

Abstract
Objective:
Recruitment of neutrophils and formation of neutrophil extracellular traps (NETs) contribute to lethality in acute mesenteric infarction. To study the impact of the gut microbiota in acute mesenteric infarction, we used gnotobiotic mouse models to study whether gut commensals prime the reactivity of neutrophils towards NETosis.
Approach and Results:
We applied a mesenteric ischemia-reperfusion (I/R) injury model to germ-free (GF) and colonized C57BL/6J mice. By intravital imaging, we quantified leukocyte adherence and NET formation in I/R-injured mesenteric venules. Colonization with gut microbiota or monocolonization with Escherichia coli augmented the adhesion of leukocytes, which was dependent on the Toll-like receptor-4 (TLR4) / TIR-domain-containing adapter-inducing interferon-β (TRIF) pathway. Although neutrophil accumulation was decreased in I/R-injured venules of GF mice, NETosis following I/R injury was significantly enhanced compared with conventionally raised (CONV-R) mice or mice colonized with the minimal microbial consortium altered Schaedler flora (ASF). Also ex-vivo, neutrophils from GF and antibiotic-treated mice showed increased lipopolysaccharide (LPS)-induced NETosis. Enhanced TLR4 signaling in GF neutrophils was due to elevated TLR4 expression and augmented IRF3 phosphorylation. Likewise, neutrophils from antibiotic-treated CONV-R mice had increased NET formation before and after ischemia. Increased NETosis in I/R injury was abolished in CONV-R mice deficient in the TLR adaptor TRIF. In support of the desensitizing influence of enteric LPS, treatment of GF mice with LPS via drinking water diminished LPS-induced NETosis in vitro and in the mesenteric I/R injury model.
Conclusions:
Collectively, our results identified that the gut microbiota suppresses NETing neutrophil hyperreactivity in mesenteric I/R injury, while ensuring immunovigilance by enhancing neutrophil recruitment.
Graphical Abstract
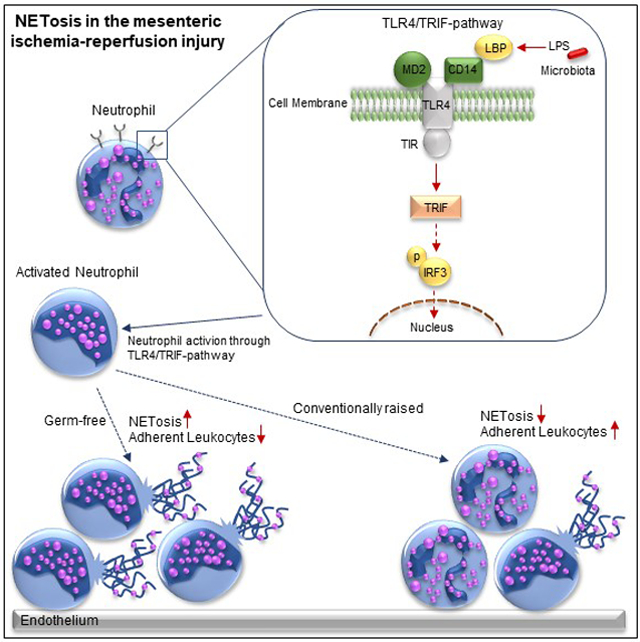
Introduction
Acute mesenteric ischemia (AMI) is a life-threatening clinical situation in which occlusion of the superior mesenteric artery reduces the blood flow to the intestine. This condition potentially leads to bowel wall necrosis with perforation, followed by peritonitis and multiple organ failure.1, 2 Necrosis triggers the sequestration of polymorphonuclear neutrophils (PMN) within affected intestinal sections, producing reactive oxygen species (ROS).3 During mesenteric infarction, the recruitment of leukocytes dictates the extent of the ischemia-reperfusion injury. Prolonged ischemia of the intestine causes tissue damage from reperfusion, mediated by the increased production of ROS due to neutrophil accumulation, leading to increased microvascular permeability.4
In AMI, the role of the commensal gut microbiota is largely unexplored. Intestinal ischemia disrupts the intestinal barrier and results in the translocation of gut bacteria and bacterial products, such as lipopolysaccharides (LPS), into the systemic circulation, thereby promoting the development of multiple organ failure.5 In support of a role of the gut microbiota in intestinal ischemia-reperfusion (I/R) injury, decontamination of gut commensals was shown to protect from organ failure.6 Microbial-associated molecular patterns (MAMPs) stimulate pattern-recognition receptors in the host, evoking various (and even opposite) biological responses. Dependent on the host colonization status, muramyl dipeptide signaling, engaging pattern recognition through nucleotide-binding oligomerization domain-containing protein 2 (NOD2), is protective against I/R-induced intestinal injury.7 This indicates that the presence of gut microbiota supports the resilience of intestinal tissues to resist I/R injury. Conversely, it was demonstrated that signaling via the adaptor molecule myeloid differentiation primary response gene 88 (MyD88) by the gut microbiota promotes acute intestinal and lung injury, inflammation and endothelial damage.8
Even if the role of gut microbiota as a trigger of TLR4 signaling is well-established, its impact in the recruitment of leukocytes and platelets to I/R damaged mesenteric venules and its influence on the formation of neutrophil extracellular traps (NETs) is unexplored. TLR signaling pathways are induced during small intestinal ischemia,9 contributing to I/R injury.10 TLR4 activation results in a complex signaling cascade, involving the recruitment of the Toll/interleukin-1 receptor (TIR) domain containing adaptor proteins (MYD88), TIR-associated protein (TIRAP), MYD88 adaptor like protein (MAL), TIR-domain-containing adapter-inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM).11 Since TRAM interacts with TRIF to phosphorylate tumor necrosis factor receptor-associated factor 3 (TRAF3), yielding in interferon regulatory factor-3 (IRF3) phosphorylation, whereas TIRAP interacts with MYD88 to phosphorylate interleukin-1 receptor-associated kinase 4 (IRAK4), TLR4 signaling is subdivided into a MYD88-dependent and a MYD88-independent TRIF-mediated pathway.11 The deficiency of the adaptor proteins MYD88 or TRIF is generally used to investigate the TLR4 receptor downstream signaling pathway.
In mesenteric I/R injury, the endothelial adhesion mechanisms of leukocytes and their extravasation into the surrounding tissue, together with platelet deposition, play an important role. During ischemia, the surface exposure of P-selectin by activated microvascular endothelial cells together with the release of von Willebrand factor from endothelial cell Weibel-Palade bodies supports platelet deposition, leukocyte rolling, adhesion, and aggregate formation in the small intestinal microcirculation.12, 13 Blockade of P-selectin strongly reduced platelet rolling and adhesion in mesenteric I/R injury.14 Since platelets from P-selectin-deficient mice adhere normally post-ischemia when transfused into WT recipient mice, but adhesion of WT platelets is nearly absent in P-selectin-deficient recipient mice, the platelet-endothelium interactions in the mesenteric I/R mouse model are primarily dependent on the P-selectin expression of the vascular endothelium.14 Furthermore, the interaction of endothelial intercellular adhesion molecule-1 (ICAM-1) with the leukocyte β2 integrin Mac-1 is a critical determinant in microcirculatory ischemia-reperfusion injury.15, 16 While the expression of ICAM-1 has been shown to be reduced in the mesenteries of germ-free (GF) mice,17 it remains elusive if the colonization status of the host influences leukocyte adhesion during AMI via the interaction of P-selectin on activated endothelial cells and platelets with its counter-receptor P-selectin glycoprotein ligand-1 (PSGL-1), expressed on leukocytes (including neutrophils and monocytes).
Activated PMNs, together with eosinophils and basophils, are the first line of the innate immune defence, undergoing a specialized form of programmed cell death. Augmented by the interaction with P-selectin of platelets with PSGL-1,18 neutrophils release granule proteins and chromatin that form extracellular DNA fibres, called neutrophil extracellular traps (NETs), which bind and prevent the dissemination of bacteria.19, 20 The activation of PMNs with LPS results in chromatin decondensation, followed by energy-dependent NET production.21, 22 In addition to their recognized role in promoting arterial and venous thrombus formation,23, 24 the formation of NETs was also observed in the microvascular I/R-injured myocardium and the I/R-injured intestine25, 26. While the role of NETs in preventing the dissemination of pathogenic bacteria during sepsis has been well defined, the contribution of the commensal microbiota in NET formation in the mesenteric microvasculature during mesenteric ischemia-reperfusion is entirely unresolved. Under this condition, resident gut microbes leak into the portal circulation and need to be sequestered by activated neutrophils to prevent spreading to the systemic circulation.
As the impact of the gut microbiota on leukocyte adhesion and NETosis in mesenteric I/R injury is unexplored, we here set out to analyse these key leukocyte functions in I/R-injured mesenteric venules of GF mice compared to conventionally raised (CONV-R) counterparts. To grasp the dynamic nature of vascular microbiota-host interactions, we studied leukocyte adherence and NETosis in gnotobiotic mouse models by using GF mice that lack colonization with a gut microbiota or CONV-R mice that were depleted from their commensal microbiota by treatment with broad-spectrum antibiotics (Abx). Taking advantage of germ-free isolator technology, we conducted colonization experiments to address the role of gram-negative (Escherichia coli JP313) and gram-positive (Bacillus subtilis PY79) colonizers as well as colonization with a minimal microbial consortium (altered Schaedler flora; ASF) on leukocyte adhesion and NETosis in mesenteric ischemia-reperfusion injury. To pinpoint the involvement of TLR4, we studied mouse models deficient in critical elements of this signaling pathway. Furthermore, to figure out if the revealed changes in LPS-induced NETosis are an inherent trait of GF neutrophils, we explored the cell-intrinsic reactivity of neutrophils from GF mice in cell culture assays with isolated bone marrow neutrophils.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals.
TLR4−/−27, MyD88−/−28, Trif−/−29, MyD88−/− x Trif−/−, altered Schaedler flora colonized (ASF) mice housed in flexible-film isolators30, and TLR4fl/fl × VE-Cdh-Cre+, TLR4wt/wt × VE-Cdh-Cre+ 31 mice on a C57BL6 background were used for the experiments. Further C57BL/6N and Swiss Webster mice were used in control experiments. C57BL/6J, C57BL/6N and Swiss Webster mice were maintained as GF mouse colonies in sterile flexible film mouse isolator systems. The germ-free status of mice was verified every second week by 16S rDNA PCR and by bacterial culture testing. Conventionally raised (CONV-R) SPF C57BL/6J, C57BL/6N and Swiss Webster mice originate from the same colonies. Conventionally derived mice (CONV-D) were born GF and recolonized with a normal gut microbiota at an age of 5–12 weeks; offsprings of these mice at weaning were used as indicated. All the compared groups were fed the same standard laboratory diet (IPS, LabDiet 5021) and were co-housed, multiple litters were represented in each group. Cecal microbiota donor mice were randomized. Cecal content was solved in 3 mL PBS and 200 μL were administered by oral gavage in each conventionalization experiment. C57BL/6J GF mice were monocolonized with the Escherichia coli strain JP 313 (provided by Evelyne Turlin, Institute Pasteur, Paris, France)32 and Bacillus subtilis PY79, a prototrophic variant engineered from the wild type 168 strain33, 34 (provided by Prof. Ezio Ricca, University of Naples, Naples, Italy) in germ-free flexible film isolators. At OD600nm = 0.6, a 200 μl-aliquot of the E. coli suspension was administered to each mouse by oral gavage. For mono-colonization with B. subtilis, a suspension of 1.33×108 B. subtilis spores in 200 μl sterile PBS was administered to each mouse by oral gavage. Mice were analyzed following 14 days of colonization. C57BL/6J mice were colonized with the minimal microbial consortium altered Schaedler flora (ASF), were kept for several generations in sterile isolators and analysed at the age of 6–8 weeks. All experimental animals were 5–12 weeks old male or female mice housed in the Translational Animal Research Center (TARC) of the University Medical Center Mainz under specific pathogen-free (SPF) or GF conditions in EU type II cages with 2–5 cage companions with standard autoclaved lab diet and water ad libitum, 22 ± 2 °C room temperature and a 12 h light/dark cycle. All groups of mice were age-matched and free of clinical symptoms. Due to limited space in isolators and restricted number of littermates, male and female germ-free mice were used for the studies. All procedures on mice were approved by the local committee on legislation of animals (Landesuntersuchungsamt Rheinland-Pfalz, Koblenz, Germany; G11-1-025, G13-1-035, G13-1-072, and G16-1-013).
Administration of broad-spectrum antibiotics (Abx) and LPS.
For depletion of gut microbes, CONV-R C57BL/6J mice were treated with broad-spectrum antibiotics (1 g/L ampicillin, Carl Roth, Germany; 1 g/L neomycin, Sigma-Aldrich, USA; 0,5 g/L vancomycin, Hikma, UK; and 1 g/L metronidazole, Sigma-Aldrich, USA) for 14 days via the drinking water according to an established protocol.35, 36 To evaluate the role of metabolic endotoxemia37 on neutrophil reactivity and NET formation, germ-free C57BL/6J mice were treated for one week with lipopolysaccharide (100 μg/mL, LPS-EB Ultrapure, E.coli 0111:B4, InvivoGen, USA) via the drinking water. Treated mice were daily monitored.
Mouse LPS ELISA kit.
EDTA-anticoagulated whole blood was collected by intracardial puncture and the assay was performed according to manufactureŕs instructions (Mouse Lipopolysaccharides (LPS) ELISA kit, Cusabio, USA).
Isolation of mouse bone marrow neutrophils.
Previously described protocols were applied and modified.38, 39 Mice were sacrificed by cervical dislocation and the skin was removed from the legs. The remaining muscles and the end of the femur and tibia were cut. The bone marrow was flushed through a 70 μm cell strainer with HBSS wash buffer (HBSS supplemented with 25 mM HEPES and 10% FCS) and the collected bone marrow suspension was centrifuged at 300xg for 10 min. The supernatant was removed and the pellet was resuspended in HBSS and HEPES. For isolating the neutrophils, the suspension was overlayed on a Histopaque gradient (4 mL Histopaque 1119 under 4 mL Histopaque 1077) and centrifuged for 30 min at 700xg without brake. The isolated neutrophils were carefully collected at the interface of the Histopaque 1077 (Sigma-Aldrich, USA) and Histopaque 1119 (Sigma-Aldrich, USA) layers. The neutrophils were washed with 20 mL washing buffer and centrifuged for 10 min at 300xg. 10 mL of the wash buffer was removed and 10 mL of fresh wash buffer was added to the cells and resuspended, following centrifugation at 300xg for 10 min. For the last washing step, the whole buffer was removed and the cells were washed with 20 mL wash buffer and centrifuged for 10 min at 300xg. Washed neutrophils were resuspended in RPMI 1640 medium (without phenol red, ThermoFisher scientific, USA) and counted.
Neutrophil activation by LPS.
Isolated neutrophils were seeded to a black 96-well plate at a density of 3×105 cells per well and stimulated with 10 ng/mL LPS (LPS-EB Ultrapure, E.coli 0111:B4, InvivoGen, USA). The plate was placed in a humidified incubator at 37°C with CO2 (5%) for 5h.
Quantification of NETs by fluorescence microplate reader.
Stimulated neutrophils were stained with 50 μL of 5 μM cell impermeable dye SYTOX orange solution (ThermoFisher Scientific, USA) and incubated in the dark for 10 min. Afterwards the cells were centrifuged at 200xg for 10 min and washed with 50 μL RPMI 1640 medium (without phenol red, ThermoFisher scientific, USA) and centrifuged at 200xg for 2 min. Quantification of NETs was performed with the SpectraMax i3x microplate-reader (Molecular Devices, UK). The NET-specificity of the detected signal was determined by incubation with DNase I (50 U/mL, Qiagen, Germany).
Immunofluorescence of in vitro LPS stimulated neutrophils.
Isolated neutrophils were seeded to glass chamber slides at a density of 3×105 cells per well and stimulated with 10 ng/mL LPS (LPS-EB Ultrapure, E.coli 0111:B4, InvivoGen, USA). The chamber slides were placed in a humidified incubator at 37°C with CO2 (5%) for 5h. After the incubation, the slides were washed with PBS. For permeabilized cells, 0,1 % Triton X-100 (Carl Roth, Germany) was added to the sample and incubated for 10 min. Cells were washed with PBS and blocked for 2h with 10% bovine serum albumin (BSA) in PBS. Primary Antibody was added to the cells and incubated overnight. Cells were washed with PBS and incubated with secondary antibody for 2h in the dark. Chamber slides were washed with PBS and incubated for 10 min. with 4’,6-Diamidino-2-phenylindole (DAPI, Sigma-Aldrich, USA). Cells were washed again with PBS. The stained NETs were analyzed with the fluorescence microscope Axio Observer (10x magnification, Zeiss, Germany). Antibodies used for immunoflourescence analysis are shown in the Major Resources Tables in the online-only Data Supplement.
MACS neutrophil isolation kit.
Briefly, mice were sacrificed and the legs were removed. The remaining muscles and the end of the femur and tibia were cut. The bone marrow was flushed through a 70 μm cell strainer with 10 mL PBS. Mouse neutrophils were isolated according to the manufactureŕs protocol (Neutrophil isolation kit (mouse), Miltenyi Biotec, Germany).
Red blood cell lysis.
EDTA-anticoagulated whole blood was collected by intracardial puncture. Cell lysis was performed according to the manufactureŕs protocol (1xRBC lysis buffer, Invitrogen, USA).
Flow cytometric analysis of neutrophils and monocytes.
Studies were conducted using a BD FACSCanto II flow cytometer with BD FACSDiva software (BD Bioscience, Germany) and FlowJo-Software (Tree Star Inc., USA). 50 μL of EDTA-anti-coagulated whole blood was incubated for 30 min (room temperature; RT) with 200 μL PBS pH 7.4 and 250 μL 0.4% (v/v) formaldehyde. Fixation was stopped by adding 1 mL PBS pH 7.4 and samples were centrifuged for 5 min at 800xg (RT). Supernatant was discarded up to 100 μL. FACS antibodies were added to the samples, briefly vortexed and incubated for 45–60 min at RT on a shaker. Red blood cells were lysed by adding 2 mL lysing-solution IO-test (Beckman Coulter, USA) and incubated for 10 min (RT) without shaking. Lysing step was stopped by adding 2 mL of PBS pH 7.4 and centrifuged for 10 min at 800xg (RT). The supernatant was discarded and 250 μL PBS was added to the tubes and measured. Antibodies used for FACS analysis are shown in the Major Resources Tables in the online-only Data Supplement.
Isolated bone marrow neutrophils and lysed blood samples were incubated with TruStain FcX (1:50, anti-mouse CD16/32, BioLegend, USA) for 20 min on ice. 100 μL of the blocked bone marrow or blood solution (collected by heart puncture) was added in each well of a 96-well plate and centrifuged for 2 min at 2000 rpm (4°C). The supernatant was discarded and FACS antibodies were added and incubated for 20 min in the dark (4°C). 4’,6-diamidin-2-phenylindol (DAPI, 1:10000, Sigma-Aldrich, USA) was added shortly before the incubation time ends. The plate was centrifuged at 2000 rpm (4°C) for 2 min and washed with 100 μL PBS. 200 μL of the final stained cell solution was added in tubes and measured. Antibodies used for FACS analysis are shown in the Major Resources Tables in the online-only Data Supplement.
Western Blot.
Western blot was performed to determine levels of total IRF-3 protein and levels of IRF-3 phosphorylated at Ser386. Bone marrow-derived neutrophils were isolated and lysed in RIPA lysis buffer (Merck, Germany) in the presence of Pierce protease inhibitor mini tablets (ThermoFisher Scientific, USA). Soluble proteins were fractionated on 8 % SDS-polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF, Invitrogen, CA, USA) membranes. Antibodies used for Western blot analysis are shown in the Major Resources Tables in the online-only Data Supplement. Membranes were developed by adding ECL (1:1 mix of two solutions, BioRad, USA). Detection of the chemiluminescence signal was performed with Fusion FX7 western blot detection device (Vilber, France).
Preparation of platelets, leukocytes and NET visualization by intravital epifluorescence microscopy.
Citrate-anticoagulated whole blood was collected by intracardial puncture on anaesthesized mice. Platelet count was determined by using an automatic cell counter (KX-21, Sysmex, Germany). Platelets were isolated and labeled with rhodamine B (20 μg/mL) (Sigma-Aldrich, USA).40 The labeled platelet suspension was adjusted to a final concentration of 150 × 103 platelets/μL and 250 μL suspension was injected i.v. via a jugular vein catheter. To characterize platelet leukocyte interactions in vivo, acridine orange (50 μg/μL, 50 μL per mouse, Sigma-Aldrich, USA) stained leukocytes and ex vivo rhodamine B labeled platelets were imaged simultaneously. In vivo neutrophil extracellular traps (NETs) were stained with SYTOX orange (5 μM, 50 μL per mouse, Thermo Fisher Scientific, USA).41 NETs and leukocytes were imaged simultaneously.
Mesenteric ischemia-reperfusion injury model.
Mice were anesthetized by i.p. injection of a solution of midazolame (5 mg/kg, Hameln pharma plus, Germany), medetomidine (0.5 mg/kg, Zoetis, USA) and fentanyl (0.5 mg/kg, Janssen-Cilag GmbH, Germany). Briefly, a polyethylene catheter (0.28mm ID, 0.61mm OD, Smiths Medical Deutschland GmbH, Germany) was implanted into the jugular vein. The abdomen was entered via a midline laparotomy incision. The superior mesenteric artery was identified and occluded with a small vascular clamp. After an ischemic interval of 60 minutes, reperfusion was allowed. Before (pre) and immediately after ischemia-reperfusion (post) the entire small intestine was carefully taken out of the abdomen. Platelets, leukocytes and NETs were visualized in situ by in vivo epifluorescence high-speed video microscopy in the mesenterial venules. At the end of the experiments, all animals were sacrificed by cervical dislocation.
Intravital high-speed video epifluorescence microscopy.
Measurements were performed with a high-speed wide-field Olympus BX51WI fluorescence microscope using a long-distance condenser and a 10x (NA 0.3) water immersion objective with a monochromator (MT 20E, Olympus Deutschland GmbH, Germany) and a charge-coupled device camera (ORCA-R2, Hamamatsu Photonics, Japan). For image acquisition and analysis, the Real-time Imaging System eXcellence RT (Olympus Deutschland GmbH, Germany) and cellSens (Olympus Deutschland GmbH, Germany) software were used. Cell recruitment was quantified in one field view of 0.06 mm2. Adherent leukocytes and adherent platelets were defined as cells that did not move or detach from the endothelial lining within an observation period of 20 seconds. Rolling leukocytes were defined as cells that at least should once shortly attach the endothelium within the 30 seconds of recording time. Leukocyte conjugates were defined as an association of at least two leukocytes, while platelet-leukocyte conjugates were defined as an association of at least one leukocyte and one platelet. Neutrophil extracellular traps were defined as an extracellular structure, which is labelled with acridine orange and/or SYTOX orange (a cell impermeable nucleic acid stain).
Statistical analysis.
Data are presented as mean ± S.E.M and analyzed with GraphPad Prism 8 (GraphPad Software Inc., San Diego, California, US). The DÁgostino-Pearson omnibus K2 test was performed to determine the normality of the data, and the F test was used to determine the Equality of Variances. The independent samples Student’s t-test was applied for comparison of two groups. If data were not normally distributed, the nonparametric Mann-Whitney test was used.
Results
Germ-free mice show reduced leukocyte adhesion in ischemia-reperfusion-injured mesenteric venules
During acute mesenteric infarction, leukocytes and platelets adhere to the activated endothelium, causing ischemia-reperfusion injury. While the response of leukocytes to pathogenic determinants during microbial invasion is well-recognized, the possible role of the gut-resident microbiota in mesenteric ischemia-reperfusion injury is unresolved. To investigate if the adherence of leukocytes and platelets in acute mesenteric infarction is influenced by the host colonization status, we compared conventionally raised (CONV-R) specific pathogen-free (SPF) mice, that were colonized from birth by environmental microbes, to germ-free (GF) mice, which lack colonization with a gut microbiota (Figure 1A). After the induction of an I/R injury in the mesentery by ceasing the blood supply of the superior mesenteric artery for 60 minutes,14 leukocyte adhesion, leukocyte rolling, and leukocyte conjugate formation were increased in the mesenteric venules of CONV-R mice compared to their GF counterparts (Figure 1B–E). Furthermore, increased platelet deposition to the I/R-injured mesenteric endothelium of CONV-R mice relative to GF mice was observed (Figure 1F). Interestingly, post-ischemia, the number of platelet-leukocyte conjugates was significantly increased in the CONV-R group (Figure 1G), demonstrating that the presence of a gut microbiota enhances heterotypic cell interactions in mesenteric I/R injury.
Figure 1. Germ-free mice show reduced leukocyte adhesion to ischemia-reperfusion injured mesenteric venules.
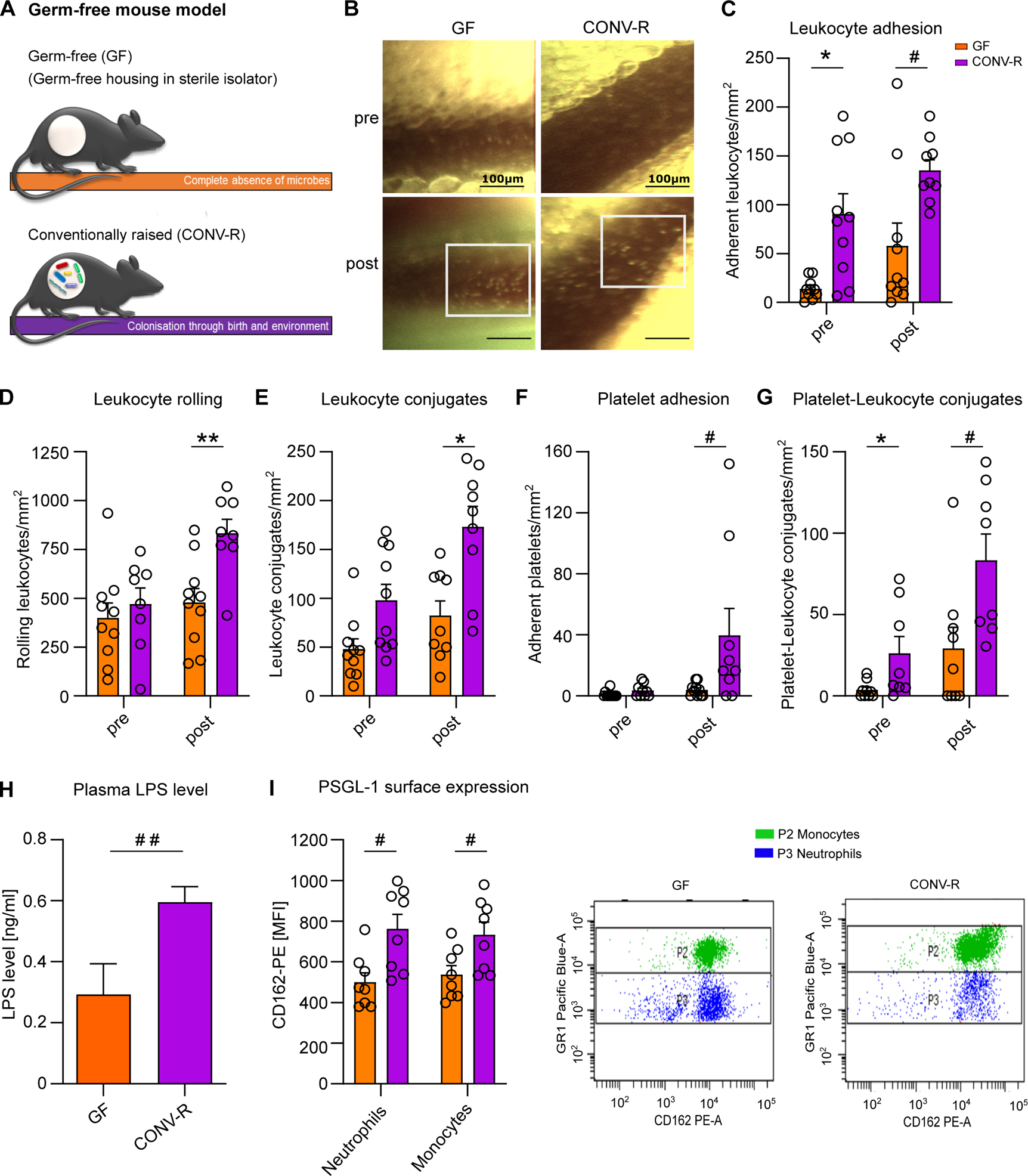
(A) Germ-free (GF) and conventionally raised (CONV-R) mouse model (schematic representation). (B) Intravital imaging of mesenteric venules of GF and CONV-R mice pre- and post-ischemia; scale bar: 100 μm; representative images; white frame: acridine orange-stained adherent leukocytes. (C) Number of adhering leukocytes in mesenteric venules of GF and CONV-R mice (10 vs 9 mice/group) pre-ischemia and one hour post-ischemia. For GF and CONV-R male and female mice were used. (D) Number of rolling leukocytes pre-ischemia and one hour post-ischemia in GF and CONV-R mice (10 vs 8 mice/group). For GF and CONV-R male and female mice were used. (E) Number of leukocyte conjugates pre-ischemia and one hour post-ischemia in GF vs CONV-R mice (10 vs 9 mice/group). For GF and CONV-R male and female mice were used. (F) Number of adhering platelets pre-ischemia and one hour post-ischemia in GF and CONV-R mice (10 vs 9 mice/group). For GF and CONV-R male and female mice were used. (G) Number of platelet-leukocyte conjugates pre-ischemia and one hour post-ischemia in GF and CONV-R (9 vs 8 mice/group). For GF and CONV-R male and female mice were used. (G) Plasma LPS levels in GF vs CONV-R mice (6 vs 10 mice/group). For GF and CONV-R male and female mice were used. (H, I) FACS analysis on PSGL-1 surface expression on monocytes (green) and neutrophils (blue) in whole blood of GF and CONV-R mice (8 vs 8 mice/group). For GF and CONV-R male and female mice were used. Results are shown as means ± s.e.m. Scale bar: 100 μm. Statistical comparisons were performed using the independent samples Studentś t-test (*) or the Mann-Whitney test (#), */#p<0.05, **/##p<0.01.
In line with previous work, demonstrating that elevated LPS plasma levels in colonized mice drive metabolic endotoxemia,37, 42 our results confirmed increased plasma LPS levels in CONV-R mice compared with their GF counterparts prior to I/R injury (Figure 1H). Most likely, LPS in the plasma of GF mice is diet-derived. As LPS is a trigger of Weibel-Palade body exocytosis yielding in the release of endothelial P-selectin43 and the functional inhibition of pro-adhesive P-selectin function suppresses microvascular leukocyte accumulation as well as platelet-leukocyte interaction in this model,14 the reduction in leukocyte adhesion to the mesenteric microcirculation and the impaired formation of platelet-leukocyte conjugates can be explained by reduced constitutive surface expression of its ligand PSGL-1 on neutrophils and monocytes in the blood of GF mice (Figure 1I). Thus, our results indicate that the presence of the gut microbiota is a decisive factor for the severity of mesenteric ischemia-reperfusion injury as evaluated by leukocyte tethering.
Leukocyte adhesion in ischemia-reperfusion-injured mesenteric venules dynamically adapts dependent on host colonization status
To experimentally test if enhanced leukocyte adhesion in mesenteric I/R-injury, evoked by the presence of a commensal microbiota, dynamically adapts to the presence or absence of gut commensals, we again took advantage of the GF mouse model. First, we analyzed whether the identified I/R-induced increase in leukocyte accumulation in CONV-R mice is re-established by colonization of GF mice with a cecal gut microbiota of a CONV-R donor mouse. Ex-GF mice, colonized for 14 days with a cecal microbiota are termed conventional-derived mice (CONV-D)44 (Figure 2A). In support of a pro-adhesive role of gut microbial colonization in mesenteric I/R injury (Figure 1A, B), the mean numbers of adhering leukocytes were also increased post-ischemia in CONV-D mice compared to GF controls (Figure 2A). In sharp contrast, antibiotic depletion of the gut microbiota by a 14 day administration protocol (Abx; cocktail of ampicillin, neomycin, vancomycin and metronidazole) on CONV-R mice efficiently prevented I/R-induced tethering of leukocytes (Figure 2B), demonstrating that this microbiota-dependent vascular phenotype is highly dynamic and fully reversible.
Figure 2. Colonization with a gut microbiota or monocolonization with Escherichia coli augments leukocyte adhesion to the ischemia-reperfusion injured endothelium of mesenteric venules.
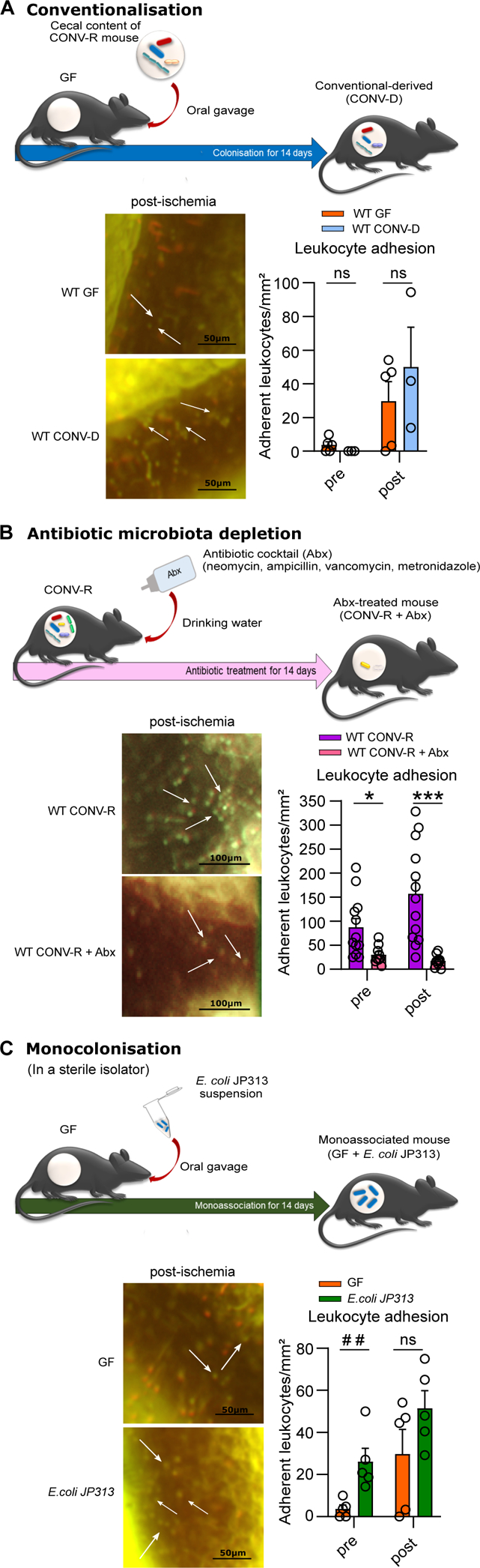
(A) Number of adhering leukocytes in mesenteric venules of germ-free (GF) and conventional-derived (CONV-D) mice (5 vs 4 mice/group) pre-ischemia and one hour post-ischemia; adhering leukocytes were stained with acridine orange. For WT GF male mice and WT CONV-D male and female mice were used. (B) Number of adhering leukocytes pre-ischemia and post-ischemia in conventionally raised (CONV-R) controls and CONV-R antibiotic (Abx)-treated mice (13 vs 9 mice/group); adhering leukocytes were stained with acridine orange. For WT CONV-R and CONV-R (Abx)-treated male and female mice were used. (C) Number of adhering leukocytes pre-ischemia and post-ischemia in GF and GF monocolonized with E. coli JP313 mice (5 vs 5 mice/group); adhering leukocytes were stained with acridine orange. For GF male mice and for GF monocolonized with E. coli JP313 male and female mice were used. Results are shown as means ± s.e.m. Scale bar: 50 μm or 100 μm. Statistical comparisons were performed using the independent samples Studentś t-test (*) or the Mann-Whitney test (#), */#p<0.05, **/##p<0.01, ***/### p<0.001.
To pinpoint if monocolonization with an individual gut-resident bacterium is sufficient to evoke increased leukocyte adherence in mesenteric I/R-injury, we performed monocolonization experiments with the gram-negative gut microbe Escherichia coli JP313 (Figure 2C). At the end of the 14 day monocolonization experiment, the mice had a fecal colonization density of 2.68×106 (colony forming units) CFU/mg. Interestingly, in this gnotobiotic approach, monocolonization with E. coli JP313 increased leukocyte adhesion to I/R-injured mesenteric venules pre- and post-ischemia (Figure 2C). In conclusion, both colonization with a gut microbiota, but also monocolonization with E. coli JP313 enhanced the extent of leukocyte adherence to I/R-injured mesenteric venules, whereas antibiotic depletion of the microbiota reversed this phenotype.
The presence of gut commensals restricts in vivo NETosis
In AMI, a proportion of the adherent activated neutrophils forms NETs19,20, contributing to inflammatory conditions of the splanchnic circulation and aggravating the outcome.26 To quantify the extent of NETosis in gnotobiotic mouse models by fluorescence intravital microscopy in vivo,41 we counted the SYTOX orange positive adhering neutrophils in the mesenteric venules prior to and after I/R injury. As expected, pre-ischemia, the number of NETing leukocytes was modest (Figure 3A). Strikingly, post-ischemia NETosis was vastly enhanced and GF mice showed 2-fold increased NET-formation in I/R-injured mesenteric venules compared with CONV-R counterparts (Figure 3A). Thus, our experiments demonstrate a strong increase in I/R-induced in vivo NETosis in GF mice compared to CONV-R mice.
Figure 3. The presence of gut commensals restricts the formation of neutrophil extracellular traps (NETosis) in ischemia-reperfusion injured mesenteric venules.
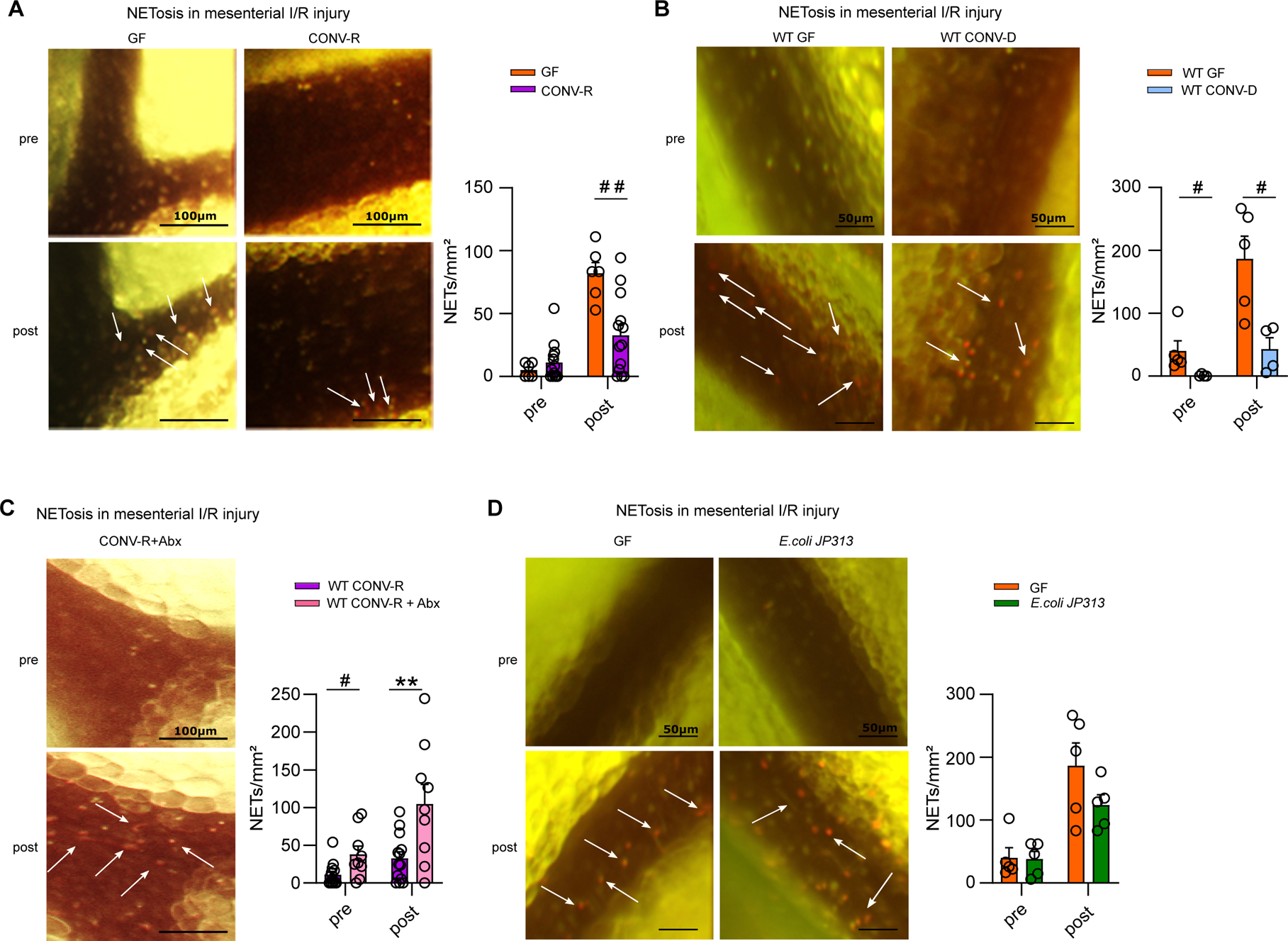
(A) NETosis in mesenteric venules of GF and CONV-R mice (6 vs 13 mice/group) pre- and post-ischemia; adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. For GF and CONV-R male and female mice were used. (B) NETosis in mesenteric venules pre- and post-ischemia in GF and CONV-D mice (5 vs 4 mice/group); adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. For WT GF male mice and WT CONV-D male and female mice were used. (C) NETosis in mesenteric venules pre- and post-ischemia in CONV-R and (Abx)-treated CONV-R mice (13 vs 9 mice/group); adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. For WT CONV-R and CONV-R (Abx)-treated male and female mice were used. (D) NETosis in mesenteric venules pre- and post-ischemia in GF and GF mice monoassociated with E.coli JP 313 (5 vs 5 mice/group); adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. For GF male mice and GF monoassociated with E.coli JP 313 male and female mice were used. Results are shown as means ± s.e.m. Scale bar: 50 μm or 100 μm. Statistical comparisons were performed using the independent samples Studentś t-test (*) or the Mann-Whitney test (#), */#p<0.05, **/##p<0.01.
To address whether the impact of gut commensals on mesenteric I/R injury-induced NETosis is a dynamic process, we colonized GF mice with a complex cecal gut microbiota from a CONV-R donor mouse over a 14 day period (conventionalization, CONV-D) and then applied the mesenteric I/R injury model. In line, the conventionalized ex-GF mice showed strongly reduced numbers of NETing neutrophils at 1 hour of I/R injury compared to the GF group, demonstrating that the microbiota dynamically regulates NETosis (Figure 3B). In contrast, depletion of commensals by antibiotic treatment significantly increased the number of NETing neutrophils in the mesenteric I/R injury model (Figure 3C). Collectively, our experiments identify the gut microbiota as an environmental factor that dynamically suppresses NETosis in I/R injured mesenteric venules.
Next, we tested whether monocolonization with the gram-negative enterobacterium E. coli JP313 is sufficient to reduce the extent of NETosis in the mesenteric I/R injury model. While the numbers of NETing leukocytes were comparable pre-ischemia, in the post-ischemic state, our gnotobiotic monocolonization experiments with E. coli JP313 showed a clear trend towards suppressed in vivo NETosis in the I/R injury model (Figure 3D). Furthermore, we monocolonized with the gram-positive gut bacterium Bacillus subtilis PY7933, 34, reaching a final colonization density of 4.36×106 CFU/mg feces, which likewise resulted in elevated leukocyte tethering and strongly reduced NETosis in I/R-injured mesenteric venules (Figure S1A, B). In addition, post-ischemic NETosis in mesenteric venules was significantly reduced in altered Schaedler flora (ASF) colonized mice30 relative to GF controls (Figure S1C). Collectively, our results demonstrate that the colonization-induced suppression of I/R injury-induced NETosis is broadly inducible even by individual gut commensals or minimal microbial consortia.
Suppressed NETosis of colonized mice is due to attenuated neutrophil Toll-like receptor-4 signaling
Since mesenteric infarction typically is accompanied by the loss of intestinal barrier function and hence leukocytes in the intestinal microcirculation face elevated levels of pro-inflammatory patterns,5, 6 we reasoned that hyperreactive TLR4 signaling in the neutrophils of GF mice could by causal for enhanced NET formation. To pinpoint whether GF neutrophils show hyperreactive LPS-induced NETosis ex vivo, we stimulated isolated bone marrow-derived neutrophils from GF and CONV-R mice (Figure 4A and Figure S2A, B) and measured the SYTOX orange signal of plated neutrophils in a fluorimetric assay. Increased LPS-induced NETosis was consistently observed ex vivo in isolated bone marrow neutrophils from GF mice, indicating that TLR4 signaling is hyperreactive at GF housing conditions (Figure 4A). Hyperreactive ex vivo NETosis was not strain-specific as we consistently observed this microbiota-regulated neutrophil phenotype in the GF C57BL/6N and the GF Swiss Webster mouse line (Figure S3). The role of chronic LPS exposure (e.g. in CONV-R mice) in attenuating NETosis of isolated bone marrow neutrophils was further corroborated by in vivo administration of LPS via the drinking water, which efficiently suppressed LPS-induced in vitro NETosis (Figure 4B). Similar to the GF mouse model, the reduction of colonizing gut bacteria by Abx resulted in exaggerated LPS-triggered ex vivo NET formation of isolated bone marrow neutrophils, controlled by DNase I digestion of expelled DNA fibers (Figure 4C, D). Indeed, fluorescence activated cell sorting (FACS) analyses on neutrophils in whole blood and on isolated bone marrow neutrophils revealed a subtle but significant increase in TLR4 surface expression on neutrophils from GF mice, suggesting that GF neutrophils may be more reactive in terms of LPS-induced TLR4 signaling (Figure 4E and Figure S2C, D). Furthermore, the IRF3 signaling axis, which is primarily triggered downstream of TRIF adaptor mediated TLR signaling cues45, was increased in the bone marrow neutrophils of GF mice (Figure 4F), demonstrating that intestinal microbiota-derived signals profoundly shape reactivity of the neutrophil-intrinsic TLR4 signaling pathway.
Figure 4. Hyperreactive TLR4 signaling promotes enhanced NETosis in the germ-free mouse model.
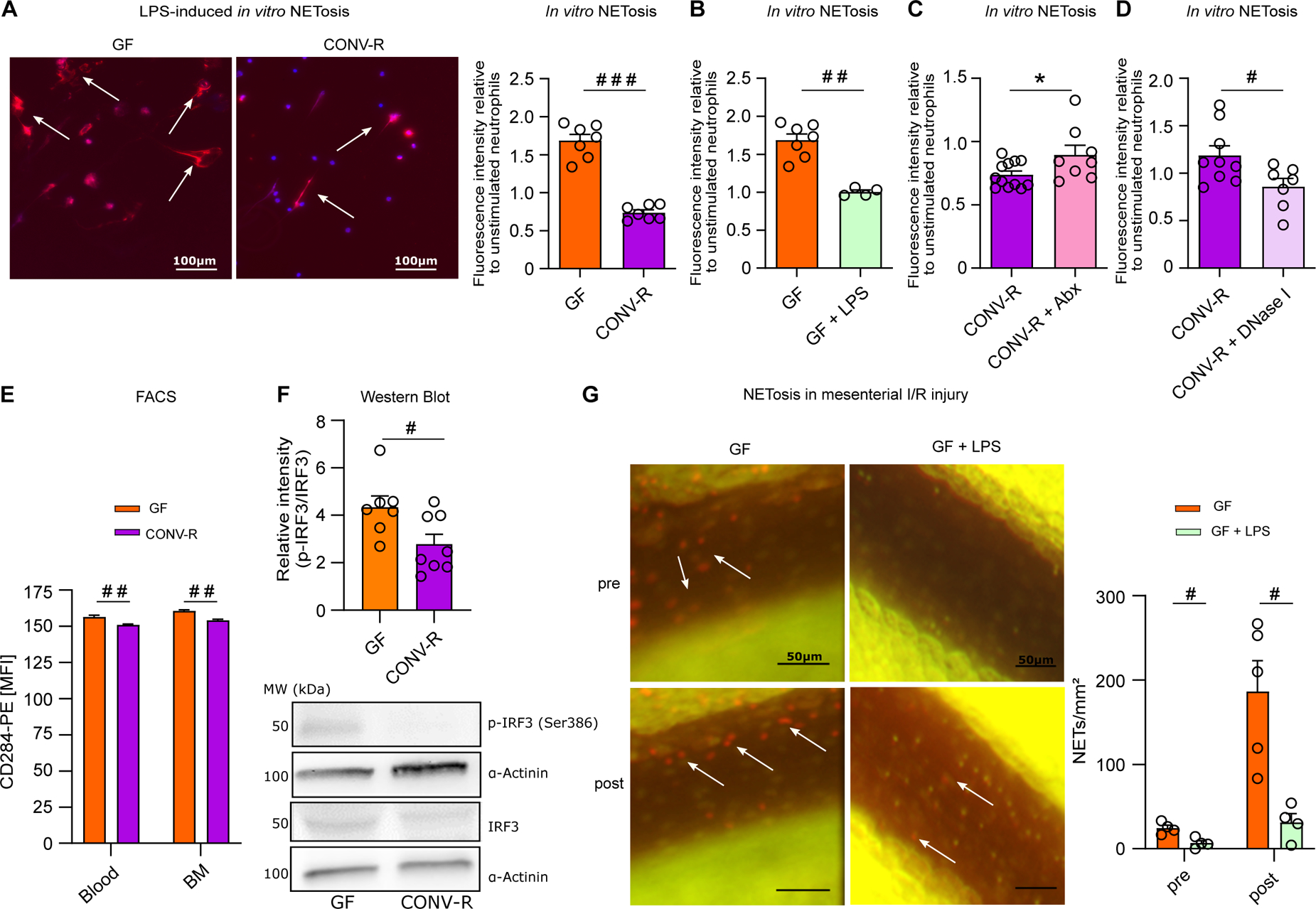
(A) Representative images of LPS-induced in vitro NET formation; nuclear staining (DAPI; blue); NETs (visualized by citrullinated histone H3 antibody staining; red); scale bar: 100 μm. LPS-induced in vitro NETosis of cultured bone marrow neutrophils from GF and CONV-R mice (7 vs 7 mice/group). For GF and CONV-R male and female mice were used. (B) LPS-induced in vitro NETosis of cultured bone marrow neutrophils from GF and LPS-treated GF mice (7 vs 4 mice/group). For GF and LPS-treated GF male and female mice were used. (C) LPS-induced in vitro NETosis of cultured bone marrow neutrophils from CONV-R and (Abx)-treated CONV-R mice (12 vs 8 mice/group). For CONV-R and (Abx)-treated CONV-R male and female mice were used. (D) LPS-induced in vitro NETosis of cultured bone marrow neutrophils from CONV-R and DNase I-treated CONV-R control mice (9 vs 7 mice/group). For CONV-R and DNase I-treated CONV-R controls male and female mice were used. (E) FACS analysis on CD284 (TLR4) surface expression on neutrophils in whole blood and bone marrow of GF and CONV-R mice. For GF and CONV-R male and female mice were used. (F) Western Blot analysis of relative intensity of phosphorylated IRF-3 level to total IRF-3 protein level in GF and CONV-R mice; results are normalized to α-actinin. For GF and CONV-R male and female mice were used. (G) NETosis in mesenteric venules pre- and post-ischemia in GF and LPS-treated GF mice (5 vs 4 mice/group). For GF male mice and LPS-treated GF male and female mice were used. Results are shown as means ± s.e.m. Scale bar: 50 μm or 100 μm. Statistical comparisons were performed using the independent samples Studentś t-test (*) or the Mann-Whitney test (#), */#p<0.05, **/##p<0.01, ***/###p<0.001.
To demonstrate in vivo that tonic TLR4 stimulation via the enteric route desensitizes neutrophils towards reduced I/R-induced NETosis, we administered E. coli LPS to GF mice via the drinking water for 7 days at sterile isolator conditions. As hypothesized, exposure of GF mice to LPS in their drinking water efficiently prevented excessive NETosis in mesenteric I/R injury (Figure 4G). Collectively, our results reveal that disabled neutrophil-intrinsic TLR4/TRIF signaling is linked to diminished LPS-triggered NETosis of the bone marrow neutrophils isolated from colonized mice or GF mice that were challenged with enteric LPS.
TLR4/TRIF signaling drives NETosis in mesenteric ischemia-reperfusion injury
As endotoxin challenge is known to augment leukocyte-vessel wall interactions through PSGL-1/P-selectin binding (Figure 1I),46 we next addressed the role of TLR4 signaling in neutrophil adhesion,10 evoked by I/R injury analyzing various genetic knock-out mouse models. As expected, the adherence of neutrophils was significantly reduced in I/R-injured mesenteric venules of TLR4−/− mice. Interestingly, in vivo NETosis was dependent on intact TLR4 signaling, as mesenteric I/R injury in TLR4−/− mice did not yield in significantly increased numbers of NETs in the post-ischemic state (Figure 5A–C). In line, in vitro LPS-stimulated bone marrow neutrophils from TLR4−/− mice showed significantly reduced NETosis compared to WT controls (Figure 5D), confirming the direct involvement of TLR4-mediated signaling cues on neutrophils. Mechanistically, I/R-induced in vivo NET formation was strongly dependent on TRIF-mediated signaling (Figure 5E–H), as Trif−/− mice did not show differences in the number of NETs (Figure 5G, H), whereas MyD88−/− mice consistently showed increased NETosis in the post-ischemic state (Figure 5E, F). To unambiguously demonstrate the role of TLR downstream signals in I/R-induced NETosis, we generated MyD88−/−xTrif−/− double-deficient mice, which also showed reduced leukocyte tethering and diminished NETosis in mesenteric venules in the post-ischemic state (Figure S4). To address if in addition to TLR4 signaling on neutrophils also signals via endothelial TLR4 contribute to neutrophil adhesion and NETosis in I/R-injured mesenteric venules, we studied conditional TLR4-flox × VE-Cadherin-Cre mice that are deficient in endothelial TLR4. Our experiments demonstrated that TLR4 signaling on the microvascular endothelium also contributes to the adherence of neutrophils in mesenteric I/R injury (Figure 5I, J), but did not significantly affect in vivo NETosis (Figure 5I, K). In conclusion, our results demonstrate that TLR4/TRIF signaling, both on neutrophils and the vascular endothelium, is critically involved in I/R injury-induced NETosis in the mesenteric microvasculature.
Figure 5. The TLR4/TRIF signaling axis is critically involved in mesenteric I/R injury-induced NETosis.
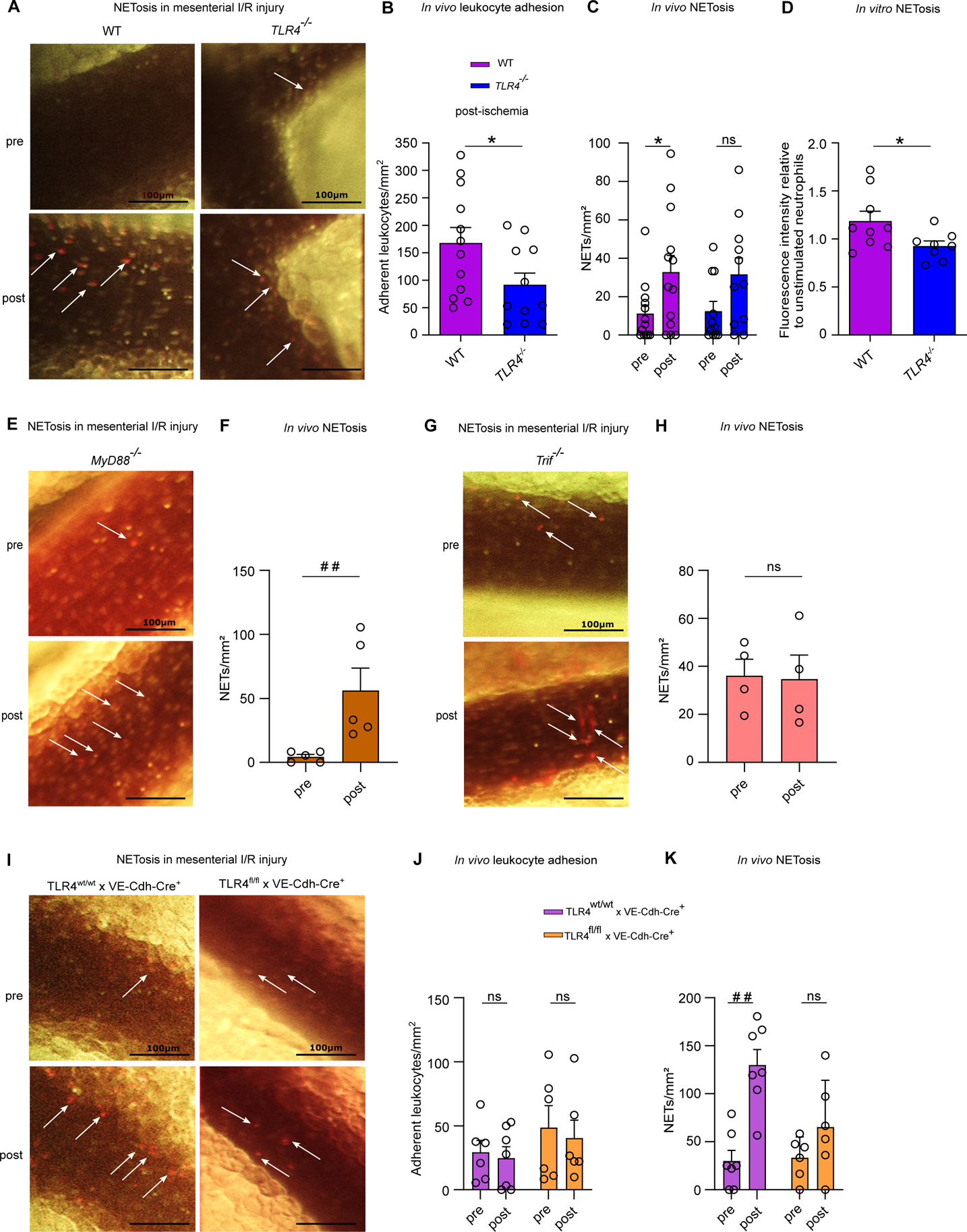
(A) Intravital imaging of mesenteric venules of CONV-R WT and CONV-R TLR4-deficient mice pre- and post-ischemia; scale bar: 100 μm; adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. (B) Number of adhering leukocytes one hour post-ischemia in CONV-R WT and CONV-R TLR4-deficient mice (12 vs 12 mice/group); adhering leukocytes were stained with acridine orange. For CONV-R WT and CONV-R TLR4−/− male and female mice were used. (C) NETosis in mesenteric venules pre- and post-ischemia in CONV-R WT and CONV-R TLR4-deficient mice (13 vs 11 mice/group). For CONV-R WT and CONV-R TLR4−/− male and female mice were used. (D) LPS-induced in vitro NETosis of cultured bone marrow neutrophils from CONV-R WT and CONV-R TLR4-deficient mice (9 vs 8 mice/group). For CONV-R WT and CONC-R TLR4−/− male and female mice were used. (E) Intravital imaging of mesenteric venules of CONV-R MyD88-deficient mice pre- and post-ischemia; scale bar: 100 μm; adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. (F) NETosis in mesenteric venules pre- and post-ischemia in MyD88-deficient mice (5 mice; male and female mice were used). (G) Intravital imaging of mesenteric venules of Trif-deficient mice pre- and post-ischemia; scale bar: 100 μm; adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. (H) NETosis in mesenteric venules pre- and post-ischemia in Trif-deficient mice (4 mice; male and female mice were used). (I) Intravital imaging of mesenteric venules of TLR4-flox × VE-Cadherin-Cre (deficient in endothelial TLR4) mice and their controls pre- and post-ischemia; scale bar: 100 μm; adhering leukocytes were stained with acridine orange; NETs were visualized by SYTOX orange. (J) Number of adhering leukocytes pre-ischemia and one hour post-ischemia in TLR4-flox × VE-Cadherin-Cre (deficient in endothelial TLR4) mice and their controls (7 vs 6 mice/group). For TLR4-flox × VE-Cadherin-Cre Controls female and for TLR4-flox × VE-Cadherin-Cre female and male mice were used. (K) NETosis in mesenteric venules pre- and post-ischemia in TLR4-flox × VE-Cadherin-Cre (deficient in endothelial TLR4) mice and their controls (7 vs 6 mice/group). For TLR4-flox × VE-Cadherin-Cre Controls female and for TLR4-flox × VE-Cadherin-Cre female and male mice were used. Results are shown as means ± s.e.m. Scale bar: 100 μm. Statistical comparisons were performed using the independent samples Studentś t-test (*) or the Mann-Whitney test (#), */#p<0.05, **/##p<0.01.
Discussion
Our experiments on the microbiota’s impact on acute mesenteric infarction revealed markedly decreased leukocyte rolling, firm adhesion, homotypic and platelet-heterotypic conjugate formation in I/R-injured mesenteric venules of GF mice compared with CONV-R mice in the post-ischemic state, demonstrating that the colonization status of the host dramatically affects the extent of I/R injury. Reduced conjugate formation was attributed to lower constitutive PSGL-1 surface expression in GF neutrophils and monocytes. By germ-free mouse isolator technology, we demonstrated that increased leukocyte deposition in the mesenteric I/R injury model dynamically adapts to the colonization status of the host. Increased leukocyte adhesion was triggered by 14 day colonization of ex-GF mice with a complex gut microbiota (CONV-D), but also by monocolonization with Escherichia coli JP313. Conversely, Abx-decimation of the gut microbiota reverted colonization-dependent leukocyte adherence.
In line with previous work on LPS-induced neutrophil recruitment in the GF mouse model,47 we observed that the presence of a gut microbiota supports the interaction of leukocytes with the I/R-injured vessel wall, yielding in increased numbers of adherent leukocytes in the post-ischemic state. Coherent with diminished vascular ICAM-1 in the mesentery of GF mice,17 we detected reduced constitutive PSGL-1 surface expression on blood neutrophils and monocytes of GF mice, which likely accounts for the reduced platelet-leukocyte conjugate formation. In addition, diminished Mac-1/ICAM-1 interaction could play a role in this process15, which is supported by the reduced expression of ICAM-1 in GF mice, specifically in the mesentery and the gastrointestinal tract, which has been reported by Komatsu and coworkers.17 However, the microbiota-dependent mechanisms that contribute to I/R-induced endothelial cell activation and the exposure of adhesion receptors await further exploration.
Demonstrating the influence of the host colonization status on a major neutrophil effector function, we found a vast increase in TLR4/TRIF-dependent NET formation as a result of mesenteric I/R injury in GF mice, whereas the I/R-induced increase in NETosis in CONV-R mice was far less pronounced. Therefore, we hypothesized that tonic exposure of neutrophils to microbiota-derived LPS (e.g. as in CONV-R mice) dampens TLR4 expression in neutrophils and NET-formation. Accordingly, increased I/R-induced NETosis could also be triggered by the depletion of the gut microbiota through Abx-treatment, demonstrating that the reactivity of neutrophils to undergo I/R-induced NETosis is dynamically regulated. By ex vivo cell culture experiments, we showed that bone marrow-derived neutrophils of GF mice are more prone to LPS-induced NETosis than their CONV-R counterparts and this was also found in the Abx-microbiota depletion model, which is in line with increased TLR4 surface expression detected on GF neutrophils.
Endotoxin tolerance is a well-characterized feature of neutrophils, which develops upon prolonged LPS exposure and is characterized by the loss of TLR4 expression.48 In line, we confirmed reduced TLR4 surface expression and identified diminished IRF3 phosphorylation in bone marrow neutrophils of CONV-R mice relative to their GF counterparts, which explains their reduced capacity of LPS-induced NET formation. Our LPS-induced neutrophil assays identified the TLR4/TRIF signaling axis as a major pathway influenced by the presence of a gut microbiota to protect against NET-mediated mesenteric I/R injury. The role of TLR4 in I/R-induced NETosis was also evident in mice that specifically lack TLR4 expression in the vascular endothelium, indicating that in addition to the direct signaling-dependent effects on neutrophils, TLR4 signaling in endothelial cells is also contributing to microvascular NETosis. However, our results do not rule out the participation of other pattern recognition receptors, such as nucleotide-binding oligomerization domain-containing protein-2 (NOD2) signaling.7 Clearly, these microbiota-dependent mechanisms need to be tightly balanced to prevent neutrophil hyperreactivity when encountering microbial challenges, but they have to educate sufficient immunovigilance to ensure host fitness.
Of note, our finding of an increased NETing of neutrophils in GF mice is consistent with a previous study on GF pigs, demonstrating that gut luminal endotoxin exposure reduces I/R injury of the gut wall.49 In the mesenteric I/R injury mouse model in vivo and in isolated in vitro bone marrow-derived neutrophils ex vivo, we clearly showed that the exposure of the host to a gut microbiota determines the extent of NETing neutrophils when stimulated with LPS.50 This finding is in accordance with a previous study, reporting a reduction in superoxide production of blood neutrophils from LPS pre-treated rats that were subjected to an I/R injury model.51 Indeed, NETosis has recently been shown as a cause of I/R injury in a neonatal rat model of midgut volvolus.26 In conclusion, the observed decrease of leukocyte adhesion, but increase of NETing neutrophils in the GF mouse model and in Abx-treated mice clearly demonstrates the pivotal role of the gut microbiota as an environmental pre-conditioning factor, contributing to the resilience to NETosis mediated I/R injury in acute mesenteric infarction.
Supplementary Material
Highlights.
The gut microbiota suppresses NETosis in mesenteric ischemia-reperfusion injury in vivo.
Isolated bone marrow-derived neutrophils from germ-free mice and broad-spectrum antibiotic-treated mice show hyperreactive LPS-induced NETosis.
Enteric LPS administraton and colonization with individual gut microbes or altered Schaedler flora suppresses hyperreactive NETosis of LPS-stimualted bone marrow neutrophils and in mesenteric ischemia-reperfusion injured venules.
Tonic stimulation of the cell-intrinsic TLR4/TRIF signaling pathway by gut commensals attenuates LPS-induced NETosis.
Acknowledgments
The authors state that no conflict of interests exists. We are grateful to Cornelia Karwot and Klaus-Peter Derreth for expert technical assistance. We thank Markus Radsak (III. Medical Clinics, University Medical Center Mainz) and Philip Wenzel (Center for Thrombosis and Hemostasis, University Medical Center Mainz) for their support with the TLR4−/−, Trif−/−, and MyD88−/− mouse lines. We are grateful to Evelyn Turlin (Institut Pasteur, Paris, France) for providing E. coli JP313 and to Ezio Ricca, (University of Naples, Naples, Italy) and Vincenzo de Filippis (University of Padua, Padua, Italy) for providing Bacillus subtilis PY79. Christoph Reinhardt is a Fellow of the Gutenberg Research College at the Johannes Gutenberg University of Mainz.
Sources of Funding
The project was funded by an intramural MAIFOR grant to K.J.L. and C.R., grant support from the Inneruniversitäre Forschungsförderung (Stufe 1) to J.M.K and C.R., the CTH Junior Group Translational Research in Thrombosis and Hemostasis (BMBF 01EO1003 and 01EO1503), DFG Individual Grants (RE 3450/3-1, RE 3450/5-1, RE 3450/5-2 BO 3482/3-3, BO 3482/4-1), the National Institutes of Health (1R01HL141513, 1R01HL139641 to M.B.), by a project grant of the Naturwissenschaflich-Medizinisches Forschungszentrum at the Johennes Gutenberg University of Mainz (NMFZ) to C.R., and a project grant from the Boehringer Ingelheim Foundation (“Novel and neglected cardiovascular risk factors”) to C.R.. C.R. is a member of Young DZHK. The authors are responsible for the content of this publication.
Nonstandard Abbreviations and Acronyms
- Abx
broad-spectrum-antibiotics
- AMI
acute mesenteric ischemia
- ASF
altered Schaedler flora
- CFU
colony forming units
- CONV-D
conventionally-derived
- CONV-R
conventionally raised
- GF
germ-free
- I/R
ischemia/reperfusion
- ICAM-1
intercellular adhesion molecule-1
- IRAK4
interleukin-1 receptor-associated kinase 4
- IRF3
interferon regulatory factor-3
- LPS
lipopolysaccharide
- MAL
MYD88 adaptor like protein
- MAMP
microbial-associated molecular pattern
- MyD88
myeloid differentiation primary response gene 88
- NET
neutrophil extracellular trap
- NOD2
nucleotide-binding oligomerization domain-containing protein 2
- PMN
polymorphonuclear neutrophils
- PSGL-1
P-selectin glycoprotein ligand-1
- ROS
reactive oxygen species
- SPF
specific pathogen free
- TIR
Toll/interleukin-1 receptor
- TIRAP
TIR-associated protein
- TLR4
Toll-like receptor 4
- TRAF3
tumor necrosis factor receptor-associated factor 3
- TRAM
TRIF-related adaptor molecule
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- WT
wild type
Footnotes
Disclosures
The authors declare no conflicts of interest.
References
- 1.Walker TG. Mesenteric ischemia. Semin Intervent Radiol. 2009; 26:175–183. doi: 10.1055/s-0029-1225662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harward TR., Brooks DL, Flynn TC, Seeger JM. Multiple organ dysfunction after mesenteric artery revascularization. J Vasc Surg. 1993; 18:459–467. doi: 10.1067/mva.1993.48586 [DOI] [PubMed] [Google Scholar]
- 3.Shigematsu T, Wolf RE, Granger DN. T-lymphocytes modulate the microvascular and inflammatory responses to intestinal ischemia-reperfusion. Microcirculation 2002; 9:99–109. doi: 10.1038/sj/mn/7800126 [DOI] [PubMed] [Google Scholar]
- 4.Schoenberg MH, Berger HG. Reperfusion injury after intestinal ischemia. Crit Care Med. 1993; 21:1376–1386. doi: 10.1097/00003246-199309000-00023 [DOI] [PubMed] [Google Scholar]
- 5.Cicalese L, Billiar TR, Rao AS, Bauer AJ. Interaction between ischemia/reperfusion-induced leukocyte emigration and translocating bacterial enterotoxins on enteric muscle function. Transplant Proc. 1997; 29:1815. doi: 10.1016/s0041-1345(97)00080-8 [DOI] [PubMed] [Google Scholar]
- 6.Sorkine P, Szold O, Halpern P, Gutmann M, Greemland M, Rudick V, Goldman G. Gut decontamination reduces bowel ischemia-induced lung injury in rats. Chest. 1997; 112:491–495. doi: 10.1378/chest.112.2.491 [DOI] [PubMed] [Google Scholar]
- 7.Perez-Chanona E, Mühlbauer M, Jobin C. The microbiota protects against ischemia/reperfusion-induced intestinal injury through nucleotide-binding oligomerization domain-containing protein 2 (NOD2) signaling. Am J Pathol. 2014; 184:2965–2975. doi: 10.1016/j.ajpath.2014.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Victoni T, Coelho FR, Soares AL, de Freitas A, Secher T, Guabiraba R, Erard F, de Oliveira-Filho RM, Vargaftig BB, Lauvaux G, Kamal MA, Ryffel B, Moser R, Tavares-de-Lima W. Local and remote tissue injury upon intestinal ischemia and reperfusion depends on the TLR/MyD88 signaling pathway. Med Microbiol Immunol. 2010; 199:35–42. doi: 10.1007/s00430-009-0134-5 [DOI] [PubMed] [Google Scholar]
- 9.Watson MJ, Ke B, Shen XD, Gao F, Busuttil RW, Kupiec-Weglinski JW, Farmer DG. Intestinal ischemia/reperfusion injury triggers activation of innate toll-like receptor 4 and adaptive chemokine programs. Transplant Protoc. 2008; 40:3339–3341. doi: 10.1016/j.transproceed.2008.07.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, He GZ, Wang YK, Zhu QK, Chen W, Guo T. TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse ischemia/reperfusion injury. World J Gastroenterol. 2015; 21:8314–8325. doi: 10.3748/wjg.v21.i27.8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007; 7:353–364. doi: 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- 12.Pinsky DJ, Naka Y, Liao H, Oz MC, Wagner DD, Mayadas TN, Johnson RC, Hynes RO, Heath M, Lawson CA, Stern DM. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. A mechanism for rapid neutrophil recruitment after cardiac preservation. J Clin Invest 1996; 97:493–500. doi: 10.1172/JCI118440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichikawa H, Flores S, Kvietys PR, Wolf RE, Yoshikawa T, Granger DN, Aw TY. Molecular mechanisms of anoxia/reoxygenation-induced neutrophil adherence to cultured endothelial cells. Circ Res. 1997; 81:922–931. doi: 10.1161/01.res.81.6.922 [DOI] [PubMed] [Google Scholar]
- 14.Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber D, Krombach F, Messmer K. Platelet-endothelial cell interactions during ischemia-reperfusion: the role of P-selectin. Blood. 1998; 92:507–515. [PubMed] [Google Scholar]
- 15.Nolte D, Hecht R, Schmid P, Botzlar A, Menger MD, Neumueller C, Sinowatz F, Vestweber D, Messmer K. Role of Mac-1 and ICAM-1 in ischemia-reperfusion injury in a microcirculation model of BALB/C mice. Am J Physiol. 1994; 267:H1320–H1328. doi: 10.1152/ajpheart.1994.267.4.H1320 [DOI] [PubMed] [Google Scholar]
- 16.Massberg S, Enders G, Matos FC, Tomic LI, Leiderer R, Eisenmenger S, Messmer K, Krombach F. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood. 1999; 94:3829–3838. [PubMed] [Google Scholar]
- 17.Komatsu S, Berg RD, Russell JM, Nimura Y, Granger DN. Enteric microflora contribute to constitutive ICAM-1 expression on vascular endothelial cells. Am J Physiol Gastrointestinal Liver Physiol. 2000; 279:G186–G191. doi: 10.1152/ajpgi.2000.279.1.G186 [DOI] [PubMed] [Google Scholar]
- 18.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015; 126:242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004; 303:1532–1535. doi: 10.1182/blood-2015-01-624023 [DOI] [PubMed] [Google Scholar]
- 20.Massberg S, Grahl L, von Bruehl M-L, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010; 16:887–896. doi: 10.1038/nm.2184 [DOI] [PubMed] [Google Scholar]
- 21.Bosmann M, Ward PA. Protein-based therapies for acute lung injury: targeting neutrophil extracellular traps. Expert Opin Ther Targets. 2014; 18:703–714. doi: 10.1517/14728222.2014.902938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: The biology of chromatin externalization. Dev Cell. 2018; 44:542–553. doi: 10.1016/j.devcel.2018.01.019 [DOI] [PubMed] [Google Scholar]
- 23.Franck G, Mawson TL, Folco EJ, et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ Res. 2018; 123:33–42. doi: 10.1161/CIRCRESAHA.117.312494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark K, Philippi V, Stockhausen S, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016; 128:2435–2449. doi: 10.1182/blood-2016-04-710632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y, Wagner DD. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014; 123:141–148. doi: 10.1182/blood-2013-07-514992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boettcher M, Eschenburg G, Mietzsch S, Jiménez-Alcázar M, Klinke M, Vincent D, Tiemann B, Bergholz R, Reinshagen K, Fuchs TA. Therapeutic targeting of extracellular DNA improves the outcome of intestinal ischemic reperfusion injury in neonatal rats. Sci Rep. 2017; 7:15377. doi: 10.1038/s41598-017-15807-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira A. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999; 162:3749–3752. [PubMed] [Google Scholar]
- 28.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-8-mediated function. Immunity. 1998; 9:143–50. doi: 10.1016/s1074-7613(00)80596-8 [DOI] [PubMed] [Google Scholar]
- 29.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003; 424:743–8. doi: 10.1038/nature01889 [DOI] [PubMed] [Google Scholar]
- 30.Dewhirst FE, Chien CC, Paster BJ, Ericson RL, Orcutt RP, Schauer DB, Fox JG. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999, 65:3287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GRS, Raparia K, Tamaki Z, Varga J. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016; 7:11703. doi: 10.1038/ncomms11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hörmann N, Brandão I, Jäckel S, Ens N, Lillich M, Walter U, Reinhardt C. Gut microbial colonization orchestrates TLR2 expression, signaling and ephithelial proliferation in the small intestinal mucosa. PLoS One. 2014; 9:e113080. doi: 10.1371/journal.pone.0113080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroeder JW and Simmons LA. Complete Genome Sequence of Bacillus subtilis strain PY79. Genome Announc. 2013, 1:e01085–13. doi: 10.1128/genomeA.01085-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youngman P, Perkins JB, Losick R. A novel method for the rapid cloning in Escherichia coli of Bacillus subtilis chromosomal DNA adjacent to Tn917 insertions. Mol Gen Genet. 1984; 195:424–433. doi: 10.1007/BF00341443 [DOI] [PubMed] [Google Scholar]
- 35.Chen LW, Chang WJ, Chen PH, Hsu CM. Commensal microflora induce host defense and decrease bacterial translocation in burn mice through toll-like receptor 4. J Biomed Sci. 2010, 17:48. doi: 10.1186/1423-0127-17-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004, 118, 229–241. doi: 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 37.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008; 57:1470–1481. doi: 10.2337/db07-1403 [DOI] [PubMed] [Google Scholar]
- 38.Saffarzadeh M, Cabrera-Fuentes HA, Veit F. Jiang D, Scharffetter-Kochanek K, Gille CG, Rooijakkers SHM, Hartl D, Preissner KT. Characterization of rapid neutrophil extracellular trap formation and its cooperation with phagocytosis in human neutrophils. Discoveries. 2014, 2:e19. doi: 10.15190/d.2014.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo Y and Dorf ME. Isolation of mouse neutrophils. Curr Protoc Immunol. 2001, 22, 3.20.1–3.20.6. doi: 10.1002/0471142735.im0320s22 [DOI] [PubMed] [Google Scholar]
- 40.Kanaji T, Russell S and Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002, 100, 2102–7. doi: 10.1182/blood-2002-03-0997 [DOI] [PubMed] [Google Scholar]
- 41.Tanaka K, Koike Y, Shimura T,Okigami M, Ide S, Toiyama Y, Okugawa Y, Inoue Y, Araki T, Uchida K, Mohri Y, Mizoguchi A, Kusunoki M. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One. 2014, 9:e111888. doi: 10.1371/journal.pone.0111888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caesar R, Reigstad CS, Bäckhed HK, Reinhardt C, Ketonen M, Lundén GÖ, Cani PD, Bäckhed F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut. 2012; 61:1701–1707. doi: 10.1136/gutjnl-2011-301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Into T, Kanno Y, Dohkan J-I, NakashimaM, Inomata M, Shibata K-I, Lowenstein CJ, Matsushita K. Pathogen recognition by Toll-like receptor 2 activates Weibel-Palade body exocytosis in human aortic endothelial cells. J Biol Chem. 2007; 282:8134–8141. doi: 10.1074/jbc.M609962200 [DOI] [PubMed] [Google Scholar]
- 44.Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Östergren-Lundén G, Petersen LC, Ruf W, Bäckhed F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 2012; 483:627–631. doi: 10.1038/nature10893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005; 7:12–19. doi: 10.1186/ar1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slotta JE, Braun OO, Menger MD, Thorlacius H. Capture of platelets to the endothelium of the femoral vein is mediated by CD62P and CD162. Platelets. 2009; 20:505–512. doi: 10.3109/09537100903215417 [DOI] [PubMed] [Google Scholar]
- 47.Fagundes CT, Souza DG, Nicoli JR, Teixeira MM. Control of host inflammatory responsiveness by indigenous microbiota reveals an adaptive component of the innate immune system. Microbes Infect. 2011; 13:1121–32. doi: 10.1016/j.micinf.2011.07.012 [DOI] [PubMed] [Google Scholar]
- 48.Parker LC, Jones EC, Prince LR, Dower SK, Whyte MK, Sabroe I. Endotoxin tolerance induces selective alterations in neutrophil function. J Leukoc Biol. 2005; 78:1301–5. doi: 10.1189/jlb.0405236 [DOI] [PubMed] [Google Scholar]
- 49.van der Hoven B, Nabuurs M, van Leengoed LA, Groeneveld AB, Thijs LG. Gut luminal endotoxin reduces ischemia-reperfusion injury of the small gut in germ-free pigs. Shock. 2001; 16:28–32. doi: 10.1097/00024382-200116010-00006 [DOI] [PubMed] [Google Scholar]
- 50.Gathiram P, Wells MT, Brock-Utne JG, Wessels BC, Gaffin SL. Oral administered nonabsorbable antibiotics prevent endotoxemia in primates following intestinal ischemia. J Surg Res. 1988; 45:187–193. doi: 10.1016/0022-4804(88)90064-9 [DOI] [PubMed] [Google Scholar]
- 51.Koike K, Moore FA, Moore EE, Trew CE, Banerjee A, Peterson VM. Endotoxin pretreatment inhibits neutrophil proliferation and function. J Surg Res. 1994; 57:49–54. doi: 10.1006/jsre.1994.1108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.