukocyte diapedesis, as reduced junctional localization of these adherens proteins enhances leukocyte transmigration (Wessel et al., 2014). A major class of extracellular matrix binding receptors expressed on the surface of endothelial cells are the β1 integrins. These heterodimeric transmembrane proteins consist of one α- and one β-subunit that binds with, and adheres to, several classes of extracellular matrix proteins, including collagen IV, perlecan, and laminins (Henry et al., 2001). Endothelial cell expression of β1 integrins are required for junctional localization of VE-cadherens (Yamamoto et al., 2015). In preclinical studies using cultured mouse brain-derived endothelial cells (bEND.5), β1 (and β3) integrin binding to laminin 511 is shown to selectively elicit RhoA-mediated localization and stabilization of VE-cadherens to cell-cell interfaces which correlated with increased TEER and reduced leukocyte paracellular transmigration (Song et al., 2017). Whether endoneurial endothelial cells cell-cell interfaces are similarly influenced by integrin signaling remains to be determined, but once again we see a penultimate role of small monomeric GTPases participating in the regulation of the BNB integrity. In this case, however, local targeted disruption of RhoA signaling (e,g., statins) in endoneurial endothelial cells would be anticipated to disrupt laminin-dependent stabilization of adherens junctions and thereby transiently alter BNB integrity. In addition to influencing endothelial cell VE-cadherin junctional localization, disrupting β1 integrin signaling also decreases claudin-5 expression in cerebral endothelial cells and increases the permeability of cerebral microvessels (Osada et al., 2011). While these preclinical findings are encouraging, development/application of localized therapeutic strategies designed to disrupt endoneurial endothelial cell β1 integrin signaling may be of limited clinical value given (i) the controversial functional role of claudin-5 at regulating the BNB permeability and (ii) restricted abluminal accessibility of therapeutic agents to the site of integrin-basal lamina matrix.
Vascular Endothelial Growth Factor
A major early hallmark in the pathogenesis of inflammatory peripheral neuropathies involves elevated circulating levels of BNB-disrupting pro-inflammatory cytokines. Among these, pericyte-produced vascular endothelial growth factor (VEGF) is particularly effective at enhancing central and peripheral paracellular permeability in association with its pro-angiogenic properties (Shimizu et al., 2010; Shimizu et al., 2011; Shimizu et al., 2008). By activating tyrosine kinase receptors (VEGFR1, VEGFR2), VEGF isoforms potently reduces the endothelial cell-cell junctional expression of the junctional proteins occludins and VE-cadherins thereby increasing vascular permeability of endoneurial microvessels (Gale and Yancopoulos, 1999; Kevil et al., 1998). As a key mediator of neovascularization, several anti-VEGF related therapies are currently used in clinical settings for the management of various neoplasms as well as several ocular disorders involving pathologic neovascularization (Khanna et al., 2019; Sitohy et al., 2012). Recently, the role of VEGF isoforms and its two neuropilin co-receptors (NRP-1, NRP-2) in the pathophysiology of chronic pain has been reviewed and discussed in the context as a potentially druggable target for the management of painful neuropathies (Llorian-Salvador and Gonzalez-Rodriguez, 2018). While preclinical studies support VEGF/VEGFR blockade as a successful strategy to alleviate nociceptive responses in various animal models (CCI, sciatic nerve ligation, diabetic neuropathy) of neuropathic pain, a retrospective cohort study found that neutralization of VEGF-A exacerbated paclitaxel-induced neuropathy (Matsuoka et al., 2016). The possibility that different VEGF isoforms exhibit distinct pro- or anti-nociceptive properties (Hulse, 2017) further complicates the use of anti-VEGF therapies for the management of neuropathic pain. Thus, the application of localized anti-VEGF therapies for the management of immune-mediated inflammatory neuropathies should be viewed with extreme caution as VEGF itself has been shown to be an effective mediator of central neurogenesis and neuroprotection (Li et al., 2017) and may be crucial for revascularization of injured peripheral nerves (eg, vasculitic neuropathy).
Integrin : Cell Adhesion Molecule Modulators
By their selective interactions with leukocyte expressed β1- or β2-integrins, several distinct endothelial cell surface expressed cell adhesion molecules (CAMs) play pivotal roles in the tightly regulated temporal and spatial “capturing” of activated leukocytes prior to transendothelial (paracellular) migration into the PNS (Mitroulis et al., 2015; Muller, 2011). Given its penultimate role of leukocyte recruitment and migration, disrupting integrin:CAM signaling remains a key therapeutic strategy for the management of inflammatory disorders (Archelos et al., 1994; Archelos et al., 1993; Archelos et al., 1999; Mitroulis et al., 2015).
In general, the leukocyte adhesion cascade begins with endothelial-expressed (E, P) and leukocyte-expressed (L)-selectin-dependent rolling followed by chemokine-induced leukocyte activation. Slow rolling allows for integrin-dependent firm adhesion/arrest of the “captured” leukocytes on the luminal surface of activated endothelium, ultimately enabling paracellular diapedesis. Both integrin affinity (integrin:ligand bond strength) and integrin valency (integrin receptor clustering) play a role in defining the overall avidity of integrin-mediated cell adhesion. In mammals, multiple integrin α-subunit isoforms (18) and β-subunit isoforms (8) have been described, forming a diverse functional array of 24 unique integrin heterodimers. Therapeutic strategies designed to limit pathogenic leukocyte paracellular migration across activated endoneurial endothelial barriers must therefore take into consideration the isoform specificity of localized leukocyte integrin:CAM interactions. Leukocyte-expressed integrins Lymphocyte Function-associated Antigen-1 (LFA-1; CD11a (αL)/CD18 (β2)) and Very Late Antigen-4 (VLA-4; CD49d (α4)/CD29 (β1)) both play essential roles in leukocyte recruitment/trafficking and in inflammatory disorders. Endothelial-expressed binding partners for these two integrins include ICAM-1 and VCAM-1, respectively. In addition, a second key β2-integrin:CAM interaction involved in recruitment of activated macrophages is Macrophage-1 Antigen (Mac-1; CD11b (αM)/CD18 (β2)) integrin:ICAM-1 (Podolnikova et al., 2016).
Despite exquisite diversity among integrin:CAM cell-cell interactions (Archelos et al., 1999), some progress has been made toward selective therapeutic application targeting integrin signaling. Blockade of leukocyte VLA-4: endothelial cell VCAM-1 interactions with the humanized monoclonal antibody natalizumab has proven clinically safe and effective at managing relapse-remitting multiple sclerosis (Brandstadter and Katz Sand, 2017). Preclinical studies suggest that blockade of αM-integrin:ICAM-1 signaling may also be an effective strategy by which to limit leukocyte trafficking across activated endoneurial endothelial cell barriers (Yosef and Ubogu, 2012). The targeting of α4-integrin receptors for the management of immune-mediated neuropathies, however, is less encouraging, as case reports of natalizumab for the treatment of CIDP are disappointing (Vallat et al., 2015; Wolf et al., 2010). These studies highlight anticipated differences in clinical responsiveness between central (BBB)- or peripheral (BNB)-targeted therapies and underscore the need to tailor therapeutic strategies towards leukocyte:endoneurial endothelial cell expressed integrin:CAM selective interactions.
FUTURE TARGETING STRATEGIES
An alternative experimental strategy that is currently under intense investigation includes selective targeting of endoneurial endothelial cell expressed intercellular adhesion molecule-1 (ICAM-1) for local delivery of proven therapeutics using nanotherapy (Langert et al., 2017). Although ICAM-1 is expressed constitutively at low levels on most microvascular endothelial cells and on some lymphocytes and monocytes, its expression can be significantly increased in the presence of pro-inflammatory cytokines (Hubbard and Rothlein, 2000). Consistent with this observation, cultured endoneurial endothelial cells were found to robustly upregulate the cell surface expression of ICAM-1 in response to a TNF-α challenge (Langert et al., 2013b). By functionalizing PLGA nanoparticles with purified membrane fragments enriched in the ICAM-1 binding partner LFA-1, efforts are underway to determine whether systemically administer nanoparticle-encapsulated therapeutics will selectively deliver their payload to activated endoneurial microvessels that express increase levels of ICAM-1 (Langert and Brey, 2018). A challenge which currently limits the clinical application of any new therapeutic strategy, including translatable nanotherapies that target integrin:CAM interactions, is the possibility of off-target effects.
Depending on the clinical application, intracellular and extracellular targeted therapies that alter the BNB integrity are showing promise as the next generation of much needed clinically viable alternatives to the current standard of care for the management of traumatic, metabolic, or immune-mediated inflammatory neuropathies. It is hoped that the many exciting articles presented within, and referenced throughout, this Experimental Neurology Special Issue will renew interest and foster further enthusiasm for the development, testing, and ultimate clinical application of novel and innovative BNB therapeutics.
FUNDING
This work was supported by grants from the Department of Veterans Affairs Biomedical Laboratory R&D Service (1I01BX003938), the Department of Veterans Affairs Rehabilitation R&D Service (I21RX001553 and I01RX000130) and the National Institutes of Health (IR21NS085420).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abe M, Sano Y, Maeda T, Shimizu F, Kashiwamura Y, Haruki H, Saito K, Tasaki A, Kawai M, Terasaki T, Kanda T, 2012. Establishment and characterization of human peripheral nerve microvascular endothelial cell lines: a new in vitro blood-nerve barrier (BNB) model. Cell Struct Funct 37, 89–100. [DOI] [PubMed] [Google Scholar]
- Adamson P, Greenwood J, 2003. How do statins control neuroinflammation? Inflamm Res 52, 399–403. [DOI] [PubMed] [Google Scholar]
- Aiman U, Najmi A, Khan RA, 2014. Statin induced diabetes and its clinical implications. J Pharmacol Pharmacother 5, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgun E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS, 2015. Inhibition of Inflammatory and Neuropathic Pain by Targeting a Mu Opioid Receptor/Chemokine Receptor5 Heteromer (MOR-CCR5). J Med Chem 58, 8647–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archelos JJ, Maurer M, Jung S, Miyasaka M, Tamatani T, Toyka KV, Hartung HP, 1994. Inhibition of experimental autoimmune neuritis by an antibody to the lymphocyte function-associated antigen-1. Lab Invest 70, 667–675. [PubMed] [Google Scholar]
- Archelos JJ, Maurer M, Jung S, Toyka KV, Hartung HP, 1993. Suppression of experimental allergic neuritis by an antibody to the intracellular adhesion molecule ICAM-1. Brain 116 ( Pt 5), 1043–1058. [DOI] [PubMed] [Google Scholar]
- Archelos JJ, Previtali SC, Hartung HP, 1999. The role of integrins in immune-mediated diseases of the nervous system. Trends Neurosci 22, 30–38. [DOI] [PubMed] [Google Scholar]
- Argall KG, Armati PJ, Pollard JD, 1994. A method for the isolation and culture of rat peripheral nerve vascular endothelial cells. Mol Cell Neurosci 5, 413–417. [DOI] [PubMed] [Google Scholar]
- Arkadash V, Yosef G, Shirian J, Cohen I, Horev Y, Grossman M, Sagi I, Radisky ES, Shifman JM, Papo N, 2017. Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J Biol Chem 292, 3481–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C, 2011a. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21, 193–215. [DOI] [PubMed] [Google Scholar]
- Armulik A, Mae M, Betsholtz C, 2011b. Pericytes and the blood-brain barrier: recent advances and implications for the delivery of CNS therapy. Ther Deliv 2, 419–422. [DOI] [PubMed] [Google Scholar]
- Bell MA, Weddell AG, 1984a. A descriptive study of the blood vessels of the sciatic nerve in the rat, man and other mammals. Brain 107 ( Pt 3), 871–898. [DOI] [PubMed] [Google Scholar]
- Bell MA, Weddell AG, 1984b. A morphometric study of intrafascicular vessels of mammalian sciatic nerve. Muscle Nerve 7, 524–534. [DOI] [PubMed] [Google Scholar]
- Bendfeldt K, Radojevic V, Kapfhammer J, Nitsch C, 2007. Basic fibroblast growth factor modulates density of blood vessels and preserves tight junctions in organotypic cortical cultures of mice: a new in vitro model of the blood-brain barrier. J Neurosci 27, 3260–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstadter R, Katz Sand I, 2017. The use of natalizumab for multiple sclerosis. Neuropsychiatr Dis Treat 13, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K, 2004. Rho and Rac take center stage. Cell 116, 167–179. [DOI] [PubMed] [Google Scholar]
- Buschbeck M, Ullrich A, 2005. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J Biol Chem 280, 2659–2667. [DOI] [PubMed] [Google Scholar]
- Chen W, Sharma R, Rizzo AN, Siegler JH, Garcia JG, Jacobson JR, 2014. Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am J Respir Cell Mol Biol 50, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov AV, Dolkas J, Hoang K, Angert M, Srikrishna G, Vogl T, Baranovskaya S, Strongin AY, Shubayev VI, 2015. The calcium-binding proteins S100A8 and S100A9 initiate the early inflammatory program in injured peripheral nerves. J Biol Chem 290, 11771–11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Dubourg O, Irinopoulou T, Vigny M, Lachkar S, Decker L, Charnay P, Denisenko N, Maisonobe T, Leger JM, Viala K, Hauw JJ, Girault JA, 2011. Nodes of ranvier and paranodes in chronic acquired neuropathies. PLoS One 6, e14533; 14510.11371/journal.pone.0014533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, Engel WK, 1980. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch Neurol 37, 637–640. [DOI] [PubMed] [Google Scholar]
- Davidson MM, Walker WF, Hernandez-Rosa E, 2009. The m.3243A>G mtDNA mutation is pathogenic in an in vitro model of the human blood brain barrier. Mitochondrion 9, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Odaka M, Yuki N, 2012. Nodal proteins are target antigens in Guillain-Barre syndrome. J Peripher Nerv Syst 17, 62–71. [DOI] [PubMed] [Google Scholar]
- Felinski EA, Cox AE, Phillips BE, Antonetti DA, 2008. Glucocorticoids induce transactivation of tight junction genes occludin and claudin-5 in retinal endothelial cells via a novel cis-element. Exp Eye Res 86, 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Yano K, Monahan-Earley R, Morgan ES, Dvorak AM, Sellke FW, Aird WC, 2007. Vascular bed-specific endothelium-dependent vasomomotor relaxation in the hagfish, Myxine glutinosa. Am J Physiol Regul Integr Comp Physiol 293, R894–900. [DOI] [PubMed] [Google Scholar]
- Firestone GL, Kapadia BJ, 2014. Minireview: Steroid/nuclear receptor-regulated dynamics of occluding and anchoring junctions. Mol Endocrinol 28, 1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieser M, Nockel H, Pausch F, Roder C, Hahn A, Deutzmann R, Sorokin LM, 1997. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem 246, 727–735. [DOI] [PubMed] [Google Scholar]
- Fujikawa S, Saeki A, Takeshita Y, Kanda T, 2017. The effect of blood-nerve barrier (BNB)-specific laminin isoforms for leukocyte transmigration at BNB. Journal of Neurological Science 381, 288–293. [Google Scholar]
- Gale NW, Yancopoulos GD, 1999. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev 13, 1055–1066. [DOI] [PubMed] [Google Scholar]
- Getter T, Margalit R, Kahremany S, Levy L, Blum E, Khazanov N, Keshet-Levy NY, Tamir TY, Ben Major M, Lahav R, Zilber S, Senderowitz H, Bradfield P, Imhof BA, Alpert E, Gruzman A, 2019. Novel inhibitors of leukocyte transendothelial migration. Bioorg Chem 92, 103250: doi: 1032 10.101016/j.bioorg.102019.103250. [DOI] [PubMed] [Google Scholar]
- Gironi M, Saresella M, Marventano I, Guerini FR, Gatti A, Antonini G, Ceresa L, Morino S, Beghi E, Angelici A, Mariani E, Nemni R, Clerici M, 2010. Distinct cytokine patterns associated with different forms of chronic dysimmune neuropathy. Muscle Nerve 42, 864–870. [DOI] [PubMed] [Google Scholar]
- Golomb BA, Evans MA, 2008. Statin adverse effects : a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs 8, 373–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greathouse KM, Palladino SP, Dong C, Helton ES, Ubogu EE, 2016. Modeling leukocyte trafficking at the human blood-nerve barrier in vitro and in vivo geared towards targeted molecular therapies for peripheral neuroinflammation. J Neuroinflammation 13, 3; 10.1186/s12974-12015-10469-12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene C, Hanley N, Campbell M, 2019. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS 16, 3: 10.1186/s12987-12019-10123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J, Mason JC, 2007. Statins and the vascular endothelial inflammatory response. Trends Immunol 28, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel D, Brack A, Fromm M, Rittner HL, 2012a. Modulation of tight junction proteins in the perineurium for regional pain control. Ann N Y Acad Sci 1257, 199–206. [DOI] [PubMed] [Google Scholar]
- Hackel D, Krug SM, Sauer RS, Mousa SA, Bocker A, Pflucke D, Wrede EJ, Kistner K, Hoffmann T, Niedermirtl B, Sommer C, Bloch L, Huber O, Blasig IE, Amasheh S, Reeh PW, Fromm M, Brack A, Rittner HL, 2012b. Transient opening of the perineurial barrier for analgesic drug delivery. Proc Natl Acad Sci U S A 109, E2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays AP, Lee SS, Latov N, 1988. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J Neuroimmunol 18, 231–244. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP, 2001. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J Cell Sci 114, 1137–1144. [DOI] [PubMed] [Google Scholar]
- Hogarth PM, 2002. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol 14, 798–802. [DOI] [PubMed] [Google Scholar]
- Horuk R, 2009a. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8, 23–33. [DOI] [PubMed] [Google Scholar]
- Horuk R, 2009b. Promiscuous drugs as therapeutics for chemokine receptors. Expert Rev Mol Med 11, e1; 10.1017/S1462399409000921. [DOI] [PubMed] [Google Scholar]
- Hubbard AK, Rothlein R, 2000. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med 28, 1379–1386. [DOI] [PubMed] [Google Scholar]
- Hulse RP, 2017. Role of VEGF-A in chronic pain. Oncotarget 8, 10775–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante E, Ridley AJ, 2013. Roles of Rho GTPases in leucocyte and leukaemia cell transendothelial migration. Philos Trans R Soc Lond B Biol Sci 368: doi: 10.1098/rstb.2013.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, 2013. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J Neurol Neurosurg Psychiatry 84, 208–212. [DOI] [PubMed] [Google Scholar]
- Kanda T, Iwasaki T, Yamawaki M, Ikeda K, 1997. Isolation and culture of bovine endothelial cells of endoneurial origin. J Neurosci Res 49, 769–777. [DOI] [PubMed] [Google Scholar]
- Kanda T, Iwasaki T, Yamawaki M, Tai T, Mizusawa H, 2000. Anti-GM1 antibody facilitates leakage in an in vitro blood-nerve barrier model. Neurology 55, 585–587. [DOI] [PubMed] [Google Scholar]
- Kanda T, Numata Y, Mizusawa H, 2004. Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. J Neurol Neurosurg Psychiatry 75, 765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Yamawaki M, Mizusawa H, 2003. Sera from Guillain-Barre patients enhance leakage in blood-nerve barrier model. Neurology 60, 301–306. [DOI] [PubMed] [Google Scholar]
- Kashiwamura Y, Sano Y, Abe M, Shimizu F, Haruki H, Maeda T, Kawai M, Kanda T, 2011. Hydrocortisone enhances the function of the blood-nerve barrier through the up-regulation of claudin-5. Neurochem Res 36, 849–855. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR, 2008. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med 14, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E, Alexander JS, 1998. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 273, 15099–15103. [DOI] [PubMed] [Google Scholar]
- Khanna S, Komati R, Eichenbaum DA, Hariprasad I, Ciulla TA, Hariprasad SM, 2019. Current and upcoming anti-VEGF therapies and dosing strategies for the treatment of neovascular AMD: a comparative review. BMJ Open Ophthalmol 4, e000398; 0003 10.001136/bmjophth-002019-000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieseier BC, Mathey EK, Sommer C, Hartung HP, 2018. Immune-mediated neuropathies. Nat Rev Dis Primers 4, 31; 10.1038/s41572-41018-40027-41572. [DOI] [PubMed] [Google Scholar]
- Kim Y, Remacle AG, Chernov AV, Liu H, Shubayev I, Lai C, Dolkas J, Shiryaev SA, Golubkov VS, Mizisin AP, Strongin AY, Shubayev VI, 2012. The MMP-9/TIMP-1 axis controls the status of differentiation and function of myelin-forming Schwann cells in nerve regeneration. PLoS One 7, e33664; 336 10.31371/journal.pone.0033664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh K, Terasawa K, Morimoto H, Nishida E, 2006. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol Cell Biol 26, 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lach B, Rippstein P, Atack D, Afar DE, Gregor A, 1993. Immunoelectron microscopic localization of monoclonal IgM antibodies in gammopathy associated with peripheral demyelinative neuropathy. Acta Neuropathol 85, 298–307. [DOI] [PubMed] [Google Scholar]
- Langert KA, Brey EM, 2018. Strategies for Targeted Delivery to the Peripheral Nerve. Front Neurosci 12, 887: doi: 8 10.3389/fnins.2018.00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Goshu B, Stubbs EB Jr., 2017. Attenuation of experimental autoimmune neuritis with locally administered lovastatin-encapsulating poly(lactic-co-glycolic) acid nanoparticles. J Neurochem 140, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Pervan CL, Stubbs EB Jr., 2014. Novel role of Cdc42 and RalA GTPases in TNF-alpha mediated secretion of CCL2. Small GTPases 5: e29260. doi: 10.4161/sgtp.29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Von Zee CL, Stubbs EB Jr., 2013a. Cdc42 GTPases facilitate TNF-alpha-mediated secretion of CCL2 from peripheral nerve microvascular endoneurial endothelial cells. J Peripher Nerv Syst 18, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langert KA, Von Zee CL, Stubbs EB Jr., 2013b. Tumour necrosis factor alpha enhances CCL2 and ICAM-1 expression in peripheral nerve microvascular endoneurial endothelial cells. ASN Neuro 5: e00104: doi: 10.1042/AN20120048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MW, Richards MP, De Vries GH, Fisher MA, Stubbs EB Jr., 2002. Antibodies to L-periaxin in sera of patients with peripheral neuropathy produce experimental sensory nerve conduction deficits. J Neurochem 83, 592–600. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S, 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7, 678–689. [DOI] [PubMed] [Google Scholar]
- Li J, Chen S, Zhao Z, Luo Y, Hou Y, Li H, He L, Zhou L, Wu W, 2017. Effect of VEGF on Inflammatory Regulation, Neural Survival, and Functional Improvement in Rats following a Complete Spinal Cord Transection. Front Cell Neurosci 11, 381: doi: 3 10.3389/fnins.2018.00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Dong C, Ubogu EE, 2018. Immunotherapy of Guillain-Barre syndrome. Hum Vaccin Immunother 14, 2568–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shaw SK, Ma S, Yang L, Luscinskas FW, Parkos CA, 2004. Regulation of leukocyte transmigration: cell surface interactions and signaling events. J Immunol 172, 7–13. [DOI] [PubMed] [Google Scholar]
- Llorian-Salvador M, Gonzalez-Rodriguez S, 2018. Painful Understanding of VEGF. Front Pharmacol 9, 1267: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux TJ, Hu X, Ben-Kraiem A, Blum R, Chen JT, Rittner HL, 2020. Regional differences in tight junction protein expression in the blood-DRG barrier and their alterations after nerve traumatic injury in rats. Int J of Mol Sci 21, 270; doi: 10.3390/ijms21010270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Hall A, 1998. Rho GTPases. J Biol Chem 273, 20685–20688. [DOI] [PubMed] [Google Scholar]
- Maiuolo J, Gliozzi M, Musolino V, Carresi C, Nucera S, Macri R, Scicchitano M, Bosco F, Scarano F, Ruga S, Zito MC, Oppedisano F, Mollace R, Paone S, Palma E, Muscoli C, Mollace V, 2019. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int J Mol Sci 20; 10.3390/ijms20123022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamdouh Z, Mikhailov A, Muller WA, 2009. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med 206, 2795–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathey EK, Park SB, Hughes RA, Pollard JD, Armati PJ, Barnett MH, Taylor BV, Dyck PJ, Kiernan MC, Lin CS, 2015. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 86, 973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka A, Maeda O, Mizutani T, Nakano Y, Tsunoda N, Kikumori T, Goto H, Ando Y, 2016. Bevacizumab Exacerbates Paclitaxel-Induced Neuropathy: A Retrospective Cohort Study. PLoS One 11, e0168707; 01687 10.0161371/journal.pone.0168707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier C, Roberts K, Steck A, Hess C, Miloni E, Tschopp L, 1984. Polyneuropathy in Waldenstrom’s macroglobulinaemia: reduction of endoneurial IgM-deposits after treatment with chlorambucil and plasmapheresis. Acta Neuropathol 64, 297–307. [DOI] [PubMed] [Google Scholar]
- Mitroulis I, Alexaki VI, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T, 2015. Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther 147, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague K, Malcangio M, 2017. The therapeutic potential of targeting chemokine signalling in the treatment of chronic pain. J Neurochem 141, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morofuji Y, Nakagawa S, So G, Hiu T, Horai S, Hayashi K, Tanaka K, Suyama K, Deli MA, Nagata I, Niwa M, 2010. Pitavastatin strengthens the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol 30, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muley SA, Kelkar P, Parry GJ, 2008. Treatment of chronic inflammatory demyelinating polyneuropathy with pulsed oral steroids. Arch Neurol 65, 1460–1464. [DOI] [PubMed] [Google Scholar]
- Muller WA, 2011. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6, 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, 2014. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol 184, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV, 2008. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8, 34–47. [DOI] [PubMed] [Google Scholar]
- Oishi M, Shimizu F, Sano Y, Takeshita Y, Maeda T, Fujikawa S, Honda M, Sato R, Kanda T, 2019. Difference in cytokines, chemokines and growth factors produced by blood-brain barrier- and blood-nerve barrier-composing cells. Clinical and Experimental Neuroimmunology 10, 132–137. [Google Scholar]
- Olsson Y, 1966. Studies on vascular permeability in peripheral nerves. I. Distribution of circulating fluorescent serum albumin in normal, crushed and sectioned rat sciatic nerve. Acta Neuropathol 7, 1–15. [DOI] [PubMed] [Google Scholar]
- Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K, 1991. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol 9, 617–648. [DOI] [PubMed] [Google Scholar]
- Osada T, Gu YH, Kanazawa M, Tsubota Y, Hawkins BT, Spatz M, Milner R, del Zoppo GJ, 2011. Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by beta-(1)integrins. J Cereb Blood Flow Metab 31, 1972–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino SP, Helton ES, Jain P, Dong C, Crowley MR, Crossman DK, Ubogu EE, 2017. The Human Blood-Nerve Barrier Transcriptome. Sci Rep 7, 17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolnikova NP, Kushchayeva YS, Wu Y, Faust J, Ugarova TP, 2016. The Role of Integrins alphaMbeta2 (Mac-1, CD11b/CD18) and alphaDbeta2 (CD11d/CD18) in Macrophage Fusion. Am J Pathol 186, 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Berg CT, 1994. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A 91, 5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto-Dominguez N, Parnell C, Teng Y, 2019. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells 8; 10.3390/cells8030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Zha GB, Yu J, Zhang HH, Yi S, 2016. Differential temporal expression of matrix metalloproteinases following sciatic nerve crush. Neural Regen Res 11, 1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querol L, Devaux J, Rojas-Garcia R, Illa I, 2017. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol 13, 533–547. [DOI] [PubMed] [Google Scholar]
- Ramesh G, MacLean AG, Philipp MT, 2013. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm 2013: 480739. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport SI, 2000. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell Mol Neurobiol 20, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy CL, Yosef N, Ubogu EE, 2013. VEGF-A165 potently induces human blood-nerve barrier endothelial cell proliferation, angiogenesis, and wound healing in vitro. Cell Mol Neurobiol 33, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold AK, Rittner HL, 2017. Barrier function in the peripheral and central nervous system-a review. Pflugers Arch 469, 123–134. [DOI] [PubMed] [Google Scholar]
- Remacle AG, Hullugundi SK, Dolkas J, Angert M, Chernov AV, Strongin AY, Shubayev VI, 2018. Acute- and late-phase matrix metalloproteinase (MMP)-9 activity is comparable in female and male rats after peripheral nerve injury. J Neuroinflammation 15, 89; 10.1186/s12974-12018-11123-12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner M, Ferreira N, Dudele A, Jensen TS, Vaegter CB, Goncalves NP, 2018. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front Neurosci 12, 1038: doi: 10 10.3389/fnins.2018.01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridiandries A, Tan JT, Bursill CA, 2016. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int J Mol Sci 17; 10.3390/ijms17111856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridiandries A, Tan JT, Ravindran D, Williams H, Medbury HJ, Lindsay L, Hawkins C, Prosser HC, Bursill CA, 2017. CC-chemokine class inhibition attenuates pathological angiogenesis while preserving physiological angiogenesis. FASEB J 31, 1179–1192. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Amasheh S, Moshourab R, Hackel D, Yamdeu RS, Mousa SA, Fromm M, Stein C, Brack A, 2012. Modulation of tight junction proteins in the perineurium to facilitate peripheral opioid analgesia. Anesthesiology 116, 1323–1334. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Hackel D, Yamdeu RS, Mousa SA, Stein C, Schafer M, Brack A, 2009. Antinociception by neutrophil-derived opioid peptides in noninflamed tissue--role of hypertonicity and the perineurium. Brain Behav Immun 23, 548–557. [DOI] [PubMed] [Google Scholar]
- Ruano-Salguero JS, Lee KH, 2020. Antibody transcytosis across brain endothelial-like cells occurs nonspecifically and independent of FcRn. Sci Rep 10, 3685; 36 10.1038/s41598-41020-60438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Shimizu F, Nakayama H, Abe M, Maeda T, Ohtsuki S, Terasaki T, Obinata M, Ueda M, Takahashi R, Kanda T, 2007. Endothelial cells constituting blood-nerve barrier have highly specialized characteristics as barrier-forming cells. Cell Struct Funct 32, 139–147. [DOI] [PubMed] [Google Scholar]
- Sarkey JP, Richards MP, Stubbs EB Jr., 2007. Lovastatin attenuates nerve injury in an animal model of Guillain-Barre syndrome. J Neurochem 100, 1265–1277. [DOI] [PubMed] [Google Scholar]
- Sauer RS, Krug SM, Hackel D, Staat C, Konasin N, Yang S, Niedermirtl B, Bosten J, Gunther R, Dabrowski S, Doppler K, Sommer C, Blasig IE, Brack A, Rittner HL, 2014. Safety, efficacy, and molecular mechanism of claudin-1-specific peptides to enhance blood-nerve-barrier permeability. J Control Release 185, 88–98. [DOI] [PubMed] [Google Scholar]
- Schafflick D, Kieseier BC, Wiendl H, Meyer Zu Horste G, 2017. Novel pathomechanisms in inflammatory neuropathies. J Neuroinflammation 14, 232; 2 10.1186/s12974-12017-11001-12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah DI, Singh M, 2006. Involvement of Rho-kinase in experimental vascular endothelial dysfunction. Mol Cell Biochem 283, 191–199. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Kanda T, 2015. Breakdown of blood-nerve barrier in immune-mediated neuropathy. Clinical & Experimental Neuroimmunology 6, 139–148. [Google Scholar]
- Shimizu F, Sano Y, Abe MA, Maeda T, Ohtsuki S, Terasaki T, Kanda T, 2010. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J Cell Physiol 226, 255–266. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Haruki H, Kanda T, 2011. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-beta and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 54, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Maeda T, Abe MA, Nakayama H, Takahashi R, Ueda M, Ohtsuki S, Terasaki T, Obinata M, Kanda T, 2008. Peripheral nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J Cell Physiol 217, 388–399. [DOI] [PubMed] [Google Scholar]
- Sitohy B, Nagy JA, Dvorak HF, 2012. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res 72, 1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM, 2001. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 153, 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, Li L, Lutke-Enking S, Zarbock A, Stadtmann A, Striewski P, Wirth B, Kuzmanov I, Wiendl H, Schulte D, Vestweber D, Sorokin L, 2017. Endothelial Basement Membrane Laminin 511 Contributes to Endothelial Junctional Tightness and Thereby Inhibits Leukocyte Transmigration. Cell Rep 18, 1256–1269. [DOI] [PubMed] [Google Scholar]
- Staat C, Coisne C, Dabrowski S, Stamatovic SM, Andjelkovic AV, Wolburg H, Engelhardt B, Blasig IE, 2015. Mode of action of claudin peptidomimetics in the transient opening of cellular tight junction barriers. Biomaterials 54, 9–20. [DOI] [PubMed] [Google Scholar]
- Stein C, 2013. Targeting pain and inflammation by peripherally acting opioids. Front Pharmacol 4, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs EB Jr., Von Zee CL, 2012. Prenylation of Rho G-proteins: a novel mechanism regulating gene expression and protein stability in human trabecular meshwork cells. Mol Neurobiol 46, 28–40. [DOI] [PubMed] [Google Scholar]
- Subileau EA, Rezaie P, Davies HA, Colyer FM, Greenwood J, Male DK, Romero IA, 2009. Expression of chemokines and their receptors by human brain endothelium: implications for multiple sclerosis. J Neuropathol Exp Neurol 68, 227–240. [DOI] [PubMed] [Google Scholar]
- Takai T, 2002. Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2, 580–592. [DOI] [PubMed] [Google Scholar]
- Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS, 2015. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol 208, 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu EE, 2011. Chemokine receptors as specific anti-inflammatory targets in peripheral nerves. Endocr Metab Immune Disord Drug Targets 11, 141–153. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, 2013. The molecular and biophysical characterization of the human blood-nerve barrier: current concepts. J Vasc Res 50, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu EE, 2015. Inflammatory neuropathies: pathology, molecular markers and targets for specific therapeutic intervention. Acta Neuropathol 130, 445–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay RK, 2014. Transendothelial Transport and Its Role in Therapeutics. Int Sch Res Notices 2014, 309404: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat JM, Mathis S, Ghorab K, Milor MA, Richard L, Magy L, 2015. Natalizumab as a Disease-Modifying Therapy in Chronic Inflammatory Demyelinating Polyneuropathy - A Report of Three Cases. Eur Neurol 73, 294–302. [DOI] [PubMed] [Google Scholar]
- Villasenor R, Lampe J, Schwaninger M, Collin L, 2019. Intracellular transport and regulation of transcytosis across the blood-brain barrier. Cell Mol Life Sci 76, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Miao Y, Ni J, Wang Y, Qian T, Yu J, Liu Q, Wang P, Yi S, 2018. Peripheral Nerve Injury Induces Dynamic Changes of Tight Junction Components. Front Physiol 9, 1519; 15 10.3389/fphys.2018.01519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZG, Cheng Y, Yu XC, Ye LB, Xia QH, Johnson NR, Wei X, Chen DQ, Cao G, Fu XB, Li XK, Zhang HY, Xiao J, 2016. bFGF Protects Against Blood-Brain Barrier Damage Through Junction Protein Regulation via PI3K-Akt-Rac1 Pathway Following Traumatic Brain Injury. Mol Neurobiol 53, 7298–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, Nottebaum AF, Vestweber D, 2014. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol 15, 223–230. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ, 2007. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A 104, 20151–20158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild R, Klems A, Takamiya M, Hayashi Y, Strahle U, Ando K, Mochizuki N, van Impel A, Schulte-Merker S, Krueger J, Preau L, le Noble F, 2017. Neuronal sFlt1 and Vegfaa determine venous sprouting and spinal cord vascularization. Nat Commun 8, 13991: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson EL, Sidaway JE, Cross MJ, 2018. Statin regulated ERK5 stimulates tight junction formation and reduces permeability in human cardiac endothelial cells. J Cell Physiol 233, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Menge T, Stenner MP, Meyer zu Horste G, Saleh A, Hartung HP, Wiendl H, Kieseier BC, 2010. Natalizumab treatment in a patient with chronic inflammatory demyelinating polyneuropathy. Arch Neurol 67, 881–883. [DOI] [PubMed] [Google Scholar]
- Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, Loser K, Beissert S, Georges-Labouesse E, Sorokin LM, 2009. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 15, 519–527. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, Zeuschner D, Nakayama M, Vestweber D, Adams RH, 2015. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun 6, 6429: doi: 64 10.1038/ncomms7429. [DOI] [PubMed] [Google Scholar]
- Yang S, Krug SM, Heitmann J, Hu L, Reinhold AK, Sauer S, Bosten J, Sommer C, Fromm M, Brack A, Rittner HL, 2016. Analgesic drug delivery via recombinant tissue plasminogen activator and microRNA-183-triggered opening of the blood-nerve barrier. Biomaterials 82, 20–33. [DOI] [PubMed] [Google Scholar]
- Yano K, Gale D, Massberg S, Cheruvu PK, Monahan-Earley R, Morgan ES, Haig D, von Andrian UH, Dvorak AM, Aird WC, 2007. Phenotypic heterogeneity is an evolutionarily conserved feature of the endothelium. Blood 109, 613–615. [DOI] [PubMed] [Google Scholar]
- Yekkirala AS, Roberson DP, Bean BP, Woolf CJ, 2017. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16, 810; 8 10.1038/nrd.2017.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Ubogu EE, 2012. alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol 227, 3857–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Ubogu EE, 2013. An immortalized human blood-nerve barrier endothelial cell line for in vitro permeability studies. Cell Mol Neurobiol 33, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Xia RH, Ubogu EE, 2010. Development and characterization of a novel human in vitro blood-nerve barrier model using primary endoneurial endothelial cells. J Neuropathol Exp Neurol 69, 82–97. [DOI] [PubMed] [Google Scholar]
- Yu J, Gu X, Yi S, 2016. Ingenuity Pathway Analysis of Gene Expression Profiles in Distal Nerve Stump following Nerve Injury: Insights into Wallerian Degeneration. Front Cell Neurosci 10, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Bogdanova N, Gao T, Sheikh KA, 2019. Elimination of activating Fcgamma receptors in spontaneous autoimmune peripheral polyneuropathy model protects from neuropathic disease. PLoS One 14, e0220250; 02202 10.0221371/journal.pone.0220250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Chappell JC, 2019. Microvascular bioengineering: a focus on pericytes. J Biol Eng 13, 26; 10.1186/s13036-13019-10158-13033. [DOI] [PMC free article] [PubMed] [Google Scholar]