

Abstract
Lysine (Lys) residues in proteins undergo a wide range of reversible post-translational modifications (PTMs), which can regulate enzyme activities, chromatin structure, protein-protein interactions, protein stability, and cellular localization. Here we discuss the “writers,” “erasers,” and “readers” of some of the common protein Lys PTMs and summarize examples of their major biological impacts. We also review chemical biology approaches, from small molecule probes to protein chemistry technologies, that have helped to delineate Lys PTM functions and show promise for a diverse set of biomedical applications.
eTOC Blurb
Zhipeng Wang and Philip Cole review major types of chemical modifications of the amino acid lysine in proteins and how they are added and removed. They also discuss a number of chemical approaches and how they have been applied to clarify the roles of lysine modifications in biology and medicine.
Graphical Abstract
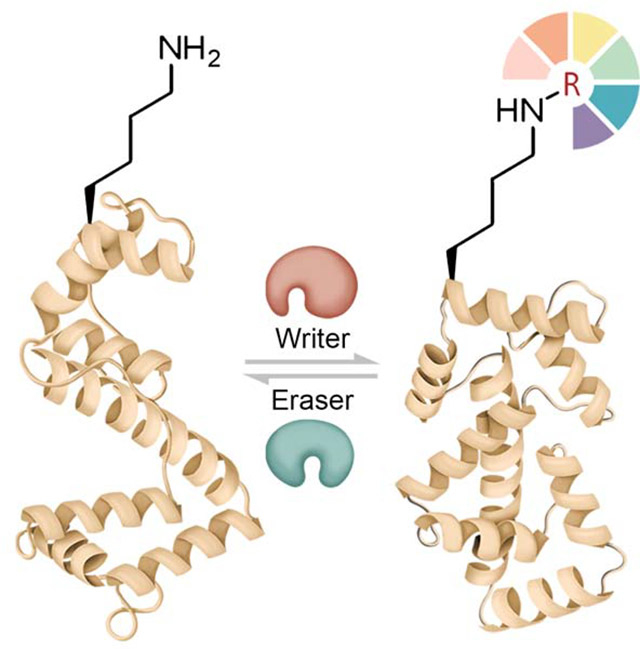
1. Introduction
The discovery of Lys sidechain acetylation and methylation over 50 years ago in the context of histone proteins initiated a transformation in our understanding of gene regulation and beyond(Ali et al., 2018)(Luo, 2018). Subsequently, protein Lys ubiquitination has revolutionized the field of protein turnover(Brooks and Gu, 2006). More recently, a wide range of protein Lys acylations including propionylation (KPr), butylation (KBu), crotonylation (KCro), malonylation (KMal), succinylation (KSucc), glutarylation (KGlu), β-hydroxybutylation (KBhb), 2-hydroxyisobutyryation (KHib), lactylation (KLac)(Zhang et al., 2019), and benzoylation (KBz)(Huang et al., 2018) have been reported although the functions of these PTMs are minimally characterized(Huang et al., 2015) (Fig. 1). Beyond ubiquitination (KUb), ubiquitin-like PTMs such as SUMOylation (KSumo), NEDDylation (KNedd), and others are becoming intensive areas of investigation. Altogether, more than 100,000 sites of Lys modifications in over 10,000 proteins have been mapped by mass spectrometry and other methods(Hornbeck et al., 2012). In many cases, particular Lys residues can be modified by a number of different PTMs allowing for the potential of a diverse pattern of biological outputs and complex regulation. Below, we provide a summary of how some of these Lys PTMs are attached and removed, examples of how they mediate their structural and functional effects on proteins, and how their dysregulation can contribute to diseases. We also cover a range of chemical approaches from small molecule probes to protein chemistry techniques that have contributed to our understanding of protein Lys modifications and their cellular effects.
Figure 1:
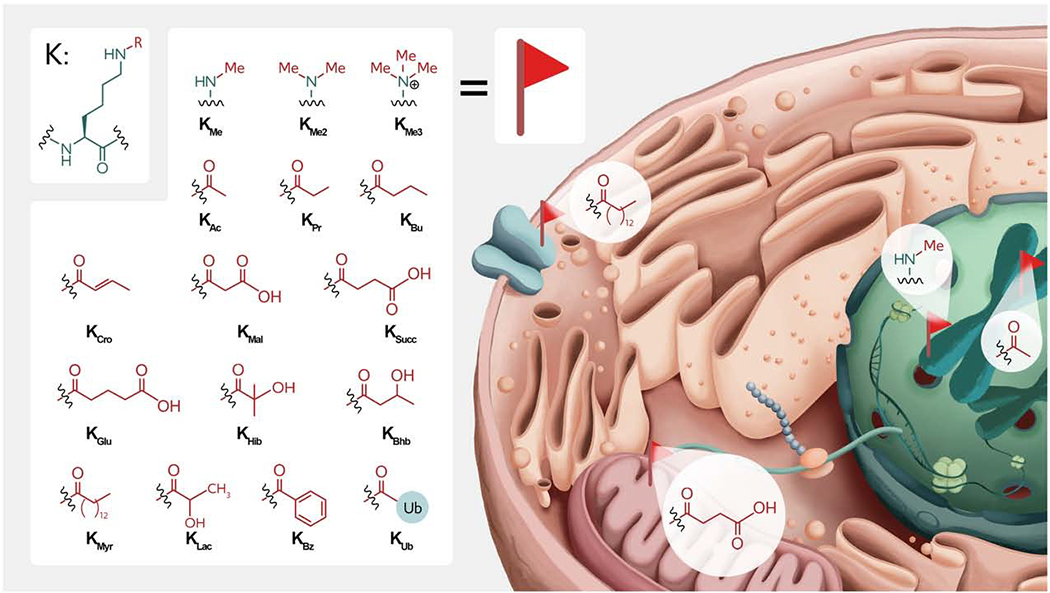
Structures and locations of various Lys PTMs in eukaryotic cells. Modifications include mono-, di-, and tri-methylation (KMe1,2,3); acetylation (KAc), propionylation (KPr), butyrylation (KBu), crotonylation (KCro), malonylation (KMal), succinylation (KSucc), glutarylation (KGlu), 2-hydroxyisobutyrylation (KHib), β-hydroxybutyrylation (KBhb), myristoylation (KMys), lactylation (KLac), benzoylation (KBz) and ubiquitination (KUb). Note that the cell structure is a cartoon to show the proposed locations of different protein Lys PTMs in the cell.
2. Writers, Erasers, and Readers for Lys PTMs
The two major classes of enzymes that add and remove Lys PTMs, also known as “writers” and “erasers,” can determine the amount, distribution, site-specificity, and pattern of particular Lys PTMs. A third group of proteins that are commonly recruited to the modified Lys residues are often called “readers” and can transmit some of the functional impacts of Lys PTMs(Lambert et al., 2019). Notably, some large proteins may contain both a “reader” domain and a “writer” or “eraser” domain for the same or different Lys PTM(s)(Cieniewicz et al., 2014). Below, we discuss well-characterized examples of writers, erasers, and readers for Lys acetylation, methylation, ubiquitination, and rare acylations.
2.1. Lys Acetylation Writers, Erasers, and Readers
Lys acetylation (KAc), exists very widely in the human proteome with more than 38,000 mapped sites and can influence many cellular activities including gene expression, cell growth, and differentiation(Hornbeck et al., 2012).
2.1.1. Lys Acetyltransferases
Lys acetyltransferases (KATs, also commonly known as histone acetyltransferases or HATs) use acetyl-coenzyme A (acetyl-CoA, Ac-CoA) as their obligatory co-substrates (Fig. 2A). Acetyl-CoA contains a reactive thioester, which facilitates acetyl transfer to Lys amino groups and the nonenzymatic rates can be appreciable. There are three main groups of well-characterized nuclear KATs: GCN5/PCAF, p300/CBP, and the MYST family(Dancy and Cole, 2015). These KAT families appear to be examples of convergent evolution, and there are little or no significant sequence or structural similarities between the three families. GCN5/PCAF are components of the SAGA multiprotein complex and are highly selective for acetylating histone H3, especially on Lys14(Nagy and Tora, 2007), whereas p300/CBP are broad spectrum acetyltransferases with over 1000 protein Lys sites mapped by mass spectrometry(Hansen et al., 2019). p300/CBP are thought to be the major enzymes for acetylating Lys18 and Lys27 in histone H3 and have been tied to regulating enhancer elements in transcription(Weinert et al., 2018). MYST enzymes are found in multiprotein complexes and can acetylate histones and other proteins. Each of these KATs has been implicated in diseases, including cancer, with both loss of function and gain of function roles in neoplasia(Dancy and Cole, 2015). p300/CBP and MYST enzymes appear to be regulated by autoacetylation and multiple protein-protein interactions(Dancy and Cole, 2015).
Fig. 2:

Writer and eraser enzymatic features for Lys acetylation, methylation, and ubiquitination. A. HATs; B. Classical HDACs, showing typical chelating Asp residues and the key Tyr residue; C. Sirtuins, ADP stands for adenosine diphosphate; D. KMTs, showing nucleophilic displacement with SAM; E. LSD1, highlighting the oxidized flavin; F. Jmj demethylase, highlighting the active site iron; G. E1-E2-RING/HECT-E3; H. DUBs, the water molecule/hydrolysis are omitted. Note that the images are cartoons of the stylized enzyme active sites instead of structurally precise crystal structures.
2.1.2. Lys Deacetylases
The removal of KAc is catalyzed by 18 different protein Lys deacetylases (KDACs, also commonly known as histone deacetylases or HDACs) in humans, which can be generally divided into four classes(Haberland, Montgomery and Olson, 2009). The eleven classical KDACs are distributed among Class I, II, and IV HDACs which are metallohydrolases that utilize a zinc(II) ion to catalyze the hydrolysis of acetyl groups (Fig. 2B). However, some members of the class II HDAC family lack a key catalytic Tyr residue which is replaced with a His and appear to be pseudoenzymes. Class I HDACs (HDAC1, 2, 3, 8) are predominantly in cell nuclei and HDAC1–3 are found in a range of multiprotein complexes. Examples of these are CoREST(Humphrey et al., 2001), Sin3(Laherty et al., 1997), NuRD(Xue et al., 1998), SMRT(Guenther et al., 2000), and MiDAC(Itoh et al., 2015). HDAC1 and HDAC2 possess about 80% sequence identity and show overlapping functions(Jamaladdin et al., 2014). Class II HDAC10 seems to be selective for hydrolyzing acetyl-polyamine metabolites rather than proteins(Hai et al., 2017). Class III HDACs are known as sirtuins(Wang, Hsu and Liu, 2017) and employ nicotinamide adenine dinucleotide (NAD+) as a co-substrate (Fig. 2C)(Jing and Lin, 2015). The sirtuins cleave acetyl-Lys by a complex mechanism that involves nucleophilic attack of the acetyl-carbonyl oxygen on the anomeric carbon of the nicotinamide ribose, displacing nicotinamide and generating O-acetyl-ADP-ribose.
2.1.3. KAc Readers
The major and best characterized class of acetyl-Lys binding domains in proteins are the bromodomains (BRDs). To date, 61 BRD-containing proteins in humans have been annotated, and these are generally divided into eight families based on their structural and sequence similarity(Filippakopoulos et al., 2012). A heavily studied group of BRDs comprise the Bromo-and-extra-terminal (BET) family, which include BRD2, BRD3, BRD4, and BRDT, possess two independent BRDs at their N-termini, and show distinct specificities for histone Lys acetylation sites(Fujisawa and Filippakopoulos, 2017). BRDs are found in several KATs, including p300/CBP and PCAF/GCN5, where they have regulatory as well as recruitment functions. BRDs are notable for their potent sensitivity to a range of small molecules which allow for acetyl-Lys protein-protein interactions to be antagonized(Filippakopoulos and Knapp, 2014).
A more recently discovered acetyl-Lys binding domain is the YEATs domain(Klein et al., 2018). This domain is found in several human proteins related to chromatin regulation including GAS41 and AF9 and displays an immunoglobulin-like fold. In addition to acetyl-Lys, YEATs domains can also bind crotonyl-Lys(Li et al., 2018) and succinyl-Lys(Yi Wang et al., 2018), potentially serving as a reader for these modifications.
2.2. Lys Methylation Writers, Erasers, and Readers
As mentioned, Lys methylation can occur as mono- (KMe1), di- (KMe2) and tri-methyl (KMe3) forms in proteins. Like Lys acetylation, Lys methylation is a common PTM, but it has been mostly closely associated with the regulation of chromatin structure and gene transcription(Luo, 2018). In histone H3, for instance, methylation exists on H3K9, H3K27, and H4K20 sites in human chromosomes and is a marker of heterochromatin and gene silencing(Huang et al., 2015). However, methylation on other sites such as H3K4, H3K36, and H3K79 sites are activation markers(Janssen et al., 2019). Lys methylation also plays roles in the regulation of different transcription factors, for example p53(Chuikov et al., 2004) and NF-κB(Lu and Stark, 2015).
2.2.1. Lys Methyltransferases
Attachment of methyl groups to Lys residues is catalyzed by Lys methyltransferases (KMTs)(Allis, 2018). KMTs utilize S-adenosyl methionine (SAM) as a methyl donor, a common chemical strategy for enzymatic methylation throughout metabolic and catabolic pathways (Fig. 2D). Mechanistically, these enzymatic methylations are accepted to occur as SN2-like displacement reactions. Two general classes of KMTs have been discovered, SET domain (about 50) and non-SET domain methyltransferases (a handful). SET domain proteins can be divided into 7 sub-families with limited sequence conservation and structural similarity(Dillon et al., 2005), and sub-family members show diverse substrate selectivity (Qian and Zhou, 2006). The first SET KMT identified was Su(Var)3–9(Ebert et al., 2004), which methylates the histone H3 Lys9 site(Schotta et al., 2002). SET methyltransferases include EZH2, which can methylate histone H3 Lys27, and MLL methyltransferase, which targets Lys4 of histone H3. Dot1L was the first reported example of a non-SET KMT, and it specifically methylates Lys79 in a globular region of histone H3 and is linked to transcriptional activation(Van Leeuwen, Gafken and Gottschling, 2002). A novel N-terminal and C-terminal domain with a unique open α/β structure for SAM binding in Dot1L is different from standard SET domain KMTs(Min et al., 2003), which show a conserved architecture that includes a pre-SET, SET and post-SET domain organization (Trievel et al., 2002). It is believed that hyperactivity of KMTs may be disease drivers, especially in cancer. There is intense interest in finding KMT inhibitors as therapeutics and there are promising candidates in development(Luo, 2018).
2.2.2. Lys Demethylases
Methyl-Lys cleavage events are catalyzed by Lys demethylases (KDMs)(Shi et al., 2004). Prior to their molecular identification beginning in 2004, the existence of KDMs was long questioned, perhaps because of the high stability of the carbon-nitrogen single bond of a methyl-Lys. We now know that there are two classes of demethylases, flavin-dependent enzymes Lysine-specific demethylase 1 (LSD1) and LSD2, and the non-heme iron dependent Jumonji (Jmj) demethylases, a family of about 10 enzymes(Kooistra and Helin, 2012). Both the flavin and Fe-dependent enzymes catalyze oxidative demethylation and use molecular oxygen as a co-substrate. LSD1 is a polyamine oxidase paralog that removes electrons from the Lys sidechain amino group to afford an imine intermediate(Culhane and Cole, 2007). Hydrolysis of the imine to the demethylated Lys residue and formaldehyde is likely to be spontaneous. In addition, in each catalytic cycle, O2 is used to regenerate the oxidized flavin, and is converted to HOOH, which is a secondary byproduct of LSD1 catalyzed demethylation (Fig. 2E). LSD1 demethylation is highly selective for histone H3 Lys4me or Lys4me2 removal but is mechanistically incapable of processing Lys4me3. More details about the catalytic mechanism can be found elsewhere (Forneris et al., 2008).
The family of Jmj KDMs employ α-oxoglutarate (2-OG) and oxygen as co-substrates and Fe as a metallocofactor. The Jmj KDMs generate a hypervalent iron oxo species by a decarboxylation of α-ketoglutarate, affording succinate as a byproduct (Fig. 2F)(Tsukada et al., 2006). In a cytochrome P450-like reaction, the iron oxo species can hydroxylate the methyl attached to Lys, affording a hemiaminal, which decomposes to formaldehyde. More details about the enzyme mechanism are described elsewhere (Klose, Kallin and Zhang, 2006). In contrast to the flavo-enzyme KDMs, Jmjs are mechanistically capable of acting on trimethyl-Lys in addition to mono-methyl- and di-methyl-Lys substrates, although specific Jmjs show different selectivities with regard to preferred methyl-Lys forms and sequences. Representative Jmj examples are FBXL11 (JHDM1A)(Tsukada et al., 2006) that demethylates histone H3K36me and RBP2 (JARID1A) that demethylates histone H3K4me. There has been outstanding success in identifying highly selective LSD1 small molecule inhibitors and more recent progress in finding pharmacologically active probes of the Jmj family(Shortt et al., 2017).
2.2.3. KMe Readers
Many methyl-Lys binding protein domains have been discovered, including Plant Homeodomain (PHD) domains (about 100), chromodomains, Tudor domains, PWWP domains, WDR domains, and MBT domains (9 in human)(Teske and Hadden, 2017). These domains are often included in epigenetic enzymes or proteins found in histone modifying complexes. Different members of these readers can show selectivity for amino acid sequence as well as the degree of methylation of Lys. The PHD domain, for example, is found in JARID1A KDM and is thought to regulate the substrate specificity of the enzyme(Wang et al., 2009). CDX7 is a drug target containing a chromodomain, whose overexpression leads to neoplastic transformation and cell proliferation(Pallante et al., 2015). Increasing numbers of inhibitors have been developed for these readers with a particular focus on the MBT domain of L3MBTL1 as an important regulatory protein that can serve as a “chromatin lock”(Trojer et al., 2007).
2.3. Lys Ubiquitination Writers, Erasers, and Readers
Lys ubiquitination is one of the most abundant PTMs in the human proteome. There is extensive protein machinery dedicated to adding, removing, and binding ubiquitin, in the range of 1000 human genes. Below we highlight key aspects of ubiquitin writers, erasers, and readers.
2.3.1. Ub Ligases
The “Writer” for Lys ubiquitination involves a cascade of three enzymes known as E1, E2, and E3 ubiquitin ligases. In a reaction with ATP, ubiquitin (Ub) is initially adenylated by the E1 enzyme and then it generates an E1 Ub-C-terminal thioester intermediate which is transferred to a catalytic Cys residue on an E2 enzyme(Scheffner, Nuber and Huibregtse, 1995). The Ub-E2 then collaborates with an E3 Ub ligase to transfer the Ub to a protein substrate Lys residue. There are two main E3 types. Members of the larger family of E3s, the RING-domain E3s, which number about 600 in humans, serve as a template that promotes the Ub-E2 to directly ubiquitinate a protein substrate Lys(Dikic, Wakatsuki and Walters, 2009). The second category of E3s including HECT-domain (28 in human) and RBR (12 multidomain enzymes)(Spratt, Walden and Shaw, 2014) form Ub-E3 thioester intermediates, and these Ub-E3 thioesters pass Ub to protein substrates (Fig. 2G)(Chen et al., 2017).
Within Ub, there are 7 Lys residues (K6, K11, K27, K29, K33, K48, and K63), as well as an N-terminal amino group, which allows the Ub to form linear or branched poly-Ub chains. Polyubiquitination on Lys48 is often a signal that leads to proteasomal targeting and degradation of the substrate. Polyubiquitination on Lys63 generally confers lysosomal targeting(Komander and Rape, 2012). Monoubiquitination of proteins is also common and can serve as a specific signal.
Interest in Ub ligases as drug targets has intensified with the discovery about a decade ago that thalidomide and related analogs act through modulating the Ub ligase cereblon as an allosteric modulator(Ito et al., 2010). Subsequently, researchers have attempted to exploit thalidomide functionality to target degradation of a protein of interest by recruitment to cereblon and other Ub ligases(Fisher and Phillips, 2018).
2.3.2. Deubiquitinases
Deubiquitinases (DUBs) can be considered as unconventional proteases, sometimes called isopeptidases, that sever the amide bond between a Lys sidechain of one protein and the Ub C-terminus. There are approximately 100 human DUBs and the overwhelming majority fall into the Cys protease family (Fig. 2H), whereas smaller subsets are Ser hydrolases and metallohydrolases(Mevissen and Komander, 2017). Many DUBs can cleave Ub-Ub amide linkages whereas others show selectivity for particular ubiquitinated proteins(Komander, Clague and Urbé, 2009). Within the DUB Cys hydrolase family, there are many sub-families which show a wide range of catalytic and regulatory properties. As knowledge about their biological functions has expanded in normal and disease states, opportunities for targeting DUBs in drug discovery programs have been identified(Schauer et al., 2020).
2.3.3. Ub Binding Domains
As mentioned, different Ub linkages may specify distinct cellular functions. Over 20 different ubiquitin binding domains (UBDs) have been described, and these are thought to convey a diverse set of outputs. UBDs may lead to protein-protein interactions, influence enzymatic activities, modulate cellular localization, and impact protein stability. One such example of a UBD occurs in HECT-E3 ligases. Many of these enzymes have a non-substrate ubiquitin binding pocket known as the exosite. These HECT Ub exosites seem to allow catalytic activity and processivity of HECT enzymes to be controlled by non-covalent ubiquitin levels(Zhang et al., 2016). In other cases, clusters of UBDs can bind concurrently to multiple Ubs via cooperativity and this can serve as a signal for cellular machinery to act on polyUb chains(Dikic, Wakatsuki and Walters, 2009).
2.4. Atypical Lys acylation Writers, Erasers, and Readers
Recently discovered “atypical” Lys acylations such as succinylation, malonylation, glutarylation, lactylation, and serotoninylation(Farrelly et al., 2019) have contributed to the idea that metabolism and protein modifications can be linked. It is not yet clear that attachment of these Lys PTMs to proteins are enzyme catalyzed although there are a few reports that this may be possible(Harmel and Fiedler, 2018). Physiological repurposing of some KATs as acyltransferases in vivo has been proposed, but this potential must be understood as difficult given the comparatively high concentrations of acetyl-CoA and more favorable catalytic parameters of KAT activity relative to the use of the atypical acyl-CoA substrates. Succinylation has been mapped on more than a thousand protein Lys sites and is abundant on mitochondrial proteins(Xie et al., 2012). It is highly likely that nearly all of this occurs non-enzymatically, as succinyl-CoA is especially reactive with amine nucleophiles due to its propensity to form a cyclic anhydride(Weinert et al., 2013). Although Lys succinylation most likely occurs non-enzymatically, its removal has been well-documented to be mediated by sirtuin 5 (Sirt5)(Du et al., 2011). Sirt5 can also act efficiently on other acyl-Lys PTMs including malonylation(Sadhukhan et al., 2016) and glutarylation(Tan et al., 2014). It seems plausible that Sirt5 helps to prevent the toxic effects of a buildup of acyl-Lys modified proteins. Lys benzoylation appears to be driven by dietary sources of benzoate, while its removal is catalyzed by Sirt2 (Huang et al., 2018). Beyond the possibility of the Yeats domain for crotonyl-Lys(Li et al., 2018), specific reader domains for atypical acyl-Lys modifications have not yet been convincingly demonstrated.
3. Chemical Biology Approaches for Installing Lysine PTMs Site-specifically into Proteins
In order to gain a detailed understanding of the structural and functional regulation of Lys PTMs, accessing pure and well-defined site-specifically modified proteins can be critical(Wang et al., 2015). Isolation of Lys modified proteins from endogenous sources typically affords heterogenous materials with sub-stoichiometric and mixed patterns of PTMs. For example, typical Lys acetylation stoichiometries across sites in the cellular proteome are less than 1 %, indicating that obtaining a pure species is out of reach(Hansen et al., 2019). Efforts to overcome this with classical site-directed mutagenesis by replacing Lys with Gln or Arg to represent acetyl-Lys and unmodified Lys, respectively, are problematic because of the dissimilarity between these mimics and natural chemical structures. Moreover, there are not even crude natural mimics for methylation or larger acylation groups, let alone ubiquitination. It is also generally not feasible to use natural writer enzymes to introduce Lys PTMs at specific protein sites because of their insufficient site-selectivity or because their catalytic powers are too low. Facing these challenges, chemical biologists have developed a range of approaches(Chin, 2017) to generate specifically modified proteins or protein complexes(Hoyt et al., 2019). Below we summarize several of the promising advances in this area.
3.1. Lysine PTM Mimics
Although the selective labeling of native Lys is accessible(Adusumalli et al., 2018), the thiol group of Cys is the most versatile among the proteinogenic amino acids and can undergo a wide range of “bio-orthogonal reactions” through alkylation as well as radical addition(Spicer and Davis, 2014). ThiaLys analogs containing N-methyl groups(Simon et al., 2007) and N-acyl modifications(Jing et al., 2018) have been generated in this fashion. These thiaLys analogs are structurally quite similar to a natural Lys, in size and geometry. In the standard alkylation procedure, a Lys modified site of interest is mutated to Cys and then reacted with a bromoethylamino substituted reagent (Fig. 3A). Alternatively, for the thiol-ene radical addition reactions, acyl-enamines are used(Trang et al., 2014). These approaches have been extensively used in the chromatin field to generate modified nucleosomes. These experiments have investigated the role of methyl-Lys to recruit particular readers and how these affect chromatin dynamics and remodeling(Voigt et al., 2012). Reductive hydrazide acyl-Lys mimics have also been generated at Cys residues and these show special promise in generating Ub-modified proteins that serve as alternative substrates for DUBs (Fig. 3B)(Bhat et al., 2018).
Fig. 3:

Chemical biology approaches for the site-specific installation of Lys PTMs and mimics into proteins. A. ThiaLys analogs generated by Cys modification; B. Introduction of Hydrazide acyl-Lys mimics; C. PTM installation by noncanonical amino acid incorporation, using acetyl-Lys as an example; D. A typical example for the “tag-and-modify” strategy: azidonorleucine (AznL) was first installed by noncanonical amino acid incorporation, followed by traceless-Staudinger ligation reactions with different phosphine reagents to generate acetyl-Lys and succinyl-Lys; E. Expressed protein ligation by chemoselective Cys/thioester reaction; F. Enzyme-catalyzed protein semisynthesis, the cartoon motif recognized by the enzyme is shown as “sequence”.
A related approach to install thiaLys analogs from Cys in proteins involves a reaction scheme of elimination generating dehydroalanine (Dha)(Bernardes et al., 2008). After Dha formation, these unsaturated residues can undergo Michael additions or radical coupling with thioethylamino groups. This approach has the potential to be quite versatile(Wright et al., 2016), but a limitation of the method is the scrambling of the stereochemistry of the α-carbon of the Lys analog upon thiol addition.
Fortunately, Cys is one of the lowest abundance amino acids in natural proteins. Nevertheless, to avoid multiple modification sites, it is common practice to replace a small number of natural Cys with Ala or Ser, depending on the structural predictions about their functions. However, it should be stated that in some cases, the sulfur atom replacement of the gamma-methylene in natural Lys to generate thiaLys may not be well-tolerated due to altered conformational and electronic effects conferred by thiol substitution(Chen et al., 2018)
3.2. Noncanonical Amino Acid Incorporation
Noncanonical amino acid incorporation (ncAA) via amber codon suppression(Wang et al., 2001) is showing increasing promise for introducing modified Lys residues at specific sites in proteins (Tharp, Ehnbom and Liu, 2018). The ncAA method has been used to install more than 100 unnatural amino acids into proteins via different tRNA-synthetase/tRNA pairs in various hosts (Fig. 3C). Included in the ncAA roster of residues installed are KAc, KPr, KBu, KCro, and KHib(Yang, Cho and Park, 2018). The direct incorporation of KAc into 5 different sites of the small GTP-binding protein, Ran, as an example, revealed how Lys acetylation can influence Ran localization, GTPase activity, signaling functions and receptor interactions(De Boor et al., 2015).
Some Lys acylations with more complicated structures or bulkier groups are not readily introduced in a direct fashion via the tRNA-synthetase/tRNA system due to the limited space and hydrophobicity of the enzyme active sites(Wan, Tharp and Liu, 2014). This limitation has been overcome using a multi-step approach, termed a “tag-and-modify” strategy(Chalker, Bernardes and Davis, 2011). In this fashion, a two-step introduction of KSucc was achieved, in which azidonorleucine (AznL) was installed and converted into different acyl-lysines via traceless-Staudinger ligation reactions(Wang et al., 2017). By switching the acyl phosphinothioester, theoretically this strategy can be applied to any Lys acylation (Fig. 3D)(Wang, Tian and Zheng, 2015).
The application of ncAA to produce methyl-lysine forms in proteins has also been pursued. Monomethyl-Lys precursors can be incorporated into proteins via amber codon suppression, and a deprotection step is needed to generate the native methyl-Lys structure(Wang, 2019). However, for dimethyl-Lys, the situation is more complicated(Nguyen et al., 2010), but has been achieved via multistep chemical procedures. The reductive amination of allysine can generate mono- and di-methyl-Lys, and this approach was used to analyze the cross-talk between K372 methylation and K120 acetylation in p53(Wang et al., 2016). To date, we are unaware of success in generating trimethyl-Lys via ncAA.
3.3. Chemoselective Ligations
Chemical preparation of proteins by total synthesis represents the ultimate method for complete control of the generation of a modified protein(Hackenberger and Schwarzer, 2008). The chemoselective reaction between peptides containing C-terminal thioesters and N-terminal Cys, known as “native chemical ligation”(Dawson and Kent, 2000) has enabled the synthesis of small to medium sized proteins(Zheng et al., 2013). Many Lys PTMs have been successfully installed, especially those two challenging groups as KMe3 and KUb, particularly in the context of histones(Qi et al., 2018).
These ligation strategies have been expanded to link recombinant protein fragments and synthetic peptides to generate larger proteins and circumvent technical challenges of protein refolding that can occur with total synthesis(Wang and Cole, 2020). The most popular version of this semisynthetic strategy, expressed protein ligation, involves using an intein to generate a recombinant protein fragment thioester that is ligated to an N-Cys synthetic peptide(Muir, Sondhi and Cole, 1998). A complementary semisynthetic approach utilizes an N-Cys recombinant protein generated by proteolysis with SUMO protease or TEV and a synthetic peptide thioester. These companion semisynthetic methods have been extensively utilized to generate acetyl-Lys, methyl-Lys, and Ub-Lys containing proteins (Fig. 3E). Several semisynthetic H2Bs with ubiquitination at Lys120 have been prepared with either native isopeptide bonds, mutated Ub(G76A), or structural mimics(Fierz and Muir, 2012). These protein chemical approaches have been employed in detailed functional and mechanistic characterization of ubiquitin-modified H2B(Müller and Muir, 2015). Examples of such analyses include the structural characterization of the cross-talk between H2B and H3K79 methylation by Dot1L (Fig. 4A)(Worden et al., 2019), H3K4 methylation by COMPASS(Hsu et al., 2018), and H3K36 methylation by Set2(Halic, 2019).
Fig. 4.

Examples of biological investigation facilitated by the chemical preparation of site-specially modified histones. A. The recognition of H2BK120ub leads to H3K79 methylation as activation marker by Dot1L; B. H3K14ac blocks LSD1 of CoREST complex catalyzed demethylation at Lys4.
Beyond histones, applications of these semisynthetic approaches include generating modified full-length tau(Haj-Yahya and Lashuel, 2018), whose K280 acetylation showed its strong stimulation of tau aggregation, and S-adenosyl homocysteine hydrolase(Wang et al., 2014), whose acetylation was shown to regulate its catalytic activity in a manner that could lead to interplay with epigenetic methylation.
3.4. Enzyme-catalyzed Protein Semisynthesis
The synthesis of thioesters and the requirement of N-Cys residues can be an impediment in some protein ligation applications. Enzymes for ligating peptides and proteins analogous to DNA ligases used commonly in molecular cloning are beginning to show promise. Several examples of such enzymes include sortase, subtiligase, butelase-1, and trypsiligase(Thompson and Muir, 2019). Sortase is a bacterial enzyme that catalyzes transpeptidation reactions involving cell wall remodeling (Wu et al., 2017). Sortase and its engineered forms have been used in protein labeling at both the C-terminus(Guimaraes et al., 2013) and N-terminus(Theile et al., 2013) of proteins of interest. Natural sortase recognizes the epitope LPXTG for catalysis and it has been engineered to expand its substrate scope to include “APXTG”(Piotukh et al., 2011). Conveniently, histone H3 contains an “APATG” sequence in aa residues 29–33, which enables the engineered sortase-catalyzed semisynthesis of H3 with Lys PTMs throughout the N-terminal tail (Fig. 3F). Using this approach, histone H3 acetylated and methylated nucleosomes have been prepared and used for analysis of the site-selectivity of the HDAC1-containing CoREST complex(Wu et al., 2018). These studies revealed that acetylation at Lys14 is especially resistant to deacetylation which is notable because this modification also blocks CoREST complex catalyzed demethylation at Lys4 (Fig. 4B). A similar approach has been applied to study all five known class I HDACs, which show different activity and selectivity over various nucleosome H3 acetylation sites(Wang et al., 2020). Histone H3 with K4 propargyl group has been prepared by this approach for the structural analysis of CoREST complex binding(Song et al., 2020). Compared with chemical ligation methods, enzyme-catalyzed protein semisynthesis strategies are generally more rapid in mild aqueous conditions and require less chemistry expertise. However, the need for specialized folded protein reagents and corresponding recognition sequences can prove an impediment to enzyme-catalyzed protein semisynthesis. For a more detailed discussion of the strengths and weaknesses of various approaches, please refer to a recent review (Wang and Cole, 2020).
4. Small Molecule Probes for Lys Writers, Erasers, and Readers
The development of small molecule pharmacological probes has greatly impacted our understanding of the function of the writers, readers, and erasers of Lys PTMs. The earliest important tool compounds, trichostatin A and trapoxin, are natural products that are potent HDAC inhibitors. These were characterized as blocking histone acetyl-Lys removal before the molecular identification of any of the reversible Lys modifying enzymes or readers(Furumai et al., 2001). Building on the understanding of these compounds, six HDAC inhibitors are now FDA approved for the treatment of various hematologic and cutaneous malignancies(Banerjee et al., 2019). Over the past decade, major progress has been made in identifying a range of small molecules that have sufficiently robust pharmacological properties to be reliable probes of writer, eraser, and reader functions of key proteins involved with Lys acetylation, methylation, and ubiquitination. Representative small molecule probes are listed in Table 1 and highlighted based on a combination of established clinical impact, perceived pharmacological utility, and chemical novelty.
Table 1:
Small Molecule Tools for Lys PTM Writers, Erasers, and Readers
| Inhibitor | Target Enzyme/Domain | Features | Reference |
|---|---|---|---|
| A485 | P300/CBP | Binds to the active site, leads to a rapid global acetylation level drop in nuclear proteins | (Lasko et al., 2017) |
| WM-1119/WM-3835 | HBO1 (KAT7 or MYST2) | Competitive inhibitor of Ac-CoA causes the decrease of H3K14ac and shows promise in leukemias | (MacPherson et al., 2020) |
| WM-8014/WM-2474 | KAT6A (MOZ) KAT6B (MORF) | Competitive inhibitor of Ac-CoA leads to histone acetylation reduction | (Baell et al., 2018) |
| SAHA (Vorinostat) | classical HDAC | Zn2+-chelating hydroxamic acid for cutaneous T-cell lymphoma treatment approved by FDA in 2006 | (Banerjee et al., 2019) |
| FK228 (Romidepsin) | classical HDAC | Cyclic peptide natural product for the treatment of cutaneous T-cell lymphoma approved by FDA in 2009 | (Banerjee et al., 2019) |
| PXD101 (Belinostat) | classical HDAC | Used to treat Peripheral T-cell lymphoma approved by FDA in 2014 | (Banerjee et al., 2019) |
| LBH589 (Panobinostat) | classical HDAC | Used for the treatment of relapsed multiple myeloma approved by FDA in 2015 | (Banerjee et al., 2019) |
| HBI-8000 (Chidamide) | classical HDAC | Benzamide functions as Zn2+-chelator approved by FDA in 2015 for relapsed or refractory peripheral T-cell lymphoma and pancreatic cancer | (Banerjee et al., 2019) |
| SB939 (Pracinostat) | classical HDAC | Drug approved by FDA in 2016 for the treatment of acute myeloid leukemia | (Banerjee et al., 2019) |
| MS275 (Entinostat) | HDAC1–3 | Benzamide, Phase III candidate for cancer therapy | (Connolly, Rudek and Piekarz, 2017) |
| PCI-34051 | HDAC8 | Selectively inhibits HDAC8, causing caspase-depend apoptosis in certain T-cell leukemia cell lines | (Balasubramanian et al., 2008) |
| Mercaptoaceami de-7a | HDAC6 | Selective inhibitor for HDAC6 with neuroprotective effects | (Kalin et al., 2012) |
| Corin | CoREST complex | A chemical combination of entinostat and tranylcypromine analog inhibiting both HDAC1 and LSD1, shows antitumor effects | (Kalin et al., 2018) |
| TM | sirtuin2 | Thioacyl functionality, causes degradation of c-Myc | (Jing et al., 2016) |
| SirReal2 | sirtuin2 | Induces enzyme active site conformational change, leads to tubulin hyperacetylation in cancer cells | (Rumpf et al., 2015) |
| NPD11033 | sirtuin2 | Binds and inhibits the deacetylase activity but not the defatty-acylase activity of sirtuin2 selectively, resulting in pancreatic cancer cell growth inhibition | (Kudo et al., 2018) |
| (+)-JQ1 | BET/BET-BRD domain | Pan-BET BRD competitive inhibitor with antiproliferation effects and widespread epigenetic effects | (Filippakopoulos et al., 2010) |
| OTX015 | BET/BET-BRD domain | Pan-BET BRD inhibitor with antiproliferative action | (Lu et al., 2016) |
| L-Moses | PCAF/GCN5/Non-BETBRD | Selective inhibitor for non-BET BRD with no toxicity, good cell permeability and high stability | (Moustakim et al., 2017) |
| I-CBP112 | P300/CBP/Non-BETBRD | Allosterically activates p300/CBP acetyltarnsferase activity; can synergize with A-485 in blocking cancer cell growth | (Zucconi et al., 2016) (Zucconi et al., 2019) |
| GNE-781 | P300/CBP/Non-BETBRD | Highly specific Inhibition of BRD in p300/CBP leading to the decrease of Foxp3 transcription and antitumor activity in tumor models | (Romero et al., 2017) |
| VTP50469 | MLL (KMT2A)/ SET domain | Selectively inhibits Menin-MLL protein-protein interaction and causes drop in chromatin occupancy and cell differentiation or apoptosis for cancer treatment | (Krivtsov et al., 2019) |
| MS453 | SETD8/SET domain | Covalently modifies a Cys near the binding site of SET domain and blocks H4K20me | (Butler et al., 2016) |
|
UNC0638/UNC0 642 |
EHMT2(G9a, KMT1C); EHMT1(GLP, KMT1D)/ SET domain | Blocks H3K9 methylation; UNC0642 shows improved pharmacokinetic properties | (Liu et al., 2013) |
| EPZ-6438 (Tazemetostat) | EZH2/SET domain | SAM competitive inhibitor, studied in clinical trials | (Italiano et al., 2018) |
| EPZ-5676 (Pinometostat) | Dot1L/Non-SET domain | Selective inhibitor for Dot1L leading to the decrease of H3K79 methylation for the treatment of acute leukemia with MLL-rearranged gene | (Campbell et al., 2017) |
| GSK-LSD1 | LSD1 (KDM1A)/ amine oxidase | Cyclopropylamine, mechanism-based irreversible inhibitor, shows antiproliferation against acute leukemia | (Mohammad et al., 2015) (Egolf et al., 2019) |
| GSK-J1 GSK-J4 (Prodrug) | JMJD3 (UTX)/Jmj domain | Chelates Fe in the active site, modulates H3K27 methylation for pharmacological applications | (Kruidenier et al., 2012) |
| UNC1215 | L3MBTL3/MBT domain | Binds to the methyl-Lys pocket competitively | (James et al., 2013) |
| UNC2170 | 53BP1/Tudor domain | Competitive active site binding ligand for methyl-Lys | (Perfetti et al., 2015) |
| UNC4976 | CBX7/ Chromodomain | A peptidomimetic as a positive allosteric modulator (PAM) that blocks H3K27me3 binding, leads to the relocation of PRC1 | (Lamb et al., 2019) |
| TAK-243 | E1 | Binds to the active site and blocks Ub transfer to the E2 enzyme, leading to antiproliferation of cancer cells | (Rabino et al., 2018) |
| (S)-thalidomide and analogs | Cereblon (CRBN)/Ring E3 | Allosteric CRBN inhibitor, reduces ubiquitination of various transcription factor substrates | (Liu et al., 2015) |
| Nutlin1/2/3 | MDM2-p53/DUB | Competitively binds to MDM2 and blocks p53 ubiquitination | (Vassilev et al., 2004) |
| P5091 | USP7/DUB | Selective inhibitor with action against multiple myeloma | (Chauhan et al., 2012) |
| XL188 | USP7/DUB | High selectivity based on structure-guided development, reversible with utility in cells and in vivo | (Lamberto et al., 2017) |
| IU1/IU1–47 | USP14/DUB | Allosteric inhibitor that blocks substrate binding, leading to an increase in proteasome substrate degradation | (Yiwei Wang et al., 2018) (Boselli et al., 2017) |
4.1. Small Molecule Probes for Lys Acetylation
4.1.1. Lys acetyltransferase inhibitors
For many years, KAT enzymes were rather recalcitrant to the kind of cell active robust chemical inhibitors that had been well-established for KDACs. An incremental advance in this regard was the identification of C646 (Fig. 5A) as a moderately potent and selective p300/CBP KAT inhibitor(Bowers et al., 2010). C646 was identified as an active site p300 inhibitor through virtual screening and, despite limitations as a reversible covalent Cys modifier, has been widely used to assess p300/CBP function. Representative studies employed C646 to analyze p300/CBP in hematologic, prostate, and cutaneous malignancies as well as basic transcriptional processes involving RNA polymerase regulation(Dancy and Cole, 2015).
Fig. 5.

Examples of small molecule inhibitors developed for Lys PTM writers, erasers, and readers: A. acetylation writer inhibitors; B. acetylation eraser inhibitors; C. acetylation reader inhibitors; D. methylation writer inhibitors; E. methylation eraser inhibitors; F. methylation reader inhibitors; G. ubiquitination regulators.
However, the landscape of p300/CBP pharmacology was transformed with the development of A485 as a highly potent and specific p300/CBP acetyltransferase through a combination of virtual screening and extensive medicinal chemistry(Lasko et al., 2017). A485, a spiro-oxazolidinedione, was determined by crystallography to bind to the active site excluding acetyl-CoA interaction. A485 has become a powerful tool in the assessment of p300/CBP acetyltransferase activity in cellular pathways. Notably, a comprehensive mass spectrometric proteomics experiments employing A485 revealed that p300/CBP behaves as an “acetyl-spray” in the nucleus, coating a vast swathe of the transcriptional machinery with Lys acetylation(Weinert et al., 2018). The kinetics analysis of these acetylation mark changes with A485 revealed that many are on minute time scales. Unexpectedly, the tail of histone H2B showed a large cluster of dynamic p300/CBP acetylation sites(Weinert et al., 2018). In a cancer related study, A485 was key in pinpointing the role of the transcription factor MITF in melanoma responsiveness to HAT inhibition(Kim et al., 2019). A485 has also helped enhance our understanding of p300/CBP in prostate cancer and other malignancies.
Another recent exciting advance in KAT inhibition has been the identification of high-quality inhibitors for the MYST family of KATs including MORF, MOZ, and HBO1(MacPherson et al., 2020). These sulfonyl hydrazide compounds like WM-1119/WM-3835 are potent against two or more MYST family members. Crystal structures have shown how they target the active sites of these enzymes by occluding acetyl-CoA binding. These compounds demonstrate the ability to block cancer cell proliferation with particular promise in hematologic malignancies and show in vivo activity in mice(Baell et al., 2018). They have been used to demonstrate that HBO1 may modulate acetylation of Lys14 of histone H3(MacPherson et al., 2020). These new MYST inhibitors promise to be great tools for analyzing a wide range of functions of these KATs (Fig. 5A).
4.1.2. Lys deacetylase Inhibitors
Although potent, pharmacologically active KDAC inhibitors have been established for three decades and have been reviewed previously, we will highlight several recent advances (Fig. 5B). HDAC8 is a nuclear deacetylase and is unusual for a KDAC in having high catalytic activity in the absence of a corepressor binding partner(Castañeda et al., 2017). Although its detailed functions are still being elucidated, a range of highly selective inhibitors containing Zn(II) binding warheads have been described in the past 5 years(Balasubramanian et al., 2008). These show effectiveness in a range of anti-tumor applications(Connolly, Rudek and Piekarz, 2017). Likewise, cytoplasmic deacetylase HDAC6 has become a popular therapeutic target that plays key roles in protein homeostasis(Kalin et al., 2012). Preclinical studies in neurologic diseases such as Charcot-Marie Tooth have been a recent direction in this area. Targeting HDAC1 in the context of a particular corepressor complex is a relatively new direction in the field. The designed CoREST complex inhibitor compound corin has dual warheads, one a phenyl-cyclopropylamine for LSD1 demethylase and the other a benzamide that engages HDAC1(Kalin et al., 2018). Corin shows high potency for both enzymatic activities in the CoREST complex and has a prolonged residence time in the HDAC1 active sites relative to monofunctional benzamide KDAC inhibitor. Corin has demonstrated promising antitumor behavior across melanoma, diffuse pontine glioma(Anastas et al., 2019), and colon cancer(Miller et al., 2020), and can enhance cancer immune surveillance(Xiong et al., 2020). These data underscore the concept that selectively targeting KDACs in the context of an individual corepressor complex may offer therapeutic advantages.
There has also been recent progress in the identification of sirtuin KDAC inhibitors. Although activators of sirtuins have long been discussed as a strategy to slow the aging process, sirtuin inhibitors are now being pursued as therapeutics for a range of applications. Thiomyristoyl derivative TM(Jing et al., 2016), thiopyrazine SirReal2(Rumpf et al., 2015) and pyridone NPD11033(Kudo et al., 2018) show high potency and selectivity as well as antitumor potential. Crystal structures have been determined that help define the binding mode of these compounds to Sirt2 via active site interactions. NPD11033 is notable for blocking deacetylase activity of Sirt2 but not its less well-studied fatty deacylase activity. As for many Lys PTM targeting compounds, the precise mechanism of how Sirt2 inhibition leads to antitumor effects is not fully understood but an impact on inhibiting Myc transcriptional activity has been described.
4.1.3. Bromodomain Inhibitors
Broad spectrum BET bromodomain inhibitors from the early compounds like (+)-JQ1(Filippakopoulos et al., 2010) and OTX015(Lu et al., 2016) (Fig. 5C) to next generation specific BET isoform inhibitors have been heavily applied in biological investigation. These agents have helped establish the role of BET bromodomain-acetyl-Lys interactions in transcriptional regulation and have shaped the concept of “superenhancers”(Lovén et al., 2013). Although they have shown tremendous promise in preclinical investigation in cancer studies and other disease models, BET bromodomain inhibitors have demonstrated toxicity in human trials, especially thrombocytopenia(Falchook et al., 2020). More recent compound discovery efforts to identify non-BET bromodomain antagonists have led to selective PCAF/GCN5 and p300/CBP bromodomain inhibitors. The triazolophthalazine compound dubbed L-Moses is a pharmacologically potent PCAF/GCN5 antagonist and interestingly shows anti-parasitic action(Moustakim et al., 2017). Selective p300/CBP bromodomain compounds including earlier generation I-CBP112(Zucconi et al., 2016) and the more potent compound GNE-781(Romero et al., 2017) have demonstrated antineoplastic activity. Interestingly, I-CBP112 has been shown to enhance p300-catalyzed nucleosome acetylation(Zucconi et al., 2019) while GNE-781 lacks this action(Morrison-Smith et al., 2020), suggesting that the allosteric regulation of p300/CBP is sensitive to precise bromodomain interactions (Fig. 5C).
4.2. Small molecule probes for Lys Methylation
4.2.1. Methyltransferase Inhibitors
Dramatic progress has been achieved in targeting a series of methyltransferases that methylate histone H3 on Lys4 (MLL), Lys9 (G9a/GLP), Lys27 (EZH2), and Lys79 (Dot1L). Many of the compounds target the SAM binding site of these enzymes, which are sufficiently distinct to engender specificity (Butler et al., 2016). The identification of VTP50469 as a Menin-MLL protein-protein interaction disruptor rather than a SAM antagonist can prevent MLL from binding to chromatin and is showing promise in leukemias characterized by MLL gene translocations(Krivtsov et al., 2019) (Fig. 5D). G9a/GLP inhibitor UNC0638/UNC0642(Liu et al., 2013) has been used in a range of applications including a study to treat the Praeder-Willi metabolic syndrome(Kim et al., 2017). EZH2 SAM antagonists such as tazemetostat have moved into advanced clinical trials for cancer and are indispensable tools for the analysis of the polycomb repressor complex 2 function(Italiano et al., 2018). Similarly, Dot1L SAM anatgonists like pinometostat(Daigle et al., 2013) have been investigated as epigenetic modulators in malignancy(Campbell et al., 2017) (Fig. 5D).
4.2.2. Lys Demethylase Inhibitors
In the context of lysine demethylases, small molecule inhibitors that are the furthest along are mechanism-based inactivators based on classical monoamine oxidase pharmacology. Selective phenyl-cyclopropylamine compounds, as exemplified by GSK-LSD1 (Fig. 5E), show time-dependent covalent adducts with the tightly bound flavin cofactor resulting in irreversible inhibition(Mohammad et al., 2015). These compounds have shown antiproliferative activity against a subset of leukemias and lung cancer(Egolf et al., 2019). It was originally believed that such LSD1 inhibitors achieved their cell growth effects exclusively through catalytic inactivation, but more recent studies indicate that the mechanism in some cases involves disruption of LSD1 and SNAG transcription factor binding(Vinyard et al., 2019).
Jmj Fe-dependent demethylases have proven more difficult to inhibit than LSD1, but there are now pharmacological tools of increasing potency and specificity for these enzymes. A combined JMJD3/UTX inhibitor GSK-J1 and its prodrug ester form GSK-J4 are pyridyl-pyrimidines(Kruidenier et al., 2012). As shown by X-ray crystallography, this family of compounds blocks H3K27me demethylase activity by coordinating the Fe and obstructing α-ketoglutarate interaction. These compounds show cellular activity. Nevertheless, further work is needed to advance chemical probe development for the family of Jmj demethylases (Fig. 5E).
4.2.3. Methyl-Lys Reader Antagonists
There are a wide range of methyl-Lys protein reader domains that show distinct preferences for mono- di- and tri-methyl-Lys levels as well as the proximal amino acid sequence surrounding the modified Lys. Small molecule probes of these domains have been more difficult to identify than for acetyl-Lys binding bromodomains, but progress is ramping up. An important early step in this direction was the identification of the pyrrolidine derivative UNC1215 (Fig. 5F) as an antagonist of the MBT domain of L3MBTL3(James et al., 2013). This domain preferentially binds mono-methyl and di-methyl Lys and is functionally associated with gene silencing. A crystal structure of UNC1215 in complex with the MBT of L3MBTL3 revealed deep engagement of the small molecule in the methyl-Lys pocket. UNC1215 was shown to block L3MBTL3 in cell culture and led to the identification of the transcriptional repressor protein BCLAF1, a methylated protein, as a binding partner of L3MBTL3(James et al., 2013). A fairly potent chemical probe UNC2170 has been developed for the methyl-Lys binding tandem tudor domain of the DNA repair related protein 53BP1(Perfetti et al., 2015). This t-butylamine was shown to be selective for 53BP1 and could block class switch recombination in cells. Most recently, a chromobox (CBX7) chromo methyl-Lys reader domain antagonist UNC4976 was reported(Lamb et al., 2019). CBX7 binds to histone H3K27me3 and helps silence gene expression as part of the polycomb repressor complex 1 (PRC1). UNC4976 is a short peptide and allosteric modulator of CBX7 that can enhance nucleic acid interaction with the protein. It was shown that UNC4976 can induce gene reactivation of PRC1 target genes(Lamb et al., 2019) (Fig. 5F).
4.3. Ub Chemical Probes
The discovery of thalidomide analogs as cereblon Ring E3 ubiquitin ligase allosteric modulators has led to an explosive interest in targeted protein degradation and has been heavily discussed in the literature(Liu et al., 2015). These glutaramide derivatives appear to steer the E3 toward certain protein substrates, such as key transcription factors, by enhancing their recruitment. Potent and selective E1 ubiquitin ligase and proteasome inhibitors have also been well-characterized(Rabino et al., 2018) and several proteasome inhibitors are in clinical use for cancer(Rulina et al., 2016). In general, it has been challenging to identify active site small molecule probes of Ring or HECT domain E3 enzymes that act in a traditional inhibitor sense. This could reflect our limited understanding of the detailed catalytic mechanisms of Ub ligases. One early success story for the E3 ligase, MDM2, was the discovery of the nutlin compounds (Fig. 5G), which blocks MDM2/p53 interaction and thereby shows anti-tumor activity(Vassilev et al., 2004). It has been hard to build on this finding, however. Moreover, there have not been specific probes reported of ubiquitin-reader interactions. However, there has been great recent progress in the development of deubiquitnase (DUB) small molecule inhibitors that show high potency and isoform selectivity. Perhaps the clearest example of this has been the development of a promising inhibitors for the DUB USP7. Highly potent and selective USP7 inhibitors, including irreversible covalent inhibitors such as P5091(Chauhan et al., 2012) and reversible compound XL188(Lamberto et al., 2017), show antineoplastic activity in a mechanism that likely involves MDM2 destabilization with rescue of p53. Selective inhibitors for other DUBs(Yiwei Wang et al., 2018), such as USP14(Boselli et al., 2017), suggest this to be a fertile field in coming years (Fig. 5G).
4.4. Chemical Complementation of Mutant Methyltransferases and Acetyltransferases
An emerging area in the chemical analysis of KMTs and KATs has involved introducing point mutations into specific co-substrate interacting residues in these enzymes which relaxes their specificities for SAM and acetyl-CoA analogs, respectively. In this way, SAM and acetyl-CoA analogs containing “clickable” groups like alkynes can be efficiently processed by these mutant KMTs and KATs in a fashion that makes them orthogonal to the natural enzymes. In this fashion, these clickable co-substrates leave a handle that allows for efficient protein substrate identification(Han et al., 2017). These mutant enzyme co-substrate pairs have been applied to the histone H3K9 KMTs G9a and GLP1, KATs GCN5, MOF, and p300 and are leading to the identification of novel substrate proteins modified by these enzymes(Luo, 2018).
5. Summary and Outlook
The biological, chemical, and therapeutics landscapes of reversible Lys PTMs have become increasingly rich and complex over the past 25 years since the molecular identification of the first KATs and KDACs(Allis and Jenuwein, 2016). Indeed, the integration of chemical tools and concepts with the functional and structural characterization of the writers, erasers, and readers of Lys PTMs has led to major advances in our understanding of the proteins and modifications involved. From the big chemistry on proteins and protein complexes to the small chemistry of probe development, we have learned how site-specific modifications of Lys regulate gene expression, cell growth, and a multitude of disease processes. It is gratifying that there are now a number of epigenetic therapies that have emerged from the marriage between the chemistry and biology of the study of Lys PTMs. However, despite this progress, we believe that we have barely scratched the surface in unraveling the details of the interconnected cellular pathways that give rise to the Lys PTM and their impacts. There are still major gaps in our knowledge surrounding Lys PTMs and the chemical tools needed to address them. We have a very limited understanding of how a Lys PTM at one site influences a PTM at a different site in the same protein, or another protein, and what this PTM interplay means biomedically. Increasingly sophisticated top down and middle down mass spectrometric approaches will be needed to help illuminate these PTM inter-relationships. We lack high quality small molecule probes for the large majority of enzymes that write and erase Lys PTMs in the multi-subunit macromolecular complexes that they reside in and are deficient in such chemical probes of most readers as well. Increasingly, protein chemical methods that can work inside the live cell will be needed to pinpoint the functional features of specific Lys PTMs. Despite these challenges, we are most sanguine that, given the creativity and commitment of the investigators who populate the epigenetics community, the next 25 years will revolutionize our understanding of reversible Lys PTMs even more than the last.
Acknowledgement
We are grateful to our many colleagues and collaborators for the work cited in this article and for sharing ideas with us over the years. We thank the NIH (GM62437, DK118266), the Melanoma Research Alliance and the Leukemia and Lymphoma Society (SCOR) for financial support. We thank Yi Zheng @Yillustration Studio LLC. for assistance with figure preparation.
Footnotes
Declaration of Interest Statement
P.A.C. is a cofounder of Acylin and has been a scientific advisor for Abbvie which have had therapeutics programs targeting p300 and CBP.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adusumalli SR et al. (2018) ‘Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology’, Journal of the American Chemical Society. American Chemical Society, 140(44), pp. 15114–15123. doi: 10.1021/jacs.8b10490. [DOI] [PubMed] [Google Scholar]
- Ali I et al. (2018) ‘Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics’, Chemical Reviews, 118(3), pp. 1216–1252. doi: 10.1021/acs.chemrev.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD (2018) ‘Pursuing the Secrets of Histone Proteins: An Amazing Journey with a Remarkable Supporting Cast’, Cell. Elsevier Inc., 175(1), pp. 18–21. doi: 10.1016/j.cell.2018.08.022. [DOI] [PubMed] [Google Scholar]
- Allis CD and Jenuwein T (2016) ‘The molecular hallmarks of epigenetic control’, Nature Reviews Genetics. Nature Publishing Group, 17(8), pp. 487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Anastas JN et al. (2019) ‘Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG’, Cancer Cell. Elsevier Inc., 36(5), pp. 528–544.e10. doi: 10.1016/j.ccell.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Baell JB et al. (2018) ‘Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth’, Nature. Springer US, 560(7717), pp. 253–257. doi: 10.1038/s41586-018-0387-5. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S et al. (2008) ‘A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas’, Leukemia, 22(5), pp. 1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- Banerjee S et al. (2019) ‘Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview’, European Journal of Medicinal Chemistry. Elsevier Masson SAS, 164, pp. 214–240. doi: 10.1016/j.ejmech.2018.12.039. [DOI] [PubMed] [Google Scholar]
- Bernardes GJL et al. (2008) ‘Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: Versatile and switchable access to functionalized proteins’, Journal of the American Chemical Society. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- Bhat S et al. (2018) ‘Hydrazide Mimics for Protein Lysine Acylation to Assess Nucleosome Dynamics and Deubiquitinase Action’, Journal of the American Chemical Society. American Chemical Society, 140(30), pp. 9478–9485. doi: 10.1021/jacs.8b03572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boor S et al. (2015) ‘Small GTP-binding protein Ran is regulated by posttranslational lysine acetylation’, Proceedings of the National Academy of Sciences of the United States of America, 112(28), pp. E3679–E3688. doi: 10.1073/pnas.1505995112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boselli M et al. (2017) ‘An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons’, Journal of Biological Chemistry, 292(47), pp. 19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM et al. (2010) ‘Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor’, Chemistry and Biology. Elsevier Ltd, 17(5), pp. 471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL and Gu W (2006) ‘p53 ubiquitination: Mdm2 and beyond’, Molecular Cell, 21(3), pp. 307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KV et al. (2016) ‘Structure-Based Design of a Covalent Inhibitor of the SET Domain-Containing Protein 8 (SETD8) Lysine Methyltransferase’, Journal of Medicinal Chemistry, 59(21), pp. 9881–9889. doi: 10.1021/acs.jmedchem.6b01244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CT et al. (2017) ‘Mechanisms of pinometostat (EPZ-5676) treatment–emergent resistance in MLL-rearranged leukemia’, Molecular Cancer Therapeutics, 16(8), pp. 1669–1679. doi: 10.1158/1535-7163.MCT-16-0693. [DOI] [PubMed] [Google Scholar]
- Castañeda CA et al. (2017) ‘HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides’, Journal of Biological Chemistry, 292(52), pp. 21568–21577. doi: 10.1074/jbc.M117.811026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker JM, Bernardes GJL and Davis BG (2011) ‘A “tag-and-modify” approach to site-selective protein modification’, Accounts of Chemical Research, 44(9), pp. 730–741. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- Chauhan D et al. (2012) ‘A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance’, Cancer Cell. Elsevier Inc., 22(3), pp. 345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z et al. (2017) ‘A Tunable Brake for HECT Ubiquitin Ligases’, Molecular Cell, 66(3), pp. 345–357.e6. doi: 10.1016/j.molcel.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z et al. (2018) ‘Quantitative and Structural Assessment of Histone Methyllysine Analogue Engagement by Cognate Binding Proteins Reveals Affinity Decrements Relative to Those of Native Counterparts’, Biochemistry, 57(3), pp. 300–304. doi: 10.1021/acs.biochem.7b00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JW (2017) ‘Expanding and reprogramming the genetic code’, Nature. Nature Publishing Group, 550(7674), pp. 53–60. doi: 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- Chuikov S et al. (2004) ‘Regulation of p53 activity through lysine methylation’, Nature, 432(7015), pp. 353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Cieniewicz AM et al. (2014) ‘The Bromodomain of Gcn5 Regulates Site Specificity of Lysine Acetylation on Histone H3’, Molecular & Cellular Proteomics, 13(11), pp. 2896–2910. doi: 10.1074/mcp.M114.038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly RM, Rudek MA and Piekarz R (2017) ‘Entinostat: A promising treatment option for patients with advanced breast cancer’, Future Oncology, 13(13), pp. 1137–1148. doi: 10.2217/fon-2016-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane JC and Cole PA (2007) ‘LSD1 and the chemistry of histone demethylation’, Current Opinion in Chemical Biology, 11(5), pp. 561–568. doi: 10.1016/j.cbpa.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR et al. (2013) ‘Potent inhibition of DOT1L as treatment of MLL-fusion leukemia’, Blood, 122(6), pp. 1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy BM and Cole PA (2015) ‘Protein lysine acetylation by p300/CBP’, Chemical Reviews, 115(6), pp. 2419–2452. doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PE and Kent SBH (2000) ‘Synthesis of Native Proteins by Chemical Ligation’, Annual Review of Biochemistry, 69(3), pp. 923–960. [DOI] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S and Walters KJ (2009) ‘Ubiquitin-binding domains from structures to functions’, Nature Reviews Molecular Cell Biology. Nature Publishing Group, 10(10), pp. 659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC et al. (2005) ‘The SET-domain protein superfamily: Protein lysine methyltransferases’, Genome Biology, 6(8). doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J et al. (2011) ‘Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase’, Science, 334(6057), pp. 806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A et al. (2004) ‘Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila’, Genes and Development, 18(23), pp. 2973–2983. doi: 10.1101/gad.323004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egolf S et al. (2019) ‘LSD1 Inhibition Promotes Epithelial Differentiation through Derepression of Fate-Determining Transcription Factors’, Cell Reports. ElsevierCompany., 28(8), pp. 1981–1992.e7. doi: 10.1016/j.celrep.2019.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook G et al. (2020) ‘Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies’, Clinical Cancer Research, 26(6), pp. 1247–1257. doi: 10.1158/1078-0432.CCR-18-4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA et al. (2019) ‘Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3’, Nature. Springer US, 567(7749), pp. 535–539. doi: 10.1038/s41586-019-1024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz B and Muir TW (2012) ‘Chromatin as an expansive canvas for chemical biology’, Nature Chemical Biology. Nature Publishing Group, 8(5), pp. 417–427. doi: 10.1038/nchembio.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P et al. (2010) ‘Selective inhibition of BET bromodomains’, Nature. Nature Publishing Group, 468(7327), pp. 1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P et al. (2012) ‘Histone recognition and large-scale structural analysis of the human bromodomain family’, Cell, 149(1), pp. 214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P and Knapp S (2014) ‘Targeting bromodomains: Epigenetic readers of lysine acetylation’, Nature Reviews Drug Discovery. Nature Publishing Group, 13(5), pp. 337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Fisher SL and Phillips AJ (2018) ‘Targeted protein degradation and the enzymology of degraders’, Current Opinion in Chemical Biology. Elsevier Ltd, 44, pp. 47–55. doi: 10.1016/j.cbpa.2018.05.004. [DOI] [PubMed] [Google Scholar]
- Forneris F et al. (2008) ‘LSD1: oxidative chemistry for multifaceted functions in chromatin regulation’, Trends in Biochemical Sciences, 33(4), pp. 181–189. doi: 10.1016/j.tibs.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Fujisawa T and Filippakopoulos P (2017) ‘Functions of bromodomain-containing proteins and their roles in homeostasis and cancer’, Nature Reviews Molecular Cell Biology. Nature Publishing Group, 18(4), pp. 246–262. doi: 10.1038/nrm.2016.143. [DOI] [PubMed] [Google Scholar]
- Furumai R et al. (2001) ‘Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin’, Proceedings of the National Academy of Sciences of the United States of America, 98(1), pp. 87–92. doi: 10.1073/pnas.98.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG et al. (2000) ‘A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness’, Genes and Development, 14(9), pp. 1048–1057. doi: 10.1101/gad.14.9.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes CP et al. (2013) ‘Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions’, Nature Protocols. Nature Publishing Group, 8(9), pp. 1787–1799. doi: 10.1038/nprot.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL and Olson EN (2009) ‘The many roles of histone deacetylases in development and physiology: Implications for disease and therapy’, Nature Reviews Genetics, 10(1), pp. 32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberger CPR and Schwarzer D (2008) ‘Chemoselective ligation and modification strategies for peptides and proteins’, Angewandte Chemie International Edition, 47(52), pp. 10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- Hai Y et al. (2017) ‘Histone deacetylase 10 structure and molecular function as a polyamine deacetylase’, Nature Communications. Nature Publishing Group, 8(May), pp. 1–9. doi: 10.1038/ncomms15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Yahya M and Lashuel HA (2018) ‘Protein Semisynthesis Provides Access to Tau Disease-Associated Post-translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States’, Journal of the American Chemical Society. American Chemical Society, 140(21), pp. 6611–6621. doi: 10.1021/jacs.8b02668. [DOI] [PubMed] [Google Scholar]
- Halic M (2019) ‘Nucleosome and ubiquitin position Set2 to methylate H3K36’, Nature Communications. Springer US, 10(3795), pp. 1–9. doi: 10.1038/s41467-019-11726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z et al. (2017) ‘Profiling Cellular Substrates of Lysine Acetyltransferases GCN5 and p300 with Orthogonal Labeling and Click Chemistry’, ACS Chemical Biology, 12(6), pp. 1547–1555. doi: 10.1021/acschembio.7b00114. [DOI] [PubMed] [Google Scholar]
- Hansen BK et al. (2019) ‘Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation’, Nature Communications. Springer US, 10(1), pp. 1–11. doi: 10.1038/s41467-019-09024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmel R and Fiedler D (2018) ‘Features and regulation of non-enzymatic post-translational modifications’, Nature Chemical Biology. Nature Publishing Group, 14(3), pp. 244–252. doi: 10.1038/nchembio.2575. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV et al. (2012) ‘PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse’, Nucleic Acids Research, 40(D1), pp. 261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt EA et al. (2019) ‘Contemporary approaches to site-selective protein modification’, Nature Reviews Chemistry, 3(3), pp. 147–171. doi: 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- Hsu PL et al. (2018) ‘Crystal Structure of the COMPASS H3K4 Methyltransferase Catalytic Module’, Cell. Elsevier Inc., 174(5), pp. 1106–1116.e9. doi: 10.1016/j.cell.2018.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H et al. (2015) ‘Quantitative proteomic analysis of histone modifications’, Chemical Reviews, 115(6), pp. 2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H et al. (2018) ‘Lysine benzoylation is a histone mark regulated by SIRT2’, Nature Communications. Springer US, 9(3374), pp. 1–11. doi: 10.1038/s41467-018-05567-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey GW et al. (2001) ‘Stable Histone Deacetylase Complexes Distinguished by the Presence of SANT Domain Proteins CoREST/kiaa0071 and Mta-L1’, Journal of Biological Chemistry, 276(9), pp. 6817–6824. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- Italiano A et al. (2018) ‘Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study’, The Lancet Oncology. Elsevier Ltd, 19(5), pp. 649–659. doi: 10.1016/S1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- Ito T et al. (2010) ‘Identification of a primary target of thalidomide teratogenicity’, Science, 327(5971), pp. 1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- Itoh T et al. (2015) ‘Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting’, Nucleic Acids Research, 43(4), pp. 2033–2044. doi: 10.1093/nar/gkv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamaladdin S et al. (2014) ‘Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells’, Proceedings of the National Academy of Sciences of the United States of America, 111(27), pp. 9840–9845. doi: 10.1073/pnas.1321330111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LI et al. (2013) ‘Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain’, Nature Chemical Biology, 9(3), pp. 184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen KA et al. (2019) ‘Quantitation of Single and Combinatorial Histone Modifications by Integrated Chromatography of Bottom-up Peptides and Middle-down Polypeptide Tails’, Journal of the American Society for Mass Spectrometry. Journal of The American Society for Mass Spectrometry, 30(12), pp. 2449–2459. doi: 10.1007/s13361-019-02303-6. [DOI] [PubMed] [Google Scholar]
- Jing H et al. (2016) ‘A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity’, Cancer Cell, 29(3), pp. 297–310. doi: 10.1016/j.ccell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H and Lin H (2015) ‘Sirtuins in epigenetic regulation’, Chemical Reviews, 115(6), pp. 2350–2375. doi: 10.1021/cr500457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y et al. (2018) ‘Site-Specific Installation of Succinyl Lysine Analog into Histones Reveals the Effect of H2BK34 Succinylation on Nucleosome Dynamics’, Cell Chemical Biology. Elsevier Ltd., 25(2), pp. 166–174.e7. doi: 10.1016/j.chembiol.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Kalin JH et al. (2012) ‘Chiral Mercaptoacetamides Display Enantioselective Inhibition of Histone Deacetylase6 and Exhibit Neuroprotection in Cortical Neuron Models of Oxidative Stress’, ChemMedChem, 7(3), pp. 425–439. doi: 10.1002/cmdc.201100522. [DOI] [PubMed] [Google Scholar]
- Kalin JH et al. (2018) ‘Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors’, Nature Communications, 9(1). doi: 10.1038/s41467-017-02242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E et al. (2019) ‘MITF expression predicts therapeutic vulnerability to p300 inhibition in human melanoma’, Cancer Research, 79(10), pp. 2649–2661. doi: 10.1158/0008-5472.CAN-18-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y et al. (2017) ‘Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome’, Nature Medicine. Nature Publishing Group, 23(2), pp. 213–222. doi: 10.1038/nm.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BJ et al. (2018) ‘Structural insights into the π-π-π stacking mechanism and DNA-binding activity of the YEATS domain’, Nature Communications. Springer US, 9(1). doi: 10.1038/s41467-018-07072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM and Zhang Y (2006) ‘JmjC-domain-containing proteins and histone demethylation’, Nature Reviews Genetics, 7(9), pp. 715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ and Urbé S (2009) ‘Breaking the chains: Structure and function of the deubiquitinases’, Nature Reviews Molecular Cell Biology. Nature Publishing Group, 10(8), pp. 550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Komander D and Rape M (2012) ‘The Ubiquitin Code’, Annual Review of Biochemistry, 81(1), pp. 203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kooistra SM and Helin K (2012) ‘Molecular mechanisms and potential functions of histone demethylases’, Nature Reviews Molecular Cell Biology, 13(5), pp. 297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV et al. (2019) ‘A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia’, Cancer Cell. Elsevier Inc., 36(6), pp. 660–673.e11. doi: 10.1016/j.ccell.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L et al. (2012) ‘A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response’, Nature. Nature Publishing Group, 488(7411), pp. 404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N et al. (2018) ‘Identification of a novel small molecule that inhibits deacetylase but not defatty-acylase reaction catalysed by SIRT2’, Philosophical Transactions of the Royal Society B: Biological Sciences, 373(1748). doi: 10.1098/rstb.2017.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty CD et al. (1997) ‘Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression’, Cell, 89(3), pp. 349–356. doi: 10.1016/S0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- Lamb KN et al. (2019) ‘Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7’, Cell Chemical Biology. Elsevier Ltd., 26(10), pp. 1365–1379.e22. doi: 10.1016/j.chembiol.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP et al. (2019) ‘Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains’, Molecular Cell, 73(3), pp. 621–638.e17. doi: 10.1016/j.molcel.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I et al. (2017) ‘Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7’, Cell Chemical Biology. Elsevier, 24(12), pp. 1490–1500.e11. doi: 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM et al. (2017) ‘Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours’, Nature. Nature Publishing Group, 550(7674), pp. 128–132. doi: 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen F, Gafken PR and Gottschling DE (2002) ‘Dot1p modulates silencing in yeast by methylation of the nucleosome core’, Cell, 109(6), pp. 745–756. doi: 10.1016/S0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- Li X et al. (2018) ‘Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking’, Nature Chemical Biology. Springer US, 14(12), pp. 1140–1149. doi: 10.1038/s41589-018-0144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F et al. (2013) ‘Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP’, Journal of Medicinal Chemistry, 56(21), pp. 8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y et al. (2015) ‘A novel effect of thalidomide and its analogs: Suppression of cereblon ubiquitination enhances ubiquitin ligase function’, FASEB Journal, 29(12), pp. 4829–4839. doi: 10.1096/fj.15-274050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J et al. (2013) ‘Selective inhibition of tumor oncogenes by disruption of super-enhancers’, Cell, 153(2), pp. 320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P et al. (2016) ‘The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb’, Scientific Reports. Nature Publishing Group, 6(March), pp. 1–13. doi: 10.1038/srep24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T and Stark GR (2015) ‘NF-κB: Regulation by methylation’, Cancer Research, 75(18), pp. 3692–3695. doi: 10.1158/0008-5472.CAN-15-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M (2018) ‘Chemical and Biochemical Perspectives of Protein Lysine Methylation’, Chemical Reviews. American Chemical Society, 118(14), pp. 6656–6705. doi: 10.1021/acs.chemrev.8b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson L et al. (2020) ‘HBO1 is required for the maintenance of leukaemia stem cells’, Nature. Springer US, 577(7789), pp. 266–270. doi: 10.1038/s41586-019-1835-6. [DOI] [PubMed] [Google Scholar]
- Mevissen TET and Komander D (2017) ‘Mechanisms of Deubiquitinase Specificity and Regulation’, Annual Review of Biochemistry, 86(1), pp. 159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- Miller SA et al. (2020) ‘Lysine-specific demethylase 1 mediates Akt activity and promotes epithelial-to-mesenchymal transition in PIK3CA-mutant colorectal cancer’, Molecular Cancer Research, 18(2), pp. 264–277. doi: 10.1158/1541-7786.MCR-19-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J et al. (2003) ‘Structure of the catalytic domain of human Dot1L, a non-SET domain nucleosomal histone methyltransferase’, Cell, 112(5), pp. 711–723. doi: 10.1016/S0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Mohammad HP et al. (2015) ‘A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC’, Cancer Cell. Elsevier Inc., 28(1), pp. 57–69. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Morrison-Smith CD et al. (2020) ‘Combined targeting of the BRD4-NUT-p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734’, Molecular Cancer Therapeutics, p. molcanther.0087.2020. doi: 10.1158/1535-7163.mct-20-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakim M et al. (2017) ‘Discovery of a PCAF Bromodomain Chemical Probe’, Angewandte Chemie International Edition, 56(3), pp. 827–831. doi: 10.1002/anie.201610816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir TW, Sondhi D and Cole PA (1998) ‘Expressed protein ligation: A general method for protein engineering’, Proceedings of the National Academy of Sciences, 95(12), pp. 6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MM and Muir TW (2015) ‘Histones: At the crossroads of peptide and protein chemistry’, Chemical Reviews, 115(6), pp. 2296–2349. doi: 10.1021/cr5003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z and Tora L (2007) ‘Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation’, Oncogene. Nature Publishing Group, 26(37), pp. 5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- Nguyen DP et al. (2010) ‘Genetically directing ε-N, N-dimethyl-l-lysine in recombinant histones’, Chemistry and Biology. Elsevier Ltd, 17(10), pp. 1072–1076. doi: 10.1016/j.chembiol.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Pallante P et al. (2015) ‘Polycomb protein family member CBX7 plays a critical role in cancer progression’, American Journal of Cancer Research, 5(5), pp. 1594–1601. [PMC free article] [PubMed] [Google Scholar]
- Perfetti MT et al. (2015) ‘Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53BP1’, ACS Chemical Biology, 10(4), pp. 1072–1081. doi: 10.1021/cb500956g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotukh K et al. (2011) ‘Directed evolution of sortase A mutants with altered substrate selectivity profiles’, Journal of the American Chemical Society, 133(44), pp. 17536–17539. doi: 10.1021/ja205630g. [DOI] [PubMed] [Google Scholar]
- Qi Y-K et al. (2018) ‘Total Chemical Synthesis of Modified Histones’, Frontiers in Chemistry, 6(February), pp. 1–11. doi: 10.3389/fchem.2018.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C and Zhou MM (2006) ‘SET domain protein lysine methyltransferases: Structure, specificity and catalysis’, Cellular and Molecular Life Sciences, 63(23), pp. 2755–2763. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabino C et al. (2018) ‘A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment’, Nature Medicine, 24(2), pp. 186–193. doi: 10.1038/nm.4474. [DOI] [PubMed] [Google Scholar]
- Romero FA et al. (2017) ‘GNE-781, A Highly Advanced Potent and Selective Bromodomain Inhibitor of Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP)’, Journal of Medicinal Chemistry, 60(22), pp. 9162–9183. doi: 10.1021/acs.jmedchem.7b00796. [DOI] [PubMed] [Google Scholar]
- Rulina AV et al. (2016) ‘Distinct outcomes of CRL-Nedd8 pathway inhibition reveal cancer cell plasticity’, Cell Death and Disease. Nature Publishing Group, 7(12), pp. 1–11. doi: 10.1038/cddis.2016.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf T et al. (2015) ‘Selective Sirt2 inhibition by ligand-induced rearrangement of the active site’, Nature Communications, 6. doi: 10.1038/ncomms7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan S et al. (2016) ‘Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function’, Proceedings of the National Academy of Sciences of the United States of America, 113(16), pp. 4320–4325. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer NJ et al. (2020) ‘Advances in Discovering Deubiquitinating Enzyme (DUB) Inhibitors’, Journal of Medicinal Chemistry, 63(6), pp. 2731–2750. doi: 10.1021/acs.jmedchem.9b01138. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Nuber U and Huibregtse JM (1995) ‘Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade’, Nature, pp. 81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- Schotta G et al. (2002) ‘Central role of Drosophila SU(VAR)3–9 in histone H3-K9 methylation and heterochromatic gene silencing’, EMBO Journal, 21(5), pp. 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Yujiang et al. (2004) ‘Histone demethylation mediated by the nuclear amine oxidase homolog LSD1’, Cell, 119(7), pp. 941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shortt J et al. (2017) ‘A chemical probe toolbox for dissecting the cancer epigenome’, Nature Reviews Cancer, 17(3), pp. 160–183. doi: 10.1038/nrc.2016.148. [DOI] [PubMed] [Google Scholar]
- Simon MD et al. (2007) ‘The Site-Specific Installation of Methyl-Lysine Analogs into Recombinant Histones’, Cell, 128(5), pp. 1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y et al. (2020) ‘Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex’, Cell Reports. ElsevierCompany., 30(8), pp. 2699–2711.e8. doi: 10.1016/j.celrep.2020.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer CD and Davis BG (2014) ‘Selective chemical protein modification’, Nature Communications. Nature Publishing Group, 5(1), p. 4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Spratt DE, Walden H and Shaw GS (2014) ‘RBR E3 ubiquitin ligases: New structures, new insights, new questions’, Biochemical Journal, 458(3), pp. 421–437. doi: 10.1042/BJ20140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M et al. (2014) ‘Lysine glutarylation is a protein posttranslational modification regulated by SIRT5’, Cell Metabolism, 19(4), pp. 605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teske KA and Hadden MK (2017) ‘Methyllysine binding domains: Structural insight and small molecule probe development’, European Journal of Medicinal Chemistry. Elsevier Masson SAS, 136, pp. 14–35. doi: 10.1016/j.ejmech.2017.04.047. [DOI] [PubMed] [Google Scholar]
- Tharp JM, Ehnbom A and Liu WR (2018) ‘tRNAPyl: Structure, function, and applications’, RNA Biology. doi: 10.1080/15476286.2017.1356561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile CS et al. (2013) ‘Site-specific N-terminal labeling of proteins using sortase-mediated reactions’, Nature Protocols, 8(9), pp. 1800–1807. doi: 10.1038/nprot.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE and Muir TW (2019) ‘Chemoenzymatic Semisynthesis of Proteins’, Chemical Reviews. doi: 10.1021/acs.chemrev.9b00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang VH et al. (2014) ‘Chemoenzymatic synthesis of bifunctional polyubiquitin substrates for monitoring ubiquitin chain remodeling’, ChemBioChem, 15(11), pp. 1563–1568. doi: 10.1002/cbic.201402059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trievel RC et al. (2002) ‘Structure and catalytic mechanism of a SET domain protein methyltransferase’, Cell, 111(1), pp. 91–103. doi: 10.1016/S0092-8674(02)01000-0. [DOI] [PubMed] [Google Scholar]
- Trojer P et al. (2007) ‘L3MBTL1, a Histone-Methylation-Dependent Chromatin Lock’, Cell, 129(5), pp. 915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- Tsukada YI et al. (2006) ‘Histone demethylation by a family of JmjC domain-containing proteins’, Nature, 439(7078), pp. 811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Vassilev LT et al. (2004) ‘In Vivo Activation of the p53 Pathway by Small-MoleculeAntagonists of MDM2.’, Science, 303(5659), pp. 844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vinyard ME et al. (2019) ‘CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML’, Nature Chemical Biology. Springer US, 15(5), pp. 529–539. doi: 10.1038/s41589-019-0263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P et al. (2012) ‘Asymmetrically modified nucleosomes’, Cell. Elsevier, 151(1), pp. 181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W, Tharp JM and Liu WR (2014) ‘Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool’, Biochimica et Biophysica Acta - Proteins and Proteomics. Elsevier B.V., 1844(6), pp. 1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG et al. (2009) ‘Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger’, Nature. Nature Publishing Group, 459(7248), pp. 847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L et al. (2001) ‘Expanding the genetic code of Escherichia coli’, Science, 292(August), pp. 408–500. doi: 10.1038/nchembio.1043. [DOI] [PubMed] [Google Scholar]
- Wang Y et al. (2014) ‘Regulation of S-adenosylhomocysteine hydrolase by lysine acetylation’, Journal of Biological Chemistry, 289(45), pp. 31361–31372. doi: 10.1074/jbc.M114.597153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Yi et al. (2018) ‘Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation’, Proceedings of the National Academy of Sciences of the United States of America, 115(10), pp. 2365–2370. doi: 10.1073/pnas.1717664115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Yiwei et al. (2018) ‘Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade’, Cell Research. Springer US, 28(12), pp. 1186–1194. doi: 10.1038/s41422-018-0091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z-P et al. (2015) ‘The Study of the Chemical Synthesis and Preparation of Histone with Post- Translational Modifications’, Current Organic Synthesis, 12(2), pp. 150–162. doi: 10.2174/1570179411666141125215343. [DOI] [Google Scholar]
- Wang ZA et al. (2016) ‘A Genetically Encoded Allysine for the Synthesis of Proteins with Site-Specific Lysine Dimethylation’, Angewandte Chemie International Edition, 56(1), pp. 212–216. doi: 10.1002/anie.201609452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA et al. (2017) ‘A Versatile Approach for Site-Specific Lysine Acylation in Proteins’, Angewandte Chemie - International Edition, 56(6), pp. 1643–1647. doi: 10.1002/anie.201611415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA (2019) ‘The Recent Progresses in Chemical Synthesis of Proteins with Site-Specific Lysine Post-translational Modifications’, Current Organic Synthesis, 16, pp. 369–384. doi: 10.2174/1570179416666190328233918. [DOI] [PubMed] [Google Scholar]
- Wang ZA et al. (2020) ‘Diverse nucleosome site-selectivity among histone deacetylase complexes’, eLife, 9, p. e57663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA and Cole PA (2020) ‘Methods and Applications of Expressed Protein Ligation’, in Methods in Molecular Biology, pp. 1–13. doi: 10.1007/978-1-0716-0434-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Hsu W and Liu WR (2017) ‘Role of SIRT1 in Epigenetics’, in Handbook of Nutrition, Diet, and Epigenetics, pp. 1–19. doi: 10.1007/978-3-319-31143-2. [DOI] [Google Scholar]
- Wang ZPA, Tian CL and Zheng JS (2015) ‘The recent developments and applications of the traceless-Staudinger reaction in chemical biology study’, RSC Advances, 5(130), pp. 107192–107199. doi: 10.1039/c5ra21496c. [DOI] [Google Scholar]
- Weinert BT et al. (2013) ‘Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation’, Cell Reports, 4(4), pp. 842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Weinert BT et al. (2018) ‘Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome’, Cell, 174(1), pp. 231–244.e12. doi: 10.1016/j.cell.2018.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden EJ et al. (2019) ‘Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L’, Cell. Elsevier Inc., 176(6), pp. 1490–1501.e12. doi: 10.1016/j.cell.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TH et al. (2016) ‘Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity’, Science, 354(6312). doi: 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- Wu M et al. (2018) ‘Lysine-14 acetylation of histone H3 in chromatin confers resistance to the deacetylase and demethylase activities of an epigenetic silencing complex’, eLife, 7. doi: 10.7554/eLife.37231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z et al. (2017) ‘Efficient expression of sortase A from Staphylococcus aureus in Escherichia coli and its enzymatic characterizations’, Bioresources and Bioprocessing. Springer Berlin Heidelberg, 4(1). doi: 10.1186/s40643-017-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z et al. (2012) ‘Lysine succinylation and lysine malonylation in histones’, Molecular and Cellular Proteomics, 11(5), pp. 100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y et al. (2020) ‘Inhibiting the coregulator CoREST impairs Foxp3+ Treg function and promotes antitumor immunity’, Journal of Clinical Investigation, 130(4), pp. 1830–1842. doi: 10.1172/JCI131375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y et al. (1998) ‘NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities’, Molecular Cell, 2(6), pp. 851–861. doi: 10.1016/S1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yang A, Cho K and Park HS (2018) ‘Chemical biology approaches for studying posttranslational modifications’, RNA Biology. Taylor & Francis, 15(4–5), pp. 427–440. doi: 10.1080/15476286.2017.1360468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D et al. (2019) ‘Metabolic regulation of gene expression by histone lactylation.’, Nature. Springer US, 574(7779), pp. 575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W et al. (2016) ‘System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes’, Molecular Cell. Elsevier Inc., 62(1), pp. 121–136. doi: 10.1016/j.molcel.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J-S et al. (2013) ‘Chemical synthesis of proteins using peptide hydrazides as thioester surrogates’, Nature Protocols. doi: 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]
- Zucconi BE et al. (2016) ‘Modulation of p300/CBP Acetylation of Nucleosomes by Bromodomain Ligand I-CBP112’, Biochemistry, 55(27), pp. 3727–3734. doi: 10.1021/acs.biochem.6b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucconi BE et al. (2019) ‘Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP’, Biochemistry. American Chemical Society, 58, pp. 2133–2143. doi: 10.1021/acs.biochem.9b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]