

Abstract
Although numerous reports conclude that nonsteroidal anti-inflammatory drugs (NSAIDs) have anticancer activity, this common drug class is not recommended for long-term use because of potentially fatal toxicities from cyclooxygenase (COX) inhibition. Studies suggest the mechanism responsible for the anticancer activity of the NSAID sulindac is unrelated to COX inhibition but instead involves an off-target, phosphodiesterase (PDE). Thus, it might be feasible develop safer and more efficacious drugs for cancer indications by targeting PDE5 and PDE10, which are overexpressed in various tumors and essential for cancer cell growth. In this review, we describe the rationale for using the sulindac scaffold to design-out COX inhibitory activity, while improving potency and selectivity to inhibit PDE5 and PDE10 that activate cGMP/PKG signaling to suppress Wnt/β-catenin transcription, cancer cell growth, and tumor immunity.
Keywords: sulindac, phosphodiesterase, cGMP/PKG, β-catenin, immunotherapy
NSAIDs, cGMP/PKG signaling, and cancer cell growth inhibitory activity
NSAIDs commonly used for relief of pain, fever, and inflammation act by inhibiting COX-1 and COX-2 to suppress proinflammatory prostaglandins [1]. NSAIDs also inhibit tumorigenesis in multiple rodent models, while epidemiological studies report reduced incidence of colorectal and other cancers in humans by as much as a 50% reduction in risk [2–4]. Although most epidemiological studies have focused on the cancer-chemopreventive activity of aspirin, prescription-strength NSAIDs, such as sulindac and its congener, indomethacin, have greater potency to inhibit cancer cell growth and offer more potential to treat either precancerous or malignant disease, if not for their toxicity. Sulindac, in particular, is the most active within the family of NSAIDs for the treatment of precancerous conditions. For example, clinical trials have reported that sulindac can reduce the size and number of precancerous colon adenomas by ~60% in patients with familial adenomatous polyposis (FAP), who are at high risk of developing colorectal cancer (CRC) [5]. Indomethacin, which is closely related chemically to sulindac, has been reported to significantly increase survival of patients with malignant disease by ~9 months [6]. Unfortunately, all NSAIDs cause potentially fatal gastrointestinal, renal, and cardiovascular toxicities resulting from COX-1 or COX-2 inhibition and suppression of physiologically important prostaglandins and are not recommended for cancer indications that tend to require long-term administration and high dosages [7,8].
However, the toxicity limitations of NSAIDs might be avoidable given multiple lines of evidence suggesting that a mechanism(s) other than COX inhibition is responsible for their anticancer activity. Thus, it might be feasible to develop safer NSAID derivatives by targeting the underlying off-target mechanism. A drug discovery approach aimed at an unrecognized anticancer target also presents an opportunity to design derivatives that not only lack COX inhibitory activity and have reduced toxicity, but also have the potential for greater anticancer efficacy. Given that NSAIDs are one of the few drugs that have been shown to effectively prevent cancer in humans, this drug discovery approach is especially significant for cancer chemoprevention. This approach is also applicable for developing new drugs for cancer treatment, given the ability of NSAIDs to selectively induce apoptosis of cancer cells, albeit at high concentrations, by a noncytotoxic mode of action.
Although the biological mechanism(s) for the antineoplastic activity of NSAIDs has been elusive and is undoubtedly complex, as reported by numerous investigators, multiple lines of evidence suggests that cGMP PDE inhibition is partially or fully responsible for the cancer cell growth inhibitory activity of sulindac, and possibly other NSAIDs [9]. For example, sulindac sulfide inhibits cancer cell growth (as measured using multiple cancer cell lines) and cGMP PDE enzymatic activity (as measured using recombinant enzymes) with nearly identical IC50 values. Intracellular levels of cGMP are physiologically induced by guanylyl cyclases (GCs) that are ubiquitous and stimulated in response to nitric oxide or natriuretic peptides from the tumor microenvironment (TME). PDE inhibition by sulindac sulfide can amplify the effects of endogenous activators of GC to increase intracellular cGMP levels and activate PKG. An important substrate of PKG appears to be the oncoprotein, β-catenin, for which PKG-mediated phosphorylation can induce ubiquitination and proteasomal degradation of the ‘oncogenic’ (nonphosphorylated, stable) pool β-catenin and suppress Wnt-induced Tcf/Lef transcription of cancer cell proliferation and survival genes, such as those encoding survivin and cyclin D [10–14].
The ability of NSAIDs to suppress Wnt/β-catenin signaling, which is well known to impact tumorigenesis, prognosis, and resistance to chemotherapy, has been previously reported by others, but the mechanism is poorly understood and not widely recognized to result from cGMP PDE inhibition. Activation of cGMP/PKG signaling from cGMP PDE inhibition and suppression of Wnt/β-catenin signaling is mechanistically different from other small-molecule inhibitors of Wnt/β-catenin signaling being developed for cancer indications and that suffer from toxicities associated with disruption of normal stem cell function, for which Wnt/β-catenin-mediated transcription is needed to maintain stemness. As described later, the overexpression of the cGMP PDE isozymes, PDE5 and PDE10, in cancer cells offers the unique opportunity to selectively block the oncogenic pool of β-catenin, thereby averting toxicities associated with nonselective blockage of Wnt signaling in normal stem cells.
NSAIDs other than sulindac might have similar, although less potent, cGMP PDE inhibitory activity, as revealed by a strong positive correlation between their potency (IC50 values) to inhibit cancer cell growth and cGMP PDE activity (as measured using lysates from HT29 colon cancer cells) [10]. Interestingly, these experiments showed no association between inhibition of colon cancer cell growth and COX-2, even though COX-2 inhibition is widely considered to be the sole mechanism responsible for the anticancer activity of NSAIDs.
Although with less potency and PDE isozyme selectivity, the non-COX inhibitory sulfone metabolite also inhibits cGMP PDE at concentrations that inhibit cancer cell growth, and showed promising anticancer activity in multiple rodent models of carcinogenesis relating to CRC, lung, bladder, and breast cancers [15–19]. Given its cancer chemopreventive activity in rodent models of colon tumorigenesis without suppressing prostaglandin levels in colon mucosa [17] and reduced potential for NSAID-like toxicity, as evident from preclinical toxicity testing, sulindac sulfone (exisulind) was evaluated in clinical trials involving patients with FAP. The results from a Phase III clinical trial, which were compromised by unexpected variability in the disease, showed only modest efficacy and unexpected hepatotoxicity, which did not support US Food and Drug Administration (FDA) approval.
Sulindac sulfide tends to show greater selectivity for PDE isozymes that degrade cGMP, such as PDE2, 3, 5, and 10, compared with PDE isozymes that degrade cAMP, such as PDE 4, 7, and 8 [14]. In addition, the activation of cAMP/PKA signaling does not appreciably affect cancer cell growth in contrast to compounds that activate cGMP/PKG signaling and have strong cancer cell growth inhibitory activity. For example, forskolin, an activator of adenylate cyclase, and rolipram, an inhibitor of the cAMP-specific PDE isozyme, PDE4, do not significantly affect the growth of human colon cancer cell lines in vitro [10,11]. By contrast, nitric oxide donors and YC-1, which activate GC, and inhibitors of certain cGMP PDE isozymes do inhibit cancer cell growth with various degrees of potency [10,11].
PDE10 is a particularly interesting anticancer target, given that both mRNA and protein for this PDE isozyme are overexpressed in colon and lung cancer cell lines compared with cells from normal tissues grown in culture under similar conditions [20,21]. PDE10 mRNA and protein were also overexpressed in colon and lung tumors obtained from patients with cancer or mouse tumor models relative to normal colon mucosa or lung. PDE10 showed similar high expression levels in colon precancerous adenomas as well as in metastatic lesions relative to normal colon, which suggests that PDE10A is induced during early stages of tumorigenesis and sustained throughout malignant progression [20].
Prior studies of PDE10 have reported high expression levels in the brain striatum and mostly focused on the role of PDE10 in cognition and motor function [22–25]. PDE10 is encoded by PDE10A in humans and is classified as a cAMP and cAMP-inhibited cGMP PDE isozyme, with a higher Vmax for cGMP compared with cAMP [26]. Unique from all other PDE isozymes, PDE10 has limited expression and no known physiological function outside the central nervous system (CNS) [27–30]. The pharmaceutical industry has developed numerous PDE10 inhibitors for the treatment of schizophrenia and Huntington’s disease, but all appeared to have failed in clinical trials, probably because of lack of efficacy.
Experiments using cultured colon and lung cancer cell lines showed that PDE10 is essential for cancer cell proliferation and survival, as determined by growth suppression from genetic [e.g., small interfering (si)RNA or short hairpin (sh)RNA] knockdown or by conventional PDE10 inhibitors [20,21]. However, concentrations of PDE10 inhibitors were required to inhibit the growth of cancer cell lines grown in culture that were appreciably higher than concentrations that inhibit recombinant PDE10, but matched the concentration that was required to induce cGMP/PKG signaling in cancer cells. This was especially true for Pfizer’s PDE10 inhibitor, Pf2545920, which was in clinical trials for schizophrenia and represents a highly potent and selective PDE10 inhibitor. Nonetheless, Pf2545920 selectively inhibited the growth of colon and lung cancer cells expressing PDE10 compared with normal colonocytes and normal airway epithelial cells, respectively, which expressed low or undetectable levels of PDE10. Conversely, overexpression of PDE10 in normal colonocytes or precancerous adenoma cells resulted in increased proliferation [20]. The growth inhibitory activity resulting from PDE10 knockdown or small-molecule inhibition was also associated with reduced levels of β-catenin and proteins, such as cyclin D and survivin, which are regulated by Tcf/Lef transcription. By contrast, the mitogenic effects resulting from ectopic expression of PDE10 in cells derived from normal colon mucosa or precancerous adenomas were associated with increased levels of β-catenin, as well as cyclin D and survivin, which are regulated by Tcf/Lef transcription.
Inhibition of the cGMP-specific PDE5 isozyme might also have anticancer activity. Similar to PDE10, PDE5 is overexpressed in multiple cancer cell lines, as well as patient-derived tumors of various histopathology [10–14]. Genetic knockdown of PDE5 in colon and breast cancer cells can also suppress growth. Also, similar to PDE10selective inhibitors, selective PDE5 inhibitors require high concentrations to inhibit cancer cell growth in vitro relative to concentrations required to inhibit recombinant PDE5. Nonetheless, PDE5 overexpression in certain cancers, especially CRC, might have therapeutic significance, as suggested by recent studies reporting that the PDE5 inhibitor, sildenafil, can suppress inflammation-induced colon tumorigenesis in rodent models at pharmacologically relevant dosages [31,32] and reduce the risk of developing CRC in humans [33]. Consistent with these reports, others have also shown that the activation of cGMP/PKG signaling can induce apoptosis of colon and renal cancer cells [34,35]. Also consistent are reports that uroguanylin, a peptide activator of GC, can inhibit tumor formation in the Min mouse model of colon tumorigenesis [36]. The discrepancy between the modest in vitro growth inhibitory potency of PDE5 inhibitors and the ability of PDE5 inhibitors to suppress colon tumorigenesis in mice and humans at pharmacologically relevant dosages could be attributed to mechanisms not captured using isolated cancer cells, possibly involving the suppression of tumor immunity, as discussed later. Nonetheless, PDE5 appears to be a valid anticancer target, at least in relation to CRC, for which there is strong evidence of efficacy with PDE5-selective inhibitors [37].
Given that both PDE5 and PDE10 function to degrade cGMP, it is not too surprising that there might be anticancer efficacy advantages from inhibiting both PDE5 and PDE10 simultaneously, especially because both PDE isozymes appear to be co-expressed, as previously reported in colon and lung cancer cells [20,21]. In support of this possibility, dual genetic silencing of PDE5 and PDE10 and combination treatment with PDE5 and PDE10 inhibitors was reported to additively inhibit colon cancer cell growth and β-catenin transcriptional activity [38]. The co-expression of PDE5 and PDE10 in cancer cells could also explain why highly specific inhibitors of PDE5 or PDE10 as a single agent have relatively modest potency to inhibit cancer cell growth in vitro relative to their ultra-high potency to inhibit enzymatic activity in cell-free assays using recombinant PDE isozymes, as discussed earlier. This might also explain why high dosages of sildenafil were required to inhibit in vivo growth of colon cancer cells known to express PDE10 in a subcutaneous mouse tumor model [39]. In this case, PDE10 overexpression in cancer cells could override the inhibitory effect of a PDE5-specific inhibitor by rapidly degrading any cGMP that arises above basal levels, resulting in the need for high dosages that act in a nonselective manner.
Immune-potentiating effects of NSAIDs
In addition to the direct effects of NSAIDs on cancer cell growth, their ability to activate cGMP/PKG signaling and block Wnt/β-catenin transcription can elicit effects on immune cells to suppress mechanisms by which cancer cells can evade immune surveillance. Although this relationship is less understood and requires further study, the immune-potentiating effects of NSAIDs are being increasingly recognized [40]. For example, aspirin and celecoxib have been reported to enhance the antitumor activity of programmed cell death protein-1 (PD-1) inhibitors in mouse tumor models [41], although neither were effective as a single agent. The investigators attributed the enhancement of antitumor activity to the reduction of prostaglandin E2 (PGE2), an immunosuppressive factor that can inhibit immune cell activation [41–43]. The COX-2/PGE2 axis has also been established as a driver to expand myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) in tumors [44–46]. COX-2/PGE2 can also regulate programmed cell death ligand-1 (PD-L1) expression in tumor-associated macrophages and MDSCs. Targeting the COX-2/PGE2 axis to restore immune surveillance using NSAIDs has shown encouraging results in preclinical models. For example, sulindac effectively inhibited the influx of M2 macrophages in a breast cancer model by reducing factors related to tumor angiogenesis, inflammation, and immunosuppression (e.g., IL-1β, TGFβ, versican, and Arg-1), thus relieving the immune suppression of cytotoxic lymphocytes [47]. In addition, a recent study demonstrated that genetic ablation of COX and inhibition of COX activity by aspirin enhanced the efficacy of PD-1 blockade in multiple mouse tumor models [41], providing a rationale for clinical testing of the combination of NSAIDs with cancer immunotherapies.
Although targeting COX-2/PGE2 can improve the immunogenicity of the TME, this effect is limited by potentially severe or fatal gastrointestinal, renal, and cardiovascular adverse effects associated with COX inhibition resulting from long-term use of NSAIDs at the high dosages that appear to be required for anticancer activity. In addition, the modest anticancer activity of known NSAIDs, such as aspirin and celecoxib, as a monotherapy limits their potential for development in the field of oncology. By contrast, the development of non-COX inhibitory sulindac derivatives that inhibit PDE5 and PDE10 to suppress cancer cell growth and tumor immunity offers the opportunity to not only enhance host antitumor immunity with reduced adverse effects, but also kill cancer cells directly.
The role of cGMP signaling in immune regulation is supported by reports from Meyer and colleagues showing the enhancement of antitumor immunity by the PDE5 inhibitor, Viagra® (sildenafil) [48]. Others have reported that PDE5 inhibition from Cialis® (tadalafil) can enhance antitumor immune responses by reducing the generation and function of tumor-promoting MDSCs [49–52]. Additionally, Cialis® increased the numbers and activation of tumor-infiltrating CD8+ and CD4+ lymphocytes in patients with melanoma or head and neck squamous cell carcinoma [52,53].
The ability of PDE10 inhibitors to suppress β-catenin activity in tumor cells [13,14,19,20] also has the potential to elicit productive antitumor immune responses, given that inhibition of oncogenic β-catenin can activate dendritic cells and promote T cell tumor infiltration [54,55]. There is increasing evidence that patients lacking sufficient T cell tumor infiltration respond poorly to immunotherapy [56–59]. These studies also suggest that the Wnt/β-catenin pathway has a crucial role in determining the immunogenicity of the TME and mediating immune exclusion. Thus, targeting PDE5 and PDE10 to suppress β-catenin could enhance the response to immunotherapeutics by affecting the TME, as previously reviewed, and illustrated in Figure 1 [60–64].
Figure 1.
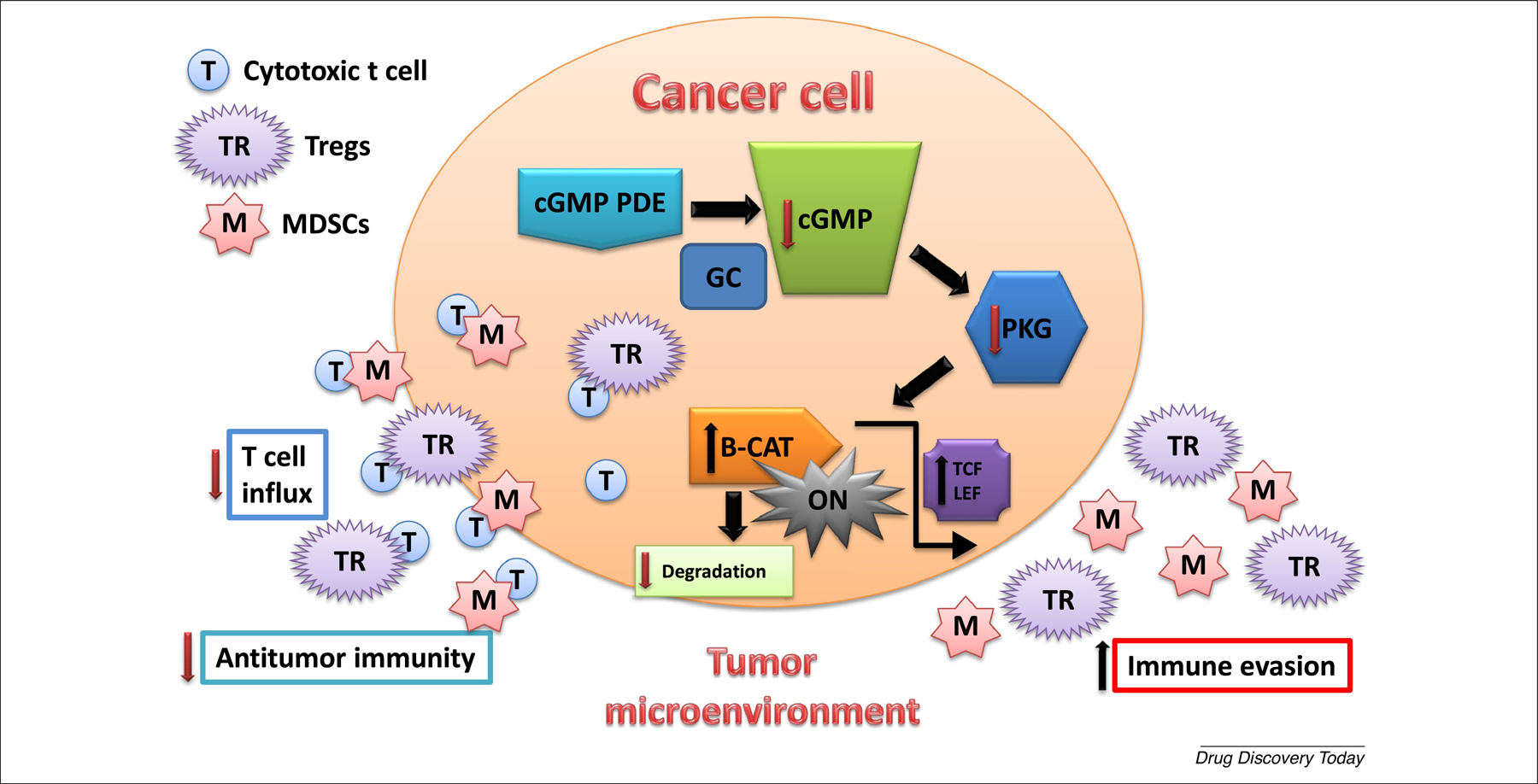
Phosphodiesterase 5 (PDE5) and PDE10 activity increase cancer cell immune evasion. PDE5 and PDE10 are co-expressed in cancer cells and function to maintain low intracellular cGMP levels that suppress PKG phosphorylation of β-catenin (B-CAT), which allows B-CAT to activate Tcf/Lef-mediated transcription as result of Wnt stimulation or mutations in B-CAT or upstream regulators (e.g., APC). Genes transcribed by Tcf/Lef encode proteins essential for proliferation and survival of cancer cells (e.g., cyclin D, survivin, and Myc). Activated B-CAT can also increase in cancer cell immune evasion by decreasing cytotoxic T cell infiltration to the tumor site resulting in the increase in the immunosuppressive activity of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) within the tumor microenvironment. cGMP is physiologically induced by guanylyl cyclases (GC), and regulated by cGMP PDE isozymes.
The suppression of β-catenin by NSAIDs and derivatives, as described earlier and reviewed previously [38], can also influence T cell differentiation and functional development [65]. For example, previous studies reported that the Wnt/β-catenin pathway promotes CD8+ T cell survival and memory formation while inhibiting terminal effector differentiation [66,67]. The role of β-catenin in CD4+ T cell differentiation appears to be more complex. One study showed that β-catenin signaling drives the development of inflammatory Th17 and Treg cells that promote colitis and CRC [61]. Majchrzak et al. reported that transient inhibition of β-catenin and PI3Kδ leads to expansion of Th17 cells, which later regain β-catenin and exhibit heightened stemness and antitumor activity [68].
Apart from activation of cGMP signaling or suppression of Wnt/β-catenin pathway, several studies suggest that various NSAIDs act as prooxidants to exhibit antineoplastic properties by inducing direct tumor cell apoptosis and cell cycle arrest via a mechanism involving induction of reactive oxidative species (ROS) [69–72]. In addition, many NSAIDs can induce the expression of death receptor 5 (DR5) in cancer cells through induction of ROS. DR5 is one of the cell surface receptors that binds to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to induce apoptosis. Several studies imply that certain NSAIDs, such as ibuprofen [71,73], sulindac [74], and indomethacin [75], can sensitize TRAIL-resistant cancer cells to TRAIL-induced apoptosis through ROS-mediated upregulation of DR5 [76]. Given that activated T cells express TRAIL [77–79], combining DR5-inducing sulindac derivatives with T cell-based cancer immunotherapy, such as immune checkpoint blockade and adoptive T cell therapy, represents a promising strategy for cancer treatment. Although NSAIDs can exhibit immune-potentiating effects that are largely attributed to reduction of the immunosuppressive PGE2 from COX inhibition [40–43], evidence as described above that their anticancer activity is attributed to mechanisms other than COX inhibition, suggests that the effects of NSAIDs on the TME also involve a COX-independent mechanism of action.
Novel non-COX-inhibitory sulindac derivatives that inhibit PDE5 and PDE10
A variety of non-COX inhibitory sulindac derivatives were synthesized and a lead compound, exisulind, was developed by Cell Pathways Inc. during the 1990s that inhibited colon cancer cell growth in vitro with corresponding potency to inhibit cGMP PDE activity and promising anticancer activity in preclinical tumor models [15–19,80]. As mentioned earlier, exisulind failed in clinical trials involving patients with FAP, as well as the more potent derivative, CP461, which was later developed by OSI Pharmaceuticals Inc [80]. These failures ultimately led to the abandonment of exisulind, CP461 (aka OSI 461) and related analogs for the treatment of precancerous conditions, as well as cancer indications where there was preclinical evidence of benefits if combined with chemotherapy [81].
Given that PDE10 was only recently been recognized as an anticancer target, with the first publication appearing in 2015 linking PDE10 with cancer [20], the development of sulindac derivatives that are optimized to selectively inhibit PDE10 or have dual PDE5 and PDE10 inhibitory activity represents a new unexplored approach for the treatment of precancerous or malignant disease. With the goal of identifying novel sulindac derivatives with greater potency and selectivity to inhibit cancer cell growth, a chemically diverse collection of ~1500 compounds sharing the indene scaffold of sulindac was synthesized and screened for PDE5 or PDE10 inhibitory activity using recombinant isozymes. As recently reported, ADT-094 represents a prototypic dual inhibitor of PDE5 and PDE10, which shows high potency to inhibit colon cancer cell growth in vitro [38]. Similar to sulindac sulfide, the mechanism resulting from PDE5/10 inhibition by ADT-094 involves the activation of cGMP/PKG signaling to phosphorylate and induce ubiquitination and proteasomal degradation of β-catenin, thereby blocking Wnt-induced β-catenin/Tcf-mediated transcriptional activity. ADT-094 is appreciably a more potent and selective inhibitor of cancer cell growth compared with conventional PDE5 and PDE10 inhibitors, possibly, as discussed earlier, because of its ability to inhibit simultaneously both cGMP-degrading isozymes, which minimizes the potential for compensation (e.g., resistance) from an unaffected PDE isozyme by using an isozyme-specific inhibitor. Other sulindac derivatives that were optimized for PDE10 with attractive pharmacological properties show promising anticancer activity in multiple extremely aggressive mouse tumor models of CRC, breast, and lung cancers by oral delivery without discernable toxicity.
Anticancer compounds derived from the sulindac indene scaffold might have additional advantages over conventional PDE5 or PDE10 inhibitors beyond efficacy. For example, conventional PDE5 inhibitors (e.g., sildenafil and tadalafil) have been reported to promote melanoma in mice [82,83] and humans [84], although this potential adverse effect requires further investigation. By contrast, sedation appears to be a major adverse effect of PDE10 inhibitors developed for CNS conditions and that readily cross the blood–brain barrier. All conventional PDE10 inhibitors also appear to be rapidly metabolized when in peripheral tissues, which is unsuitable for cancer therapy, which requires systemic exposure. Sulindac derivatives clearly inhibit the growth of melanoma cells and do not cause sedation.
Concluding remarks
In conclusion, novel sulindac derivatives lacking COX inhibitory activity that act as either PDE10 selective or dual PDE5/PDE10 inhibitors have the potential to treat or prevent cancer as a monotherapy and enhance or broaden the anticancer activity of immunotherapy. However, additional research is warranted to determine how cGMP/PKG signaling can suppress tumor immunity and whether the mechanisms by which novel sulindac derivatives inhibit the Wnt/β-catenin pathway and induce apoptosis of cancer cells are the same as the mechanisms that is responsible for suppressing tumor immunity.
Figure 2.
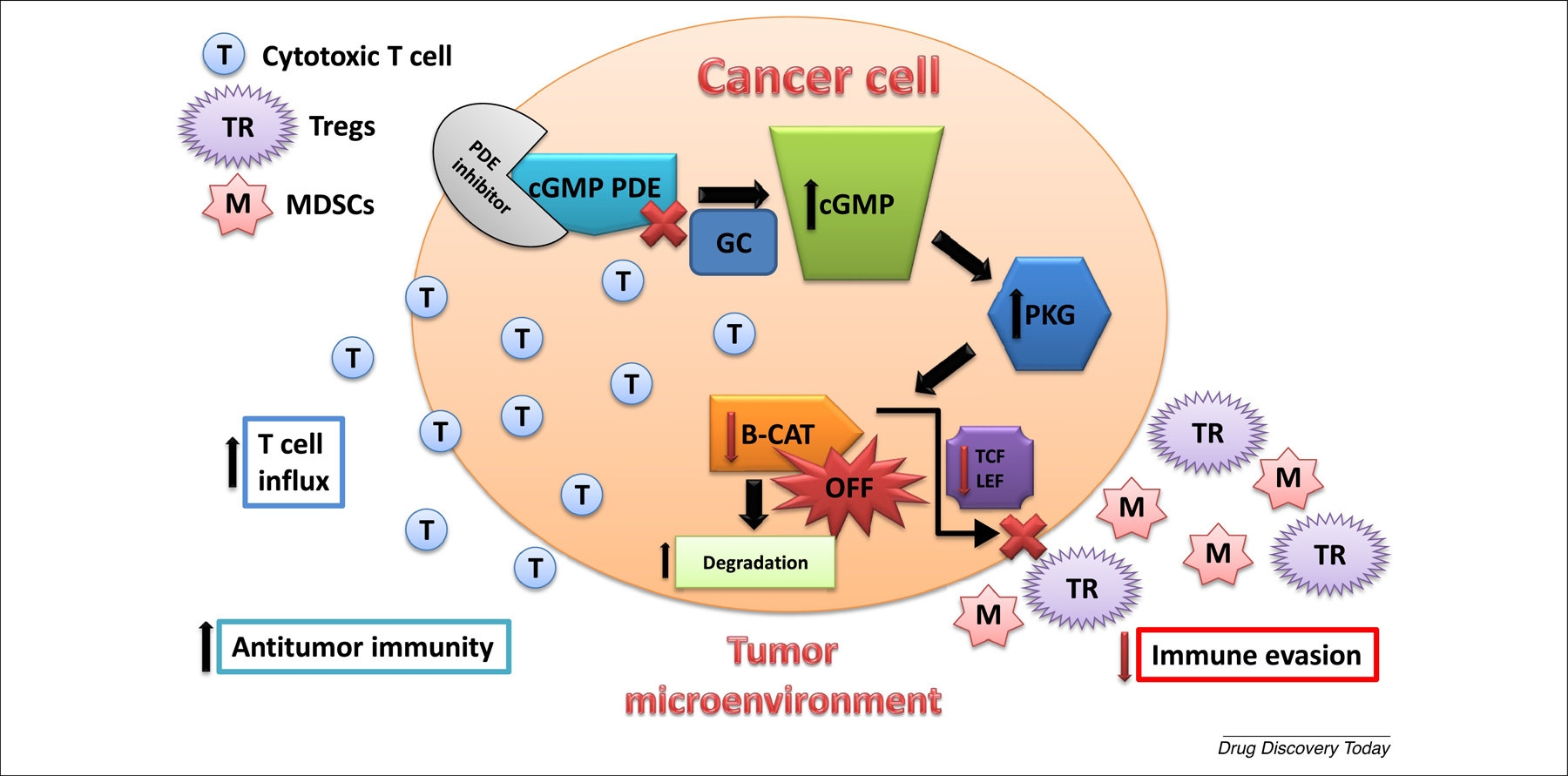
Phosphodiesterase 5 (PDE5) and PDE10 inhibition suppress tumor immunity. PDE5 and PDE10 inhibition increase intracellular cGMP levels to activate PKG that phosphorylates β-catenin (B-CAT) to induce ubiquitination and proteasomal degradation of B-CAT, thereby suppressing Tcf/Lef-mediated gene transcription that results in direct killing of cancer cells as well as greater cytotoxic T cell infiltration to the tumor site by decreasing the immunosuppressive activity of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), therefore enhancing the immunogenic milieu within the tumor microenvironment. cGMP is physiologically induced by guanylyl cyclases (GC) and acts in concert with cGMP PDE inhibitors to increase intracellular cGMP levels.
Research Highlights.
NSAIDs have anticancer activity but have toxicities resulting from COX inhibition.
The mechanism of NSAID anticancer activity may not require COX inhibition.
PDE5 and PDE10 are overexpressed in tumors and essential for cancer cell growth.
Novel sulindac derivatives targeting PDE5 or PDE10 have strong anticancer activity.
Acknowledgment
This publication was supported, in part, by the National Cancer Institute of the National Institutes of Health under awards R01CA155638, R01CA197147, and R01CA131378.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
G.A.P., A.B.K., X.C., and M.B. are co-founders of ADT Pharmaceuticals Inc.
References
- 1.Brune K and Patrignani P New insights into the use of currently available non-steroidal anti–inflammatory drugs. J. pain research 8 (2015) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jänne PA and Mayer RJ Chemoprevention of colorectal cancer. New England J. Medicine 342, (2000) 1960–1968 [DOI] [PubMed] [Google Scholar]
- 3.Rao CV and Reddy SB NSAIDs and chemoprevention. Current cancer drug targets 4, (2004) 29–42 [DOI] [PubMed] [Google Scholar]
- 4.Keller J and Giardello F Chemopreventive strategies using NSAIDs and COX-2 inhibitors. Cancer biology & therapy 2, (2003) (Suppl.) 139–148 [PubMed] [Google Scholar]
- 5.Giardiello FM et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. New England J. Medicine 328, (1993) 1313–1316 [DOI] [PubMed] [Google Scholar]
- 6.Lundholm K et al. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. Cancer research 54, (1994) 5602–5606 [PubMed] [Google Scholar]
- 7.Wolfe MM et al. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. New England J. Medicine 340, (1999) 1888–1899 [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee D et al. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 286, (2001) 954–959 [DOI] [PubMed] [Google Scholar]
- 9.Gurpinar E et al. NSAIDs inhibit tumorigenesis, but how? Clinical cancer research 20, (2014) 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tinsley HN et al. Colon tumor cell growth–inhibitory activity of sulindac sulfide and other nonsteroidal anti-inflammatory drugs is associated with phosphodiesterase 5 inhibition. Cancer Prevention Research 3, (2010) 1303–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tinsley HN et al. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Molecular cancer therapeutics 8, (2009) 3331–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tinsley HN et al. Inhibition of PDE5 by Sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin-mediated transcription in human breast tumor cells. Cancer prevention research 4, (2011) 1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li N et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Molecular cancer therapeutics 12, (2013) 1848–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitt JD et al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and β-catenin transcriptional activity. Cancer prevention research 5, (2012) 822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piazza GA et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer research 61, (2001) 3961–3968 [PubMed] [Google Scholar]
- 16.Malkinson AM et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung tumor formation by FGN-1 (sulindac sulfone). Carcinogenesis 19, (1998) 1353–1356 [DOI] [PubMed] [Google Scholar]
- 17.Piazza GA et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer research 57, (1997) 2909–2915 [PubMed] [Google Scholar]
- 18.Thompson HJ et al. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer research 57, (1997) 267–271 [PubMed] [Google Scholar]
- 19.Thompson WJ et al. Exisulind induction of apoptosis involves guanosine 3′, 5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated β-catenin. Cancer research 60, (2000) 3338–3342 [PubMed] [Google Scholar]
- 20.Li N et al. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and β-catenin-dependent TCF transcriptional activity. Oncogene 34, (2015) 1499–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu B et al. Phosphodiesterase 10A is overexpressed in lung tumor cells and inhibitors selectively suppress growth by blocking β–catenin and MAPK signaling. Oncotarget 8, (2017) 69264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie Z et al. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience 139, (2006) 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bollen E and Prickaerts J Phosphodiesterases in neurodegenerative disorders. IUBMB life 64, (2012) 965–970 [DOI] [PubMed] [Google Scholar]
- 24.Siuciak JA The role of phosphodiesterases in schizophrenia. CNS drugs 22, (2008) 983–993 [DOI] [PubMed] [Google Scholar]
- 25.Saavedra A et al. Regulation of hippocampal cGMP levels as a candidate to treat cognitive deficits in Huntington’s disease. PLoS ONE 8, (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujishige K et al. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biological Chemistry 274, (1999) 18438–18445 [DOI] [PubMed] [Google Scholar]
- 27.Coskran TM et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J. Histochemistry Cytochemistry 54, (2006) 1205–1213 [DOI] [PubMed] [Google Scholar]
- 28.Soderling SH et al. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proceedings of the National Academy of Sciences 96, (1999) 7071–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loughney K et al. Isolation and characterization of PDE10A, a novel human 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 234, (1999) 109–117 [DOI] [PubMed] [Google Scholar]
- 30.Seeger TF et al. Immunohistochemical localization of PDE10A in the rat brain. Brain research 985, (2003) 113–126 [DOI] [PubMed] [Google Scholar]
- 31.Lin S et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. American J. cancer research 7, (2017) 41. [PMC free article] [PubMed] [Google Scholar]
- 32.Islam BN et al. Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prevention Research 10, (2017) 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W et al. Use of phosphodiesterase 5 inhibitors is associated with lower risk of colorectal cancer in men with benign colorectal neoplasms. Gastroenterology 157, (2019) 672–681 [DOI] [PubMed] [Google Scholar]
- 34.Deguchi A et al. Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer research 64, (2004) 3966–3973 [DOI] [PubMed] [Google Scholar]
- 35.Ren Y et al. Essential role of the cGMP/PKG signaling pathway in regulating the proliferation and survival of human renal carcinoma cells. International J. molecular medicine 34, (2014) 1430–1438 [DOI] [PubMed] [Google Scholar]
- 36.Shailubhai K et al. Uroguanylin treatment suppresses polyp formation in the ApcMin/+ mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer research 60, (2000) 5151–5157 [PubMed] [Google Scholar]
- 37.Piazza GA Validation of PDE5 as a chemoprevention target. Cancer prevention research 10, (2017) 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li N et al. Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget 6, no. 29 (2015) 27403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mei X-L et al. Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. American J. cancer research 5, (2015) 3311. [PMC free article] [PubMed] [Google Scholar]
- 40.Hussain M et al. Non-steroidal anti-inflammatory drugs, tumour immunity and immunotherapy. Pharmacological research 66, (2012) 7–18 [DOI] [PubMed] [Google Scholar]
- 41.Zelenay S et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, (2015) 1257–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalinski P Regulation of immune responses by prostaglandin E2. J. Immunology 188, (2012) 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakanishi M and Rosenberg DW Multifaceted roles of PGE2 in inflammation and cancer. Seminars immunopathology 35, 123–137 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez PC et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. experimental medicine 202, (2005) 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinha P et al. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer research 67, (2007) 4507–4513 [DOI] [PubMed] [Google Scholar]
- 46.Wang Di. and DuBois RN Eicosanoids and cancer. Nature Reviews Cancer 10, (2010) 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin T et al. Sulindac, a non-steroidal anti-inflammatory drug, mediates breast cancer inhibition as an immune modulator. Scientific reports 6 (2016) 19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer C et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proceedings of the National Academy of Sciences 108, no. 41 (2011) 17111–17116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serafini P et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. experimental medicine 203, (2006) 2691–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noonan KA et al. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer immunology research 2, (2014) 725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weed DT et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clinical Cancer Research 21, (2015) 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Califano JA et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clinical Cancer Research 21, (2015) 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hassel JC et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 6, (2017) e1326440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spranger S et al. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, (2015) 231–235 [DOI] [PubMed] [Google Scholar]
- 55.Spranger S et al. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer cell 31, (2017) 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trujillo JA et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. Journal immunotherapy of cancer 7, (2019) 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luke JJ et al. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clinical Cancer Research 25, (2019) 3074–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galluzzi L et al. WNT signaling in cancer immunosurveillance. Trends cell biology 29, (2019) 44–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teng MWL et al. Classifying cancers based on T-cell infiltration and PD-L1. Cancer research 75, (2015) 2139–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pai SG et al. Wnt/beta–catenin pathway: modulating anticancer immune response. J. hematology oncology 10, (2017) 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keerthivasan S et al. β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Science translational medicine 6, (2014) 225ra28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang B et al. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends pharmacological sciences 39, (2018) 648–658 [DOI] [PubMed] [Google Scholar]
- 63.Li X et al. WNT/β-catenin signaling pathway regulating T cell-inflammation in tumor microenvironment. Frontiers Immunology 10 (2019) 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin-Orozco E et al. WNT signaling in tumors: the way to evade drugs and immunity. Frontiers Immunology 10 (2019), 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gattinoni L et al. Wnt/β-catenin signaling in T-cell immunity and cancer immunotherapy. Clinical Cancer Research 16, no. 19 (2010) 4695–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao D-M et al. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunology 184, (2010) 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nature medicine 17, (2011) 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Majchrzak K et al. β-catenin and PI3Kδ inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI insight 2, e90547 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ralph SJ et al. Hitting the bull’s-eye in metastatic cancers—NSAIDs elevate ROS in mitochondria, inducing malignant cell death. Pharmaceuticals 8, (2015) 62–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adachi M et al. Nonsteroidal anti-inflammatory drugs and oxidative stress in cancer cells. Histology and histopathology (2007) 22, 437–442 [DOI] [PubMed] [Google Scholar]
- 71.Raza H et al. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. European J. pharmacology 668, (2011) 15–24 [DOI] [PubMed] [Google Scholar]
- 72.Ralph S et al. NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. J Cancer Metastasis Treat 4, (2018) 1–26 [Google Scholar]
- 73.Todo M et al. Ibuprofen enhances TRAIL-induced apoptosis through DR5 upregulation. Oncology reports 30, (2013) 2379–2384 [DOI] [PubMed] [Google Scholar]
- 74.Leibowitz B et al. BID mediates selective killing of APC-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proceedings of the National Academy of Sciences 111, (2014) 16520–16525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tse AK-W et al. Indomethacin sensitizes TRAIL-resistant melanoma cells to TRAIL-induced apoptosis through ROS-mediated upregulation of death receptor 5 and downregulation of survivin. J. Investigative Dermatology 134, (2014) 1397–1407 [DOI] [PubMed] [Google Scholar]
- 76.Huang Y et al. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer research 61, (2001) 6918–6924 [PubMed] [Google Scholar]
- 77.Falschlehner C et al. Following TRAIL’s path in the immune system. Immunology 127, (2009) 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kayagaki N et al. Involvement of TNF-related apoptosis-inducing ligand in human CD4+ T cell-mediated cytotoxicity. J. Immunology 162, (1999) 2639–2647 [PubMed] [Google Scholar]
- 79.Mirandola P et al. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 104, (2004) 2418–2424 [DOI] [PubMed] [Google Scholar]
- 80.Sun W et al. Phase I and pharmacokinetic trial of the proapoptotic sulindac analog CP-461 in patients with advanced cancer. Clinical cancer research 8, (2002) 3100–3104 [PubMed] [Google Scholar]
- 81.Haanen C Sulindac and its derivatives: a novel class of anticancer agents. Current opinion investigational drugs 2, (2001) 677–683 [PubMed] [Google Scholar]
- 82.Arozarena I et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer cell 19, (2011) 45–57 [DOI] [PubMed] [Google Scholar]
- 83.Dhayade S et al. Sildenafil potentiates a cGMP-dependent pathway to promote melanoma growth. Cell reports 14, (2016) 2599–2610 [DOI] [PubMed] [Google Scholar]
- 84.Li W-Q et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA internal medicine 174, (2014) 964–970 [DOI] [PMC free article] [PubMed] [Google Scholar]