

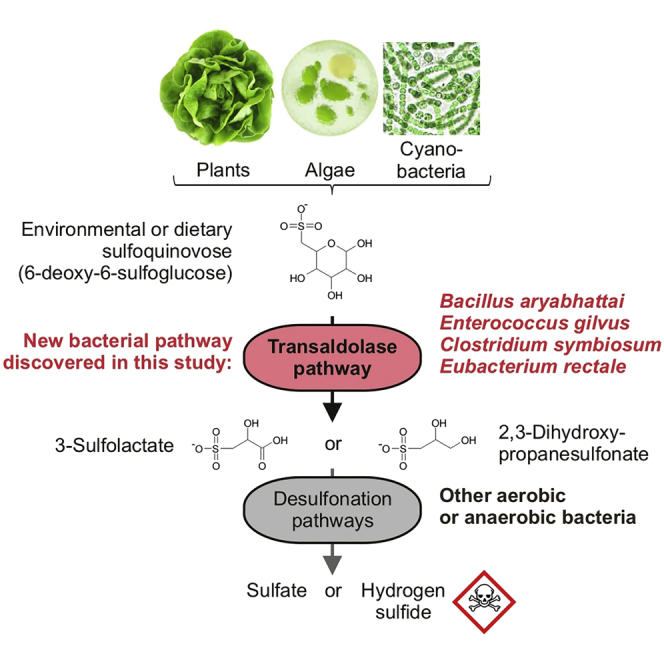
Summary
Bacterial degradation of the sugar sulfoquinovose (SQ, 6-deoxy-6-sulfoglucose) produced by plants, algae, and cyanobacteria, is an important component of the biogeochemical carbon and sulfur cycles. Here, we reveal a third biochemical pathway for primary SQ degradation in an aerobic Bacillus aryabhattai strain. An isomerase converts SQ to 6-deoxy-6-sulfofructose (SF). A novel transaldolase enzyme cleaves the SF to 3-sulfolactaldehyde (SLA), while the non-sulfonated C3-(glycerone)-moiety is transferred to an acceptor molecule, glyceraldehyde phosphate (GAP), yielding fructose-6-phosphate (F6P). Intestinal anaerobic bacteria such as Enterococcus gilvus, Clostridium symbiosum, and Eubacterium rectale strains also express transaldolase pathway gene clusters during fermentative growth with SQ. The now three known biochemical strategies for SQ catabolism reflect adaptations to the aerobic or anaerobic lifestyle of the different bacteria. The occurrence of these pathways in intestinal (family) Enterobacteriaceae and (phylum) Firmicutes strains further highlights a potential importance of metabolism of green-diet SQ by gut microbial communities to, ultimately, hydrogen sulfide.
Subject Areas: Microbiology, Microbial Metabolism
Graphical Abstract
Highlights
-
•
First known SQ-degradation pathway in phylum Firmicutes (Gram-positive) bacteria
-
•
New enzyme, sulfofructose:D-glyceraldehyde-3-phosphate glyceronetransferase
-
•
SQ fermentation pathway in human gut Enterococcus, Clostridium, and Eubacterium strains
-
•
Novel degradation route for green-diet SQ to H2S by intestinal microbial communities
Microbiology; Microbial Metabolism
Introduction
6-Deoxy-6-sulfoglucose (sulfoquinovose, SQ) is the polar head group of the plant sulfolipids sulfoquinovosyl diacylglycerols (SQDGs) (Benson, 1963). These lipids are constituents of photosynthetically active membranes in, essentially, all phototrophic organisms (Benning, 1998). Furthermore, certain archaea contain SQ in their surface-layer glycoproteins (Meyer et al., 2011). More on the role of SQ in the biosphere can be found in a recent review (Goddard-Borger and Williams, 2017). Microbial degradation of the large amounts of SQ produced by phototrophs is an important yet largely understudied part of the biogeochemical sulfur cycle (Denger et al., 2012, 2014; Felux et al., 2015; Burrichter et al., 2018). SQ is introduced into the digestive systems of all herbivores and omnivores through their green-vegetable diet and, thus, SQ may be utilized as a substrate also by intestinal microbes (Burrichter et al., 2018).
Complete degradation (mineralization) of the SQ into, ultimately, inorganic sulfate (Denger et al., 2012, 2014) or hydrogen sulfide (Burrichter et al., 2018) can be achieved in two tiers by bacterial communities through metabolite cross-feeding. In the first tier, bacteria catalyze a primary degradation of SQ to C3-organosulfonates, either 3-sulfolactate (SL) or 2,3-dihydroxypropane-1-sulfonate (DHPS). Such a primary SQ-degradation pathway is the subject of this study. In the second tier, SL- and DHPS-mineralizing bacteria release the inorganic sulfur. Notably, specialized anaerobic bacteria can utilize these SQ degradation intermediates (SL and DHPS) for sulfite respiration (e.g., some Desulfovibrio and Bilophila spp.) producing potentially harmful hydrogen sulfide (H2S) (Burrichter et al., 2018), which implies that a complete, anaerobic two-step degradation of SQ to H2S may be a trait of the intestinal microbiota that is relevant to human health (see ref. Burrichter et al., 2018 and Discussion).
We have previously described two types of biochemical pathways (depicted in Figure 1A) for a primary degradation of SQ to DHPS or SL, in Escherichia coli K-12 and Pseudomonas putida SQ1, respectively (Denger et al., 2014; Felux et al., 2015).
Figure 1.

Pathways for Primary SQ Degradation in Bacteria
Illustration of (A) the two known SQ-degradation pathways in Escherichia coli K12 and Pseudomonas putida SQ1, respectively, and (B) the third SQ-degradation pathway identified in this study in aerobic Bacillus aryabhattai SOS1 and in strictly anaerobic, SQ-fermenting bacteria such as Clostridium symbiosum LT0011. (A) E. coli uses an SQ-Embden-Meyerhof-Parnas (sulfo-EMP) pathway for acquisition of carbon and energy from SQ under both aerobic (Denger et al., 2014) and fermentative growth conditions (Burrichter et al., 2018). It employs a 6-deoxy-6-sulfofructose-1-phosphate (SFP) aldolase (indicated in orange), and it excretes 2,3-dihydroxypropanesulfonate (DHPS) as degradation product during growth with SQ. P. putida uses an SQ-Entner-Doudoroff (sulfo-ED) pathway (Felux et al., 2015). It employs a 2-keto-3,6-dideoxy-6-sulfogluconate (KDSG) aldolase (yellow), and it excretes 3-sulfolactate (SL) as degradation product during growth with SQ. See the main text (Introduction) for more detailed descriptions of the pathways and of the abbreviations used for intermediates and enzymes (roman numerals). (B) In this study, a third pathway for SQ was discovered in B. aryabhattai SOS1, which excretes SL during SQ degradation. It employs a newly discovered 6-deoxy-6-sulfofructose (SF) transaldolase enzyme (red) that derives fructose-6-phosphate (F6P) for growth (or sedoheptulose-7-phosphate [S7P]; not depicted in Figure 1B) through transfer of the non-sulfonated C3-(glycerone) moiety of SF onto GAP as acceptor molecule (or erythrose-4-phosphate [E4P]; not depicted in Figure 1B). The pathway was identified by differential proteomics to be present also in SQ-fermenting, human gut bacteria, for example, in C. symbiosum LT0011, which excretes DHPS during SQ fermentation.
Escherichia coli K-12 catalyzes an SQ-degradation pathway analogous to the Embden-Meyerhof-Parnas (EMP) pathway for glucose-6-phosphate (thus, an sulfo-EMP pathway) under aerobic (Denger et al., 2014) as well as fermentative growth conditions (Burrichter et al., 2018). The sulfo-EMP pathway starts with an aldose/ketose isomerase (enzyme I in Figure 1A; YihS) converting SQ to 6-deoxy-6-sulfofructose (SF), which is phosphorylated by an ATP-dependent SF kinase (enzyme II in Figure 1A; YihV) to 6-deoxy-6-sulfofructosephosphate (SFP). The SFP is then cleaved by an SFP aldolase (enzyme III in Figure 1A; YihT) into 3-sulfolactaldehyde (SLA) and dihydroxyacetone phosphate (DHAP) (Denger et al., 2014). The DHAP supports energy conservation and growth, whereas the SLA cannot be catabolized further but is reduced via an NADH-dependent SLA reductase (enzyme IV in Figure 1A; YihU) to DHPS, which is excreted (Denger et al., 2014). Notably, this SLA reduction step is beneficial for anaerobic, SQ-fermenting E. coli as an additional fermentation step to recover NAD+ (Burrichter et al., 2018). The sulfo-EMP gene cluster is part of the core genome of commensal and pathogenic E. coli strains and can also be found in genomes of other facultatively anaerobic Enterobacteriaceae that can thrive in the gastrointestinal tract, e.g., Salmonella, Klebsiella, Cronobacter, and Citrobacter spp. (Denger et al., 2014; Burrichter et al., 2018). Hence, until today, metabolism of dietary SQ by Enterobacteriaceae via the sulfo-EMP pathway has been considered most relevant in intestinal microbiomes of herbivorous animals and humans (Denger et al., 2014; Burrichter et al., 2018), although this has never been assessed in situ.
Aerobic Pseudomonas putida SQ1 catalyzes a second pathway, analogous to the Entner-Doudoroff (ED) pathway for glucose-6-phosphate (thus, an sulfo-ED pathway) (Felux et al., 2015). The corresponding gene cluster was found in genomes of a wide range of aerobic and nitrate-reducing (respiring) terrestrial, freshwater, and marine Proteobacteria (Felux et al., 2015). Starting with an NAD+-dependent SQ dehydrogenase (in Figure 1A, enzyme V), the SQ is oxidized to 6-sulfogluconolactone (SGL). The lactone is hydrolyzed by an SGL lactonase (enzyme VI) to 6-deoxy-6-sulfogluconate (SG), which is converted by an SG dehydratase (enzyme VII) to 2-keto-3,6-deoxy-6-sulfo-gluconate (KDSG). The KDSG is then cleaved by a KDSG aldolase (enzyme VIII) into pyruvate and SLA. The pyruvate supports energy conservation and growth. The SLA is also not catabolized further but, in contrast to its reduction to DHPS as in E. coli, is oxidized by a NAD+-dependent SLA dehydrogenase (enzyme IX in Figure 1A) to SL, which is excreted. Notably, this SLA oxidation step is beneficial for respiring bacteria, in order to gain an additional NADH (Felux et al., 2015).
In this study, we revealed a third pathway for primary SQ degradation in a newly isolated aerobic bacterium, Bacillus aryabhattai SOS1, which represents the first known SQ-utilizing Gram-positive (phylum Firmicutes) bacterium. The pathway was identified by differential proteomics and reconstituted by heterologously produced enzymes, and all key intermediates were identified by mass spectrometry, using 13C6-isotopically labeled SQ as substrate. Additionally, we isolated an anaerobic, SQ-fermenting strain, Clostridium symbiosum LT0011, from human feces, and we demonstrate that this strain and two common human gut bacteria, Enterococcus gilvus DSM15689 and Eubacterium rectale DSM17629, harbor and express the newly discovered pathway during SQ fermentation. Hence, metabolism of dietary SQ in intestinal microbiomes may be catalyzed by members of the family Enterobacteriaceae via the known sulfo-EMP pathway, as well as by members of the phylum Firmicutes via the SQ pathway discovered in this study.
Results
Isolation and Examination of an SQ-Degrading Bacillus aryabhattai Strain
Sulfo-EMP and sulfo-ED pathway gene clusters can be found frequently in genomes of Proteobacteria but not in Gram-positive (phylum Firmicutes) bacteria. Therefore, we started new aerobic enrichment cultures with SQ as sole carbon and energy source (Denger et al., 2012; Felux et al., 2015) and using soil samples, pond water, or plant leaves as inocula. Replicate soil inocula were pasteurized in order to aid in the enrichment of spore-forming Firmicutes. All enrichments grew, degraded SQ as confirmed by HPLC, and were sub-cultivated until pure cultures were obtained, which were identified by 16S rRNA gene sequencing. From the unpasteurized samples, proteobacterial strains of Pseudomonas sp. (plant leaf), Rhanella sp. (non-pasteurized soil), and Aeromonas sp. (pond) were obtained but not examined further, as they degrade SQ most likely via one of the previously investigated SQ pathways (Figure 1A). However, one of the unpasteurized enrichments (maple leaf) yielded a Bacillus sp. strain (termed strain SOS1) with a 16S rRNA gene sequence identity of 99.9% to the B. aryabhattai type strain. Two more Bacillus sp. strains (strains AF1 and AF2) were isolated from the pasteurized soil enrichment cultures. They utilized the substrate SQ concomitantly with stoichiometrical SL excretion, as determined by HPLC during growth experiments (a linearized growth plot is shown for strain SOS1 in the Supplementary files, Figure S1), but they did not excrete DHPS or sulfate.
We chose B. aryabhattai SOS1 (deposited as DSM 104036) as our model organism for further examination of its SQ-degradation pathway. Cell extracts of SQ-grown cells were examined for enzyme activities and metabolites of the two known SQ-degradation pathways, under the reaction conditions used previously (Denger et al., 2014; Felux et al., 2015). Upon addition of SQ, formation of SF was detectable by HPLC-MS (see below), indicative of an SQ isomerase activity. However, addition of known co-substrates (see Figure 1A, ATP and/or NAD+) yielded no traces of the key intermediate(s) of the sulfo-EMP pathway (SFP; Figure 1A) (Denger et al., 2014) nor of the sulfo-ED pathway (SG, KDSG; Figure 1A) (Felux et al., 2015). We concluded that strain SOS1 may harbor a different pathway that branches from the sulfo-EMP pathway at the level of SF as first intermediate. Furthermore, the SQ isomerase activity was not detectable in cell extract of glucose-grown cells, suggesting that also this SQ pathway is inducibly expressed (Denger et al., 2014; Felux et al., 2015).
Identification of a Metabolic Gene Cluster that Is Highly Expressed during SQ Degradation in B. aryabhattai SOS1
An annotated draft-genome sequence of strain SOS1 was generated for proteomics using Illumina HiSeq sequencing and JGI's Integrated Microbial Genomes (IMG) annotation pipeline (Felux et al., 2015; Burrichter et al., 2018); the annotation is available at IMG (GOLD Analysis Project Id, Ga0111075). Two-dimensional protein gel electrophoresis (2D-PAGE) as well as total proteomics were performed (Figure 2) for extract of cells grown with SQ in comparison with cells grown with glucose, in order to identify the inducible pathway enzymes/genes (e.g., Denger et al., 2014; Felux et al., 2015; Burrichter et al., 2018). For the 2D-PAGE (Figure 2A), all major protein spots visible only for SQ-grown cells, indicative of abundant proteins specifically induced during growth with SQ, were excised and identified by peptide fingerprinting-mass spectrometry. These results were confirmed by the total proteomic analyses (Figure 2B).
Figure 2.

Proteomic Analysis of B. aryabhattai SOS1 Cells Identified a Single Gene Cluster for SQ Degradation
(A) Proteins in cell extracts of SQ- or glucose-grown cells were separated via 2D gel electrophoresis (2D-PAGE), and all prominent protein spots visible only for SQ-grown cells were excised and submitted to peptide fingerprinting-mass spectrometry. These spots are labeled according to their IMG gene locus tag numbers (A) and are encoded in the same gene cluster (illustrated in C). The results were replicated once when starting from an independent growth experiment.
(B) Relative abundances of the proteins encoded in this gene cluster as observed by total proteomic analysis in SQ-versus glucose-grown cells. For comparison, the relative abundances of constitutively expressed proteins (GroL, RpoB, AtpA) are also shown, as well as of glucose-inducible enzymes (Entner-Doudoroff/pentose phosphate pathway enzymes). Data from an independent growth experiment and proteomic analysis are shown (n = 1).
(C) Illustration of the identified SQ degradation gene cluster in B. aryabhattai SOS1. The locus tag numbers and gene annotations shown refer to the IMG-draft genome annotation of strain SOS1; the IMG locus tag prefix is indicated. These genes were termed sftATXGIFDE (6-deoxy-6-sulfofructose transaldolase pathway gene cluster; SFT pathway). The blue coloration indicates genes identified by proteomics (A and/or B).
Six identified, strongly produced proteins (Figure 2A) were encoded in the same cluster (Figure 2C), for which the predicted proteins/genes had been auto-annotated by the IMG pipeline as follows (gene numbers; the IMG locus tag prefix, Ga0111075_1003, is omitted): 1319 as predicted MFS-type sugar:cation symporter; 1320 as predicted transaldolase; 1321 as protein domain of unknown function (DUF4867) gene; 1322 as glucoside hydrolase (alpha-glucosidase); 1323 as aldose/ketose isomerase; 1324 as transcriptional regulator (GntR family); 1325 as aldehyde dehydrogenase; and 1326 as predicted sulfite/organosulfonate exporter (TauE-type) (Figure 2C). The total proteomic analysis (Figure 2B) confirmed a strong expression of these six proteins (Figure 2A), and identified an additional protein (1319) encoded in the same gene cluster, for a predicted glycoside symporter in SQ-grown B. aryabhattai SOS1.
Identification of a 6-Deoxy-6-sulfofructose Transaldolase Enzyme in Cell Extracts
The identified, predicted transaldolase 1320 (termed SftT) showed 41.7% amino acid identity (full length) to a characterized fructose-6-phosphate transaldolase (from Thermoplasma acidophilum; Lehwess-Litzmann et al., 2011a; Lehwess-Litzmann et al., 2011b) but much lower identity to characterized fructose-6-phosphate aldolases (e.g., from E. coli, 25.2%; ref. Schurmann and Sprenger, 2001), suggesting that an acceptor molecule, such as glyceraldehyde-3-phosphate (GAP) or erythrose-4-phosphate (E4P), may be crucial for catalysis. We refined our enzyme reaction conditions (see Transparent Methods) and used gel-filtered cell extract, in which potential co-substrates and acceptor molecules had been removed (size exclusion, >1–5 kDa). Gel-filtered cell extract and SQ alone produced only SF, but a conversion of the SQ via SF further to SLA was detectable after addition of GAP or E4P to the assay, i.e., as acceptor molecules for a transaldolase reaction. Furthermore, with addition of NAD+ for a predicted SLA dehydrogenase reaction, also a formation of SL was detectable. The transfer of a dihydroxyacetone moiety of SF onto an acceptor molecule by a transaldolase enzyme (glyceronetransferase; EC 2.2.1.2) concomitant with SLA formation (see Figure 1B), as strongly suggested by the results described above, was unequivocally confirmed when we used fully isotopically (13C)-labelled SQ (13C6-SQ) as substrate and when we observed the corresponding mass shifts of the intermediates in MS/MS while the HPLC retention times were unaffected in comparison with authentic, unlabeled standards. As shown in Figure 3, the 13C6-SQ was converted to 13C6-SF, and through the presence of unlabeled E4P, accumulation of [1,2,3-13C3]-SLA and of [1,2,3-13C3]-sedoheptulose-7-phosphate (S7P) was detectable, and in presence of NAD+, [1,2,3-13C3]-SL accumulated. Hence, the mass shifts occurring for the molecular-ion of S7P (Figure 3) and its fragmentation products (see Figure S2) identified 13C6-labeled SF as the origin of the transferred C3-(dihydroxyacetone) moiety. When unlabeled GAP instead of E4P was used together with NAD+, the reaction products were 13C3-SLA and 13C3-SL, and [1,2,3-13C3]-hexose phosphates but not [1,2,3-13C3]-S7P (Figure S3), confirming that also GAP served as acceptor for the SF transaldolase reaction.
Figure 3.

HPLC Mass Spectrometry Confirmed a Transaldolase Reaction in Cell Extracts of SQ-grown B. aryabhattai SOS1 Using Fully 13C-Labeled SQ as Substrate and Erythrose-4-Phosphate as Acceptor Molecule
In the presence of unlabeled erythrose-4-phosphate (E4P) and of 13C6-SQ, a transient formation of 13C6-SF and 13C3-SLA, and an accumulation of [1,2,3-13C3]-sedoheptulose-7-phosphate (S7P) and of 13C3-SL, was detected. The 13C3-SL formation resulted from an SLA dehydrogenase reaction because of the additional presence of NAD+ in the reaction mixture. The reaction contained 2 mM 13C6-SQ, 6 mM E4P (with G6P as impurity; not shown), and 6 mM NAD+ and was sampled before addition of the gel-filtered cell extract (exclusion, >5 kDa) and after 10, 30, and 240 min of incubation. For the organosulfonates, the total-ion chromatograms (TIC) of the MS/MS-fragmentation of the quasi-molecular ions ([M-H]-) are shown. For the sugar phosphates E4P and [1,2,3-13C3]-S7P, the MS/MS-ion traces of the phosphate group ([H2PO4]-, m/z = 97) are shown. Note that 13C3-SLA was separated by HPLC as trident peak. The results were replicated twice when starting from independent growth experiments. When E4P was exchanged by GAP as the acceptor, [1,2,3-13C3]-F6P/G6P accumulation was detected (see Figure S3). MS/MS fragmentation mass spectra are shown as Supplementary files for [1,2,3-13C3]-S7P (Figure S2), 13C6-SF and 13C6-SQ (Figure S5), 13C3-SLA (Figure S6), 13C3-SL (Figure S7), and E4P (Figure S13).
From the results of these enzyme tests with cell extract (Figures 2, S2, and S3) in combination with the proteomic and genomic results (Figures 2A–2C), we concluded that the SQ catabolic pathway in strain SOS1 involves only three inducible enzymes, as depicted in Figure 1B: the isomerase gene product 1323 (Figure 2), catalyzing the interconversion of SQ and SF (Figure 3), and the co-encoded, co-induced transaldolase gene product 1320 (Figure 2), catalyzing the transfer reaction with SF and E4P to SLA and S7P (Figure 3), or with SF and GAP to SLA and F6P (Figure S3). The F6P/S7P powers energy conservation and growth of B. aryabhattai SOS1 and serves for regeneration of the corresponding acceptor molecule (as depicted in Figure 1B). The SLA is oxidized to SL (Figures 3 and S3) by the co-encoded, co-induced NAD+-dependent dehydrogenase 1325 (Figure 2).
In Vitro Reconstitution of the SQ-Degradation Pathway
The candidate genes 1320 (sftT), 1323 (sftI), and 1325 (sftD) were cloned into plasmids, heterologously overexpressed in E. coli, and the proteins purified via His6-Tag affinity chromatography (Figure S4). The purified proteins were added sequentially to 13C6-SQ-containing reaction mixtures, and samples were taken for HPLC-MS/MS (Figure 4).
Figure 4.

In Vitro Reconstitution of the SQ Transaldolase Pathway by Three Recombinant Enzymes
For the organosulfonates, the total-ion chromatograms (TICs) of the MS/MS fragmentation of the quasi-molecular ions ([M-H]-) are depicted, and for the sugar phosphates F6P and G6P, the characteristic ion traces of the phosphate group ([H2PO4]-, m/z = 97). The reaction mixture initially contained 2 mM 13C6-SQ and 12 mM GAP (0 min). SQ isomerase SftI (protein 1323) was added (100 μg/mL) and the reaction sampled after 10 min. Then, the SF transaldolase SftT (protein 1320) (50 μg/mL) was added and the reaction sampled after 1 min. Finally, SLA dehydrogenase SftD (protein 1325) (100 μg/mL) and 6 mM NAD+ was added and the reaction sampled after 17 h. Note that GAP and NAD+ were added in excess (12 and 6 mM) because the SLA dehydrogenase oxidizes also GAP in the presence of NAD+; therefore, the SQ conversion was incomplete. Furthermore, the reaction mixture converted the [1,2,3-13C3]-F6P to [1,2,3-13C3]-G6P, owing to an activity of the SQ-isomerase also with F6P. The results were replicated twice using independently produced enzyme preparations. A reaction sequence with E4P instead of GAP as the acceptor is shown in the Supplementary files, Figure S8. A chromatogram depicting the HPLC separation of G6P and F6P in more detail is shown in Figure S11, and MS/MS fragmentation mass spectra for G6P and [1,2,3-13C3]-G6P, and for F6P and [1,2,3-13C3]-F6P, are shown in Figures S12 and S13, respectively.
With addition of protein 1323 (SftI), formation of a peak showing the same mass as 13C6-SQ but a slightly earlier HPLC retention time (Denger et al., 2014) was detected (Figure 4), identified as 13C6-SF by its MS/MS fragmentation pattern (Denger et al., 2014) including corresponding 13C mass shifts (see Figure S5). Thus, protein SftI is an SQ isomerase. After addition of protein 1320 (SftT) together with unlabeled GAP as acceptor molecule, 13C3-SLA was formed (Figure 4), as identified by its fragmentation pattern (Figure S6) (Felux et al., 2015). Furthermore, [1,2,3-13C3]-F6P formation was observed (Figure 4), confirming that the non-sulfonated 13C3-dihydroxyacetone moiety of SQ is transferred onto the unlabeled GAP as acceptor. The enzyme catalyzed the SF-cleavage also with E4P as acceptor, forming 13C3-SLA and [1,2,3-13C3]-S7P (see Figure S8), but not with F6P as acceptor. Thus, protein SftT is an SF-cleaving, SLA-forming, GAP/E4P-dependent transaldolase; it showed activity also as sedoheptulose-7-phosphate transaldolase (see Figure S9). Finally, after addition of protein 1325 (SftD) and NAD+ to the reaction mixture, the 13C3-SLA was converted to 13C3-SL (Figure 4), as compared with authentic SL standard (Figure S7). Thus, protein SftD is another NAD+-dependent SLA dehydrogenase, in addition to the one identified in the P. putida sulfo-ED pathway (Figure 1A); SftD oxidized also GAP and E4P as substrates but showed no activity with SL and DHPS, as tested in photometrical assays. Note that specific activities for SLA dehydrogenase could not be determined, because no authentic SLA was available as substrate; the SLA for the enzyme tests was generated in a coupled assay with E. coli SLA-reductase (YihU), converting DHPS with NAD+ to SLA (Felux et al., 2015; Burrichter et al., 2018). Finally, in reaction mixtures in which GAP served as the acceptor (Figure 5), the F6P formed by the SF transaldolase was converted further to G6P, and SQ isomerase SftI was responsible for this activity (see Figure S10).
Figure 5.

Homologous SF-Transaldolase Pathway Gene Clusters are Found Widespread Particularly in Genomes of Aerobic and Anaerobic (Phylum) Firmicutes (Class) Bacilli and Clostridia
Shown are selected gene-cluster architectures as retrieved from the IMG Ortholog Neighborhood Viewer when using the transaldolase SftT as query. In total, 183 orthologous gene clusters were identified in IMG. The gene clusters shown encode also candidate genes for SQ-glyceride hydrolases (alpha-glucosidases), transport systems (import of SQ and excretion of SL or DHPS), and regulation. Importantly, most of the genes clusters of the strictly anaerobic Firmicutes encode SLA reductase candidate genes (termed SftR) homologous to YihU of E. coli, instead of SLA dehydrogenases (SftD) genes, suggesting that these strains most likely catalyze an SQ-fermentation pathway that results in DHPS instead of SL as degradation product (see text). Indicated by underlined letters are three representative strains that were examined in this study for their ability to ferment SQ, excrete SL or DHPS, and produce the SFT pathway proteins specifically during growth with SQ, as confirmed by proteomics for all three strains (Figure 6).
Enterococcus gilvus, Clostridium symbiosum, and Eubacterium rectale Strains Employ the SFT Pathway for SQ Fermentation
We searched for bacterial genomes that harbor sft-gene clusters using IMG's (https://img.jgi.doe.gov) Ortholog Neighborhood Viewer and Cluster Scout (Hadjithomas et al., 2017) and retrieved a total of 189 candidate clusters, predominantly in members of the classes Bacilli and Clostridia (phylum Firmicutes), e.g., in members of the genera Bacillus, Enterococcus, Lactobacillus, Butyrivibrio, Pseudobutyrivibrio, Eubacterium, and Clostridium, and also in individual genomes of members of the phyla Fusobacteria, Chloroflexi, Actinobacteria, Spirochaetes, and Thermotogae. Representative gene-cluster architectures comprising the key genes for enzymes SftITD as well as for SQ-glyceride cleavage of the sulfolipid (SftG, alpha-glucosidases), and predicted genes for transport systems (import of SQ and excretion of SL or DHPS), regulation and metabolism of glycerol, if present (Discussion), are illustrated in Figure 5. For a list of all genomes retrieved via IMG (as of 3 July 2020) that contain candidate SFT-gene clusters, see Table S2. For example, we found candidate gene cluster in approximately 1% of all Bacillus genomes; hence, the gene cluster may be descriptive of specific ecotypes, e.g., plant or intestine associated, but it is not part of the core genome (pan-genome) of Bacillus species, in contrast to the sulfo-EMP pathway gene cluster in E. coli (Denger et al., 2014).
Interestingly, the gene clusters of the strictly anaerobic Clostridia encoded, instead of an SLA dehydrogenase (SftD; dark green in Figures 5 and 1A), exclusively an NADH-dependent DHPS-forming SLA reductase (SftR, indicated in light green in Figures 5 and 1A), such as the characterized SLA reductases of E. coli (YihU; ref. Denger et al., 2014; Sharma et al., 2020) and of Desulfovibrio sp. DF1 (DhpA, physiologically catalyzing the reverse reaction; see ref. Burrichter et al., 2018). This strongly suggested that these strains catalyze an SQ fermentation pathway that may result in DHPS as degradation product, instead of SL, hence, as an additional fermentation step to recover NAD+, as with the sulfo-EMP pathway of E. coli (Figure 1A; ref. Burrichter et al., 2018).
Hence, we additionally confirmed as part of this study for three strictly anaerobic, human-gut Firmicutes strains with sft-gene clusters, that they (1) indeed are capable of growing through SQ fermentation and (2) excrete DHPS or SL (Figure 6A) and (3) produce the Sft proteins specifically during growth with SQ, as observed by differential proteomics (Figure 6B). Enterococcus gilvus DSM15689 (order Lactobacillales) (Tyrrell et al., 2002) and Eubacterium rectale (Agathobacter rectalis) DSM17629 (order Clostridiales) (Duncan and Flint, 2008) were examined, and Clostridium symbiosum LT0011, which we isolated from human feces by streaking on SQ-containing agar plates and picking of colonies into SQ-containing liquid medium. Strain LT0011 (deposited as DSM108250) was genome sequenced (IMG Genome ID, 2802428835) for the proteomic analysis (Figure 6B); SQ fermentation was confirmed also for C. symbiosum type-strain DSM934 (Kaneuchi et al., 1976). For growth of the strains in anaerobic liquid culture, a carbonate-buffered minerals salts medium with Ti(III)NTA as reducing agent (Burrichter et al., 2018) supplemented with SQ (10 mM) and yeast extract (0.1% w/v) was used.
Figure 6.

Fermentation of SQ by Human Intestinal Bacteria via the SF-transaldolase Pathway
(A and B) Fermentation of SQ to SL or DHPS by human intestinal bacterial strains Enterococcus gilvus DSM15689, Clostridium symbiosum LT0011, and Eubacterium rectale DSM17629 (A) and proteomic confirmation of a strong, inducible expression of the Sft-proteins during growth with SQ (B). (A) E. gilvus produced SL during SQ fermentation and C. symbiosum and E. rectale produced DHPS but not SL. The used mineral salts medium had to be supplemented with yeast extract (0.1% w/v) for observing growth, and no complete turnover of the SQ provided (10 mM) was obtained, presumably due to depletion of supplements, except for E. gilvus. Triplicates (n = 3) are shown; the error bars indicate standard deviations. From replicate cultures, cellular biomass was collected at the end of the growth phase and submitted to total proteomic analyses in comparison to glucose-fermenting cells. (B) Differential proteomics confirmed a strongly inducible expression of the Sft-pathway genes during fermentation of SQ but not when the strains were grown with glucose. Each result was replicated at least once when starting from independent growth experiments. Abbreviations used: SftA, predicted SQ importer; SftG, SQG hydrolase (alpha-glucosidase); SftI, SQ isomerase; SftT, SF transaldolase; SftR, SLA reductase; SftD, SLA dehydrogenase; SftX, DUF4867; SftE, predicted SL/DHPS exporter; GroL, chaperon; TF, translation factor; EF, elongation factor; Fba, fructose bisphosphate aldolase; Tpi, triosephosphate isomerase; Gap, GAP dehydrogenase; Pyk, pyruvate kinase; n.d., not detected; n/e, not encoded in the respective sft-gene cluster (see Figure 5).
As shown in Figure 6A, E. gilvus produced SL but not DHPS during fermentation of SQ under our growth conditions, whereas C. symbiosum and E. rectale produced DHPS but not SL. The predicted inducible expression of the SFT pathway genes during SQ fermentation was confirmed by the differential proteomics results, as shown in Figure 6B.
Discussion
In this study, we established the first primary SQ-degradation pathway known in the ecologically and industrially important group of Gram-positive (phylum Firmicutes) bacteria, by using a newly isolated environmental B. aryabhattai strain as model system, by differential proteomics, by in vitro reconstruction of the pathway with recombinant enzymes, and by identification of all intermediates using fully 13C-labeled SQ as substrate. Surprisingly, homologous pathway gene cluster can be found widespread particularly in genomes of aerobic and anaerobic (phylum) Firmicutes, (class) Bacilli and Clostridia, and also in individual genomes of members of other important bacterial phyla, for example, Fusobacteria, Actinobacteria, Spirochaetes, Chloroflexi, and Thermotogae. For three representative strains of the strictly anaerobic and important human-gut (class) Bacilli and Clostridia, we confirmed their predicted ability to grow with SQ and that they express the pathway enzymes inducibly during fermentative growth with SQ.
Our previous additions to the known glycolytic (monosaccharide degradation) pathways in bacteria were the two “sulfoglycolytic” pathways (Denger et al., 2014; Felux et al., 2015) for catabolism of the 6-sulfonated-6-deoxy hexose SQ, which are analogous to the Embden-Meyerhof-Parnas (e.g., Kresge et al., 2005; Barańskaa et al., 2007) and the Entner-Doudoroff (Entner and Doudoroff, 1952) pathways for glucose-6-phosphate (G6P) (Figure 1A), given the structural similarity of the substrates SQ and G6P. All these pathways employ aldolases (aldehyde lyases, EC 4.1.2.x) for cleavage of the C6-monosaccharides into C3 fragments. The third bacterial pathway for primary SQ degradation uncovered in this study reminds somewhat of another glycolytic pathway for G6P, the pentose-phosphate pathway (Gunsalus et al., 1955; Horecker, 2002), in that a transaldolase enzyme (aldehyde/ketone transferase, EC 2.2.1.x) is involved in acquisition of monosaccharide carbon, as depicted in Figure 1B. The SF transaldolase (proposed systematic name, 6-deoxy-6-sulfofructose:D-glyceraldehyde-3-phosphate glyceronetransferase) excises a sulfonated C3 fragment (SLA), and it acquires a non-sulfonated C3 fragment from SQ by transferring a glycerone-moiety onto an acceptor molecule, GAP or E4P, yielding F6P or S7P (Figures 3, 4, S3, and S8). The F6P and S7P is used for regeneration of the acceptor and as carbon and energy source for cellular biomass formation (Figure 1B). Differences of the newly discovered SQ pathway to the pentose phosphate pathway, however, are absence of both NADH generation and decarboxylation reaction and, thus, that not a pentose intermediate is cleaved.
The biochemical and genetic identity of this third bacterial pathway for SQ catabolism, as established in this study, is adding to a surprisingly high, but yet mostly underappreciated, diversity of microbial pathways for utilization of organosulfonates, as has probably first been noted for taurine (e.g., Cook and Denger, 2006; see also MetaCyc [https://biocyc.org], taurine degradation I–IV, and Peck et al., 2019). The genetic and biochemical identification of this third SQ pathway is also adding valuable insight, as well as future experimental access, to important questions regarding physiology, enzymology, and pathway energetics of SQ degradation, regarding the environmental microbiology of SQ degradation and its contributions to the biogeochemical carbon and sulfur cycles, and also regarding its potential roles in intestinal microbiomes and for human health.
The glycolytic pathways for G6P are optimized according to their overall thermodynamic and biochemical properties of the pathway, necessity of investing in enzymes (protein) to maintain sufficient pathway fluxes, and their molar yields of energy equivalents (ATP) and reducing equivalents (NADH) relative to the electron acceptors used (e.g., Bar-Even et al., 2012; Flamholz et al., 2013). Given the structural similarity of SQ and G6P, similar energetic and ecophysiological constraints have obviously led to the evolution of also several biochemical strategies for degradation of SQ. However, a common theme for all known SQ-degradation pathways is that the “waste product” SLA is employed either as additional electron donor or as additional electron acceptor (Figure 1). One of the many interesting questions arising is whether the benefits of this type of SLA utilization, either as electron donor or acceptor, might in many environmental settings outweigh the benefit of further catabolizing the SLA-carbon (for example, by desulfonation of SL to pyruvate through sulfolactate sulfo-lyase, SuyAB; ref. Burrichter et al., 2018) and whether this might be an explanation for the division of labor (Tsoi et al., 2018; Giri et al., 2019) observed thus far in complete SQ degradation, that is, primary SQ degradation and cross-feeding of the DHPS or SL to different microorganisms that mediate the desulfonation reactions (Denger et al., 2012, 2014; Burrichter et al., 2018) for closing the sulfur cycle. Further interesting questions are arising around the different forms non-sulfonated C3-carbon acquired through the three different SQ pathways, that is DHAP by the sulfo-EMP pathway, pyruvate by the sulfo-ED pathway, and a C3-(glycerone)-moiety incorporated into F6P/S7P by the SFT pathway (Figure 1). Although the yields of reducing equivalents and ATP of the different pathways appear well traceable (see below), the particular benefits and costs of funneling the SQ-carbon into the anabolic reactions and fermentations at these different levels is unclear. For example, the particular advantages of the different SQ-pathway strategies may become fully visible only if degradation of the sulfolipid metabolite SQ-glyceride (SQG) is explored further as substrate, instead of only SQ: every SQ-degradation gene cluster examined thus far co-encodes an SQG glucosidase (sulfoquinovosidase) homolog (SftG in Figures 2 and 5; see also refs. Denger et al., 2014; Felux et al., 2015) for cleaving SQG to SQ and glycerol (Speciale et al., 2016), and every SQ-degrading strain examined thus far by proteomics strongly co-induced this sulfoquinovosidase (Figures 2 and 6B; refs. Denger et al., 2014; Felux et al., 2015; Burrichter et al., 2018; Li et al., 2020). Furthermore, E. coli is able to grow with SQG (Abayakoon et al., 2018, and our own observation) as well as P. putida SQ1, B. aryabhattai SOS1 (our own observations), and SQ-degrading Rhizobium leguminosarum SRDI565 (Li et al., 2020). We suggest that for the SFT pathway, the glycerol acquired from SQG cleavage may directly feed into generation of the acceptor molecule GAP for the SF transaldolase, through glycerol kinase and glycerol-3-phosphate dehydrogenase, some of which are co-encoded in the predicted gene clusters (see Figure 5). This needs to be addressed in future, as well as the directly related questions whether indeed SQG rather than SQ might be the more environmentally relevant substrate for SQ-catabolic microorganisms (Abayakoon et al., 2018) and whether the SQG glucosidase gene may be a universal genetic marker for SQ(G) metabolism across all types of pathways.
Overall, the now three known pathways for SQ with either SLA oxidation to SL or its reduction to DHPS, seem to reflect adaptations to the aerobic, facultatively anaerobic, or strictly anaerobic ecophysiology of the different environmental and intestinal microorganisms. SQ utilization by the SL-producing, aerobic P. putida SQ1 through the sulfo-ED pathway inclusive of the SLA dehydrogenase reaction (Figure 1A) (Felux et al., 2015), is represented by the equation
| SQ + 2 NAD+ → pyruvate + SL + 2 NADH +2 H+. |
The pyruvate-carbon is assimilated and oxidized for energy generation through aerobic respiration, and the two NADH are also driving respiration. SQ utilization by DHPS-producing, facultatively anaerobic E. coli K12 through the sulfo-EMP pathway including the SLA reductase reaction (Denger et al., 2014), and including substrate-level phosphorylation of the DHAP released (see Figure 1A), is represented by the equation
| SQ + ADP + Pi → pyruvate + DHPS + ATP. |
The pyruvate-carbon is also assimilated and oxidized for energy generation through aerobic respiration (Denger et al., 2014), or it is metabolized and reduced as electron acceptor in mixed-acid fermentation (Burrichter et al., 2018), as has been discussed previously (Felux et al., 2015; Burrichter et al., 2018). The SQ utilization by the SL-producing, aerobic B. aryabhattai SOS1 through the oxidative SFT pathway including the SLA dehydrogenase reaction, as described in this study, and including of the regeneration of the transaldolase-acceptor molecule and substrate-level phosphorylation of the DHAP acquired from SQ (see Figure 1B), is represented by the equation
| SQ + 2 NAD+ + ADP + Pi → pyruvate + SL + 2 NADH + ATP +2 H+. |
Hence, as with the sulfo-ED pathway in P. putida, this pathway generates NADH for respiration; however, in addition, also one ATP is conserved from DHAP further downstream (see Figure 1B), as with the sulfo-EMP pathway of E. coli. Furthermore, the acquisition of the SQ-carbon requires only two enzymes in the SFT pathway, in comparison with the sulfo-ED pathway with four enzymes and the sulfo-EMP pathway with three enzymes (Figure 1). Hence, explanations on the particular advantages of the different SQ-pathway strategies may be provided not only based on pathway energetics and the ATP and NADH yields in relation to the electron acceptor used but also on the level of investment in protein synthesis. Finally, the SQ utilization through the reductive SFT pathway by the fermenting C. symbiosum and E. rectale strains that encode (Figure 5) and express SLA reductases (Figure 6B) and excrete DHPS (Figure 6A) is represented by the equation
| SQ + ADP + Pi → pyruvate + DHPS + ATP, |
thus, as with the sulfo-EMP pathway of E. coli. However, why Enterococcus gilvus DSM15689 is excreting SL instead of DHPS during SQ fermentation under our cultivation conditions (Figure 6A), which is counterintuitive, remains yet unresolved. Note that its gene cluster (Figure 5) encodes both SLA dehydrogenase (SftD) and reductase (SftR) candidates and that both are expressed (Figure 6B). Thus, E. gilvus may be able to switch between SL and DHPS production depending on its growth conditions, e.g., in presence of alternative electron acceptors or in dependence on a metabolic partner organism. In addition, future work will also have to corroborate the yet only tentatively identified non-sulfonated fermentation products formed, that is, for E. gilvus apparently acetate and formate, and for C. symbiosum and E. rectale apparently acetate and H2, but not formate, butyrate, or butanol. Their identification and thorough quantification relative to total biomass formation will establish carbon and electron balances (e.g., Burrichter et al., 2018) and, thus, allow for an overall quantitative, stoichiometrical comparison of the SQ fermentations employing the SFT pathway (Figure 6) relative to that of E. coli via the sulfo-EMP pathway (Burrichter et al., 2018). Such detailed examination might also provide answers to the intriguing questions why the sulfo-EMP pathway is especially widespread among Enterobacteriaceae (Denger et al., 2014), the sulfo-ED pathway across (other) Proteobacteria (Felux et al., 2015), and why the (oxidative and reductive) SFT pathway(s) are found so prominently across genomes of aerobic as well as anaerobic Gram-positive (phylum Firmicutes) bacteria (Figure 5).
Environmental studies of SQ-degrading microorganisms, as well as of SL- or DHPS-degrading microorganisms, are needed to fully reveal their diversity, ecological niches, and significance in the carbon and sulfur cycles in all habitats where SQ is produced and/or degraded. The sulfo-ED pathway genes are found in marine- or saline-associated bacteria (e.g., Halomonas, Salinarimonas), freshwater-, soil-, and plant rhizosphere-associated bacteria (e.g. Microvirga, Ensifer, Herbaspirillum, Rhizobium strains), and also in human-gut symbionts (e.g., Hafnia, Leminorella, and Serratia strains) and/or potential pathogens (Vibrio, Plesiomonas, and Halomonas strains). The sulfo-EMP pathway gene cluster is a feature of the E. coli core genome and also of other Enterobacteriaceae genomes (e.g., Salmonella; Rhanella sp. isolated in this study) as trait relevant for their intestinal as well as environmental lifestyles. The SFT pathway gene cluster is found (Figure 5) in Firmicutes of the soil and plant environment (e.g., Bacillus and Lactobacillus strains) and also among strains of the human intestinal tract and rumen of livestock (e.g., Clostridium, Eubacterium, Enterococcus, Butyrivibrio, and Faecalibacterium strains). Given SQ is a relevant constituent of the green-diet of all herbivores and omnivores (Denger et al., 2014; Goddard-Borger and Williams, 2017) and given the wide distribution of SQ degradation gene clusters in phylogenetically diverse intestinal bacteria, SQ and its degradation products DHPS and/or SL might represent yet unappreciated substrates for the intestinal microbiota of humans and animals. For the primary degradation (fermentation) of SQ, it is probably most interesting to explore whether commensal or pathogenic Enterobacteriaceae, or whether intestinal Firmicutes, utilize the dietary SQ. The genetic signatures of these metabolic features are now available, e.g., for their assessments using omics methods. Importantly, organosulfonates such as the SL and DHPS produced from primary SQ degradation can be substrates for sulfite respiration and sulfide (H2S) production by the intestinal pathobiont Bilophila wadsworthia and by Desulfovibrio spp. (Burrichter et al., 2018; Peck et al., 2019). H2S is a key microbial and host-cell-derived metabolite in the intestine and has many detrimental as well as beneficial contributions to human health and disease (e.g., Attene-Ramos et al., 2010; Carbonero et al., 2012; Singh and Lin, 2015; Ijssennagger et al., 2016). Thus, the genetic and biochemical information provided in this study will help to disentangle also effects of diet-derived SQ on the microbiota composition, and on H2S homeostasis in the gut, each contributing to an improved understanding of the complex interactions between diet, microbiota, and host health (Heintz-Buschart and Wilmes, 2018).
Limitations of the Study
The insights gained via our approach using pure bacterial cultures and classical microbiological and biochemical methods must be complemented with empirical data on SQ degradation processes examined directly in the environment and gut microbiomes, e.g., by omics methods. Hence, environmental studies of SQ-degrading microorganisms, as well as of SL- or DHPS-degrading microorganisms, are urgently needed to fully reveal their diversity, ecological niches, and significance in the carbon and sulfur cycles in all habitats where SQ is produced and/or degraded. A thorough determination of carbon and electron balances and, thus, an overall stoichiometric and energetic comparison of, e.g., the SQ fermentations via the SFT pathway relative to that of E. coli via the sulfo-EMP pathway, must also await future, more detailed studies, as well as further descriptions of the enzymes and their kinetic parameters once authentic substrates are available. In summary, our study highlights the importance of establishing the genetic identity of physiological and biochemical features in isolated bacterial strains in the laboratory, for evaluating their role and importance directly in the environment by omics methods. In this case, for adding valuable insight to important questions regarding the environmental microbiology of SQ degradation and its contribution to the biogeochemical carbon and sulfur cycles, and also regarding its potential roles in intestinal microbiomes and for human health.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. David Schleheck (david.schleheck@uni-konstanz.de).
Materials Availability
Data related to this paper may be requested from the lead author. The bacterial strains isolated and examined in this study were deposited at the Leibniz Institute-German Collection of Microorganisms and Cell Cultures (https://www.DSMZ.de) under reference numbers DSM 104036 for B. aryabhattai SOS1 and DSM108250 for Clostridium symbiosum LT0011. Their (draft) genome sequences are available at IMG (https://img.jgi.doe.gov), for strain SOS1 under GOLD Project Id. Ga0111075 and for strain LT0011 under IMG Genome Id. 2802428835.
Data and Code Availability
This study did not generate code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We like to thank Andreas Marquardt for the proteomic analyses, the AGs Schink and Schleheck, especially Anna Burrichter, Karin Denger, Jasmin Frey, and Sylke Wiechmann for their support, and Bernhard Schink and Valentin Wittmann for fruitful discussions. We also thank Maraike Mühleck and Christof Mayer for their unfortunately unsuccessful attempts for establishing a transformation and knockout system for B. aryabhatta SOS1, and we thank Magnus Schmidt and Markus Ringwald. This work was funded by the University of Konstanz, the Konstanz Young Scholar Fund (YSF), the Konstanz Research School Chemical Biology (KoRS-CB), the Austrian Science Fund (FWF project grant I2320-B22), and the Deutsche Forschungsgemeinschaft (DFG grants SCHL1936/3 and 4).
Author Contributions
D. Schleheck conceived the study. B.F. did the enzymic and analytical-chemical work. S.R.O. enriched and isolated strain SOS1 and S.R.O. and A.W.F. other aerobic SQ-degrading strains, and they characterized their growth. A.W.F. performed the proteomics and growth experiments with strain SOS1 and the SQ-fermenting strains. P.F. established the strain SOS1 genome. B.F. and D Spiteller performed the HPLC-MS analysis. B.T.H. and A.L. isolated C. symbiosum LT0011 and established its genome sequence. B.F. and D. Schleheck wrote the manuscript and all authors revised and approved it.
Declaration of Interests
The authors declare they have no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101510.
Supplemental Information
References
- Abayakoon P., Jin Y., Lingford J.P., Petricevic M., John A., Ryan E., Wai-Ying Mui J., Pires D.E.V., Ascher D.B., Davies G.J. Structural and biochemical insights into the function and evolution of sulfoquinovosidases. ACS Cent. Sci. 2018;4:1266–1273. doi: 10.1021/acscentsci.8b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attene-Ramos M.S., Nava G.M., Muellner M.G., Wagner E.D., Plewa M.J., Gaskins H.R. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ. Mol. Mutagen. 2010;51:304–314. doi: 10.1002/em.20546. [DOI] [PubMed] [Google Scholar]
- Bar-Even A., Flamholz A., Noor E., Milo R. Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat. Chem. Biol. 2012;17:509–517. doi: 10.1038/nchembio.971. [DOI] [PubMed] [Google Scholar]
- Barańskaa J., Dzugajb A., Kwiatkowska-Korczakc J. Embden-Meyerhof-Parnas, the first metabolic pathway: the fate of prominent polish biochemist Jakub Karol Parnas. Compr. Biochem. 2007;45:157–207. [Google Scholar]
- Benning C. Biosynthesis and function of the sulfolipid sulfoquinovosyl diacylglycerol. Annu. Rev. Plant Biol. 1998;49:53–75. doi: 10.1146/annurev.arplant.49.1.53. [DOI] [PubMed] [Google Scholar]
- Benson A. The plant sulfolipid. Adv. Lipid Res. 1963;1:387–394. doi: 10.1016/b978-1-4831-9937-5.50016-8. [DOI] [PubMed] [Google Scholar]
- Burrichter A., Denger K., Franchini P., Huhn T., Müller N., Spiteller D., Schleheck D. Anaerobic degradation of the plant sugar sulfoquinovose concomitant with H2S production: Escherichia coli K-12 and Desulfovibrio sp. strain DF1 as co-culture model. Front. Microbiol. 2018;9:2792. doi: 10.3389/fmicb.2018.02792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonero F., Benefiel A.C., Alizadeh-Ghamsari A.H., Gaskins H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook A.M., Denger K. Metabolism of taurine in microorganisms: a primer in molecular biodiversity? In: Oja S.S., Saransaari P., editors. Taurine 6. Vol. 583. Springer; 2006. pp. 3–13. [DOI] [PubMed] [Google Scholar]
- Denger K., Huhn T., Hollemeyer K., Schleheck D., Cook A.M. Sulfoquinovose degraded by pure cultures of bacteria with release of C3-organosulfonates: complete degradation in two-member communities. FEMS Microbiol. Lett. 2012;328:39–45. doi: 10.1111/j.1574-6968.2011.02477.x. [DOI] [PubMed] [Google Scholar]
- Denger K., Weiss M., Felux A.K., Schneider A., Mayer C., Spiteller D., Huhn T., Cook A.M., Schleheck D. Sulphoglycolysis in Escherichia coli K-12 closes a gap in the biogeochemical sulphur cycle. Nature. 2014;507:114–117. doi: 10.1038/nature12947. [DOI] [PubMed] [Google Scholar]
- Duncan S.H., Flint H.J. Proposal of a neotype strain (A1-86) for Eubacterium rectale. Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2008;58:1735–1736. doi: 10.1099/ijs.0.2008/004580-0. [DOI] [PubMed] [Google Scholar]
- Entner N., Doudoroff M. Glucose and gluconic acid oxidation of Pseudomonas saccharophila. J. Biol. Chem. 1952;196:853–862. [PubMed] [Google Scholar]
- Felux A.-K., Spiteller D., Klebensberger J., Schleheck D. Entner-Doudoroff pathway for sulfoquinovose degradation in Pseudomonas putida SQ1. Proc. Natl. Acad. Sci. U S A. 2015;112:E4298–E4305. doi: 10.1073/pnas.1507049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamholz A., Noor E., Bar-Even A., Liebermeister W., Milo R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc. Natl. Acad. Sci. U S A. 2013;110:10039–10044. doi: 10.1073/pnas.1215283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S., Waschina S., Kaleta C., Kost C. Defining division of labor in microbial communities. J. Mol. Biol. 2019;431:4712–4731. doi: 10.1016/j.jmb.2019.06.023. [DOI] [PubMed] [Google Scholar]
- Goddard-Borger E.D., Williams S.J. Sulfoquinovose in the biosphere: occurrence, metabolism and functions. Biochem. J. 2017;474:827–849. doi: 10.1042/BCJ20160508. [DOI] [PubMed] [Google Scholar]
- Gunsalus I.C., Horecker B.L., Wood W.A. Pathways for carbohydrate metabolism in microorganisms. Bacteriol. Rev. 1955;19:79–128. doi: 10.1128/br.19.2.79-128.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjithomas M., Chen I.A., Ken Chu K., Huang J., Ratner A., Palaniappan K., Andersen E., Markowitz V.M., Kyrpides N.C., Ivanova N.N. IMG-ABC: new features for bacterial secondary metabolism analysis and targeted biosynthetic gene cluster discovery in thousands of microbial genomes. Nucleic Acids Res. 2017;45:D560–D565. doi: 10.1093/nar/gkw1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz-Buschart A., Wilmes P. Human gut microbiome: function matters. Trends Microbiol. 2018;26:563–574. doi: 10.1016/j.tim.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Horecker B.L. The pentose phosphate pathway. J. Biol. Chem. 2002;277:47965–47971. doi: 10.1074/jbc.X200007200. [DOI] [PubMed] [Google Scholar]
- Ijssennagger N., van der Meer R., van Mil S.W. Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol. Med. 2016;22:190–199. doi: 10.1016/j.molmed.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Kaneuchi C., Watanabe K., Terada A., Benno Y., Mitsuoka T. Taxonomic study of Bacteroides clostridiiformis subsp. clostridiiformis (Burri and Ankersmit) Holdeman and Moore and of related organisms: proposal of Clostridium clostridiiformis (Burri and Ankersmit) comb. nov. and Clostridium symbiosum (Stevens) comb. nov. Int. J. Syst. Evol. Microbiol. 1976;26:195–204. [Google Scholar]
- Kresge N., Simoni R.D., Hill R.L. Otto Fritz Meyerhof and the elucidation of the glycolytic pathway. J. Biol. Chem. 2005;280:e3. [PubMed] [Google Scholar]
- Lehwess-Litzmann A., Neumann P., Golbik R., Parthier C., Tittmann K. Crystallization and preliminary X-ray diffraction analysis of transaldolase from Thermoplasma acidophilum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2011;67:584–586. doi: 10.1107/S1744309111009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehwess-Litzmann A., Neumann P., Parthier C., Lüdtke S., Golbik R., Ficner R., Tittmann K. Twisted Schiff base intermediates and substrate locale revise transaldolase mechanism. Nat. Chem. Biol. 2011;7:678–684. doi: 10.1038/nchembio.633. [DOI] [PubMed] [Google Scholar]
- Li J., Epa R., Scott N.E., Skoneczny D., Sharma M., Snow A.J.D., Lingford J.P., Goddard-Borger E.D., Davies G.J., McConville M.J., Williams S.J. A sulfoglycolytic Entner-Doudoroff pathway in Rhizobium leguminosarum bv. trifolii SRDI565. Appl. Environ. Microbiol. 2020;86:e00720–e00750. doi: 10.1128/AEM.00750-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.H., Zolghadr B., Peyfoon E., Pabst M., Panico M., Morris H.R., Haslam S.M., Messner P., Schäffer C., Dell A., Albers S.-V. Sulfoquinovose synthase – an important enzyme in the N-glycosylation pathway of Sulfolobus acidocaldarius. Mol. Microbiol. 2011;82:1150–1163. doi: 10.1111/j.1365-2958.2011.07875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck S.C., Denger K., Burrichter A., Irwin S.M., Balskus E.P., Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc. Natl. Acad. Sci. U S A. 2019;116:3171–3176. doi: 10.1073/pnas.1815661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurmann M., Sprenger G.A. Fructose-6-phosphate aldolase is a novel class I aldolase from Escherichia coli and is related to a novel group of bacterial transaldolases. J. Biol. Chem. 2001;276:11055–11061. doi: 10.1074/jbc.M008061200. [DOI] [PubMed] [Google Scholar]
- Sharma M., Abayakoon P., Lingford J.P., Epa R., John A., Jin Y., Goddard-Borger E.D., Williams S.J. Dynamic structural changes accompany the production of dihydroxypropanesulfonate by sulfolactaldehyde reductase. ACS Catal. 2020;10:2826–2836. [Google Scholar]
- Singh S.B., Lin H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms. 2015;3:866–889. doi: 10.3390/microorganisms3040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speciale G., Jin Y., Davies G.J., Williams S.J., Goddard-Borger E.D. YihQ is a sulfoquinovosidase that cleaves sulfoquinovosyl diacylglyceride sulfolipids. Nat. Chem. Biol. 2016;12:215–217. doi: 10.1038/nchembio.2023. [DOI] [PubMed] [Google Scholar]
- Tsoi R., Wu F., Zhang C., Bewick S., Karig D., You L. Metabolic division of labor in microbial systems. Proc. Natl. Acad. Sci. U S A. 2018;115:2526–2531. doi: 10.1073/pnas.1716888115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell G.J., Turnbull L., Teixeira L.M., Lefebvre J., Carvalho M.d.G.S., Facklam R.R., Lovgren M. Enterococcus gilvus sp. nov. and Enterococcus pallens sp. nov. isolated from human clinical specimens. J. Clin. Microbiol. 2002;40:1140–1145. doi: 10.1128/JCM.40.4.1140-1145.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code.