

Abstract
The main protease (Mpro) of SARS-CoV-2 is responsible for the cleavage of viral replicase polyproteins 1a and 1ab into their mature form and is highly specific and exclusive in its activity. Many studies have targeted this enzyme by small molecule inhibitors to develop therapeutics against the highly infectious disease Covid-19. Our diet contains many natural antioxidants which along with providing support for proper growth and functioning of the body, pose additional health benefits. Present in-silico analysis depicted that natural antioxidants like sesamin, ellagic acid, capsaisin, and epicatechin along with galangin, exhibited significant binding at the catalytic site of the Mpro enzyme. They interacted with excellent efficiency with the chief active site residue Cys145 and thus seem to possess the remarkable potential to act as drug candidates for the treatment of Covid-19. Such dietary compounds can be easily administered orally with least toxicity related concern and thus yell for urgent exhaustive research to develop into efficient therapies.
Communicated by Ramaswamy H. Sarma
Keywords: Covid-19, SARS-CoV-2 main protease, natural antioxidants, molecular docking, pharmacophore modeling
Introduction
Covid-19, the ongoing pandemic has exposed the world to extreme vulnerability. SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2), the causative agent of corona virus disease comprises a single stranded RNA genome of 30 kb. The two essential polyproteins translated from first ORF (Open Reading Frame) of its genome namely 1a and 1ab are significant contributors to viral replication mechanism (Jin et al., 2020a). The main protease (Mpro) of SARS-CoV-2, also known as 3CLpro, together with papain like proteases, process these polyproteins to non-structural mature forms. The Mpro, process on around 11 sites for cleavage in the polyprotein 1ab, a 790 kDa replicase enzyme, to makes it functional. The leading Mpro recognition sequence for cleavage in polyprotein is L-Q | (S, A, G) (| indicates the site of cleavage) and recent researches suggest that the cleavage specificity of Mpro is exclusive and is not found in any human protease (Zhang et al., 2020). Hence, targeting Mpro for the development of therapeutics seems to be a highly promising approach. Structural inhibition of Mpro at active site can give rise to an extremely selective treatment with least damage to human host (Joshi et al., 2020). Recently, Jin et al., in their in-vitro and crystallography analysis reported significantly effective inhibition of Mpro by a synthetic antineoplastic agent, carmofur, which forms covalent bonds with Cys145 residues at the active site of the enzyme (Jin et al., 2020a).
Natural antioxidants like lignans, polyphenols and capsiacinoids, usually found in oil seeds, fruits and vegetables comprise a vital part of our daily diet, and possess exceptional pharmacological properties (Pandey & Rizvi, 2009; Pathak et al., 2014; Rosa et al., 2002). Presence of numerous acceptors, donors, hydrophobic and aromatic functional groups in these compounds leadingly contribute to their medicinal effects (Diniz do Nascimento et al., 2020; Tsao, 2010) .
Utilization of such natural compounds to develop Mpro targeted therapies against the dreaded disease like Covid-19 can stretch effective outcomes with least toxicity. Along with this oral administration of such therapeutics through regular diet will in turn increase the ease of treatment.
Hence, the present in-silico analysis deals with in depth evaluation of effective structural inhibition potential of natural antioxidants against the main protease Mpro of SARS-CoV-2 and hence can be developed into effectual treatment against the prevalent deadly disease, Covid-19.
Method
To evaluate our hypothesis, we performed pharmacophore modeling of several natural antioxidants present in regular diet with a native inhibitor carmofur of Mpro (PDB id: 7BUY) by using PharmaGist webserver tool (Pinto et al., 2019; Schneidman-Duhovny et al., 2008a, 2008b). This tool predicts the pharmacophoric features of a molecule essential to interact with receptor of interest based on multiple flexible alignment of molecules provided as input (da Costa et al., 2019; Schneidman-Duhovny et al., 2008a). The 5 top scoring compounds having substantial overlap with the reference compound were selected for molecular docking with main protease Mpro (PDB ID 7BUY), to evaluate their structural inhibition potential against the enzyme (Ramos et al., 2019; Vale et al., 2020). Raccoon VS Autodock software (http://autodock.scripps.edu/resources/raccoon) was used to perform molecular docking analysis (Forli et al., 2016). The 3D structures of protein and ligands were prepared using AutoDock Tools. Gasteiger charges were added to the ligands. Kollman charge of −149.064 was added to the protein structure. Polar hydrogens were added and non polar hydrogens were merged. The grid parameter file was generated with number of grid point in X,Y and Z direction as 126,126 and 126 respectively. The grid centre was set to −26.479, 12.595, and 58.704 along X, Y and Z axis. The grid spacing was selected as 0.475 Ǻ to cover the whole protein. The docking parameter file was generated by utilizing Lamarckian GA docking parameters as number of GA runs 10, population size = 150, maximum number of evaluations as 2500000, gene mutation rate of 0.02, number of generations as 27000, and cross over rate of 0.8 (Morris et al., 1996, 1998). The 3D protein and ligands structure files and parameter files were then subjected to Raccoon VS Autodock software to perform multiple dockings of the considered natural compounds with the main protease Mpro enzyme. Selective filtering of the compounds based on Lipinski’s rule of 5 was also done by the software where all the compounds passed the crieteria with out any violation (Lipinski et al., 2001).
Further, to analyse the effectiveness of interactions of these compounds with the important active site residue Cys145, C145G mutation was incorporated in the protein structure by using SDM (Site Directed Mutator) webserver tool and then again molecular docking of the 5 lead molecules with the mutated C145G Mpro protease was performed by using same parameters (Pandurangan et al., 2017). Site directed mutator works on analysing the changes that occur in stability and functionality of protein upon replacement of amino aicds and transform them into substitution probabilities. It also generates the energy minimized mutated protein structure which was utilized for docking analysis (Pandurangan et al., 2017).
The efficacy of interactions was evaluated based on the changes observed in predicted binding energies and hydrogen bonds formed, with the active site residues of Mpro along with the predicted inhibition constant (Ki value) in the molecular docking analysis. The Ki value or the inhibition constant is in fact the dissociation constant of docked enzyme inhibitor complex. The smaller the value of Ki the lower will be the probability of dissociation and hence higher will be the inhibiton. It is calculated as Ki = exp(deltaG/(R*T)) where deltaG is the free energy of binding, R is the gas constant (1.987 cal K−1 mol−1) and T is the temperature (298.15 K) (Morris et al., 1998).
Further the docking interaction poses were generated by using Pymol, Discovery studio visualizer 2019 and LigPlot software where Pymol exhibited the 3D ligand binding cavities, Discovery Studio displayed different types of interactions including weak and strong hydrogen bonding interactions between the natural compounds and Mpro enzyme residues whereas Ligplot revealed only the strongest hydrogen bonds formed depending on the standard criteria (McDonald & Thornton, 1994).
Results
Our results depicted that natural antioxidants, sesamin, galangin, ellagic acid, capsaicin and epicatechin, which are usually found in various dietary sources (Table 1) contain numerous pharmacophoric features (Table 2) that are relevant for their application as drug like compounds.
Table 1.
Dietary sources of the top 5 lead natural molecules.
| S.no. | Molecule | Dietary source | Structures |
|---|---|---|---|
| 1 | Sesamin | Sesame (Sesamum indicum) seed | 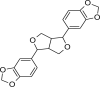 |
| 2 | Galangin | Lasser galangal (Alpinia officinarum), Honey | 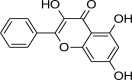 |
| 3 | Ellagic acid | Raspberries (Rubus idaeus), strawberries (Fragaria ananassa), cherries (Prunus avium), blackberries (Rubus fruticosa), and walnuts (Juglans regia) | 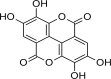 |
| 4 | Capsaicin | Chilli pepper (Capsicum annuum) | 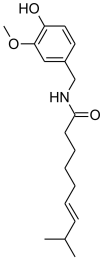 |
| 5 | Epicatechin | Green tea (Camellia sinensis), apple (Malus domestica) |  |
Table 2.
Pharmacophore features of the considered natural molecules by PharmaGist.
| S.no. | Molecule | Atoms | Features | Spatial Features | Aromatic | Hydrophobic | Donors | Acceptors | Negatives | Positives |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sesamin | 44 | 9 | 9 | 2 | 1 | 0 | 6 | 0 | 0 |
| 2 | Galangin | 30 | 11 | 8 | 3 | 0 | 3 | 5 | 0 | 0 |
| 3 | Ellagic acid | 28 | 14 | 10 | 2 | 0 | 4 | 8 | 0 | 0 |
| 4 | Capsaicin | 49 | 16 | 15 | 1 | 10 | 2 | 3 | 0 | 0 |
| 5 | Epicatechin | 35 | 14 | 9 | 2 | 1 | 5 | 6 | 0 | 0 |
| 6 | Carmofur* | 34 | 11 | 11 | 1 | 5 | 2 | 3 | 0 | 0 |
*Native active site inhibitor used as reference for pharmacophore modelling.
Alignment of these antioxidant compounds over the native inhibitor (carmofur) displayed significant overlap and similarity (Figure 1). Several features of these compounds which were found to match with the reference inhibitor have been denoted in Table 3. Thus, such similarities made it evident that these 5 natural antioxidants can act as active inhibitors of Mpro.
Figure 1.

Alignment pose of A) sesamin (red) B) galangin (mauve) C) ellagic acid (yellow) D) capsaicin (orange) E) epicatechin (green) upon the reference compound carmofur (blue).
Table 3.
Overlapping pharmacophore features of considered natural compounds with the native inhibitor carmofur.
| S.no. | Score | Features | Spatial features | Aromatic | Hydrophobic | Donors | Acceptors | Negatives | Positives | Molecules |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6.02432 | 3 | 3 | 1 | 0 | 0 | 2 | 0 | 0 | Carmofur-sesamin |
| 2 | 7.53452 | 4 | 4 | 1 | 0 | 0 | 3 | 0 | 0 | Carmofur-galangin |
| 3 | 7.53153 | 4 | 4 | 1 | 0 | 0 | 3 | 0 | 0 | Carmofur-ellagic acid |
| 4 | 7.55099 | 8 | 8 | 1 | 5 | 1 | 1 | 0 | 0 | Carmofur-capsaicin |
| 5 | 7.51928 | 4 | 4 | 1 | 0 | 1 | 2 | 0 | 0 | Carmofur-epicatechin |
Further validation of this assumption by performing molecular docking of Mpro with these compounds gave exceptionally significant negative binding energies of −8.93, −6.47, −7.9, −7.16 and −6.68 kcal/mol for sesamin, galangin, ellagic acid, capsaicin and epicatechin respectively which were significantly higher than the binding energy of carmofur (Table 4). The binding of these compounds with Mpro displayed effective interactions and formation of hydrogen bonds with Leu141, Gly143, Ser144, Cys145, His164, His164, Glu166, Arg188, Gln189, Thr190 and Gln192 residues (Table 4 and Figure 2) where four out of 5 compounds i.e. sesamin ellagic acid, capsaicin and epicatechin, were found to be specifically enclosed within the active site while galangin was laid in close vicinity of active site. Additionally, interactions like van der waals, pi-pi T shaped, pi-alkyl, pi sulphur, pi-sigma, pi donor hydrogen bonds and carbon hydrogen bonds in turn facilitated the proper positioning and contact of these compounds with the active site residues and strengthened our analysis to a much greater extent (Figure 2). The strongest hydrogen bonding interactions were analysed also using standard criteria (Supplementary Figure 1).
Table 4.
Docking results of the lead hits with SARS CoV-2 main protease.
| S.no. | Molecules | Binding energy (kcal/mol) | Interaction energy (kcal/mol) | Inhibition constant (Ki value) | No. of H-bonds | Residues involved in H-bonding |
|---|---|---|---|---|---|---|
| 1 | Sesamin | −8.93 | −10.21 | 285.02nM | 4 | Ser144, Cys145, Gln189, Gln192 |
| 2 | Galangin | −6.47 | −9.24 | 18.23μM | 3 | Glu166 (1), Gln192(2) |
| 3 | Ellagic acid | −7.9 | −11.17 | 1.62μM | 5 | His164, Cys145, Glu166, Arg188, Thr190 |
| 4 | Capsaicin | −7.16 | −11.715 | 5.61μM | 4 | Glu166, Gln189, Thr190, Gln192 |
| 5 | Epicatechin | −6.68 | −10.631 | 12.77μM | 8 | Leu141, Gly143, Cys145, His163, His164, Glu166, Gln189(2) |
| 6 | Carmofur | −5.93 | −7.564 | 45.32 μM | 4 | Glu166, Arg188, Thr190, Gln192 (significant interaction with Cys145) |
Figure 2.

3D Docking pose and 2D interactions of A) sesamin (red) B) galangin (mauve) C) ellagic acid (yellow) D) capsaicin (orange) E) epicatechin (green) F) carmofur (blue) with wild type SARS-CoV-2 main protease (Mpro) (green).
Furthermore, the in-silico incorporation of mutation C145G in the Mpro enzyme by site-directed mutagenesis mildly increased the stability of enzyme with a positive change in ΔG (ΔΔG) value of 0.2 but decreased the solvent accessibility form 24.3% of wild type to 16.4% of mutant type. Moreover, the molecular docking of the mutant Mpro enzyme with the 5 lead hit compounds displayed significant differences in binding energies and interactions with respect to the binding energies and interaction observed with the wild type Mpro (Table 4, Figure 2, Table 5, Figure 3, Supplementary Figure 1, Supplementary Figure 2). This revealed that Cys145, which is an important active site residue and contribute significantly for catalytic activity of enzyme, was effectively interacting with the considered natural inhibitors thus proving that these inhibitors will pose significant hinderance in the enzyme’s activity to perform the cleavage of viral replicase polyproteins. An exceptional reduction in negative binding energy was observed when capsaicin was docked with mutant Mpro, from −7.16 kcal/mol for wild type to −5.7 kcal/mol for mutant type. In wild type interaction, capsaicin formed 2 bonds, one alkyl and other pi-alkyl bond, with Cys145 residue while in its interaction with the mutant type it formed single hydrogen bond with Gly145 that led to a large reduction in its binding potential for the enzyme thus proving specific and effective inhibiting potential of capsaicin against wild Mpro enzyme via Cys145 residue. Moreover, the reduction suffered by capsaicin was significantly large compared to that suffered by reference compound carmofur, where the binding energy of carmofur with native enzyme was −5.93 Kcal/mol and with the mutant type was −5.23 Kcal/mol. Sesamin, ellagic acid and epicatechin also suffered mild reductions in negative binding potentials when interacted with the mutant type due to loss of hydrogen bonding with Cys145 which was operative in interaction with wild type enzyme (Figures 2 and 3). In contrast, galangin showed insignificant increase in negative binding energy when interacted with the mutant type thus displayed no mutation effect on its interactions with the enzyme which can be beneficial in case of viral proteins which frequently suffer point mutations.
Table 5.
Docking results of lead hits with C145G mutated SARS CoV-2 main protease.
| S.no. | Molecules | Binding Energy (Kcal/mol) | No. of H bonds | Residues involved in H bonds |
|---|---|---|---|---|
| 1 | Sesamin | −8.66 | 5 | His41, Ser144, Gln189, Thr190, Gln192, |
| 2 | Galangin | −6.54 | 3 | Glu166(1), Gln192(2) |
| 3 | Ellagic acid | −7.7 | 4 | His164, Glu166, Arg188, Thr190 |
| 4 | Capsaicin | −5.7 | 5 | Leu141, Gly143, Ser144, Gly145, His164 |
| 5 | Epicatechin | −6.27 | 4 | His164, Glu166, Gln189(2) |
| 6 | Carmofur | −5.23 | 4 | Thr190(2), Gln192(2) |
Figure 3.

3D Docking pose and 2D interactions of A) sesamin (red) B) galangin (mauve) C) ellagic acid (yellow) D) capsaicin (orange) E) epicatechin (green) F) carmofur (blue) with C145G mutant SARS-CoV-2 main protease (Mpro) (green).
Henceforth, our hypothesis of Mpro inhibition with potential considered natural compounds seems to be exceptionally potent one and demands exhaustive further research to develop effective natural treatment against Covid-19.
Discussion
SARS-CoV-2 main protease Mpro plays noteworthy role in regulating viral replication by cleaving and thus converting the major replicase polyproteins 1a and 1ab into their mature form. The high specificity of the enzyme for its cleavage site and lack of similarity with the human proteases make it a highly operative target for the treatment of corona virus disease-19. Structural inhibition of this enzyme due to the binding of small molecule inhibitors in its active site have been investigated by many researchers (Jin et al., 2020a; Joshi et al., 2020; Zhang et al., 2020). Jin et al. identified an effective inhibitor N3 which conveys mechanism-based inhibition against Mpro, by using in-silico approach and then resolved the crystallographic structure of N3-Mpro complex that exposed modification of Cys145 residue via addition of vinyl group by N3. They further performed an exhaustive analysis of over 10,000 compounds to analyse their structural inhibition potential against Mpro and discovered 7 potential synthetic drug-like compounds (Jin et al., 2020b). In continuation to this, additional studies were conducted for the evaluation of structural interactions of one of the seven identified compounds, carmofur, with Mpro. Carmofur with IC50 value of 1.82 μM presented vigorous Mpro inhibition in in-vitro studies. Determination of its complex with Mpro by crystallography revealed interaction of the carbonyl group of carmofur with Cys145 residue. Mpro contain 2 protomers A and B and thus forms a homodimer where each protomer has 3 domains and a long connecting loop between domain I and III. The substrate interacting pocket is present at the interface of domain I and II which adjoin the catalytic dyad of Cys145 and His41. Carmofur covalently modifies the Cys145 residue by forming C-S bond, further the fatty acid tail of the drug extends and interact with His41, Met49, Tyr54, Met165 and Asp187 (Jin et al., 2020a). In the present study we found that epicatechin with significantly high interaction and binding energy lie in the same active site of the enzyme and formed 8 hydrogen bonds with residues Leu141, Gly143, Cys145, His163, His164, Glu166, and Gln189. Other residues of Mpro including His41, Met49, Met165, Asn142, Ser144, Phe140, His172, Gln192, Thr190, and Arg188 took part in van der waals interactions, carbon hydrogen, pi-alkyl and pi-donor hydrogen bonds with epicatechin thus proving it an effective candidate for inhibition of Mpro. Another considered antioxidant sesamin with the highest binding energy among the considered compounds interacted with Mpro to formed 4 hydrogen bonds with Ser144, Cys145, Gln189, and Gln192 and showed significant interactions with effective residues His41, Met49, and Met165. Similarly, ellagic acid and capsaicin interactions with operative catalytic site residues of Mpro were extremely significant, this fact was not only evident by the docking results with the wild type Mpro but was also depicted by the reduction in the interaction energies confronted by the docking complexes upon mutation at Cys145 residue. The mutation at C145 not only changed the binding energies but also modified the interactions of the compounds with the target enzyme. In case of sesamin and capsaicin though the number of hydrogen bonds formed with the mutated Mpro was higher than that of native form but the negative binding energies were significantly reduced indicating reduction in stability of the bonds formed with the mutated enzyme compared to the native form, this makes it evident that the compounds were forming high stability complexes by interacting with Cys145 and are more likely to inhibit the cleavage catalysis by the native enzyme. By losing the hydrogen bond with C145, sesamin in case of mutated form C145G, formed hydrogen bonds with His41 and Thr190. Also, the transformation from pi-alkyl to pi-sulphur bond was noticed with residue Met165 which might be a probable reason for reduction in docking energy. In case of capsaicin, the alkyl bond with His163 formed upon interaction with native protein was transformed to pi-cation bond, also the hydrogen bond with Glu166 in native structure was converted to van der waal’s interaction in mutated structure, thus a high decrease in binding energy was encountered. In contrary, no effect of mutation was noticed on the interaction of galangin thus providing scope for a consistent treatment for such a frequently mutating viral system. Similar to galangin, ellagic acid interacted in the same cavity in both native and mutated forms but the mutation at Csy145 residue reduced one hydrogen bond which was being formed by ellagic acid with Cys145 when interacted with native form of enyme. This reduction in hydrogen bond seems to be the only reason for the reduction of binding enrgy and thus proves significant interaction of ellagic acid with Cus145 showing the high potential of compond to pose inhibition at the active site. Interaction of epicatechin with Cys145 consisted three bond types including hydrogen bond, pi-donor hydrogen bond and alkyl bond but mutation on this residue posed significant change in the electronic configuration of the compound and lead to reduction of 4 hydrogen bonds which were being formed with Cys145, Leu141, Gly143 and His163 residues in native form of protein. Moreover Zhang et al., in their Mpro inhibitor analysis developed an alpha-ketoamide inhibitor with enhanced plasma half life and revealed its X-ray crystallographic structure in complex with Mpro. Their crystallographic structure displayed interaction of the ketoamide inhibitor with the same active site residues including Ser1, His41, Met49, Phe140, Gly143, Ser144, Cys145, His164, Glu166, Pro168, and Gln189 (Zhang et al., 2020). Hence all the above facts prove that natural antioxidants like sesamin, ellagic acid, capsiasin and epicatechin can be considered as compelling inhibitors of the principal target of SARS-CoV-2 (Mpro) and thus can deliver effective treatment to the highly infectious viral diseases Covid-19, through special diet or can also be formulated into drugs having least toxicity for oral administration.
Conclusion
Our in-silico analysis proved that natural antioxidants like sesamin, ellagic acid, epicatechin and capsaicin can specifically bind and interact with the most vital active site residue Cys145 of SARS-CoV-2 main protease with great significance and can inhibit its activity in an extreamely effective manner. Such inhibition of main protease will profoundly down regulate the viral replication process and will fortunately contribute to fight the highly infectious mortal disease Covid-19. Hence, further research in this concern is demanded to develop an easy, safe and sound treatment against the current viral pandemic.
Supplementary Material
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- da Costa, K. S., Galúcio, J. M., da Costa, C., Santana, A. R., Dos Santos Carvalho, V., do Nascimento, L. D., Lima E Lima, A. H., Neves Cruz, J., Alves, C. N., & Lameira, J. (2019). Exploring the potentiality of natural products from essential oils as inhibitors of odorant-binding proteins: A structure- and ligand-based virtual screening approach to find novel mosquito repellents. ACS Omega, 4(27), 22475–22486. 10.1021/acsomega.9b03157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz do Nascimento, L., Moraes, A., Costa, K., Pereira Galúcio, J. M., Taube, P. S., Costa, C., Neves Cruz, J., de Aguiar Andrade, E. H., & Faria, L. (2020). Bioactive natural compounds and antioxidant activity of essential oils from spice plants: New findings and potential applications. Biomolecules, 10(7), 935–988. 10.3390/biom10070988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905–919. 10.1038/nprot.2016.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020. a). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Jin, Z., Zhao, Y., Sun, Y., Zhang, B., Wang, H., Wu, Y., Zhu, Y., Zhu, C., Hu, T., Du, X., Duan, Y., Yu, J., Yang, X., Yang, X., Yang, K., Liu, X., Guddat, L. W., Xiao, G., Zhang, L., Yang, H., & Rao, Z. (2020. b). Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nature Structural & Molecular Biology, 27(6), 529–532. 10.1038/s41594-020-0440-6 [DOI] [PubMed] [Google Scholar]
- Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure & Dynamics, 38, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 46(1–3), 3–26. 10.1016/S0169-409X(00)00129-0 [DOI] [PubMed] [Google Scholar]
- McDonald, I. K., & Thornton, J. M. (1994). Satisfying hydrogen bonding potential in proteins. Journal of Molecular Biology, 238(5), 777–793. https:// 10.1006/jmbi.1994.1334 [DOI] [PubMed] [Google Scholar]
- Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., & Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. [DOI] [Google Scholar]
- Morris, G. M., Goodsell, D. S., Huey, R., & Olson, A. J. (1996). Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. Journal of Computer-Aided Molecular Design, 10(4), 293–304. 10.1007/BF00124499 [DOI] [PubMed] [Google Scholar]
- Pandey, K. B., & Rizvi, S. I. (2009). Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Medicine and Cellular Longevity, 2(5), 270–278. 10.4161/oxim.2.5.9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandurangan, A. P., Ochoa-Montaño, B., Ascher, D. B., & Blundell, T. L. (2017). SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Research, 45(W1), W229–W235. 10.1093/nar/gkx439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak, N., Rai, A. K., Kumari, R., & Bhat, K. V. (2014). Value addition in sesame: A perspective on bioactive components for enhancing utility and profitability. Pharmacognosy Reviews, 8(16), 147–155. 10.4103/0973-7847.134249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, V. S., Araújo, J., Silva, R. C., da Costa, G. V., Cruz, J. N., De A Neto, M. F., Campos, J. M., Santos, C., Leite, F., & Junior, M. (2019). In silico study to identify new antituberculosis molecules from natural sources by hierarchical virtual screening and molecular dynamics simulations. Pharmaceuticals ( Pharmaceuticals), 12(1), 19–36. 10.3390/ph12010036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, R. S., Macêdo, W., Costa, J. S., da Silva, C., Rosa, J., da Cruz, J. N., de Oliveira, M. S., de Aguiar Andrade, E. H., E Silva, R., Souto, R., & Santos, C. (2019). Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. Journal of Biomolecular Structure & Dynamics, 37, 1–23. 10.1080/07391102.2019.1688192 [DOI] [PubMed] [Google Scholar]
- Rosa, A., Deiana, M., Casu, V., Paccagnini, S., Appendino, G., Ballero, M., & Dessi, M. A. (2002). Antioxidant activity of capsinoids. Journal of Agricultural and Food Chemistry, 50(25), 7396–7401. 10.1021/jf020431w [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Dror, O., Inbar, Y., Nussinov, R., & Wolfson, H. J. (2008. a). Deterministic pharmacophore detection via multiple flexible alignment of drug-like molecules. Journal of Computational Biology : A Journal of Computational Molecular Cell Biology, 15(7), 737–754. 10.1089/cmb.2007.0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Dror, O., Inbar, Y., Nussinov, R., & Wolfson, H. J. (2008. b). PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Research, 36, W223–W228. 10.1093/nar/gkn187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, R. (2010). Chemistry and biochemistry of dietary polyphenols. Nutrients, 2(12), 1231–1246. 10.3390/nu2121231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, V. V., Cruz, J. N., Viana, G. M. R., Póvoa, M. M., do Socorro Barros Brasil, D., & Dolabela, M. F. (2020). Naphthoquinones isolated from Eleutherine plicata herb: In vitro antimalarial activity and molecular modeling to investigate their binding modes. Medicinal Chemistry Research, 29(3), 487–494. 10.1007/s00044-019-02498-z [DOI] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.).), 368(6489), 409–412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.