

Abstract
Background:
Increased fatty acid oxidation (FAO) has long been considered a culprit in the development of obesity/diabetes induced cardiomyopathy. However, enhancing cardiac FAO by removing the inhibitory mechanism of long-chain fatty acids transport into mitochondria via deletion of acetyl-CoA carboxylase 2 (ACC2) does not cause cardiomyopathy in non-obese mice, suggesting that high FAO is distinct from cardiac lipotoxicity. We hypothesize that cardiac pathology associated obesity is attributable to the imbalance of fatty acid supply and oxidation. Thus, we here seek to determine whether further increasing FAO by inducing ACC2 deletion prevents obesity induced cardiomyopathy, and if so, to elucidate the underlying mechanisms.
Methods:
We induced high FAO in adult mouse hearts by cardiac-specific deletion of ACC2 using a tamoxifen inducible model (ACC2 iKO). Control (Con) and ACC2 iKO mice were subjected to high fat diet (HFD) feeding for 24 weeks to induce obesity. Cardiac function, mitochondria function and mitophagy activity were examined.
Results:
Despite both Con and ACC2 iKO mice exhibiting similar obese phenotype, increasing FAO oxidation by deletion of ACC2 prevented HFD induced cardiac dysfunction, pathological remodeling as well as mitochondria dysfunction. Similarly, increasing FAO by knock down of ACC2 prevented palmitate induced mitochondria dysfunction and cardiomyocyte death in vitro. Furthermore, HFD suppressed mitophagy activity and caused damaged mitochondria to accumulate in the heart, which was partially attenuated in ACC2 iKO heart. Mechanistically, ACC2 iKO prevented HFD induced downregulation of parkin. During stimulation for mitophagy, mitochondria localized parkin was severely reduced in Con HFD-fed mouse heart, which was partially restored in ACC2 iKO HFD-fed mice.
Conclusions:
These data show that increasing cardiac FAO alone does not cause cardiac dysfunction but protect against cardiomyopathy in chronically obese mice. The beneficial effect of enhancing cardiac FAO in HFD induced obesity is mediated, in part, by maintenance of mitochondria function through regulating parkin mediated mitophagy. Our findings also suggest that targeting the parkin dependent mitophagy pathway could be an effective strategy against the development of obesity induced cardiomyopathy.
Keywords: Fatty acid oxidation, Obesity, Cardiomyopathy, Mitophagy
Introduction
In the setting of obesity and/or diabetes, cardiac substrate metabolism shifts toward increased fatty acid oxidation (FAO) while lipid accumulates in the heart.1, 2 Cardiac dysfunction developed under these conditions has thus been linked to the abnormal lipid metabolism, referred to as lipotoxic cardiomyopathy.3 The molecular mechanisms for lipotoxic cardiomyopathy appear to be complex; a number of pathogenic pathways have been proposed, e.g. mitochondrial dysfunction and oxidative stress, endoplasmic reticulum (ER) stress, toxic lipid induced apoptosis etc. However, the specific link between cardiac metabolism and the proposed cause of cardiomyopathy remains elusive. There are no specific therapies available for the condition.
A number of studies have shown that the strategies for reducing excess fatty acid supply or promoting fatty acid storage in neutral lipids can alleviate lipotoxic phenotypes and be beneficial for cardiac function,4–6 suggesting that excessive fatty acid utilization could be the culprit. Consistent with the notion, increased cardiac FAO has long been considered to cause elevated reactive oxygen species (ROS) production in the mitochondria and subsequent oxidative damage and mitochondrial dysfunction, which contribute to cardiac dysfunction in obese models.7–9 However, our previous study has demonstrated that increasing FAO by deletion of Acetyl-CoA Carboxylase 2 (ACC2) in the heart, did not cause any mitochondrial or cardiac dysfunction in non-obese mice.10 On the other hand, intervention that prevents the upregulation of FAO in obesity causes lipid accumulation and cardiac dysfunction.11 Taken together, these observations suggest that cardiac lipotoxicity is not attributable to increased FAO per se but an imbalance of fatty acid supply, storage and utilization.1 It would therefore be very interesting to determine whether directing fatty acids into the mitochondria for β-oxidation, via ACC2 deletion, prevents the development of cardiac dysfunction under obese condition.
Accumulation of damaged mitochondria has been observed in cardiomyopathy associated with obesity and diabetes.12 Clearance of damaged mitochondria through the autophagy pathway (referred to as mitophagy) plays an essential role in maintaining mitochondria homeostasis. Previous studies have shown that autophagy and mitophagy are compromised in hearts of obesity and/or diabetes,13–15 but whether the change is a cause or consequence of lipotoxic cardiomyopathy is unclear. Chronologically, altered lipid metabolism develops before mitochondrial function deteriorates during the development of obesity.16, 17 Furthermore, suppression of mitophagy preceded the impairment of mitochondrial or cardiac function, and activation of mitophagy prevents myocardial damages in acute and chronic stress conditions.18, 19 These observations collectively lead us to hypothesize that excess and toxic lipid accumulation may contribute to mitochondrial dysfunction through suppression of mitophagy. Therefore, we sought to test whether correcting the mismatch between fatty acid supply and oxidation by enhancing mitochondrial FAO will be able to prevent mitochondrial dysfunction through normalizing mitophagy activity.
Using a diet induced obesity (DIO) model in the present study, we demonstrate that enhancing FAO through deletion of ACC2 is sufficient to prevent obesity induced cardiomyopathy. The cardioprotective effect of increasing FAO in obese mice is through maintenance of parkin mediated mitophagy and thus preventing mitochondrial dysfunction. These findings indicate that impaired mitophagy contributes to mitochondrial dysfunction in obese mice, and that targeting the parkin dependent mitophagy pathway is a viable therapeutic intervention for obesity induced cardiomyopathy.
Methods
An expanded methods section is provided online as supplemental materials. The data that support the findings of this study are available from the corresponding author upon reasonable request. The authors declare that all supporting data are available within the article.
Animal Model
ACC2 flox/flox-MerCreMer+ (ACC2−f/f−MCM+) mice were mated with ACC2f/f to produce both study mice and control littermates on the C57BL/6 background carrying homozygous nicotinamide nucleotide transhydrogenase (Nnt) mutations.20 The alpha myosin heavy chain-MerCreMer (αMHC-MCM) mice is applied as separate control for ACC2f/f−MCM+ mice. A transgenic mouse expressing mitochondria targeted Keima (designated as mt-Keima) was generated on an FVB background as described previously21. Mt-Keima was crossed with ACC2−f/f−MCM+ four times to generate ACC2f/f, mt-Keima and ACC2f/f−MCM+, mt-Keima bigenic mice on a mixed background carrying homozygous Nnt mutations. At 8 weeks of age, all mice received an intraperitoneal injection of tamoxifen (20mg/kg) for 5 days, which was sufficient to induce ACC2 deletion in ACC2f/f−MCM+. Four weeks after the last injection of tamoxifen, the alpha myosin heavy chain-MerCreMer (designated as αMHC-MCM), ACC2f/f (designated as Con), ACC2f/f−MCM+ (designated as iKO), ACC2f/f, mt-Keima (designated as Con-mt-Keima) and ACC2f/f−MCM+, mt-Keima (designated as iKO-mt-Keima) mice were subjected to high fat diet (Research Diets, D12492) feeding for 24 weeks. All mice were housed at 22°C with a 12-hour light, 12-hour dark cycle with free access to water and standard chow or high fat diet (HFD). Experiments included in this study were performed with male mice. All protocols concerning animal use were approved by the Institutional Animal Care and Use Committee at the University of Washington.
Isolated Heart Perfusion Experiments and Nuclear Magnetic Resonance (NMR) Spectroscopy
Langendorff, isolated mouse heart experiments were conducted as previously described.10, 20 Changes in cardiac high-energy phosphate content were monitored by 31P nuclear magnetic resonance (NMR) spectroscopy. Hearts were perfused with 13C-labeled substrates (1,6-13C glucose and U-13C fatty acids) to determine substrate oxidation. 13C NMR spectroscopy was performed on lyophilized heart extracts. The contribution of each labeled substrate and the total unlabeled substrates were determined by modeling the tricarboxylic acid (TCA) cycle metabolism using peak areas of C3 and C4 13C isotopomers of glutamate as previously reported.10 (Online supplemental materials for expanded methods)
Mouse Heart Mitochondria Isolation and Measurement of Mitochondria Respiration
Mitochondria were isolated by differential centrifugation using ice-cold mitochondria isolation buffer (MIB; 70 mM Sucrose, 200 mM D-Mannitol, 5mM MOPS, 2mM Taurine, 1.6 mM Carnitine Hydrochloride, 1 mM EDTA, 0.025% BSA). Mitochondrial respiration was assessed by oxygen consumption rates (OCRs) under specific conditions using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). (Online supplemental materials for expanded methods)
Assessing Cardiac Mitophagy using the mt-Keima Transgenic Mice
Cardiomyocytes isolated from mice harboring mt-Keima transgene were plated on a glass slide and analyzed using a Leica TCS SP8 confocal laser scanning microscope. Fluorescence of mt-Keima was imaged in two channels via two sequential excitations (458 nm, green; 561 nm, red) and using a 570 to 695-nm emission range. High (561/458) ratio areas were segmented and quantified with the Analyze Particles plugin in ImageJ. The parameter (high [561/458] ratio area/total cell area) was used as an index of mitophagy, as described.22 (Online supplemental materials for expanded methods)
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed with a one-way analysis of variance (ANOVA) or two-way ANOVA with a Tukey test for multiple comparisons. Two group comparisons were made using Student’s t-test. All analyses were performed using Prism 7.0 (GraphPad). The value of p<0.05 was considered to be significant.
Results
Deletion of ACC2 Increased Fatty Acid Oxidation but Maintained Cardiac Energetics in Obese Mice
As we previously reported, deletion of ACC2 in adult mouse heart caused a significant increase in the contribution of fatty acids to oxidative metabolism with no adverse effects.20 To determine the consequence of ACC2 deletion in the heart of obese mice, both Con and ACC2 iKO mice were subjected to chow or high fat diet (HFD) feeding for 24 weeks. From here, we will use Con-chow, iKO-chow, Con-HFD and iKO-HFD to represent the four groups of mice in this study. Compared with chow-fed mice, both Con-HFD and iKO-HFD mice developed similar obese phenotype, evidenced by comparable body weight gain, accumulation of fat content, increased liver triglyceride (TG) content and higher fasting blood glucose level (Figure IA–IC, IE–IF in the Supplement). There is no difference in serum TG concentration in all the groups (Figure ID in the Supplement). Immunoblot results revealed a near complete deletion of ACC2 protein in the heart. Up to 6-months post tamoxifen (TAM) injection, ACC2 protein level was barely detected in iKO mice compared with Con mice under both chow- and HFD-fed conditions (Figure 1A). Chronic HFD treatment did not alter the expression of total and phosphorylated ACC2 in Con hearts (Figure 1A).
Figure 1. Increasing FAO attenuated HFD induced cardiomyopathy.
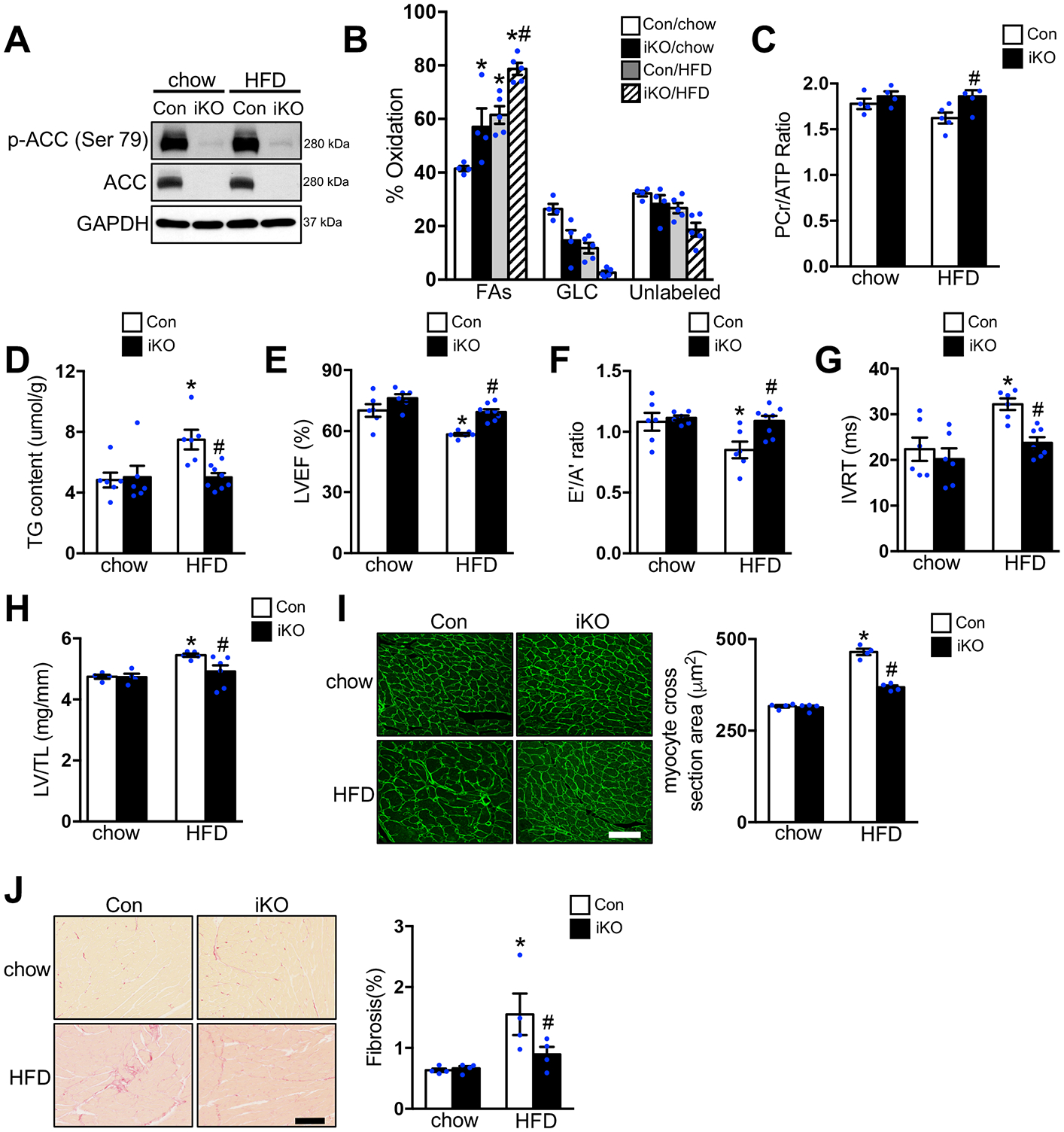
Con and ACC2 iKO mice were subjected to chow or HFD feeding for 24 weeks. (A) Representative immunoblots of heart lysates of p-ACC (Ser 79), ACC and GAPDH are shown. (B-C) Hearts were perfused with a buffer containing 13C labeled fatty acids, 13C labeled glucose, lactate, and insulin (mixed substrate). (B) Contribution of 13C labeled substrates to tricarboxylic acid (TCA) cycle was determined by 13C NMR spectroscopy in heart extracts. Relative contribution of fatty acids (FAs), glucose (GLC), and other unlabeled substrates (lactate, endogenous) (Unlabeled) is shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–5). (C) Phosphocreatine to ATP (PCr/ATP) ratio was measured by 31P NMR spectroscopy in isolated perfused hearts (#p<0.05 vs. Con/HFD, n=4–5). (D) Cardiac triglyceride (TAG) content normalized to tissue weight (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6–8). (E-G) Left ventricular ejection fraction (LVEF), E’/A’ ratio and isovolumic relaxation time (IVRT) were measured by echocardiography (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6–8). (H) The left ventricle weight to tibia length (LV/TL) ratio of ACC2 iKO and Con mice under chow- or HFD-fed conditions (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–6). (I) Representative wheat germ agglutinin (WGA) staining and quantification of cardiomyocyte cross-sectional area in indicated hearts after 24 weeks’ chow or HFD feeding (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4), Scale bar, 50μm. (J) Representative picric acid sirius red (PASR) staining and quantification of the percentage of fibrosis in indicated hearts after 24 weeks’ chow or HFD feeding (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4), Scale bar, 100μm.
Since deletion of ACC2 shifted cardiac metabolism towards FAO in normal mice, we then asked whether increasing fatty acid supply with HFD feeding would further alter cardiac substrate metabolism in iKO mice. In isolated perfused hearts with comparable contractile function (Figure IIA–IIC in the Supplement), iKO-chow demonstrated a ~40% increase in FAO as compared to Con-chow, that was accompanied by a reciprocal reduction in the contribution of glucose oxidation (Figure 1B, Figure IID in the Supplement). A similar increase in FAO was also observed in Con-HFD (Figure 1B, Figure IID in the Supplement). These changes are consistent with previous observations.20, 23, 24 Interestingly, HFD feeding further increased FAO in iKO compared with Con-HFD, associated with reduced oxidation of both glucose and unlabeled substrates (lactate and endogenous) (Figure 1B, Figure IID in the Supplement). Real time PCR (RT-PCR) and immunoblot analysis revealed that the expression of cluster of differentiation 36 (CD36), the major fatty acid transporter in the heart, was increased in both Con-HFD and iKO-HFD hearts (Figure IIIA–IIIB in the Supplement). Consistently, the upregulation of pyruvate dehydrogenase kinase 4 (PDK4), the negative regulator of glucose oxidation, was also observed in both Con-HFD and iKO-HFD mouse hearts (Figure IIIA in the Supplement). Transcriptional activation of other genes involved in fatty acid metabolism, mitochondrial respiration and uncoupling proteins was rather modest in Con hearts after 6 months of HFD (Figure IIIA and IIIC in the Supplement). A similar change was also observed in iKO-HFD hearts (Figure IIIA and IIIC in the Supplement).
To determine whether altered substrate utilization observed in iKO hearts affected myocardial energetics in obese mice, we measured the high-energy phosphate content in isolated hearts with 31P NMR spectroscopy. Chronic HFD feeding caused a moderate decrease of the phosphocreatine/ATP (PCr/ATP) ratio in Con hearts (p=0.054 vs. Con-chow). However, iKO-HFD hearts maintained the PCr/ATP ratio demonstrating an improved myocardial energetics status compared to Con-HFD (Figure 1C, Figure IIE in the Supplement). HFD induced TG accumulation was also completely abolished in iKO hearts compared with Con hearts (Figure 1D). Taken together, these data suggested that deletion of ACC2 further enhanced FAO, maintained cardiac energetics and prevented lipid accumulation in obese mouse hearts.
ACC2 iKO Prevented Cardiomyopathy Caused by Long-term HFD Feeding
Previously, we have demonstrated that 12 weeks of HFD feeding does not induce systolic dysfunction but contributes to the development of diastolic dysfunction in obese mice.25 However, extending the feeding period to 24 weeks in the present study caused a mild impairment of systolic function. We found a 15–20% reduction of the left ventricular ejection fraction or fractional shortening in Con-HFD hearts, which was attenuated in iKO-HFD hearts (Figure 1E, Table I in the Supplement). Tissue Doppler analysis showed that HFD feeding caused a decrease in the E′/A′ ratio and a prolonged isovolumic relaxation time (IVRT) in Con hearts, indicative of diastolic dysfunction (Figure 1F and 1G, Table II in the Supplement). Conversely, no significant changes in these measures were observed in iKO-HFD hearts (Figure 1F and 1G, Table II in the Supplement). In order to exclude the possibility that the beneficial effects observed in iKO heart is due to the transient induction of CRE by tamoxifen, in a separate cohort we injected the same dose of tamoxifen (20mg/kg/day for 5 days) in the alpha myosin heavy chain-MerCreMer (αMHC-MCM) mice and subjected them to 24 weeks of HFD feeding. These mice also developed systolic and diastolic dysfunction compared to chow-fed αMHC-MCM (Table III and IV in the Supplement), suggesting that the protective effect in iKO mice is not due to taxomifen administration and the presence of MCM system. Moreover, Con-HFD hearts developed cardiac hypertrophy, assessed by the left ventricular weight to tibia length ratio (LV/TL), which was significantly attenuated in iKO-HFD hearts (Figure 1H, Table V in the Supplement). This observation was consistent with the reduced myocyte cross section area in iKO-HFD hearts compared with Con-HFD hearts (Figure 1I). Histological analysis also showed a reduction of fibrosis in iKO-HFD hearts (Figures 1J). In total, these findings revealed that increasing cardiac FAO through deletion of ACC2 protected against the development of cardiac dysfunction and the pathological remodeling of the heart in mice with diet induced obesity.
HFD Feeding Induced Mitochondrial Dysfunction Was Attenuated in ACC2 iKO Hearts
Mitochondrial dysfunction, associated with changes in mitochondria morphology and accumulation of damaged mitochondria, is a pathogenic hallmark of obesity and diabetes induced cardiomyopathy.26, 27 Given the fact that myocardial energetics and cardiac function was maintained in iKO-HFD hearts, we speculated that cardiac mitochondrial function was preserved in iKO mice after chronic HFD feeding. To test our hypothesis, we examined the respiratory activity of isolated mitochondria from Con-chow, iKO-chow, Con-HFD and iKO-HFD hearts. Respiration driven by Complex I substrates or fatty acids, but not by complex II substrates was significantly decreased in Con-HFD, indicative of mitochondrial dysfunction. These changes were partially restored in iKO-HFD (Figure 2A–2B, Figure IVA in the Supplement). Using the increase of pH in respiration media as a readout of ATP synthesis,28, 29 we observed that the estimated ATP synthesis was decreased in isolated mitochondria from Con-HFD hearts compared to Con-chow hearts (Figure 2C, Figure IVB in the Supplement). On the other hand, the estimated ATP synthesis was sustained in iKO-HFD group (Figure 2C, Figure IVB in the Supplement). We also observed increased protein carbonylation, a hallmark of oxidative damage, in the mitochondria isolated from Con-HFD hearts, but not in iKO-HFD hearts (Figure 2D). Collectively, these findings suggested that increased oxidative stress and mitochondria dysfunction in the hearts of obese mice were unlikely caused by high FAO as previously proposed. Instead, increasing cardiac FAO in iKO mice prevented HFD induced mitochondrial dysfunction. Excess endoplasmic reticulum (ER) stress triggered by lipid overloading has been implicated as an important mechanism for the development of obesity induced cardiomyopathy.30, 31 Particularly, transcriptional activation of the C/EBP homologous protein (CHOP) and parallel activation of caspase 12 is essential for mediating apoptotic response to ER stress.30, 31 However, chronic HFD feeding did not cause any changes in the expression of CHOP and caspase 12 in either Con or iKO hearts (Figure V in the Supplement). The expression of binding immunoglobulin protein (BiP), a chaperone helping to restore ER homeostasis during unfolded protein response, remained the same in all groups (Figure V in the Supplement). These data suggested that the cardioprotective effect of ACC2 deletion was unlikely through regulating ER stress response in this model.
Figure 2. Increasing FAO prevented HFD induced mitochondria dysfunction.
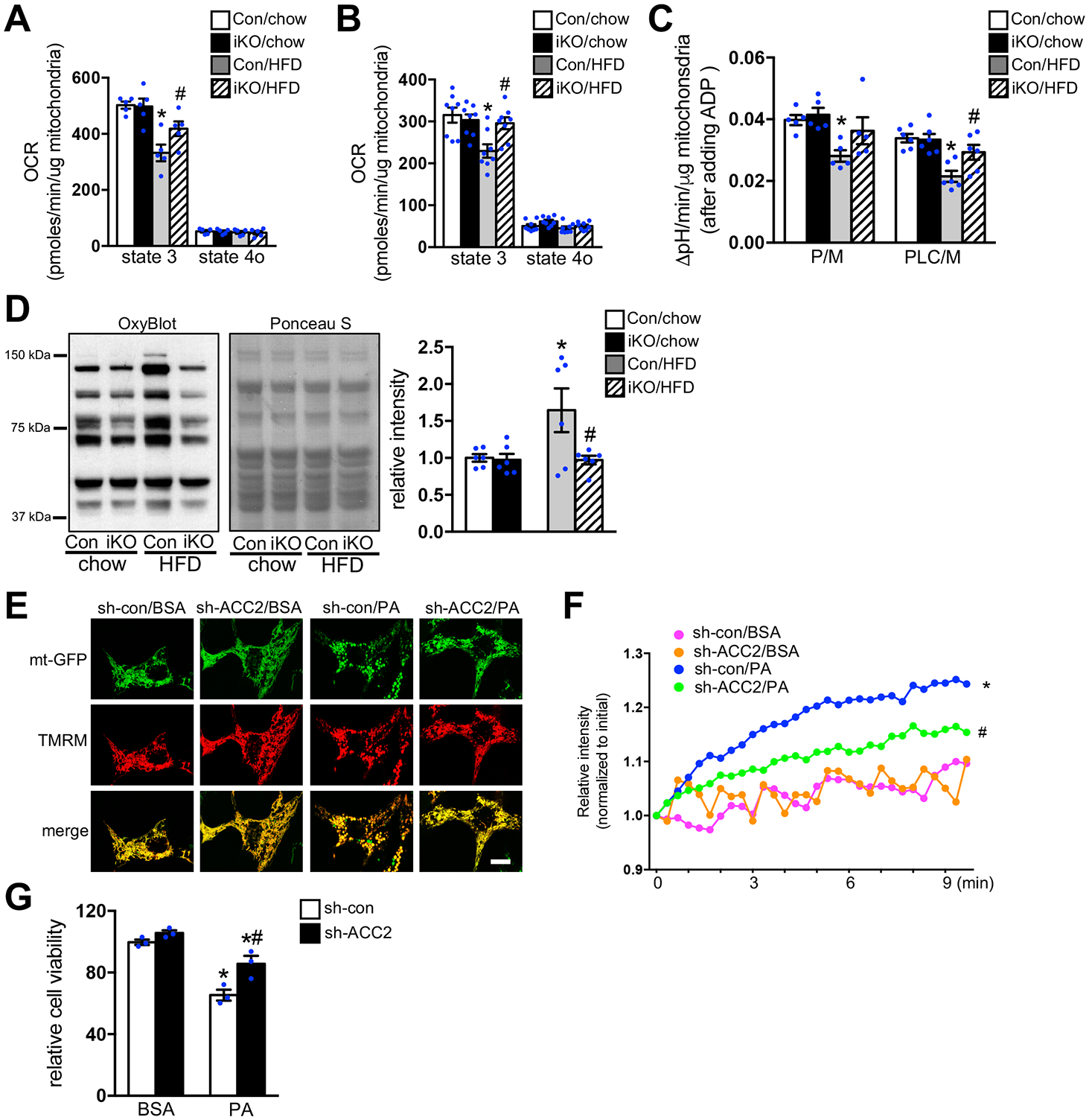
(A-D) Mitochondria were isolated from Con and ACC2 iKO mouse hearts after 24 weeks’ chow or HFD feeding. (A) Pyruvate/malate (P/M) and (B) palmitoyl-L-carnitine/malate (PLC/M) driven respiration was measured by Seahorse XF24 Analyzer. State 3 (+ADP), State 4o (+oligomycin) (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=5–8). (C) Absolute pH level change per minute after adding ADP in the presence of pyruvate/malate (P/M) or palmitoyl-L-carnitine/malate (PLC/M) as substrates (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=5–6). (D) Representative immunoblot of mitochondria carbonylated proteins (left), the loading control stained with Ponceau S (middle) and statistical analysis of densitomeric measurement of carbonylated proteins to total proteins (right) are shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6). (E) NRCMs transfected with indicated adenovirus were incubated with DMEM containing BSA or 0.4 mM PA for 12 h. After the cells were stained with TMRM, mitochondria morphology and membrane potential were imaged using the confocal microscopy. Results are representative of three independent experiments. Scale bar, 10μm. (F) NRCMs transfected with indicated adenovirus for 72 h were incubated with DMEM containing BSA or 0.4 mM PA for 12 h. After incubation with MitoSOX red, mitochondria superoxide production was examined using the confocal microscopy (*p<0.05 vs. sh-con/BSA, #p<0.05 vs. sh-con/PA, 10–15 cells were analyzed per condition, n=4). (G) NRCMs transfected with indicated adenovirus for 72 h were incubated with DMEM containing BSA or 0.4 mM PA for 24 h. The cell viability was examined (*p<0.05 vs. sh-con/BSA, #p<0.05 vs. sh-con/PA, n=3).
We next examined whether increasing FAO by ACC2 knocking down (KD) in a cell autonomous mode protected cardiomyocytes challenged by palmitate (PA), a major saturated fatty acid species in HFD and a known mediator of lipotoxicity.3 We first constructed an adenovirus harboring ACC2 short hairpin RNA (Ad-sh-ACC2) (Figure VIA–VIB in the Supplement). We have observed a greater oxidation of lipids in cardiomyocytes transfected with Ad-sh-ACC2 (ACC2-KD) compared with cells transfected with adenovirus harboring scramble shRNA (Ad-sh-con) (Figure VIC–VID in the Supplement). These data confirmed that knocking down ACC2 in cultured cardiomyocytes also increased FAO. To visualize the mitochondrial morphology, cardiomyocytes were transfected with the mitochondria targeted GFP (mt-GFP) adenovirus. The live cell image revealed that the normal mitochondrial network in cultured cardiomyocytes appeared as tubular mitochondria (Figure 2E). Palmitate supplementation changed the mitochondrial morphology from a tubular to the more rounded shape and some of the mitochondria lost their membrane potential when co-stained with etramethylrhodamine methyl ester (TMRM) (Figure 2E). Interestingly, these morphological changes were partially prevented by knocking down ACC2 (Figure 2E). Furthermore, PA induced mitochondrial superoxide production and cardiomyocyte death were also attenuated in ACC2-KD cardiomyocytes (Figure 2F–2G). Taken together, these data suggested that increasing FAO by knocking down of ACC2 prevented mitochondria dysfunction and improved cell survival under chronic PA treatment.
ACC2 iKO Prevented the Accumulation of Damaged Mitochondria during Long-term HFD Feeding
We performed electron microscopy (EM) analysis to examine whether chronic HFD feeding alters cardiac ultrastructure in Con and iKO mice. EM images showed that sarcomere arrangement and mitochondria distribution are preserved in both Con-HFD and iKO-HFD hearts. However, we noticed that the morphology of mitochondria in Con-HFD hearts displayed notable heterogeneity with some mitochondria showing reduced matrix density or swelling. These changes were not found in iKO-HFD hearts (Figure 3A). There was an increase in mitochondrial volume density and mitochondria DNA content in Con-HFD hearts, which was attenuated in iKO-HFD hearts (Figure 3B–3C). Higher magnification images also revealed the presence of damaged mitochondria in Con-HFD hearts with an increase of disarrayed cristae and a reduced electron density in the matrix (Figure 3D). However, mitochondrial morphology and ultrastructure were unaltered in iKO-HFD hearts and fewer damaged mitochondria was noted (Figure 3D). In addition, HFD feeding increased the lipid droplet density in Con hearts but not in iKO hearts (Figure 3E), consistent with the notion that a greater fraction of FAs was directed to FAO in iKO-HFD hearts. Taken together, these data suggested that iKO prevented the accumulation of damaged mitochondria during long-term HFD feeding.
Figure 3. Increasing FAO prevented the accumulation of damaged mitochondria in vivo.
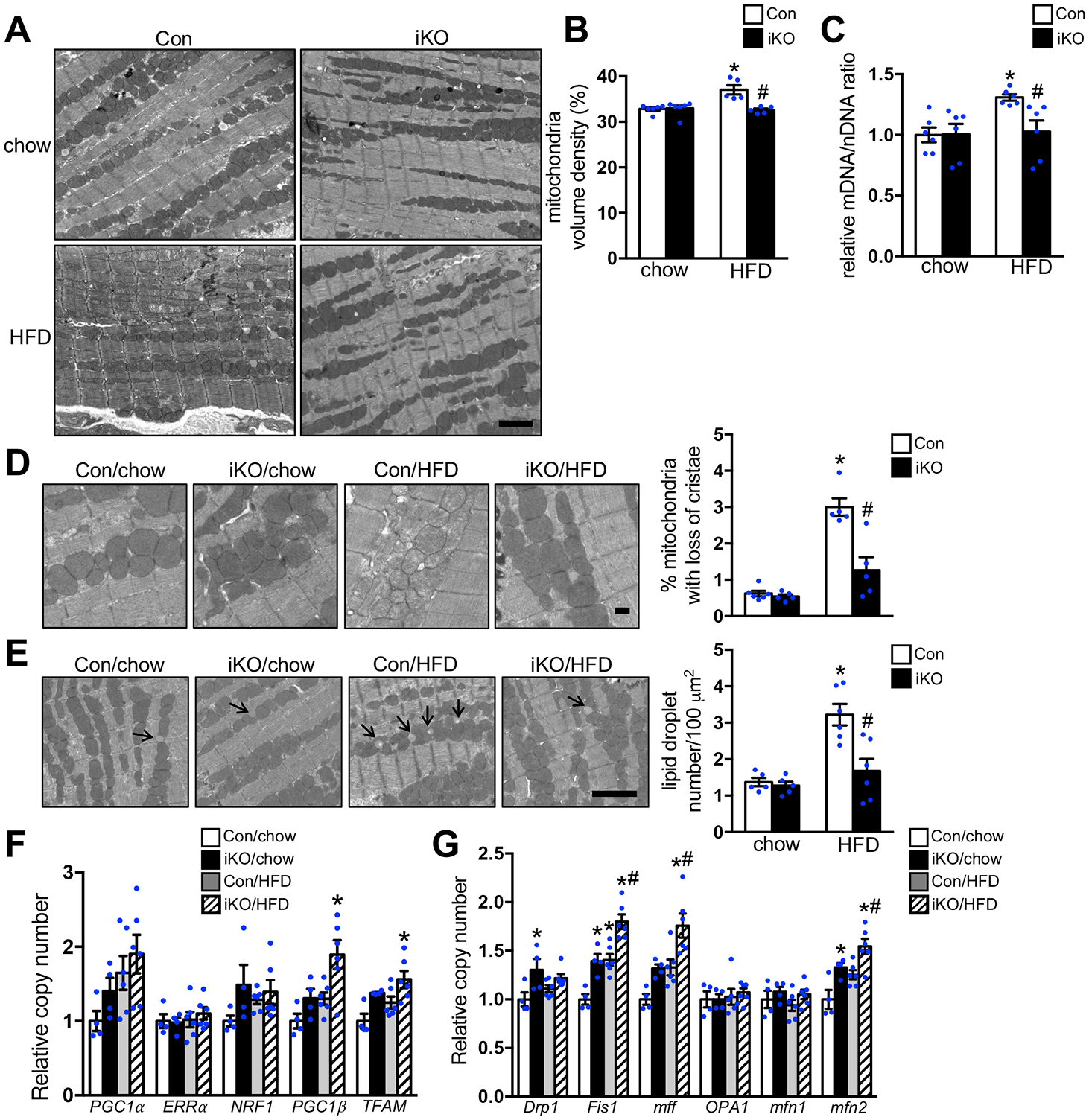
Con and ACC2 iKO mice were subjected to chow or HFD feeding for 24 weeks. (A) Representative images of heart sections from electron microcopy, Scale bar 2μm. (B) Mitochondria density was determined by the percentage of mitochondria area per field area. (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, 10–15 random fields (10000x) per heart, n=5–6). (C) The ratio of mitochondrial DNA (mDNA) to nuclear DNA (nDNA) was determined by RT-PCR and represented as fold change compared with Con-chow, arbitrarily defined as 1. (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6). (D) (Left) Representative EM image showing disarrayed cristae and reduced electron density of the matrix in the mitochondria of Con-HFD heart but not ACC2 iKO-HFD heart. Scale bar 500nm. (Right) Quantification of the percent of damaged mitochondria. About 12–15 random fields with 1500–1800 mitochondria were analyzed per heart sample (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=5–6). (E) (Left) Representative EM image showing lipid droplet accumulation in the Con-HFD heart but not in ACC2 iKO-HFD heart, arrows indicate lipid droplets. Scale bar 2μm. (Right) Quantification of the lipid droplet number (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, 10–15 random fields (10000x) per heart sample, n=5–6). (F) qRT-PCR measurements of indicated genes involved in mitochondria biogenesis (*p<0.05 vs. Con/chow, n=4–6). (G) qRT-PCR measurements of indicated genes involved in mitochondria fusion and fission process (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–6).
Alteration of mitochondrial biogenesis, dynamics or turnover could lead to changes in mitochondrial mass, size or accumulation of damaged mitochondria. RT-PCR analysis demonstrated that the expression of genes involved in mitochondria biogenesis in Con-HFD hearts was similar to Con-chow hearts (Figure 3F). These data suggested that mitochondria biogenesis pathway was not activated and thus unlikely contributed to the increased mitochondria volume density in Con-HFD hearts. We then examined whether HFD altered the expression of proteins regulating mitochondrial dynamics. RT-PCR analysis revealed that the expression of the majority of the genes involved in the regulation of mitochondria fusion and fission were not altered in Con-HFD hearts, except a slight increase in mitochondria fission 1 (Fis1) and mitofusin 2 (mfn2) (Figure 3G). Interestingly, iKO-HFD hearts showed not only upregulation of Fis1 and mfn2 but also multiple genes for mitochondria biogenesis and dynamics, such as peroxisome-proliferator-activated receptor-gamma co-activator 1 beta (PGC1β), transcription factor A, mitochondrial (TFAM), mitochondria fission factor (mff) (Figure 3F–3G). Since the mitochondrial volume density did not change in iKO-HFD, we speculated that increased gene expression observed here suggested a potential increase of mitochondrial turnover activity.
Long-term HFD Feeding Suppressed Basal Mitophagy Activity in Con but not in ACC2 iKO Hearts
Damaged mitochondria can be cleared via mitophagy during which the organelle is enveloped by autophagosomes and eventually fused with lysosomes for degradation. Upon stimulation, the autophagosomal docking proteins are recruited to damaged mitochondria and linked to microtubule-associated protein 1A/1B-light chain 3 (LC3) on autophagosomes.32 Therefore, we measured the level of LC3, and autophagy adapter protein, p62/SQSTM1 (Sequestosome 1), that were directly associated with mitochondria, as a biochemical marker for mitophagy. The protein expressions of a lipidated form of LC3 (LC3 II) and p62 were significantly lower in the mitochondrial fraction of Con-HFD hearts compared with Con-chow hearts. On the other hand, the downregulation of LC3 II and p62 by HFD feeding was completely abolished in ACC2 iKO hearts (Figure 4A). In order to assess mitophagy in vivo, we crossed Con and iKO with mt-Keima transgenic mice.21 The mt-Keima reporter is a mitochondrial targeted form of Keima, a fluorescent protein that resists degradation by lysosomal proteases. The peak of the mt-Keima excitation spectrum shifts from 458 nm (neutral pH) to 561 nm (acidic pH), when mitochondria are delivered to lysosomes for degradation. Therefore, the ratio of the 561 nm/458 nm excited Keima fluorescence is used to reflect the mitophagy activity.22 In isolated cardiomyocytes, the signal indicative of lysosome-localized mt-Keima was significantly reduced in Con-HFD but was maintained in iKO-HFD when compared to chow fed groups (Figure 4B).
Figure 4. Increasing FAO attenuated HFD induced suppression of mitophagy.
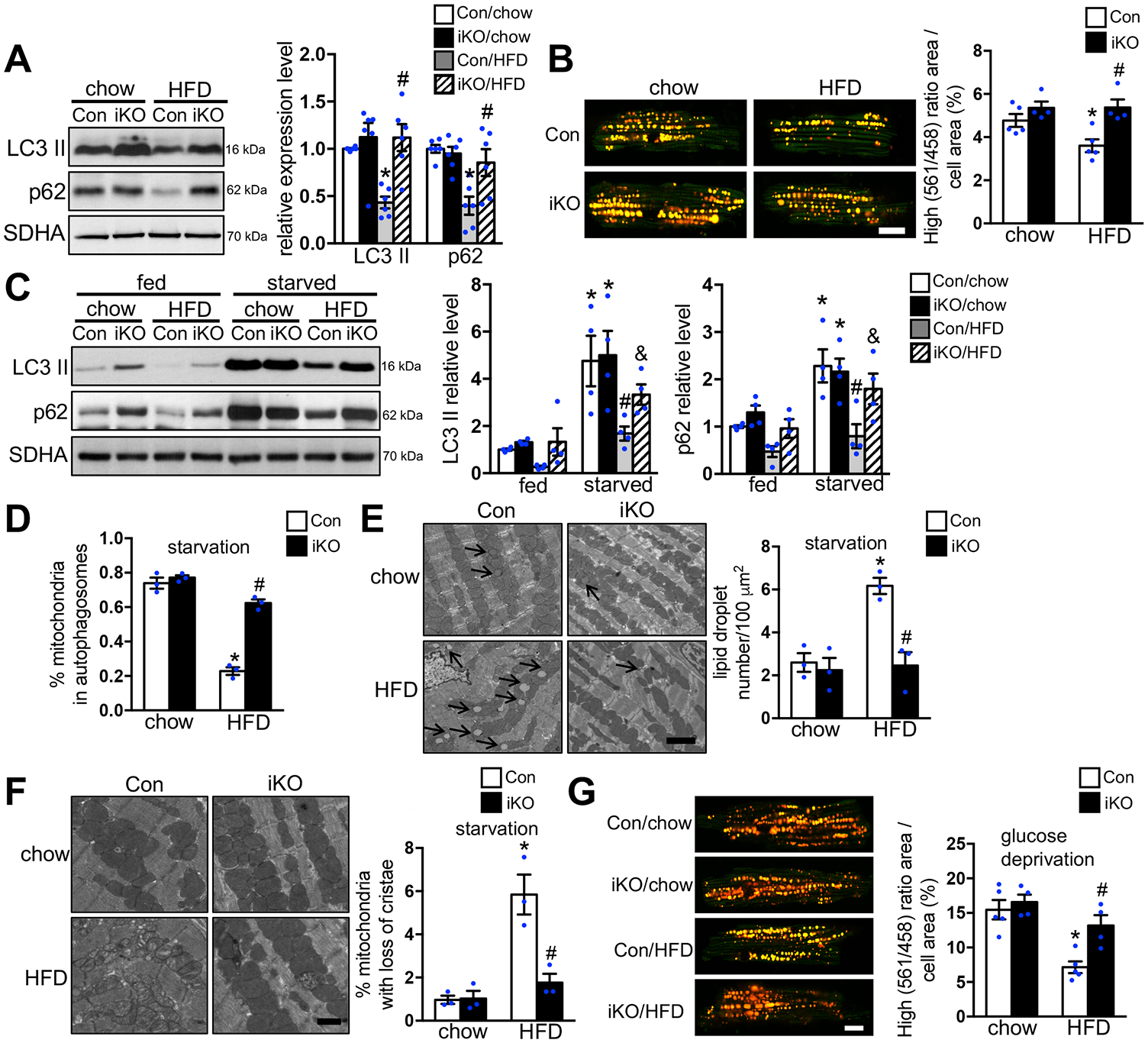
(A) Mitochondria isolated from Con and ACC2 iKO mouse hearts after 24 weeks’ chow or HFD feeding were subjected to immunoblot analysis. Representative immunoblots of LC3 II, p62 and SDHA in mitochondria homogenates (left) and statistical analyses of densitometric measurements of LC3 II and p62 (right) are shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6). (B) Cardiomyocytes were isolated from Con/mt-Keima and ACC2 iKO/mt-Keima bigenic mice after 24 weeks’ chow or HFD feeding. High (561/458) ratio area/total cell area was quantified as an index of mitophagy. About 25 to 35 cardiomyocytes per heart were analyzed (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–5), Scale bar 15μm. (C-F) Con and ACC2 iKO mice after 24 weeks’ chow or HFD feeding were subjected to starvation for 24 hours. (C) Representative immunoblots of LC3 II, p62 and SDHA in mitochondria homogenates (left) and statistical analyses of densitomeric measurements of LC3 II and p62 (right) are shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/chow/starvation, &p<0.05 vs. Con/HFD/starvation, n=4). (D) Quantification of the percentage of mitochondria sequestered in autophagosomes under starvation condition. About 15–20 random fields with 2000–2500 mitochondria were analyzed per heart sample (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=3). (E) (Left) Representative EM image showing lipid droplet accumulation in the Con-HFD but not in ACC2 iKO-HFD heart under starvation condition, arrows indicate lipid droplets. Scale bar 2μm. (Right) Quantification of the lipid droplet number (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, 10–15 random fields (10000x) per heart sample, n=3–4). (F) (Left) Representative EM image showing more disarrayed cristae and reduced electron density of the matrix in the Con-HFD than in ACC2 iKO-HFD heart under starvation condition. Scale bar 500nm. (Right) Quantification of damaged mitochondria percentage. About 15–20 random fields with 2000–2500 mitochondria were analyzed per heart sample (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=3). (G) Cardiomyocytes isolated from Con/mt-Keima and ACC2 iKO/mt-Keima bigenic mice after 24 weeks’ chow or HFD feeding were incubated with no glucose DMEM for 4 hours. High (561/458) ratio area/total cell area was quantified as an index of mitophagy. About 25 to 35 cardiomyocytes per heart were analyzed (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–5), Scale bar 15μm.
We also evaluated the mitochondria enriched autophagosome accumulation in response to a short-term (4-hour) chloroquine treatment to assess the role of autophagy flux.33 As expected, chloroquine treatment caused a greater accumulation of LC3 II and p62 in mitochondria fraction of both Con-chow and iKO-chow hearts compared with vehicle treated condition, indicating that the autophagy flux was intact in these hearts (Figure VIIA in the Supplement). The chloroquine induced LC3 II and p62 accumulation in mitochondria fraction was suppressed in Con-HFD hearts, whereas it was restored in iKO-HFD hearts (Figure VIIA in the Supplement). Chloroquine treatment did not change the mt-keima signal in all the groups compared with vehicle treatment (Figure VIIB in the Supplement), suggesting that the formation of mitochondria enriched autophagosome is likely the main defect in Con-HFD hearts. Taken together, these results suggested that chronic HFD feeding inhibited mitophagy activity in the heart, which was prevented in iKO mice.
Failure to Activate Mitophagy during Stress Contributed to the Accumulation of Damaged Mitochondria in the Heart of Obese Mice
In order to determine whether HFD suppresses mitophagy activation in the heart during stress, Con and iKO mice were subjected to fasting, an energy starvation condition which can effectively stimulate mitophagy in vivo. After 24 hours of fasting, the expression of LC3 II and p62 in the mitochondria fraction of Con-chow hearts was significantly increased compared with the ones under the fed condition. The iKO-chow hearts showed a similar upregulation pattern of LC3 II and p62 as Con-chow in response to fasting, indicating that deletion of ACC2 did not affect starvation induced mitophagy under chow-fed condition (Figure 4C). On the other hand, chronic HFD feeding completely blocked fasting mediated upregulation of LC3 II and p62 in the mitochondria fraction of Con-HFD hearts, which was partially prevented in iKO-HFD hearts (Figure 4C). Moreover, EM analysis revealed that HFD feeding significantly reduced the percentage of mitochondria in autophagasomes in Con-HFD hearts compared with Con-chow hearts in response to fasting (Figure 4D). On the contrary, the percentage of mitochondria in autophagasomes was similar in iKO-chow and iKO-HFD hearts upon fasting (Figure 4D). Taken together with biochemical studies, these experiments demonstrated that HFD feeding inhibited starvation induced mitophagy activation in Con hearts, but not in iKO hearts. In addition, more lipid droplets and damaged mitochondria accumulated in Con-HFD than iKO-HFD hearts in response to fasting (Figure 4E–4F). In cell culture subjected to glucose deprivation condition, high 561 nm/458 nm ratio, indicative of mitophagy activity, was lower in cardiomyocytes isolated from Con-HFD hearts than Con-chow hearts. However, high 561 nm/458 nm ratio did not differ between cardiomyocytes from iKO-HFD and that from iKO-chow when subjected to glucose deprivation (Figure 4G).
Starvation induced mitophagy is triggered by activation of macroautophagy, which is considered a consequential, non-selective process of energy deprivation. Therefore, we examined whether mitophagy triggered by a specific mitochondria stress is also altered in the hearts of HFD-fed mice. We evaluated the mitophagy activity in hearts that were treated with or without the mitochondrial uncoupler, carbonyl cyanide p-trichloromethoxyphenylhydrazone (CCCP), which can induce mitochondria depolarization and activate mitophagy without affecting overall nutrient status. Heart samples were collected 12 hours after an intraperitoneal injection of 5 mg kg−1 CCCP in mice. Immunoblot analysis revealed that injection of CCCP significantly increased the expression of LC3 II and p62 in the mitochondria fraction of both Con-chow and iKO-chow hearts (Figure 5A). Similar to the fasting condition, HFD feeding completely abolished CCCP induced upregulation of LC3 II and p62 in the mitochondria fraction isolated from Con hearts, which was prevented in iKO hearts (Figure 5A). EM and mt-Keima analysis provided additional evidence demonstrating that CCCP induced activation of mitophagy was inhibited in Con-HFD hearts but was maintained in iKO-HFD hearts, which was also associated with reduced accumulation of damaged mitochondria in the presence of CCCP treatment (Figure 5B–5D). Taken together, these results suggested that maintaining mitophagy activity in iKO hearts was critical for preventing the accumulation of damaged mitochondria in obese mice.
Figure 5. HFD suppressed CCCP induced mitophagy activation in Con but not in ACC2 iKO mice.
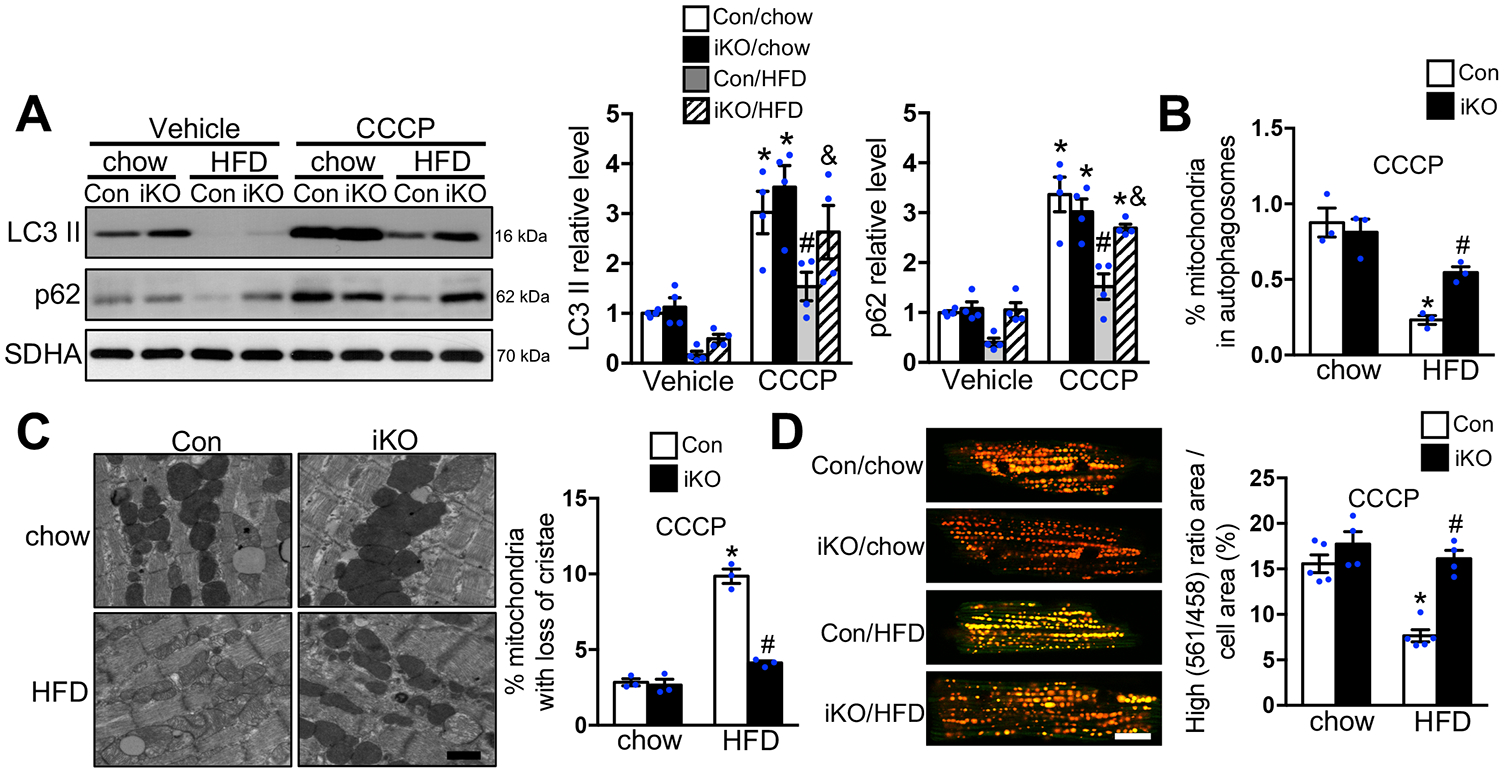
(A-C) Con and ACC2 iKO mice after 24 weeks’ chow or HFD feeding were subjected to CCCP (5mg/kg) injection for 12 hours. (A) Representative immunoblots of LC3 II, p62 and SDHA in mitochondria homogenates (left) and statistical analyses of densitomeric measurements of LC3 II and p62 (right) are shown (*p<0.05 vs. Con/chow/Vehicle, #p<0.05 vs. Con/chow/CCCP, &p<0.05 vs. Con/HFD/CCCP, n=4). (B) Quantification of the percentage of mitochondria sequestered in autophagosomes with CCCP injection. About 15–20 random fields with 2000–2500 mitochondria were analyzed per heart sample (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=3). (C) (Left) Representative EM image showing more disarrayed cristae and reduced electron density of the matrix in the Con-HFD than in ACC2 iKO-HFD heart with CCCP injection. Scale bar 500nm. (Right) Quantification of damaged mitochondria percentage. About 15–20 random fields with 2000–2500 mitochondria were analyzed per heart sample (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=3). (D) Cardiomyocytes isolated from Con/mt-Keima and ACC2 iKO/mt-Keima bigenic mice after 24 weeks’ chow or HFD feeding were incubated with 2μM CCCP for 4 hours. High (561/458) ratio area/total cell area was quantified as an index of mitophagy. About 25 to 35 cardiomyocytes per heart were analyzed (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4–5), Scale bar 15μm.
ACC2 iKO Prevented HFD Induced Downregulation of Parkin
Translocation of parkin to the outer membrane of depolarized mitochondria plays an essential role for initiating selective mitophagy through recruitment of autophagy receptor proteins, such as p62, and further recruitment of LC3 positive autophagosomes.34 Immunoblot analysis revealed that chronic HFD feeding significantly reduced the expression of parkin in the mitochondria fraction of Con hearts, which was attenuated in iKO hearts (Figure 6A). Moreover, we observed that Con-HFD failed to upregulate mitochondria localized parkin expression in response to fasting or CCCP injection (Figure 6B–6C). On the other hand, iKO-HFD had a significantly higher level of parkin expression in the mitochondria fraction of the heart compared with the Con-HFD group. These data suggested that chronic HFD feeding attenuated parkin mediated mitophagy in the heart, which was prevented in ACC2 iKO mice. PTEN-induced kinase 1 (Pink1) can act as a molecular sensor of damaged mitochondria and its activity is required for recruiting parkin to the outer membrane of damaged mitochondria.34, 35 As expected, CCCP treatment in mice caused Pink1 accumulation in mitochondria fraction from both Con-chow and iKO-chow hearts (Figure VIIIA in the Supplement). On the other hand, Pink1 accumulation in mitochondria fraction induced by CCCP was not affected by HFD treatment, suggesting that Pink1 activity was maintained and unlikely responsible for the reduced parkin accumulation on mitochondria of Con-HFD hearts (Figure VIIIA in the Supplement). Consistent with the observation made in the mitochondria fraction shown in Figure 6A, immunoblot analysis using whole heart lysates revealed that HFD feeding significantly decreased the expression of parkin in Con-HFD but not iKO-HFD (Figure 6D). The RT-PCR analysis revealed that HFD treatment did not alter parkin mRNA level in both Con and iKO hearts, suggesting that the downregulation of parkin was through a transcription independent mechanism (Figure VIIIB in the Supplement). Incubation of cardiomyocytes with PA also caused downregulation of parkin expression, which was restored in ACC2-KD cells (Figure 6E). More importantly, overexpression of parkin significantly reduced PA induced cell death (Figure 6F); the protective effect of ACC2-KD on cell survival in response to PA treatment was abolished in cardiomyocytes co-transfected with adenovirus harboring parkin short hairpin RNA (Ad-sh-parkin) (Figure 6G, Figure VIIIC–VIIID in the Supplement). Taken together, these data suggested that the protective effect of increasing FAO by ACC2 deletion, in part, was mediated by regulating parkin mediated mitophagy under obese condition.
Figure 6. Increasing FAO prevented lipid induced downregulation of parkin.
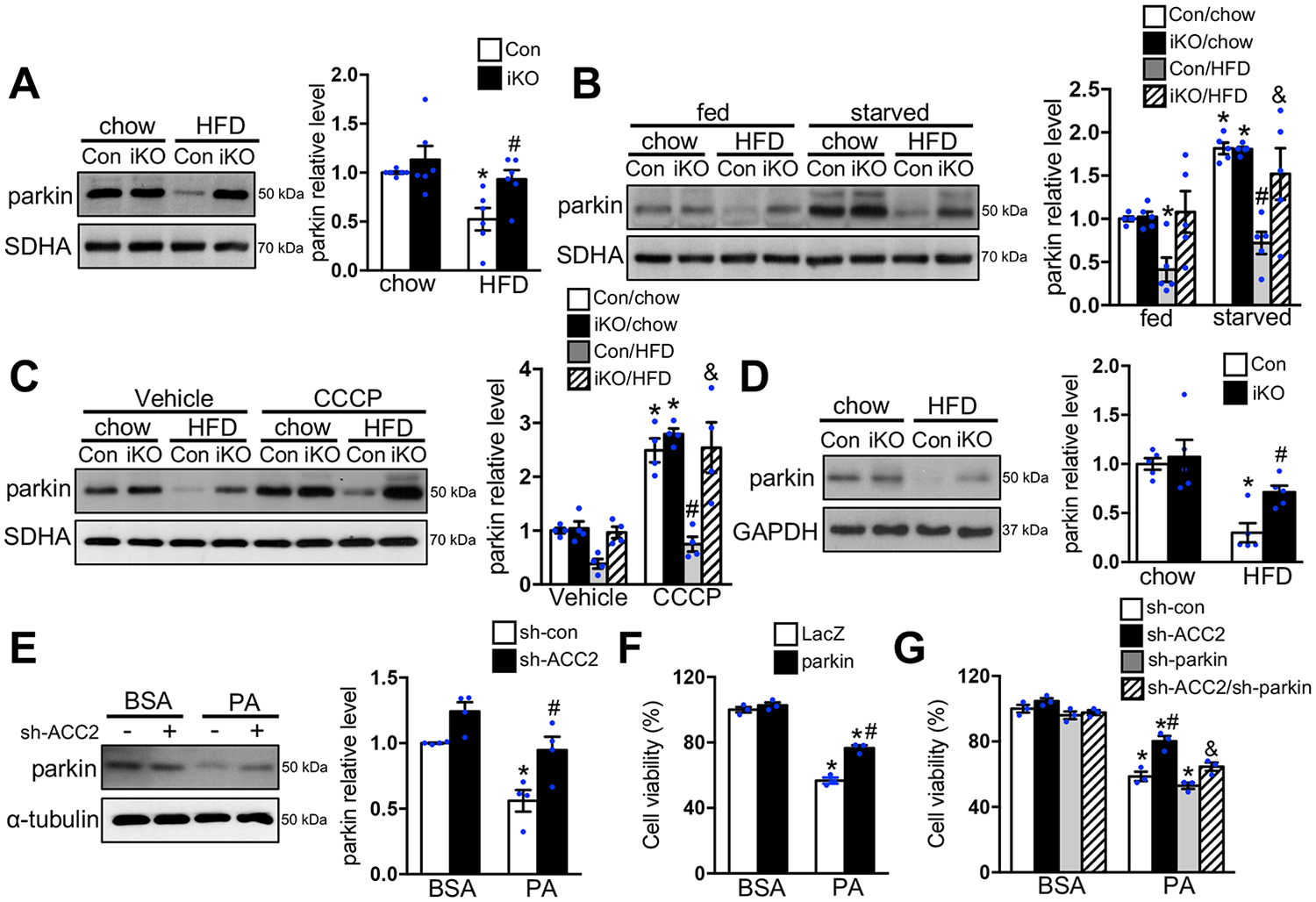
(A-D) Con and ACC2 iKO mice were subjected to chow or HFD feeding for 24 weeks. (A) Representative immunoblots of parkin and SDHA in mitochondria homogenates (left) and statistical analysis of densitomeric measurement of parkin (right) is shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=6). After 24-hour starvation (B) or 12-hour CCCP (5mg/kg) injection (C), mitochondria were isolated and subjected to immunoblot analysis. Representative immunoblots of parkin and SDHA in mitochondria homogenates (left) and statistical analysis of densitomeric measurement of parkin (right) is shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/chow/starvation or CCCP, &p<0.05 vs. Con/HFD/starvation or CCCP, n=4–5). (D) Representative immunoblots of parkin and SDHA in total heart lysates (left) and statistical analysis of densitometric measurement of parkin (right) is shown (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=5). (E) NRCMs transfected with indicated adenovirus for 72 hours were incubated with DMEM containing BSA or 0.4 mM PA for 12 hours. Representative immunoblots of parkin and SDHA in total cell lysates (left) and statistical analysis of densitometric measurement of parkin (right) is shown (*p<0.05 vs. sh-con/BSA, #p<0.05 vs. sh-con/PA, n=4). (F-G) NRCMs transfected with indicated adenovirus for 48 hours (F) or 72 hours (G) were incubated with DMEM containing BSA or 0.4 mM PA for 24 hours. The cell viability was examined (*p<0.05 vs. LacZ/BSA or sh-con/BSA, #p<0.05 vs. LacZ/PA or sh-con/PA, &p<0.05 vs. sh-ACC2/PA, n=3).
Discussion
In the present study, we find that promoting cardiac FAO via ACC2 deletion prevents the development of mitochondrial dysfunction and pathological remodeling (i.e. hypertrophy and fibrosis) of the heart in the face of chronic HFD feeding, and consequently sustains systolic and diastolic function in obese mice. Mechanistically, we show that long-term HFD feeding inactivates parkin mediated mitophagy resulting in accumulation of damaged mitochondria in the heart, which can be ameliorated by enhancing FAO. Our results thus identify a novel mechanism for mitochondrial dysfunction in the heart under obese conditions. Furthermore, we suggest that interventions leading to restoration of mitophagy activity has potential therapeutic implications for obesity induced cardiomyopathy.
By deleting ACC2 in the adult mouse heart, we are able to increase cardiac FAO in a lean mouse to the level comparable to that of an obese mouse, and achieve a further increase of FAO by HFD feeding. The change in FAO is associated with reduced oxidation of other substrates with no change of cardiac energetics and function,10, 20 demonstrating that the adult heart has a high capacity and flexibility for increases of FAO. Previous studies have suggested that increased cardiac FAO in obesity and/or diabetes is correlated with the reduced cardiac efficiency and increased ROS production, eventually leading to impaired myocardial energetics and contractile dysfunction.2, 27, 36 In the present study, a similar scenario is observed in control mice subjected to long-term HFD but not in ACC2 deficient mouse hearts fed with either chow or HFD, although cardiac FAO is equally increased or greater in ACC2 deficient hearts. We found ROS-mediated damage evidenced by the increasing mitochondrial oxidative protein modification in Con-HFD hearts. Meanwhile, upregulation of uncoupling protein 3 (UCP3) is observed in Con-HFD hearts, which may serve as a compensatory mechanism to lower ROS production at the expense of efficiency.37 On the other hand, we did not observe any alteration in mitochondrial oxidative protein modification or UCP3 expression level in ACC2 deficient hearts. These results strongly suggest that high FAO is not the cause of cardiac damage in obesity but instead insufficient FAO under conditions of increased fatty acids supply contributes to lipotoxicity. In support of this notion, strategies that reduce fatty acid uptake or promote fatty acid storage is accompanied by cardiac functional improvements in models of obesity or cardiac PPAR activation.4, 6, 38 In contrast, blocking the increase of myocardial FAO exacerbates cardiac damage in obese mice.11 Our current observations suggest that directing more fatty acid toward oxidation in mitochondria represents another effective way to protect the heart from toxic lipids during the development of obesity.
Results of the present study show that mitochondrial dysfunction is a culprit of cardiomyopathy associated with obesity. Inhibition of mitophagy activity in lipid overloaded heart is a contributing mechanism for the accumulation of damaged mitochondria in these hearts. Mitophagy plays an essential role for mitochondrial quality control through clearance of damaged mitochondria by autophagy/lysosome pathway. A recent study reports that short-term HFD feeding (2 months) activates mitophagy in the heart.39 This is consistent with increased mitochondrial biogenesis in the heart during early stage of obesity.36 Increased turnover of mitochondria likely serves as an adaptive response to maintain mitochondrial protein quality as well as mitochondrial and cardiac function in the early stage of obesity. This is in agreement with the literature showing minimum change of cardiac function with short-term (up to 12 weeks) HFD feeding despite significant obesity.25, 40, 41 Moreover, in support of a compensatory role of increased mitophagy, genetic ablation of Atg7 which encodes a critical player in the autophagy pathway, impaired mitophagy activity leading to cardiac dysfunction with short-term HFD feeding.39 Notably, increased mitochondria turnover is no longer observed in the hearts after long-term HFD feeding (6 months). Instead, these hearts fail to activate mitophagy during nutritional or mitochondrial stress suggesting that the early compensatory mechanisms are obliterated by chronic obesity. Intriguingly, our observations reveal that enhancing FAO is sufficient to prevent the inactivation of mitophagy caused by chronic HFD feeding, thus directly linking cardiac lipid metabolism with mitophagy activity.
The present study shows that the pathogenesis of mitochondria dysfunction in obesity is linked to a chronic imbalance of lipid supply and consumption in the heart, which impairs mitophagy, an important mechanism for maintaining mitochondria quality. Our study further identifies parkin expression level as an important mechanism linking cardiac lipid metabolism and mitophagy activity. Downregulation of parkin expression has been observed in the hearts of type 1 diabetes animal model, which correlates with increased lipid accumulation.14 These data suggest that altered lipid metabolism affects parkin activity as well as mitophagy activity. On the other hand, Pink1 protein in the mitochondrial fraction of HFD-fed hearts is maintained. CCCP treatment is able to induce mitochondria pink1 accumulation even under the HFD feeding condition, suggesting that HFD treatment does not affect pink1 expression and/or its activity. We speculate that the impaired mitophagy activity in Con-HFD hearts is mainly due to a reduction of the parkin pool that supposed to be recruited by pink1. Moreover, HFD feeding did not alter the parkin mRNA level, suggesting that transcriptional independent mechanism(s) are involved. For potential mechanisms, posttranslational modifications and ubiquitination-mediated protein degradation pathway has been implicated to regulate parkin expression and activity.42 We speculate that accumulation of toxic lipid species contribute to the downregulation of parkin and subsequently inhibit mitophagy activity, while expanding the neural lipid pool, such as TG helps to buffer the toxic lipid species.6 Identification of lipid species that is responsible for HFD mediated downregulation of parkin warrants more investigation. In non-cardiac tissues, restoration of parkin expression ameliorates diabetic nephropathy and HFD induced neurologic damages.43, 44 Forced expression of parkin in cardiomyocyte confers resistance to cardiac aging through maintaining the mitophagy activity.45 Lack of parkin mediated mitophagy in perinatal mouse hearts renders the heart intolerant of the switch to fatty acid oxidation after birth, which leads to premature death.46 It is thus likely that parkin-mediated mitophagy also protects mitochondria during postnatal physiological lipid loading when energy substrates shift from carbohydrates to fatty acids in the heart. We therefore propose that parkin mediated mitophagy is a critical mechanism to maintain mitochondria homeostasis during changes of cardiac lipid metabolism. During perinatal stage, parkin mediated mitophagy replaces the immature mitochondria with mature ones to accommodate for the substrate switch from carbohydrates to fatty acids. Under obese condition, parkin mediated mitophagy is essential to replace the “damaged” mitochondria with healthy ones to accommodate for increased lipid challenge in the mitochondria.
A limitation of the study is its dependence on murine echocardiography for assessment of cardiac function, especially diastolic function. While tissue doppler echocardiography provides a non-invasive mode to assess diastolic function, the fast heart rate in mice makes it technically challenging. Furthermore, echocardiographic measurements are prone to variations caused by anesthesia and operator preference resulting in batch effects, which is also observed in the present study. Thus, for subtle changes in diastolic function, it would be ideal to validate the results with the gold standard testing by invasive hemodynamic measurements.
In summary, we demonstrate that increasing cardiac FAO through targeting ACC2 in the adult mouse heart is protective against obesity induced cardiomyopathy resulting in sustained cardiac function and attenuated pathological remodeling during chronic HFD feeding. Furthermore, the findings of the present study suggest that inhibition of parkin mediated mitophagy contributes to mitochondrial dysfunction under chronic lipid overloading condition, which can be reversed through increasing cardiac FAO. Targeting the parkin mediated mitophagy pathway may have potential therapeutic implications for obesity induced cardiomyopathy.
Supplementary Material
Clinical Perspective.
1). What is new?
The study demonstrates that enhancing mitochondrial fatty acid oxidation (FAO) prevents obesity induced cardiomyopathy in mice.
The result highlights the importance of impaired mitophagy in the pathogenesis of mitochondrial dysfunction and cardiomyopathy in obese mice.
Lipid overloading reduces parkin expression and subsequently inhibits parkin-dependent mitophagy activity, both can be reversed by increasing FAO.
2). What are the clinical implications?
Results of the study imply that impairments of mitophagy is a key mechanism for the development of mitochondrial dysfunction in the heart of obesity and diabetic patients.
Targeting the parkin mediated mitophagy pathway represents a therapeutic opportunity for lipotoxic cardiomyopathy associated with obesity and diabetes.
Acknowledgments
We thank Dr. Julia Ritterhoff for her invaluable discussion and assistance with the animal models. We thank Dr. Toren Finkel (National Institutes of Health) for generously providing the mt-Keima transgenic mice. We thank Dr. Nuo Sun (National Institutes of Health) for the technical support for the mt-Keima imaging. We thank Edward Parker and the UW Vision Core facility for helping with EM sample preparation and image acquisition.
Sources of Funding
This work was supported in part by U.S. National Institutes of Health Grants HL-088634, HL-118989, HL-129510 (to R. Tian), HL-137266 (to W. Wang), HL-128368, AR-074900 (to M. Regnier), S10RR029021 (to UW 14T High Resolution NMR Core Facility), P30 EY01730 (to UW Vision Research Center Core) and the American Heart Association Grants 14SDG18590020 (to SC. Kolwicz), 18EIA33900041 (to W. Wang), 18CDA34080486 (to D. Shao). K. Nishi is supported by the fellowship for research abroad from Japan Heart Foundation. D. Shao is also supported by the pilot and feasibility program from the Diabetes Research Center at the University of Washington (P30 DK017047).
Non-standard Abbreviations and Acronyms
- ACC2
Acetyl-CoA Carboxylase 2
- αMHC-MCM
alpha myosin heavy chain-MerCreMer
- ATP
adenosine triphosphate
- BiP
binding immunoglobulin protein
- CCCP
carbonyl cyanide p-trichloromethoxyphenylhydrazone
- CD36
cluster of differentiation 36
- CHOP
C/EBP homologous protein
- DIO
diet induced obesity
- EF
ejection fraction
- EM
electron microscopy
- ER
endoplasmic reticulum
- FAO
fatty acid oxidation
- Fis 1
mitochondria fission 1
- FS
fractional shortening
- GFP
green fluorescent protein
- HFD
high fat diet
- IVRT
isovolumic relaxation time
- KD
knocking down
- LV
left ventricle
- LC3
microtubule-associated protein 1A/1B-light chain 3
- Mff
mitochondria fission factor
- Mfn2
mitofusin 2
- Nnt
nicotinamide nucleotide transhydrogenase
- NMR
nuclear magnetic resonance
- ANOVA
one-way analysis of variance
- OCR
oxygen consumption rate
- PA
palmitate
- PCr/ATP
phosphocreatine/ATP
- PCR
polymerase chain reaction
- PDK4
pyruvate dehydrogenase kinase 4
- PGC1β
peroxisome-proliferator-activated receptor-gamma co-activator 1 beta
- Pink1
PTEN-induced kinase 1
- ROS
reactive oxygen species
- RT-PCR
real time PCR
- SEM
standard error of the mean
- SQSTM1
Sequestosome 1
- TAM
tamoxifen
- TFAM
transcription factor A, mitochondrial
- TG
triglyceride
- TL
tibia length
- TMRM
etramethylrhodamine methyl ester
- UCP3
uncoupling protein 3
Footnotes
References
- 1.Goldberg IJ, Trent CM and Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukushima A and Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta. 2016;1861:1525–1534. [DOI] [PubMed] [Google Scholar]
- 3.Wende AR and Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN and Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. [DOI] [PubMed] [Google Scholar]
- 5.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS and Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH and Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S and Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. [DOI] [PubMed] [Google Scholar]
- 8.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L and Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boudina S and Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. [DOI] [PubMed] [Google Scholar]
- 10.Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE and Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R and Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrauwen P, Schrauwen-Hinderling V, Hoeks J and Hesselink MK. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801:266–271. [DOI] [PubMed] [Google Scholar]
- 13.Kubli DA and Gustafsson AB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1527–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi S and Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:252–261. [DOI] [PubMed] [Google Scholar]
- 16.Chanseaume E, Tardy AL, Salles J, Giraudet C, Rousset P, Tissandier A, Boirie Y and Morio B. Chronological approach of diet-induced alterations in muscle mitochondrial functions in rats. Obesity (Silver Spring). 2007;15:50–59. [DOI] [PubMed] [Google Scholar]
- 17.Laurent D, Yerby B, Deacon R and Gao J. Diet-induced modulation of mitochondrial activity in rat muscle. Am J Physiol Endocrinol Metab. 2007;293:E1169–E1177. [DOI] [PubMed] [Google Scholar]
- 18.Saito T and Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R and Kolwicz SC Jr. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. 2016;100:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA, et al. Measuring In Vivo Mitophagy. Mol Cell. 2015;60:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katayama H, Kogure T, Mizushima N, Yoshimori T and Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. [DOI] [PubMed] [Google Scholar]
- 23.Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N and Clarke K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS and Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen S, Shao D, Tomasi LC, Braun A, de Mattos ABM, Choi YS, Villet O, Roe N, Halterman CR, Tian R, et al. The effects of fatty acid composition on cardiac hypertrophy and function in mouse models of diet-induced obesity. J Nutr Biochem. 2017;46:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bugger H and Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. 2010;88:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schilling JD. The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid Redox Signal. 2015;22:1515–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA and Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das KC and Muniyappa H. Age-dependent mitochondrial energy dynamics in the mice heart: role of superoxide dismutase-2. Exp Gerontol. 2013;48:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L, Zhao D, Ren J and Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:209–218. [DOI] [PubMed] [Google Scholar]
- 31.Han J and Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. 2016;57:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickles S, Vigie P and Youle RJ. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 2018;28:R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L and Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ and Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- 35.Jin SM and Youle RJ. PINK1-and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. [DOI] [PubMed] [Google Scholar]
- 37.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL and Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. [DOI] [PubMed] [Google Scholar]
- 38.Steinbusch LK, Luiken JJ, Vlasblom R, Chabowski A, Hoebers NT, Coumans WA, Vroegrijk IO, Voshol PJ, Ouwens DM, Glatz JF, et al. Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice. Am J Physiol Endocrinol Metab. 2011;301:E618–E627. [DOI] [PubMed] [Google Scholar]
- 39.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A and Sadoshima J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raher MJ, Thibault HB, Buys ES, Kuruppu D, Shimizu N, Brownell AL, Blake SL, Rieusset J, Kaneki M, Derumeaux G, et al. A short duration of high-fat diet induces insulin resistance and predisposes to adverse left ventricular remodeling after pressure overload. Am J Physiol Heart Circ Physiol. 2008;295:H2495–H2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. [DOI] [PubMed] [Google Scholar]
- 42.Durcan TM and Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khang R, Park C and Shin JH. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience. 2015;294:182–192. [DOI] [PubMed] [Google Scholar]
- 44.Zhan M, Usman IM, Sun L and Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. 2015;26:1304–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T and Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. [DOI] [PubMed] [Google Scholar]
- 46.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ and Dorn GW 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saupe KW, Spindler M, Tian R and Ingwall JS. Impaired cardiac energetics in mice lacking muscle-specific isoenzymes of creatine kinase. Circ Res. 1998;82:898–907. [DOI] [PubMed] [Google Scholar]
- 49.Malloy CR, Sherry AD and Jeffrey FM. Analysis of tricarboxylic acid cycle of the heart using 13C isotope isomers. Am J Physiol. 1990;259:H987–H995. [DOI] [PubMed] [Google Scholar]
- 50.Tu LN, Zhao AH, Hussein M, Stocco DM and Selvaraj V. Translocator Protein (TSPO) Affects Mitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology. 2016;157:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kabaeva Z, Zhao M and Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1667–H1674. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. [DOI] [PubMed] [Google Scholar]
- 54.Narendra D, Tanaka A, Suen DF and Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.