

Abstract
Intramembrane enzymes are often difficult for biochemical characterization. Human vitamin K epoxide reductase (VKOR) is the target of warfarin. However, this intramembrane enzyme becomes insensitive to warfarin inhibition in vitro, preventing the characterization of inhibition kinetics for decades. Here we employ structural biology methods to identify stable VKOR and VKOR-like proteins and purify them to near homogeneity. We find that the key to maintain their warfarin sensitivity is to stabilize their native protein conformation in vitro. Reduced glutathione drastically increases the warfarin sensitivity of a VKOR-like protein from Takifugu rubripes, presumably through maintaining a disulfide-bonded conformation. Effective inhibition of human VKOR-like requires also the use of LMNG, a mild detergent developed for crystallography to increase membrane protein stability. Human VKOR needs to be preserved in ER-enriched microsomes to exhibit warfarin sensitivity, whereas human VKOR purified in LMNG is stable only with pre-bound warfarin. Under these optimal conditions, warfarin inhibits with tight-binding kinetics. Overall, our studies show that structural biology methods are ideal for stabilizing intramembrane enzymes. Optimizing toward their inhibitor-binding conformation enables characterization of enzyme kinetics in difficult cases.
Introduction
Vitamin K epoxide reductases (VKOR) represent a large family of intramembrane thiol oxidoreductases [1]. These enzymes catalyze disulfide-bond formation in bacteria [2], archaea [3] and plants [4] to facilitate the oxidative folding of many proteins [5]. In vertebrates, however, the major function of VKOR changes to support blood coagulation through the vitamin K cycle [6–8]. This cycle begins with the γ-carboxylation of selected glutamic acids in several coagulation factors, a posttranslational modification required for their activity. The γ-carboxylation is driven by the epoxidation of the vitamin K hydroquinone, which is regenerated by VKOR to complete the vitamin K cycle (Fig. 1A).
Figure 1. Scheme of VKOR and VKORL catalysis and purification of stable proteins.
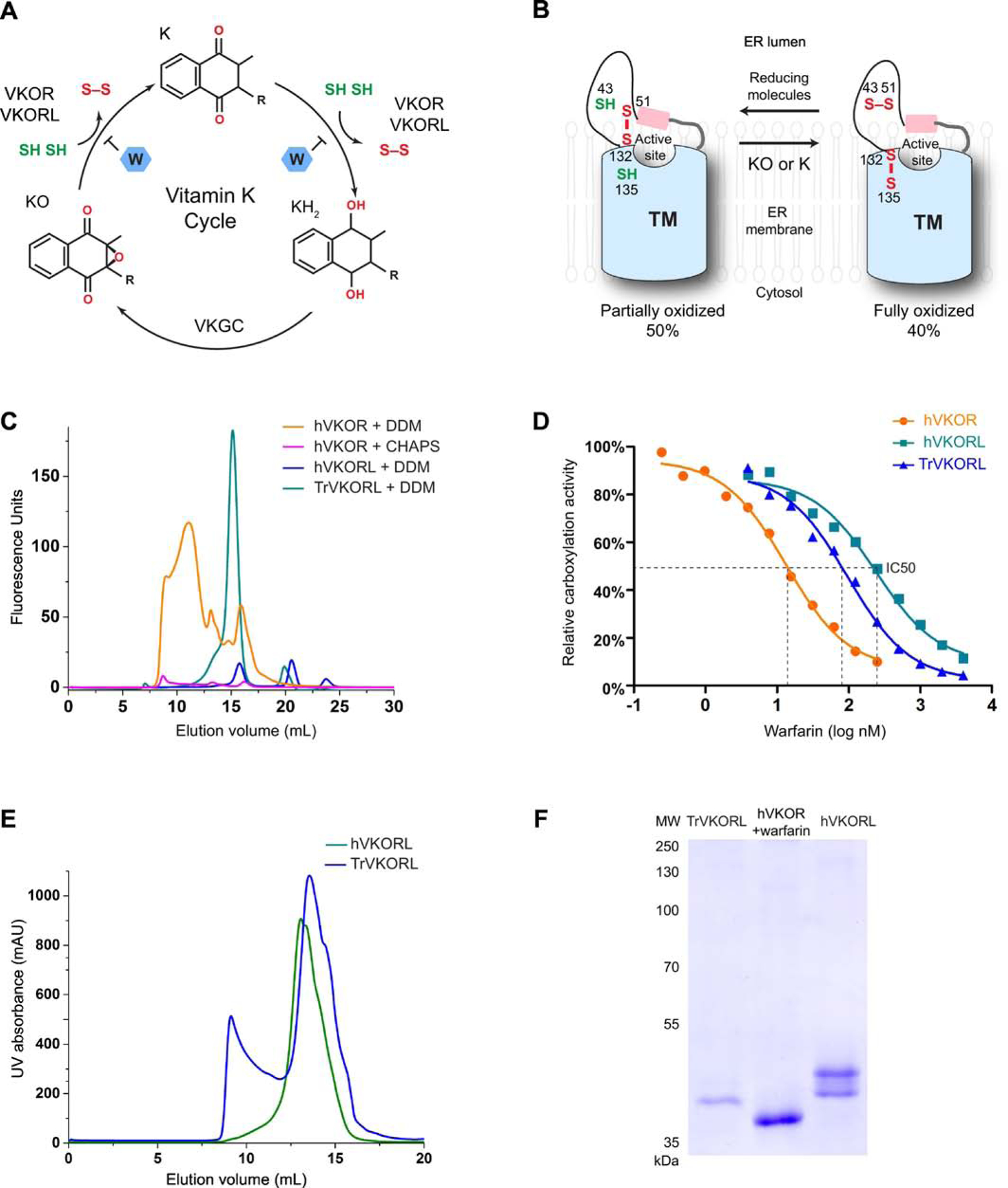
A, The vitamin K cycle. VKOR and VKORL reduce KO to K and then to KH2, which is converted back to KO by vitamin K dependent γ-carboxylase (VKGC). Each reduction step requires two free cysteines, resulting in the formation of a disulfide bond. Warfarin (W) inhibits both reduction steps. B, Catalysis of VKOR and VKORL in cells. The active site of these endoplasmic membrane enzymes is located at the membrane interface facing the ER lumen. Most of the cellular proteins, VKOR and VKORL, are in the partially (~50%) or fully oxidized state (40%) [26]. The partially oxidized enzyme (with two free cysteines) reduces the substrates and becomes fully oxidized. Reducing molecules in the ER converts the enzymes back to the partially oxidized state. C, FSEC profiles of TrVKORL, hVKORL and hVKOR. D, Warfarin inhibition curves using cell-based activity assay. E, Size exclusion chromatography during protein purification. The major elution peak around 15 mL (24 mL Superdex 200 column) corresponds to monomeric proteins with DDM micelles. F, SDS-PAGE of the purified proteins with Coomassie staining.
The human genome encodes two paralogs, VKOR and VKOR-like, that exhibit the epoxide reductase activity [8–14]. Both enzymes can reduce vitamin K epoxide (KO) to the quinone (K) and then to the hydroquinone (KH2). Each step of the reduction is coupled to the oxidation of a cysteine pair (CXXC motif) at the active site. To restore the catalytic activity, the enzymes are further reduced by protein partners or small molecules. These reducing molecules are located in the endoplasmic reticulum (ER) lumen, and VKOR and VKOR-like are embedded in the ER membrane with their active site facing the ER lumen (Fig. 1B). The electrons are transferred from the reducing molecules through another conserved cysteine pair in VKORs, thereby driving the catalysis at the membrane interface.
Human VKOR (hVKOR) is the target of warfarin, an oral anticoagulant commonly used to treat and prevent thromboembolic diseases. At the therapeutic dose, free warfarin concentration in blood plasma is in the nM range [15]. Warfarin shows similar inhibition range in cell-based assays that measure the γ-carboxylation of coagulation factors [10,16–18]. However, the warfarin IC50 measured in vitro is few magnitudes higher (in μM range) [19,20]. These in vitro assays typically use DTT to reduce hVKOR in cells or microsomes that have been dissolved in CHAPS, a detergent found to temporarily maintain the hVKOR activity. Due to the quick loss of activity, purification of hVKOR has been difficult, which has prevented the biochemical identification of the hVKOR protein before the human genome is known [9,21]. Later studies discovered that adding back lipids restores the hVKOR activity [22,23]. However, the purified enzyme remains poorly inhibited by warfarin. These various problems highlight the difficulties of studying intramembrane enzymes that are often unstable in vitro.
Crystal structures have been determined for a bacterial VKOR homolog from Synechococcus sp. [24,25] The structures show that the active site is surrounded by a bundle of four transmembrane helices (TM) and capped by a short helix. This helix connects to TM1 through an extramembrane loop, which contains the cysteine pair designated for electron transfer. Sequence alignment shows that vertebrate VKOR proteins have a much longer extramembrane region that can be highly flexible, explaining the instability of vertebrate VKORs.
Here we systematically investigated the in vitro stability of VKOR and VKOR-like enzymes. Under the conditions stabilizing their protein conformation, these enzymes can be effectively inhibited by warfarin with tight binding kinetics.
Results
Analyses of the factors potentially affecting warfarin inhibition in vitro
The poor warfarin inhibition in vitro could possibly originate from several factors in the traditional assay systems. This relatively simple assay generally starts by dissolving cells or microsomes containing VKOR in CHAPS and preincubating the mixture with warfarin. Subsequently, the KO substrate and DTT are added to initiate the reaction. However, the use of DTT can be problematic because it is an artificial reductant not found in cells. In fact, previous studies find that VKOR pre-reduced by DTT becomes less inhibited by warfarin, suggesting that warfarin preferably inhibits oxidized VKOR and DTT reduction interferes with this inhibition process [26,27]. On the other hand, the cells and microsomes contain many other proteins and small molecules; these impurities may also interfere with warfarin inhibition. Moreover, the choice of detergent is well known to affect the stability of membrane proteins. Although CHAPS is traditionally used to temporarily maintain VKOR activity, the ligand-binding ability of this membrane protein may be better preserved by an optimal detergent. Finally, human VKOR is an intrinsically unstable protein [22], whose ligand-binding property may be compromised in vitro. Notably, maintaining the protein stability and optimizing detergent usage are the major focus of structural biology methods, which are, so to speak, specialized in generating membrane proteins with the best quality. Thus, we employ membrane crystallography methods and systematically investigate these potential problems, with the goal of elucidating the inhibition kinetics of this group of intramembrane enzymes.
Identification of stable homologous proteins
To identify proteins stable in detergent solution, we selected 18 VKOR homologs and VKOR-like homologs from vertebrate species and expressed these proteins in Pichia pastoris. These proteins were tagged with a C-terminal GFP to allow detection by fluorescence-detection size-exclusion chromatography (FSEC), a well-accepted method in the structural studies of membrane proteins [28]. We dissolved whole cells expressing the homologous proteins in a buffer containing n-dodecyl β-D-maltoside (DDM), a mild detergent commonly used in membrane structural biology. The solubilized mixture was subsequently passed through a size-exclusion column and analyzed by a fluorescence detector. Most of the GFP-tagged homologs show low expression levels and/or broad peaks (Fig. S1). In particular, hVKOR shows poor expression and broad FSEC peaks in DDM (Fig. 1C). Because CHAPS is used traditionally in activity assays, we tested this detergent for the solubilization and FSEC analysis of hVKOR (Fig. 1C). The low FSEC peaks indicate that protein extraction by CHAPS is highly inefficient. Only a small portion of the hVKOR protein shows an unaggregated peak at ~15 mL elution volume on a Superdex 200 column, whereas most of the protein is aggregated (8–10 mL) in CHAPS. Because the CHAPS extraction is heavily contaminated with denatured hVKOR, previous reports involving CHAPS solubilization need to be interpreted with caution. On the other hand, human VKOR-like protein (hVKORL) shows a monodispersed FSEC peaks at ~15 mL elution volume, as expected for a monomeric protein in DDM micelles (Fig. 1C). However, the expression level of hVKORL is relatively low. In contrast, the VKOR-like homologs from Xenopus tropicalis and Takifugu rubripes show both good expression and monodispersed peaks on FSEC (Fig. S1 and Fig. 1C). Taken together, FSEC rapidly assesses the expression level and detergent stability of these intramembrane enzymes without the need for protein purification.
Warfarin inhibition in cells
We confirmed that these homologous proteins are sensitive to warfarin inhibition in the cellular environment. For the activity assay, we used a HEK293 cell line that has endogenous VKOR and VKORL knocked out and a chimeric FIXGla-PC introduced [5]. The γ-carboxylation of FIXGla-PC requires KO reductase activity of the transfected homologs, allowing subsequent measurements of warfarin IC50; this assay is however unable to monitor enzyme kinetics because the diffusion rates of substrate and warfarin into ER is unknown and VKOR activity is indirectly measured. With the same amount of DNA transfection, we found that the IC50 of warfarin inhibition against hVKOR, hVKORL and Takifugu rubripes VKOR-like (TrVKORL) are 13 nM, 238 nM and 97 nM, respectively (Fig. 1D). Despite the variation in IC50s, all these homologs can be inhibited by warfarin in the nM range. Therefore, we could start with the stable homologous proteins to improve their warfarin sensitivity in vitro, and ultimately resolve this long-standing issue for hVKOR.
Glutathione reduction restores warfarin sensitivity of purified TrVKORL
Given the high stability and expression level of TrVKORL, we first purified this protein for biochemical analyses. During the purification, size-exclusion chromatography showed that the major protein peak is monodispersed at the expected elution volume (Fig. 1E), suggesting that the TrVKORL protein is correctly folded and without aggregation. From 1 L yeast cell culture, about 10 mg of TrVKORL protein could be purified to near homogeneity (Fig. 1F). This high protein quality avoids the potential interference from protein misfolding and impurities, both of which can cause misinterpretation of enzyme kinetics [29].
The purified TrVKORL was analyzed using a DTT-driven fluorometric assay [24], in which fluorescence of the reaction product, KH2, is followed in real time (Fig. S2A, B). In absence of warfarin, TrVKORL shows high activity of catalyzing the KO or K reduction to KH2. However, purified TrVKORL is still poorly inhibited by warfarin. With 500 nM enzyme, the warfarin IC50 is 4.7 μM and 3.8 μM in presence of KO or K, respectively (Fig. 2A, B). In contrast, the cell-based assay shows that warfarin inhibits TrVKORL with an IC50 about 40 times lower (Fig. 1D). Similar difference between cellular assay and in vitro assay has been observed for hVKOR. Therefore, improving the protein purity or intrinsic stability is insufficient to restore warfarin sensitivity in vitro.
Figure 2. Tight-binding warfarin inhibition of TrVKORL requires GSH as the reductant.
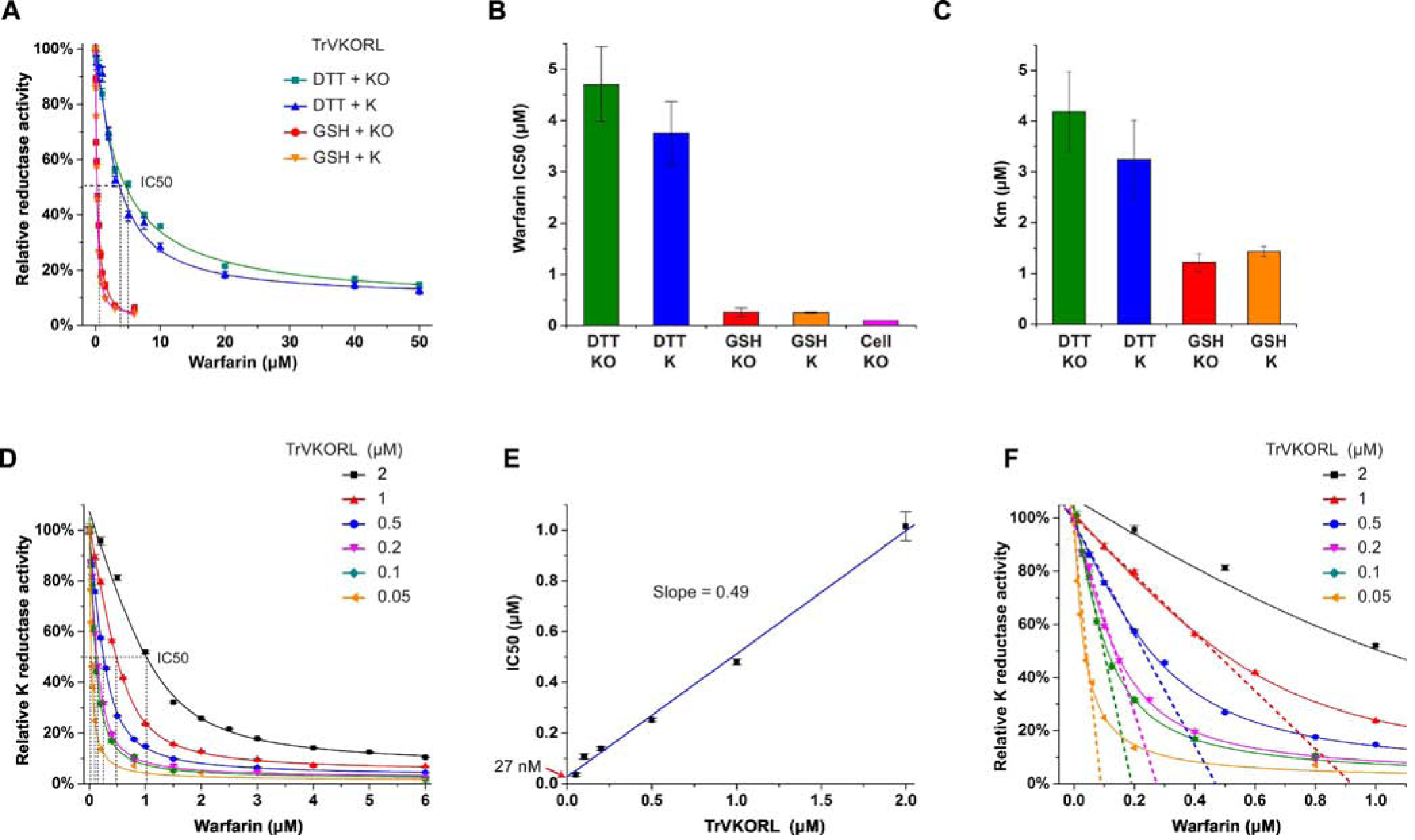
A, Warfarin inhibition curves with DTT and GSH. B, Comparison of IC50s obtained from in vitro and cellular assays. C, Comparison of KM values. D, Warfarin inhibition curves at different TrVKORL concentrations with K as substrate. E, Linear correlation between warfarin IC50 and TrVKORL concentration. F, Extrapolation from the linear section of the tight-binding inhibition curves (zoom-in view from D) gives the effective enzyme concentration.
We next investigated whether the choice of reductant causes the poor warfarin inhibition in vitro. We substituted DTT with glutathione (GSH), a native reductant that is presented at high concentration (~ 5 mM) in the ER lumen. Because the active site of VKOR faces also the ER lumen, GSH may serve as a reductant to VKOR in the cellular environment. Indeed, we found that GSH can act as the reducing equivalent in vitro to promote the KO and K reductase activities of TrVKORL (Fig. S2C, D). Moreover, the GSH-driven reaction drastically lowers the IC50 of warfarin. For 500 nM TrVKORL, the apparent IC50 is about 260 nM and 250 nM in presence of KO or K, respectively (Fig. 2A, B). The IC50s with GSH reduction are close to that from the cell-based assay (97 nM) and much lower than those with DTT reduction (Fig. 2B), suggesting that the warfarin-binding conformation of TrVKORL is better maintained in presence of GSH.
Given that GSH stabilizes warfarin binding, we investigated whether GSH also improves substrate binding. Compared to DTT reduction, the KM with GSH reduction decreased by 3.4 and 2.3-fold when KO and K is used as the substrate, respectively (Fig. 2C). Thus, GSH also improves the apparent affinities of substrates, although the effects are not as drastic as warfarin binding.
Warfarin is a tight-binding inhibitor
With TrVKORL effectively inhibited by warfarin, we could characterize the inhibition kinetics. Fig. 2A shows that, in presence of GSH, the IC50 of warfarin (250–260 nM) is about half of the enzyme concentration (500 nM). Because inhibitor and enzyme concentrations are close, the general assumption of a largely free inhibitor is no longer valid. Instead, the enzyme binds inhibitor stoichiometrically, a scenario known as tight-binding inhibition. With K as the substrate, we systematically varied the TrVKORL concentrations and found that raising the enzyme concentration ([E]) also increases the warfarin IC50 (Fig. 2D). Due to the stoichiometric inhibition, the IC50 and [E] follow a linear correlation (Fig. 2E) that is characteristic of tight-binding inhibition, which defines that IC50 = ½ [E] + Kiapp. Thus, extrapolation from the linear correlation gives the Kiapp (apparent Ki), which is estimated to about 27 nM. The slope is about 0.5, indicating that the measured enzyme concentration is close to the effective concentration, which can be obtained also from the extrapolation from linear part of the tight-binding inhibition curves (Fig. 2F). Thus, the amount of dysfunctional or denatured enzyme is negligible under the assay condition. At low enzyme concentration, the inhibition curves can be well fitted by the Morrison equation that defines tight-binding inhibition. The Kiapp estimated from the Morrison equation is 29 nM, close to that estimated from the linear correlation. This nM range of Kiapp is consistent with the IC50 obtained from the cellular activity assay (Fig. 2B).
Human VKORL requires a mild detergent
To compare with TrVKORL, we investigated the warfarin inhibition kinetics of hVKORL, which could also be purified in milligram amount and to near homogeneity (Fig. 1F). As expected, hVKORL is much better inhibited by warfarin in GSH than in DTT, with both KO and K as the substrate (Fig. 3A, B). However, the IC50s are several times higher than the enzyme concentration (250 nM), an observation inconsistent with the mechanism of stoichiometric inhibition, in which one molecule warfarin binds to one molecule of the enzyme. This discrepancy suggests that GSH alone cannot fully maintain the native conformation of the hVKORL.
Figure 3. LMNG stabilizes hVKORL for tight-binding warfarin inhibition.
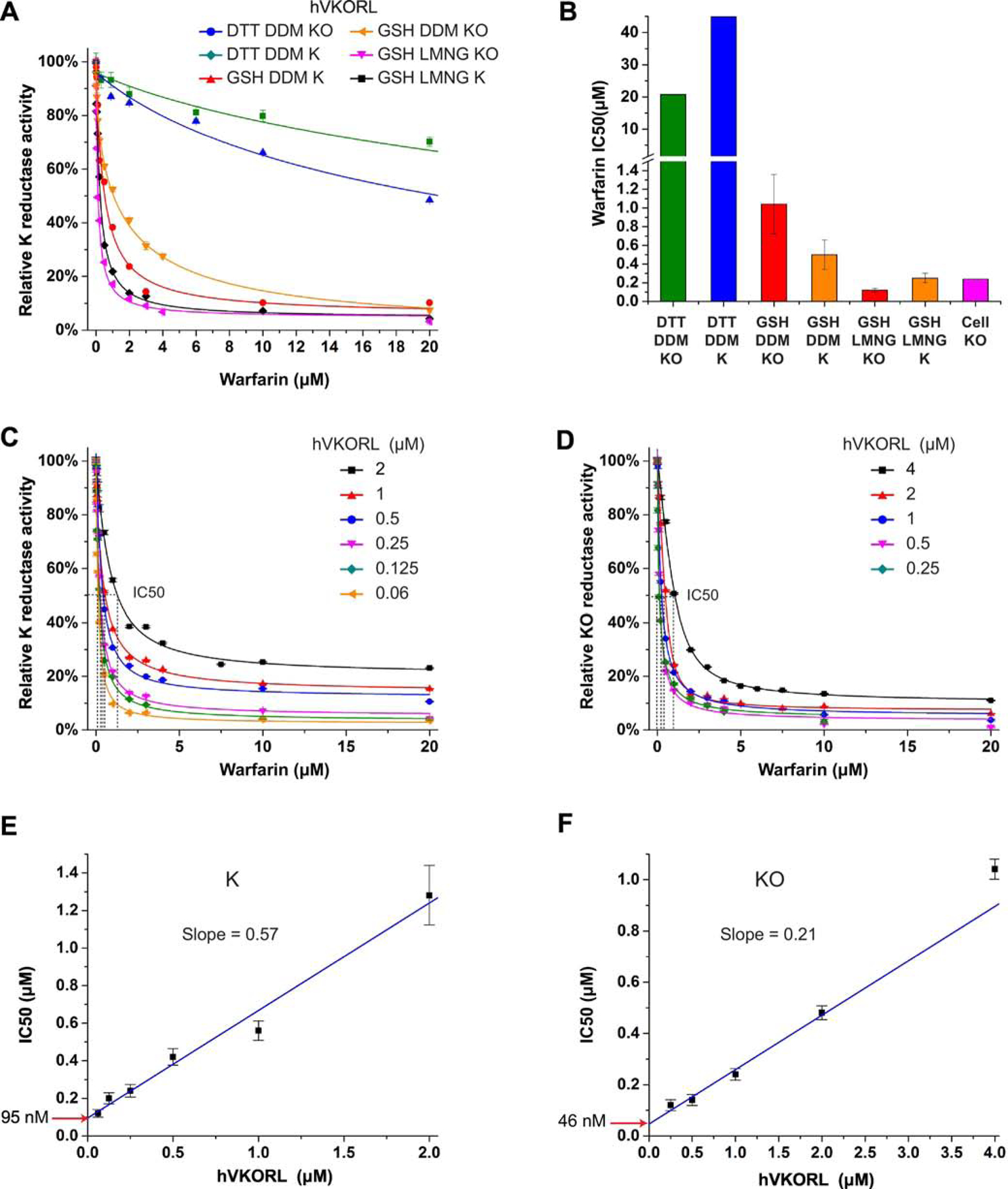
A, Comparison of warfarin inhibition curves with DTT or GSH as the reductant, with K or KO as the substrate and with different detergents, DDM or LMNG. B, Comparison of IC50s obtained from in vitro and cellular assays. C-D, Warfarin inhibition curves at different TrVKORL concentrations with K (C) and KO (D) as substrate. E-F, Linear correlation between warfarin IC50 and TrVKORL concentration with K (E) and KO (F) as the substrate.
The choice of detergent is a major factor affecting membrane protein stability. Therefore, we sought to use a different detergent, lauryl maltose neopentyl glycol (LMNG), to stabilize the hVKORL protein. The chemical structure of LMNG is essentially two linked DDM molecules; the increased sizes of head and tail groups better stabilize membrane proteins and allowed structure determination in difficult cases [30]. We found that hVKORL purified and analyzed in LMNG solution is inhibited by warfarin with the IC50 (120 nM in KO and 250 nM in K) close to half of the enzyme concentration (250 nM) (Fig. 3B). With the enzyme concentrations systematically varied (Fig. 3C, D), we found that plotting IC50 with [E] gives a slope close to 0.5 (Fig. 3E) with K as the substrate, as expected for stoichiometric inhibition. However, the KO substrate gives a slope of 0.21 (Fig. 3F), indicating that the effective enzyme concentration is about half as that observed for K. This is probably because the KO to KH2 reduction is a two-step reaction and involves two enzyme molecules for the catalysis. The Kiapp values estimated from the intercepts of the linear fittings are 46 nM and 95 nM for KO and K, respectively, close to the IC50 from cellular assay (Fig. 3B). Taken together, the hVKORL protein requires both the proper reductant and the proper detergent to maintain its native conformation and warfarin binding affinity.
Characterization of hVKOR
Because hVKOR is unstable in detergent (Fig. 1C), we used microsomes to provide a native-like lipid bilayer environment. There are previous reports using microsomal VKOR that is not subjected to detergent solubilization [20,31], but these warfarin inhibition studies are always conducted with DTT. Therefore, we changed the reductant to GSH and measured the warfarin inhibition of KO to K conversion catalyzed by microsomal hVKOR. A high IC50 (650 nM), however, is required to inhibition hVKOR in crude microsomes (Fig. 4A). To overcome this problem, we prepared the ER-enriched microsomes. After rough purification, the warfarin IC50 is lowered to 130 nM (Fig. 4A) with similar amount of the hVKOR protein (quantified by the fluorescence of fused GFP). Morrison fitting of the inhibition curve of ER-enriched microsomes gives a Kiapp of 16 nM, as expected for tight-binding inhibition. Thus, removing the impurities in crude microsomes and using GSH as the reductant restores the in vitro warfarin sensitivity of hVKOR in the membrane environment.
Figure 4. Warfarin stabilizes hVKOR in cells and in vitro.
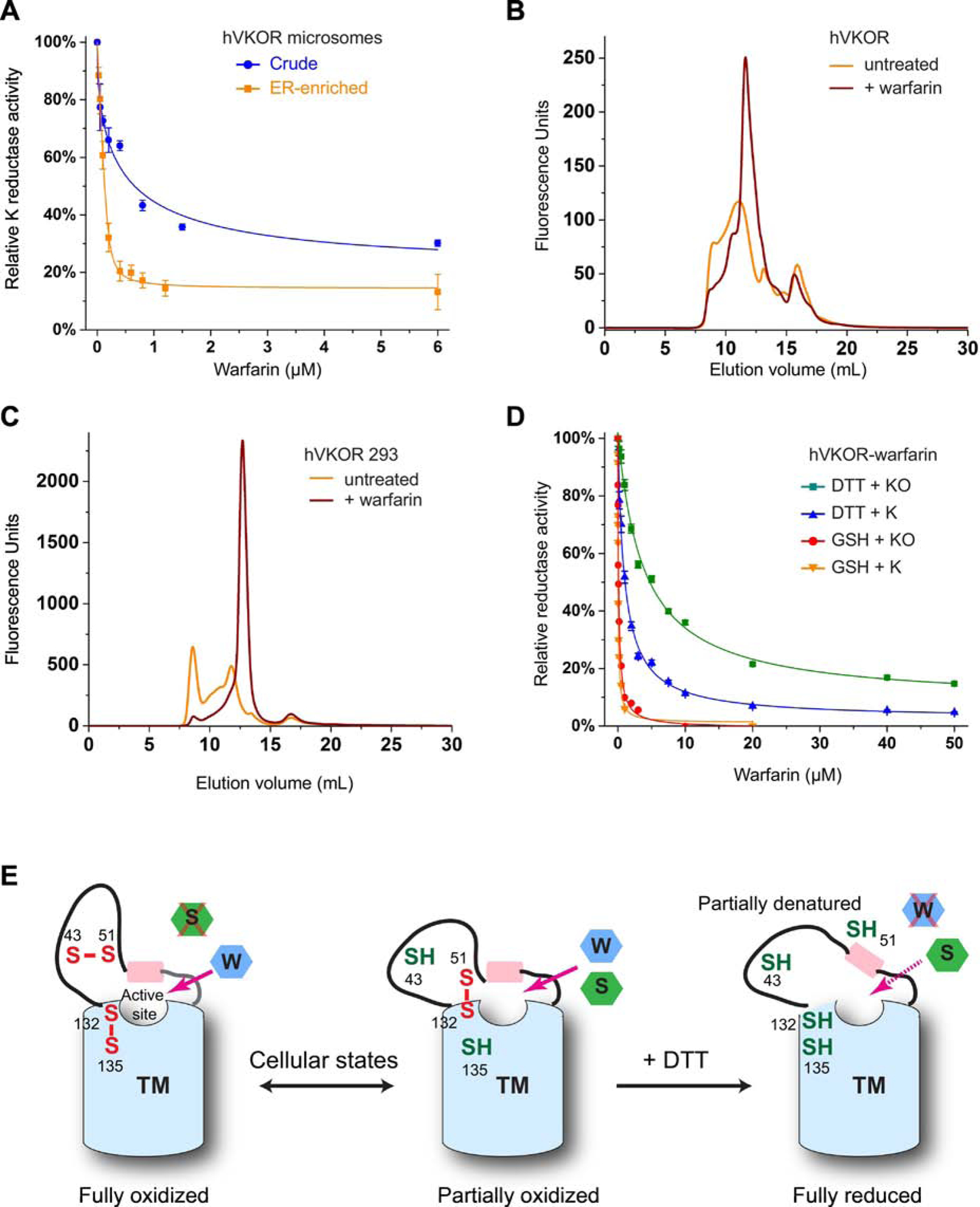
A, Warfarin inhibition curves of VKOR in microsomes. GSH is used as the reductant. B, FSEC profile of hVKOR pre-bound with warfarin (before detergent solubilization of microsomes). C, FSEC profile of hVKOR expressed in HEK293 cells treated with warfarin. D, Inhibition curves of hVKOR-warfarin complex with different reductants. E, Cartoon shows that DTT disrupts the native conformation of hVKOR through reducing the fully and partially oxidized states. W: warfarin; S: substrates. The TM domain is shown in blue and luminal helix in pink.
In order to study purified proteins, we sought to further stabilize hVKOR. Although none of the detergents appear effective in generating hVKOR free of aggregation (Fig. 1C), we found that hVKOR pretreated with warfarin before detergent solubilization results in a symmetrical peak on size-exclusion chromatography (Fig. 1F and 4B). Most of the protein is shifted to this peak at the unaggregated elution volume. Moreover, when HEK293 cells were incubated with warfarin, hVKOR was expressed in much larger amount and with a good FSEC profile (Fig. 4C); the stabilization probably has overcome the cell quality control system that normally degrades much of the unstable hVKOR molecules [32]. Thus, warfarin appears to stabilize the native conformation of hVKOR in the cellular environment.
We purified this stable complex of hVKOR and warfarin, which are bound to each other probably at a 1:1 molar ratio. Similar to the hVKORL and TrVKORL, this complex is poorly inhibited by warfarin when DTT is used as the reductant (Fig. 4D), which is known to compete with warfarin for hVKOR binding [20]. In contrast, with LMNG and GSH, the apparent warfarin IC50 are 40 nM in K and 100 nM in KO with 250 nM purified complex, consistent with the mechanism of tight binding inhibition.
Discussion
A key commandment in enzymology is “do not waste clean thinking on dirty enzymes” [29]. The ‘dirty’ enzymes can be either contaminated by impurities or can be misfolded or aggregated proteins. Particular caution needs to be taken for intramembrane enzymes, as they often become unstable after being solubilized in detergent. Even a mild detergent, such as DDM, cannot guarantee that native protein conformation is maintained. For instance, only three out of the 18 VKOR and VKORL homologs we investigated appear monodispersed after DDM solubilization. The hVKOR protein is mostly aggregated in CHAPS, although this detergent has been used for decades to study VKOR enzymology. Not surprisingly, hVKOR becomes insensitive to warfarin inhibition in vitro, preventing meaningful characterization of its inhibition kinetics.
A more worrisome fact is that hVKOR retains reductase activity despite the disturbance of its native conformation. The active site of this thiol oxidoreductase contains a CXXC motif (Cys132 and Cys135) at membrane interface, which can be directly reduced by DTT [24,33], a small and membrane-permeable molecule. With this strong reductant provided in large excess, hVKOR retains activity probably as long as its disturbed conformation binds substrates to some extent. Based on the structure of the bacterial homolog [24–26], the active site of hVKOR is surrounded by a four-TM bundle and capped by a short helix. This cap helix connects to a flexible ER luminal region that contains the cysteine pair (Cys43 and Cys51) designated for electron transfer. With these cysteines, the flexible region is either held together by a disulfide bond (Cys43-Cys51) or anchored to the TMs by an alternative disulfide (Cys51-Cys132), conformations occurring at the fully and partially oxidized state, respectively (Fig. 4E); the majority (~90%) of hVKOR molecules are found to be in one of these oxidized states in the cellular environment [34]. However, DTT reduction clearly can disrupt both states, as well as the protein conformations stabilized by the corresponding disulfide bonds. On the other hand, conformation of the four TM bundle is unlikely to change with DTT reduction. Therefore, the native conformation of hVKOR is probably disrupted only in the luminal region including the short helix capping the active site. The substrates may remain bound to hVKOR at this partially denatured conformation. A similar scenario can occur when VKOR conformation is disturbed in detergent solution. Consequently, the reductase activity appears to be maintained in vitro. This apparent activity, however, does not indicate that native protein conformation is maintained. Thus, in vitro ‘activity’ of intramembrane enzymes can be misleading and must be interpreted with extra caution.
Compared to the apparent activity of an intramembrane enzyme, its inhibitor-binding affinity can be more sensitive to the change of native protein conformation. In the case of hVKOR, warfarin inhibition is compromised with the use of nonoptimal reductant and detergents that partially denature the protein, whereas the enzyme activity is relatively insensitive to the partial denaturation. However, a careful comparison shows that both the warfarin IC50 and the KM of substrate catalysis of TrVKORL can be improved when DTT is substituted by GSH (Fig. 2B, C), a reductant natively presented in the ER lumen. Unlike DTT, GSH is a monothiol that reduces one cysteine at a time. Moreover, GSH has a much higher redox potential than DTT does. These chemical properties allow GSH to maintain the partially oxidized state of vertebrate VKOR and VKORL paralogs and retain one of their native conformations.
Optimizing the ligand binding of membrane proteins is an effective approach towards identifying conditions maintaining their native conformation. This is because the binding of a ligand generally requires many side-chain interactions, including those from multiple TMs and from extramembrane regions. Thus, ligands are exquisite indicators of whether a protein is correctly folded. In fact, binding of radiolabeled ligands is frequently used for improving the protein stability of G-protein coupled receptors [35]. The same principle applies to the study of intramembrane enzymes. Here, preincubation with warfarin clearly stabilizes the folding and prevents the aggregation of hVKOR during cellular expression (Fig. 4C) and after detergent solubilization (Fig. 4B). Through monitoring the IC50 of warfarin, we find that the GSH reductant is required for various epoxide reductases to retain their native conformation. The hVKORL is less stable than TrVKORL, and therefore requires a milder detergent, LMNG, to maintain warfarin sensitivity. This mild detergent however is ineffective for the highly unstable hVKOR protein. Instead, hVKOR needs to be kept in microsomes that provide a lipid-bilayer environment. Additionally, the microsomes need to be purified to remove unknown factors that interfere with the warfarin inhibition. These studies illustrate that IC50 of a known inhibitor is highly useful to probe the stability of intermembrane enzymes; maintaining in vitro stability is the key to the studies of these difficult enzymes.
Maintaining protein stability is a central goal of structural biology. Numerous tools have been developed to stabilize and crystallize membrane proteins, and applying these tools can also significantly facilitate their biochemical characterization. Here we use FSEC as a rapid readout to identify homologous proteins that are sufficiently stable after detergent solubilization. The most stable homolog, TrVKORL, allows us to identify that the use of reductant is the key to maintain warfarin sensitivity of this difficult group of intramembrane enzymes. With such knowledge, subsequent experiments can be focused on identifying suitable detergents to maintain protein stability. Indeed, if FSEC were previously available and known to biochemists, they could have realized that CHAPS causes protein aggregation and is therefore problematic for activity assays. In our opinion, mild detergent such as DDM, which is the first choice in structural biology, should also be more frequently used in the biochemical study of intermembrane enzymes. Newer and milder detergents and lipid mimics that have been developed for structural studies, such as LMNG, GDN [36] and cholesteryl hemisuccinate (CHS) [37], should be introduced into the biochemical field. Some intramembrane enzymes may require the addition of lipids for stabilization, and the even less stable ones may need to be kept in lipid bilayers, such as keeping hVKOR in ER-enriched microsomes. To this end, a newly developed co-polymer, diisobutylene maleic acid (DIBMA), allows protein purification within the lipid bilayer and without the involvement of detergent solubilization [38]. Lipid nanodiscs provide another means for replacing detergents, but a mechanism for exchanging lipid substrates within nanodiscs to support assays of membrane enzymes of lipid metabolism remains challenging. Overall, structural biology tools should be exquisitely used to study intramembrane enzymes due to their intrinsic instability, and protein quality and stability should always be examined before the biochemical characterization.
Methods
Cloning
The DNA sequences of the VKOR and VKORL proteins were codon optimized and synthesized by Genscript. These constructs were cloned into a modified pPICZ-B expression vector (Invitrogen). For FSEC analyses, a PreScission protease site, an eGFP and a 10X His tag were added to C-terminal of the constructs. For activity assays, a split superfolder GFP was fused to the N- and C-termini of the constructs, followed by a C-terminal 10X His tag; the split GFP constructs were used due to their improved enzymatic activities in detergent solutions. For cell-based assay, the constructs without these tags were cloned into an engineered pBudCE4.1 expression vector.
Protein expression
The constructs were linearized and transformed into Pichia pastoris by electroporation. Transformants were selected by Zeocin resistance on YPDS-agar (1% yeast extract, 2% peptone, 2% glucose, 1.2 M sorbitol, and 2% agar) plates. Resistant colonies were inoculated to 5 mL BMG media (1% glycerol, 0.34% yeast nitrogen base, 1% ammonium sulfate, 0.4 μg/mL biotin, and 100 mM potassium phosphate pH 6.0) and grown at 30°C for 20 h. Subsequently, the growth media was exchanged to 5 mL BMM (0.34% yeast nitrogen base, 1% ammonium sulfate, 0.4 μg/mL biotin, and 200 mM potassium phosphate pH 6.0). The protein expression was induced with 0.7% methanol for 24 h at 25°C. The cells were harvested by centrifugation.
FSEC
The cells were resuspended in a buffer containing 150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl pH 8.0, and protease inhibitor cocktail. The cell suspension was lysed with a mixer mill (Retsch). The cell lysate was extracted with 2% n-dodecyl-β-D-maltopyranoside (DDM) in the resuspension buffer for 1 h. After centrifugation at 20,000 × g for 1 h, the supernatant was analyzed on a Superdex 200 column. The FSEC system is equipped with an online fluorescence detector and an autosampler (Shimadzu).
Protein purification
Frozen Pichia cells (20 g) were applied to a ball mill (Retsch) to break the cell wall. The cell powder was resuspended in 40 mL lysis buffer containing 225 mM NaCl, 75 mM Tris-HCl pH 8.0, 10 μg/mL DNase I and protease inhibitor cocktail. Cell membranes were subsequently solubilized in a buffer containing 2% n-dodecyl-β-d-maltopyranoside (DDM), 150 mM NaCl, 100 mM Tris-HCl pH 8.0, 10 μg/mL DNase I, and protease inhibitor cocktail. After stirring at 4°C for 3 h, the suspension was centrifuged at 30,000 g for 45 min. Subsequently, the supernatant was incubated with 3 mL TALON metal-affinity resin (Clontech) for 3 h at 4°C. The resin was collected on a gravity-flow column and washed with 60 mL wash buffer (20 mM imidazole, 2 mM DDM, 150 mM NaCl, and 20 mM Tris pH 8.0). The protein was eluted with 10 mL elution buffer (250 mM imidazole, 1 mM DDM, 150 mM NaCl, and 20 mM Tris pH 8.0). The eluted protein was concentrated and applied to Superdex 200 for size exclusion chromatography (SEC) in a buffer containing 0.1% DDM or 0.05% lauryl maltose neopentyl glycol (LMNG), 150 mM NaCl, and 20 mM Tris pH 8.0. The peak fractions were collected and concentrated to ~40 mg/mL. The purified protein was immediately used for the activity assay.
For the purification of hVKOR, the suspension of broken cells was incubated with 1 mM warfarin at 4°C for 20 min. The affinity chromatography and size exclusion chromatography were carried out in same buffers with the addition of a lipid mixture that contains 0.1 mg/mL total of POPC, POPE and POPG (Avanti) at a 3:1:1 molar ratio.
Preparation of ER-enriched microsomes
The frozen Pichia pastoris cells (20 g) were applied to a ball mill (Retsch) to break the cell wall. The cell powder was resuspended in 200 mL buffer containing 150 mM KCl and 50 mM potassium phosphate pH 7.5. The cell membrane was subsequently disrupted by sonication and centrifuged at 15,000g for 30 min. The supernatant was centrifuged at 42,000 rpm in a Ti45 rotor to collect crude microsomes. The microsomes were washed and resuspended in 10 mL low-salt buffer (10mM NaCl and 10 mM HEPES pH 7.5). To enrich the ER fraction, the microsomes were purified by sucrose gradient using a reported protocol[20] with modifications. Briefly, the 10 mL membrane suspension was laid on 25 mL 35% (w/v) sucrose and 25 mL 60% sucrose in the low-salt buffer, and centrifuged at 40,000 rpm in a Ti45 rotor overnight. The ER membranes enriched between the two sucrose layers were carefully taken out and collected by ultracentrifugation. The purified microsomes were washed and resuspended in 50 mM HEPES pH 7.5, 150 mM KCl. The concentration of total proteins was determined by bicinchoninic acid (BCA) assay and adjusted to 5 mg/mL.
Activity assay of microsomal human VKOR
Microsomes at different concentrations were preincubated with different concentrations of warfarin for 1 h on ice. The VKOR catalysis was initiated in 500 μL reaction buffer containing 80 mM GSH, 10 μM KO, 150 mM KCl and 200 mM HEPES pH 7.5. The reaction was carried out at 30 °C for 1.5 h and quenched by mixing with 1 mL isopropanol/hexane (3:2 v/v). After brief centrifugation, the hexane fraction was extracted, dried in air, and dissolved in methanol. The KO to K conversion was analyzed by HPLC using a reverse-phase C8 column for HPLC separation (Thermo) with methanol as the mobile phase. Alternatively, used liquid chromatography–mass spectrometry (LC-MS), which identifies KO and K by their masses and quantifies the extracted peaks with these masses, with following modifications from a reported assay[31]. The samples were loaded onto a C8 column and the elution was analyzed by a Bruker MaXis 4G quadrupole time-of-flight mass spectrometer equipped with atmospheric pressure chemical ionization (APCI) source under positive mode. The m/z corresponding to KO and K are 467.3520 and 451.3571 (z = 1, within 10 ppm), respectively. The areas of extracted ion chromatogram for KO and K were integrated. The fraction of KO to K conversion was calculated as K / (K + KO) and used to determine relative VKOR activities.
Fluorometric based activity assay
We modified a previous reported fluorometric assay [22,24] to study the inhibition kinetics. The VKOR and VKORL proteins were preincubated warfarin for 1 h on ice. The reduction of KO was initiated in a reaction buffer containing 20 μM KO, 40 mM GSH, 0.1% LMNG, 0.1 M NaCl, and 20 mM Tris pH 7.6. The reduction of K was initiated in a reaction buffer containing 50 μM K, 40 mM GSH, 1% LMNG or DDM, 0.1 M NaCl, and 20 mM Tris pH 7.6. For the comparison, GSH was substituted by 6.25 mM DTT. To detect the fluorescence of KH2 product in all these assays, excitation was carried out at 243 nm and excitation was followed at 430 nm in a plate reader.
Cell-based activity assay
The VKOR and VKORL in pBudCE4.1 containing a luciferase gene were transfected into a cell line established with a chimeric FIXgla-Protein C gene and with endogenous VKORC1 and VKORL1 genes knocked out [10] The carboxylation level of secreted FIXgla-PC was measured by a sandwich ELISA by using the cell-culture medium, with luciferase activity serving as the control for transfection efficiency. To measure warfarin IC50, the transfected cells were treated with 11 different concentrations of warfarin. The IC50 of each construct was analyzed using GraphPad Prism.
Supplementary Material
Supplementary Figure 1. FSEC profiles of VKOR and VKORL homologs.
Supplementary Figure 2. Catalytic activity and KM of purified TrVKORL and hVKORL. A, TrVKORL with KO. B, TrVKORL with K. C, hVKORL with KO. D, hVKORL with K. Left, progressive reaction curves. Right, Michaelis-Menten plots.
Highlights.
Structural biology methods enable stabilization of intramembrane enzymes for biochemical characterization.
Systematic optimization of protein stability through monitoring inhibitor efficacy allows the kinetics studies of an intramembrane thiol oxidoreductase family.
Effective warfarin inhibition requires glutathione as the reductant.
Warfarin stabilizes the native conformation of human vitamin K epoxide reductase.
Acknowledgment
W.L. is supported by NHLBI (R01 HL121718), W. M. Keck Foundation (Basic Science @ the Forefront of Science), Children’s Discovery Institute (MC-II-2020–854), NEI (R21 EY028705), and NIGMS (R01 GM131008).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Goodstadt L, Ponting CP, Vitamin K epoxide reductase: homology, active site and catalytic mechanism, Trends Biochem. Sci 29 (2004) 2002–2005. [DOI] [PubMed] [Google Scholar]
- [2].Dutton RJ, Boyd D, Berkmen M, Beckwith J, Bacterial species exhibit diversity intheir mechanisms and capacity for protein disulfide bond formation., Proc. Natl. Acad. Sci. U. S. A 105 (2008) 11933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hatahet F, Ruddock LW, Topological plasticity of enzymes involved in disulfide bond formation allows catalysis in either the periplasm or the cytoplasm, J. Mol. Biol 425 (2013) 3268–76. [DOI] [PubMed] [Google Scholar]
- [4].Feng W-K, Wang L, Lu Y, Wang X-Y, A protein oxidase catalysing disulfide bond formation is localized to the chloroplast thylakoids., FEBS J. 278 (2011) 3419–30. [DOI] [PubMed] [Google Scholar]
- [5].Sevier CS, a Kaiser C, Formation and transfer of disulphide bonds in living cells., Nat. Rev. Mol. Cell Biol 3 (2002) 836–47. [DOI] [PubMed] [Google Scholar]
- [6].Stafford DW, The vitamin K cycle., J. Thromb. Haemost 3 (2005) 1873–8. [DOI] [PubMed] [Google Scholar]
- [7].Oldenburg J, Marinova M, Müller-Reible C, Watzka M, The vitamin K cycle., Vitam. Horm 78 (2008) 35–62. [DOI] [PubMed] [Google Scholar]
- [8].Van Horn WD, Structural and functional insights into human vitamin K epoxide reductase and vitamin K epoxide reductase-like1., Crit. Rev. Biochem. Mol. Biol 48 (2013) 357–72. [DOI] [PubMed] [Google Scholar]
- [9].Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2, Nature. 427 (2004) 537–541. [DOI] [PubMed] [Google Scholar]
- [10].Tie JK, Jin DY, Tie K, Stafford DW, Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells, J. Thromb. Haemost 11 (2013) 1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tie JK, Jin DY, Stafford DW, Conserved Loop Cysteines of Vitamin K Epoxide Reductase Complex Subunit 1-Like 1 (VKORC1L1) Are Involved in Its Active Site Regeneration., J. Biol. Chem 289 (2014) 9396–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, Müller J, Bevans CG, Müller CR, Oldenburg J, Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function., J. Biol. Chem 286 (2011) 15085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lacombe J, Rishavy MA, Berkner KL, Ferron M, VKOR paralog VKORC1L1 supports vitamin K-dependent protein carboxylation in vivo., JCI Insight. 3 (2018) 96501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hammed A, Matagrin B, Spohn G, Prouillac C, Benoit E, Lattard V, VKORC1L1, an enzyme rescuing the vitamin K 2,3-epoxide reductase activity in some extrahepatic tissues during anticoagulation therapy, J. Biol. Chem 288 (2013) 28733–28742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Huang C, Yang J, Du Y, Miao L, Measurement of free concentrations of highly protein-bound warfarin in plasma by ultra performance liquid chromatography-tandem mass spectrometry and its correlation with the international normalized ratio, Clin. Chim. Acta 393 (2008) 85–9. [DOI] [PubMed] [Google Scholar]
- [16].Fregin A, Czogalla KJ, Gansler J, Rost S, Taverna M, Watzka M, Bevans CG, Müller CR, Oldenburg J, A new cell culture-based assay quantifies vitamin K 2,3-epoxide reductase complex subunit 1 function and reveals warfarin resistance phenotypes not shown by the dithiothreitol-driven VKOR assay, J. Thromb. Haemost 11 (2013) 872–880. [DOI] [PubMed] [Google Scholar]
- [17].Haque JA, McDonald MG, Kulman JD, Rettie AE, A cellular system for quantitation of vitamin K cycle activity: Structure-activity effects on vitamin K antagonism by warfarin metabolites, Blood. 123 (2014) 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tie J-K, Jin D-Y, Straight DL, Stafford DW, Functional study of the vitamin K cycle in mammalian cells., Blood. 117 (2011) 2967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hodroge A, Longin-Sauvageon C, Fourel I, Benoit E, Lattard V, Biochemical characterization of spontaneous mutants of rat VKORC1 involved in the resistance to antivitamin K anticoagulants., Arch. Biochem. Biophys 515 (2011) 14–20. [DOI] [PubMed] [Google Scholar]
- [20].Bevans CG, Krettler C, Reinhart C, Tran H, Koßmann K, Watzka M, Oldenburg J, Determination of the warfarin inhibition constant Ki for vitamin K 2,3-epoxide reductase complex subunit-1 (VKORC1) using an in vitro DTT-driven assay., Biochim. Biophys. Acta 1830 (2013) 4202–10. [DOI] [PubMed] [Google Scholar]
- [21].Li T, Chang C-YY, Jin D-YY, Lin P-JJ, Khvorova A, Stafford DW, Identification of the gene for vitamin K epoxide reductase, Nature. 427 (2004) 541–544. [DOI] [PubMed] [Google Scholar]
- [22].Chu P-H, Huang T-Y, Williams J, Stafford DW, Purified vitamin K epoxide reductase alone is sufficient for conversion of vitamin K epoxide to vitamin K and vitamin K to vitamin KH2., Proc. Natl. Acad. Sci. U. S. A 103 (2006) 19308–19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rishavy MA, Hallgren KW, Wilson LA, Usubalieva A, Runge KW, Berkner KL, The vitamin K oxidoreductase is a multimer that efficiently reduces vitamin K epoxide to hydroquinone to allow vitamin K-dependent protein carboxylation., J. Biol. Chem 288 (2013) 31556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA, Structure of a bacterial homologue of vitamin K epoxide reductase., Nature. 463 (2010) 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu S, Cheng W, Fowle Grider R, Shen G, Li W, Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer, Nat. Commun 5 (2014) 3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W, Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer, Nat. Struct. Mol. Biol 24 (2017) 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fasco MJ, Principe LM, a Walsh W, a Friedman P, Warfarin inhibition of vitamin K 2,3-epoxide reductase in rat liver microsomes., Biochemistry. 22 (1983) 5655–60. [DOI] [PubMed] [Google Scholar]
- [28].Hattori M, Hibbs RE, Gouaux E, A fluorescence-detection size-exclusion chromatography-based thermostability assay to identify membrane protein expression and crystallization conditions, Structure. 20 (2012) 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kornberg A, Ten commandments: Lessons from the enzymology of DNA replication, J. Bacteriol 182 (2000) 3613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH, Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins, Nat. Methods 7 (2010) 1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Matagrin B, Hodroge A, Montagut-Romans A, Andru J, Fourel I, Besse S, Benoit E, Lattard V, New insights into the catalytic mechanism of vitamin K epoxide reductase (VKORC1) - The catalytic properties of the major mutations of rVKORC1 explain the biological cost associated to mutations., FEBS Open Bio. 3 (2013) 144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ellgaard L, Helenius A, Quality control in the endoplasmic reticulum, Nat. Rev. Mol. Cell Biol 4 (2003) 181–191. [DOI] [PubMed] [Google Scholar]
- [33].a Rishavy M, Usubalieva A, Hallgren KW, Berkner KL, Novel insight into the mechanism of the vitamin K oxidoreductase (VKOR): electron relay through Cys43 and Cys51 reduces VKOR to allow vitamin K reduction and facilitation of vitamin K-dependent protein carboxylation., J. Biol. Chem 286 (2011) 7267–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W, Guomin S, Weidong C, Hao Z, Fengbo Z, Wei H, Qian L, Yihu Y, Bowman GR, Evan SJ, Gross ML, Weikai L, Warfarin prevents blood coagulation by trapping human vitamin K epoxide reductase in an intermediate state during electron transfer, Nat. Struct. Mol. Biol 24 (2016) 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Magnani F, Serrano-Vega MJ, Shibata Y, Abdul-Hussein S, Lebon G, Miller-Gallacher J, Singhal A, Strege A, Thomas JA, Tate CG, A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies, Nat. Protoc 11 (2016) 1554–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Kruse AC, Manglik A, Cho KH, Nurva S, Gether U, Guan L, Loland CJ, Byrne B, Kobilka BK, Gellman SH, A new class of amphiphiles bearing rigid hydrophobic groups for solubilization and stabilization of membrane proteins, Chem. - A Eur. J 18 (2012) 9485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weiß HM, Grisshammer R, Purification and characterization of the human adenosine A2a receptor functionally expressed in Escherichia coli, Eur. J. Biochem 269 (2002) 82–92. [DOI] [PubMed] [Google Scholar]
- [38].Oluwole AO, Danielczak B, Meister A, Babalola JO, Vargas C, Keller S, Solubilization of Membrane Proteins into Functional Lipid-Bilayer Nanodiscs Using a Diisobutylene/Maleic Acid Copolymer, Angew. Chemie - Int. Ed 56 (2017) 1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. FSEC profiles of VKOR and VKORL homologs.
Supplementary Figure 2. Catalytic activity and KM of purified TrVKORL and hVKORL. A, TrVKORL with KO. B, TrVKORL with K. C, hVKORL with KO. D, hVKORL with K. Left, progressive reaction curves. Right, Michaelis-Menten plots.