

Summary
Aminoacyl-tRNA synthetases (aaRSs) serve a dual role in charging tRNAs. Their enzymatic activities both provide protein synthesis flux and reduce uncharged tRNA levels. Although uncharged tRNAs can negatively impact bacterial growth, substantial concentrations of tRNAs remain deacylated even under nutrient-rich conditions. Here we show that tRNA charging in Bacillus subtilis is not maximized due to optimization of aaRS production during rapid growth, which prioritizes demands in protein synthesis over charging levels. The presence of uncharged tRNAs is alleviated by precisely tuned translation kinetics and the stringent response, both insensitive to aaRS overproduction but sharply responsive to underproduction, allowing for just-enough aaRS production atop a “fitness cliff”. Notably, we find that the stringent response mitigates fitness defects at all aaRS underproduction levels even without external starvation. Thus, adherence to minimal, flux-satisfying protein production drives limited tRNA charging and provides a basis for the sensitivity and setpoints of an integrated growth-control network.
Keywords: Aminoacylation, aminoacyl-tRNA synthetase, uncharged tRNA, translation, growth control, bacteria, gene expression, growth optimization
Graphical Abstract
eTOC Blurb:
An integrated analysis of tRNA charging, translation elongation, and bacterial growth rates reveals that the production of aminoacyl-tRNA synthetases is growth-optimized atop a ‘fitness cliff’ in Bacillus subtilis. The optimized production rates satisfies the flux required for peptide chain elongation without minimizing the levels of uncharged tRNAs, whose detrimental effects are alleviated by precisely tuned stringent response and autoregulation.
Introduction
Aminoacyl-tRNA synthetases (aaRS) catalyze a universal step in the synthesis of proteins by ligating amino acids to the appropriate tRNA, a processed termed ‘tRNA charging’. Their enzymatic activities determine two interrelated but distinct properties: the flux of charged tRNAs to the ribosome and the steady-state levels of uncharged tRNAs, both of which are important for regulating cell growth (Figure 1A). First, the flux of tRNA aminoacylation is directly related to the overall rate of peptide chain growth and hence the rates of mass accumulation and growth in bacteria (Bremer and Dennis, 2008; Dai et al., 2016). Second, the steady-state level of charged (acylated) tRNAs is inversely related to the level of uncharged (deacylated) tRNAs, which have several negative effects on cell physiology. On a global scale, uncharged tRNAs trigger stress responses that slow down cell growth in both bacteria and eukaryotes (Hinnebusch, 2005; Potrykus and Cashel, 2008; Srivatsan and Wang, 2008). In bacteria, uncharged tRNA levels are sensed by the (p)ppGpp synthetase RelA, whose activity reduces growth via a variety of pathways collectively referred to as the stringent response (Gourse et al., 2018; Hauryliuk et al., 2015; Liu et al., 2015). On the molecular scale, reduced tRNA charging is thought to cause ribosome pausing, which has adverse effects on mRNA stability and co-translational protein folding (Gloge et al., 2014; Hanson and Coller, 2018; Rauscher and Ignatova, 2018). Given the importance of aaRS’s dual roles, it is plausible that aaRS production is tuned to both minimize uncharged tRNA levels and provide ample capacity for charging flux during rapid cell growth.
Figure 1. Fitness landscape of tRNA synthetase production shows growth optimization near a fitness cliff.
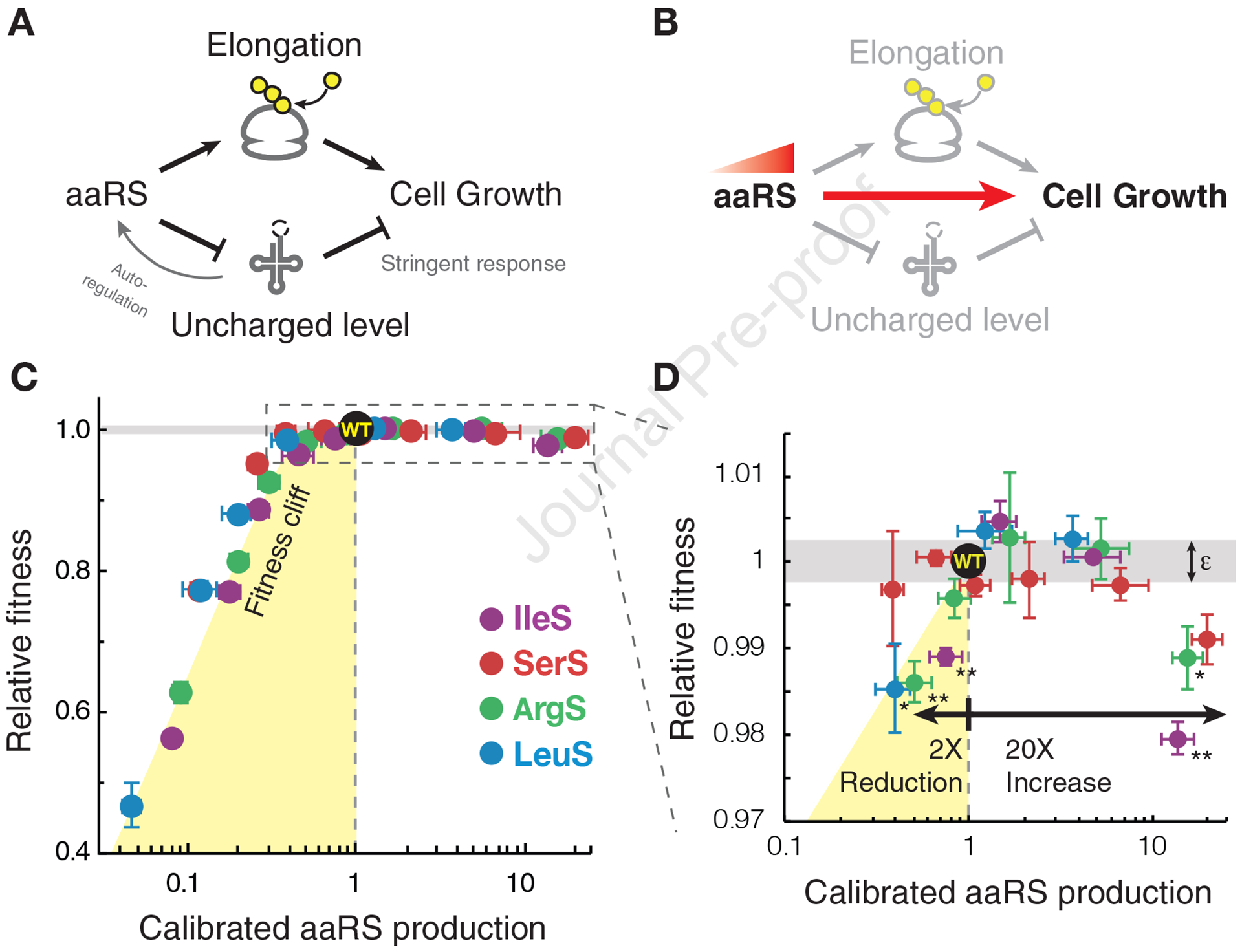
(A) Model describing the dual roles of aminoacyl-tRNA synthetase (aaRS) concentrations and their relation to the cellular growth rate (black lines). The production of aaRS is subject to negative feedback regulation by uncharged tRNAs in Bacillus subtilis (grey line). (B) Fitness landscape defined as the effective relationship between aaRS production and exponential-phase population growth rate. To measure the fitness landscape, we removed the negative feedback regulation to allow direct control of aaRS production. (C-D) Empirical fitness landscape of aaRS production. For each aaRS, gene expression is tuned by IPTG and the effective protein production is calibrated to its native levels using a method based on ribosome profiling (STAR Methods). Relative fitness is defined as the relative growth rate compared to barcoded control strains in pooled competition experiments. Yellow shaded regions indicate the approximate location of the fitness cliff. Data near wildtype fitness levels (dashed box) are depicted in (D). Error bars indicate standard error of fitness measurements for individually barcoded, biological replicates. Grey area indicates experimental uncertainty in fitness values relative to wildtype (ε=0.5%). Asterisks indicate p-value <0.05 (one asterisk) and <0.01 (two asterisks) from two-sample one-tailed Student’s t test comparing barcoded replicates between control and aaRS induction experiment. See also Figure S1.
However, a substantial concentration of uncharged tRNAs has been frequently observed in vivo even under rich nutrient conditions across divergent bacteria (Avcilar-Kucukgoze et al., 2016; Dittmar et al., 2005; McClain et al., 1999; Tockman and Vold, 1977; Yegian et al., 1966). For a wide spectrum of tRNAs – both within the same organism as well as across divergent bacterial species, the concentrations of uncharged tRNAs and total tRNAs are close to the same order of magnitude. This is despite the fact that the production of many aaRSs is directly set by uncharged tRNA levels (and not charging flux) via various negative autoregulatory mechanisms (Giegé and Springer, 2016; Gutiérrez-Preciado et al., 2009; Putzer et al., 1995; Vitreschak et al., 2008), suggesting that the setpoints of these feedback loops have evolved to avoid high levels of aaRSs and low levels of uncharged tRNAs. The potential tension between high concentrations of uncharged tRNA and their pleiotropic effects points to a global optimization problem of aaRS production that prevents maximal tRNA charging by cells.
Understanding this potential optimization in aminoacylation requires a systems-level characterization of the molecular and physiological consequences of altering aaRS production rates. A priori, it is unclear whether charging levels are sensitive to small perturbations in aaRS production, as multiple enzymes have been shown to be produced with excess capacities (Davidi and Milo, 2017; O’Brien et al., 2016; Peters et al., 2016; Sander et al., 2019). Even if charging levels can be increased with aaRS overproduction, whether higher charging leads to faster translation depends on additional biochemical parameters in vivo, such as the kinetics of ribosome translocation and availability of EF-Tu. Furthermore, elevated aaRS production may incur benefit-offsetting burdens on cell fitness, including cost of synthesis or synthetase specific overproduction defects (Dekel and Alon, 2005; Dong et al., 1995; Scott and Hwa, 2011; Sherman et al., 1992; Swanson et al., 1988). These considerations contribute to the fitness landscape of cells relative to aaRS production, which is potentially modified by the stringent response that connects growth rates to charging levels. In general, detailed quantitative relationships between protein production, biochemical activities, and phenotypic outcomes have been difficult to establish.
Here we determined the dependency and mechanistic underpinnings of bacterial cell growth on aminoacyl-tRNA synthetase production. High-precision measurements of cellular fitness and aaRS production rates show that these proteins are produced with little excess in order to optimize the growth rate of Bacillus subtilis in nutrient-rich conditions. Although tRNA charging is not maximized at the growth-optimized aaRS levels, the charging capacity matches the flux required to support translation elongation. At native aaRS production levels, the substantial pool of uncharged tRNAs does not lead to pleiotropic effects, due in part to a lack of strong activation of the stringent response. Nevertheless, cells remain exquisitely sensitive to even slight aaRS underproduction despite being relatively insensitive to overproduction. Notably, although the stringent response is known for curbing growth during external amino acid starvation, we find that it partially alleviates growth defects during aaRS underproduction – a condition in which de novo amino acid synthesis does not provide benefits. Ribosome profiling reveals that stringent null cells display both a lack of coordination of gene expression and exacerbated translation kinetic defects during aaRS underproduction. Together, these results show that the circuits for both controlling and sensing tRNA aminoacylation are collectively tuned to optimize bacterial growth, leading to unexpected properties of cellular mechanisms that are elucidated only with a fully integrated analysis.
Results
aaRS production is growth optimized on an acutely asymmetric landscape
We first mapped the quantitative relationship between cell proliferation rates and individual aaRS protein production in B. subtilis, a relationship we refer to as the fitness landscape (Figure 1B). Four essential synthetase genes were chosen as models (argS, ileS, leuS, and serS) for several key reasons. First, they reside in their own monocistronic operons, which allow for targeted perturbations. Second, these aaRSs are not heteromeric, therefore eliminating the need to maintain stoichiometric production of multiple subunits exogenously. Third, the regulation of these genes is representative of aaRS regulation in B. subtilis, which includes both feedback-regulation by T Box elements (ileS, leuS, and serS) and constitutive expression (argS). Although it is expected that cell growth would be affected at extreme levels of aaRS underproduction, the precise mapping of aaRS production onto fitness has not been determined.
In order to systematically vary aaRS production, we first moved each gene outside its native context and placed it under the control an IPTG-inducible promoter (Britton et al., 2002). Using this system, the effective aaRS production relative to the wildtype level ranges from tenfold underproduction to tenfold overproduction, as estimated by considering the IPTG-dependent promoter strength and the translation efficiency of the ectopic mRNA (measured by ribosome profiling) (Figure S1A, STAR Methods). At each IPTG concentration, the competitive disadvantage during exponential growth was measured using a pooled competition assay with DNA barcoding, which can reliably detect 0.5% differences in the net steady-state growth rate (Figure S1B). Fitness for aaRS inducible strains is defined as the growth rate compared to barcoded control strains with the same antibiotic resistance markers. The combination of these methods provides a generalizable approach for mapping the precise dependence of fitness on individual protein production.
The fitness landscapes for all synthetases showed a strong asymmetry between low and high levels of production (Figure 1C). At low levels of aaRS, fitness is acutely sensitive to aaRS production. This ‘fitness cliff’ may arise from insufficient flux of tRNA charging for cell growth or premature activation of the stringent response. By contrast, fitness is much less sensitive to high levels of aaRS, which may be due to a lack of changes in tRNA charging or reduced dependence of fitness on tRNA charging. The mechanistic basis underlying these behaviors is central to understanding bacterial growth regulation and is examined in further detail below.
We found that the wildtype production rates of aaRS are growth-optimized on the fitness landscape, placing cells near the edge of the fitness cliff. For all synthetases tested, no other aaRS expression level leads to a measurable increase in growth rate. Near the growth-optimized levels, cells with twofold less aaRS production have an average fitness defect of 1% (Figure 1D) and are steadily lost in the competition assay. In contrast, a greater than tenfold overproduction is required to produce a similar fitness defect (Figure 1D). The observed fitness landscape is quantitatively similar across different aaRSs, including SerS whose tRNA charging levels are often low and could potentially benefit from overproduction (Avcilar-Kucukgoze et al., 2016; Dittmar et al., 2005; Ferro et al., 2017). The asymmetric sensitivity is further confirmed by measuring mass doubling times at selected inducer concentrations (Figure S1C). Despite the comparatively small fitness effects of overproduction, cells appear to produce just enough aaRS proteins, leaving limited buffering capacity for tRNA charging for each of the aaRSs. Together, the asymmetric fitness landscape surrounding the growth optimum suggest that there is a non trivial transition in the level of tRNA charging or dynamics of mRNA translation near native aaRS production.
aaRS overproduction boosts tRNA charging despite a lack of fitness benefit
We next determined whether tRNA charging ceases to increase with aaRS overproduction, which could explain a lack of benefit in the overproduction regime (Figure 2A). Using Northern blotting to quantify aminoacylated and deacylated tRNAs, we found that the charging levels in wildtype B. subtilis cells were between 50–65% (Figure 2B), near the levels observed in other bacterial species (Avcilar-Kucukgoze et al., 2016; Dittmar et al., 2004; Yegian et al., 1966). For all aaRSs tested, ectopic overproduction (5 to 20-fold above native levels) increased the charging levels of cognate tRNAs, in some cases to nearly 90% (Figure 2B). These results indicate that the lack of fitness benefit beyond native aaRS levels is not due to cessation in tRNA charging increase.
Figure 2. aaRS overproduction reduces uncharged tRNA levels.
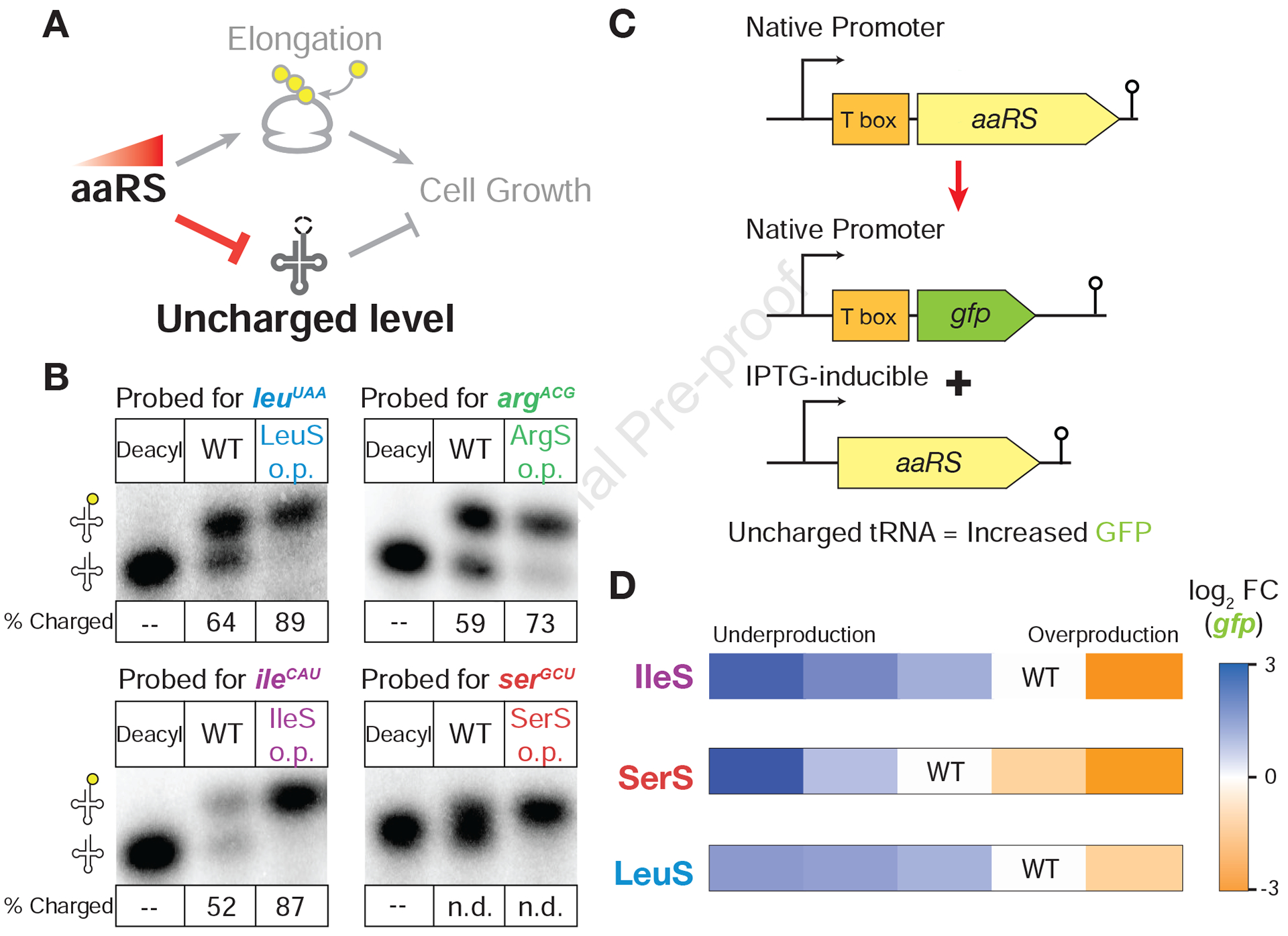
(A) Simplified relationship between aaRS production and uncharged tRNA levels. In practice, overproduction of aaRS may not reduce uncharged tRNA levels if there is already excess charging capacity at native levels of aaRS production. (B) Representative northern blots to determine tRNA charging levels in wildtype and aaRS overproduction strains. A control sample of deacylated tRNA was used to determine location of uncharged tRNAs. The middle lane of each blot contains tRNA extracted from wildtype cells (strain 168), and the final lane (o.p.) contains tRNA from aaRS induction strains with 500 μM IPTG (3.7x, 16x, 14x, and 20x overproduction for LeuS, ArgA, IleS, and SerS, respectively). Fraction of charged tRNA was calculated as the ratio between the lower band and the sum of the upper and lower band. Due to a lack of sufficient separation of aminoacylated serine tRNA, quantification was not determined (n.d.). (C) Schematic of reporter used to measure changes in uncharged tRNA levels. The native copy of the aaRS gene was replaced with a gfp gene and moved under the control of an IPTG-inducible promoter (pSpankHy) at the amyE locus in the genome. (D) T box reporter activity over range of aaRS production. For each inducible aaRS strain, the change in uncharged tRNA levels was reported by the fold change (FC) of gfp compared to induction conditions with near native aaRS production (indicated). Induction conditions with both under and over native production was tested and gfp mRNA measured by qRT-PCR. The native argS locus does not contain a T box and therefore was not tested in this assay. See also Figure S2.
To independently confirm these results, we created reporter systems utilizing the native autoregulation for aaRS genes (Figure 2C). In B. subtilis, most aaRS genes have T box leader elements in their 5’ UTR, which bind to uncharged tRNA and activate transcription in a concentration-dependent manner (Green et al., 2010; Gutiérrez-Preciado et al., 2009). Taking advantage of these sensors, we fused the T Box with a gene encoding GFP at the locus that originally harbors the native copy of synthetase gene. In this broken autoregulatory system, increases in uncharged tRNA levels can be read out by increases in gfp expression as measured by qRT-PCR (Figure 2C). These reporters confirmed that aaRS overproduction leads to higher charging (decreased gfp expression) (Figure 2D). They also showed that the riboswitches (and consequently charging level) are continuously responsive to a wide range of aaRS production, i.e. both under and overproduction (Figure 2D, S2A). We note that the magnitude of changes in gfp expression is dependent upon the sensitivity of the T Box elements and is not a quantitative readout of uncharged tRNA levels. Together with the fitness landscape profiling, these results suggest that the benefits of reducing uncharged tRNA levels during aaRS overproduction are limited and potentially negated by other processes in the cells.
Translation elongation is sensitive to decreased but not increased aaRS production
We next show that a transition in translation elongation kinetics underlie the saturated growth response associated with elevated aaRS production (Figure 3A). To estimate the relative ribosome transit time at each codon, we performed ribosome profiling on wildtype cells and the inducible aaRS strains across the expression landscape. The mean ribosome occupancy for each codon accounts for the total transit time across a full elongation cycle (Wu et al., 2019), including the waiting times for ternary complex binding and ribosome translocation, that determines the net rates of peptide chain synthesis and cell growth (Figure 3B).
Figure 3. Translation elongation kinetics change only with aaRS underproduction.
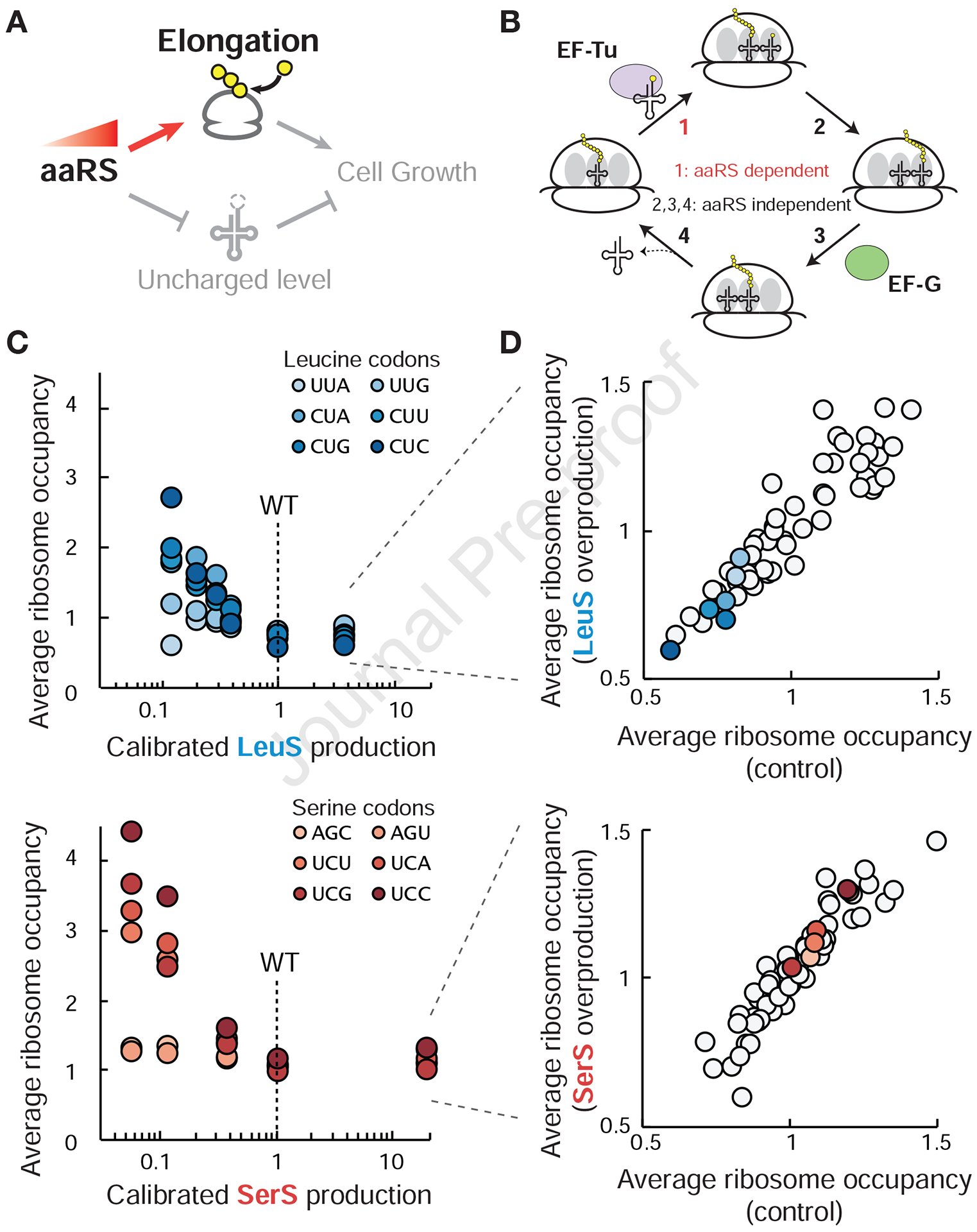
(A) Relationship between aaRS production and translation elongation rate to be tested. (B) Simplified schematic of the translation elongation cycle. The step of ternary complex binding (1) depends on aaRS production, but may not be rate-limiting for the elongation cycle. (C) Changes in codon occupancy across aaRS production. Average codon occupancy for indicated codons was acquired by ribosome profiling at the specific aaRS induction levels for LeuS and SerS (STAR methods). 81 genes for LeuS induction and 218 genes for SerS induction passed the depth threshold in all experiments and were used for analysis. (D) Average ribosome occupancy for all 61 sense codons for aaRS overproduction and control. See also Figure S3.
We found that ribosome transit time is sensitive to aaRS underproduction but not overproduction. aaRS reduction leads to increased ribosome occupancy for the cognate codons (Figure 3C and S3A,C), and even a two-fold reduction from the native production rates has a noticeable effect. Although the slower growth rates in these conditions may reduce global tRNA expression, such a global decrease is not expected to lead to the amino acid specific ribosome pausing that we observed when the corresponding aaRS is underproduced. Furthermore, we found that the levels of tRNA cognate to the underproduced aaRS are not reduced compared to other tRNAs, demonstrating that decreased tRNA charging is the major mechanism leading to increased ribosome pausing (Figure S3E). Consistent with previous reports in the context of amino acid limitation (Elf et al., 2003; Subramaniam et al., 2013a; 2013b), we observed a hierarchy of sensitivity among the cognate codons for both LeuS and SerS, where only four of the six codons saw a substantial increase in ribosome occupancy upon aaRS reduction. Such differential sensitivity may arise from differences in the ratio of codon usage and tRNA abundance (Elf et al., 2003; Subramaniam et al., 2013a; 2013b). These results show that slower translation due to reduced charging is associated with the acute sensitivity on the fitness landscape upon aaRS underproduction.
In contrast to underproduction, aaRS overproduction does not lead to substantial changes in the elongation kinetics. For codons cognate to the inducible synthetase, we observed similar ribosome occupancy during aaRS overproduction compared to wildtype (Figure 3D and S3B, D), suggesting that the elongation cycle is limited by other processes or that the increase in charged tRNA levels is not sufficient to accelerate translation elongation. Although it is possible that the transit time decreases below our limit of detection, the lack of sensitivity of elongation kinetics at elevated tRNA charging levels is consistent with several lines of evidence in the literature suggesting that the ternary complex concentration is near saturation for the elongation cycle (Dai et al., 2016; Gouy and Grantham, 1980; Klumpp et al., 2013; Subramaniam et al., 2014). Consequently, elevated aaRS production has little to no benefit on translation, even though tRNA charging is increased. In addition to this limited benefit, the observed decrease in fitness with elevated aaRS production may arise from the costs associated with unnecessary protein synthesis (~2% of global protein synthesis capacity with 10-fold overproduction for individual aaRS) (Dong et al., 1995; Scott et al., 2010). Together, these results show that the wildtype levels of aaRS production are set to provide just enough capacity for supporting translational flux instead of minimizing uncharged tRNA levels.
The stringent response is protective during aaRS underproduction in rich media
The omnipresence of uncharged tRNAs, even at native aaRS production, suggests that (p)ppGpp and the stringent response may play an important role in shaping the fitness landscape. The physiological consequences of the stringent response are well-studied under conditions of external amino acid starvation, but how the response affects aaRS optimization under nutrient-rich conditions is unknown. Gene expression profiling by RNA sequencing across the aaRS production landscape showed signatures of elevated (p)ppGpp levels only during aaRS underproduction, including branch chain amino acid biosynthesis and CodY-controlled gene upregulation (Figure 4A, S4A) (Eymann et al., 2002; Kriel et al., 2014; Sonenshein, 2005). This upregulation was prominent despite the fact that excess amino acids are provided in the media. A major role of the stringent response during amino acid limitation is growth reduction by diverting gene expression away from genes involved in protein synthesis and towards those involved in de novo amino acid biosynthesis. However, since amino acids are readily available in the media, spending resources on creating biosynthetic enzymes is unnecessary and could be wasteful, thereby amplifying the effects of insufficient tRNA charging during aaRS underproduction. Alternatively, the redistribution of resources may instead be beneficial due to reduction in ribosome production. These two models predict opposite growth effects upon removal of the stringent response (Figure 4B).
Figure 4. Removal of the stringent response negatively impacts cell growth during aaRS underproduction.
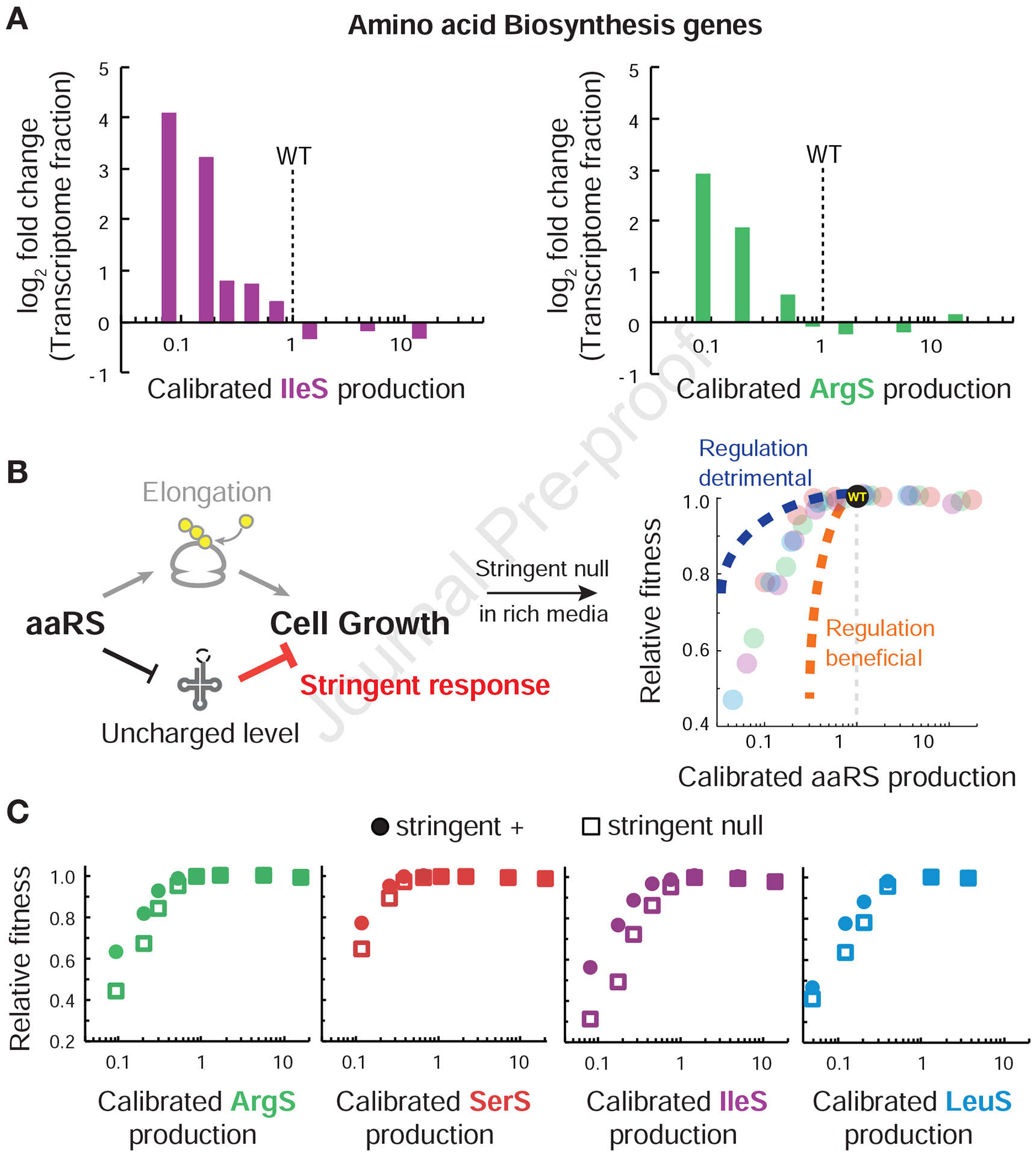
(A) Induction of CodY-controlled amino acid biosynthetic genes as a proxy for stringent response activation across aaRS expression. RNA-seq was performed on strains producing IleS and ArgS over a range of induction conditions. The fold change in transcriptome fraction (RPKM) for genes related to amino acid metabolism was compared to wildtype. See Table S1 for list of genes. aaRS production was calibrated in the same way as Figure 1C. (B) Potential models for aaRS fitness landscape for cells lacking the stringent response. The stringent response may amplify the effects of insufficient tRNA charging (Regulation detrimental), or reduce adverse effects (Regulation beneficial). (C) Effects on aaRS fitness landscape of stringent response removal. Strains with stringent response intact (circles) were competed against stringent null cells (squares) across the aaRS production landscape. Measurements of fitness and aaRS production were calculated as in Figure 1. See also Figure S4.
To test the effect of the stringent response on cell growth when aaRS is underproduced in rich media, we measured fitness of B. subtilis strains with inducible aaRS but lack the ability to activate RelA-dependent (p)ppGpp synthesis. In these strains, we introduced a point mutation to the native copy of relA (relA D264G) that has been previously shown to abolish the (p)ppGpp synthetase activity while retaining the ability to hydrolyze background levels of (p)ppGpp, as evidenced by no detectable (p)ppGpp during serine hydroxamate treatment (Nanamiya et al., 2008). As a control, we found that this mutation has only a mild effect on growth at native aaRS levels (0.6 ± 0.3% relative fitness) and few gene expression changes (Figure S4B, S4C). However, upon aaRS underproduction, strains without the stringent response showed further exacerbated fitness cliffs as measured by the competition assay (Figure 4C). Across all aaRSs tested, removal of (p)ppGpp synthetase activity leads to an additional decrease in fitness of ~2.5% at ~2-fold aaRS underproduction, and ~25% at ~10-fold underproduction. The severe growth defects were further confirmed by measuring mass doubling of individual cultures (Figure S4C). These results indicate that, instead of curtailing bacterial growth rates, the stringent response in fact enhances growth when there is insufficient aminoacyl-tRNA synthetase activity in the absence of external starvation.
We next characterized the molecular defects in mRNA translation associated with the reduced fitness in stringent-null cells during aaRS underproduction. In E. coli, the stringent response enforces a linear relationship between growth rate and the production of translation-related proteins (Scott et al., 2010; Zhu and Dai, 2019), such that ribosomes are operating near capacity at most growth rates. Using ribosome profiling to quantify the allocation of global protein synthesis to translation-related factors, we found that B. subtilis cells with aaRS underproduction adhere to a similar linear relationship when the stringent response is present (Figure 5A). This applies to both core ribosomal proteins and translation factors, such as other aaRSs that are not externally controlled. However, the coordinated reduction is lost in cells without the stringent response, which overproduce ribosomal proteins and translation factors without matching production of supporting aaRSs (Figure 5A and S5A).
Figure 5. Stoichiometry and kinetics of translation machinery are perturbed without stringent response.
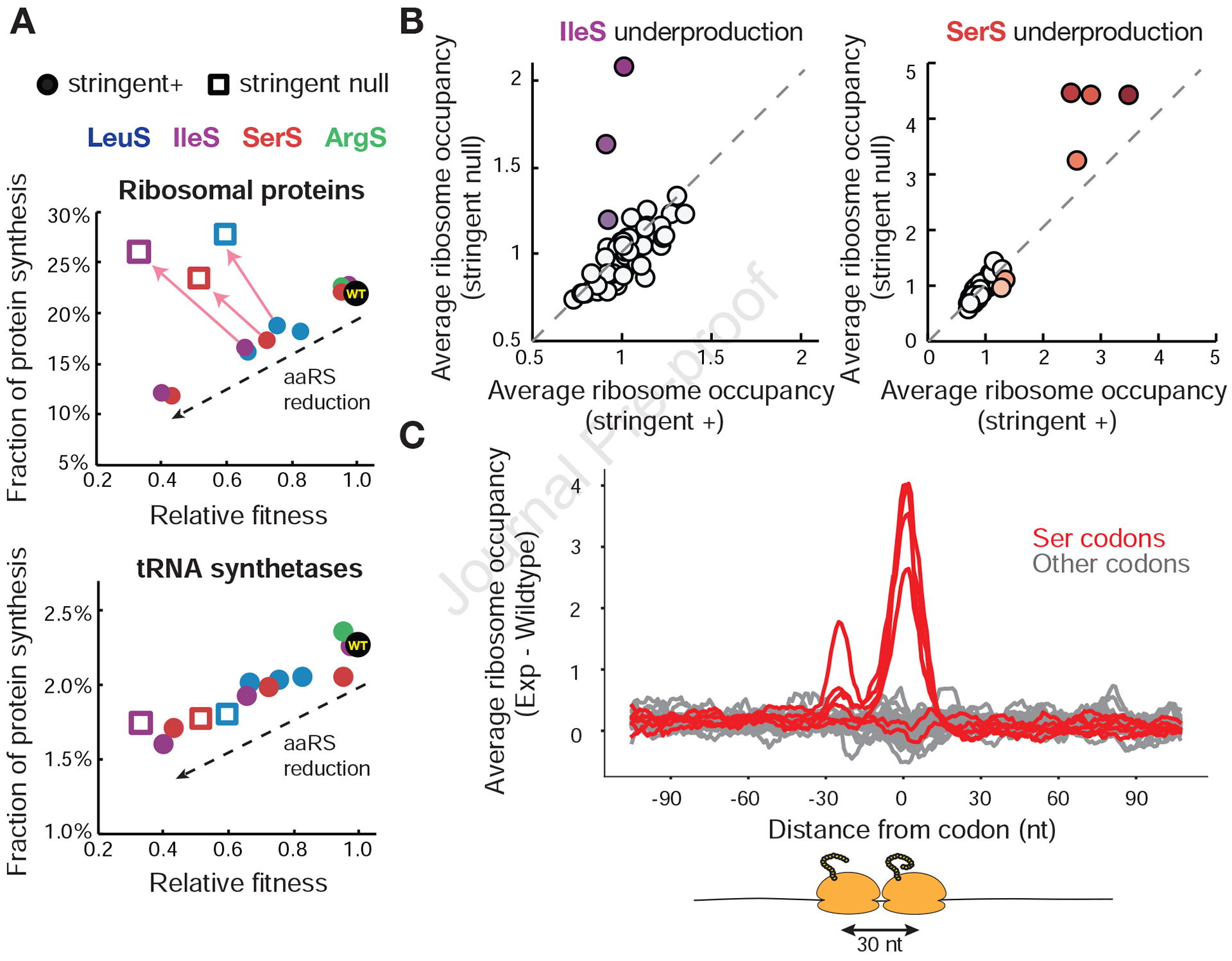
(A) Fraction of protein synthesis devoted to translation-related genes as a function of relative fitness in cells with and without the stringent response. For each inducible aaRS strain at each induction level profiled, the relative fitness as measured by the competition assay was plotted against the fraction of protein synthesis (rpkm) as measured by ribosome profiling for ribosomal proteins and aminoacyl-tRNA synthetases (STAR Methods, Table S1). Stringent positive cells are plotted as closed circles, stringent null cells as open squares. Arrows point to changes when the stringent response is removed at identical induction conditions. (B) Ribosome occupancy changes due to stringent response removal for IleS and SerS underproduction. Average codon occupancy for 61 sense codons with matched induction conditions is plotted for cells with and without the stringent response. Induction conditions lead to 0.18X and 0.12X native production levels of IleS and SerS based on calibration. Colored codons indicate cognate codons to the inducible aaRS. 310 genes for IleS induction and 218 genes for SerS induction passed the depth threshold in paired experiments and were used for analysis. (C) Metagene analysis of ribosome pausing and queuing during SerS underproduction in strains lacking the stringent response. Genes were aligned from each of the 61 codons and averaged across them. Difference in ribosome occupancy between SerS underproducing cells without the stringent response and wildtype is plotted. Red traces indicate serine codons. For full calculation see STAR Methods. See also Figure S5.
We also found that the uncoordinated production of translation machinery is accompanied with exacerbated ribosome pausing and can even lead to ribosome queuing during translation. For cells that cannot activate the stringent response, ribosome profiling data showed higher ribosome occupancy at the starved codons compared to cells with an intact stringent response (Figure 5B). In the case of SerS, underproduction leads to elevated ribosome occupancy at regularly spaced intervals upstream of severely starved codons, suggesting that additional ribosomes are queued behind the waiting ribosome (Figure 5C). This effect is only observed when the stringent response is removed (Figure S5B). Ribosome queuing is potentially a consequence of overcapacity of protein synthesis in strains deficient in the stringent response, which could in turn further exacerbate the growth defect of the cell. In summary, these findings show that RelA-dependent (p)ppGpp synthesis helps avoid adverse effects on translation during forced reduction in tRNA charging.
Importantly, we found that the majority of gene expression changes during aaRS underproduction are mediated through the stringent response. In addition to the translation machinery, the bulk of the changes between wildtype and aaRS underproduction involved increased expression of known genes that promote survival under starvation, such as those regulated by CodY and SigB that fail to increase in the stringent null cells (Eymann et al., 2002; Zhang and Haldenwang, 2003) (Figure S5C). Overall, stringent null cells during aaRS underproduction show relatively few expression changes compared to WT (with intact stringent response and native aaRS levels) (Figure S5C). Of the few increases in expression selectively in the stringent null cells, we find that clpE, a AAA+ unfoldase and subunit of the ClpE-ClpP protease, is massively increased in expression, indicating potential protein folding stress due to the observed translation pausing. However, despite the few changes and lack of wasteful amino acid biosynthesis expression due to growth in rich media, stringent null cells still display lower fitness. Taken together, we find that the benefits of the coordinated production of translation components outweigh the unnecessary production of amino acid biosynthetic enzymes, making the stringent responsive protective against perturbations in aaRS levels in rich media.
Native levels of uncharged tRNAs cause limited stringent response in rich media
If the stringent response remains activated by the native levels of uncharged tRNAs, it could adversely modify the optimal levels aaRS production. However, we found that in the absence of aaRS perturbations, the stringent null cells have similar gene expression patterns as wildype cells, including ribosomal protein genes, amino acid biosynthetic genes, and CodY-controlled genes (Fig. S4C). At the level of translation, eliminating the stringent response with native aaRS production does not lead to substantial differences in ribosome pausing (Fig. S5D). These results corroborate the limited fitness effects of the relA D264G mutation in otherwise wildtype cells under nutrient-rich conditions (Fig. S4B). Further increasing aaRS production from native levels also showed limited differences in (p)ppGpp-regulated genes, ribosome pausing, and fitness (Fig. 3D, S3B, 4A and S4A), which is in stark contrast to aaRS underproduction in the same media. Therefore, the stringent response is sensitively programmed to activate with slight aaRS underproduction but not at native levels (Figure 4A and S4A), even though the latter state is also associated with substantial amounts of uncharged tRNAs. Taken together, these data show that the stringent response participates in ameliorating the fitness cliff without affecting optimal aaRS production rates.
Discussion
Here we thoroughly characterized the mechanistic underpinnings of bacterial growth optimization with respect to aminoacyl-tRNA synthetase production. By profiling the molecular and physiological effects that contribute to the overall global fitness landscape, we showed that the production of aaRS proteins is tuned to satisfy the requirement of tRNA charging flux and not to minimize the steady-state levels of uncharged tRNAs during growth in rich media. Moreover, our work yielded insights into several fundamental biological processes that have been well studied in isolation but are nevertheless extensively cross-regulated in living cells.
First, we found that native levels of aaRS production provide limited excess capacity to support translation. For the synthetases examined, a slight decrease from native production rates results in slower translation at cognate codons and a fitness cliff (Figure 6), suggesting that the enzymes are normally operating near saturation in fast growth conditions. This ‘just enough’ aaRS production stands in contrast to the situations of enzymatic over-capacity proposed for other metabolic enzymes, which are thought to be advantageous by providing a buffer against environmental or stochastic fluctuations (Davidi and Milo, 2017; O’Brien et al., 2016; Peters et al., 2016; Sander et al., 2019). Our findings corroborate a recent study on the origin of haploinsufficient genes in yeast suggesting that the expression of haploinsufficient genes are limited by fitness costs associated with their overexpression (Morrill and Amon, 2019). In the case of aaRSs, we found that even a small fitness defect of overproduction, by comparison to underproduction, could drive evolution towards an expression that is next to the fitness cliff. In addition, the homeostatic feedback of uncharged tRNAs on aaRS production may further ensure that cells maintain sufficient tRNA charging capacity for translation elongation (Figure 6). Whether or not these same principles apply to growth in nutrient poor conditions when amino acids must be synthesized de novo remains to be explored. Finally, given that all four aaRSs tested here show limited excess capacity, our findings demonstrate that bacterial growth may be limited simultaneously by multiple enzymes in the terms of their underproduction and not overproduction, akin to ribosomal production.
Figure 6. An integrated view of growth optimization for aaRSs.
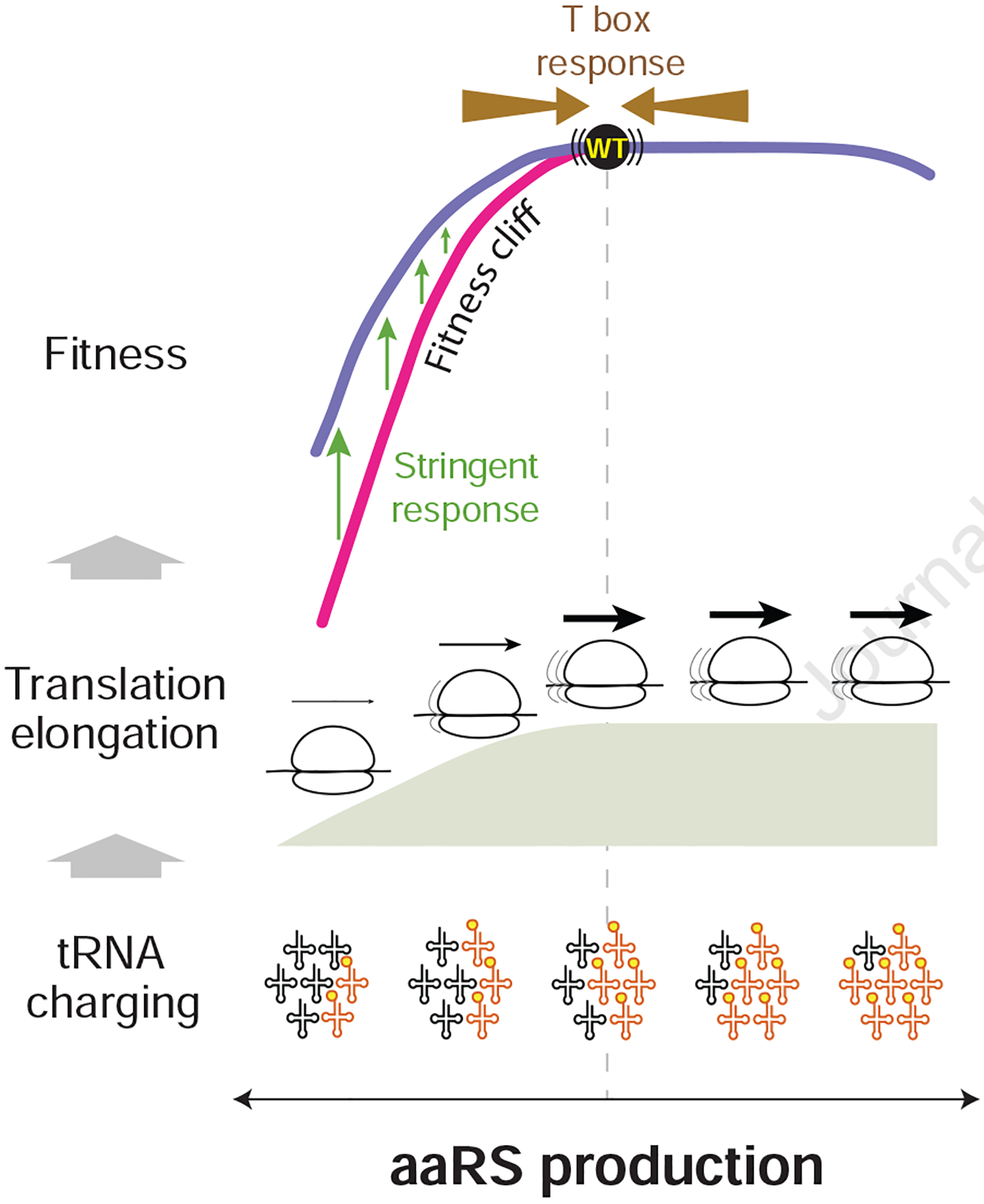
At growth optimized levels of aaRS production (vertical dashed line), tRNA charging is not yet maximized. However, further increasing aaRS production and tRNA charging does not result in faster translation elongation, leading to no fitness benefit during overproduction. The native levels of aaRS production, with just enough capacity for the amino acid flux into protein synthesis, are positioned near the edge of the fitness cliff in the highly asymmetric landscape. The sharp sensitivity to aaRS underproduction is partially alleviated by the stringent response despite the unnecessary activation of amino acid biosynthesis genes in rich media. Furthermore, the T box-based autoregulation buffers perturbations to aaRS production from both directions, which is enabled by having non-maximized tRNA charging at the native levels.
Second, our results show that the feedback regulation for aaRS synthesis is programmed to prevent both overproduction and underproduction despite the highly asymmetric fitness landscape (Figure 6). As a homeostatic mechanism, negative feedback regulation may not be constantly active. In one design scenario, the feedback may be constitutively on and inactivated only during reduced output to ensure sufficient charging capacity. Alternatively, it may be constitutively off and activated only during excessive output to prevent wasteful aaRS production. Although the extreme sensitivity to aaRS underproduction suggests a priority to ensure sufficient charging capacity, our T box reporters show that the native autoregulation remains responsive to both under and overproduction, which is consistent with a previous report for thrS and valS (Gendron et al., 1994; Luo et al., 1997). Therefore, the negative feedback loop for aaRS has not evolved to be a merely single-purpose device that is protective in only one direction. Importantly, this ability to respond in both regimes is rooted in the fact that the signal for feedback, i.e. tRNA charging, is not fully saturated at the native aaRS production (Figure 6).
Finally, similar to how translation speed is only sensitive to aaRS underproduction, global growth regulation by the stringent response is activated with mild aaRS reduction while having limited effects at native uncharged tRNA levels. Although it is possible that some (p)ppGpp is produced with native aaRS production or even in the relA D264G mutant used here, our gene expression data for aaRS overproduction suggest that these basal uncharged tRNA levels do not substantially activate the stringent response. Given that the native uncharged fraction is estimated to be 10–50% for most tRNAs, the concentration of uncharged tRNAs cannot increase drastically with a small decrease in aaRS production. How the stringent response is programmed to be sharply activated by small changes in uncharged tRNA levels remains to be resolved, and recent structural analyses on how RelA is activated on the ribosome may provide insights (Loveland et al., 2016; Winther et al., 2018). Upon activation, the stringent response curbs ribosome biogenesis and promotes de novo amino acid biosynthesis. However, the latter likely leads to wasteful production of anabolic enzymes when aaRS is limiting in the presence of externally supplied amino acids. It is therefore striking that we observed a strong protective effect of the stringent response during aaRS limitation (Figure 6). We propose that the maintenance of protein stoichiometry for the translation apparatus contributes to the protective effect. Without the stringent response, ribosomal protein and some translation factors remain highly expressed, whereas most aaRSs (in addition to the limiting one) have reduced expression in line with the growth rate. The resulting imbalance between ribosomes and aaRSs, together with defects in other (p)ppGpp-dependent regulation (Srivatsan and Wang, 2008; Trinquier et al., 2019), may lead to the observed translation defects, including exacerbated ribosome pausing and queuing. This result underscores the importance of proper stoichiometry among functionally associated proteins and offers an explanation for the broadly conserved protein stoichiometry across a wide range of species (Lalanne et al., 2018).
Elucidating the interdependence between gene expression, protein activities, and cellular fitness is a fundamental problem in systems biology. With the rapidly expanding knowledge of gene expression and protein functions, a key challenge is to understand how these molecular properties are integrated in vivo to define the behavior of a cell. Our work outlines an approach to connect the fitness landscape to its mechanistic understandings, providing powerful insight into the optimization of protein abundance and regulation.
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gene-Wei Li (gwli@mit.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The Ribosome profiling and RNA sequencing datasets generated during this study are available at the Gene Expression Omnibus with accession number GSE141389. The raw sequencing files of the amplicons generated from competition experiments is available in the NCBI Sequencing Read Archive (SRA) under accession number PRJNA593245. Full code is available upon request. The raw image for Northern blots was deposited at Mendeley Data and can be viewed at https://data.mendeley.com/datasets/f9kkszrtch/draft?a=1fcb4abc-bd3d-452a-94c1-caa43ee378ca.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strain construction
Bacillus subtilis 168 strain was used as the wild-type strain and base strain for transformations. All further strains were constructed using natural competence as described below. For all assays, cells were grown in a defined rich minimal media (MCCG) described below. All inducible aaRS strains were created by first cloning individual aaRS ORFs under the pSpankHY promoter system in a plasmid containing the lac repressor, a spectinomycin resistance cassette, and flanked by 500 nt homology arms of the amyE locus (Britton et al., 2002). After creation, plasmids were linearized, transformed into B. subtilis 168, and selected with 100 μg/mL spectinomycin. To remove the native copy of the aaRS gene, a transcriptional fusion of GFP and a chloramphenicol resistance cassette (CmR) was assembled using Gibson assembly with additional flanking 500-nucleotide homology arms to the aaRS locus of interest (Gibson et al., 2009). The complete linearized DNA product were directly transformed into the inducible aaRS strains and selected on LB-agar plates with 5 μg/mL chloramphenicol and 100 μM IPTG. To the generate stringent null strains, a gBlock DNA fragment (Integrated DNA technologies) was purchased containing 378 nt of the relA gene sequence: 186 nt upstream and 189 nt downstream of codon 264 while changing codon 264 GAT (Asp) to GGT (Gly). The full sequence can be found in Table S2. Fragments containing further upstream and downstream arms of the relA locus as well as a Kanamycin resistance gene (KanR) were assembled. Upon full product amplification by PCR, DNA was transformed into strains and selected on LB plates with 10 μg/mL Kanamycin.
METHOD DETAILS
Strain construction
Competent cells were prepared by following protocol; Cells to be transformed were streaked onto LB plates with the appropriate selection marker. The following morning a single colony was picked into 5 mL of LM media (LB + 3 mM MgSO4). Cultures were incubated at 37°C with shaking until OD600 was between 0.8 and 1.2. 300 μL of cells were then transferred to 3 mL competence media (1% Glucose, 3 mM MgSO4, 2.5 mg/mL Potassium Aspartate, 11 μg/mL Ferric Ammonium Citrate, 40 μg/mL Tryptophan, 10.7 g/L Dipotassium Phosphate, 6 g/L Monopotassium Phosphate, 1 g/L Trisodium Citrate Dihydrate) and incubated with shaking for 3.5 hours at 37°C. A 200 μL aliquot of competent cells was mixed with 100–1000 ng of DNA and shaken at 37°C for an additional 1.5 hours before plating on the appropriate selection plates. Selection was performed on LB agar plates using the following concentrations of antibiotics; 10 μg /mL Kanamycin, 100 ng/mL Spectinomycin, 5 μg /mL Chloramphenicol, and/or 10 μg/mL Tetracycline.
Growth media
For all experiments, a defined rich media was used for growth of cells (MCCG). See Table S3 for a complete recipe. Given the essentiality of the aaRS genes tested, all strain maintenance, overnight growth conditions, or LB-agar plates included 100 μM IPTG. For competition experiments “conditioned MCCG” media was used instead of fresh MCCG media. To create conditioned MCCG, wildtype B. subtilis 168 was back-diluted 2000-fold from an overnight culture into 0.5 L of fresh MCCG media. Cells were grown with vigorous shaking at 37°C until OD600 = 0.2. The culture was then immediately poured over a 0.2 μm vacuum filtration flask, collecting the sterilized “conditioned” MCCG media. We found that conditioned MCCG media nearly eliminates a lag phase (0–5 minutes) upon back-dilution during the competition experiment while fresh MCCG induces a 10–15 minute lag phase before the resumption of exponential growth (data not shown).
qRT-PCR
For qRT-PCR measurements, overnight cultures of cells were back-diluted 1000-fold and grown in fresh MCCG media with the appropriate concentration of IPTG. When the culture reached OD600 = 0.3, cells were collected by mixing 5 mL of culture with 5 mL of ice-cold methanol. After 10 minutes on ice, cells were spun down at 4°C for 10 minutes, decanted of excess liquid and resuspended in 100 μL of a solution containing 10 mg/mL Lysozyme, 10 mM Tris-HCl pH 7, and 1 mM EDTA. RNA was extracted using RNeasy Mini Kits (Qiagen) according to the manufacturers protocols for Gram-positive Bacteria. Random hexamer reverse transcription with Moloney Murine Leukemia Virus (M-MuLV) (M0253; NEB) was performed according to the manufacturer’s specifications. Gene expression changes were normalized to sigA. For a list of primers used see Table S2.
Ribosome Profiling
For ribosome profiling experiments, cells were grown in fresh MCCG media, using a protocol outlined previously (Lalanne et al., 2018; Li et al., 2014). An overnight liquid culture, started from a single colony from a fresh plate, was diluted about 5,000-fold into 250 mL fresh MCCG media in a 2.8 L flask at 37°C with aeration (200 rpm) until OD600 = 0.4. The cell culture was rapidly filtered over a nitrocellulose filter and scraped into liquid nitrogen at 37°C. Cell pellets were combined with 650 mL of frozen droplets of lysis buffer 10 mM MgCl2 , 100 mM NH4 Cl, 20 mM Tris pH 8.0, 0.1% NP-40, 0.4% Triton X-100, 100 U/mL DNase I, and 1 mM chloramphenicol. Cells and lysis buffer were pulverized in 10 mL canisters prechilled in liquid nitrogen using a TissueLyser II (Qiagen) for 5 cycles of 3 min at 15 Hz. Pulverized lysate was thawed on ice and clarified by centrifugation at 20,000 rcf. for 10 min at 4°C. 0.5 mg of clarified lysate was then digested with 5 mM CaCl2 and 750 U of micrococcal nuclease (Roche) at 25°C for 1 hr before quenching with 6 mM EGTA.
Following nuclease digestion, the monosome fraction was collected using sucrose gradient and the RNA extracted by hot-phenol extraction. Ribosome-protected mRNA fragments (footprints) with size ranging from 15 to 45 nucleotides were size selected on a polyacrylamide gel. Footprint RNA was dephosphorylated using T4 polynucleotide kinase (NEB) at 37°C for one hour. Three picomoles of footprints were ligated to 100 pmole of 5’ adenylated and 3’ end blocked DNA oligo (Linker-1, Table S2) using truncated T4 RNA ligase 2 K277Q at 37°C for 2.5 hr. The ligated product was purified by using polyacrylamide gel. cDNA was generated using Superscript III (ThermoFisher) and custom primer oCJ485 (Table S2) and isolated by size excision on a polyacrylamide gel.
Single-stranded cDNA was circularized using 100 U of CircLigase (Lucigen) at 60°C for 2 hr. Ribosomal cDNA fragments were removed using biotin-linked custom DNA oligos (Table S2) and MyOne Streptavidin C1 Dynabeads (ThermoFisher). Final cDNA was amplified using Q5 DNA polymerase (NEB) with o231 primer and indexing primers (Table S2). Sequencing was performed on an Illumina HiSeq 2000 or NextSeq500.
RNA sequencing
For RNA-seq experiments, overnight cultures of cells were back-diluted 1000-fold and grown in fresh MCCG media with the appropriate concentration of IPTG. When the culture reached OD600 = 0.3, cells were collected by mixing 9 mL of culture with 1 mL of ice-cold stop solution (95% Ethanol, 5% Phenol). Cells were spun down for 10 minutes at 20,000 rcf in a 4°C centrifuge. Excess solution was decanted and removed by pipette. Cell pellets were resuspended in 200 μL 10mM Tris-HCL 1mM EDTA and split into two 100 μL fractions.
To one fraction, cells were lysed with 10 mg/mL lysozyme and RNA extracted using RNeasy column (Qiagen #74104). Purified RNA was ribosomal RNA depleted with MICROBExpress Bacterial mRNA Enrichment Kit (ThermoFischer #AM1905) according to the manufacturer’s instructions. RNA from these samples was to be used for gene expression experiments having been depleted for rRNA and tRNA.
To the other fraction, the 100 μL aliquot was re-spun and liquid was removed by pipette. RNA from cell pellet was extracted using RNASnap (Stead et al., 2012). Briefly, cell pellets were resuspended in 500 μL Extraction solution (95% Formamide, 1% BME, 0.025% SDS, 18 mM EDTA) and 200 μL chilled zirconia beads (0.1 mm diameter). Resuspensions were vortexed for 10 minutes using a horizontal vortex adapter, incubated for 7 minutes at 95°C, and 300 μL of the aqueous solution was transferred to a fresh tube. Samples were spun for 5 minutes at 16,000g and supernatant was diluted with four volumes of water in a fresh tube and precipitated with five volumes of Isopropanol. After freezing and centrifugation, RNA was resuspended in 10 mM Tris pH 7. RNA from these samples was to be used for total RNA expression experiments and was not depleted for rRNA or tRNA.
Each RNA sample was then fragmented (ThermoFischer #AM8740), dephosphorylated, polyadenlyated, and reverse transcribed using Superscript III and custom oligo-dT reverse transcription primers. Resulting cDNA was ligated with Illumina sequencing adapters and PCR amplified. Single end (50 bp) sequencing was performed on an Illumina NextSeq.
AUPAGE & Northern Blot
tRNA charging analysis was performed similarly to (Darnell et al., 2018; Kimura and Waldor, 2019; Varshney et al., 1991). Cells were grown to OD600 0.3, mixed 50:50 with pre-warmed 10% Trichloroacetic Acid, and placed on ice. Mixtures were spun down at 3000 × g for 10 minutes at 4°C and the supernatant was poured off. Cell pellets were then resuspended in 750 μL of AE buffer (0.3 M NaOAc pH 4.5, 10 mM EDTA) and either frozen at -80°C or immediately mixed with 750 μL ice cold Phenol pH 4.5 before transferring to a 2 mL tube. Mixtures were vortexed for 10 min, rested on ice for 5 min, and spun for 10 min at 4°C. Aqueous supernatant was removed and mixed with 750 μL cold Isopropanol. Another 750 μL of AE buffer was then added to the remaining organic layer before another round of vortexing and separation was performed. The aqueous layer was transferred to another tube with 750 μL cold Isopropanol and all samples were vortexed and precipitated for more than two hours at -20°C. After spinning down precipitations, pellets were washed with 70:30::Ethanol:10 mM NaOAc pH 4.5 and dried. Pellets were re-dissolved in SA Buffer (10 mM NaOAc pH 4.5, 1 mM EDTA) and quantified using Qubit BR RNA kit. To mark gel mobility shift of uncharged tRNA, total RNA samples were deacylated in 100 mM Tris pH 9.5 at 37° for 1 hour then precipitated and re-dissolved in SA buffer.
To perform the acid urea gel electrophoresis, 0.5 – 1 μg of each sample was mixed with 2X loading buffer (0.1 M NaOAc pH 5.0, 9 M urea, 0.05% bromophenol blue, and 0.05% xylene cyanol) and loaded onto a gel containing 6.5% polyacrylamide, 7 M Urea, and 0.1 M NaOAc pH 5.0. Following electrophoresis, the region between the bromophenol blue and xylene cyanol bands was excised and washed in 1X TBE. Gels were transferred to BrightStar-Plus positively charged nylon membrane in 1X TBE using a semi-dry transfer apparatus at 3 mA/cm2 for 30 min. The blots were crosslinked twice with 1,200 μJ UV light, prehybridized in PerfectHyb buffer for 30 min at 42°C, and hybridized at 42°C with 5 pmol of the appropriate probe (see Table S2). Probes were end-labeled with [γ-32P] ATP using T4 PNK and purified with G25 Sepharose columns. Blots were twice washed with Wash Buffer (2X SSC and 0.5% SDS) at 60°C, exposed to a Phosphor-Imaging screen for 1 hour, and imaged using a FLA-5000 phosphoimager (Fuji). To properly assess the absolute charging level for samples with very high charging fractions, we first extracted raw intensities from the northern blot using ImageJ (Schneider et al., 2012). Using the x-coordinates of positions of maximal peak heights in wildtype lanes, we fit two gaussians around the uncharged and charged fractions and calculated the area under the curve. The charged fraction was calculated as the area under the charged band divided by the sum of the charged and uncharged bands. These peak positions were used to fit curves for both bands in the aaRS overproduction samples, calculating the charging ratio in the same way.
Competition Experiments
To barcode strains for competition analysis a barcoding plasmid was created to integrate at the lacA locus with a tetracycline resistance gene tetM (genotype lacA::BC-tetM). Each plasmid has a random 12 nucleotide sequence near the end of lacA integration site to replace the endogenous sequence and differentiate the strain from the others by DNA sequence. All “strain-types” (i.e. argS inducible strain or ileS inducible with relA D254G mutation strain) used in the competition experiment except for wildtype 168 were transformed with the barcoding plasmid. For all strain-types used in the experiment, 2–4 independent clones after transformation with verified unique barcodes were kept and used in the competition experiment as biological replicates and termed unique “strain-BC”. Prior to the experiment, all strains were streaked out and single colonies of each strain type and barcode were picked and grown overnight to saturation. The following morning, 5 μL of the overnight culture for each strain was mixed together thoroughly before back-dilution into fresh conditioned MCCG media with specified inducer concentration (see ‘Media’ for details). Eight concentrations of inducer (IPTG) was tested: 10, 15, 20, 25, 30, 40, 70, and 500 μM. When the culture reached OD600 = 0.1, 156 μL of culture was transferred to 10 mL fresh, pre-warmed, shaking conditioned MCCG to continue exponential growth (1/64 or 26 back-dilution). The remainder of the cells were spun down at 4°C, decanted of media and immediately frozen for eventual DNA extraction. Subsequently, when cells again reached OD600 = 0.1 another back-dilution was performed and cells were frozen for DNA. This process continued until reaching roughly 40 generations total. Due to the extensive experiment time some later rounds of back-dilution were decreased (312.5 or 625 μL OD600=0.1 culture into 10 mL fresh culture: 1/32 or 1/16 back-dilution). This decrease in generation time was noted during quantification & final analysis.
To generate amplicons for sequencing and barcode counting frozen cell pellets were first thawed and resuspended in 480 μL 50 mM EDTA and 120 μL 10 mg/mL lysozyme. Genomic DNA was harvested using the Wizard Genomic DNA Purification Kit (Promega) according to the manufacturers protocol for Gram Positive Bacteria. Barcode amplicons were created using a two round PCR protocol. During the first round of PCR (4 cycles), primers added specific “timepoint barcodes” for multiplexing, 17 random nucleotides for collapsing PCR duplicates, and partial Illumina sequencing adaptors. Samples were pooled by induction condition and cleaned up using Select-a-Size DNA Clean & Concentrator columns (Zymo). During the second round (12–16 cycles), full Illumina sequencing adaptors were added using standard library prep primers (o231 & indexing primers). Illumina indices were used to differentiate samples on the sequencer. Final PCR products were run on an 8% TBE gel (Novex) for size selection, excised, extracted, and precipitated. Libraries were sequenced on an Illumina HiSeq 2000 or NextSeq500. For a list of primers see Table S2 and for details on amplicon structure and barcodes see Table S4.
QUANTIFICATION & STATISTICAL ANALYSIS
Fitness calculation
The method to calculate the fitness of strains during the competition experiment is as follows. First, sequencing reads were checked for correct amplicon structure, matched to the appropriate experiment, timepoint, barcode, and duplicates in the 17N random region were collapsed. At each timepoint, the log2 ratio of the counts of each strain-BC to the average counts of the four strain-BC strains for the ‘no insert control’ strain-type was calculated. The ‘no insert control’, is a strain derived from wildtype 168 with an insertion at the amyE locus including the pSpankHY promoter system and spcR resistance gene similar to the inducible aaRS constructs. However, instead of a protein-coding open reading frame inserted under the pSpankHY promoter, the ORF was removed and no protein product was produced (hence ‘no insert’). This strain was then barcoded with the barcoding plasmid (lacA::BC-tetM) producing four independent strain-BC biological replicates of this strain-type. This strain-type was used as the point of comparison to control for the addition of antibiotic markers and deletion of amyE ORF. This control showed a ~1% growth defect compared to wildtype at all induction levels. After calculating the log2 ratio for each timepoint for a given strain-BC, the slope of the linear regression of this ratio over the number of generations of the full experiment was then calculated. The number of generations was calculated as the number of doublings of the wildtype since the prior backdilution (20 minute per doubling). In general, a 64-fold backdilution was used and therefore 6 generations had passed since the previous timepoint. When the backdilution was reduced (32-fold or 16-fold) the number of generations from the previous timepoint was adjusted accordingly (5 or 4 respectively). The fitness for the strain-BC was then computed as 1 + slope. The fitness of the 2–4 strain-BC of each strain-type was then averaged to report the final fitness for that strain-type. The fitness difference between all pairwise comparisons of strain-BCs of equivalent strain-type is plotted as a distribution to demonstrate strain-BC “biological” noise in Figure S1B.
In our analysis, we found a floor of read counts for each strain-BC of around 200 reads, potentially due to PCR crosstalk, which could skew the calculated fitness. For example, if the slope of the linear regression was calculated over all timepoints for a particularly unfit strain, this read count floor would artificially decrease the slope and artificially increase the fitness calculated. To circumvent this problem, we only considered generation timepoints prior to the first timepoint with <200 reads when calculating the slope of the linear regression. For particularly unfit strains such as low induction of leuS, this meant the slope and fitness were calculated with the first two or three timepoints. We also noticed that two timepoints in some induction experiments (40 μM Timepoint 1, 20 μM Timepoint 0) had extremely low read counts across all strain-BC. We attribute this to loss of DNA during collection or a failed PCR reaction in amplicon preparation. To circumvent this problem, we simply removed the affected timepoint from the slope calculation, calculating using the remaining times that passed the read floor threshold.
Calibration of aaRS production
Protein production from the ectopic locus was estimated by first performing ribosome profiling for each aaRS strain fully induced with IPTG (500 μM), which showed limited expression changes except for the induced aaRS. The fully induced aaRS production rate relative to the rest of the cell, as measured by ribosome footprint density, was compared to that in the wildtype strain 168. At other IPTG concentrations, aaRS production relative to the fully induced situation was calibrated by the relative promoter strength, which we measured by qRT-PCR for an independent strain that has the same inducible promoter (pSpankHY) driving gfp expression from the same amyE locus. For example, ribosome profiling data showed that the level of IleS production at full induction is 14-fold higher than in wildtype cells. At 40 μM IPTG, the promoter activity, determined from RT-qPCR of the gfp construct, is 9.4-fold lower than at 500 μM, meaning that IleS production is inferred through calibration to be 1.5-fold higher than the wildtype level.
This calibration provides an estimate for the relative promoter strength compared to the wildtype level and should not be taken as an absolute measure of expression levels. Because aaRS production affects growth and growth affects global gene expression even for a constitutive promoter (Klumpp et al., 2009; Liang et al., 1999; Zhu and Dai, 2019), we expect the absolute output of the promoter to vary with aaRS production. Although our ribosome profiling data could be used to empirically measure the aaRS expression at different IPTG concentrations, such an analysis is limited to providing the expression relative to other genes, which themselves are affected by growth rate changes. For example, a low IPTG concentration not only reduces aaRS production, but also leads to reduced global gene expression. Indeed, in this case, the apparent ‘knockdown’ in aaRS as a fraction of protein synthesis (as measured by ribosome profiling) is less than the calibrated fold-change in promoter strength from the wildtype level because most other genes have reduced expression (Figure S6A). If the fitness landscape is plotted for aaRS production in terms of fraction of protein synthesis instead of calibrated promoter strength, this would lead to an artificially sharper fitness cliff at underproduction (Figure S6B), although in reality the promoter is much more repressed than the apparent knockdown. We therefore chose to report the calibrated production to avoid such confounding effects.
Ribosome profiling and RNA-seq data analysis
Processing of ribosome profiling sequencing reads included removal of the linker sequence and mapping with Bowtie (Langmead et al., 2009) (options −v 2 −k 1) to the NC_000964.3 reference genome obtained from NCBI Reference Sequence Bank. Footprint reads with sizes between 20 to 40 nucleotides were stripped of the first and last 8 nucleotides and a count of 1/n where n equals the remaining read length was assigned to the remaining positions. To calculate proteome fractions for regulons, the total read counts for a subset of genes in the regulon was compared to all ORF mapping reads excluding start and stop codons. Processing of RNA sequencing reads included removal of PolyA tails and mapping with Bowtie (Langmead et al., 2009) (options −v 2 −k 1) to the NC_000964.3 reference genome. The five prime ends of reads mapping to open reading frames was counted excluding the first and last five positions. Read counts were normalized by total mapped reads and gene length (RPKM) or total mapped reads (RPM).
The average ribosome occupancy at the 61 sense codons was computed by first normalizing read counts at each nucleotide in a coding sequence by the mean read count for that sequence (excluding the first and last 15 nucleotides) using the Ribosome profiling data. This normalized ribosome occupancy was averaged across the three nucleotides of each codon, and then averaged across all occurrences of that codon within the coding region. Final values were computed by averaging across all coding sequences meeting a threshold of five sequencing reads per nucleotide. Only genes reaching this threshold in all compared experiments were considered in the final analysis. Ribosome queuing was calculated by similarly normalizing read counts at each nucleotide in a coding region by the mean read count for that region. For each instance of a codon, a window of 100 nucleotides upstream and downstream of the second codon position were obtained and windows were averaged across all instances of the codon within that gene. Only codons over 115 nucleotides away from a start and stop codon were considered. Final values were computed by averaging across all coding sequences meeting a threshold of five sequencing reads per nucleotide. For comparison between experiments, the average density at each position in the window from the wildtype was subtracted from the average density in the SerS underproduction experiment.
Supplementary Material
Table S1. List of genes in each proteome fraction category, related to Figures 4, 5, and STAR Methods.
Table S2. List of oligonucleotides used, related to STAR Methods.
Table S3. Table of Components & Recipe for MCCG media, related to STAR Methods.
Table S4. Full Amplicon Structure with associated barcodes & primers for competition experiment, related to STAR Methods.
Table S5. Table of strains & associated genotypes, related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| Bacillus subtilis strains | This Paper | See Table S5 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chemicals for MCCG Media | This Study | See Table S3 |
| Trichloroacetic Acid | Sigma | T6399 |
| Isopropanol | Macron | 3032–16 |
| 1M Tris pH 9.5 | Teknova | T1095 |
| Qubit BR RNA | ThermoFisher | Q10210 |
| Superase-IN RNase | ThermoFisher | AM2696 |
| PerfectHyb Plus Hybridization buffer | Sigma | H7033 |
| Glycoblue | ThermoFisher | AM9516 |
| Corning® 500 mL 0.2 μm Pore filter | Corning | 430773 |
| Select-a-size DNA Clean & Concentrator | Zymo | D4080 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621S |
| DNase I recombinant, RNase-free (Roche) | Sigma-Aldrich | 4716728001 |
| Micrococcal nuclease | Roche | No longer available |
| T4 Polynucleotide Kinase (PNK) | New England Biolabs | M0201S |
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080093 |
| CircLigase ssDNA ligase | Lucigen | CL4115K |
| Dynabeads MyOne Streptavidin C1 | ThermoFisher Scientific | 65001 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | M0491L |
| T4 RNA ligase 2 truncated K277Q | J. Weissman | N/A |
| ATP, [γ-32P] | PerkinElmer | BLU502A250UC |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | 18080044 |
| G25 Sepharose columns | GE Healthcare Life Sciences | 27532501 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | QIAGEN | 74104 |
| MICROBExpress Bacterial mRNA Enrichment Kit | ThermoFisher Scientific | AM190 |
| Oligo Clean & Concentrator | Zymo Research | D4060 |
| Deposited Data | ||
| Raw .fastq and processed .wig files from Ribosome profiling | This paper | GEO: GSE141389 |
| Northern blots raw data | This paper; Mendeley data | http://dx.doi.org/10.17632/f9kkszrtch.1 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Bacillus subtilis strains | This Paper | See Table S5 |
| Oligonucleotides | ||
| Northern Blot assay probes | This paper | See Table S2 |
| RT-qPCR primers | This paper | See Table S2 |
| Library preparation primers | This paper | See Table S2 |
| Strain creation primers | This paper | See Table S2 |
| Competition experiment primers | This paper | See Table S2 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Bowtie v2.0.0b3 | Langmead et al. (2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| Python 2.7 | Python Software Foundation | http://www.python.org |
| Other custom python scripts | This paper | Available upon request |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Other | ||
| Super Membrane Disc Filters, Pall Laboratory | VWR | 28147–980 |
| Grinding Jar Set, Stainless Steel | Qiagen | 69985 |
| TissueLyser II | Qiagen | 85300 |
| Novex TBE-Urea gels, 15% | ThermoFisher | EC6885BOX |
| Novex TBE-Urea gels, 10% | ThermoFisher | EC6875BOX |
| Novex TBE gels, 8% | ThermoFisher | EC62152BOX |
Highlights:
Bacteria produce just enough aaRSs to support the amino acid fluxes for translation
tRNA charging is not maximized at growth-optimized levels of aaRS production
Native levels of uncharged tRNAs have limited impacts on cell fitness
Stringent response alleviates fitness defects of aaRS underproduction in rich media
Acknowledgments
We thank members of the MIT BioMicro Center for help in design and completion of the high-throughput sequencing; members of the Li lab and Grossman lab for discussions. This research is supported by NIH R35GM124732, Pew Biomedical Scholars Program, a Sloan Research Fellowship, Searle Scholars Program, the Smith Family Award for Excellence in Biomedical Research, an NIH Pre-Doctoral Training Grant (T32 GM007287, to D.J.P. and G.E.J.), an NSF graduate research fellowship (to G.E.J.), an NSERC graduate fellowship (to J.B.L.), and an HHMI International Student Fellowship (to J.B.L.). S.K.’s work in the laboratory of M.K.W. is supported by the HHMI and NIH Grant R01-AI-042347.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Avcilar-Kucukgoze I, Bartholomäus A, Cordero Varela JA, Kaml RF-X, Neubauer P, Budisa N, and Ignatova Z (2016). Discharging tRNAs: a tug of war between translation and detoxification in Escherichia coli. Nucleic Acids Res. 44, 8324–8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer H, and Dennis PP (2008). Modulation of Chemical Composition and Other Parameters of the Cell at Different Exponential Growth Rates. EcoSal Plus 3. [DOI] [PubMed] [Google Scholar]
- Britton RA, Eichenberger P, Gonzalez-Pastor JE, Fawcett P, Monson R, Losick R, and Grossman AD (2002). Genome-wide analysis of the stationary-phase sigma factor (sigma-H) regulon of Bacillus subtilis. J. Bacteriol 184, 4881–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhu M, Warren M, Balakrishnan R, Patsalo V, Okano H, Williamson JR, Fredrick K, Wang Y-P, and Hwa T (2016). Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat Microbiol 2, 16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell AM, Subramaniam AR, and O’Shea EK (2018). Translational Control through Differential Ribosome Pausing during Amino Acid Limitation in Mammalian Cells. Mol. Cell 71, 229–243.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidi D, and Milo R (2017). Lessons on enzyme kinetics from quantitative proteomics. Curr. Opin. Biotechnol 46, 81–89. [DOI] [PubMed] [Google Scholar]
- Dekel E, and Alon U (2005). Optimality and evolutionary tuning of the expression level of a protein. Nature 436, 588–592. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Mobley EM, Radek AJ, and Pan T (2004). Exploring the regulation of tRNA distribution on the genomic scale. Journal of Molecular Biology 337, 31–47. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Sørensen MA, Elf J, Ehrenberg M, and Pan T (2005). Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Reports 6, 151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nilsson L, and Kurland CG (1995). Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol 177, 1497–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J, Nilsson D, Tenson T, and Ehrenberg M (2003). Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300, 1718–1722. [DOI] [PubMed] [Google Scholar]
- Eymann C, Homuth G, Scharf C, and Hecker M (2002). Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J. Bacteriol 184, 2500–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro I, Liebeton K, and Ignatova Z (2017). Growth-Rate Dependent Regulation of tRNA Level and Charging in Bacillus licheniformis. Journal of Molecular Biology 429, 3102–3112. [DOI] [PubMed] [Google Scholar]
- Gendron N, Putzer H, and Grunberg-Manago M (1994). Expression of both Bacillus subtilis threonyltRNA synthetase genes is autogenously regulated. J. Bacteriol 176, 486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Giegé R, and Springer M (2016). Aminoacyl-tRNA Synthetases in the Bacterial World. EcoSal Plus 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloge F, Becker AH, Kramer G, and Bukau B (2014). Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol 24, 24–33. [DOI] [PubMed] [Google Scholar]
- Gourse RL, Chen AY, Gopalkrishnan S, Sanchez-Vazquez P, Myers A, and Ross W (2018). Transcriptional Responses to ppGpp and DksA. Annu. Rev. Microbiol 72, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, and Grantham R (1980). Polypeptide elongation and tRNA cycling in Escherichia coli: a dynamic approach. FEBS Letters 115, 151–155. [DOI] [PubMed] [Google Scholar]
- Green NJ, Grundy FJ, and Henkin TM (2010). The T box mechanism: tRNA as a regulatory molecule. FEBS Letters 584, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, and Merino E (2009). Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol. Mol. Biol. Rev 73, 36–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson G, and Coller J (2018). Codon optimality, bias and usage in translation and mRNA decay. Nature Reviews Molecular Cell Biology 19, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, and Gerdes K (2015). Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nature Reviews Microbiology 13, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG (2005). Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol 59, 407–450. [DOI] [PubMed] [Google Scholar]
- Kimura S, and Waldor MK (2019). The RNA degradosome promotes tRNA quality control through clearance of hypomodified tRNA. Proc. Natl. Acad. Sci. U.S.a 116, 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Scott M, Pedersen S, and Hwa T (2013). Molecular crowding limits translation and cell growth. Proc. Natl. Acad. Sci. U.S.a 110, 16754–16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Zhang Z, and Hwa T (2009). Growth rate-dependent global effects on gene expression in bacteria. Cell 139, 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriel A, Brinsmade SR, Tse JL, Tehranchi AK, Bittner AN, Sonenshein AL, and Wang JD (2014). GTP dysregulation in Bacillus subtilis cells lacking (p)ppGpp results in phenotypic amino acid auxotrophy and failure to adapt to nutrient downshift and regulate biosynthesis genes. J. Bacteriol 196, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalanne J-B, Taggart JC, Guo MS, Herzel L, Schieler A, and Li G-W (2018). Evolutionary Convergence of Pathway-Specific Enzyme Expression Stoichiometry. Cell 173, 749–761.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-W, Burkhardt D, Gross C, and Weissman JS (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Bipatnath M, Xu Y, Chen S, Dennis P, Ehrenberg M, and Bremer H (1999). Activities of constitutive promoters in Escherichia coli. Journal of Molecular Biology 292, 19–37. [DOI] [PubMed] [Google Scholar]
- Liu K, Bittner AN, and Wang JD (2015). Diversity in (p)ppGpp metabolism and effectors. Curr. Opin. Microbiol 24, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland AB, Bah E, Madireddy R, Zhang Y, Brilot AF, Grigorieff N, and Korostelev AA (2016). Ribosome•RelA structures reveal the mechanism of stringent response activation. Elife 5, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Leautey J, Grunberg-Manago M, and Putzer H (1997). Structure and regulation of expression of the Bacillus subtilis valyl-tRNA synthetase gene. J. Bacteriol 179, 2472–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain WH, Jou YY, Bhattacharya S, Gabriel K, and Schneider J (1999). The reliability of in vivo structure-function analysis of tRNA aminoacylation. Journal of Molecular Biology 290, 391–409. [DOI] [PubMed] [Google Scholar]
- Morrill SA, and Amon A (2019). Why haploinsufficiency persists. Proc. Natl. Acad. Sci. U.S.a 116, 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanamiya H, Kasai K, Nozawa A, Yun C-S, Narisawa T, Murakami K, Natori Y, Kawamura F, and Tozawa Y (2008). Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol. Microbiol 67, 291–304. [DOI] [PubMed] [Google Scholar]
- O’Brien EJ, Utrilla J, and Palsson BO (2016). Quantification and Classification of E. coli Proteome Utilization and Unused Protein Costs across Environments. PLoS Comput. Biol 12, e1004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo B-M, Marta E, et al. (2016). A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrykus K, and Cashel M (2008). (p)ppGpp: still magical? Annu. Rev. Microbiol 62, 35–51. [DOI] [PubMed] [Google Scholar]
- Putzer H, Laalami S, Brakhage AA, Condon C, and Grunberg-Manago M (1995). Aminoacyl-tRNA synthetase gene regulation in Bacillus subtilis: induction, repression and growth-rate regulation. Mol. Microbiol 16, 709–718. [DOI] [PubMed] [Google Scholar]
- Rauscher R, and Ignatova Z (2018). Timing during translation matters: synonymous mutations in human pathologies influence protein folding and function. Biochem. Soc. Trans 46, 937–944. [DOI] [PubMed] [Google Scholar]
- Sander T, Farke N, Diehl C, Kuntz M, Glatter T, and Link H (2019). Allosteric Feedback Inhibition Enables Robust Amino Acid Biosynthesis in E. coli by Enforcing Enzyme Overabundance. Cell Syst 8, 66–75.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M, and Hwa T (2011). Bacterial growth laws and their applications. Curr. Opin. Biotechnol 22, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M, Gunderson CW, Mateescu EM, Zhang Z, and Hwa T (2010). Interdependence of cell growth and gene expression: origins and consequences. Science 330, 1099–1102. [DOI] [PubMed] [Google Scholar]
- Sherman JM, Rogers MJ, and Söll D (1992). Competition of aminoacyl-tRNA synthetases for tRNA ensures the accuracy of aminoacylation. Nucleic Acids Res. 20, 2847–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenshein AL (2005). CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr. Opin. Microbiol 8, 203–207. [DOI] [PubMed] [Google Scholar]
- Srivatsan A, and Wang JD (2008). Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol 11, 100–105. [DOI] [PubMed] [Google Scholar]
- Stead MB, Agrawal A, Bowden KE, Nasir R, Mohanty BK, Meagher RB, and Kushner SR (2012). RNAsnap™: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 40, gks680–e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Deloughery A, Bradshaw N, Chen Y, O’Shea E, Losick R, and Chai Y (2013a). A serine sensor for multicellularity in a bacterium. Elife 2, e01501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Pan T, and Cluzel P (2013b). Environmental perturbations lift the degeneracy of the genetic code to regulate protein levels in bacteria. Proc. Natl. Acad. Sci. U.S.a 110, 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Zid BM, and O’Shea EK (2014). An Integrated Approach Reveals Regulatory Controls on Bacterial Translation Elongation. Cell 159, 1200–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, and Söll D (1988). Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science 242, 1548–1551. [DOI] [PubMed] [Google Scholar]
- Tockman J, and Vold BS (1977). In vivo aminoacylation of transfer ribonucleic acid in Bacillus subtilis and evidence for differential utilization of lysine-isoaccepting transfer ribonucleic acid species. J. Bacteriol 130, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinquier A, Ulmer JE, Gilet L, Figaro S, Hammann P, Kühn L, Braun F, and Condon C (2019). tRNA Maturation Defects Lead to Inhibition of rRNA Processing via Synthesis of pppGpp. Mol. Cell 74, 1227–1238.e3. [DOI] [PubMed] [Google Scholar]
- Varshney U, Lee CP, and RajBhandary UL (1991). Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem 266, 24712–24718. [PubMed] [Google Scholar]
- Vitreschak AG, Mironov AA, Lyubetsky VA, and Gelfand MS (2008). Comparative genomic analysis of T-box regulatory systems in bacteria. Rna 14, 717–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther KS, Roghanian M, and Gerdes K (2018). Activation of the Stringent Response by Loading of RelA-tRNA Complexes at the Ribosomal A-Site. Mol. Cell 70, 95–105.e4. [DOI] [PubMed] [Google Scholar]
- Wu CC-C, Zinshteyn B, Wehner KA, and Green R (2019). High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell 73, 959–970.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegian CD, Stent GS, and Martin EM (1966). Intracellular condition of Escherichia coli transfer RNA. Proc. Natl. Acad. Sci. U.S.a 55, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, and Haldenwang WG (2003). RelA is a component of the nutritional stress activation pathway of the Bacillus subtilis transcription factor sigma B. J. Bacteriol 185, 5714–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, and Dai X (2019). Growth suppression by altered (p)ppGpp levels results from non-optimal resource allocation in Escherichia coli. Nucleic Acids Res. 47, 4684–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of genes in each proteome fraction category, related to Figures 4, 5, and STAR Methods.
Table S2. List of oligonucleotides used, related to STAR Methods.
Table S3. Table of Components & Recipe for MCCG media, related to STAR Methods.
Table S4. Full Amplicon Structure with associated barcodes & primers for competition experiment, related to STAR Methods.
Table S5. Table of strains & associated genotypes, related to STAR Methods.
Data Availability Statement
The Ribosome profiling and RNA sequencing datasets generated during this study are available at the Gene Expression Omnibus with accession number GSE141389. The raw sequencing files of the amplicons generated from competition experiments is available in the NCBI Sequencing Read Archive (SRA) under accession number PRJNA593245. Full code is available upon request. The raw image for Northern blots was deposited at Mendeley Data and can be viewed at https://data.mendeley.com/datasets/f9kkszrtch/draft?a=1fcb4abc-bd3d-452a-94c1-caa43ee378ca.