

Abstract
Colorectal cancer (CRC) is a worldwide health concern with respect to both incidence and mortality, and as a result, CRC tumorigenesis, progression and metastasis have been heavily studied, especially with respect to identifying genetic, epigenetic, transcriptomic and proteomic profiles of disease. DNA methylation alterations are hallmarks of CRC, and epigenetic driver genes have been identified that are thought to be involved in early stages of tumorigenesis. Moreover, distinct CRC patient subgroups are organized based on DNA methylation profiles. CRC tumors displaying CpG island methylator phenotypes (CIMPs), defined as DNA hypermethylation at specific CpG islands in subsets of tumors, show high concordance with specific genetic alterations, disease risk factors and patient outcome. This review details the DNA methylation alterations in CRC, the significance of CIMP status, the development of treatments based on specific molecular profiles and the application of epigenetic therapies for CRC patient treatment.
INTRODUCTION TO CRC
Colorectal cancer (CRC) is a significant global health burden, with an incidence of 1.4 million cases and ~700 000 deaths worldwide in 2012.1 CRC is the third leading cause of cancer mortality in the United States and the second leading cause of cancer mortality in Europe. Owing to more widely implemented screening modalities, such as colonoscopy and image-based detection, as well as effective therapies, CRC mortality has decreased in many countries,1 and the median survival of patients with metastatic disease now approaches 30 months.2,3 In contrast to survival rates of 65% for patients with localized disease, < 10% of metastatic CRC patients survive 5 years after diagnosis. Owing to its prevalence in the population and occurrence as both sporadic and familial diseases, CRC has been well studied at the molecular level in order to characterize the genetic, epigenetic, transcriptomic and proteomic changes for the purposes of disease detection, surveillance and ultimately to develop novel therapeutic approaches to improve patient outcome and survival.
GENETIC ALTERATIONS GUIDING CRC TUMOR DEVELOPMENT AND PROGRESSION
The majority of CRCs (70%) develop sporadically, whereas the remaining cases develop through genetic predisposition or familial influence. Genetic predisposition, or genetic susceptibility, describes the increased risk of developing disease owing to inherited genetic alterations. Only a small percentage (5%) of all CRCs are hereditary, in which family members develop cancer via germline transmission of genetic alterations. Approximately 2–4% of CRCs are characterized as Lynch Syndrome (formerly described as hereditary non-polyposis colorectal cancer), an autosomal dominant disease that arises due to mutations in the DNA mismatch repair genes MLH1 (42%), MSH2 (33%), MSH6 (18%) and PMS2 (7–8%) (reviewed in reference 4).
Fearon and Vogelstein first proposed a multistep model of colon cancer tumorigenesis in 1990,5 in which chromosomal instability, namely mutations and deletions of key oncogenes and tumor suppressors, correlate with disease progression. APC mutations or deletions are thought to be among the earliest events in CRC tumorigenesis in which normal colonic mucosa transitions to hyperproliferative epithelium. Subsequent development of adenomas occurs mainly in the distal colon and involves activating KRAS mutations (KRAS-mut), losses of DCC on chromosome 18q, and inactivation of TGF-β response by SMAD2/SMAD4 changes.6 Finally, TP53 mutations and/or losses correlate with the development of adenocarcinomas, whereas additional alterations are associated with tumor metastases.
Sottoriva et al.7 recently provided evidence for a Big Bang model of CRC tumorigenesis, challenging the Vogelstein model, after sequencing multiple regions of individual tumor glands. Instead of stepwise accumulation of genomic alterations described in the Vogelstein model, the Big Bang model suggests that CRC tumors grow as a single expansion after the initial cellular transformation. The expanded cells contain the genomic alterations present in the initial transformed cell, and accumulate more alterations as a result of cellular growth and expansion. Although these additional changes provide growth advantages, the earliest changes are prevalent, and latter changes are present only in small tumor subpopulations.
There does seem to be room for both models in cancer biology. The stepwise Vogelstein model is supported by epidemiological data of CRC incidence, and is generally thought to describe the accumulation of somatic driver mutations with selection and clonal expansion, but does not support intratumor heterogeneity, a frequent confounder in cancer genomics. The Big Bang model, although may not apply to all tumors, does support intratumor heterogeneity and its occurrence early and continuously during tumorigenesis.7 Moreover, the Big Bang model corroborates with observations that clonal selection is infrequent after the tumor becomes advanced.7
Irrespective of the tumorigenic model, APC mutations are hallmarks of CRC, are present in up to 70% of all CRCs, and result in the failure to block the G1–S phase of the cell cycle. In addition, wild-type APC functions to negatively regulate WNT signaling by degrading beta catenin. A report from The Cancer Genome Atlas Research Network showed that over 90% of all CRCs involve WNT signaling pathway alterations, especially with respect to the presence of APC alterations in CRCs.8 The Cancer Genome Atlas Research Network has identified other high-frequency driver alterations for CRC, including TP53, KRAS, PIK3CA, ACVR2A, TGFBR2, BRAF, MSH3 and MSH6. Integrated analyses of mutation, copy number and gene expression data show that in addition to WNT signaling alterations, CRCs show activation of TGF-β and p53 signaling, as well as inactivation of RAS and PI3K pathways.
Although most (70%) CRC tumors are thought to develop from the traditional adenoma pathway, ~ 30% of CRCs develop from the serrated pathway9 and include hyperplastic polyps, traditional serrated adenomas and sessile serrated adenomas. Hyperplastic polyps are mainly located in the distal (left) side of the colon, can be stratified into microvesicular, goblet cell and mucin poor subgroups, but do not progress past the adenoma state. Traditional serrated adenomas are the least frequent serrated polyps, present in the distal colon with a saw-tooth like appearance and are enriched for KRAS mutations. Sessile serrated adenomas are mainly located in the proximal (right) colon, and are highlighted by large size, pronounced serration, flattened appearance as well as dilated and horizontal colon crypts. Sessile serrated adenomas are mainly enriched for BRAF mutations and unique DNA methylation alterations.
EPIGENETIC ALTERATIONS INVOLVED IN CRC DEVELOPMENT AND PROGRESSION
Introduction to epigenetics
Models of CRC tumorigenesis and progression are mainly based on genetic alterations, however, epigenetic changes are highly prevalent in CRCs. Epigenetics is defined as changes in gene expression that are not due to changes in gene sequence, and include DNA methylation, histone modifications, microRNAs (miRNAs) and nucleosome positioning. Unlike genetic alterations, epigenetic changes are reversible due to enzymatic activity and via pharmacological treatment with small molecule inhibitors, namely those that target DNA methylation and chromatin modifications.
DNA methylation
DNA methylation in mammalian organisms mostly occurs by the addition of a methyl group to the C-5 position of cytosine in a 5′-CG-3′ or CpG sequence context. Non-CpG DNA methylation occurs at low levels in somatic cells and is generally found in embryonic stem cells.10 CpG methylation is evolutionary unstable as methylated CpGs spontaneously deaminate to thymine faster than the rate at which unmethylated CpGs deaminate to uracil. As a result, CpG content in the human genome is only 20% of what is expected by sequence context alone (reviewed in reference 11). The human genome is CpG depleted, and ~70% of all CpGs are methylated,11,12 mostly in transposable elements and intergenic regions of the human genome. However, there are regions of the genome, CpG islands, that contain their expected CpG content, are unmethylated in normal somatic tissues and are more often (>50%) located in gene promoter regions.
Cytosine DNA methylation marks are placed by the enzymatic activities of DNA methyltransferases (DNMTs) using S-adenosylmethionine as a co-factor. DNMT1, DNMT3A and DNMT3B are largely responsible for catalyzing DNA methylation in human tissues. DNMT1 is mainly involved in maintenance DNA methylation to copy DNA methylation patterns from parental DNA onto daughter DNA strands in conjunction with cellular DNA replication.13 DNMT3A and DNMT3B are classified as de novo DNMTs and place new DNA methylation marks at CpG sites that were previously unmethylated. DNMT3A and DNMT3B are predominantly expressed in embryonic stem cells and are responsible for placing de novo DNA methylation marks during development. Although DNMT3A and DNMT3B are expressed at low levels in somatic tissues, both are overexpressed in human cancers, including CRC, and are thought to be involved in generating cancer-specific DNA methylation profiles.
HCT116 colon cancer cells have been instrumental in determining the mechanisms of DNA methylation in human cancers. HCT116 colon cancer cells harboring hypomorphic knockdown of DNMT1 (DNMT1Δ2-5) and/or knockout of DNMT3B (DNMT3B−/−) showed that down regulating DNMT1 or DNMT3B alone did not substantially alter global DNA methylation levels. However, DNMT1/DNMT3B double knockout (DKO, DNMT1−/− DNMT3B−/−) cells display near complete (95%) DNA demethylation, suggesting that DNMT1 and DNMT3B work in concert to maintain DNA methylation marks.14-16 DNMT3B is expressed as ~ 30 alternatively spliced variants that play important roles by: (1) serving as an accessory protein to recruit DNMT3A to sites requiring DNA methylation and (2) maintaining or restoring DNA methylation of CpGs located in gene bodies or transcribed regions by recognition of histone H3 lysine 36 trimethylation (H3K36me3) marks,17,18 which are positively correlated with actively expressed genes.
DNA methylation alterations in human cancers
DNA methylation changes are hallmarks of CRC and virtually all tumor types, highlighted by gene-specific DNA hypermethylation occurring together with DNA hypomethylation of repetitive elements and CpG-poor regions.19-21 DNA methylation alterations may result in gene expression changes, including gene silencing via CpG island promoter DNA hypermethylation and gene activation owing to DNA hypomethylation of CpG-poor gene promoters. Gene body DNA hypermethylation is associated with oncogene overexpression,18 suggesting that genes regulated by DNA methylation are driving events in tumorigenesis. In addition, DNA methylation alterations can also be exploited for use as diagnostic, predictive and prognostic biomarkers for CRC tumorigenesis and metastasis.
DNA methylation-based driver genes in CRC
The search for genetic-based changes in human cancers is ultimately aimed at identifying a select set of alterations that are essential for tumorigenesis. These elements are linked to the concept of oncogenic addiction, defined as the dependence on a single oncogenic pathway for cancer cell survival.22 Oncogenic addiction supports the idea that targeting these pathways will lead to effective therapeutic treatments, as these pathways are generally not constitutively active in normal cells. Examples of addicted oncogenes are BRAF, EGFR, HER2, MYC and RAS, as well as others, across CRC and several other forms of human cancer.23 Addiction can also be applied to cancer epigenetics, specifically retained DNA hypermethylation of selected genes is essential for cancer cell growth and survival after evaluation of DNA methylation in human colon cancer cells deficient for one or more DNMTs (DNMT1−/−, DNMT3B−/−).24 Indeed, DNA hypermethylation of ADAM2, ARMCX1, BCHE, CDO1, ESX1, IRAK3, P2RY14 and SYCP3 are required for cancer cell growth and survival, and DNA demethylation of these genes resulted in cell death and apoptosis.
Attesting to their importance in CRC tumorigenesis, promoter DNA hypermethylation results in the silencing of genes essential for DNA repair, cell cycle progression, signaling pathway checkpoints, among others and include: (1) MLH1 (mut-L homolog 1); (2) CDKN2A(INK4A) (p16, cyclin-dependent kinase inhibitor 2A), (3) MGMT (O-6-methylguanine methyltransferase), (4) RUNX3 (runt-related transcription factor 3), (5) TPEF (transmembrane protein with EGF-like and two follistatin-like domains 2), (6) VIM (vimentin), (7) SFRP1/2/4/5 (secreted frizzled-related protein) family, and others. Of note, SFRPs inhibit WNT signaling, and their silencing is one mechanism of WNT signaling alterations that are plentiful in colorectal tumors.25
miRNA epigenetic silencing
miRNAs are short RNA sequences of 20–22 nucleotides in length that are transcribed from their own promoters or from intronic gene regions (reviewed in reference 26,27). MiRNAs form double-stranded complexes with target mRNAs, which signals either the degradation of the mRNA-miRNA complex or translational inhibition. As a result, a single miRNA can regulate multiple mRNAs, implicating miRNAs in substantially altering translation and enzymatic signaling. miRNAs are frequently altered in human cancers,26,27 and miRNA expression is also altered in human cancers via enhancer function, binding of hormones and growth factors at individual miRNA promoters, and miRNA promoter DNA hypermethylation.
Indeed, miRNA promoter DNA hypermethylation is prevalent in CRCs and virtually every cancer type, implicating these miRNAs as tumor suppressors. Saito et al.28 provided the first evidence of miRNA epigenetic silencing in cancer cells (miR-127), and additional miRNAs silenced by DNA hypermethylation have been described in CRC, the first of which was miR-124a,29 which allows for cyclin D kinase 6 activation and subsequent RB phosphorylation. Activated cyclin D kinase 6 acts in an oncogenic capacity in phosphorylating RB, thereby inactivating the enzyme, thus leading to loss of cell cycle control and tumor progression.30 Additional examples of miRNA silencing by DNA hypermethylation include: (1) miR34b and miR34c, which share a CpG island with the tumor suppressor gene B-cell translocation gene 4;31 (2) miR-137, which regulates the lysine specific demethylase KDM1A/LSD1,32 miR-342, which targets DNMT1, thereby leading to activation of RASSF1, ADAM23, RECK and HINT1 as a result of promoter DNA hypomethylation,33 as well as several others (reviewed in references 34-36). miRNA silencing via promoter DNA hypermethylation has downstream effects owing to the inability of the specific miRNA to regulate gene expression and cellular programming.
DNA methylation biomarkers of CRC
Cancer-specific DNA methylation can be identified not only in tumors, but also in adenomas, circulating tumor DNA, cfDNA (cell-free DNA) in patient blood (plasma/serum), urine and fecal matter as sensitive (early) detection protocols (reviewed in reference 37). DNA hypermethylation of key CRC epigenetic driver genes has been identified in human stool/fecal matter (MLH1, CDKN2A, MGMT, VIM, SFRP2),38-44 urine (VIM, WIF1, ALX4, NDRG4 (reviewed in reference 45) and blood (MLH1, APC, MGMT, RASSF2A, TMEFF2 (reviewed in reference 37). DNA methylation markers of CRC tumor recurrence and patient survival have also been identified46-49 and include CDKN2A, HLTF and TPEF. Finally, CRC patients with promoter DNA hypermethylation of p14ARF, RASSF1 or APC1A showed poor prognosis, while patients with MGMT promoter DNA methylation show improved prognosis.50
DNA methylation-based biomarkers with unknown biological relevance to CRC have also been recently identified through genome-wide and genome-scale approaches.51,52 Recent examples include THBS1, C9ORF50 and SEPT9. THBS and C9ORF50 were identified by Lange et al.52 from publicly-available TCGA DNA methylation data, and displayed CRC-specific DNA hypermethylation after comparison with 14 other tumor types. Both markers validated in cfDNA in pre-therapeutic plasma and serum samples from CRC patients, and outperformed the carcino-embryonic antigen blood test used in the clinic with respect to tumor detection sensitivity and specificity.
SEPT9 was first identified from a genome-wide screen of CRC-specific DNA methylation profiles.53 SEPT9 DNA methylation of the v2 variant promoter region occurs in nearly all colorectal tumors and adenomas, but not in normal colonic mucosa. The first release of the cfDNA SEPT9 DNA methylation assay showed high sensitivity (72%) and specificity (86%) of CRC detection in plasma,53 and evaluations of an updated version of the assay (Epi proColon 2.0) showed similar sensitivities (68–95%) and specificities (80–99%) in detecting CRCs in blood plasma. Interestingly, the Epi proColon 2.0 assay identified stage I disease in 60–84% of cases and 80–100% of stage II disease (reviewed in reference 54). Importantly, DNA methylation of SEPT9 and TAC1 in post-operative serum blood samples serves as independent predictors of CRC recurrence and patient survival.55 The SEPT9 assays outperform fecal occult blood and carcino-embryonic antigen tests with respect to detection sensitivity and specificity. The SEPT9 DNA methylation assays, unlike colonoscopy, are noninvasive, cost effective and do not require outpatient medical procedures. SEPT9 is one example of the power of DNA methylation biomarkers as clinically important and effective means of CRC detection.
CPG ISLAND METHYLATOR PHENOTYPES (CIMPS) IN CRC
DNA methylation aberrancies substantially outnumber somatic mutations in human cancers,56 and individual tumor types can be stratified into subgroups based on DNA methylation profiles. In 1999, Toyota and colleagues first identified a unique subset of colorectal tumors positive for a CIMP (now classified as CIMP-high (CIMP-H)) that display extensive DNA hypermethylation at a unique set of CpG islands that remained unmethylated in other colorectal tumors and normal tissues57 (Figure 1). Follow-up experiments showed that CIMP-H tumors are preferentially located in the proximal (right) colon, are enriched in women of older age, patients with a family history of CRC, and harbor the BRAF V600E (BRAF-mut) point mutation, as well as MLH1 epigenetic silencing due to promoter DNA hypermethylation, microsatellite instability (MSI), diploid copy number and the absence of TP53 mutations51,58 (Figure 2). Moreover, tumors with CIMP-H, MSI and BRAF-mut are positively associated with smoking51,59,60 and body mass index in women.51
Figure 1.
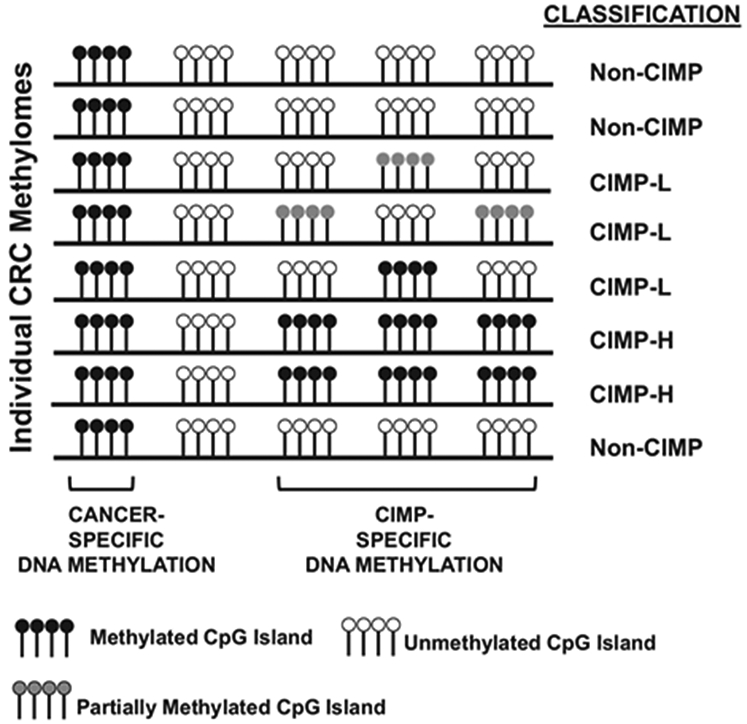
Description of CIMP-H, CIMP-L and non-CIMP CpG island DNA methylation in CRCs. Each row represents a CRC methylome, and lollipop clusters indicate CpG islands. Black lollipops indicate methylated CpG islands, white lollipops indicate unmethylated CpG islands and gray lollipops indicate partially methylated CpG islands. Classification of each methylome as CIMP-H, CIMP-L or non-CIMP are indicated to the right of each methylome. Tumor and CIMP-specific DNA methylation profiles are indicated in the figure.
Figure 2.
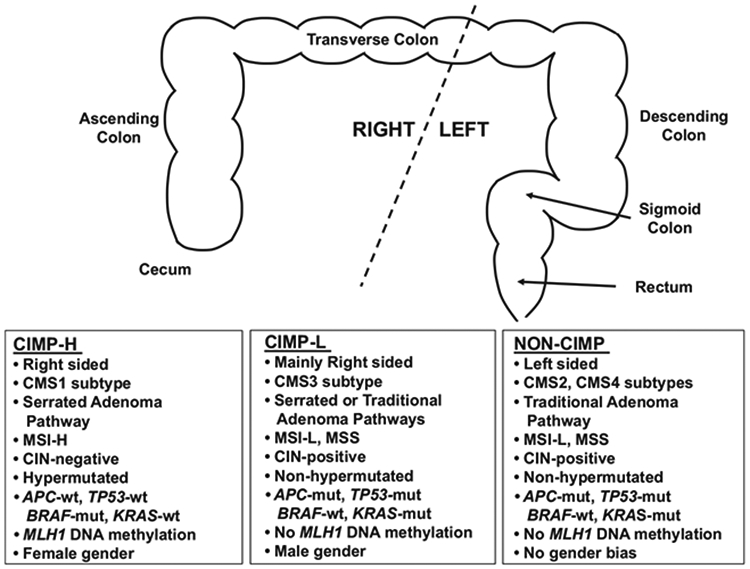
Description of CIMP-H, CIMP-L and non-CIMP tumors. Top section, graphic representation of the colorectum, stratified by location as left or right sides. Bottom section, correlations of each CIMP subgroup with location, CMS subtype, adenoma pathway, mutation status, MLH1 DNA methylation status and gender bias.
A second CIMP subgroup, CIMP-low (CIMP-L) was identified as another distinct subgroup of CRCs.61 CIMP-L tumors display an attenuated and partial DNA methylation status at CIMP-defining regions, both with respect to the number of methylated CIMP loci and their DNA methylation levels (Figure 1). In support of this, a genome-scale DNA methylation analysis of primary CRCs showed that 20% of CIMP-H sites are also methylated in CIMP-L tumors.62 CIMP-L tumors are enriched in KRAS and TP53 mutations and male gender (Figure 2). Although generally located on the right side of the colon, CIMP-L tumors are not like CIMP-H tumors as they are non-hypermutated, chromosomal instability positive and do not harbor MLH1 DNA hypermethylation.
Shen et al.63 reported the CIMP2 subgroup along with CIMP1 (CIMP-H) and non-CIMP subgroups. Like non-CIMP tumors, CIMP2 tumors also harbor KRAS and TP53 mutations, but are generally located in the proximal colon. Yagi et al.64 categorized three CRC subgroups, with the high-methylation epigenotype enriched for BRAF mutations and the intermediate-methylation epigenotype harboring KRAS mutations. Finally, Hinoue et al62 and TCGA8 categorized CRCs into four groups based on unsupervised clustering: CIMP-H, CIMP-L and two non-CIMP groups.
An analysis of the consequences of CIMP DNA methylation on gene expression showed that only a small percentage (7%) of genes with DNA hypermethylation in CIMP-H tumors were downregulated in expression.62 However, these appear to be enriched for genes essential for CRC tumorigenesis. Indeed, a search for epigenetic driver genes in colorectal tumors identified a handful of genes displaying both DNA hypermethylation and gene expression reduction in CIMP-H and non-CIMP tumors. Interestingly, these include SFRP1, SFRP2, FOXD2 and TMEFF2/HPP1, and are key regulators of the WNT pathway (SFRP1, SFRP2), have roles in transcription factor associated gene regulation (FOXD2) and coordinate cellular proliferation, differentiation and apoptosis (TMEFF2/HPP1). These epigenetic driver genes are attractive therapeutic targets, such that their activation by treatment with DNA methylation inhibitors may result in resetting of cellular programs.
CONSENSUS MOLECULAR SUBGROUP (CMS) CLASSIFICATION OF CRC BASED ON GENETIC AND EPIGENETIC ALTERATIONS
The CRC Subtyping Consortium performed comprehensive cross-comparative analyses of tumor subtype assignments based on publicly-available molecular data sets and six existing algorithms for determining CRC subgroups to assess whether the subtype assignments correlated with patient outcome, and ultimately, to institute a translational approach for the use of molecular subtypes in the clinic.65 The classifier identified four CMS of CRC: CMS1 (MSI immune), CMS2 (Canonical), CMS3 (Metabolic) and CMS4 (Mesenchymal) (Figure 2). CMS1 tumors display CIMP-H, MSI, BRAF-mut, DNA hypermutation, as well as immune infiltration and activation, and CMS1 patients show poor survival after relapse. CMS2 tumors are non-CIMP, with SCNAs (somatic copy number alterations), as well as activation of WNT and MYC signaling. CMS3 tumors are characterized by CIMP-L, MSI/microsatellite stable (MSS) status, low SCNAs, KRAS-mut and metabolic dysregulation. Finally, CMS4 patients are also non-CIMP and display SCNAs, infiltration of stromal cells, as well as activated TGF-β and angiogenic signaling. In addition, CMS4 patients show worse relapse-free survival and overall survival (OS).
CRC THERAPEUTICS
The intersection of CIMP with EGFR and VEGFR signaling pathway status
The EGFR and VEGFR pathways are instrumental for determining appropriate and effective CRC treatments. EGFR, upon activation by binding EGF ligand, activates KRAS, which activates BRAF. Activated BRAF then stimulates MEK1 and MEK2, and subsequently ERK signaling. ERK signaling activates oncogenic transcription factors (MYC, ELK1, FOS, JUN) that in turn activate genes that drive cell proliferation, cell cycle progression and differentiation (Figure 3). The VEGFR pathway also overlaps with the EGFR pathway via RAS and BRAF signaling, but also activates transcription factors through PI3K/AKT/mTOR signaling to promote cell growth, differentiation and angiogenesis (Figure 3).
Figure 3.

EGFR and VEGFR signaling in CRCs. Protein signaling from EGF and VEGF binding to their respective receptors. Black arrows indicate traditional signaling, whereas green arrows indicate constitutive signaling. Red indicates inhibition of specific aspects of the pathways.
Even though large numbers of CRC genomes have been sequenced to identify potential novel drug targets, this has not yet resulted in the identification of new and highly penetrant mutations or novel therapeutics. Only RAS-mut status exists to guide CRC therapeutic decisions, however, 40–60% of RAS-wt tumors are resistant to EGFR-based treatments.66 The CRYSTAL and OPUS phase III clinical trials67,68 showed that adding cetuximab to FOLFOX (5-florouracil (5-FU), folinic acid and oxaliplatin) or FOLFIRI (5-FU, folinic acid and irinotecan) increased patient OS, progression-free survival (PFS) and objective response in first-line treatment of RAS-wt (KRAS or NRAS) metastatic CRCs, whereas patients with RAS mutations did not benefit from these treatment schemes. KRAS mutations predict resistance to EGFR-based antibodies (panitumumab and cetuximab), as KRAS is involved in signal transduction from ligand-bound EGFR from the cell membrane to the nucleus (Figure 3). DNA methylation alterations in KRAS-mut CRCs, especially those related to the CIMP-L CRC subset, may play important roles in EGFR resistance; however, CIMP-L-specific DNA methylation signatures have not yet been identified.
EGFR silencing may also be involved in drug resistance, especially in CIMP-H tumors that display extensive DNA hypermethylation and correlated gene repression. In support of this, Scartozzi et al.69 demonstrated EGFR promoter DNA hypermethylation in ~ 60% of primary colon tumors, and showed that patients with EGFR promoter DNA methylation have 5 month shorter PFS and ~ 12 month shorter OS than patients without EGFR promoter DNA methylation. This was supported by a recent report by Demurtas and colleagues in which 88 CRC patients were evaluated.70 In contrast, Geißler et al.71 showed that EGFR promoter DNA methylation, CIMP status and MSI status are not correlated with patient response after treatment with cetuximab and/or panitumumab. However, it should be noted that only a small number (n = 25) of CRC patients were studied in the Geißler report, and only two were CIMP-positive, and highlights the importance of CIMP status in selecting patients for treatment.
Interestingly, the Geißler report also showed that treatment responses were linked to PIK3CA mutations, whereas non-responders were associated with ATM mutations and low CDH1 expression.71 ATM forms a protein complex with EGFR and causes AKT phosphorylation. E-cadherin is involved in recruiting and activating EGFR. In contrast, cells with low or no E-cadherin expression can bypass EGFR signaling and become resistant to EGFR-based antibodies. Therefore, restoring E-cadherin expression is an important facet of tumor sensitivity to EGFR-targeted antibodies.
Tumor location and CIMP status as predictive and prognostic tool
As CIMP-H tumors are mainly found in the right side of the colon, tumor location may be an important determinant of CRC patient outcome. Left-sided tissues derive from the hindgut, whereas right-sided tissues arise from the midgut. Indeed, major differences in embryonic patterning genes and crypt stem cell populations begin in normal colonic tissue in both mouse and human systems.72-74 Left- and right-sided colon cancers differ extensively in terms of gene expression, DNA mutation and DNA methylation profiles,72 however, how these relate to embryological origin or other site-associated factors, is still unknown. Clinically, left- and right-sided colon tumors have different epidemiologic trends and outcomes. Driver germline genetic alterations in hereditary syndromes show non-random propensity to develop left- or right-sided polyps and tumors. Lynch syndrome patients, for example, predominantly develop right-sided tumors. Most sporadic colon cancers are left-sided, whereas a minority of sporadic tumors are right-sided and have unique molecular profiles.
In the Cancer and Leukemia Group B and Southwest Oncology Group 80 405 trial, 1104 metastatic colon cancer patients with a KRAS-wt genotype were treated with either FOLFIRI or FOLFOX prior to adjuvant therapy with cetuximab or bevacizumab. Although overall patient survival rates were not significantly different between treatment arms, significant differences in OS and PFS were evident after stratification by tumor location (left vs right),75,76 suggesting that tumor location is an important predictor of patient outcome. Tejpar et al.77 supported this observation by showing that RAS-wt patients with left-sided tumors had improved PFS and OS compared with patients with right-sided tumors. Moreover, patients with right-sided RAS-wt tumors showed comparable to nominally improved treatment efficacy by adding cetuximab to FOLFIRI, but a marked improvement of the same treatment regimen in CRC patients with left-sided tumors and RAS-wt status.
Loupakis et al.78 showed that patients with right-sided (generally CIMP-H) tumors exhibit shorter OS and PFS, as well as higher mortality rates than patients with left-sided tumors (generally non-CIMP). An analysis of CIMP data from over 10 000 patients79 showed that CIMP-H CRC patients display shorter disease-free survival (DFS) and OS than non-CIMP patients, both with MSI or MSS disease. These results were supported by a recent study from the HE6C/05 trial of 441 patients with stage II/III disease treated with either XELOX (capecitabine, oxaliplatin) or FOLFOX.80 No differences in survival were found between CIMP-H and non-CIMP patients, however, stage II patients showed lower risk of relapse, and patients with lower stage and left-sided tumors displayed a lower risk of death.
The prognostic value of CIMP is not well understood. In agreement with other clinical trials, CIMP-H, stage III CRC patients had shorter OS after surgical resection than CIMP-negative patients,81 and Ahn et al.82 showed that CIMP-H, BRAF-mut and proximal location correlated with a significantly worse DFS. However, the prognostic utility of CIMP status in mixtures of stage II and III patients shows either a decreased DFS in CIMP-positive patients or no difference in DFS between CIMP and non-CIMP patients (summarized in reference83).
CIMP-positive patients show improved survival after 5-FU treatment. DNA hypermethylation-associated silencing of DPYD (dihydropyrimidine dehydrogenase), a gene specific for 5-FU degradation, is prevalent in CIMP-positive CRC patients, and may explain the correlation between CIMP status and 5-FU sensitivity84 (reviewed in83). Moreover, DNA hypermethylation-based silencing of GGH (gamma-glutamyl hydrolase), a regulator of folate levels for methyl-transfer and nucleotide biosynthesis, may also help explain this association. However, this may also be confounded by MSI, which although enriched in CIMP-positive tumors, is also present in non-CIMP tumors.83 Nonetheless, the CIMP-specific DNA methylation and silencing of DPYD may impede the tumor cells ability to degrade and deactivate 5-FU, thereby resulting in drug sensitivity, whereas epigenetic silencing of GGH may result in increased cellular folate concentrations and dysregulated nucleotide synthesis.85
The association of CIMP with positive response to therapeutic agents also appears to be limited to 5-FU adjuvant therapy. In one study,86 CIMP patients showed improved outcome after 5-FU treatment and improved OS after FOLFIRI treatment as compared with non-CIMP patients. A report from Van Rijnsoever showed that CIMP-positive patients show shorter OS as compared with non-CIMP patients, yet CIMP patients showed improved OS after 5-FU adjuvant treatment.81 A survival benefit was also shown for stage II CRC patients after 5-FU treatment.87 Finally, Min et al.88 showed that CIMP-positive patients had improved DFS after 5-FU treatment.
DNA methylation of candidate genes and repetitive elements can be used for prognostic and predictive purposes. DNA hypomethylation of LINE-1-repetitive elements, a surrogate marker for global DNA methylation, is associated with poor patient outcome, and worse PFS and OS following FOLFOX-based chemotherapy. LINE-1 DNA hypomethylation is independently associated with poor prognosis as well as resistance to FOLFOX treatment.89 In addition, DNA methylation of HYLA2 (hyaluronoglucosaminidase 2) was associated with positive response of 5-FU in stage II and III CRC patients.90
Mechanisms of CIMP-specific DNA methylation in BRAF-mutant CRCs
BRAF-mut results in a constitutively active BRAF protein and resistance to MEK and EGFR inhibitors. In addition, the relationship of BRAF-mut and CIMP, whereas highly associated, has not been understood until recently, nor are the reasons as to why CIMP-H tumors are almost entirely KRAS-wt and CIMP-L tumors are mainly KRAS-mut. Fang et al.91 identified the transcriptional repressor MAFG as essential for CIMP-H DNA hypermethylation and silencing in BRAF-mut tumors. MAFG binds to CIMP-H genes, and recruits co-repressor proteins including BACH1, the chromatin remodeler CHD8 and DNMT3B to methylate and silence CIMP-H target genes (Figure 4). MAFG is overexpressed in CRCs, and MAFG-binding sites are located in the majority of CIMP-H-defining genes.
Figure 4.

Effects of KRAS-mut and BRAF-mut on EGFR signaling. Models are based on previously described reports.91,92 Left panel: BRAF-mut constitutively activates MEK and ERK signaling. The ERK enzyme phosphorylates MAFG, stabilizing the protein and allowing MAFG to bind to CIMP-H target regions. MAFG recruits BACH1 and CHD8 co-repressors, as well as DNMT3B to place DNA methylation marks at CIMP-H loci. Right panel: KRAS-mut activates PKRD1, which phosphorylates USP28 (PUSP28), thereby activating the protein. PUSP28 removes ubiquitin moieties from ZNF304, thus allowing ZNF304 to bind to CIMP-L-defining loci. ZNF304 binding recruits KAP1, SETDB1 co-repressors, as well as DNMT1, which is thought to methylate CIMP-L loci. Black arrows indicate traditional signaling, whereas green arrows indicate constitutive signaling. Red indicates inhibition of specific aspects of the pathways.
MAFG is a substrate for the BRAF/MEK/ERK pathway. The BRAF-mut protein is constitutively active in CIMP-H tumors, leading to increased BRAF/MEK/ERK signaling. ERK then phosphorylates MAFG, resulting in decreased MAFG ubiquitination and subsequent MAFG protein stability (Figure 4). Ultimately, this leads to high expression of MAFG protein, resulting in MAFG binding to CIMP-H defining loci. This model suggests a direct connection between BRAF-mut activity, MAFG levels and CIMP-H-specific DNA methylation. This signaling system appears to be specific for BRAF-mut CRCs, as MAFG knockdown in KRAS-mut tumors did not show an effect. However, KRAS-mut CRCs utilize a unique set of co-repressors and include ZNF304, which recruits KAP1, SETDB1 and DNMT1 to CIMP-L target regions, resulting in DNA hypermethylation and gene silencing92 (Figure 4). ZNF304 does not have a role in BRAF-mut CRCs, thus indicating two separate pathways for DNA methylation in CIMP-H and CIMP-L CRCs (Figure 4).
DNA METHYLATION INHIBITION AS A THERAPEUTIC APPROACH FOR CANCER TREATMENT
5-Azacytidine-based DNA methylation inhibitors
The extensive DNA methylation alterations in CRC patients suggest that a substantial number of patients may benefit from epigenetic therapies, especially with DNA methylation inhibitors. Small molecule DNA methylation inhibitors, such as nucleoside and non-nucleoside based molecules, have played important roles in understanding human methylomes in normal and tumor cells. The first DNA methylation inhibitors, 5-azacytidine (5-Aza-CR) and 5-aza-2’-deoxycytidine (5-Aza-CdR), were designed and synthesized in the 1960s by Sorm and colleagues (reviewed in reference 93) as cytotoxic anticancer drugs, akin to 5-FU. Aza-substituted analogs are converted to Aza-triphosphates after entering the nucleus, and are then incorporated into newly synthesized DNA during DNA replication. Aza-incorporated DNA traps DNMTs to genomic DNA, leading to their depletion and passive DNA demethylation.94,95
A modification of 5-Aza-CdR, Guadecitabine (SGI-110), was more recently developed, and consists of 5-Aza-CdR followed by deoxyguanosine.96,97 SGI-110 shows promising clinical utility, displays improved stability and lower toxicity over 5-Aza-CdR alone, is better tolerated upon delivery, and is effective in mice and patient-derived xenograft models of cancer.96 Currently, SGI-110 is under evaluation in clinical trials for CRC patients, as well as patients with other malignancies.
DNA methylation as a therapeutic target
The DNA demethylation and gene activation aspects of 5-Aza-CdR have been well characterized with respect to tumor suppressor and DNA repair systems. However, 5-Aza-CdR treatment also reduces the overexpression of genes through DNA demethylation of gene bodies and transcribed regions, which are normally methylated in actively expressed genes.18 Genes whose expression is downregulated after 5-Aza-CdR treatment include oncogenes and those involved in c-MYC regulated processes (Figure 3), suggesting that combining 5-Aza-CdR with EGFR and VEGFR therapies may have synergistic anti-tumor effects.
In addition, ERVs (endogenous retroviruses) and other repetitive elements located in transcribed (gene body) regions, normally silenced by DNA methylation in cancer cells, are reactivated after treatment with DNA methylation inhibitors.98-101 Demethylated and activated ERVs trigger activation of an interferon response and essentially mislead the cancer cell to operate in a viral-infected state, and therefore susceptible to immuno-modulating drugs that have shown success in the clinic (Figure 5). Li et al.102 showed that treatment of human colon cancer cell lines with 5-Aza-CR resulted in activation of immunomodulatory pathways, namely interferon, inflammation, cytokine/chemokine and cancer testis antigen-signaling pathways. The activation of an immune response, coupled with activation of tumor suppressors (p14, p15, p16), DNA repair genes (MLH1, MGMT) and the reduction in oncogene (MYC) expression, provide substantial evidence of the efficacy of DNA methylation inhibition as a treatment option for CRC patients (Figure 5).
Figure 5.

Potential efficacy of DNA methylation inhibition for CRC therapy. Top, promoter and gene body DNA methylation in normal somatic cells. Black lollipops indicate methylated CpG islands, white lollipops indicate unmethylated CpG islands. Middle, promoter (left) or gene body (right) DNA hypermethylation in human cancers. Bottom, promoter DNA hypermethylation may correlate with gene silencing, whereas gene body DNA methylation is associated with actively expressed genes. Treatment with DNA methylation inhibitors results in demethylation of gene promoters and gene body regions, resulting in activation of tumor suppressors, DNA repair response, miRNAs and ERVs, with suppression of oncogenes.
TET enzyme-based DNA demethylation
Although DNMTs and DNA methylation inhibitors are well described, DNA demethylases have only been recently characterized. The Ten Eleven Translocase (TET) family of enzymes (TET1, TET2, TET3) were shown to convert 5-methylcytosine to 5-hydroxymethylcytosine using ascorbic acid (vitamin C) as a co-factor.103,104 TETs can further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine,105,106 with both 5-formylcytosine and 5caC marks removed and replaced with an unmethylated cytosine residue via DNA glycosylase-involved base excision repair.
The correlation between TET activity and cancer-specific DNA methylation was first shown in human glioblastoma. A specific heterozygous point mutation in isocitrate dehydrogenase 1 results in the catalysis of an oncometabolite that inhibits DNA demethylation. Although isocitrate dehydrogenase 1 functions as a dimer in the citric acid cycle by converting isocitrate to alpha-ketoglutarate,107 mutant isocitrate dehydrogenase 1 further converts alpha-ketoglutarate to D-2-hydroxyglutarate,108 which is an inhibitor of TET activity109 and DNA demethylation, resulting in DNA hypermethylation.
Interestingly, only low frequency IDH and TET mutations were identified in CRCs,8 suggesting that CRC DNA methylation profiles are also generated independent of TET and IDH mutations alone. In addition to alpha-ketoglutarate, TET enzymes also require oxygen as a substrate for activity, as well as Fe(II) and vitamin C as cofactors.110 Many tumor types, including CRC, display hypoxia, described as decreased cellular oxygen levels, which inhibit TET function, thereby retaining DNA methylation profiles. In addition, cancer patients commonly present with vitamin C deficiency,111 also resulting in TET enzyme inhibition and retained DNA methylation.
Vitamin C is an effector of 5-Aza-CdR based DNA demethylation. Combining vitamin C with 5-Aza-CdR treatment of cancer cells results in a synergistic boost in DNA demethylation, as both active (TET) and passive (Aza) mechanisms of DNA demethylation are activated.102 Vitamin C enhances the activation of ERVs, tumor suppressors, DNA repair genes and other genes silenced by DNA promoter hypermethylation. Vitamin C is orally available, cost-effective and only physiological concentrations (57 μm) are required for synergistic DNA demethylation.
DNA METHYLATION INHIBITION AS A TREATMENT OPTION FOR CRC PATIENTS
Treatment of human colon cancer cell lines with DNA methylation inhibitors has provided evidence for potential treatment efficacy. In one report, human colon cancer cell lines were treated with combinations of conventional chemotherapies (5-FU, irinotecan, oxaliplatin), DNA methylation inhibitors (5-Aza-CR, 5-Aza-CdR, zebularine) and histone deacetylase inhibitors (Trichostatin A, SAHA, valproic acid) to determine whether epigenetic therapies improve tumor toxicity.112 The addition of DNA methylation inhibitors resulted in synergistic effects incurred by chemotherapy, and in particular, 5-Aza-CdR showed the most potent synergistic effect and enhanced oxaliplatin cytotoxicity. Moreover, 5-Aza-CdR added to 5-FU or oxaliplatin treatments of CRC cell lines showed synergism based on cell viability and cell counts.113
Early clinical trials evaluating efficacy of 5-Aza based DNA methylation inhibitors in CRC patients have shown inconsistent findings. A total of 11 clinical trials involving 5-Aza-CR (Vidaza), 5-Aza-CdR (Decitabine) or SGI-110 are active or have been completed (Table 1; clinicaltrials.gov). A recent phase II study of 5-Aza-CR and the histone deacetylase inhibitor entinostat,114 although tolerated, did not result in clinical activity. However, DNA demethylation occurred in a subset of patients and was correlated with improved PFS. A separate phase I/II trial115 was performed by treating CIMP-H CRC patients who are resistant to 5-FU and oxaliplatin with 5-Aza-CR and CAPOX (capecitabine and oxaliplatin). DNA demethylation was detected, but did not correlate with occurrence of stable disease. Moreover, CIMP status did not correlate with stable disease or PFS, suggesting that evaluating additional drug combinations, both in the clinic and in the laboratory, are required to determine treatment efficacy for CRC patients. Finally, a phase I/II trial116 to assess the performance of 5-Aza-CdR and panitumumab in metastatic CRC patients with KRAS-wt tumors showed tolerance and activity to this drug combination. Partial responses were observed in 2/20 (10%) of patients and stable disease was observed in 10/20 (50%) of patients, suggesting that this drug combination may improve survival and quality of life in patients with metastatic colon cancer.
Table 1.
Clinical trials involving DNA methylation inhibitors listed on www.clinicaltrials.gov
| Number | Trial ID | Drug | Phase | Clinical test |
|---|---|---|---|---|
| 1 | NCT01193517 | 5-Azacitidine (Vidaza), capecitabine and oxaliplatin | Phase I/II | Phase I: Find the highest tolerable dose of 5-Azacitidine with capecitabine and oxaliplatin (CAPOX). Phase II: Determine whether 5-Azacitidine with CAPOX is effective in metastatic colon cancer patients |
| 2 | NCT02260440 | 5-Azacitidine and Pembrolizumab | Phase II | To determine the anti-tumor, safety and tolerability of 5-Azacitidine and Pembrolizumab in patients with chemo-resistant metastatic CRC |
| 3 | NCT01105377 | 5-azacitidine and entinostat | Phase II | Efficacy of 5-azacitidine and entinostat in metastatic CRC patients |
| 4 | NCT02316028 | Decitabine | Phase I | Efficacy of Decitabine delivered by hepatic arterial infusion in patients with non-resectable liver metastases |
| 5 | NCT02959437 | 5-Azacitidine with Pembrolizumab and Epacadostat | Phase I/II | Phase I: Dose escalation of 5-Azacitidine with Pembrolizumab and Epacadostat. Phase II: Expansion cohort of MSS CRC patients with recommended dose from Phase I. |
| 6 | NCT02811497 | 5-Azacitidine and Durvalumab | Phase II | To determine the anti-tumor activity of 5-Azacitidine and durvalumab in MSS CRC patients |
| 7 | NCT02512172 | Romidepsin and/or 5-Azacitidine with MK-3475 | Phase I | To evaluate the anti-tumor activity of Romidepsin and/or 5-Azacitidine with MK-3475 in MSS CRC patients |
| 8 | NCT00879385 | Ddcitabine and panitumumab | Phase I | To determine the clinical utility of Decitabine and panitumumab in KRAS-wt in second or third line treatment of metastatic CRC patients |
| 9 | NCT01966289 | SGI-110 with Allogeneic Colon Cancer Cell Vaccine (GVAX) and Cyclophosphamide (CY) | Phase I | SGI-110 with Allogeneic Colon Cancer Cell Vaccine (GVAX) and Cyclophosphamide (CY) in metastatic CRC patients |
| 10 | NCT01896856 | SGI-110 with irinotecan or regorafenib | Phase I/II | Phase I: SGI-110 and/or irinotecan. Phase II: SGI-110 and/or irinotecan vs regorafenib |
| 11 | NCT01882660 | Decitabine | To determine whether: (1) pre-operative decitabine treatment increases Wnt-target gene expression; (2) DNA demethylation indices more favorable tumor characteristics, and (3) to measure Wnt signaling, DNA methylation and tumor characteristics comparing Wnt methylated vs Wnt unmethylated tumors |
CONCLUSIONS AND FUTURE DIRECTIONS
DNA methylation alterations are not only abundant in CRCs, but also have clinical importance. The correlation of CIMP-H with BRAF-mut involves cell-signaling aberrancies that dictate the types of effective treatments for CRC patients. However, DNA methylation inhibition is only at the clinical trial phase for treating CRC patients. There is substantial evidence that DNA methylation inhibition by 5-Aza-CdR/SGI-110 and vitamin C sensitizes the tumor cells to traditional chemotherapies, immune-based therapies and DNA repair inhibitors. These attributes, as well as the activation of tumor suppressors and miRNAs and the down regulation of oncogenes by DNA demethylating agents, make DNA methylation inhibition an attractive therapeutic strategy. Combining DNA methylation inhibitors with EGFR antibodies may also show clinical promise, as the EGFR promoter is hypermethylated in a substantial proportion of CRCs, and DNA methylation inhibition may boost efficacy of EGFR inhibitors by blocking BRAF-mut signaling and reducing MYC signaling to inhibit cellular proliferation.
Combining DNA methylation inhibition with targeted agents, cytotoxic agents and immuno-modulating drugs in CIMP-H/CIMP-L patients will help determine the prognostic utility of CIMP status on response to treatment and patient outcome. Determining the DNA methylation status of gene regions and their correlation with clinical outcome is also important to determine whether CIMP or candidate gene regions are better predictive and prognostic biomarkers. These DNA methylation-based signatures, such as DNA methylation of MMP9 and RASSF1, can be used to determine specific patients who may benefit from epigenetic therapies.
One aspect of epigenetics that has been overlooked is the role of tumor heterogeneity in epigenetic targeting. Although this has been a focus of genetic and mutation-based analyses, the extent of DNA methylation heterogeneity is not fully understood in primary colorectal tumors. Only cellular-based tumor cell contamination, especially white blood cells, has been documented in DNA methylation-based analyses.117
Additional future directions involve the development of next-generation DNA methylation inhibitors, especially those that result in sustained DNA methylation inhibition concurrent with low cellular toxicity, as well as addressing drug activity and patient response using both quantitative and qualitative methods. There is a need for targeted delivery of DNA methylation inhibitors to specific tissues and/or tumor cells so as to achieve an optimal response and to avoid off-target effects. This is exceptionally challenging and requires engineering to not only target tumor cells but also ensure their delivery into the cell and nucleus. Combining highly focused epigenetic therapies with immune-modulating therapies, for instance, may be an effective strategy for targeted cancer treatment. Exploiting expressed cell surface markers for targeted therapy of specific tissue types may also provide an efficacious drug delivery system.
Determining treatment efficacy and patient response are also challenges that require the development of highly sensitive and specific biomarkers, and/or the ability to obtain quantitative data on circulating tumor DNA, cfDNA and small amounts of primary or metastatic tissues. These methods need to be time-effective so that treatments can be fine-tuned and tailored for each patient in order to achieve a durable and prolonged effect.
ACKNOWLEDGEMENTS
This work was supported by the Vicky Joseph Cancer Research Lab (to GL), NIH 5R21 CA201865 (to GL and DJW) and NIH/NCI P30 CA014089 (to DJW).
ABBREVIATIONS
- 2-HG
2-hydroxyglutarate
- 5-Aza-CR
5-aza-cytidine
- 5-Aza-CdR
5- aza-2’-deoxycytidine
- 5-FU
5-fluorouracil
- 5caC
5-carboxylcytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- α-KG
alpha-ketoglutarate
- ADAM2
ADAM metallopeptidase domain 2
- ADAM23
ADAM metallopeptidase domain 23
- APC
adenomatous polyposis coli
- ARMCX1
armadillo repeat containing, X-linked 1
- ATM
ataxia telangiectasia mutated
- BACH1
BTB domain and CNC homolog 1
- BCHE
butyrylcholinesterase
- BRAF
B-Raf proto-oncogene, serine/threonine kinase
- BRAF-mut
mutant BRAF
- BTG4
BTG anti-proliferation factor 4
- C9ORF50
chromosome 9 open reading frame 50
- CALGB
aancer and leukemia group B
- CAPOX
capecitabine and oxaliplatin
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- CDH1
E-cadherin
- CDO1
cysteine dioxygenase type 1
- CEA
carcino-embryonic antigen
- cfDNA
cell-free DNA
- CHD8
chromodomain-helicase-DNA-binding protein 8
- CIN
chromosomal instability
- CRC
colorectal cancer
- CIMP
CpG island methylator phenotype
- CIMP-H
CIMP-high
- CIMP-L
CIMP-low
- CMS
Consensus molecular subgroup
- CRCSC
CRC subtyping condortium
- ctDNA
circulating tumor DNA
- DCC
deleted in colorectal cancer
- DKO
DNMT1/DNMT3B double knockout
- DNMT
DNA Methyltransferase
- DNMT1
DNA methyltransferase 1
- DNMT3A
DNA methyltransferase 3A
- DNMT3B
DNA methyltransferase 3B
- DPYD
dihydropyrimidine dehydrogenase
- EGFR
epidermal growth factor receptor
- ES
embryonic stem
- ESX1
ESX homeobox 1
- EPCAM
epithelial cell adhesion molecule
- FOLFIRI
5-FU, folinic acid and irinotecan
- FOLFOX
folinic acid, 5-FU and oxaliplatin
- FOXD2
forkhead box D2
- GGH
gamma-glutamyl hydrolase
- H3K36me3
histone H3 lysine 36 trimethylation
- HER2
ERBB2 (erb-b2 receptor tyrosine kinase 2)
- HINT1
histidine triad nucleotide-binding protein 1
- HNPCC
hereditary non-polyposis colorectal cancer
- HPP
hyperplastic polyp
- IDH1
isocitrate dehydrogenase 1
- IME
intermediate methylation epigenotype
- IRAK3
interleukin receptor associated kinase 3
- KAP1
KRAB-associated protein 1
- KRAS
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- KRAS-mut
mutant KRAS
- MAFG
MAF BZIP transcription factor G
- MEK1
mitogen-activated protein kinase 1
- MEK2
mitogen-activated protein kinase 2
- MGMT
O-6-methylguanine-DNA methyltransferase
- miRNA
microRNA
- MMP9
matrix metallopeptidase 9
- MSH2
MutS homolog 2
- MSI
microsatellite instability
- MSI-H
MSI-high
- MSI-L
MSI-low
- MLH1
Mut-L homolog 1
- MSH6
MutS homolog 6
- MSS
microsatellite stable
- MYC
MYC proto-oncogene, bHLH transcription factor
- OS
overall survival
- P2RY14
purigenic receptor P2Y14
- PFS
progression-free survival
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PMS1
post-meiotic separation increased 1
- RASSF1
RAS association domain family member 1
- RB
retino-blastoma
- RECK
reversion inducing cysteine-rich protein with kazal motifs
- RFS
relapse-free survival
- RUNX3
Runt-related transcription factor 3
- SAHA
suberoylanilide hydroxamic acid
- SAM
S-adenosylmethionine
- SCNA
somatic copy number alteration
- SEPT9
septin 9
- SETDB1
SET domain bifurcated 1
- SFRP
secreted frizzled-related protein 1
- SGI-110
Guadecitabine
- SMAD2
SMAD family member 2
- SMAD4
SMAD family member 4
- SSA
sessile serrated sdenoma
- SWOG
southwest oncology group
- SYCP3
synaptonemal complex protein 3
- TAC1
techykinin precursor 1
- TCGA
The cancer genome atlas
- TET
Ten eleven translocase
- TGF-β
transforming growth factor beta
- THBS1
thrombospondin 1
- TMEFF2
transmembrane protein with EGF-like and two follistatin-like domains 2
- TP53
Tumor protein 53
- TPEF
transmembrane protein with EGF-like and two follistatin-like domains 2
- TSA
traditional serrated adenoma
- VEGFR
vascular endothelial growth factor receptors
- VIM
vimentin
- WNT
wingless-related integration site
- ZNF304
zinc finger protein 304
Footnotes
CONFLICT OF INTEREST
DJW is a consultant for Zymo Research Corporation. Zymo did not contribute to this report, nor has an interest in this research.
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, Stintzing S. FOLFIRI with cetuximab or bevacizumab: FIRE-3-authors' reply. Lancet Oncol 2014; 15: e583–584. [DOI] [PubMed] [Google Scholar]
- 3.Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med 2014; 371: 1609–1618. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 2015; 15: 181–194. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. [DOI] [PubMed] [Google Scholar]
- 6.Zarate R, Boni V, Bandres E, Garcia-Foncillas J. MiRNAs and LincRNAs: could they be considered as biomarkers in colorectal cancer? Int J Mol Sci 2012; 13: 840–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas Research Network, Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathol 2013; 62: 367–386. [DOI] [PubMed] [Google Scholar]
- 10.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA 2000; 97: 5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13: 484–492. [DOI] [PubMed] [Google Scholar]
- 12.Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis 2000; 21: 461–467. [DOI] [PubMed] [Google Scholar]
- 13.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009; 10: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egger G, Jeong S, Escobar SG, Cortez CC, Li TW, Saito Y et al. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci USA 2006; 103: 14080–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000; 404: 1003–1007. [DOI] [PubMed] [Google Scholar]
- 16.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002; 416: 552–556. [DOI] [PubMed] [Google Scholar]
- 17.Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun 2016; 7: 11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014; 26: 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science 1981; 212: 1350–1357. [DOI] [PubMed] [Google Scholar]
- 20.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301: 89–92. [DOI] [PubMed] [Google Scholar]
- 21.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 1983; 11: 6883–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinstein IB. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis 2000; 21: 857–864. [DOI] [PubMed] [Google Scholar]
- 23.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21: 3214–3231. [DOI] [PubMed] [Google Scholar]
- 24.De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012; 21: 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36: 417–422. [DOI] [PubMed] [Google Scholar]
- 26.Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol 2014; 24: R762–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kita Y, Vincent K, Natsugoe S, Berindan-Neagoe I, Calin GA. Epigenetically regulated microRNAs and their prospect in cancer diagnosis. Exp Rev Mol Diagn 2014; 14: 673–683. [DOI] [PubMed] [Google Scholar]
- 28.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443. [DOI] [PubMed] [Google Scholar]
- 29.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007; 67: 1424–1429. [DOI] [PubMed] [Google Scholar]
- 30.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006; 25: 5220–5227. [DOI] [PubMed] [Google Scholar]
- 31.Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68: 4123–4132. [DOI] [PubMed] [Google Scholar]
- 32.Balaguer F, Link A, Lozano JJ, Cuatrecasas M, Nagasaka T, Boland CR et al. Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res 2010; 70: 6609–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Wu J, Meng X, Ying X, Zuo Y, Liu R et al. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 2011; 32: 1033–1042. [DOI] [PubMed] [Google Scholar]
- 34.Kaur S, Lotsari-Salomaa JE, Seppanen-Kaijansinkko R, Peltomaki P. MicroRNA methylation in colorectal ancer. Adv Exp Med Biol 2016; 937: 109–122. [DOI] [PubMed] [Google Scholar]
- 35.Menigatti M, Staiano T, Manser CN, Bauerfeind P, Komljenovic A, Robinson M et al. Epigenetic silencing of monoallelically methylated miRNA loci in precancerous colorectal lesions. Oncogenesis 2013; 2: e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan H, Choi AJ, Lee BH, Ting AH. Identification and functional analysis of epigenetically silenced microRNAs in colorectal cancer cells. PloS ONE 2011; 6: e20628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toth K, Bartak BK, Tulassay Z, Molnar B. Circulating cell-free nucleic acids as biomarkers in colorectal cancer screening and diagnosis. Exp Rev Mol Diagn 2016; 16: 239–252. [DOI] [PubMed] [Google Scholar]
- 38.Bosch LJ, Carvalho B, Fijneman RJ, Jimenez CR, Pinedo HM, van Engeland M et al. Molecular tests for colorectal cancer screening. Clin Colorectal Cancer 2011; 10: 8–23. [DOI] [PubMed] [Google Scholar]
- 39.Chen WD, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst 2005; 97: 1124–1132. [DOI] [PubMed] [Google Scholar]
- 40.Kisiel JB, Yab TC, Taylor WR, Chari ST, Petersen GM, Mahoney DW et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer 2012; 118: 2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenhard K, Bommer GT, Asutay S, Schauer R, Brabletz T, Goke B et al. Analysis of promoter methylation in stool: a novel method for the detection of colorectal cancer. Clin Gastroenterol Hepatol 2005; 3: 142–149. [DOI] [PubMed] [Google Scholar]
- 42.Mansour H Cell-free nucleic acids as noninvasive biomarkers for colorectal cancer detection. Front Genet 2014; 5: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberwalder M, Zitt M, Wontner C, Fiegl H, Goebel G, Zitt M et al. SFRP2 methylation in fecal DNA--a marker for colorectal polyps. Int J Colorectal Dis 2008; 23: 15–19. [DOI] [PubMed] [Google Scholar]
- 44.Petko Z, Ghiassi M, Shuber A, Gorham J, Smalley W, Washington MK et al. Aberrantly methylated CDKN2A, MGMT, and MLH1 in colon polyps and in fecal DNA from patients with colorectal polyps. Clin Cancer Res 2005; 11: 1203–1209. [PubMed] [Google Scholar]
- 45.Altobelli E, Angeletti PM, Latella G. Role of urinary biomarkers in the diagnosis of adenoma and colorectal cancer: a systematic review and meta-analysis. J Cancer 2016; 7: 1984–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbst A, Wallner M, Rahmig K, Stieber P, Crispin A, Lamerz R et al. Methylation of helicase-like transcription factor in serum of patients with colorectal cancer is an independent predictor of disease recurrence. Eur J Gastroenterol Hepatol 2009; 21 : 565–569. [DOI] [PubMed] [Google Scholar]
- 47.Kou CH, Zhou T, Han XL, Zhuang HJ, Qian HX. Downregulation of mir-23b in plasma is associated with poor prognosis in patients with colorectal cancer. Oncol Lett 2016; 12: 4838–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lecomte T, Berger A, Zinzindohoue F, Micard S, Landi B, Blons H et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer 2002; 100: 542–548. [DOI] [PubMed] [Google Scholar]
- 49.Nakayama H, Hibi K, Takase T, Yamazaki T, Kasai Y, Ito K et al. Molecular detection of p16 promoter methylation in the serum of recurrent colorectal cancer patients. Int J Cancer 2003; 105: 491–493. [DOI] [PubMed] [Google Scholar]
- 50.Nilsson TK, Lof-Ohlin ZM, Sun XF. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. Int J Oncol 2013; 42: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weisenberger DJ, Levine AJ, Long TI, Buchanan DD, Walters R, Clendenning M et al. Association of the colorectal CpG island methylator phenotype with molecular features, risk factors, and family history. Cancer Epidemiol Biomark Prev 2015; 24: 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lange CP, Campan M, Hinoue T, Schmitz RF, van der Meulen-de Jong AE, Slingerland H et al. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PloS ONE 2012; 7: e50266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.deVos T, Tetzner R, Model F, Weiss G, Schuster M, Distler J et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin Chem 2009; 55: 1337–1346. [DOI] [PubMed] [Google Scholar]
- 54.Payne SR. From discovery to the clinic: the novel DNA methylation biomarker (m)SEPT9 for the detection of colorectal cancer in blood. Epigenomics 2010; 2: 575–585. [DOI] [PubMed] [Google Scholar]
- 55.Tham C, Chew M, Soong R, Lim J, Ang M, Tang C et al. Postoperative serum methylation levels of TAC1 and SEPT9 are independent predictors of recurrence and survival of patients with colorectal cancer. Cancer 2014; 120: 3131–3141. [DOI] [PubMed] [Google Scholar]
- 56.Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3: 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–793. [DOI] [PubMed] [Google Scholar]
- 59.Limsui D, Vierkant RA, Tillmans LS, Wang AH, Weisenberger DJ, Laird PW et al. Cigarette smoking and colorectal cancer risk by molecularly defined subtypes. J Natl Cancer Inst 2010; 102: 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samowitz WS, Albertsen H, Sweeney C, Herrick J, Caan BJ, Anderson KE et al. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst 2006; 98: 1731–1738. [DOI] [PubMed] [Google Scholar]
- 61.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn 2006; 8: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA 2007; 104: 18654–18659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yagi K, Akagi K, Hayashi H, Nagae G, Tsuji S, Isagawa T et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res 2010; 16: 21–33. [DOI] [PubMed] [Google Scholar]
- 65.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21: 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26: 374–379. [DOI] [PubMed] [Google Scholar]
- 67.Bokemeyer C, Kohne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer 2015; 51: 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Cutsem E, Lenz HJ, Kohne CH, Heinemann V, Tejpar S, Melezinek I et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015; 33: 692–700. [DOI] [PubMed] [Google Scholar]
- 69.Scartozzi M, Bearzi I, Mandolesi A, Giampieri R, Faloppi L, Galizia E et al. Epidermal growth factor receptor (EGFR) gene promoter methylation and cetuximab treatment in colorectal cancer patients. Br J Cancer 2011; 104: 1786–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demurtas L, Puzzoni M, Giampieri R, Ziranu P, Pusceddu V, Mandolesi A et al. The role of primary tumour sidedness, EGFR gene copy number and EGFR promoter methylation in RAS/BRAF wild-type colorectal cancer patients receiving irinotecan/cetuximab. Br J Cancer 2017; 117: 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geissler AL, Geissler M, Kottmann D, Lutz L, Fichter CD, Fritsch R et al. ATM mutations and E-cadherin expression define sensitivity to EGFR-targeted therapy in colorectal cancer. Oncotarget 2017; 8: 17164–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Missiaglia E, Jacobs B, D'Ario G, Di Narzo AF, Soneson C, Budinska E et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol 2014; 25: 1995–2001. [DOI] [PubMed] [Google Scholar]
- 73.Powell AE, Vlacich G, Zhao ZY, McKinley ET, Washington MK, Manning HC et al. Inducible loss of one Apc allele in Lrig1-expressing progenitor cells results in multiple distal colonic tumors with features of familial adenomatous polyposis. Am J Physiol Gastrointest Liver Physiol 2014; 307: G16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Poulin EJ, Coffey RJ. LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br J Cancer 2013; 108: 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Venook AP, Niedzwiecki D, Lenz H-J, Innocenti F, Mahoney MR, O'Neil BH et al. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). J Clin Oncol 2014; 32: 5s (suppl; abstr LBA3). [Google Scholar]
- 76.Venook AP, Niedzwiecki D, Innocenti F, Fruth B, Greene C, O'Neil BH et al. Impact of primary (1°) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016; 34: (suppl; abstr 3504). [Google Scholar]
- 77.Tejpar S, Stintzing S, Ciardiello F, Tabernero J, Van Cutsem E, Beier F et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: retrospective analyses of the crystal and fire-3 trials. JAMA Oncol 2016; epub ahead of print 10 October 2016; doi: 10.1001/jamaoncol.2016.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loupakis F, Yang D, Yau L, Feng S, Cremolini C, Zhang W et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst 2015; 107: pii: dju427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol 2014; 25: 2314–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cohen SA, Wu C, Yu M, Gourgioti G, Wirtz R, Raptou G et al. Evaluation of CpG island methylator phenotype as a biomarker in colorectal cancer treated with adjuvant oxaliplatin. Clin Colorectal Cancer 2016; 15: 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res 2003; 9: 2898–2903. [PubMed] [Google Scholar]
- 82.Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011; 117: 1847–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gallois C, Laurent-Puig P, Taieb J. Methylator phenotype in colorectal cancer: a prognostic factor or not? Crit Rev Oncol Hematol 2016; 99: 74–80. [DOI] [PubMed] [Google Scholar]
- 84.Iacopetta B, Kawakami K, Watanabe T. Predicting clinical outcome of 5-fluorouracil-based chemotherapy for colon cancer patients: is the CpG island methylator phenotype the 5-fluorouracil-responsive subgroup? Int J Clin Oncol 2008; 13: 498–503. [DOI] [PubMed] [Google Scholar]
- 85.Kim SE, Hinoue T, Kim MS, Sohn KJ, Cho RC, Cole PD et al. gamma-Glutamyl hydrolase modulation significantly influences global and gene-specific DNA methylation and gene expression in human colon and breast cancer cells. Genes Nutr 2015; 10: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shiovitz S, Bertagnolli MM, Renfro LA, Nam E, Foster NR, Dzieciatkowski S et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterol 2014; 147: 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Donada M, Bonin S, Barbazza R, Pettirosso D, Stanta G. Management of stage II colon cancer - the use of molecular biomarkers for adjuvant therapy decision. BMC Gastroenterol 2013; 13: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Min BH, Bae JM, Lee EJ, Yu HS, Kim YH, Chang DK et al. The CpG island methylator phenotype may confer a survival benefit in patients with stage II or III colorectal carcinomas receiving fluoropyrimidine-based adjuvant chemotherapy. BMC Cancer 2011; 11:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaneko M, Kotake M, Bando H, Yamada T, Takemura H, Minamoto T. Prognostic and predictive significance of long interspersed nucleotide element-1 methylation in advanced-stage colorectal cancer. BMC Cancer 2016; 16: 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pfutze K, Benner A, Hoffmeister M, Jansen L, Yang R, Blaker H et al. Methylation status at HYAL2 predicts overall and progression-free survival of colon cancer patients under 5-FU chemotherapy. Genomics 2015; 106: 348–354. [DOI] [PubMed] [Google Scholar]
- 91.Fang M, Ou J, Hutchinson L, Green MR. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol Cell 2014; 55: 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Serra RW, Fang M, Park SM, Hutchinson L, Green MR. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. eLife 2014; 3: e02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raynal NJ-M, Issa JP. DNA methyltransferase inhibitors In: Egger G, Arimondo P (eds). Drug Discovery in Cancer Epigenetics. Academic Press: Waltham, MA, USA, 2016, pp 169–190. [Google Scholar]
- 94.Christman JK. 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21: 5483–5495. [DOI] [PubMed] [Google Scholar]
- 95.Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA 1984; 81: 6993–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chuang JC, Warner SL, Vollmer D, Vankayalapati H, Redkar S, Bearss DJ et al. S110, a 5-Aza-2'-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther 2010; 9: 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C et al. Delivery of 5-aza-2'-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res 2007; 67: 6400–6408. [DOI] [PubMed] [Google Scholar]
- 98.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2016; 164: 1073. [DOI] [PubMed] [Google Scholar]
- 99.Kasinathan S, Henikoff S.5-Aza-CdR delivers a gene body blow. Cancer Cell 2014; 26: 449–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu M, Ohtani H, Zhou W, Orskov AD, Charlet J, Zhang YW et al. Vitamin C increases viral mimicry induced by 5-aza-2'-deoxycytidine. Proc Natl Acad Sci USA 2016; 113: 10238–10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014; 5: 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013; 500: 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336: 934–937. [DOI] [PubMed] [Google Scholar]
- 106.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 2013; 13: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010; 465: 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 2017. [DOI] [PubMed]
- 111.Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med 2005; 19: 17–20. [DOI] [PubMed] [Google Scholar]
- 112.Ikehata M, Ogawa M, Yamada Y, Tanaka S, Ueda K, Iwakawa S. Different effects of epigenetic modifiers on the cytotoxicity induced by 5-fluorouracil, irinotecan or oxaliplatin in colon cancer cells. Biol Pharm Bull 2014; 37: 67–73. [DOI] [PubMed] [Google Scholar]
- 113.Flis S, Gnyszka A, Flis K. DNA methyltransferase inhibitors improve the effect of chemotherapeutic agents in SW48 and HT-29 colorectal cancer cells. PloS ONE 2014; 9: e92305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Azad NS, El-Khoueiry A, Yin J, Oberg AL, Flynn P, Adkins D et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat; a phase 2 consortium/stand Up 2 cancer study. Oncotarget 2017; 8: 35326–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Overman MJ, Morris V, Moinova H, Manyam G, Ensor J, Lee MS et al. Phase I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in refractory CIMP-high metastatic colorectal cancer: evaluation of circulating methylated vimentin. Oncotarget 2016; 7: 67495–67506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Garrido-Laguna I, McGregor KA, Wade M, Weis J, Gilcrease W, Burr L et al. A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Invest New Drugs 2013; 31: 1257–1264. [DOI] [PubMed] [Google Scholar]
- 117.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]