

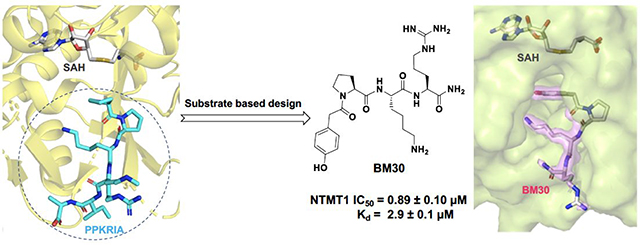
Abstract
Protein N-terminal methyltransferases (NTMTs) methylate the α-N-terminal amines of proteins starting with the canonical X-P-K/R motif. Genetic studies imply that NTMT1 regulates cell mitosis and DNA damage repair. Herein, we report the rational design and development of the first potent peptidomimetic inhibitors for NTMT1/2. Biochemical and co-crystallization studies manifest that BM30 (IC50 of 0.89 ± 0.10 μM) is a competitive inhibitor to the peptide substrate and noncompetitive to the cofactor S-adenosylmethionine. BM30 exhibits over 100-fold selectivity to NTMT1/2 among a panel of 41 methyltransferases, indicating the potential to achieve high selectivity when targeting the peptide substrate binding site of NTMT1/2. Its cell-permeable analog DC432 (IC50 of 54 ± 4 nM) decreases the N-terminal methylation level of regulator of chromosome condensation 1 and SET proteins in HCT116 cells. This proof-of principle study provides valuable probes for NTMT1/2 and highlights the opportunity to develop more cell-potent inhibitors to elucidate the function of NTMTs in the future.
Graphical Abstract
INTRODUCTION
Protein methylation is an important epigenetic modification that regulates dynamic chromatin states and transcription.1,2 Therefore, potent and specific inhibitors of protein methyltransferases can serve as valuable chemical probes to elucidate the function in physiological and pathological contexts. For example, recently approved Tazverik (tazemetostat) for epithelioid sarcoma treatment is an inhibitor for protein lysine methyltransferase (PKMT) EZH2. Another PKMT inhibitor, Pinometostat targeting DOT1L, is in a phase I trial for combination use for patients with acute myeloid leukemia.1 In addition, a few protein arginine methyltransferase 5 (PRMT5) inhibitors, including JNJ64619178, GSK3326595, PF06939999, and PRT811, are also in clinical trials for advanced solid tumors.3
Protein α-N-terminal methylation has been known for nearly four decades since it was first uncovered on bacteria ribosomal proteins L33.4,5 The identification of N-terminal methyltransferase 1 (NTMT1/NRMT1) unveiled the first methylation writer for human protein α-N-termini.6,7 In addition to NTMT1, its close homolog NTMT2/NRMT2 that shares over 50% sequence similarity has been identified as another writer for human protein α-N-terminal methylation.8 Both NTMT1 and 2 share the same X-P-K/R canonical recognition motif, where X can be any amino acid although D and E are not preferred.9–11 Recently, METTL13 has been discovered to solely methylate the elongation factor 1A (EF1A) on both the α-N-terminus and lysine side chain.12,13 However, the N-terminal sequence of EF1A is different from the X-P-K/R motif that NTMT1/2 prefer. Structural studies on NTMT1/2 in complex with their substrate peptides reveal that the peptide substrates bind at a defined binding pocket that juxtapose with the cofactor S-adenosyl methionine (SAM) binding site.9–11 This is distinct from PKMTs and PRMTs, which normally have a narrow channel to accommodate only the side chain of the Lys or Arg. The structural information gained from the co-crystal structures substantiated the unique substrate selectivity of NTMT1/2.9–11
Protein α-N-terminal methylation was originally proposed to regulate protein-protein interactions since the methylated proteins initially identified were involved in large macromolecular complexes including histone proteins, cytochrome c-557, and myosin light chain proteins.4,14 Recent discoveries have demonstrated its relevance in protein-DNA interactions, as shown in strengthening the interactions of chromatin with centromere protein A (CENPA) and regulator of chromosome condensation 1 (RCC1), as well as DNA damage-binding protein 2 (DDB2) to DNA damage foci.15–17 Meanwhile, newly identified substrates of NTMT1 also include the tumor suppressor retinoblastoma 1 (RB1), oncoprotein SET, and poly(ADP-ribose) polymerase 3.6,18 In addition, previous genetic studies have inferred the importance of NTMT1 in mitosis, DNA damage repair, aging, and a variety of cancers.6,16,17,19–21 Despite the aforementioned progresses, our understanding of protein α-N-terminal methylation function is still in its infancy. Therefore, it is imperative to discover specific and potent inhibitors for NTMTs to probe their biological functions. Guided by the kinetic mechanism of NTMT1, bisubstrate analogs that covalently link both SAM and peptide substrate were designed to mimic the transition state and displayed high selectivity and potency for NTMT1.22–26 Nevertheless, intrinsic properties of those bisubstrate analogs impose a challenge to penetrate cell membranes, which limit their applications in cell-based studies. Therefore, there is a need for a new strategy to discover inhibitors for NTMTs in order to probe the downstream process of protein α-N-terminal methylation. Given that the NTMT family has a unique peptide substrate binding site, we hypothesize that targeting the peptide binding site should achieve higher selectivity for NTMTs. Herein, we report the design and synthesis of the first potent and selective peptidomimetic inhibitors for the NTMT1/2.
RESULTS AND DISCUSSION
Design
According to the crystal structures of NTMT1 in complex with peptide substrates (XPKRIA), the backbone carbonyl group of the first amino acid (X1) interacts with a conserved Asn168 residue NTMT1 through H-bonding. 9,27 Mutation of Asn168 to Lys resulted in a ~36-fold loss in Km, indicating the importance of this interaction between NTMT1 and its peptide substrate9. However, the tolerance at the first position of the substrate suggests a possible spacious binding pocket surrounding the first position. The N-terminal amine of the peptide substrate points toward the methyl group of the SAM and serves as a nucleophile to receive the methyl group from the SAM. The second residue P2 forms a stacking interaction with the indole of Trp136. The third residue K3 forms two key hydrogen bonds with side chains of two aspartic acids.9–11 Either a Lys or Arg residue has been discovered at the third position of known protein substrates. The fourth amino acid is adjacent to a negatively charged substrate binding channel, where the fifth and sixth residues are primarily exposed to the solvent.9–11 Hence, we speculate that the α-N-terminal amine is essential for methyl transfer by acting as a nucleophile to attack SAM, but it is not essential for binding. Therefore, we hypothesized that the removal of the N-terminal amine of NTMT1/2 peptide substrates would convert the substrate into an inhibitor. Since the first four amino acids contribute significantly to the interaction with NTMT1/2, we initiated our efforts by incorporating different substitutions (R1) onto a tripeptide (PKR) through an amid bond (Figure 1) to retain as much interaction as the peptide substrates. PPKRIA and YPKRIA are two peptide substrates for NTMT1 with a Kd value at 0.7+0.2 nM and 0.1+0.08 μM, respectively.9 Hence, compounds 1-5 were designed to mimic the Pro or Tyr moiety by introducing cyclopentane, cyclohexane, furan, phenyl, and a benzyl moiety as the R1 group.
Figure 1.

Design inhibitors based on the structure of NTMT1 peptide substrates. H-bond interactions are shown in red dotted lines.
Synthesis
The peptides were synthesized following the standard Fmoc solid-phase synthesis on Rink amide resin using a CEM Liberty microwave peptide synthesizer. The peptidomimetics that have carboxylic acids at the first position were prepared through the standard amino acid coupling reaction (Scheme 1). All compounds were cleaved from the solid support in a cleavage cocktail consisting of trifluoroacetic acid/2,2’-(ethylenedioxy)diethanethiol/H2O/triisopropylsilane (94:2.5:2.5:1), and followed by purification through reverse-phase HPLC and characterized through mass spectrometry (Figure S1).
Biochemical Characterization
All peptidomimetics were first subject to a methylation progression MALDI-MS study to examine if they were NTMT1 substrates.22,28 None of the peptidomimetics displayed any methylation, indicating that they were not NTMT1 substrates. Next, the synthesized peptidomimetics were evaluated in a SAH hydrolase (SAHH)-coupled fluorescence assay at 100 μM of SAM and at the Km value of the peptide substrate (SPKRIA or GPKRIA).22 Initial screening was carried out at the concentration of 5 μM, 30 μM, and 100 μM of each compound. Compound 5 exhibited more than 50% of inhibition at 30 μM, and was then subjected to subsequent IC50 studies by ranging the inhibitor concentration from 0 – 100 μM in a three-fold dilution. Compound 5 showed an IC50 of 6.85+2.82 μM (Figure S2). A summary of the IC50 values for the peptidomimetics is shown in Table 1.
Table 1.
SAR of the first position of R1-PKR*
| Compound | Structure | IC50 (μM) | Compound | Structure | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 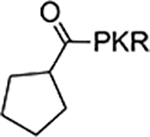 |
>100 | 7 | 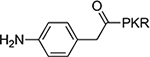 |
4.3±0.28 |
| 2 | 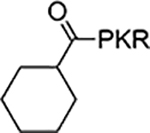 |
>100 | 8 | 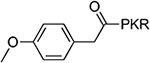 |
>100 |
| 3 | 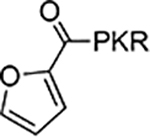 |
>100 | 9 | 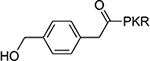 |
1.5±0.1 |
| 4 | 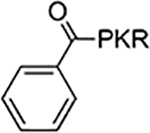 |
>100 | 10 | 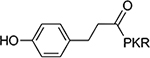 |
4.5±0.5 |
| 5 | 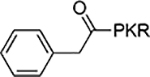 |
6.85+2.82 | 11 | 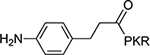 |
12±1.4 |
| 6 (BM30) | 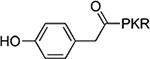 |
0.89±0.10 | 12 | >100 |
IC50 values were determined in triplicates (n = 3) and presented as mean ± standard deviation (SD).
Structure-Activity Relationship (SAR) Studies
In order to improve the inhibitory activity of the peptidomimetics, SAR studies were conducted by using 5 as the lead compound (Tables 1–2).
Table 2.
SAR of the 2nd-4th position of R1-PKR*
| Compound | Structure | IC50 (μM) | Compound | Structure | IC50 (μM) |
|---|---|---|---|---|---|
| 13 | 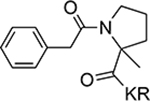 |
>100 | 19 | 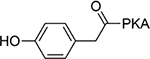 |
4.8±0.71 |
| 14 | 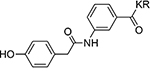 |
>100 | 20 | 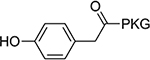 |
13±1.6 |
| 15 | 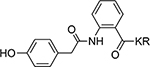 |
>100 | 21 | 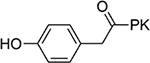 |
16±4.7 |
| 16 | 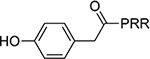 |
21±3.3 | 22 | 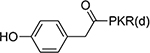 |
4.2±1.2 |
| 17 | 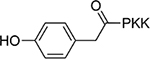 |
1.0±0.22 | 23 | 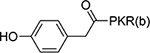 |
6.4±0.5 |
| 18 | 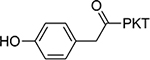 |
4.8±0.31 | 24 | 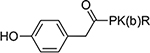 |
32.9±8.3 |
IC50 values were determined in triplicates (n = 3) and presented as mean ± SD.
The first (R1) position
The benzoyl substitution of compound 5 resembled the side chain of Tyr; therefore, we focused our SAR investigation on introducing various para-substitutions on the phenyl ring, including a hydroxy, an amino, and a methoxy group at the para-position of the phenyl ring to yield compounds 6-8 (Table 1). Among all synthesized peptidomimetics, compound 6 (BM30) which contained a hydroxyl group at the para position showed the greatest inhibitory activity with an IC50 of 0.89+0.10 μM (Figure S3), approximately 7-fold increased activity compared to 5. However, a methoxy substitution led to a loss of inhibition of 8 (IC50 > 100 μM), while its constitutional isomer 9 rescued the inhibitory activity (IC50 = 1.5+0.1 μM) with a 4-hydroxymethyl substitution. This suggests the preference of a H-bond donor at the first position. Given that introducing a methylene linker between the phenyl ring and the carbonyl group significantly increased the inhibition of 5 compared to 4, an ethylene linker was introduced in 10 and 11 to probe the optimal linker length (Table 1). Both 10 and 11 displayed a 5-fold and 3-fold loss in inhibition compared to their respective parent compounds 6 and 7, suggesting a methylene group as the optimal linker for the NTMT1 inhibitor. Lastly, compound 12 was prepared to explore the importance of the carbonyl group at the first position. Nevertheless, 12 did not show any inhibitory activity at 100 μM, implying the significance of the carbonyl group at the first position.
The second position
All available co-crystal structures of NTMT1/2 in complex with X-P-K/R substrates reveal that Pro2 only exhibits the trans conformation, suggesting that the trans conformation is favored to interact with NTMT1/2.9,11,27 To verify the preference of this trans-conformation, an alpha-methyl-Pro was introduced at the 2nd position since the predominant conformation of alpha-methyl-Pro is cis.29 The resulting compound 13 completely lost the inhibitory activity for NTMT1 (Table 2). These results substantiate previous findings that the Pro2 prefers the trans conformation; however, the additional methyl group may also introduce certain steric hindrances. Next, ortho- and meta-amino benzoic acids were also introduced at Pro2 to generate 14 and 15, respectively. Both compounds did not exhibit any inhibitory activity, denoting the importance of Pro2 in the canonical X-P-K/R motif. The 3rd and 4th position. Next, we explored the modifications at the third and fourth position in an attempt to increase inhibitory activity, as well as to enhance stability since peptide-based inhibitors are susceptible to degradation (Table 2). Arg has been detected at the 3rd position in the validated NTMT1 substrate CENP-A (GPRR),10,15 thus 16 was synthesized with Arg at the 3rd position. However, 16 displayed over a 20-fold loss of inhibition compared to 6. This result signified the preference for Lys over Arg at the third position, potentially due to its important role in interacting with two acidic residues (Asp177 and Asp180 in NTMT1; Asp232 and Asp 235 in NTMT2).9–11 Although Arg and Lys are two prevalent amino acids at the 4th position of the NTMT1 substrate peptides, additional amino acids have been observed in reported NTMT1 substrates. Therefore, Lys, Thr, Ala, and Gly were introduced to replace the Arg at the 4th position of BM30 to generate 17-21 to examine the contribution of side chain at this position because it is pointed to the solvent in the co-crystal structures.9 Compound 17, which included a Lys in the 4th position, exhibited negligible difference in inhibition compared to BM30 which contains an Arg. Introducing Thr or Ala at the 4th position led to a 5-fold of loss as shown in 18-19, while incorporating Gly produced almost a 14-fold loss in inhibition as shown in 20. When the 4th position was eliminated to yield 21, inhibitory activity decreased 18-fold compared to BM30. The result indicated that the side chain of the 4th amino acid does contribute to the NTMT1 interaction although it points towards to the solvent in the co-crystal structures. Additionally, non-natural amino acid was also made at either the 3rd or 4th position to produce 22-24 in attempts to increase the stability against proteases. However, loss of inhibition ranging from nearly 5- to 37-fold was observed in compounds 22-24 compared to BM30, although 22 was the most potent one among these three protease-stable analogues.
Selectivity Studies
Given that many methyltransferases exist, selectivity of the inhibitor is a critical requirement for its potential as a valuable probe. We first investigated the selectivity of BM30 and 22 against one representative member from the protein lysine methyltransferase PKMT (G9a) and protein arginine methyltransferase PRMT (PRMT1) families. Both enzymes share a cofactor SAM binding site despite having different substrate peptide binding sites from NTMT1. As shown in Table S1, BM30 demonstrated >100 μM IC50 values against both G9a and PRMT1, but 22 also exhibited inhibition for G9a. Thus, we proceeded with BM30 to evaluate its selectivity against a panel of 41 MTases (Reaction Biology) that include NTMT1/2, PKMTs, PRMTs, and DNMTs (Figure 2, Table S2). The results indicated that BM30 displays comparable inhibition against both NTMT1 and NTMT2, which was not surprising since NTMT1/2 share the canonical X-P-K/R recognition motif. Strikingly, BM30 exhibited very marginal inhibition against the remaining 39 MTases even at 100 μM. The over 100-fold selectivity that BM30 displays for NTMT1/2 compared to PKMTs, PRMTs, and DNMTs evinces the unique substrate specificity of NTMT1/2.
Figure 2.

Selectivity of NTMT inhibitor BM30. Selectivity of BM30 against a panel of 41 PKMTs, PRMTs, DNMTs and NTMT1/2 was determined at concentration of 100 μM in duplicates (n=2).
Inhibition Mechanism
Kinetic characterization of BM30 was performed to examine its mechanism of action using the SAHH-coupled fluorescence-based assay (Figure 3). As shown in Figure 3, BM30 exhibited a competitive inhibition pattern with respect to the NTMT1 peptide substrate RCC1–6 (SPKRIA). This is demonstrated by an ascending and linear dependence of the IC50 values relative to the increasing peptide substrate concentration of RCC1–6. On the other hand, BM30 displayed a noncompetitive inhibition pattern with respect to the cofactor SAM, indicated by a line that is parallel to the x-axis. These results verify that BM30 competitively binds to the peptide substrate binding site, supporting our design strategy.
Figure 3.

Inhibition mechanism of BM30. All experiments were run in triplicate (n=3). (A) IC50 curves of BM30 at varying concentrations of NTMT1 peptide substrate, RCC1–6 (SPKRIA), with fixed concentration of SAM; (B) linear regression plot of IC50 values with corresponding concentrations of RCC1–6; (C) IC50 curves of BM30 at varying concentrations of SAM with fixed concentration of RCC1–6; (D) Plot of IC50 values with corresponding concentrations of SAM.
Crystal Structure of the NTMT1-BM30-SAH Complex
To further confirm that BM30 binds to the peptide substrate binding site of NTMT1, we solved the X-ray co-crystal structure of NTMT1-BM30-SAH (PDB ID: 6WH8) (Figure 4A–C). BM30 occupies the peptide substrate binding site of NTMT1. Superimposition of our NTMT1-BM30-SAH structure with the published NTMT1-YPKRIA-SAH ternary complex (PDB ID: 5E1D) gave an RMSD value of 0.12 Å (across all residues of chain A, Figure 4D–E). The BM30-NTMT1 complex retains the same interactions as previously observed with YPKRIA-NTMT1 in the ternary complex of substrate peptide/SAH.9 For instance, the carbonyl oxygen of the 4-hydroxyl phenyl acetic acid interacts with the side chain of Asn168 through hydrogen bonding. The second Pro occupies a hydrophobic pocket that is formed by Leu31, Ile37, and Ile214. In addition, the ε-amine of the third Lys forms electronic interactions with carboxylate groups of Asp177 and Asp180. In addition, a direct H-bond exists between the carbonyl oxygen of the fourth Arg and Try215. The 4-hydroxyl group of the phenyl ring also forms a H-bond with Asp180 and one water molecule.
Figure 4.

Co-crystal structure of NTMT1(gray cartoon)-BM30 (green stick)-SAH (green stick) (PDB ID: 6WH8). (A) Fo-Fc omit density map of ligands. (B) Zoomed view of interactions of BM30 with NTMT1. H-bond interactions are shown in yellow dotted lines. (C) BM30 interaction diagram (Schrödinger Maestro) with NTMT1. (D) and (E) Structural alignment of NTMT1(gray)-BM30 (green stick)-SAH (green stick) and NTMT1(yellow cartoon)-YPKRIA (yellow stick)-SAH (yellow stick) (PDB ID: 5E1D)
Design and characterization of DC431 and DC432
We tested the cell permeability of BM30 through MS detection of its cellular levels.30 However, BM30 was minimally detected at 100 μM inside the cells (Figure S4). In order to increase the cell permeability, we designed and synthesized DC431 and DC432 by attaching cell-permeable peptide TAT (GRKKRRQRRR-NH2) and five Arg at the C-terminus of BM30, respectively. It is known that incorporating six Arg into compounds can increase the cell-permeability.31 Hence, five additional Arg were added to BM30 to generate DC432 because BM30 already had one Arg at its C-terminus. Given that the first four amino acids contribute significantly to the binding for NTMT1/2, we hypothesized that DC431 and DC432 would have slightly increased inhibitory activities than BM30, since they contained additional amino acids at the C-terminus of BM30. To confirm our hypothesis, IC50 values of DC431 and DC432 were determined to be 0.23+0.03 μM and 0.054+0.004 μM, respectively (Figure 5A&B). DC431 demonstrated a 4-fold increased inhibition for NTMT1 than BM30 as predicted. Surprisingly, DC432 displayed a 4-fold higher activity than DC431 and 16-fold inhibitory activity compared to BM30.
Figure 5.

Biochemical characterization of DC431 and DC432. (A,B) IC50 curve of DC431 and DC432. IC50 values were determined in triplicates (n = 3) and presented as mean ± SD. (C) Representative western blots showing N-terminal dimethylation (Me2RCC1) and trimethylation (Me3RCC1) of full-length recombinant human RCC1 by full-length recombinant human NTMT1 in the presence of increasing concentrations of BM30, DC431 and DC432 (n=3). NTMT1 and RCC1 blots are shown as loading controls.
In addition, we tested the levels of N-terminal methylation by directly incubating the inhibitors with NTMT1, SAM and full-length substrate protein RCC1. At 2 μM of BM30 or DC432, there is a slight increase of me2-RCC1 levels and decrease of me3-RCC1 levels as compared to untreated samples (Figure 5C). An increase in me2-RCC1 levels indicates inhibition of NTMT1 trimethylation, as NTMT1 is a distributive enzyme that normally reaches saturated trimethylation levels under these conditions.22 The appearance and persistence of me2-RCC1 signals enzymatic inhibition. At 10 and 50 μM, there was a considerable decrease in me3-RCC1 levels and increase in me2-RCC1 levels for all three inhibitors (Figure 5C). To further validate their interactions with both NTMT1 and 2, we determined binding affinities of both cell-permeable analogs and BM30 via isothermal titration calorimetry (Figure 6A&B, Table S4). BM30 exhibited a comparable Kd value of 3.0 μM and 3.7 μM to NTMT1 and 2, respectively. But DC431 showed a 4-fold increase in binding to NTMT1. Furthermore, DC432 displayed an 8-fold and 4-fold tighter binding affinity to NTMT1 and NTMT2, respectively. Increased inhibitory activities and binding affinities of both DC431 and DC432 corroborated the contributing roles of C-terminal amino acids to XPKR, as demonstrated before in the NTMT1-SPKRIA-SAH and NTMT2-SPKRIA-SAH ternary complexes.9,11 In addition, there is 3- to 6-fold difference in DC431 and DC432 for NTMT1 over NTMT2 based on the data from ITC, but unmodified BM30 showed similar Kd values in both NTMT1/2. This change of the selectivity for NTMT1/2 demonstrated the contribution of C-terminal sequence to the selectivity, suggesting the potential of development of selective inhibitors for NTMT1 over its homolog NTMT2. Furthermore, enhanced cell penetration of DC431 and DC432 were also confirmed through a cellular MS assay. Both DC431 and DC432 exhibited cellular penetration even at 1 μM, while BM30 did not display any even at 100 μM (Figure S4).
Figure 6.

Binding affinity for NTMT1(A) (n=2) and NTMT2 (B) (n=1).
Cellular N-terminal Methylation Level
NTMT1 can catalyze the trimethylation of its substrate starting with an SPK motif. Therefore, a cell permeable inhibitor of NTMT1 would be expected to decrease this trimethylation level. Then we proceeded to assess the inhibitory effects of DC432 on the N-terminal trimethylation level via western blotting with a specific antibody to the N-terminal me3-SPK motif. As shown in Figure 7, treatment with DC432 for 72 h in HCT116 cells did result in a decrease in the me3-SPK level at 100 μM. Although the impact on mono- and di-methylation levels were not examined for this investigation, cellular inhibition was over 100-fold lower than the value obtained from the enzymatic assay (Figure 5B). The discrepancy between these results may be due to low efficiency of DC432 to reach the target since NTMT1 is predominantly in the nucleus as reported before.6,8,27
Figure 7.

Cellular N-terminal methylation assay of compound DC432 in HCT116 cells. (A) Structure of DC432. (B) Representative western blot results of effects of DC432 (0–1 mM) on cellular methylation level (n=2). RCC1, SET, and Lamin B1 blots are shown as loading controls.
CONCLUSION
In summary, this study describes the discovery of the first potent and selective peptidomimetic inhibitor BM30 for NTMT1/2 reported to date. Furthermore, BM30 exhibits over 100-fold selectively against NTMT1/2 among a panel of 41 MTases. The cell-permeable derivative DC432 represses N-terminal methylation levels of NTMT1/2 substrates in HCT116 cells, demonstrating that DC432 would be a valuable probe for mechanistic studies of NTMT1/2 in cellular contexts. In addition, the crystal structure of BM30 in complex with NTMT1 revealed the structural basis for its potency and selectivity, which paves the way for the future design and development of inhibitors with improved cell potency.
MATERIALS AND METHODS
Chemistry General Information
All reagents and solvents were purchased from commercial sources (Fisher, Aldrich, and Chem-Impex) and used directly. Final compounds were purified on preparative high-pressure liquid chromatography (RP-HPLC) was performed on either Waters 1525 or Agilent 1260 Series system. Systems were run with 0–20% methanol/water gradient with 0.1% TFA. Matrix-assisted laser desorption ionization mass spectra (MALDI-MS) data were acquired in positive-ion mode using a Sciex 4800 MALDI TOF/TOF MS. The peptides (PKR, PKRIA and PRRRS) were synthesized on a CEM Liberty Blue Automated Microwave Peptide Synthesizer with the manufacturers standard coupling cycles at 0.1 mmol scale. All compounds were cleaved from the resin in a cocktail of trifluoroacetic acid/2,2’-(ethylenedioxy)diethanethiol/H2O/triisopropylsilane (94:2.5:2.5:1) and confirmed by mass spectrometry. The purity of final compounds was confirmed through Agilent 1260 Series HPLC system by running with 5% to 30% methanol/water gradient with 0.1% TFA. All the purity of target compounds showed >95%.
Cell Culture and Antibodies
HCT116 cells were obtained from ATCC and were cultured in McCoy’s 5A (modified) medium (Gibco, #16600082), supplemented with 10% FBS (Gibco, #16000044) and 1% antibiotic-antimycotic (Gibco, #15240062). Antibodies against following proteins were used: Me3-SPK (Schaner Tooley lab 6), RCC1 (Proteintech, #22142–1-AP), SET (Proteintech, #55201–1-AP), Lamin B1 (Proteintech, # 12987–1-AP), Anti-rabbit IgG, HRP-linked (Cell Signaling, #7074S).
NTMT1 Biochemical Assays
Expression and purification of NTMT1 was performed as previously described.25 Kinetic characterization of the peptide inhibitors was determined in a fluorescence-based SAHH-coupled assay as described before. The inhibitors were added at concentrations ranging from 0 to 100 μM following a three-fold dilution. The methylation assay was performed under the following conditions: 25 mM Tris (pH = 7.5), 50 mM KCl, 0.01% Triton X-100, 5 μM SAHH, 0.2 μM NTMT1, 100 μM SAM, and 15 μM ThioGlo1. After 10 min of incubation at 37 °C, the reaction was initiated with 10 μL of 50 μM RCC1–6 for a total volume of 100 μL. Fluorescence intensity was monitored using a BMG ClarioStar microplate reader (Ex = 370 nm, Em=500 nm) at 37 °C for 15 min. The rates were fit to the log[inhibitor] vs response-variable slope (four parameters) model using least squares nonlinear regression using GraphPad Prism software 7 or 8. All experiments were performed in triplicate.
Inhibition Mechanism of BM30 was performed using the fluorescent-based assay described above. Six independent IC50 studies of BM30 were performed in triplicate with varying concentrations of substrate peptide, RCC1–6 and SAM at its Km value. The inhibitors ranging in concentration (1 μL of 0 – 10 mM) and following a three-fold dilution were incubated in the well-solution that was added in the following order: ddH2O, buffer (10 μL of 250 mM Tris, pH 7.4 and 500 mM KCl), SAM (1 μL of 10 μM), SAH hydrolase (5 μL of 10 mg/mL), NTMT1 (0.5 μL of 40 μM) and ThioGlo1 (1 μL of 1.5 mM). After 10 min of incubation at 37 °C, the reaction was initiated with each concentration of RCC1–6 (10 μL of 5, 10, 15, 20, 40, 80 μM) for a total volume of 100 μL. The final concentrations are buffer (25 mM Tris, pH 7.4 and 50 mM KCl), SAM (1 μM), SAH hydrolase (10 μM), NTMT1 (0.2 μM), inhibitors (0 – 100 μM), RCC1–6 (0.25Km, 0.5Km, 0.75Km, Km, 2Km, and 4Km, 0.5, 1, 1.5, 2, 4, 8 μM, respectively) and Thioglo1 (15 μM). Fluorescence intensity was monitored using a CLARIOstar microplate reader (Ex = 370 nm, Em=500 nm) at 37 °C for 15 min. The rates were fit to the log[inhibitor] vs response model using least squares nonlinear regression through GraphPad Prism 7 software. The average IC50 value of each independent triplicate study was then plotted against the concentration of the [RCC1–6]/Km. Next, the same experiment was repeated with six IC50 studies of BM30 in triplicate at varying concentrations of SAM (1 μL of 31.25, 62.5, 125, 250, 500, and 1000 μM) and RCC1–6 at its Km value. The final concentrations are buffer (25 mM Tris, pH 7.4 and 50 mM KCl), SAM (0.3125Km, 0.625Km, 1.25Km, 2.5Km, 5Km, and 10Km, 0.3125, 0.625, 1.25, 2.5, 5, 10 μM, respectively), SAH hydrolase (10 μM), NTMT1 (0.2 μM), inhibitors (0 – 100 μM), RCC1–6 (2 μM) and Thioglo1 (15 μM). The average IC50 value of each independent triplicate study were plotted against the concentration of the [SAM]/Km.
Selectivity Studies
To determine selectivity, kinetic analysis of the peptide inhibitors was initially carried out using the SAH hydrolase-coupled fluorescence assay against G9a and PRMT1, respectively. The inhibitors were added at concentrations ranging from 0 to 100 μM following a three-fold dilution. For G9a, the assay was performed in 1x PBS, pH7.4, 100 μM SAM, 10 μM SAH hydrolase, 25 nM G9a, inhibitors (0 – 100 μM), 20 μM H3–15 peptide, and 15 μM Thioglo1. For PRMT1, the assay was carried out in 2.5 mM HEPES (pH 7.0), 25 mM NaCl, 25 μM EDTA, 50 μM TCEP, 100 μM SAM, 10 μM SAH hydrolase, 200 nM PRMT1, 100 μM H4–12 peptide, and 15 μM Thioglo1. Fluorescence intensity was monitored using a CLARIOstar microplate reader (Ex = 370 nm, Em=500 nm) at 37 °C for 15 min. The rates were fit to the log[inhibitor] vs response model using least squares nonlinear regression through GraphPad Prism 7 software. All experiments were performed in triplicate.
Co-crystallization and Structure Determination
Purified NTMT1 at 30mg/ml was mixed with BM30 at a molar ratio of 1:2, incubated for 1 hr at 4°C. Broad matrix crystallization screening was performed using a Mosquito-LCP high throughput crystallization robot (TTP LabTech) using hanging-drop vapor diffusion method at 20°C. Crystals containing BM30 were grown in 0.1 M Na-HEPES, pH 7.5, 1.6 M ammonium sulfate, and 2% PEG 1000; all crystals were harvested directly from the drop and flash cooled into liquid nitrogen. Data were collected on single crystals at 12.0 keV at the GM/CA-CAT ID-D beamline at the Advanced Photon Source, Argonne National Laboratory. The data were processed using the on-site automated pipeline with Fast DP and molecular replacement performed with Phaser-MR (PHENIX).32,33 All model building was performed using COOT and subsequent refinement done using phenix.refine (PHENIX).33,34 The structure has been solved, refined and deposited into the Protein Data Bank with ID: 6WH8. Structure-related figures were made with PyMOL (Schrödinger) and annotated and finalized with Adobe Photoshop and Illustrator.
In Vitro Methylation Assays with Full-length Recombinant Protein
All methylation assays were conducted at 30°C for 1 hour in methyltransferase buffer (50 mM potassium acetate, 50 mM Tris, pH 8.0). Recombinant NTMT1 (1 μg) was mixed with 1.0 μg full-length recombinant RCC1 and 100 μM SAM and inhibitors added to give the indicated concentrations (2, 10, and 50 μM). All His6-tagged recombinant proteins were expressed in E. coli and purified as previously described. The same batches of NTMT1 and RCC1 were used in all experiments. Positive controls contained no inhibitor, negative controls contained no NTMT1. One tenth of each reaction was mixed with 5x loading dye and loaded onto a 10% SDS–PAGE gel. Gels were transferred to nitrocellulose membranes using a Biorad Trans-Blot SD Semi-Dry Transfer Cell. Membranes were blocked for 1 h in 5% milk/TBST, then incubated with anti-me3-SPK (1:10,000), anti-me2-SPK (1:5000), or NTMT1 (1:1000) antibodies (add Tooley CE et al Nature 2010) overnight at 4 °C. They were then washed three times with 1x TBST and incubated with Rabbit IgG-HRP antibody (Jackson ImmunoResearch) for 1 h at room temperature. The membrane was washed three times with 1x TBST and detected using a Biorad ChemiDoc imaging system. Image quantification was done using ImageJ software (NIH). For Me3RCC1, all bands were compared to the untreated control, which was set at 1.0. For Me2RCC1, all bands were compared to the highest intensity band, which was set at 1.0. For NTMT1 and RCC1 loading controls, all bands were compared to the average band intensity, which was set at 1.0.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were performed at 25 °C by VP-ITC MicroCal calorimeter. Purified NTMT1 and NTMT2 were diluted with the ITC buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5 mM TECP) to the final concentration of 30–50 μM, and the compounds were dissolved to 0.5–1mM in the same buffer. The compound was titrated into the protein solution with 26 injections of 10 μl each. Injections were spaced 180 sec with a reference power of 15 μcal/sec. The ITC data were processed with Origin 7.0 software (Microcal).
Cell Permeability Evaluation by MALDI
The colon cancer cell line HCT116 was cultured in Mccoy’s media supplemented with 10% fetal bovine serum and 1% Antibiotic-Antimycotic. The cells were cultured in tissue culture dish (Falcon 353003). Cells were maintained in cell culture flasks until seeding into a 24-well tissue culture plate (Falcon 353047). Media was removed and the cells were washed with 1xPBS (1 mL) twice followed by treating with TrypLE Express (1 mL) into a 100 × 20 mm culture flask. The reaction was quenched by addition of 4 mL of media and the cells were counted. Cells were seeded into a 24-well tissue culture plate (Falcon 353047) at a density of 0.1 × 106 cells/mL and incubated overnight at 37 °C, 5% CO2, and 95% humidity, with the lid on. They were then treated with the inhibitors at different concentrations and incubation was continued for the specified time. TAT peptide (GRKKRRQRRR-NH2) was also incubated as a control experiment. The media was removed, and the cells were washed with 1x PBS three times to remove any residual compound or peptide attached to the cell surface. Then 100 μL of 1 x PBS was added and the cells were snap freeze in liquid nitrogen twice. The cell lysate was then analyzed with MALDI using DHB matrix to identify the presence of the compound inside the cell.
Cellular N-methylation Level
HCT116 cells were seeded 20,000 cells/well on 24-well plates in the presence of 1x PBS or DC432 at different concentrations for 3 days. Then cells were lysed in 1x RIPA cell lysis buffer (25mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, protease inhibitors) and incubated for 30 min on ice. The cell lysates were centrifuged at 15000 rpm for 10 min and the precipitates were removed. The concentration of total protein was quantified by bicinchoninic acid (BCA) protein assay kit (ThermoFisher, #23228). Equal amounts of total protein were mixed with 4x loading dye and loaded onto a 12.5% SDS–PAGE gel and separated. The gel was transferred to a polyvinylidene difluoride (PVDF) membrane using Biorad Trans-Blot Turbo system. The membrane was then blocked for 1 h in 5% milk TBST solution and washed with 1x TBST solution three times. The membrane was incubated with anti-me3-SPK antibody (1:5000), Lamin B1 antibody (1:2000), SET (1:1000) and RCC1 antibody (1:1000) at 4 °C for 12 h, washed with 1x TBST solution three times and then incubated with Rabbit IgG-HRP antibody (1:1000) for 1 h at room temperature. The membrane was again washed with 1x TBST solution three times and detected using a Protein Simple FluorChem imaging system. The data was analyzed in GraphPad prism software.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the support from NIH grants R01GM117275 (RH), 1R01GM127896 (NN), 1R01AI127793 (NN), R01GM112721 (CST), P30 CA023168 (Purdue University Center for Cancer Research), and Natural Sciences and Engineering Research Council Grant RGPIN-2016-06300 (JM). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI-055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Ontario Ministry of Research, Innovation and Science (MRIS), Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We also thank supports from the Department of Medicinal Chemistry and Molecular Pharmacology (RH) and Department of Biological Sciences (NN) at Purdue University.
ABBREVIATIONS
- NTMT
protein N-terminal methyltransferase
- SAM
S-5’-adenosyl-L-methionine
- SAH
S-5’-adenosyl-L-homocysteines
- SAHH
SAH hydrolase
- PKMT
protein lysine methyltransferase
- PRMT
protein arginine methyltransferase
- rt
room temperature
- TFA
trifluoroacetic acid
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Figure S1. MALDI-MS and HPLC spectra of compounds 1-24, DC431 and DC432; Figure S2. IC50 curve of compound 5; Figure S3. IC50 curve of BM30; Figure S4. Cell permeability evaluation; Table S1. Selectivity study BM30 and 22 against G9a and PRMT1; Table S2. Selectivity study of BM30 against a panel of 41 MTases (Reaction Biology); Table S3. Crystallography data and refinement statistics (PDB ID: 6WH8); Table S4. IC50 values and binding affinities of BM30 and its cell-permeable analogs.
Molecular formula strings (CSV)
Accession Codes
The coordinates for the structure of human NTMT1 in complex with BM30 have been deposited under PDB ID 6WH8. Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- (1).Ribich S; Harvey D; Copeland RA Review Drug Discovery and Chemical Biology of Cancer Epigenetics. Cell Chem. Biol. 2017, 24, 1120–1147. [DOI] [PubMed] [Google Scholar]
- (2).Scheer S; Ackloo S; Medina TS; Schapira M; Li F; Ward JA; Lewis AM; Northrop JP; Richardson PL; Kaniskan HÜ; Shen Y; Liu J; Smil D; McLeod D; Zepeda-Velazquez CA; Luo M; Jin J; Barsyte-Lovejoy D; Huber KVM; De Carvalho DD; Vedadi M; Zaph C; Brown PJ; Arrowsmith CH A Chemical Biology Toolbox to Study Protein Methyltransferases and Epigenetic Signaling. Nat. Commun. 2019, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chan-Penebre E; Kuplast KG; Majer CR; Boriack-Sjodin PA; Wigle TJ; Johnston LD; Rioux N; Munchhof MJ; Jin L; Jacques SL; West KA; Lingaraj T; Stickland K; Ribich SA; Raimondi A; Scott MP; Waters NJ; Pollock RM; Smith JJ; Barbash O; Pappalardi M; Ho TF; Nurse K; Oza KP; Gallagher KT; Kruger R; Moyer MP; Copeland RA; Chesworth R; Duncan KW A Selective Inhibitor of PRMT5 with in Vivo and in Vitro Potency in MCL Models. Nat. Chem. Biol. 2015, 11, 432–437. [DOI] [PubMed] [Google Scholar]
- (4).Huang R Chemical Biology of Protein N-Terminal Methyltransferases. ChemBioChem 2019, 20, 976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen R; Brosius J; Wittmann-Liebold B Occurrence of Methylated Amino Acids as N-Termini of Proteins from Escherichia Coli Ribosomes. J. Mol. Biol. 1977, 111, 173–181. [DOI] [PubMed] [Google Scholar]
- (6).Schaner Tooley CE; Petkowski JJ; Muratore-Schroeder TL; Balsbaugh JL; Shabanowitz J; Sabat M; Minor W; Hunt DF; Macara IG NRMT Is an Alpha-N-Methyltransferase That Methylates RCC1 and Retinoblastoma Protein. Nature 2010, 466, 1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Webb KJ; Lipson RS; Al-Hadid Q; Whitelegge JP; Clarke SG Identification of Protein N-Terminal Methyltransferases in Yeast and Humans. Biochemistry 2010, 49, 5225–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Petkowski JJ; Bonsignore LA; Tooley JG; Wilkey DW; Merchant ML; Macara IG; Schaner Tooley CE NRMT2 Is an N-Terminal Monomethylase That Primes for Its Homologue NRMT1. Biochem. J. 2013, 456, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dong C; Mao Y; Tempel W; Qin S; Li L; Loppnau P; Huang R; Min J Structural Basis for Substrate Recognition by the Human N-Terminal Methyltransferase 1. Genes Dev. 2015, 29, 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wu R; Yue Y; Zheng X; Li H Molecular Basis for Histone N-Terminal Methylation by NRMT1. Genes Dev. 2015, 29, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dong C; Dong G; Li L; Zhu L; Tempel W; Liu Y; Huang R; Min J An Asparagine/Glycine Switch Governs Product Specificity of Human N-Terminal Methyltransferase NTMT2. Commun. Biol 2018, 1, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jakobsson ME; Małecki JM; Halabelian L; Nilges BS; Pinto R; Kudithipudi S; Munk S; Davydova E; Zuhairi FR; Arrowsmith CH; Jeltsch A; Leidel SA; Olsen JV; Falnes PØ The Dual Methyltransferase METTL13 Targets N Terminus and Lys55 of EEF1A and Modulates Codon-Specific Translation Rates. Nat. Commun. 2018, 9, 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu S; Hausmann S; Carlson SM; Fuentes ME; Francis JW; Pillai R; Lofgren SM; Hulea L; Tandoc K; Lu J; Li A; Nguyen ND; Caporicci M; Kim MP; Maitra A; Wang H; Wistuba II; Porco JA Jr.; Bassik MC; Elias JE; Song J; Topisirovic I; Van Rechem C; Mazur PK; Gozani O METTL13 Methylation of EEF1A Increases Translational Output to Promote Tumorigenesis. Cell 2019, 176, 491–504.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nomoto Minoru, Kyogoku Yoshimasa, and K. I. N-Trimethylalanine, a Novel Bloacked N-Terminal Residue of Tetrahymena Histone H2B. J. Biochem 1982, 92, 1675–1678. [DOI] [PubMed] [Google Scholar]
- (15).Sathyan KM; Fachinetti D; Foltz DR α-Amino Trimethylation of CENP-A by NRMT Is Required for Full Recruitment of the Centromere. Nat. Commun. 2017, 8, 14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen T; Muratore TL; Schaner Tooley CE; Shabanowitz J; Hunt DF; Macara IG N-Terminal Alpha-Methylation of RCC1 Is Necessary for Stable Chromatin Association and Normal Mitosis. Nat. Cell Biol. 2007, 9, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cai Q; Fu L; Wang Z; Gan N; Dai X; Wang Y α-N-Methylation of Damaged DNA-Binding Protein 2 (DDB2) and Its Function in Nucleotide Excision Repair. J. Biol. Chem. 2014, 289, 16046–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dai X; Rulten SL; You C; Caldecott KW; Wang Y Identification and Functional Characterizations of N-Terminal Alpha-N-Methylation and Phosphorylation of Serine 461 in Human Poly(ADP-ribose) Polymerase 3. J Proteome Res 2015, 14, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shields KM; Tooley JG; Petkowski JJ; Wilkey DW; Garbett NC; Merchant ML; Cheng A; Schaner Tooley CE Select Human Cancer Mutants of NRMT1 Alter Its Catalytic Activity and Decrease N-Terminal Trimethylation. Protein Sci. 2017, 26, 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bonsignore LA; Tooley JG; Van Hoose PM; Wang E; Cheng A; Cole MP; Schaner Tooley CE NRMT1 Knockout Mice Exhibit Phenotypes Associated with Impaired DNA Repair and Premature Aging. Mech. Ageing Dev. 2015, 146, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hitakomate E; Hood FE; Sanderson HS; Clarke PR The Methylated N-Terminal Tail of RCC1 Is Required for Stabilisation of Its Interaction with Chromatin by Ran in Live Cells. BMC Cell Biol. 2010, 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Richardson SL; Mao Y; Zhang G; Hanjra P; Peterson DL; Huang R Kinetic Mechanism of Protein N-Terminal Methyltransferase 1. J. Biol. Chem. 2015, 290, 11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang G; Richardson SL; Mao Y; Huang R Design, Synthesis, and Kinetic Analysis of Potent Protein N-Terminal Methyltransferase 1 Inhibitors. Org. Biomol. Chem. 2015, 13, 4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang G; Huang R Facile Synthesis of SAM-Peptide Conjugates through Alkyl Linkers Targeting Protein N-Terminal Methyltransferase 1. RSC Adv. 2016, 6, 6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen D; Dong G; Noinaj N; Huang R Discovery of Bisubstrate Inhibitors for Protein N-Terminal Methyltransferase 1. J. Med. Chem. 2019, 62, 3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen D; Dong C; Dong G; Srinivasan K; Min J; Noinaj N; Huang R Probing the Plasticity in the Active Site of Protein N-Terminal Methyltransferase 1 Using Bisubstrate Analogs. J. Med. Chem. 2020, 63, 8419–8431. 10.1021/acs.jmedchem.0c00770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Petkowski JJ; Schaner Tooley CE; Anderson LC; Shumilin IA; Balsbaugh JL; Shabanowitz J; Hunt DF; Minor W; Macara IG Substrate Specificity of Mammalian N-Terminal α-Amino Methyltransferase. Biochemistry 2012, 51, 5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Richardson SL; Hanjra P; Zhang G; Mackie BD; Peterson DL; Huang R A Direct, Ratiometric, and Quantitative MALDI-MS Assay for Protein Methyltransferases and Acetyltransferases. Anal. Biochem. 2015, 478, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).De Poli M; Moretto A; Crisma M; Peggion C; Formaggio F; Kaptein B; Broxterman QB; Toniolo C Is the Backbone Conformation of Cα-Methyl Proline Restricted to a Single Region? Chem. - A Eur. J 2009, 15, 8015–8025. [DOI] [PubMed] [Google Scholar]
- (30).Chen D; Li L; Diaz K; Iyamu ID; Yadav R; Noinaj N; Huang R Novel Propargyl-Linked Bisubstrate Analogues as Tight-Binding Inhibitors for Nicotinamide N-Methyltransferase. J. Med. Chem. 2019, 62, 10783–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Qian Z; Liu T; Liu Y-YY; Briesewitz R; Barrios AM; Jhiang SM; Pei D Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS Chem. Biol. 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Winter G; Ashton A Fast_dp. Methods Zenodo. 2014, 55, 81–93. [Google Scholar]
- (33).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.