

Abstract
DNA Encoded Libraries (DEL) have shown promise as a valuable technology for democratizing the hit discovery process. Although DEL provides relatively inexpensive access to libraries of unprecedented size, their production has been hampered by the idiosyncratic needs of the encoding DNA tag relegating DEL compatible chemistry to dilute aqueous environments. Recently Reversible Adsorption to Solid Support (RASS) has been demonstrated as a promising method to expand DEL reactivity using standard organic synthesis protocols. Here we demonstrate a suite of on-DNA chemistries to incorporate medicinally relevant and C–S, C–P and N–S linkages into DELs, which are underrepresented in the canonical methods.
Keywords: DNA Encoded Library, Reversible Adsorption to Solid Support, Nickel Cross-Coupling, Sulfonamide, Sulfone
DNA encoded libraries (DEL) have become a valuable part of the hit identification repertoire. The power of DEL lies in the ability to screen billions of small molecules, for affinity, against a molecular target of interest, all at once, in a single tube.1–9 This technology has the potential to democratize the processes of drug discovery by providing an alternate route to high-throughput screening which currently requires cost intensive, specialized facilities.1–9 Although DEL has proven valuable, the idiosyncratic nature of the DNA tag (which is not soluble in organic solvents and rich in functional groups) limit both the synthetic toolbox available to elaborate the small molecule library members and the resulting molecular diversity of the libraries produced.10–13 As such, the development of chemistries that are compatible with DEL synthesis, and could be used to increase molecular diversity is of great interest.
Many of the inherent limitations of DNA as the encoding element of DEL can be overcome through immobilization,14–18 and an approach termed Reversible Adsorption to Solid Support (RASS)14–18 has been developed to facilitate the covalent manipulation of macromolecules such as proteins and DNA in organic solvents.14–18 Non-DNA encoding strategies, namely peptide nucleic acids (PNAs) can also be employed to overcome the inherent limitations of DNA as a encoding tag.19 Recently our lab has shown that DNA can be easily immobilized (through a simple mixing step) onto a highly crosslinked mixed mode polystyrene anion exchange resin bearing quaternary ammonium moieties,14 and the approach has been further supported utilizing a quaternary amine functionalized ChemMatrix (PEG) resin.17 Once adsorbed, the DNA can be readily transferred into organic solvents, where near anhydrous chemistry can be performed.14 Previously this process has been used to construct high value Csp2-Csp3, and Csp2-N bonds through a low valent nickel mediator that was shown to be water sensitive.14 These results inspired the pursuit of a suite of reactions for the robust incorporation of medicinally relevant C-S and C-N bonds on-DNA.
Currently, on-DNA sulfonamides are synthesized by treating DNA bound amines in a slightly basic aqueous solution with a sulfonyl chloride (Figure 1).29, 30 Although this method is efficient and straightforward, the practical aspects of library building limit its utility. DEL libraries which may contain billions of individual members, can take hundreds of 96 well plates and months to synthesize. Unfortunately, sulfonyl chlorides are unstable and have proven unsuitable for storage and utilization during this protracted process, presenting a challenge to their utilization. Also, synthesizing the “reverse sulfonamide” (the -SO2- group is proximal to the DNA) has been challenging or intractable, as bifunctional sulfonyl chloride building blocks are rare, and the on-DNA sulfonyl chloride moiety is not stable to conventional library building manipulations, such as aqueous enzymatic encoding, and ethanol precipitations (Figure 1). As a result, few conditions for the creation of on-DNA C-S bonds, including sulfones and sulfides, have been reported. Low valent nickel has been proven to be a powerful mediator of multiple cross-coupling reactions and its utility has been recently realized and exploited in the DEL field.13, 14, 31–33
Figure 1:
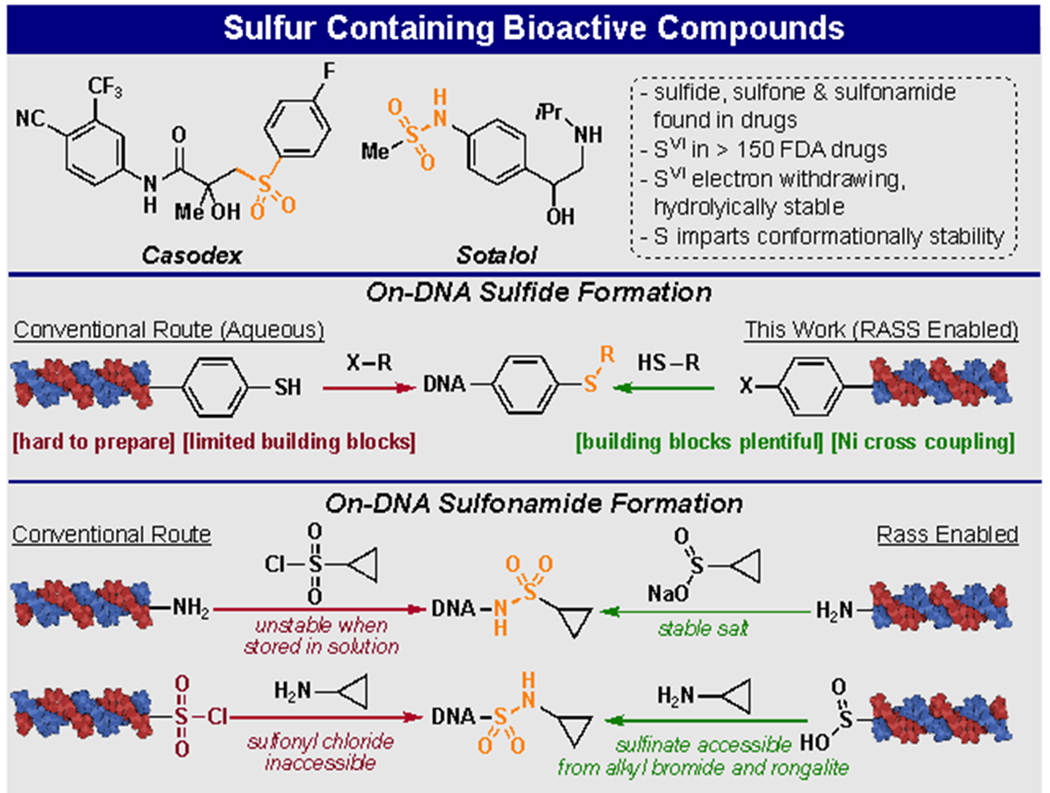
Sulfur containing bioactive compounds and routes to sulfur containing moiety incorporation into DELs
These ideas have spurred our labs interest in using a homogeneous silane based reductant in nickel mediated RASS-based cross couplings.34–36 We found the Ni/silane system in DMA to be flexible in its utility. Thus, a Ni(II) 4,4’-dimethoxybipyridine precursor, and RubenSilane (isopropoxy(phenyl)silane) was able to furnish a thioether bond between an on-DNA aryl iodide and 4-methoxythiophenol 12 in 83% yield (Figure 2A). These conditions proved competent with numerous aryl and heteroaryl thiols of varying nucleophilicity in synthetically useful yields (as defined by Berst et. al. for DEL synthesis).17 For the reactions presented typical side-product are predictable and easily identified, in this case the side products are exclusively starting material and proto-dehalogenation.
Figure 2:
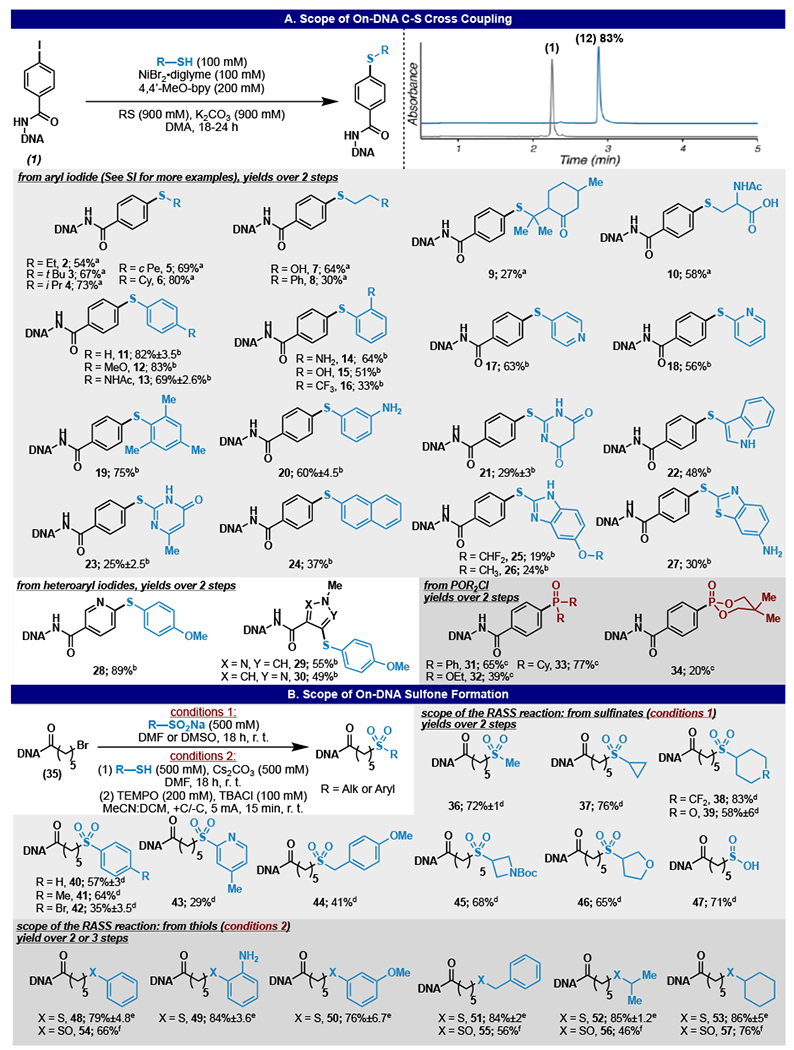
On-DNA sulfide and sulfone synthesis: (A) nickel mediated C-S cross coupling, conditions, HPLC chromatogram of reaction, and reaction scope. Deviation from above reaction conditions: a alkyl thiol (500 mM), 36 h, b none, c phosphonic chloride (100 mM), 4,4’ ditertbutyl bipyridine (200 mM). (B) on-DNA sulfone formation reaction conditions and reaction scope. Deviation from stated reaction conditions: d none; e thiol (500 mM), Cs2CO3 (500 mM), DMF, 18 h. f Electrochemical oxidation: tetrabutyl ammonium chloride (100 mM), TEMPO (200 mM), +C/−C, 5 mA, 15 min, alternating polarity every 30 sec
Various heteroaryl iodides, pyrazole and pyridine cores, were competent. Finally, these conditions were also extended to accept alkyl thiols, which had previously been challenging substrates in our hands, by increasing the concentration (500 mM vs 100 mM) (Figure 2A). Thus, the developed conditions allow facile access to diaryl and aryl-alkyl thioethers. In accord with our previous study14, the analogous reaction using conventional solution-based DEL protocols (aqueous, dilute) did not furnish any products.
During the course of this study, it was also found that phosphinic chlorides are competent coupling partners for aryl iodides creating C-P linkages that are increasingly desired as pharmacophores (Figure 2A).37, 38 Notably, these cross-coupling conditions are intolerant to water, making them incompatible with traditional DEL formats. While the reaction is facile in dry DMA, the presence of even 1% water quenches the reaction on the RASS platform and, as with C–S bond formation, no product is recovered under a standard aqueous format without the support (see SI).
Dialkyl sulfones were easily accessed through a nucleophilic displacement of an on-DNA alkyl halide and a stable alkyl or aryl sodium sulfinate salt in DMF or DMSO. This reaction yields the dialkyl sulfone in good to acceptable yields (Figure 2B). Hydrolysis was observed when performing this reaction in an aqueous solution without the RASS support. With the addition of Cs2CO3, alkyl-alkyl thioethers could be constructed in good yields (Figure 2B). All substantial side products in this reaction corresponded to the starting materials.
Sulfones and sulfoxides are highly desired pharmacophores that can be generated from sulfides.39 To increase the molecular diversity accessible from the C-S bond forming reactions, the oxidation of sulfides was explored. An initial screen revealed that employing sodium perborate (SPB) in methylene chloride as an insoluble oxidant would exclusively yield the sulfoxide oxidized product (see SI). This initial hit was further optimized, acetic acid and 1% water were found to be necessary additives to push this reaction to completion (see SI). Interestingly no DNA was recovered when using this system in an aqueous solution without RASS support, presumably due to destructive oxidation of the encoding molecule.40
The immobilization of the DNA in RASS can be leveraged to facilitate electrochemical transformations.14 An electrochemical oxidation of a model substrate was found to yield the sulfone product. The system was optimized (see SI) and TEMPO was found to be a necessary electrochemical mediator,41 with tetrabutylammonium chloride in dry DCM/MeCN. This oxidation was driven by electrochemical stimulation at a constant current of 5 mA (+C/−C) for 15 minutes. Alternating the polarity every 30 seconds proved critical to prevent fouling at the electrode and subsequent stalling of the reaction. This reaction was used to oxidize on-DNA sulfides to a mixture of sulfone and sulfoxide as a secondary manipulation (Figure 2B). Electrochemical reactions in DEL are unique to the RASS platform in which the adsorbed DNA is protected from direct oxidative damage at the anode, as well as irreversible adsorption to the cathode.14 In the context of DEL, these oxidative secondary manipulations can be envisioned as a useful step after pooling a resin bound library to convert all, or a subset of the recently created thioethers to sulfones or sulfoxides. The primary side product was generally starting material, importantly, over-oxidation was not observed.
Sodium sulfinate salts are attractive alternative sulfonamide precursors because they exhibit greater stability than the corresponding sulfonyl chloride.42 Fortunately, multiple examples of sulfonamide formation between sulfinates and amines have been reported in the synthetic organic literature and conditions were easily identified that would furnish an aryl sulfonamide product.42–45 These conditions utilized molecular iodine as oxidant and ethanol as solvent to couple the sulfinate with an on-DNA amine (Figure 3A).
Figure 3:
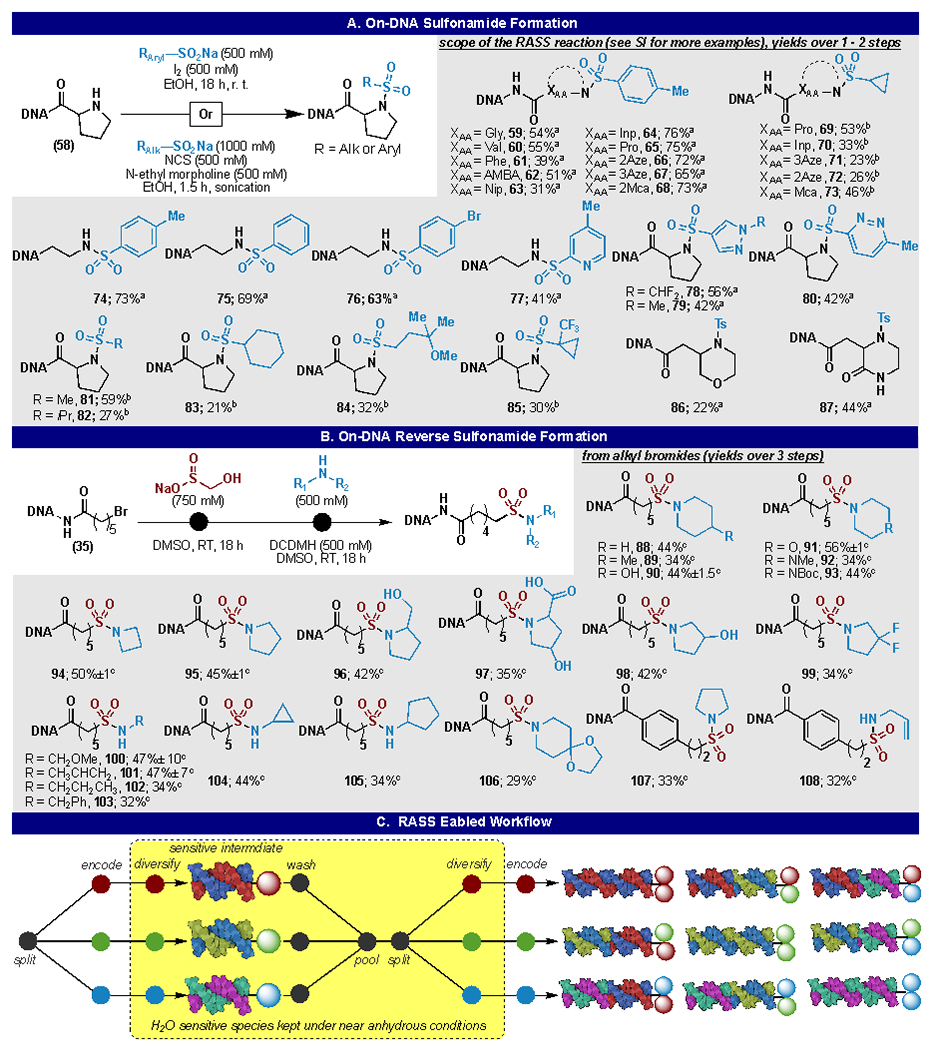
On-DNA sulfonamide and reverse sulfonamide synthesis: (A) on-DNA sulfonamide reaction conditions and reaction scope. a aryl sulfonamide formation, aryl sulfinate (500 mM), iodine (500 mM), in EtOH, 18 h. b alkyl sulfonamide formation: alkyl sulfinate (1000 mM), NCS (500 mM), in EtOH, 1.5 h sonication. (B) reverse sulfonamide formation reaction conditions and reaction scope. c reaction conditions: rongalite (750 mM), DMSO, 18 h. wash with DMSO. amine (500 mM), DCDMH (500 mM), DMSO, 18 hr. (C) proposed RASS enabled workflow for performing multiple encoded diversification steps using a sensitive intermediate.
During our reaction development process, the traditional, non-resin bound, aqueous reaction was also explored. In our hands the RASS reaction was immediately fruitful, providing >50% yield in the initial screen while the aqueous reaction provided <10% for the same system. Recently, it was reported that the iodine/sulfinate system can furnish sulfonamides from aryl sodium sulfinates in an aqueous reaction.46 This report is consistent with pilot studies at Pfizer that confirmed that sulfonamides can indeed be constructed in an aqueous environment (see SI). Interestingly, under those conditions it is acknowledged that only simple aryl sulfinates proved competent coupling partners. 46 Indeed, alkyl sulfinates and 5-membered heteroaryl sulfinates proved incompetent in the recent report, and in our efforts, the aqueous reaction afforded zero to trace yields (see SI). With this in mind, we were excited to observe heteroaryl sulfinates gave synthetically useful yields (40-60%) in the RASS system (Figure 3A).17 Unfortunately, these conditions still precluded the use of alkyl sulfinates, which resulted in low yields (<10%). The conditions were reoptimized for use of alkyl sulfinates. Employing NCS, N-ethylmorpholine, and subjecting the reaction mixture to sonication for 90 minutes furnished alkyl sulfinates in synthetically useful yields. (20-50%).17 The primary side product encountered was starting material, which would lead to a predicable deletion or termination during a real library build.
Interestingly, during the development of the dialkylsulfone reaction, it was observed that when rongalite was employed as the coupling partner the product resulted in the corresponding on-DNA sulfinic acid (47). This fortuitous result provides another handle from which to create on-DNA sulfonamides. A one-pot two addition protocol was developed to create “reverse sulfonamides” from on-DNA alkyl halides (Figure 3B). Utilizing the on-DNA sulfinic acid in situ, the addition of 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) and an aliphatic amine, in DMSO resulted in creation of the “reverse sulfonamide.” Ùntil recently the construction of on-DNA “reverse sulfonamides” was unreported and a recent report utilized bifunctional sulfinate-benzoic acids.46 Although this strategy permits the construction of “reverse sulfonamides” the building block set of bifunctional sulfinic acids is limited. In contrast our method permits the use of readily available alkyl halides and amines in the construction of “reverse sulfonamides.” This expansion of the on-DNA sulfonamide chemistry permits the design of libraries containing sulfonamides in either direction, allowing for increased molecular complexity and building block diversity.
Another advantage the RASS system presents is the ability to use unstable intermediates through two chemical elaboration cycles (Figure 3C). This process would be potentially useful as many building blocks or chemistries that one could employ might not be stable to enzymatic encoding or ethanol precipitation. Thus, it is possible to consider a library cycle in which the DEL is split and encoded, the library member elaborated, the DEL pooled and split again (while remaining on beads, in organic solvent) a second chemical transformation performed, and finally encoded in the last step (Figure 3C). This process could prove useful in situations where a condition change is required, or a reactive intermediate is involved.
For the RASS platform to find widespread utility in real world library building, the binding and elution characteristics of the larger DNA to the solid support must be efficient. Duplexed DNA, in the 30-60 base pair (bp) range, is more relevant to a multicycle DEL build that includes multiple encoding steps. Strata-XAL resin (100 μm particles, 300 Å pore size) is designed to adsorb and release negatively charged macromolecules greater than 10,000 MW. DNA adsorption to Strata-XAL was monitored by loss of absorbance at 260 nm using 6 different DNA head pieces (HP) of length varying from 6 to 57 bp (Figure 4A). When incubated in PBS, complete adsorption is realized in 30 minutes for all DNA species tested. In contrast, when a 40 bp DNA duplex is adsorbed to the small molecule optimize Strata-XA resin (30 μm particles, 90 Å pore size), or adsorbed to either of these resins in unbuffered water, the adsorption kinetics are severely affected (see SI). These results suggest that resin pore size, pH, and ionic strength of the adsorbing solution are critical, but all the DNA constructs examined can be utilized with the previously published conditions (Figure 4A).14
Figure 4:
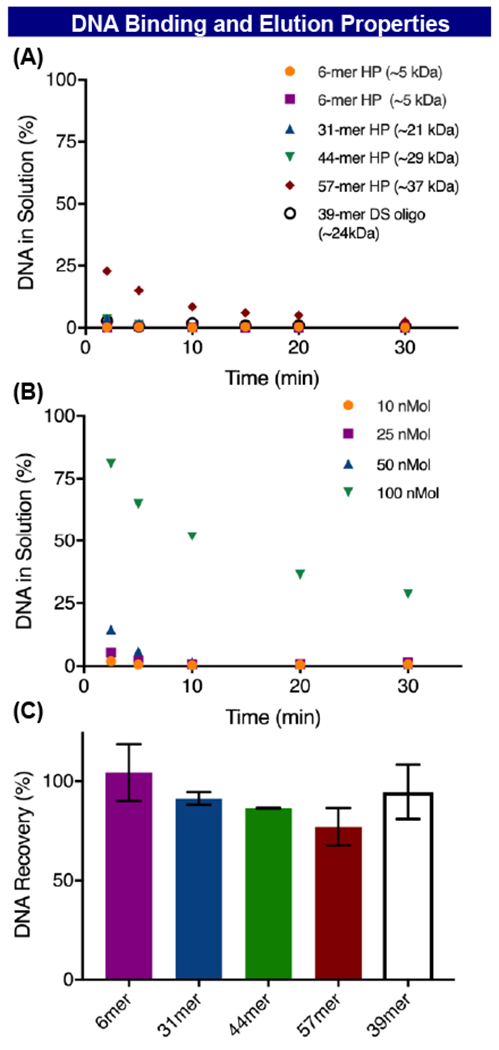
DNA binding and elution: (A) DNA adsorption kinetics to strata XAL with various DNA constructs. (B) loading capacity of DNA onto Strata XAL using 40mer dsDNA. (C) DNA elution from strata XAL using various DNA constructs.
The resin loading capacity was also studied. During any step in a DEL build it is desirable to perform reactions on between 10-50 nmol of DNA per well (~1 mL). The resin (100 μL, equilibrated in 1:1 MeCN:PBS) was loaded with multiple aliquots of 40 bp dsDNA in PBS and the amount of DNA that remained in solution after 30 minutes was quantified (Figure 4B). Unsurprisingly, it was found that when 100 μL of resin was incubated to 10, 25, and 50 nmols of 40mer dsDNA, complete DNA adsorption was observed after 30 minutes (Figure 4B). When the resin was incubated with 100 nmol of DNA, about 25% of the DNA remained in solution (not adsorbed to the resin). With this in mind, we believe that 50 nmol of DNA can be safely adsorbed to 100 μL of resin. The ability to adsorb 50 nmol of DNA to 100 μL of resin should ensure sufficient DNA loading on a sufficiently small volume of resin to be useful in any DEL step.
The elution kinetics were also monitored by A260 for various DNA headpieces (Figure 4C). Unsurprisingly the original elution buffer (1 M NaClO4, 20% MeOH, 40 mM Tris pH 8.5) proved competent to elute all tested DNA constructs from the Strata-XAL resin (100 μm particle, 300 Å pore) in acceptable quantities (77->95%). Although this recovery might be sufficient for some, the elution buffer was then optimized. Different elution buffers were screened for their ability to elute a 40 bp duplex from Strata AXL resin with two superior buffers being identified (see SI). It was also observed that performing a single 300 μL elution wash resulted in superior DNA recovery than the previously reported strategy of performing 3 elution washes at 100 μL (see SI). This new strategy has proven to be very efficient as DNA elution occurs almost instantaneously, with maximum elution occurring after only five minutes of exposure to the elution buffer (see SI). The ease and efficiency of the single wash is now the default elution protocol in all RASS reactions in our labs.
In conclusion we have presented a suite of reactions for the construction of C–S, N–S, and C–P bonds on DNA. We describe a low valent Ni mediated cross coupling between thiols and on-DNA aryl iodides to furnish diaryl or alkyl-aryl thioethers. The resulting thioethers can be subsequently oxidized (chemically or electrochemically) to the corresponding sulfoxide or sulfone. Dialkylsulfones are accessed through the nucleophilic displacement of on-DNA alkyl bromides by alkyl or aryl sodium sulfinates. Sulfonamides are formed in both directions by oxidative couplings between sulfinates and amines. Finally binding and elution kinetics of longer DNA HPs are outlined and alternative elution buffers and protocols are presented.
Supplementary Material
Acknowledgements
Financial support for this work was provided by NIH (1S10OD025208) and Pfizer. We are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy. The authors would like to acknowledge Yong Chang and Zhao Wang for their work in DNA oligomer synthesis. DTF was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR002551 and linked award TL1 TR002551. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. All DNA illustrations created using Goodsell’s Illustrate.47
References
- 1.Kowalczyk A, A Handbook for DNA-Encoded Chemistry: Theory and Applications for Exploring Chemical Space and Drug Discovery (Ed.: Goodnow RE) Wiley-VCH: Weinheim, Germany, 2014, pp. 1–18. [Google Scholar]
- 2.Belyanskaya SL, Ding Y, Callahan JF, Lazaar AL and Israel DI, ChemBioChem 2017, 18, 837–842. [DOI] [PubMed] [Google Scholar]
- 3.Favalli N, Bassi G, Scheuermann J and Neri D, FEBS Letters 2018, 592, 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lerner RA and Brenner S, Angew. Chem. Int. Ed 2017, 56, 1164–1165. [DOI] [PubMed] [Google Scholar]
- 5.Mannocci L, Leimbacher M, Wichert M, Scheuermann J and Neri D, Chem. Commun 2011, 47, 12747–12753. [DOI] [PubMed] [Google Scholar]
- 6.Neri D, ChemBioChem, 2017, 18, 827–828. [DOI] [PubMed] [Google Scholar]
- 7.Neri D and Lerner RA, Ann. Rev. of Biochem, 2018, 87, 479–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao G, Huang Y, Zhou Y, Li Y and Li X, Expert Opin. Drug Discov 2019, 14, 735–753. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann G and Neri D, Drug Discov. Today 2016, 21, 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satz AL, ACS Med. Chem. Lett 2018, 9, 408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franzini RM and Randolph C, J. Med. Chem 2016, 59, 6629–6644. [DOI] [PubMed] [Google Scholar]
- 12.Fraley i A. W., A Handbook for DNA-Encoded Chemistry: Theory and Applications for Exploring Chemical Space and Drug Discovery (Ed.: Goodnow RE) Wiley-VCH: Weinheim, Germany, 2014, pp. 153–169. [Google Scholar]
- 13.Wang J, Lundberg H, Asai S, Martin-Acosta P, Chen JS, Brown S, Farrell W, Dushin RG, O’Donnell CJ, Ratnayake AS, Richardson P, Liu Z, Qin T, Blackmond DG and Baran PS, Proc. Natl. Acad. Sci. U.S.A 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flood DT, Asai S, Zhang X, Wang J, Yoon L, Adams ZC, Dillingham BC, Sanchez BB, Vantourout JC, Flanagan ME, Piotrowski DW, Richardson P, Green SA, Shenvi RA, Chen JS, Baran PS and Dawson PE, J. Am. Chem. Soc 2019, 141, 9998–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flood DT, Yan NL and Dawson PE, Angew. Chem. Int. Ed 2018, 57, 8697–8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cistrone PA and Dawson PE, ACS Comb. Sci 2016, 18, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruffs Y, Martinez R, Pellé X, Nimsgern P, Fille P, Ratnikov M, Berst F, An Amphiphilic Polymer-Supported Strategy Enables Chemical Transformations Under Anhydrous Conditions for DNA-Encoded Library Synthesis, Chemrxiv 2019. [DOI] [PubMed] [Google Scholar]; The team at Novartis assert that the production of predicable side products, is critical for DEL library reaction development and under these conditions, a yield of >25% is synthetically useful.
- 18.Halpin DR, Lee JA, Wrenn SJ and Harbury PB, PLoS Biol. 2004, 2, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zambaldo C, Barluenga S and Winssinger N, Curr. Opin. Chem. Biol 2015, 26, 8–15. [DOI] [PubMed] [Google Scholar]
- 20.Feng M, Tang B, Liang SH and Jiang X, Curr. Top. Med. Chem 2016, 16, 1200–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beno BR, Yeung KS, Bartberger MD, Pennington LD and Meanwell NA, J. Med. Chem 2015, 58, 4383–4438. [DOI] [PubMed] [Google Scholar]
- 22.Lücking U, Org. Chem. Front 2019, 6, 1319–1324. [Google Scholar]
- 23.Chen X, Hussain S, Parveen S, Zhang S, Yang Y and Zhu C, Curr. Med. Chem 2012, 19, 3578–3604. [DOI] [PubMed] [Google Scholar]
- 24.Shoaib Ahmad Shah S, Rivera G and Ashfaq M, Mini-Rev. Med. Chem 2012, 13, 70–86. [PubMed] [Google Scholar]
- 25.Politzer P, Murray JS and Clark T, Phys. Chem. Chem. Phys 2013, 15, 11178–11189. [DOI] [PubMed] [Google Scholar]
- 26.Hale SP, A Handbook for DNA-Encoded Chemistry: Theory and Applications for Exploring Chemical Space and Drug Discovery (Ed.: Goodnow RE) Wiley-VCH: Weinheim, Germany, 2014, pp. 281–317 [Google Scholar]
- 27.Franzini RM and Randolph C, J. Med. Chem 2016, 59, 6629–6644. [DOI] [PubMed] [Google Scholar]
- 28.Malone ML and Paegel BM, ACS Comb. Sci 2016, 18, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buller F, Steiner M, Frey K, Mircsof D, Scheuermann J, Kalisch M, Buhlmann P, Supuran CT and Neri D, ACS Chem. Biol 2011, 6, 336–344. [DOI] [PubMed] [Google Scholar]
- 30.Satz AL, Cai J, Chen Y, Goodnow R, Gruber F, Kowalczyk A, Petersen A, Naderi-Oboodi G, Orzechowski L and Strebel Q, Bioconjugate Chem. 2015, 26, 1623–1632. [DOI] [PubMed] [Google Scholar]
- 31.Phelan JP, Lang SB, Sim J, Berritt S, Peat AJ, Billings K, Fan L and Molander GA, J. Am. Chem. Soc 2019, 141, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kölmel DK, Loach RP, Knauber T and Flanagan ME, ChemMedChem 2018, 13, 2159–2165. [DOI] [PubMed] [Google Scholar]
- 33.Kölmel DK, Meng J, Tsai M-H, Que J, Loach RP, Knauber T, Wan J and Flanagan ME, ACS Comb. Sci 2019, 21, 588–597. [DOI] [PubMed] [Google Scholar]
- 34.Green SA, Vasquez-Cespedes S and Shenvi RA, J. Am. Chem Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obradors C, Martinez RM and Shenvi RA, J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shevick SL, Obradors C and Shenvi RA, J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang WS, Liu S, Zou D, Thomas M, Wang Y, Zhou T, Romero J, Kohlmann A, Li F, Qi J, Cai L, Dwight TA, Xu Y, Xu R, Dodd R, Toms A, Parillon L, Lu X, Anjum R, Zhang S, Wang F, Keats J, Wardwell SD, Ning Y, Xu Q, Moran LE, Mohemmad QK, Jang HG, Clackson T, Narasimhan NI, Rivera VM, Zhu X, Dalgarno D and Shakespeare WC, J. Med. Chem 2016, 59, 4948–4964. [DOI] [PubMed] [Google Scholar]
- 38.Janczewski L, Psurski M, Switalska M, Gajda A, Goszczynski TM, Oleksyszyn J, Wietrzyk J and Gajda T, ChemMedChem 2018, 13, 105–115. [DOI] [PubMed] [Google Scholar]
- 39.Sato K, Hyodo M, Aoki M, Zheng X-Q and Noyori R, Tetrahedron 2001, 57, 2469–2476. [Google Scholar]
- 40.Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De Deken X, Van Sande J, Dumont JE, Miot F and Corvilain B, Endocr. Relat. Cancer. 2009, 16, 845–856. [DOI] [PubMed] [Google Scholar]
- 41.Studer A and Vogler T, Synthesis 2008, 2008, 1979–1993. [Google Scholar]
- 42.Aziz J, Messaoudi S, Alami M and Hamze A, Org. Biomol. Chem 2014, 12, 9743–9759. [DOI] [PubMed] [Google Scholar]
- 43.Jiang YY, Wang QQ, Liang S, Hu LM, Little RD and Zeng CC, J. Org. Chem 2016, 81, 4713–4719. [DOI] [PubMed] [Google Scholar]
- 44.Yang K, Ke M, Lin Y and Song Q, Green Chem. 2015, 17, 1395–1399. [Google Scholar]
- 45.Shyam PK and Jang HY, J. Org. Chem 2017, 82, 1761–1767. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Deng W, Sun S, Yu C, Su X, Wu A, Yuan Y, Ma Z, Li K, Yang H, Peng X and Dietrich J, Org. Lett 2019, DOI: 10.1021/acs.orglett.9b03843. [DOI] [PubMed] [Google Scholar]
- 47.Goodsell DS, Autin L and Olson AJ, Structure, 2019, DOI: 10.1016/j.str.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.