

Abstract
Simultaneous inhibition of MDM2 and CDK4 may be an effective treatment against glioblastoma. A collection of chiral spirocyclic tetrahydronaphthalene (THN)-oxindole hybrids for this purpose have been developed. Appropriate stereochemistry in THN-fused spirooxindole compounds is key to their inhibitory activity: selectivity differed by over 40-fold between the least and most potent stereoisomers in time-resolved FRET and KINOMEscan® in vitro assays. Studies in glioblastoma cell lines showed that the most active compound ent-4g induced apoptosis and cell cycle arrest by interfering with MDM2 -P53 interaction and CDK4 activation. Cells treated with ent-4g showed up-regulation of proteins involved in P53 and cell cycle pathways. The compound showed good anti-tumor efficacy against glioblastoma xenografts in mice. These results suggested that rational design, asymmetric synthesis and biological evaluation of novel tetrahydronaphthalene fused spirooxindoles could generate promising MDM2-CDK4 dual inhibitors in glioblastoma therapy.
KEY WORDS: Chiral tetrahydronaphthalene-fused spirooxindoles, Synthesis, MDM2-CDK4 dual inhibitors, Glioblastoma, Structure–activity relationship, Apoptosis, Cell cycle arrest
Graphical abstract
Simultaneous inhibition of MDM2 and CDK4 can be an effective treatment against glioblastoma. Rational design, asymmetric synthesis and biological evaluation of novel chiral tetrahydronaphthalene-fused spirooxindoles can generate promising MDM2/CDK4 dual inhibitors in targeted glioblastoma therapy.

1. Introduction
Glioblastoma is a malignant disease associated with poor prognosis, with few treatment possibilities. The disease involves deregulation of P53 and cell cycle signalling pathways1, 2, 3, 4. Our analysis of genomic alterations in glioblastoma according to data in the Cancer Genome Atlas (TCGA) identified the q13–15 region of chromosome 12 as one of the regions that most often rearranged in the disease (Fig. 1A and B)5, 6, 7. This region encodes the P53-interactor murine double minute 2 protein (MDM2) and cyclin-dependent kinase 4 (CDK4). We also verified both genes to be significantly overexpressed at the mRNA and protein levels in patients with glioblastoma, regardless of P53 mutation status (Fig. 1C–E).
Figure 1.

The mRNA and protein expression levels of MDM2 and CDK4 in various type of cancers. (A) Transcripts per million reads of MDM2 in every type of cancers retrieved from GEPIA database. (B) The differential expression and mutation status of MDM2, TP53 and CDKs in glioblastoma cohort retrieved from cBioportal and TCGA database. (C) The boxplot of MDM2 and CDK4 expression in glioblastoma cohort retrieved from TCGA database (∗P < 0.05). (D) The expression correlation of MDM2 and CDK4 in both tumor and normal samples from TCGA and GTEx database (left panel) and only tumor samples in TCGA database (right panel). (E) The protein expression levels of MDM2 and CDK4 determined by IHC in glioblastoma and normal cerebral cortex tissues, retrieved from ProteinAtlas database, scale bar: 200 μm.
Extensive efforts have already been made to develop small molecules that can disrupt the interaction between MDM2 and P53 in order to unleash the latter's anti-tumor activity. A diverse array of privileged scaffolds has been discovered, including derivatives of imidazoline, piperidinone, benzodiazepine, chromenotriazolopyrimidine, terphenyl, isoindolinone and pyrrolidine8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. Some of these derivatives have advanced to clinical trials for the treatment of breast cancer, leukemia, lymphoma and glioblastoma. Spirocyclic oxindoles have recently been patented as a newly identified type of P53–MDM2 inhibitor (Fig. 2A)21, 22, 23, 24, 25, 26. While N-, O- and S-containing heterocyclic substitutions have been extensively explored to generate novel C3-spirooxindole inhibitors of P53–MDM2 interaction, the investigation of all-carbocycle modifications at the C3 position as potent MDM2 inhibitors are underdeveloped.
Figure 2.

(A) Typical model of how the C3-spirooxindole framework interacts with MDM2. (B) Naphthalene derivatives are an effective framework for CDK4 inhibitors. (C) The combination of C3-spirooxindole and tetrahydronaphthalene (THN) may generate a novel dual-inhibitor of MDM2 and CDK4.
CDK4, one of the main controllers of cell cycle entry, is substantially overexpressed in glioblastoma, breast and ovarian cancers, making it an attractive therapeutic target27, 28, 29. Some recent efforts have generated promising leads by targeting compounds to allosteric binding sites in CDKs30, 31, 32, 33. The allosteric pocket varies among CDKs, in contrast to the highly conserved ATP-binding site. Planar naphthalene derivatives can dock well into the narrow allosteric binding site of CDK4, making them a privileged scaffold for generating subtype selective inhibitors (Fig. 2B)34,35.
Analysis of CDK expression and mutations in glioblastoma samples in TCGA database indicates that CDK4 is the most often overexpressed CDKs in the disease, and it is overexpressed in over half of patients with glioblastoma associated with mutations in P53 (Fig. 1D). These results suggest that simultaneous inhibition of both MDM2 and CDK4 may be effective against glioblastoma36, 37, 38, 39.
Moreover, the co-amplification of MDM2 and CDK4 has been reported in several type of cancers including sarcoma, glioblastoma, bladder cancer, gastric cancer, etc.40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50 Although the simple combination therapy of MDM2 and CDK4 inhibitors in preclinical experiments were reported recently, the two independent reports demonstrated the paradoxical results in sarcoma51,52. In addition, Klein et al.53 reported palbociclib-induced senescence resulted MDM2 downregulation in cancer cells. These results indicated that the regulation mechanisms between MDM2 and CDK4 may be more complicated than previously thought.
Therefore, we aimed to develop scaffolds for dual inhibitors of both proteins that could avoid resistance due to P53 mutation and that could bind CDK4 selectively to avoid off-target effects. After analysing the binding modes of known MDM2 inhibitors and CDK4 inhibitors, we speculated that fusion of the planar tetrahydronaphthalene (THN) ring at the C3-position of oxindole might generate a scaffold that could bind at the P53-binding site in MDM2 as well as at the allosteric site in CDK4. We started with THN54, 55, 56, 57, 58, 59, 60, 61 and spirooxindole derivatives62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72 because they are privileged drug-like architectures, so the resulting THN-fused C3-spirooxindoles should possess good druglikeness (Fig. 2C).
Here we rationally designed and asymmetrically synthesized a series of chiral THN-spirooxindole-based MDM2/CDK4 dual inhibitors, which showed promising anti-glioblastoma activity in vitro and in vivo. In particular, compound ent-4g displayed good CDK selectivity: it showed nanomolar IC50 against CDK4, micromolar IC50 against CDK2, and no appreciable inhibition of other CDKs or kinases. The novel compound inhibited proliferation and induced apoptosis in glioblastoma cell lines expressing wild-type or mutated P53. The novel compound inhibited the growth of glioblastoma xenografts expressing mutant P53 better than the MDM2 inhibitor nutlin-3a alone or together with palbociclib.
2. Results and discussion
2.1. Rational design and synthesis of chiral THN-fused spirooxindoles as dual inhibitors of MDM2 and CDK4
In spirocyclic oxindole-based MDM2 inhibitors, the oxindole fragment occupies the Trp23-containing cleft of P53, and appropriate stereochemistry is critical for good binding affinity73,74. Therefore, we focused on asymmetric synthesis of optically pure C3-spirooxindoles75, 76, 77, 78. We started from hydronaphthalene79, 80, 81, 82, 83, 84 and spirooxindole10,22,23,25,65 because they are privileged frameworks occurring in many anti-tumor natural products and pharmaceuticals. Combination of privileged frameworks can facilitate molecular diversity and discovery of lead compounds85,86.
We knew that the spirocyclic oxindole inhibitor would have to fit within the flat, narrow allosteric pocket of CDK4. Preliminary docking studies and integrative molecular simulations suggested that an inhibitor bearing a planar THN would bind well to CDK4 and MDM2. In the CDK4 allosteric site, the scaffold could interact with surrounding hydrophobic residues and residues in the DFG-loop, avoiding interactions with the highly conserved ATP-binding site that might reduce selectivity for CDK487. At the P53-binding site in MDM2, the THN-fused C3-spirooxindole could form hydrogen bonds and hydrophobic interactions mimicking Phe19, Trp23 and Leu25 of P53. In fact, introducing a hydrogen bond acceptor and electron-withdrawing group (EWG) onto the THN would allow formation of a hydrogen bond with Thr16, which could strengthen MDM2 binding.
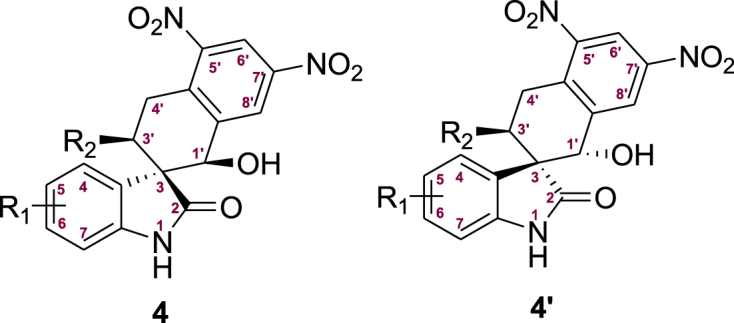
Hence, we used the 3-ylideneoxindoles88, 89, 90 (1 and 2) and 2-methyl-3,5-dinitrobenzaldehyde (3a) as substrates, to prepare the THN-fused spirooxindole derivatives91 int-4 and int-4′ through Michael-aldol cascade reaction, promoted by the bifunctional hydrogen-bonding catalyst (1R,2R-catalyst). Next, the protecting groups of int-4 and int-4′ were removed to afford the compounds 4 and the diastereoisomer 4′ (Scheme 1). The screening of reaction conditions, synthetic methods and detailed data of int-4, int-4′, 4 and 4′ are contained in the Supporting Information. To explain the diastereodivergence of the organocatalytic Michael-aldol cascade, we also proposed plausible transition-state models based on the observed stereochemistry of the products (Supporting Information Scheme S4).
Scheme 1.

Preparation of 4 and 4′ for bioactivity screening. Int = intermediate.
2.2. Structure–activity relationships in chiral THN-fused spirooxindoles based on cytotoxicity and enzymatic inhibition assays
We assessed the ability of 4a–4p and 4a′–4p′ to inhibit MDM2 and CDK4 using time-resolved fluorescence resonance energy transfer (TR-FRET). As positive control drugs, we used the MDM2 inhibitor nutlin-3a92 and the CDK4/6 inhibitor palbociclib93. The inhibition rates for each compound at 1.0 μmol/L were determined (Table 1). At concentrations below 1.0 μmol/L, the inhibition caused by nutlin-3a, 4a–4c and 4i–4p dropped from about 40% to 20%, while palbociclib, 4d and 4g still showed inhibition of 40%–60%. The IC50 values of most active compounds 4a–4j were also measured (Fig. 3A and Supporting Information Table S1). Compounds 4a–4p worked better than compounds 4a′–4p′ at inhibiting the activity of MDM2 and CDK4 as well as the proliferation of glioblastoma cell lines. Among the more active compounds 4a–4j, derivatives 4d and 4g with a halogen at the 5-position of the oxindole showed the greatest MDM2 inhibition and cytotoxicity. Although 4d and 4g inhibited MDM2 and CDK4 less than nutlin-3a and palbociclib, all compounds showed similar cytotoxicity against the tested glioblastoma cell lines based on the MTT assay. At high concentrations, all compounds showed good inhibition of two cell lines expressing mutated P53 (T98G and U251) and one cell line expressing wild-type P53 (U87MG, Fig. 3B, C). It was notable that the cell proliferation inhibitory potencies of compounds 4d–4h in U87MG cells were better than that of T98G and U251 cells, which suggested that only the activation of wild-type P53 should suppress the glioblastoma cell proliferation.
Table 1.
In vitro biological evaluation of compounds 4a–4p and 4a′–4p′a.
| Compd. | R1 | R2 | Inhibition rate at 1.0 μmol/L (%, 4/4′) |
Anti-proliferative activity (IC50, μmol/L, 4/4′) |
|||
|---|---|---|---|---|---|---|---|
| MDM2 | CDK4 | U87MG | U251 | T98G | |||
| 4a/4a′ | H | COOEt | 71.6 ± 3.8/47.8 ± 7.3 | 42.7 ± 4.3/26.9 ± 6.2 | 25.3 ± 2.7/64.7 ± 10.9 | 43.6 ± 5.5/>50 | 62.4 ± 7.9/>50 |
| 4b/4b′ | 5-F | COOEt | 81.4 ± 10.2/55.6 ± 10.6 | 53.9 ± 8.3/32.6 ± 5.1 | 25.7 ± 1.4/>50 | 32.6 ± 3.5/>50 | 49.0 ± 6.3/>50 |
| 4c/4c′ | 7-F | COOEt | 76.5 ± 13.7/45.3 ± 6.4 | 52.6 ± 7.7/32.6 ± 5.1 | 19.4 ± 2.3/59.9 ± 9.8 | 34.3 ± 4.0/>50 | 51.6 ± 6.2/>50 |
| 4d/4d′ | 5-Cl | COOEt | 82.6 ± 8.4/54.2 ± 6.7 | 70.2 ± 7.8/45.2 ± 3.5 | 8.3 ± 1.5/60.4 ± 4.8 | 17.8 ± 3.2/>50 | 21.6 ± 4.5/>50 |
| 4e/4e′ | 6-Cl | COOEt | 76.4 ± 6.0/48.3 ± 5.1 | 61.2 ± 8.2/38.6 ± 2.9 | 19.2 ± 4.4/>50 | 28.6 ± 2.9/>50 | 22.5 ± 4.2/>50 |
| 4f/4f′ | 4-Br | COOEt | 66.9 ± 9.1/54.8 ± 7.2 | 45.6 ± 4.2/29.4 ± 3.6 | 20.7 ± 3.5/69.8 ± 6.2 | 25.8 ± 4.2/>50 | 27.2 ± 3.0/>50 |
| 4g/4g′ | 5-Br | COOEt | 92.3 ± 11.2/51.9 ± 6.5 | 76.3 ± 8.7/34.1 ± 2.9 | 4.9 ± 0.5/>50 | 8.6 ± 0.6/>50 | 9.5 ± 0.7/>50 |
| 4h/4h′ | 6-Br | COOEt | 76.7 ± 5.2/61.2 ± 5.2 | 59.0 ± 5.7/32.9 ± 2.4 | 20.4 ± 3.3/47.6 ± 5.7 | 18.3 ± 1.5/>50 | 14.4 ± 2.8/>50 |
| 4i/4i′ | 5-NO2 | COOEt | 47.7 ± 4.1/39.3 ± 2.6 | 42.8 ± 5.4/37.9 ± 5.7 | 25.6 ± 2.8/47.9 ± 8.8 | 36.2 ± 4.6/>50 | 41.2 ± 6.1/>50 |
| 4j/4j′ | 5-Me | COOEt | 64.4 ± 7.2/49.2 ± 4.7 | 67.8 ± 6.9/27.1 ± 1.5 | 31.1 ± 1.76/>50 | 38.9 ± 4.5/>50 | 39.3 ± 3.6/>50 |
| 4k/4k′ | H | Bz | 46.4 ± 4.9/35.4 ± 2.4 | 39.5 ± 0.83/30.8 ± 5.1 | 38.73 ± 6.5/>50 | 53.8 ± 9.0/>50 | 43.7 ± 6.6/>50 |
| 4l/4l′ | H | 2-FBz | 47.2 ± 5.71/31.6 ± 2.4 | 38.9 ± 6.4/29.3 ± 2.7 | 32.5 ± 4.7/>50 | 38.7 ± 6.1/>50 | 43.6 ± 4.8/>50 |
| 4m/4m′ | H | 4-FBz | 44.6 ± 3.7/36.2 ± 2.5 | 48.2 ± 4.3/23.2 ± 3.0 | 47.2 ± 3.8/>50 | 39.5 ± 3.8/>50 | 37.1 ± 3.2/>50 |
| 4n/4n′ | H | 3,4-Cl2Bz | 38.4 ± 2.7/36.3 ± 5.2 | 35.6 ± 3.9/18.5 ± 2.1 | 34.9 ± 2.62/>50 | 25.9 ± 3.5/>50 | 32.5 ± 2.9/>50 |
| 4o/4o′ | H | 4-BrBz | 47.1 ± 5.8/29.0 ± 3.5 | 29.5 ± 2.3/18.6 ± 0.9 | 41.6 ± 4.3/>50 | 44.2 ± 4.6/>50 | 45.7 ± 6.7/>50 |
| 4p/4p′ | H | 4-MeOBz | 35.7 ± 4.9/26.9 ± 1.1 | 39.3 ± 5.2/18.4 ± 2.2 | 39.9 ± 4.2/>50 | 43.2 ± 5.9/>50 | 45.9 ± 8.1/>50 |
| Nutlin-3a | – | – | 86.9 ± 9.3 | N.D. | 25.3 ± 2.5 | >50 | >50 |
| Palbociclib | – | – | N.D. | 95.9 ± 6.4 | 26.4 ± 3.8 | 18.9 ± 2.4 | 29.5 ± 4.5 |
Each compound was tested in triplicate; data are mean ± SD (n = 3). The IC50 value of anti-proliferation assays was obtained after 24-h incubation. –Not applicable.
Figure 3.

Inhibitory activity of 4a–4p. (A) IC50 values against MDM2 and CDK4 in TR-FRET assays. Values are mean ± SD (n = 3). Cytotoxicity against glioblastoma cell lines U87MG (B) and U251 (C) based on the MTT method. (D) Selectivity of ent-4g as a kinase inhibitor against a panel of 99 kinases at 100 nmol/L. The size of colored circles reflects relative inhibitory potency of ent-4g to corresponding protein. (E) The superposition of average conformations of compound 4g (black), 4g′ (blue), ent-4g (yellow) and ent-4g′ (orange) to the P53-binding site in MDM2 and the allosteric site in CDK4. Analysis of the average conformations of compound 4g/4g′ and ent-4g/ent-4g′ binding to MDM2 and CDK4 after 100-ns molecular dynamics simulations. (F) Comparison of the average conformation of compound ent-4g (yellow) with the experimental conformation of SAR405838 (grey) bound to MDM2 or of ANS (grey) bound to CDK4. (G) Schematic depiction of the potential interaction modes of compound ent-4g in the P53-binding in MDM2 and allosteric site in CDK4.
Focusing on 4d and 4g as the most active compounds in these bioactivity screens, we explored their structure–activity relationships and bioactive mechanisms. According to the above methodology (Scheme 1), the corresponding enantiomers ent-4d/ent-4g and ent-4d′/ent-4g′ were synthesized using the 1S,2S-catalyst, and four of the eight possible stereoisomers for 4d and 4g were obtained with high stereoselectivities (Scheme 2). Selected isomers of these compounds were serially diluted from 50 μmol/L to 5 nmol/L and tested against MDM2 and CDK4 in TR-FRET assays (Supporting Information Fig. S1). We also tested isomers against glioblastoma cell lines expressing wild-type P53 (U87MG) or mutated P53 (U251, Table 2). The isomers ent-4d and ent-4g inhibited growth of U87MG cells to a greater extent than nutlin-3a or palbociclib, and they inhibited growth of U251 cells better than palbociclib. The strong cytotoxicity of ent-4g against glioblastoma cells expressing mutated P53 is consistent with its low IC50 values against MDM2 and CDK4. Compound ent-4g was chosen for further bioassays and mechanistic studies.
Scheme 2.

The preparation of compounds ent-4d, ent-4g, ent-4d′, and ent-4g′. Ent = enantiomer.
Table 2.
IC50 of stereoisomers against proteins and glioblastoma cell linesa.
| Cell line | IC50 (μmol/L) |
|||||||
|---|---|---|---|---|---|---|---|---|
| R1 = 5-Cl |
R1 = 5-Br |
|||||||
| 4d | Ent-4d | 4d′ | Ent-4d′ | 4g | Ent-4g | 4g′ | Ent-4g′ | |
| MDM2 | 0.52 ± 0.07 | 0.13 ± 0.04 | 1.8 ± 0.12 | 1.5 ± 0.20 | 0.28 ± 0.06 | 0.08 ± 0.02 | 2.5 ± 0.3 | 1.3 ± 0.4 |
| CDK4 | 0.78 ± 0.11 | 0.49 ± 0.13 | 3.9 ± 0.42 | 2.6 ± 0.37 | 0.32 ± 0.05 | 0.25 ± 0.08 | 10.6 ± 0.9 | 3.7 ± 0.6 |
| U87MG | 8.3 ± 1.5 | 6.1 ± 1.3 | 60.4 ± 4.8 | 41.6 ± 3.9 | 4.9 ± 0.5 | 3.6 ± 0.7 | >50 | 28.6 ± 2.5 |
| U251 | 17.8 ± 3.2 | 12.6 ± 2.3 | >50 | >50 | 8.6 ± 0.8 | 5.8 ± 1.1 | >50 | >50 |

Each compound was tested in triplicate; data are mean ± SD (n = 3). The IC50 value of anti-proliferation assays was obtained after 24-h incubation.
The KINOMEscan® method was used to determine the kinase selectivity of ent-4g against a panel of 99 kinases in parallel (Fig. 3D and Supporting Information Table S2)94. The compound caused negligible or minimal inhibition to most kinases other than CDK4-cyclinD1, CDK4 and CDK2. In the case of CDK2, 4% of control protein remained after competitive binding of 100 nmol/L ent-4g (0.8% to CDK4-cyclinD1 and 2.2% to CDK4), which is probably because CDK2 possesses 66% of sequence identities to CDK4. These results suggest that ent-4g can be regarded as a specific MDM2/CDK4 inhibitor.
2.3. Structural basis of 4g isomer binding to MDM2 and CDK4
Molecular docking and dynamics studies were conducted to gain potential insights into how 4g/4g′ and ent-4g/ent-4g′ bind to MDM2 and CDK4 (Supporting Information Fig. S3). Molecular simulations were conducted for 100 ns, and binding free energies were calculated using the MM/GBSA method (Supporting Information Table S3)95. As references, we examined the co–crystal structure of MDM2 with SAR405838 (PDB ID: 5TRF)96 and a homology model of CDK4 complexed with the allosteric inhibitor 8-anilino-1-naphthalene sulfonate (ANS), based on the crystal structure of CDK2 with ANS (PDB ID: homology model generated from 3PXZ)97. Fig. 3E reveals differences in how 4g/4g′ and ent-4g/ent-4g′ are predicted to bind to their target sites. Binding conformation differed substantially between 4g/4g′ and ent-4g/ent-4g′; during the dynamic's simulation, 4g moved to another ANS binding site, 4g′ moved to the ATP binding site and ent-4g′ moved to the hydrophobic pocket. Fig. 3F compares how ent-4g is predicted to bind to the target sites with how SAR405838 and ANS bind. These analyses suggest that ent-4g mimics P53 residues Phe19, Trp23 and Leu25 in interacting with MDM2, and that the compound forms a stable hydrogen bond with MDM2 residue Thr16 (Fig. 3G), which has never been reported before. In our simulations, compound ent-4g formed hydrophobic interactions with a pocket formed by Val57, Gly160, Leu161 and Ile164, maintaining the DFG-loop in an “out” conformation98, 99, 100. Binding of ent-4g to the CDK4 allosteric pocket is predicted to depend on π–π stacking between the oxindole ring of ent-4g and Phe93, as well as electrostatic interactions between the nitro group of ent-4g and Arg61 (Fig. 3F and G)101.
The contributions of single amino acid residues in MDM2 substrate binding pocket were decomposed by using a computational alanine-scanning which was dependent on the assumption that local changes of the protein do not influence the whole conformation of the complex significantly. The 14 residues covering the walls of MDM2 substrate binding pocket were alternatively mutated to alanine from the simulation trajectory of the wild-type MDM2–inhibitor complex and results were shown in Fig. S3. As was expected, the mutation of key binding residues resulted significant increase of binding free energies, which suggested the disrupted inhibitor–residue interactions. The highest binding free energy changes were the mutation of Leu54 to alanine in both ent-4g and SAR405838 complexed to MDM2, the Thr16 in ent-4g complex and Lys94 in SAR405838 complex were also stronger than the other residues (>4.0 kcal/mol). The computational alanine scanning results also confirmed that binding modes of ent-4g suggested by molecular docking and MD simulation.
2.4. Ent-4g inhibits U251 glioblastoma cell proliferation by altering cell cycle progression and P53 signalling
To further elucidate the molecular mechanism of ent-4g, U251 glioblastoma cells were incubated with the compound, and then changes in gene expression were analysed globally using an Illumina Hiseq4000 platform (Novogene Co., Ltd., Beijing, China, Fig. 4A and Supporting Information Fig. S4)102. Enrichment analysis using integrated GO103, KEGG104 and Biocarta105 revealed significant alteration in the cell cycle and P53 signalling pathways, as shown in the KEGG pathway enrichment results (Fig. 4B). To identify the subroutine of programmed cell death induced by ent-4g, we treated the two glioblastoma cell lines with the compound, then assessed their cell cycle distribution via propidium iodide staining with flow cytometry, as well as apoptotic levels using Annexin V-FITC/PI dual staining (Keygen, Nanjing, China). The compound induced significant apoptosis and cell cycle arrest in G1 phase in both cell lines (Fig. 4C and D). In addition, ent-4g increased the proportion of glioblastoma cells showing hyper-condensed, apoptotic nuclei based on Hoechst 33,342 staining (Beyotime, Shanghai, China, Supporting Information Fig. S5).
Figure 4.

(A) List of enriched KEGG terms of differentially expressed genes in U251 cells after incubation with ent-4g. (B) Analysis of enriched KEGG terms based on statistical significance (logarithmic Q-value) and enrichment factors. (C) Apoptosis in U251 cells treated with ent-4g based on Annexin V/PI dual staining. (D) Cell cycle analysis of U251 cells treated with ent-4g. All values are the average from three independent experiments. (E) Western blotting and analysis of CDK4-cyclin D1 and downstream proteins in U251 cells after 24-h incubation with ent-4g. (F) Western blotting and analysis of MDM2, P53 and apoptosis-related proteins in U251 cells after 24-h incubation with ent-4g. All values are the average of three experiments. NS, normal saline.
The compound treatment triggered an increase in MDM2, P53 and P21 levels (Fig. 4E and F). Like palbociclib, ent-4g inhibited autophosphorylation of CDK4 and phosphorylation of retinoblastoma (RB) in U251 cells (Fig. 4E). In fact, the compound stimulated BAX to a greater extent than nutlin-3a did, and it activated more cleavage of caspase-3 than palbociclib did.
To complement these in vitro assays, we treated U251 glioblastoma xenografts in mice with ent-4g. Animals were analysed at 21 days after oral administration of ent-4g, nutlin-3a or palbociclib (Fig. 5A–C). All treatments potently inhibited tumor growth, with ent-4g showing significantly greater effects than the reference drugs. This anti-tumor activity was associated with the up-regulated expression of MDM2, P53 and P21, as well as phosphorylation inhibition of CDK4 and RB (Fig. 5D and E). Treatment with ent-4g was also associated with significantly reduced Ki-67, which serves as a proliferation marker with prognostic and predictive potential in glioblastoma, and a significantly higher number of TUNEL-positive apoptotic nuclei. Despite these anti-tumor effects of ent-4g, hematoxylin and eosin staining of tissue sections from main organs after treatment indicated no severe toxic effects (Supporting Information Fig. S6).
Figure 5.

In vivo anti-cancer effect of ent-4g on U251 xenografts in mice. (A) Tumor volume after administration of ent-4g (30 mg/kg), nutlin-3a (30 mg/kg) or palbociclib (30 mg/kg). (B) Tumor weight at the end of therapy. (C) Body weight at different times after treatment. (D) Representative images of immunohistochemical analysis of MDM2, P53, P21, pCDK4, pRB, and Ki-67, as well as of immunofluorescence analysis of TUNEL staining for apoptosis. (E) Percentages of cells staining positively for target proteins or TUNEL stain. NS, normal saline; N + P, nutlin-3a + palbociclib. Data were expressed as mean ± SD, ∗P < 0.05, ∗∗P < 0.01.
Moreover, compound ent-4g displayed good stability in human liver microsomes assay, with over 90% of ent-4g remained after 10 min incubation of 1 mg/mL proteins at 37 °C, and its half-life period in human liver microsomes assay was 46.5 min. The tumor and plasma concentrations of compound ent-4g in mice xenograft models were measured after four daily dosage of i.p. administration (30 mg/kg per day). The results in Supporting Information Fig. S7 reveal the enhanced tumor exposure of ent-4g compared to plasma. The low plasma concentration of ent-4g (lower than 100 nmol/L after 1-h administration) suggested a potential low toxicity profile of ent-4g in vivo. The pharmacokinetic studies of p.o. administration ent-4g in rats (Table 3) indicated that ent-4g distributed well into tissues (apparent Vss of 7.36 L/kg) with a moderate plasma clearance rate (1.21 L·kg/h) after i.v. injection of 7.5 mg/kg dosage, and the absolute oral bioavailability of ent-4g was around 30%.
Table 3.
Pharmacokinetic properties of ent-4g in ratsa.
| i.v. (rat, 7.5 mg/kg) |
p.o. (rat, 15 mg/kg) |
||||||
|---|---|---|---|---|---|---|---|
| CL (L·kg/h) |
t1/2 (h) |
AUC0–t (μg/h·L) |
Vss (L/kg) |
Tmax (h) |
Cmax (μg/L) |
AUC0–t (μg/h·L) |
F |
| 1.21 | 1.85 | 21,092.4 | 7.36 | 4.03 | 5897.5 | 12,463.8 | 29.5% |
Male SD rats (6–8 weeks old, 3 animals per group) were used for pharmacokinetics study.
3. Conclusions
In summary, we have discovered THN-fused spirooxindole derivative ent-4g as a potent inhibitor through rational drug design and asymmetric synthesis of the designed compounds. The compound ent-4g showed strong ability to inhibit both MDM2 and CDK4 in glioblastoma cells expressing wild-type or mutant P53. Molecular dynamics simulations indicate that the compound ent-4g tightly binds to MDM2 and CDK4. Ent-4g could induce significant apoptosis and cell cycle arrest in G1 phase by up-regulating MDM2, P53 and P21 levels, reduced Ki-67, phosphorylation inhibition of CDK4 and RB, as well as higher number of TUNEL-positive apoptotic nuclei. The compound also strongly inhibited the growth of glioblastoma xenografts in mice. The approach presented here may be useful for discovering novel MDM2/CDK4 dual inhibitors and generating leads for the treatment of glioblastoma and many other cancers.
4. Experimental
4.1. Chemistry
Nuclear Magnetic Resonance (NMR, Bruker-400 MHz, Bruker Corporation, Karlsruhe, Germany and JEOL-600 MHz, JEOL, Tokyo, Japan) data were obtained for 1H at 400 MHz and for 13C at 100 MHz or for 1H at 600 MHz and for 13C at 150 MHz. Chemical shifts were reported in parts per million (ppm) with tetramethylsilane resonance as the internal standard in CDCl3 or DMSO-d6 solution. Data are reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br s = broad singlet), coupling constant(s) (Hz), integration]. ESI high resolution mass spectra (HR-MS) were recorded using electrospray ionization on a Waters SYNAPT G2 (Q-TOF) instrument (Milford, MA, USA). The enantiomeric ratio was determined by High Performance Liquid Chromatography (HPLC, Dionex, Sunnyvale, CA, USA, and Shimadzu, Kyoto, Japan) analysis on chiral column in comparison with authentic racemates, using the Daicel Chiralpak AD/OD/IE (250 mm × 4.6 mm). UV detection was monitored at 254 nm. Purity of compounds 4 and 4′ was determined by reverse-phase HPLC analysis to be >95% at 254 nm. HPLC instrument: Dionex Summit HPLC (column: Thermo Scientific Technologies, Hypersil GOLD™, 5 μm, 250 mm × 4.6 mm), detector: PDA-100 photodiode array, injector: ASI-100 auto sample injector, pump: p-680A LPG-4. A flow rate of 1.0 mL/min was used with mobile phase of MeOH in H2O. Optical rotation data were examined in CH2Cl2 solution at 25 °C and λ = 589 nm. Column chromatography was performed on a silica gel (200–300 mesh) using an eluent of ethyl acetate and petroleum ether. TLC was performed on glass backed silica plates; products were visualized using UV light. Melting points were determined on a Mel-Temp apparatus (Shanghai, China).
4.1.1. General procedure for the asymmetric synthesis of 4a
The reaction was carried out with 3-ylideneoxidole (1a, 95.2 mg, 0.3 mmol), 2-methyl-3,5-dinitrobenzaldehyde (3a, 75.7 mg, 0.36 mmol) and cat. (24.8 mg, 0.06 mmol) with 4 Å MS in anhydrous CH2Cl2 (4.0 mL) at −10 °C for 48 h under N2. The reaction mixture was direct purified by flash chromatography on a silica gel to afford the unprotected intermediate.
The protection hydroxyl group of the intermediate gave the corresponding easily separable THN-fused spirooxindole derivative int-4a. To a solution of the unprotected intermediate in CH2Cl2 (4 mL) was added TMSCl (25.9 μL, 0.3 mmol) and imidazole (45.8 mg, 0.6 mmol). The mixture was stirred at 0 °C until the reaction was completed based on TLC. The reaction was quenched with aqueous NaHCO3 (aq.) and CH2Cl2. The organic layer was dried by Na2SO4 and concentrated. The residue was purified by chromatography on silica gel to give the major isomer product int-4a.
Next, to a solution of int-4a (50 mg) in CH2Cl2 was added HCl/EtOAc (5–10 mL) at room temperature until the reaction was completed based on TLC. The reaction was quenched by EtOAc and water. The organic layer was dry by Na2SO4 and concentrated, which was purified by chromatography on silica gel to give the deprotected spiro-oxindole derivative 4a as a white solid in 84% yield (30.1 mg, 0.07 mmol) after flash chromatography, = +58.26 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.43 (s, 1H, NH), 8.68 (s, 1H, ArH), 8.46 (s, 1H, ArH), 7.31 (d, J = 7.6 Hz, 1H, ArH), 7.22 (t, J = 7.6 Hz, 1H, ArH), 6.95 (t, J = 7.6 Hz, 1H, ArH), 6.84 (d, J = 7.6 Hz, 1H, ArH), 6.40 (d, J = 6.4 Hz, 1H, CHOH), 5.11 (d, J = 6.0 Hz, 1H, CHOH), 3.64–3.55 (m, 3H, CH3CH2, COCH), 3.40 (dd, J = 16.4, 12.0 Hz, 1H, H of CH2), 3.06 (dd, J = 11.6, 4.4 Hz, 1H, H of CH2), 0.66 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (100 MHz, DMSO-d6): δ 178.4, 171.2, 148.3, 146.0, 145.5, 144.1, 138.9, 131.3, 128.9, 124.2, 123.2, 121.6, 118.3, 109.5, 71.6, 60.5, 55.0, 46.4, 25.7, 13.7; HR-MS (ESI): m/z Calcd. for C20H17N3O8Na [M+Na]+: 450.0913, Found 450.0916. HPLC analysis: MeOH/H2O (60:40), 11.27 min, HPLC purity 99.7%.
The compounds 4b–4q were prepared according to the synthetic method of 4a.
4.1.1.1. Ethyl (1′R,3R,3′S)-5-fluoro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4b)
White solid, 85% yield (30.8 mg, 0.07 mmol), = –18.61 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.60 (s, 1H, NH), 8.72 (s, 1H, ArH), 8.43 (s, 1H, ArH), 7.07 (td, J = 9.6, 2.4 Hz, 1H, ArH), 6.94 (dd, J = 9.0, 3.0 Hz, 1H, ArH), 6.83 (dd, J = 8.4, 4.2 Hz, 1H, ArH), 6.54 (d, J = 6.6 Hz, 1H, CHOH), 4.78 (d, J = 6.6 Hz, 1H, CHOH), 3.91−3.84 (m, 2H, CH3CH2), 3.61−3.56 (m, 2H, COCH, H of CH2), 3.48−3.42 (m, 1H, H of CH2), 0.87 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 176.4, 171.3, 157.3 (d, JCF = 234.2 Hz), 148.0, 145.4, 142.1, 138.6, 137.3, 131.2 (d, JCF = 8.3 Hz), 127.6, 118.7, 114.6 (d, JCF = 23.0 Hz), 113.7 (d, JCF = 24.8 Hz), 109.6 (d, JCF = 7.7 Hz), 68.7, 60.3, 52.2, 41.2, 25.5, 13.3; HR-MS (ESI): m/z Calcd. for C20H16N3O8FNa [M+Na]+: 468.0819, Found 468.0821. HPLC analysis: MeOH/H2O (60:40), 12.80 min, HPLC purity 97.5%.
4.1.1.2. Ethyl (1′R,3R,3′S)-7-fluoro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4c)
White solid, 82% yield (29.5 mg, 0.07 mmol), = +18.87 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.23 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.38 (s, 1H, ArH), 8.19 (s, 1H, CHOH), 7.09 (t, J = 7.8 Hz, 1H, ArH), 6.87 (d, J = 6.6 Hz, 1H, ArH), 6.67 (td, J = 7.8, 4.8 Hz, 1H, ArH), 6.25 (d, J = 7.8 Hz, 1H, CHOH), 5.35 (d, J = 7.2 Hz, 1H, COCH), 4.15−4.10 (m, 2H, CH3CH2), 3.40−3.30 (m, 2H, CH2), 1.16 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.8, 163.8, 147.6, 146.7, 146.3 (d, JCF = 240.8 Hz), 143.1, 136.8, 130.3, 129.9 (d, JCF = 12.3 Hz), 129.7, 128.5 (d, JCF = 2.1 Hz), 123.4, 121.7 (d, JCF = 5.1 Hz), 119.5, 119.2, 116.0 (d, JCF = 17.1 Hz), 72.0, 61.5, 56.1, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8FNa [M+Na]+: 468.0819, Found 468.0822. HPLC analysis: MeOH/H2O (60:40), 14.20 min, HPLC purity 99.3%.
4.1.1.3. Ethyl (1′R,3R,3′S)-5-chloro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4d)
White solid, 87% yield (31.6 mg, 0.07 mmol), ee 99%, = –75.99 (c 0.10 in CH2Cl2), ent-4d: +67.89, m.p.>220 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.48 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.53 (s, 1H, ArH), 7.60 (d, J = 2.0 Hz, 1H, ArH), 7.26 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 6.79 (d, J = 8.0 Hz, 1H, ArH), 6.62 (d, J = 6.8 Hz, 1H, CHOH), 5.25 (d, J = 6.8 Hz, 1H, CHOH), 3.90−3.72 (m, 4H, CH3CH2, COCH, H of CH2), 3.33 (dd, J = 15.2, 2.4 Hz, 1H, H of CH2), 0.83 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (100 MHz, DMSO-d6): δ 176.3, 171.1, 148.3, 146.2, 144.3, 142.9, 137.4, 133.2, 128.6, 125.7, 125.4, 124.2, 118.6, 110.7, 72.1, 60.8, 52.8, 44.6, 26.3, 13.9; HR-MS (ESI): m/z Calcd. for C20H16N3O8ClNa [M+Na]+: 484.0524, Found 484.0526. HPLC analysis: MeOH/H2O (60:40), 9.80 min, HPLC purity 99.7%. (ent-4d, 9.67 min, HPLC purity 98.3%).
4.1.1.4. Ethyl (1′R,3R,3′S)-6-chloro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4e)
White solid, 86% yield (31.2 mg, 0.07 mmol), = –76.28 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.73 (s, 1H, NH), 8.72 (s, 1H, ArH), 8.42 (s, 1H, ArH), 7.08 (d, J = 7.8 Hz, 1H, ArH), 6.99 (dd, J = 7.8, 1.8 Hz, 1H, ArH), 6.85 (d, J = 1.8 Hz, 1H, ArH), 6.52 (d, J = 6.6 Hz, 1H, CHOH), 4.78 (d, J = 6.6 Hz, 1H, CHOH), 3.92−3.85 (m, 2H, CH3CH2), 3.60−3.53 (m, 2H, COCH, H of CH2), 3.45 (dd, J = 15.0, 3.0 Hz, 1H, H of CH2), 0.90 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 176.7, 171.4, 148.1, 145.5, 144.0, 142.3, 137.4, 132.8, 128.6, 127.6, 127.4, 120.7, 118.9, 109.1, 68.8, 60.5, 51.7, 41.5, 25.7, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8ClNa [M+Na]+: 484.0524, Found 484.0523. HPLC analysis: MeOH/H2O (60:40), 14.07 min, HPLC purity 99.4%.
4.1.1.5. Ethyl (1′R,3R,3′S)-4-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4f)
White solid, 89% yield (33.2 mg, 0.07 mmol), = +42.31 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.65 (s, 1H, NH), 8.66 (s, 1H, ArH), 8.54 (s, 1H, ArH), 7.20−7.15 (m, 2H, ArH), 6.81 (t, J = 6.6 Hz, 2H, ArH, CHOH), 5.61 (d, J = 6.6 Hz, 1H, CHOH), 4.13 (dd, J = 11.4, 6.0 Hz, 1H, COCH), 3.87−3.76 (m, 3H, CH3CH2, H of CH2), 3.31 (dd, J = 18.0, 6.0 Hz, 1H, H of CH2), 0.83 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 174.9, 170.4, 147.6, 145.8, 145.5, 143.4, 137.2, 130.6, 127.0, 125.3, 124.9, 118.0, 117.9, 108.5, 67.9, 60.3, 54.3, 41.6, 25.3, 13.2; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0020. HPLC analysis: MeOH/H2O (60:40), 16.87 min, HPLC purity 98.6%.
4.1.1.6. Ethyl (1′R,3R,3′S)-5-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4g)
White solid, 88% yield (32.8 mg, 0.06 mmol), ee 95%, = –99.97 (c 0.10 in CH2Cl2), ent-4g: +109.98, m.p.>220 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.48 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.52 (s, 1H, ArH), 7.72 (d, J = 2.0 Hz, 1H, ArH), 7.39 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 6.75 (d, J = 8.4 Hz, 1H, ArH), 6.63 (d, J = 6.8 Hz, 1H, CHOH), 5.25 (d, J = 6.8 Hz, 1H, CHOH), 3.90−3.71 (m, 4H, CH3CH2, COCH, H of CH2), 3.32 (dd, J = 16.0, 3.2 Hz, 1H, H of CH2), 0.83 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (100 MHz, DMSO-d6): δ 176.2, 171.1, 148.3, 146.2, 144.3, 143.2, 137.4, 133.5, 131.4, 126.9, 125.4, 118.5, 113.3, 111.2, 72.1, 60.8, 52.8, 44.6, 26.2, 13.9; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0019. HPLC analysis: MeOH/H2O (60:40), 10.73 min, HPLC purity 98.5%. (ent-4g, 10.73 min, HPLC purity 98.4%).
4.1.1.7. Ethyl (1′R,3R,3′S)-6-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4h)
White solid, 86% yield (32.1 mg, 0.06 mmol), = –50.22 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.87 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.38 (s, 1H, ArH), 8.20 (s, 1H, CHOH), 6.98 (d, J = 1.8 Hz, 1H, ArH), 6.84−6.82 (m, 2H, ArH), 6.34 (d, J = 7.8 Hz, 1H, CHOH), 5.33 (d, J = 7.2 Hz, 1H, COCH), 4.16−4.09 (m, 2H, CH3CH2), 3.41−3.31 (m, 2H, CH2), 1.17 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.8, 163.8, 147.5, 146.8, 144.7, 143.0, 136.7, 130.3, 129.8, 124.9, 123.4, 121.6, 119.5, 112.3, 71.8, 61.5, 55.6, 30.9, 22.0, 13.6; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0020. HPLC analysis: MeOH/H2O (60:40), 32.13 min, HPLC purity 98.9%.
4.1.1.8. Ethyl (1′R,3R,3′S)-1′-hydroxy-5,5′,7′-trinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4i)
White solid, 84% yield (30.8 mg, 0.06 mmol), = –24.57 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.15 (s, 1H, NH), 8.69 (s, 1H, ArH), 8.55 (d, J = 2.4 Hz, 1H, ArH), 8.53 (s, 1H, ArH), 8.24 (dd, J = 8.4, 2.4 Hz, 1H, ArH), 7.00 (d, J = 8.4 Hz, 1H, ArH), 6.72 (d, J = 6.6 Hz, 1H, CHOH), 5.40 (d, J = 6.6 Hz, 1H, CHOH), 4.03 (dd, J = 12.0, 6.0 Hz, 1H, COCH), 3.87−3.82 (m, 2H, CH3CH2), 3.74 (dd, J = 18.0, 12.0 Hz, 1H, H of CH2), 3.42 (dd, J = 18.6, 6.0 Hz, 1H, H of CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 176.5, 170.5, 150.0, 147.6, 145.6, 143.1, 141.7, 136.5, 131.6, 125.8, 124.7, 119.3, 118.1, 108.8, 71.3, 60.4, 52.1, 43.5, 25.5, 13.2; HR-MS (ESI): m/z Calcd. for C20H16N4O10Na [M+Na]+: 495.0764, Found 495.0763. HPLC analysis: MeOH/H2O (60:40), 6.80 min, HPLC purity 97.9%.
4.1.1.9. Ethyl (1′R,3R,3′S)-1′-hydroxy-5-methyl-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4j)
White solid, 83% yield (29.8 mg, 0.07 mmol), = –62.96 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.25 (s, 1H, NH), 8.66 (s, 1H, ArH), 8.52 (s, 1H, ArH), 7.24 (s, 1H, ArH), 7.01 (d, J = 7.8 Hz, 1H, ArH), 6.67 (d, J = 7.8 Hz, 1H, ArH), 6.53 (d, J = 7.2 Hz, 1H, CHOH), 5.18 (d, J = 7.2 Hz, 1H, CHOH), 3.82−3.75 (m, 3H, CH3CH2, COCH), 3.70 (dd, J = 11.4, 4.8 Hz, 1H, H of CH2), 3.27 (dd, J = 17.4, 4.8 Hz, 1H, H of CH2), 2.28 (s, 3H, CH3), 0.78 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 179.1, 164.0, 147.5, 146.7, 143.5, 140.6, 137.7, 130.0, 129.9, 129.7, 129.3, 125.7, 123.7, 123.5, 119.6, 109.4, 72.0, 61.4, 56.0, 20.5, 13.6; HR-MS (ESI): m/z Calcd. for C21H19N3O8Na [M+Na]+: 464.1070, Found 464.1071. HPLC analysis: MeOH/H2O (60:40), 8.73 min, HPLC purity 98.1%.
4.1.1.10. (1′R,3R,3′S)-3′-Benzoyl-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4k)
White solid, 87% yield (31.2 mg, 0.07 mmol), = +35.53 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.52 (s, 1H, NH), 8.72 (s, 1H, ArH), 8.48 (s, 1H, ArH), 7.86 (d, J = 7.2 Hz, 2H, ArH), 7.63 (t, J = 7.2 Hz, 1H, ArH), 7.49 (t, J = 7.2 Hz, 2H, ArH), 7.14−7.11 (m, 1H, ArH), 7.09 (d, J = 7.2 Hz, 1H, ArH), 6.79 (d, J = 7.8 Hz, 1H, ArH), 6.76 (t, J = 7.2 Hz, 1H, ArH), 6.49 (s, 1H, CHOH), 4.90 (dd, J = 11.4, 6.0 Hz, 1H, COCH), 4.71 (s, 1H, CHOH), 3.55 (dd, J = 18.0, 12.0 Hz, 1H, H of CH2), 3.48 (dd, J = 18.0, 5.4 Hz, 1H, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 199.7, 177.1, 147.9, 145.4, 142.5, 142.4, 137.8, 135.6, 133.7, 129.7, 128.8 (2C), 128.2 (2C), 128.0, 125.5, 120.6, 118.6, 108.9, 69.2, 51.3, 41.8, 26.7; HR-MS (ESI): m/z Calcd. for C24H17N3O7Na [M+Na]+: 482.0964, Found 482.0966. HPLC analysis: MeOH/H2O (60:40), 13.53 min, HPLC purity 98.7%.
4.1.1.11. (1′R,3R,3′S)-3′-(2-Fluorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4l)
White solid, 83% yield (30.5 mg, 0.06 mmol), = +40.28 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.57 (s, 1H, ArH), 7.61−7.57 (m, 1H, ArH), 7.53 (td, J = 7.8, 1.2 Hz, 1H, ArH), 7.29−7.21 (m, 3H, ArH), 7.10 (td, J = 7.8, 1.2 Hz, 1H, ArH), 6.78−6.73 (m, 2H, ArH), 6.54 (d, J = 7.2 Hz, 1H, CHOH), 5.33 (d, J = 7.2 Hz, 1H, CHOH), 4.78 (dd, J = 12.0, 5.4 Hz, 1H, COCH), 3.77 (dd, J = 18.6, 12.2 Hz, 1H, H of CH2), 3.36 (dd, J = 18.0, 5.4 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.8, 176.5, 160.1 (d, JCF = 253.1 Hz), 147.9, 145.7, 144.5, 143.5, 137.3, 135.1 (d, JCF = 8.7 Hz), 130.4, 130.2, 128.1, 125.7 (d, JCF = 11.1 Hz), 125.1, 124.7, 122.6, 120.8, 118.0, 116.7 (d, JCF = 21.9 Hz), 108.8, 72.1, 51.9, 48.9, 26.0; HR-MS (ESI): m/z Calcd. for C24H16N3O7FNa [M+Na]+: 500.0870, Found 500.0868. HPLC analysis: MeOH/H2O (60:40), 10.07 min, HPLC purity 99.7%.
4.1.1.12. (1′R,3R,3′S)-3′-(4-Fluorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4m)
White solid, 85% yield (31.4 mg, 0.07 mmol), = +29.87 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.60 (s, 1H, ArH), 8.04−8.01 (m, 2H, ArH), 7.37 (d, J = 7.2 Hz, 1H, ArH), 7.31 (t, J = 8.4 Hz, 2H, ArH), 7.07 (t, J = 7.8 Hz, 1H, ArH), 6.78 (t, J = 7.8 Hz, 1H, ArH), 6.73 (d, J = 7.2 Hz, 1H, ArH), 5.34 (s, 1H, CHOH), 5.09 (dd, J = 12.0, 4.8 Hz, 1H, COCH), 3.69 (dd, J = 17.4, 12.6 Hz, 1H, H of CH2), 3.51 (s, 1H, CHOH), 3.33 (dd, J = 18.0, 4.8 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.6, 176.9, 165.4 (d, JCF = 251.4 Hz), 147.8, 145.7, 144.6, 143.7, 137.7, 132.3, 131.7 (d, JCF = 9.3 Hz, 2C), 131.0, 128.0, 125.2, 122.4, 120.8, 118.0, 115.9 (d, JCF = 21.6 Hz, 2C), 108.8, 72.3, 51.6, 45.0, 26.9; HR-MS (ESI): m/z Calcd. for C24H16N3O7FNa [M+Na]+: 500.0870, Found 500.0869. HPLC analysis: MeOH/H2O (60:40), 8.40 min, HPLC purity 99.2%.
4.1.1.13. (1′R,3R,3′S)-3′-(3,4-Dichlorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4n)
White solid, 87% yield (32.8 mg, 0.06 mmol), = +21.42 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.59 (s, 1H, ArH), 8.15 (d, J = 1.8 Hz, 1H, ArH), 7.79 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.74 (d, J = 8.4 Hz, 1H, ArH), 7.38 (d, J = 7.2 Hz, 1H, ArH), 7.08 (t, J = 7.2 Hz, 1H, ArH), 6.79 (t, J = 7.8 Hz, 1H, ArH), 6.73 (d, J = 7.8 Hz, 1H, ArH), 6.55 (d, J = 6.6 Hz, 1H, CHOH), 5.34 (d, J = 7.2 Hz, 1H, CHOH), 5.07 (dd, J = 12.0, 4.8 Hz, 1H, COCH), 3.70 (dd, J = 18.0, 12.0 Hz, 1H, H of CH2), 3.38 (dd, J = 18.0, 6.0 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.4, 176.6, 147.8, 145.7, 144.5, 143.5, 137.6, 136.7, 135.9, 132.0, 131.1, 130.6, 130.5, 128.3, 128.2, 125.1, 122.6, 120.9, 118.0, 108.8, 72.1, 51.7, 45.2, 26.5; HR-MS (ESI): m/z Calcd. for C24H15N3O7Cl2Na [M+Na]+ 550.0185, Found 550.0186. HPLC analysis: MeOH/H2O (60:40), 26.93 min, HPLC purity 99.1%.
4.1.1.14. (1′R,3R,3′S)-3′-(4-Bromobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4o)
White solid, 88% yield (33.2 mg, 0.06 mmol), = +27.73 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.37 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.59 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 2H, ArH), 7.69 (d, J = 8.4 Hz, 2H, ArH), 7.36 (d, J = 7.2 Hz, 1H, ArH), 7.08 (t, J = 7.8 Hz, 1H, ArH), 6.78 (t, J = 7.8 Hz, 1H, ArH), 6.73 (d, J = 7.2 Hz, 1H, ArH), 6.54 (d, J = 7.2 Hz, 1H, CHOH), 5.33 (d, J = 7.2 Hz, 1H, CHOH), 5.07 (dd, J = 12.0, 4.8 Hz, 1H, COCH), 3.69 (dd, J = 18.0, 12.0 Hz, 1H, H of CH2), 3.34 (dd, J = 18.0, 5.4 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 198.2, 176.6, 147.6, 145.3, 144.4, 143.5, 137.5, 134.4, 131.8, 130.7, 130.4, 128.0, 127.9, 125.0, 122.2, 120.7, 117.8, 108.6, 72.1, 51.4, 44.9, 26.6; HR-MS (ESI): m/z Calcd. for C24H16N3O7BrNa [M+Na]+: 560.0069, Found 560.0067. HPLC analysis: MeOH/H2O (60:40), 15.60 min, HPLC purity 99.9%.
4.1.1.15. (1′R,3R,3′S)-1′-Hydroxy-3′-(4-methoxybenzoyl)-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4p)
White solid, 86% yield (31.8 mg, 0.06 mmol), = +30.99 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.32 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.59 (s, 1H, ArH), 7.94 (d, J = 9.0 Hz, 2H, ArH), 7.34 (d, J = 7.2 Hz, 1H, ArH), 7.07 (t, J = 7.8 Hz, 1H, ArH), 6.99 (d, J = 9.0 Hz, 2H, ArH), 6.79 (t, J = 7.8 Hz, 1H, ArH), 6.72 (d, J = 7.8 Hz, 1H, ArH), 6.51 (d, J = 6.6 Hz, 1H, CHOH), 5.32 (d, J = 7.2 Hz, 1H, CHOH), 5.03 (dd, J = 12.0, 5.4 Hz, 1H, COCH), 3.82 (s, 3H, OCH3), 3.67 (dd, J = 18.0, 12.6 Hz, 1H, H of CH2), 3.28 (dd, J = 18.0, 5.4 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.1, 177.1, 163.7, 147.8, 145.7, 144.7, 143.8, 137.9, 131.3, 131.1 (2C), 128.3, 127.9, 125.1, 122.2, 120.7, 118.0, 114.1 (2C), 108.7, 72.4, 55.7, 51.5, 44.7, 27.3; HR-MS (ESI): m/z Calcd. for C25H19N3O8Na [M+Na]+: 512.1070, Found 512.1068. HPLC analysis: MeOH/H2O (60:40), 7.67 min, HPLC purity 99.0%.
4.1.2. General procedure for the asymmetric synthesis of 4a′106
The reaction was carried out with 3-ylideneoxidole (2a, 92.2 mg, 0.3 mmol), 2-methylbenzaldehyde (3a, 75.7 mg, 0.36 mmol) and cat. (24.8 mg, 0.06 mmol) with 4 Å MS in anhydrous CH2Cl2 (4.0 mL) at −10 °C for 7 days under N2. The reaction mixture was direct purified by flash chromatography on a silica gel to afford the unprotected intermediate.
The protection hydroxyl group of the unprotected intermediate gave the corresponding easily separable THN-fused spirooxindole derivative int-4a′. To a solution of the unprotected intermediate in CH2Cl2 (4 mL) was added TMSCl (25.9 μL, 0.3 mmol) and imidazole (45.8 mg, 0.6 mmol). The mixture was stirred at 0 °C until the reaction was completed based on TLC. The reaction was quenched with aqueous NaHCO3 (aq) and CH2Cl2. The organic layer was dried by Na2SO4 and concentrated. The residue was purified by chromatography on silica gel to give the major isomer product int-4a′.
Next, To a solution of int-4a′ (50 mg) in CH2Cl2 was added CF3COOH (5 eq.) at room temperature until the reaction was completed based on TLC. The reaction was quenched by EtOAc and water. The organic layer was dry by Na2SO4 and concentrated, which was purified by chromatography on silica gel to give the deprotection TMS intermediate product.
Further, potassium tert-butoxide (1.03 mL of a 1 mol/L solution in THF) was added to an aerated solution of the Bn-protecting THN-fused spirooxindole derivatives intermediate product, in DMSO (3.0 mL) at room temperature. After 20 min, 1 mol/L HCl was added and further followed by sodium hydrogen carbonate solution to give a pH neutral solution. The solution was then diluted with brine, extracted with ethyl acetate (4 × 20 mL) and evaporated under reduced pressure105. The residue was purified by flash chromatography to give the deprotected spiro-oxindole derivative 4a′ as a white solid in 76% yield (27.7 mg, 0.06 mmol) after flash chromatography, = +70.25 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.60 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.44 (s, 1H, ArH), 7.11 (t, J = 8.4 Hz, 1H, ArH), 6.81 (d, J = 7.8 Hz, 1H, ArH), 6.65−6.62 (m, 2H, ArH), 6.06 (d, J = 7.2 Hz, 1H, CHOH), 5.06 (d, J = 6.6 Hz, 1H, CHOH), 3.77 (q, J = 7.2 Hz, 2H, CH3CH2), 3.70−3.62 (m, 2H, COCH, H of CH2), 3.55 (dd, J = 18.8, 7.2 Hz, 1H, H of CH2), 0.85 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.6, 170.3, 148.2, 145.8, 144.5, 143.9, 136.6, 128.2, 126.7, 124.2, 124.0, 120.8, 118.6, 109.2, 71.3, 60.5, 53.9, 43.1, 25.3, 13.4; HR-MS (ESI): m/z Calcd. for C20H17N3O8Na [M+Na]+: 450.0913, Found 450.0914. HPLC analysis: MeOH/H2O (60:40), 11.80 min, HPLC purity 99.0%.
The compounds 4b′–4q′ was prepared according to the synthetic method of 4a′.
4.1.2.1. Ethyl (1′S,3S,3′S)-5-fluoro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4b′)
White solid, 74% yield (27.2 mg, 0.06 mmol), = +104.87 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.38 (s, 1H, NH), 8.67 (s, 1H, ArH), 8.53 (s, 1H, ArH), 7.42 (dd, J = 9.0, 3.0 Hz, 1H, ArH), 7.06−7.01 (m, 1H, ArH), 6.76 (dd, J = 8.4, 4.8 Hz, 1H, ArH), 6.60 (d, J = 6.6 Hz, 1H, CHOH), 5.24 (d, J = 7.2 Hz, 1H, CHOH), 3.86−3.81 (m, 2H, CH3CH2), 3.79−3.74 (m, 2H, COCH, H of CH2), 3.33−3.30 (m, 1H, H of CH2), 0.82 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 176.0, 170.6, 157.9 (d, JCF = 234.3 Hz), 147.8, 145.7, 143.9, 139.7, 137.0, 132.4 (d, JCF = 8.4 Hz), 124.9, 118.1, 114.4 (d, JCF = 23.0 Hz), 111.4 (d, JCF = 24.6 Hz), 109.5 (d, JCF = 7.8 Hz), 71.7, 60.3, 52.5, 44.2, 25.8, 13.4; HR-MS (ESI): m/z Calcd. for C20H16N3O8FNa [M+Na]+: 468.0819, Found 468.0822. HPLC analysis: MeOH/H2O (60:40), 8.40 min, HPLC purity 99.8%.
4.1.2.2. Ethyl (1′S,3S,3′S)-7-fluoro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4c′)
White solid, 72% yield (26.4 mg, 0.06 mmol), = +68.26 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.14 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.44 (s, 1H, ArH), 7.06 (t, J = 9.6 Hz, 1H, ArH), 6.75 (d, J = 6.6 Hz, 1H, ArH), 6.69−6.66 (m, 1H, ArH), 5.97 (d, J = 7.8 Hz, 1H, CHOH), 5.08 (d, J = 6.6 Hz, 1H, CHOH), 3.86−3.78 (m, 2H, CH3CH2), 3.73 (t, J = 8.4 Hz, 1H, COCH), 3.62 (d, J = 8.4 Hz, 2H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.4, 170.0, 148.1, 146.3 (d, JCF = 240.0 Hz), 145.7, 144.1, 136.3, 130.9 (d, JCF = 12.3 Hz), 129.8 (d, JCF = 3.5 Hz), 124.2, 121.6 (d, JCF = 5.6 Hz), 120.0, 118.6, 115.3 (d, JCF = 16.8 Hz), 71.3, 60.5, 54.2, 42.9, 25.2, 13.3; HR-MS (ESI): m/z Calcd. for C20H16N3O8FNa [M+Na]+: 468.0819, Found 468.0820. HPLC analysis: MeOH/H2O (60:40), 10.27 min, HPLC purity 98.8%.
4.1.2.3. Ethyl (1′S,3S,3′S)-5-chloro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4d′)
White solid, 76% yield (28.1 mg, 0.06 mmol), ee 95%, = +48.72 (c 0.10 in CH2Cl2), ent-4d′: –37.99, m.p.>220 °C. 1H NMR (400 MHz, CDCl3): δ 9.05 (s, 1H, NH), 8.82 (s, 1H, ArH), 8.67 (s, 1H, ArH), 7.15 (dd, J = 8.4, 2.0 Hz, 1H, ArH), 6.84 (d, J = 8.4 Hz, 1H, ArH), 6.33 (d, J = 2.0 Hz, 1H, ArH), 5.14 (d, J = 7.6 Hz, 1H, CHOH), 4.79 (d, J = 7.6 Hz, 1H, CHOH), 4.09−3.97 (m, 2H, CH3CH2), 3.79−3.77 (m, 2H, COCH, H of CH2), 3.61 (dd, J = 10.4, 8.0 Hz, 1H, H of CH2), 1.12 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (100 MHz, DMSO-d6): δ 178.4, 170.1, 148.3, 145.9, 143.9, 143.0, 136.2, 128.9, 128.3, 124.8, 124.5, 123.9, 118.8, 110.6, 71.3, 60.7, 54.1, 42.9, 25.3, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8ClNa [M+Na]+: 484.0524, Found 484.0523. HPLC analysis: MeOH/H2O (60:40), 16.33 min, HPLC purity 98.8%. (ent-4dʹ, 16.27 min, HPLC purity 99.2%)
4.1.2.4. Ethyl (1′S,3S,3′S)-6-chloro-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4e′)
White solid, 75% yield (27.9 mg, 0.06 mmol), = +71.44 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.44 (s, 1H, ArH), 6.83 (d, J = 1.8 Hz, 1H, ArH), 6.71−6.69 (m, 2H, ArH), 6.11 (d, J = 7.8 Hz, 1H, CHOH), 5.06 (d, J = 6.6 Hz, 1H, CHOH), 3.85−3.80 (m, 2H, CH3CH2), 3.71 (dd, J = 9.6, 7.8 Hz, 1H, COCH), 3.64–3.58 (m, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.7, 170.2, 148.2, 145.9, 145.6, 144.1, 136.5, 132.7, 125.8, 125.4, 124.4, 120.5, 118.8, 109.3, 71.3, 60.7, 53.7, 42.9, 25.4, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8ClNa [M+Na]+: 484.0524, Found 484.0522. HPLC analysis: MeOH/H2O (60:40), 18.53 min, HPLC purity 99.9%.
4.1.2.5. Ethyl (1′S,3S,3′S)-4-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4f′)
White solid, 79% yield (30.0 mg, 0.06 mmol), = –69.97 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.65 (s, 1H, NH), 8.66 (s, 1H, ArH), 8.54 (s, 1H, ArH), 7.20−7.16 (m, 2H, ArH), 6.82−6.80 (m, 2H, ArH, CHOH), 5.61 (d, J = 6.6 Hz, 1H, CHOH), 4.13 (q, J = 5.4 Hz, 1H, COCH), 3.87−3.76 (m, 3H, CH3CH2, H of CH2), 3.31 (dd, J = 18.0, 5.4 Hz, 1H, H of CH2), 0.83 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 174.9, 170.4, 147.6, 145.8, 145.5, 143.3, 137.2, 130.6, 127.0, 125.3, 124.9, 118.0, 117.9, 108.5, 67.9, 60.3, 54.2, 41.6, 25.3, 13.2; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0020. HPLC analysis: MeOH/H2O (60:40), 16.40 min, HPLC purity 99.1%.
4.1.2.6. Ethyl (1′S,3S,3′S)-5-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4g′)
White solid, 78% yield (29.5 mg, 0.06 mmol), ee 95%, = +38.53 (c 0.10 in CH2Cl2), ent-4g′: –31.00, m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1H, NH), 8.78 (s, 1H, ArH), 8.46 (s, 1H, ArH), 7.33 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 6.79 (d, J = 8.4 Hz, 1H, ArH), 6.74 (d, J = 7.2 Hz, 1H, CHOH), 6.21 (d, J = 1.8 Hz, 1H, ArH), 5.06 (d, J = 6.6 Hz, 1H, CHOH), 3.87−3.81 (m, 2H, CH3CH2), 3.69 (dd, J = 9.6, 7.8 Hz, 1H, COCH), 3.61−3.59 (m, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.2, 170.1, 148.3, 145.9, 143.9, 143.4, 136.1, 131.2, 129.4, 126.5, 124.5, 118.8, 112.5, 111.2, 71.3, 60.7, 54.2, 42.9, 25.2, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0019. HPLC analysis: MeOH/H2O (60:40), 18.03 min, HPLC purity 99.7%. (ent-4gʹ, 18.07 min, HPLC purity 98.1%)
4.1.2.7. Ethyl (1′S,3S,3′S)-6-bromo-1′-hydroxy-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4h′)
White solid, 77% yield (29.1 mg, 0.06 mmol), = +50.85 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1H, NH), 8.78 (s, 1H, ArH), 8.44 (s, 1H, ArH), 6.95 (d, J = 1.8 Hz, 1H, ArH), 6.83 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 6.70 (d, J = 6.6 Hz, 1H, CHOH), 6.06 (d, J = 7.8 Hz, 1H, ArH), 5.05 (d, J = 6.6 Hz, 1H, CHOH), 3.86−3.80 (m, 2H, CH3CH2), 3.71 (dd, J = 9.6, 7.8 Hz, 1H, COCH), 3.61−3.59 (m, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.6, 170.2, 148.2, 145.9, 145.7, 144.1, 136.5, 126.2, 125.8, 124.3, 123.5, 121.1, 118.8, 112.0, 71.2, 60.7, 53.7, 42.9, 25.4, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N3O8BrNa [M+Na]+: 528.0018, Found 528.0016. HPLC analysis: MeOH/H2O (60:40), 21.53 min, HPLC purity 99.8%.
4.1.2.8. Ethyl (1′S,3S,3′S)-1′-hydroxy-5,5′,7′-trinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4i′)
White solid, 73% yield (27.2 mg, 0.06 mmol), = –35.26 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.43 (s, 1H, NH), 8.83 (s, 1H, ArH), 8.46 (s, 1H, ArH), 8.14 (dd, J = 9.0, 2.4 Hz, 1H, ArH), 7.05 (d, J = 9.0 Hz, 1H, ArH), 6.86–6.85 (m, 2H, ArH, CHOH), 5.14 (d, J = 6.6 Hz, 1H, CHOH), 3.87–3.81 (m, 2H, CH3CH2), 3.80 (d, J = 9.0 Hz, 1H, COCH), 3.64 (d, J = 8.4 Hz, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 179.3, 170.0, 150.8, 148.5, 146.1, 143.7, 141.4, 135.7, 127.9, 126.1, 124.5, 119.2, 118.8, 109.4, 71.4, 61.0, 54.1, 43.0, 25.2, 13.5; HR-MS (ESI): m/z Calcd. for C20H16N4O10Na [M+Na]+: 495.0764, Found 495.0766. HPLC analysis: MeOH/H2O (60:40), 9.33 min, HPLC purity 99.2%.
4.1.2.9. Ethyl (1′S,3S,3′S)-1′-hydroxy-5-methyl-5′,7′-dinitro-2-oxo-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalene]-3′-carboxylate (4j′)
White solid, 70% yield (25.7 mg, 0.06 mmol), = +68.52 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.47 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.46 (s, 1H, ArH), 6.92 (d, J = 7.8 Hz, 1H, ArH), 6.70 (d, J = 7.8 Hz, 1H, ArH), 6.58 (d, J = 6.6 Hz, 1H, CHOH), 5.90 (s, 1H, ArH), 5.04 (d, J = 6.6 Hz, 1H, CHOH), 3.81–3.76 (m, 2H, CH3CH2), 3.66–3.61 (m, 2H, COCH, H of CH2), 3.57–3.52 (m, 1H, H of CH2), 1.98 (s, 3H, CH3), 0.87 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (150 MHz, DMSO-d6): δ 178.5, 170.2, 148.2, 145.8, 144.5, 141.5, 136.5, 129.3, 128.5, 126.8, 124.7, 124.3, 118.5, 108.9, 71.3, 60.4, 53.9, 43.0, 25.3, 20.7, 13.4; HR-MS (ESI): m/z Calcd. for C21H19N3O8Na [M+Na]+: 464.1070, Found 464.1071. HPLC analysis: MeOH/H2O (60:40), 12.67 min, HPLC purity 98.5%.
4.1.2.10. (1′S,3S,3′S)-3′-Benzoyl-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4k′)
White solid, 77% yield (28.4 mg, 0.06 mmol), = +101.84 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.51 (s, 1H, NH), 8.77 (s, 1H, ArH), 8.48 (s, 1H, ArH), 7.77 (d, J = 7.8 Hz, 2H, ArH), 7.60 (t, J = 7.8 Hz, 1H, ArH), 7.44 (t, J = 7.8 Hz, 2H, ArH), 7.05 (dd, J = 8.4, 7.2 Hz, 1H, ArH), 6.69 (d, J = 7.8 Hz, 1H, ArH), 6.63 (dd, J = 15.6, 7.2 Hz, 2H, ArH), 6.06 (d, J = 7.2 Hz, 1H, CHOH), 5.21 (d, J = 6.6 Hz, 1H, CHOH), 4.77 (dd, J = 9.0, 7.2 Hz, 1H, COCH), 3.60–3.52 (m, 2H, CH2); 13C NMR (150 MHz, DMSO-d6): δ 198.6, 178.8, 148.2, 145.9, 145.1, 143.7, 137.5, 135.8, 133.5, 128.7 (2C), 128.4 (2C), 127.8, 127.5, 124.6, 124.2, 121.0, 118.7, 109.3, 71.8, 54.1, 45.8, 27.0; HR-MS (ESI): m/z Calcd. for C24H17N3O7Na [M+Na]+: 482.0964, Found 482.0967. HPLC analysis: MeOH/H2O (60:40), 18.00 min, HPLC purity 99.8%.
4.1.2.11. (1′S,3S,3′S)-3′-(2-Fluorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4l′)
White solid, 71% yield (26.5 mg, 0.06 mmol), = +87.99 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.57 (s, 1H, NH), 8.73 (s, 1H, ArH), 8.47 (s, 1H, ArH), 7.61 (dd, J = 13.8, 7.2 Hz, 1H, ArH), 7.50 (t, J = 7.8 Hz, 1H, ArH), 7.28 (dd, J = 11.4, 8.4 Hz, 1H, ArH), 7.23 (t, J = 7.8 Hz, 1H, ArH), 7.14 (t, J = 7.8 Hz, 1H, ArH), 7.05 (d, J = 7.8 Hz, 1H, ArH), 6.80 (d, J = 7.8 Hz, 1H, ArH), 6.75 (t, J = 7.8 Hz, 1H, ArH), 6.55 (d, J = 6.0 Hz, 1H, CHOH), 4.75 (d, J = 6.6 Hz, 1H, CHOH), 4.69 (dd, J = 10.8, 6.0 Hz, 1H, COCH), 3.60 (dd, J = 18.6, 11.4 Hz, 1H, H of CH2), 3.47 (dd, J = 18.6, 5.4 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 198.7, 176.8, 160.4 (d, J = 252.6 Hz), 148.2, 145.5, 142.7, 142.5, 137.7, 135.3 (d, JCF = 9.3 Hz), 130.3, 129.3, 128.3, 128.1, 125.8, 125.7 (d, JCF = 11.3 Hz), 124.8 (d, JCF = 2.0 Hz), 120.8, 118.8, 116.8 (d, JCF = 22.5 Hz), 109.2, 69.1, 51.8, 45.9, 25.9; HR-MS (ESI): m/z Calcd. for C24H16N3O7FNa [M+Na]+: 500.0870, Found 500.0872. HPLC analysis: MeOH/H2O (60:40), 13.80 min, HPLC purity 99.3%.
4.1.2.12. (1′S,3S,3′S)-3′-(4-Fluorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4m′)
White solid, 75% yield (27.8 mg, 0.06 mmol), = –129.98 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.53 (s, 1H, NH), 8.72 (s, 1H, ArH), 8.48 (s, 1H, ArH), 7.94 (dd, J = 9.0, 6.0 Hz, 2H, ArH), 7.31 (t, J = 9.0 Hz, 2H, ArH), 7.13 (t, J = 7.8 Hz, 1H, ArH), 7.08 (d, J = 7.2 Hz, 1H, ArH), 6.79 (d, J = 7.2 Hz, 1H, ArH), 6.76 (t, J = 7.8 Hz, 1H, ArH), 6.50 (d, J = 6.6 Hz, 1H, CHOH), 4.87 (dd, J = 12.0, 6.0 Hz, 1H, COCH), 4.72 (d, J = 6.6 Hz, 1H, CHOH), 3.55 (dd, J = 18.0, 11.4 Hz, 1H, H of CH2), 3.47 (dd, J = 18.0, 6.0 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 198.2, 176.9, 165.1 (d, JCF = 251.3 Hz), 147.9, 145.3, 142.4, 142.3, 137.7, 132.4, 131.2 (d, JCF = 9.5 Hz, 2C), 129.5, 128.1, 128.0, 125.4, 120.5, 118.5, 115.7 (d, JCF = 21.8 Hz, 2C), 108.8, 69.0, 51.3, 41.8, 26.5; HR-MS (ESI): m/z Calcd. for C24H16N3O7FNa [M+Na]+: 500.0870, Found 500.0868. HPLC analysis: MeOH/H2O (60:40), 12.60 min, HPLC purity 99.3%.
4.1.2.13. (1′S,3S,3′S)-3′-(3,4-Dichlorobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4n′)
White solid, 77% yield (29.3 mg, 0.06 mmol), = –157.34 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.55 (s, 1H, NH), 8.74 (s, 1H, ArH), 8.48 (s, 1H, ArH), 7.96 (s, 1H, ArH), 7.74 (br s, 2H, ArH), 7.14 (t, J = 7.8 Hz, 1H, ArH), 7.00 (d, J = 7.2 Hz, 1H, ArH), 6.77 (dd, J = 13.8, 7.8 Hz, 2H, ArH), 6.52 (d, J = 7.2 Hz, 1H, CHOH), 4.80–4.76 (m, 2H, CHOH, COCH), 3.54–3.49 (m, 2H, CH2); 13C NMR (150 MHz, DMSO-d6): δ 198.0, 176.8, 147.9, 145.3, 142.6, 142.3, 137.6, 136.4, 136.0, 131.7, 131.0, 129.9, 129.3, 128.1, 128.0, 127.6, 125.4, 120.7, 118.5, 108.9, 69.0, 51.8, 42.6, 26.1; HR-MS (ESI): m/z Calcd. for C24H15N3O7Cl2Na [M+Na]+: 550.0185, Found 550.0187. HPLC analysis: MeOH/H2O (60:40), 14.87 min, HPLC purity 99.8%.
4.1.2.14. (1′S,3S,3′S)-3′-(4-Bromobenzoyl)-1′-hydroxy-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4o′)
White solid, 78% yield (29.9 mg, 0.06 mmol), = –139.98 (c 0.10 in CH2Cl2), m.p.>220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.53 (s, 1H, NH), 8.78 (s, 1H, ArH), 8.47 (s, 1H, ArH), 7.66 (br s, 4H, ArH), 7.06 (t, J = 7.8 Hz, 1H, ArH), 6.69 (d, J = 7.8 Hz, 1H, ArH), 6.64 (t, J = 7.8 Hz, 2H, ArH), 6.02 (d, J = 7.8 Hz, 1H, CHOH), 5.21 (d, J = 6.6 Hz, 1H, CHOH), 4.76 (dd, J = 9.0, 6.6 Hz, 1H, COCH), 3.59 (dd, J = 18.6, 9.0 Hz, 1H, H of CH2), 3.53 (dd, J = 18.6, 6.6 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.6, 178.3, 148.0, 145.6, 144.7, 143.3, 137.1, 134.6, 131.4 (2C), 130.1 (2C), 127.6, 127.4, 127.0, 124.2, 124.0, 120.7, 118.4, 109.0, 71.4, 53.9, 45.5, 26.3; HR-MS (ESI): m/z Calcd. for C24H16N3O7BrNa [M+Na]+: 560.0069, Found 560.0070. HPLC analysis: MeOH/H2O (60:40), 30.73 min, HPLC purity 99.5%.
4.1.2.15. (1′S,3S,3′S)-1′-Hydroxy-3′-(4-methoxybenzoyl)-5′,7′-dinitro-3′,4′-dihydro-1′H-spiro[indoline-3,2′-naphthalen]-2-one (4p′)
White solid, 75% yield (28.2 mg, 0.06 mmol), = –108.68 (c 0.10 in CH2Cl2), m.p. >220 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.55 (s, 1H, NH), 8.78 (s, 1H, ArH), 8.54 (s, 1H, ArH), 7.93 (d, J = 9.0 Hz, 2H, ArH), 7.17 (dd, J = 7.8, 3.6 Hz, 2H, ArH), 7.05 (d, J = 9.0 Hz, 2H, ArH), 6.83 (dd, J = 18.0, 7.8 Hz, 2H, ArH), 6.53 (d, J = 7.2 Hz, 1H, CHOH), 4.92 (dd, J = 12.0, 6.0 Hz, 1H, COCH), 4.73 (d, J = 7.2 Hz, 1H, CHOH), 3.88 (s, 3H, OCH3), 3.59 (dd, J = 18.0, 12.0 Hz, 1H, H of CH2), 3.48 (dd, J = 18.6, 6.0 Hz, 1H, H of CH2); 13C NMR (150 MHz, DMSO-d6): δ 197.6, 177.1, 163.4, 147.8, 145.2, 142.4, 142.4, 137.9, 130.6 (2C), 129.8, 128.3, 128.3, 127.8, 125.3, 120.4, 118.5, 113.9 (2C), 108.8, 69.1, 55.4, 51.1, 41.2, 26.9; HR-MS (ESI): m/z Calcd. for C25H19N3O8Na [M+Na]+: 512.1070, Found 512.1069. HPLC analysis: MeOH/H2O (60:40), 14.20 min, HPLC purity 97.9%.
4.2. Biological assays
4.2.1. Biochemical assays
The in vitro MDM2 and CDK4 assays were performed based on the HTRF method according to the previous reports and manufacturer's protocols. The protein kinase selectivity of compound ent-4g (100 nmol/L) was detected in a high-throughput binding assay (KINOMEscan, DiscoveRx, Fremont, CA, USA) against a panel of 99 kinases. The experimental procedures were described in Supporting Information.
4.2.2. Computational
The Accelrys Discovery Studio (DS3.5, Accelrys, San Diego, CA, USA) was utilized for the homology modeling of CDK4 binds to allosteric inhibitors. Then optimization of initial model, equilibration, interaction free energy calculation (MM-GBSA) and computational alanine scanning were optimized by the standard molecular dynamics protocol in AMBER12 package with the MMFF94 force field. The detailed computational procedures and parameters were provided in Supporting Information.
4.2.3. Cell proliferation, apoptosis and Western blotting assays
The glioblastoma cell lines U87MG, U251 and T98G were obtained from the ATCC (American Type Culture Collection, Virginia, VA, USA) and cultured in the state key laboratory of biotherapy, west china hospital, Sichuan University, China. The cell proliferation and apoptosis assay were measured by using the MTT method and Annexin V/PI staining kit (Keygen, Nanjing, China), respectively. The Western blot (WB) analysis was performed according to the previous reports and manufacturer's protocols. The experimental procedures were described in Supporting Information.
4.2.4. RNA sequencing and bioinformatics analysis
Total RNA from U251 cells with or without ent-4g incubation were extracted using the Trizol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol. Only the samples with RNA integrity values of >8.0 were used for mRNA sequencing on the Illumina HiSeq4000 platform by Novogene Co., Ltd. (Beijing, China). The experimental procedures were described in Supporting Information.
4.2.5. Xenograft models and in vivo evaluations
The in vivo antitumor activity and preliminary safety of ent-4g were carried out according to the Guidelines for the Care and Use of Laboratory Animals that were approved by the Committee of Ethics of Animal Experimentation of Sichuan University, Chengdu, China. The six to eight weeks old SPF (specific pathogen-free) nude mice were purchased from Beijing Huafukang Biotechnology Co., Ltd. The in vivo antitumor activity preliminary safety of ent-4g were performed on the U251 subcutaneous xenograft models, and the pharmacokinetics evaluation of ent-4g were assessed in Sprague–Dawley rats (350–400 g). The detailed experimental procedures were provided in the supplementary materials.
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (81573588, 81630101, 81773889 and 21772131), the Science & Technology Department of Sichuan Province (2017JZYD0001, 2017JQ0002, 2017JY0323 and 2019YFSY0004, China).
Author contributions
Gu He and Bo Han conceived and designed the study. Biao Wang, Fu Peng, Jin Zhou and Nan Zhang performed the experiments. Biao Wang, Wei Huang, Jia Sheng, Phensinee Haruehanroengra, Gu He and Bo Han analysed all the data. Biao Wang, Fu Peng, Gu He and Bo Han wrote the manuscript. All the authors approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.12.013.
Contributor Information
Gu He, Email: hegu@scu.edu.cn.
Bo Han, Email: hanbo@cdutcm.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173:321–337. doi: 10.1016/j.cell.2018.03.035. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Messaoudi K., Clavreul A., Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today. 2015;20:899–905. doi: 10.1016/j.drudis.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Kim H., Zheng S., Amini S.S., Virk S.M., Mikkelsen T., Brat D.J. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25:316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruan C., Liu L., Lu Y., Zhang Y., He X., Chen X. Substance P-modified human serum albumin nanoparticles loaded with paclitaxel for targeted therapy of glioma. Acta Pharm Sin B. 2018;8:85–96. doi: 10.1016/j.apsb.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G. A pathology atlas of the human cancer transcriptome. Science. 2017;357 doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 6.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espadinha M., Barcherini V., Lopes E.A., Santos M.M. An update on MDMX and dual MDM2/X inhibitors. Curr Top Med Chem. 2018;18:647–660. doi: 10.2174/1568026618666180604080119. [DOI] [PubMed] [Google Scholar]
- 9.Twarda-Clapa A., Krzanik S., Kubica K., Guzik K., Labuzek B., Neochoritis C.G. 1,4,5-Trisubstituted imidazole-based P53–MDM2/MDMX antagonists with aliphatic linkers for conjugation with biological carriers. J Med Chem. 2017;60:4234–4244. doi: 10.1021/acs.jmedchem.7b00104. [DOI] [PubMed] [Google Scholar]
- 10.Lemos A., Leão M., Soares J., Palmeira A., Pinto M., Saraiva L. Medicinal chemistry strategies to disrupt the P53–MDM2/MDMX interaction. Med Res Rev. 2016;36:789–844. doi: 10.1002/med.21393. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y., Aguilar A., Bernard D., Wang S. Small-molecule inhibitors of the MDM2–P53 protein–protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58:1038–1052. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B., Golding B.T., Hardcastle I.R. Small-molecule MDM2–P53 inhibitors: recent advances. Future Med Chem. 2015;7:631–645. doi: 10.4155/fmc.15.13. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y., Wolf S., Beck B., Köhler L.M., Khoury K., Popowicz G.M. Discovery of highly potent P53–MDM2 antagonists and structural basis for anti-acute myeloid leukemia activities. ACS Chem Biol. 2014;9:802–811. doi: 10.1021/cb400728e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W., Cao H., Wolf S., Camacho-Horvitz M.S., Holak T.A., Dömling A. Benzimidazole-2-one: a novel anchoring principle for antagonizing P53–MDM2. Biorg Med Chem. 2013;21:3982–3995. doi: 10.1016/j.bmc.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W., Hu Y. Small molecule agents targeting the P53–MDM2 pathway for cancer therapy. Med Res Rev. 2012;32:1159–1196. doi: 10.1002/med.20236. [DOI] [PubMed] [Google Scholar]
- 16.Popowicz G.M., Dömling A., Holak T.A. The structure-based design of MDM2/MDMX–P53 inhibitors gets serious. Angew Chem Int Ed. 2011;50:2680–2688. doi: 10.1002/anie.201003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardcastle I.R., Liu J., Valeur E., Watson A., Ahmed S.U., Blackburn T.J. Isoindolinone inhibitors of the murine double minute 2 (MDM2)–P53 protein–protein interaction: structure–activity studies leading to improved potency. J Med Chem. 2011;54:1233–1243. doi: 10.1021/jm1011929. [DOI] [PubMed] [Google Scholar]
- 18.Czarna A., Beck B., Srivastava S., Popowicz G.M., Wolf S., Huang Y. Robust generation of lead compounds for protein–protein interactions by computational and MCR Chemistry: P53/HDM2 antagonists. Angew Chem Int Ed. 2010;49:5352–5356. doi: 10.1002/anie.201001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardcastle I.R., Ahmed S.U., Atkins H., Farnie G., Golding B.T., Griffin R.J. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction based on an isoindolinone scaffold. J Med Chem. 2006;49:6209–6221. doi: 10.1021/jm0601194. [DOI] [PubMed] [Google Scholar]
- 20.Ding K., Lu Y., Nikolovska-Coleska Z., Qiu S., Ding Y., Gao W. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 21.Yang M.C., Peng C., Huang H., Yang L., He X.H., Huang W. Organocatalytic asymmetric synthesis of spiro-oxindole piperidine derivatives that reduce cancer cell proliferation by inhibiting MDM2–p53 interaction. Org Lett. 2017;19:6752–6755. doi: 10.1021/acs.orglett.7b03516. [DOI] [PubMed] [Google Scholar]
- 22.Gupta A.K., Bharadwaj M., Kumar A., Mehrotra R. Spiro-oxindoles as a promising class of small molecule inhibitors of p53–MDM2 interaction useful in targeted cancer therapy. Top Curr Chem. 2016;375:3. doi: 10.1007/s41061-016-0089-0. [DOI] [PubMed] [Google Scholar]
- 23.Yu B., Zheng Y.C., Shi X.J., Qi P.P., Liu H.M. Natural product-derived spirooxindole fragments serve as privileged substructures for discovery of new anticancer agents. Anti Cancer Agents Med Chem. 2016;16:1315–1324. doi: 10.2174/1871520615666151102093825. [DOI] [PubMed] [Google Scholar]
- 24.Zhou R., Wu Q., Guo M., Huang W., He X., Yang L. Organocatalytic cascade reaction for the asymmetric synthesis of novel chroman-fused spirooxindoles that potently inhibit cancer cell proliferation. Chem Commun. 2015;51:13113–13116. doi: 10.1039/c5cc04968g. [DOI] [PubMed] [Google Scholar]
- 25.Yu B., Yu D.Q., Liu H.M. Spirooxindoles: promising scaffolds for anticancer agents. Eur J Med Chem. 2015;97:673–698. doi: 10.1016/j.ejmech.2014.06.056. [DOI] [PubMed] [Google Scholar]
- 26.Ding K., Lu Y., Nikolovska-Coleska Z., Wang G., Qiu S., Shangary S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2−P53 interaction. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 27.Patnaik A., Rosen L.S., Tolaney S.M., Tolcher A.W., Goldman J.W., Gandhi L. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6:740–753. doi: 10.1158/2159-8290.CD-16-0095. [DOI] [PubMed] [Google Scholar]
- 28.O'Leary B., Finn R.S., Turner N.C. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–430. doi: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- 29.Goel S., Wang Q., Watt A.C., Tolaney S.M., Dillon D.A., Li W. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255–269. doi: 10.1016/j.ccell.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stetz G., Verkhivker G.M. Functional role and hierarchy of the intermolecular interactions in binding of protein kinase clients to the Hsp90–Cdc37 chaperone: structure-based network modeling of allosteric regulation. J Chem Inf Model. 2018;58:405–421. doi: 10.1021/acs.jcim.7b00638. [DOI] [PubMed] [Google Scholar]
- 31.Qie S., Diehl J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med. 2016;94:1313–1326. doi: 10.1007/s00109-016-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rastelli G., Anighoro A., Chripkova M., Carrassa L., Broggini M. Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2. Cell Cycle. 2014;13:2296–2305. doi: 10.4161/cc.29295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z., Ding L., Li Z., Wang Z., Suo F., Shen D. Development of the triazole-fused pyrimidine derivatives as highly potent and reversible inhibitors of histone lysine specific demethylase 1 (LSD1/KDM1A) Acta Pharm Sin B. 2019;9:794–808. doi: 10.1016/j.apsb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pratt D.J., Bentley J., Jewsbury P., Boyle F.T., Endicott J.A., Noble M.E. Dissecting the determinants of cyclin-dependent kinase 2 and cyclin-dependent kinase 4 inhibitor selectivity. J Med Chem. 2006;49:5470–5477. doi: 10.1021/jm060216x. [DOI] [PubMed] [Google Scholar]
- 35.Ikuta M., Kamata K., Fukasawa K., Honma T., Machida T., Hirai H. Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J Biol Chem. 2001;276:27548–27554. doi: 10.1074/jbc.M102060200. [DOI] [PubMed] [Google Scholar]
- 36.Verreault M., Schmitt C., Goldwirt L., Pelton K., Haidar S., Levasseur C. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res. 2016;22:1185–1196. doi: 10.1158/1078-0432.CCR-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ke B., Tian M., Li J., Liu B., He G. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med Res Rev. 2016;36:983–1035. doi: 10.1002/med.21398. [DOI] [PubMed] [Google Scholar]
- 38.Joshi S., Singh A.R., Durden D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem. 2014;289:22785–22797. doi: 10.1074/jbc.M114.587493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karkare S., Chhipa R.R., Anderson J., Liu X., Henry H., Gasilina A. Direct inhibition of retinoblastoma phosphorylation by nimbolide causes cell-cycle arrest and suppresses glioblastoma growth. Clin Cancer Res. 2014;20:199–212. doi: 10.1158/1078-0432.CCR-13-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He J., Zhao X., Huang C., Zhou X., You Y., Zhang L. Double amplifications of CDK4 and MDM2 in a gastric inflammatory myofibroblastic tumor mimicking cancer with local invasion of the spleen and diaphragm. Cancer Biol Ther. 2018;19:967–972. doi: 10.1080/15384047.2018.1480290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ricciotti R.W., Baraff A.J., Jour G., Kyriss M., Wu Y., Liu Y. High amplification levels of MDM2 and CDK4 correlate with poor outcome in patients with dedifferentiated liposarcoma: a cytogenomic microarray analysis of 47 cases. Cancer Genet. 2017;218–219:69–80. doi: 10.1016/j.cancergen.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Hirose K., Okura M., Sato S., Murakami S., Ikeda J.I., Noda Y. Gnathic giant-cell-rich conventional osteosarcoma with MDM2 and CDK4 gene amplification. Histopathology. 2017;70:1171–1173. doi: 10.1111/his.13141. [DOI] [PubMed] [Google Scholar]
- 43.Righi A., Gambarotti M., Benini S., Gamberi G., Cocchi S., Picci P. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum Pathol. 2015;46:549–553. doi: 10.1016/j.humpath.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Mariño-Enríquez A., Hornick J.L., Dal Cin P., Cibas E.S., Qian X. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol. 2014;122:128–137. doi: 10.1002/cncy.21362. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida A., Ushiku T., Motoi T., Beppu Y., Fukayama M., Tsuda H. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am J Surg Pathol. 2012;36:423–431. doi: 10.1097/PAS.0b013e31824230d0. [DOI] [PubMed] [Google Scholar]
- 46.Muthusamy V., Hobbs C., Nogueira C., Cordon-Cardo C., McKee P.H., Chin L. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 2006;45:447–454. doi: 10.1002/gcc.20310. [DOI] [PubMed] [Google Scholar]
- 47.Simon R., Struckmann K., Schraml P., Wagner U., Forster T., Moch H. Amplification pattern of 12q13–q15 genes (MDM2, CDK4, GLI) in urinary bladder cancer. Oncogene. 2002;21:2476–2483. doi: 10.1038/sj.onc.1205304. [DOI] [PubMed] [Google Scholar]
- 48.Berner J.M., Forus A., Elkahloun A., Meltzer P.S., Fodstad Ø., Myklebost O. Separate amplified regions encompassing CDK4 and MDM2 in human sarcomas. Genes Chromosomes Cancer. 1996;17:254–259. doi: 10.1002/(SICI)1098-2264(199612)17:4<254::AID-GCC7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 49.Reifenberger G., Reifenberger J., Ichimura K., Meltzer P.S., Collins V.P. Amplification of multiple genes from chromosomal region 12q13–14 in human malignant gliomas: preliminary mapping of the amplicons shows preferential involvement of CDK4, SAS, and MDM2. Cancer Res. 1994;54:4299–4303. [PubMed] [Google Scholar]
- 50.Khatib Z.A., Matsushime H., Valentine M., Shapiro D.N., Sherr C.J., Look A.T. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res. 1993;53:5535–5541. [PubMed] [Google Scholar]
- 51.Sriraman A., Dickmanns A., Najafova Z., Johnsen S.A., Dobbelstein M. CDK4 inhibition diminishes p53 activation by MDM2 antagonists. Cell Death Dis. 2018;9:918. doi: 10.1038/s41419-018-0968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laroche-Clary A., Chaire V., Algeo M.P., Derieppe M.A., Loarer F.L., Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. 2017;10:123. doi: 10.1186/s13045-017-0482-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein M.E., Dickson M.A., Antonescu C., Qin L.X., Dooley S.J., Barlas A. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene. 2018;37:5066–5078. doi: 10.1038/s41388-018-0332-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang S.C., Adhikari S., Afroze R., Brewer K., Calderwood E.F., Chouitar J. Optimization of tetrahydronaphthalene inhibitors of Raf with selectivity over hERG. Bioorg Med Chem Lett. 2016;26:1156–1160. doi: 10.1016/j.bmcl.2016.01.049. [DOI] [PubMed] [Google Scholar]
- 55.Renneberg D., Hubler F., Rey M., Hess P., Delahaye S., Gatfield J. Discovery of novel bridged tetrahydronaphthalene derivatives as potent T/L-type calcium channel blockers. Bioorg Med Chem Lett. 2015;25:3941–3946. doi: 10.1016/j.bmcl.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 56.Jung M.E., Chamberlain B.T., Koch P., Niazi K.R. Synthesis and bioactivity of a brasilicardin a analogue featuring a simplified core. Org Lett. 2015;17:3608–3611. doi: 10.1021/acs.orglett.5b01712. [DOI] [PubMed] [Google Scholar]
- 57.Chang C.F., Ke C.Y., Wu Y.C., Chuang T.H. Structure-activity relationship of synthetic 2-phenylnaphthalenes with hydroxyl groups that inhibit proliferation and induce apoptosis of MCF-7 cancer cells. PLoS One. 2015;10 doi: 10.1371/journal.pone.0141184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haiba M.E., Abd El-Karim S.S., Gouhar R.S., El-Zahar M.I., El-Awdan S.A. Synthesis and evaluation of anti-inflammatory and analgesic activity of some substituted thiazolyl and thaizolidinonyl tetrahydronapthalene derivatives. Med Chem Res. 2014;23:3418–3435. [Google Scholar]
- 59.Gamal-Eldeen A.M., Hamdy N.A., Abdel-Aziz H.A., El-Hussieny E.A., Fakhr I.M. Induction of intrinsic apoptosis pathway in colon cancer HCT-116 cells by novel 2-substituted-5,6,7,8-tetrahydronaphthalene derivatives. Eur J Med Chem. 2014;77:323–333. doi: 10.1016/j.ejmech.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 60.Sanphui P., Pramanik S.K., Chatterjee N., Moorthi P., Banerji B., Biswas S.C. Efficacy of cyclin dependent kinase 4 inhibitors as potent neuroprotective agents against insults relevant to alzheimer's disease. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto O., Sardinha J., Vaz P.D., Piedade F., Calhorda M.J., Abramovitch R. Synthesis of tetrahydronaphthalene lignan esters by intramolecular cyclization of ethyl p-azidophenyl-2-phenylalkanoates and evaluation of the growth inhibition of human tumor cell lines. J Med Chem. 2011;54:3175–3187. doi: 10.1021/jm101182s. [DOI] [PubMed] [Google Scholar]
- 62.Mei G.J., Shi F. Catalytic asymmetric synthesis of spirooxindoles: recent developments. Chem Commun. 2018;54:6607–6621. doi: 10.1039/c8cc02364f. [DOI] [PubMed] [Google Scholar]
- 63.Dalpozzo R. Recent catalytic asymmetric syntheses of 3,3-disubstituted indolin-2-ones and 2,2-disubstituted indolin-3-ones. Adv Synth Catal. 2017;359:1772–1810. [Google Scholar]
- 64.Dalpozzo R. Catalytic asymmetric synthesis of hetero-substituted oxindoles. Org Chem Front. 2017;4:2063–2078. [Google Scholar]
- 65.Santos M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron. 2014;70:9735–9757. [Google Scholar]
- 66.Cheng D., Ishihara Y., Tan B., Barbas C.F., III Organocatalytic asymmetric assembly reactions: synthesis of spirooxindoles via organocascade strategies. ACS Catal. 2014;4:743–762. [Google Scholar]
- 67.Hong L., Wang R. Recent advances in asymmetric organocatalytic construction of 3,3′-spirocyclic oxindoles. Adv Synth Catal. 2013;355:1023–1052. [Google Scholar]
- 68.Singh G.S., Desta Z.Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem Rev. 2012;112:6104–6155. doi: 10.1021/cr300135y. [DOI] [PubMed] [Google Scholar]
- 69.MacDonald J.P., Badillo J.J., Arevalo G.E., Silva-García A., Franz A.K. Catalytic stereoselective synthesis of diverse oxindoles and spirooxindoles from isatins. ACS Comb Sci. 2012;14:285–293. doi: 10.1021/co300003c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ball-Jones N.R., Badillo J.J., Franz A.K. Strategies for the enantioselective synthesis of spirooxindoles. Org Biomol Chem. 2012;10:5165–5181. doi: 10.1039/c2ob25184a. [DOI] [PubMed] [Google Scholar]
- 71.Badillo J.J., Hanhan N.V., Franz A.K. Enantioselective synthesis of substituted oxindoles and spirooxindoles with applications in drug discovery. Curr Opin Drug Discov Devel. 2010;13:758–776. [PubMed] [Google Scholar]
- 72.Trost B.M., Brennan M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis. 2009:3003–3025. [Google Scholar]
- 73.Gicquel M., Gomez C., Garcia Alvarez M.C., Pamlard O., Guérineau V., Jacquet E. Inhibition of P53-murine double minute 2 (MDM2) interactions with 3,3′-spirocyclopentene oxindole derivatives. J Med Chem. 2018;61:9386–9392. doi: 10.1021/acs.jmedchem.8b01137. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Y., Liu L., Sun W., Lu J., McEachern D., Li X. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J Am Chem Soc. 2013;135:7223–7234. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao Q., Peng C., Huang H., Liu S.J., Zhong Y.J., Huang W. Asymmetric synthesis of tetrahydroisoquinoline-fused spirooxindoles as Ras-GTP inhibitors that inhibit colon adenocarcinoma cell proliferation and invasion. Chem Commun. 2018;54:8359–8362. doi: 10.1039/c8cc04732d. [DOI] [PubMed] [Google Scholar]
- 76.Xie X., Huang W., Peng C., Han B. Organocatalytic asymmetric synthesis of six-membered carbocycle-based spiro compounds. Adv Synth Catal. 2018;360:194–228. [Google Scholar]
- 77.Han B., Huang W., Ren W., He G., Wang J.H., Peng C. Asymmetric synthesis of cyclohexane-fused drug-like spirocyclic scaffolds containing six contiguous stereogenic centers via organocatalytic cascade reactions. Adv Synth Catal. 2015;357:561–568. [Google Scholar]
- 78.Xie X., Peng C., He G., Leng H.J., Wang B., Huang W. Asymmetric synthesis of a structurally and stereochemically complex spirooxindole pyran scaffold through an organocatalytic multicomponent cascade reaction. Chem Commun. 2012;48:10487–10489. doi: 10.1039/c2cc36011j. [DOI] [PubMed] [Google Scholar]
- 79.Han N.N., Zhou Q., Huang Q., Liu K.J. Carnosic acid cooperates with tamoxifen to induce apoptosis associated with caspase-3 activation in breast cancer cells in vitro and in vivo. Biomed Pharmacother. 2017;89:827–837. doi: 10.1016/j.biopha.2017.01.084. [DOI] [PubMed] [Google Scholar]
- 80.Zurlo D., Ziccardi P., Votino C., Colangelo T., Cerchia C., Dal Piaz F. The antiproliferative and proapoptotic effects of cladosporols A and B are related to their different binding mode as PPARγ ligands. Biomed Pharmacother. 2016;108:22–35. doi: 10.1016/j.bcp.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Y., Yang B., Jiang Y., Liu Z., Liu Y., Wang X. Studies on cytotoxic activity against HepG-2 cells of naphthoquinones from green walnut husks of juglans mandshurica maxim. Molecules. 2015;20:15572–15588. doi: 10.3390/molecules200915572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.González M.A. Aromatic abietane diterpenoids: their biological activity and synthesis. Nat Prod Rep. 2015;32:684–704. doi: 10.1039/c4np00110a. [DOI] [PubMed] [Google Scholar]
- 83.Hatfield M.J., Tsurkan L.G., Hyatt J.L., Edwards C.C., Lemoff A., Jeffries C. Modulation of esterified drug metabolism by tanshinones from Salvia miltiorrhiza (“Danshen”) J Nat Prod. 2013;76:36–44. doi: 10.1021/np300628a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang N.Y., Liu L., Tao W.W., Duan J.A., Tian L.J. Diterpenoids from Pinus massoniana resin and their cytotoxicity against A431 and A549 cells. Phytochemistry. 2010;71:1528–1533. doi: 10.1016/j.phytochem.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 85.Xu Y., Liu X., Wang Y., Zhou N., Peng J., Gong L. Combinatorial pharmacophore modeling of multidrug and toxin extrusion transporter 1 inhibitors: a theoretical perspective for understanding multiple inhibitory mechanisms. Sci Rep. 2015;5:13684. doi: 10.1038/srep13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oh S., Park S.B. A design strategy for drug-like polyheterocycles with privileged substructures for discovery of specific small-molecule modulators. Chem Commun. 2011;47:12754–12761. doi: 10.1039/c1cc14042f. [DOI] [PubMed] [Google Scholar]
- 87.Hernandez Maganhi S., Jensen P., Caracelli I., Zukerman Schpector J., Fröhling S., Friedman R. Palbociclib can overcome mutations in cyclin dependent kinase 6 that break hydrogen bonds between the drug and the protein. Protein Sci. 2017;26:870–879. doi: 10.1002/pro.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Manoni F., Connon S.J. Catalytic asymmetric tamura cycloadditions. Angew Chem Int Ed. 2014;53:2628–2632. doi: 10.1002/anie.201309297. [DOI] [PubMed] [Google Scholar]
- 89.Cao Y., Jiang X., Liu L., Shen F., Zhang F., Wang R. Enantioselective michael/cyclization reaction sequence: scaffold-inspired synthesis of spirooxindoles with multiple stereocenters. Angew Chem Int Ed. 2011;50:9124–9127. doi: 10.1002/anie.201104216. [DOI] [PubMed] [Google Scholar]
- 90.Cao S.H., Zhang X.C., Wei Y., Shi M. Chemoselective reduction of isatin-derived electron-deficient alkenes using alkylphosphanes as reduction reagents. Eur J Org Chem. 2011;2011:2668–2672. [Google Scholar]
- 91.Wang B., Leng H.J., Yang X.Y., Han B., Rao C.L., Liu L. Efficient synthesis of tetrahydronaphthalene- or isochroman-fused spirooxindoles using tandem reactions. RSC Adv. 2015;5:88272–88276. [Google Scholar]
- 92.Kojima K., Konopleva M., McQueen T., O'Brien S., Plunkett W., Andreeff M. MDM2 inhibitor Nutlin-3a induces P53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108:993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Finn R.S., Crown J.P., Lang I., Boer K., Bondarenko I.M., Kulyk S.O. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 94.Treiber D.K., Shah N.P. Ins and outs of kinase DFG motifs. Chem Biol. 2013;20:745–746. doi: 10.1016/j.chembiol.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 95.Salomon-Ferrer R., Case D.A., Walker R.C. An overview of the Amber biomolecular simulation package. WIREs Comput Mol Sci. 2013;3:198–210. [Google Scholar]
- 96.Wang S., Sun W., Zhao Y., McEachern D., Meaux I., Barrière C. SAR405838: an optimized inhibitor of MDM2–P53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Betzi S., Alam R., Martin M., Lubbers D.J., Han H., Jakkaraj S.R. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem Biol. 2011;6:492–501. doi: 10.1021/cb100410m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y., Luo X., Guo Q., Nie Y., Wang T., Zhang C. Discovery of N1-(4-((7-cyclopentyl-6-(dimethylcarbamoyl)-7H-pyrrolo[2,3-d]pyrimidin-2-yl)amino)phenyl)-N8-hydroxyoctanediamide as a novel inhibitor targeting cyclin-dependent kinase 4/9 (CDK4/9) and histone deacetlyase1 (HDAC1) against malignant cancer. J Med Chem. 2018;61:3166–3192. doi: 10.1021/acs.jmedchem.8b00209. [DOI] [PubMed] [Google Scholar]
- 99.Guo Q., Li Y., Zhang C., Huang Z., Wang X., Nie Y. Selective and novel cyclin-dependent kinases 4 inhibitor: synthesis and biological evaluation. Med Chem Res. 2018;27:1666–1678. [Google Scholar]
- 100.Zou J., Xie H.Z., Yang S.Y., Chen J.J., Ren J.X., Wei Y.Q. Towards more accurate pharmacophore modeling: multicomplex-based comprehensive pharmacophore map and most-frequent-feature pharmacophore model of CDK2. J Mol Graph Model. 2008;27:430–438. doi: 10.1016/j.jmgm.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 101.Park H., Yeom M.S., Lee S. Loop flexibility and solvent dynamics as determinants for the selective inhibition of cyclin-dependent kinase 4: comparative molecular dynamics simulation studies of CDK2 and CDK4. Chembiochem. 2004;5:1662–1672. doi: 10.1002/cbic.200400214. [DOI] [PubMed] [Google Scholar]
- 102.Minoche A.E., Dohm J.C., Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 104.Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kong S.W., Pu W.T., Park P.J. A multivariate approach for integrating genome-wide expression data and biological knowledge. Bioinformatics. 2006;22:2373–2380. doi: 10.1093/bioinformatics/btl401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arnott G., Brice H., Clayden J., Blaney E. Electrophile-Induced dearomatizing spirocyclization of N-arylisonicotinamides: a route to spirocyclic piperidines. Org Lett. 2008;10:3089–3092. doi: 10.1021/ol801092s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.