

Abstract
Protein–protein interactions (PPIs) are key mechanisms in the maintenance of biological regulatory networks. Herein, we characterize PPIs within ToxR and its co‐activator, ToxS, to understand the mechanisms of ToxR transcription factor activation. ToxR is a key transcription activator that is supported by ToxS for virulence gene regulation in Vibrio cholerae. ToxR comprises a cytoplasmic DNA‐binding domain that is linked by a transmembrane domain to a periplasmic signal receiver domain containing two cysteine residues. ToxR‐ToxR and ToxR‐ToxS PPIs were detected using an adenylate‐cyclase‐based bacterial two‐hybrid system approach in Escherichia coli. We found that the ToxR‐ToxR PPIs are significantly increased in response to ToxR operators, the co‐activator ToxS and bile salts. We suggest that ToxS and bile salts promote the interaction between ToxR molecules that ultimately results in dimerization. Upon binding of operators, ToxR‐ToxR PPIs are found at the highest frequency. Moreover, disulfide‐bond‐dependent interaction in the periplasm results in homodimer formation that is promoted by DNA binding. The formation of these homodimers and the associated transcriptional activity of ToxR were strongly dependent on the oxidoreductases DsbA/DsbC. These findings show that protein and non‐protein partners, that either transiently or stably interact with ToxR, fine‐tune ToxR PPIs, and its associated transcriptional activity in changing environments.
The membrane‐bound transcription regulator ToxR controls housekeeping and virulence gene expression in V. cholerae. The activity and stability of ToxR strongly depends on its operon partner ToxS, the redox state of its periplasmic cysteine residues and environmental stimuli. Here, ToxR‐ToxR PPIs are the key for its activity, which is influenced by ToxS, DNA binding capabilities, bile and the disulfide bond forming enzymes DsbA/C, which induce intra‐ or intermolecular disulfide bonds.
1. INTRODUCTION
Prokaryotes are unicellular organisms that require sensory networks for their survival in rapidly changing habitats. In the course of evolution, transmembrane signaling systems have evolved to transmit signals from the extracellular environment across the cytoplasmic membrane into the cell. One‐component signaling systems represent the oldest and simplest solution for such signal transmission, whereas two‐component systems are evolutionarily younger (Ulrich et al., 2005). Although one‐component systems are widely distributed among bacteria, only 3% are directly integrated into cytoplasmic membranes (Ulrich et al., 2005). A literature search revealed a non‐exhaustive list of signaling molecules that includes ToxRS, TcpPH, and TfoS in Vibrio cholerae and other Vibrio spp. (Miller et al., 1987; Miller et al., 1989; Hase and Mekalanos, 1998; Dalia et al., 2014); CadC in Escherichia coli (Tetsch et al., 2011); PsaE in Yersinia tuberculosis (Yang and Isberg, 1997); WmpR in Pseudoalteromonas (Stelzer et al., 2006); and ArnR and ArnR1 in Sulfolobus acidocaldarius (Bischof et al., 2019). These bitopic ToxR‐family transcription regulators consist of a single protein molecule with one input and one output domain. They share the same modular architecture—an N‐terminal winged‐helix‐turn‐helix (w‐HTH) motif located in the cytoplasm, a single inner membrane‐spanning alpha‐helical domain and a C‐terminal periplasmic signal receiver domain (Miller et al., 1987; Martinez‐Hackert and Stock, 1997).
The dimerization of w‐HTH transcription factors is critical for their activation. It leads to enhanced DNA‐binding specificity and affinity, as well as increased cooperativity between the monomers (Littlefield and Nelson, 1999). The w‐HTH domain of ToxR consists of an N‐terminal ß‐sheet; three α‐helixes which include the DNA‐binding helix α3; and a C‐terminal winged helix. Interestingly, within the w‐HTH OmpR/ToxR regulator family, the ß‐sheet structure is involved in the PPIs needed for the formation of head‐to‐head or head‐to‐tail dimers (Martinez‐Hackert and Stock, 1997; Kenney, 2002; Maris et al., 2005). Moreover, the wing of the w‐HTH is involved in tail‐to‐tail dimerization (Littlefield and Nelson, 1999). This was shown in HSF (heat shock transcription factor) in Kluyveromyces lactis using crystallography. Reports also indicate that DNA‐binding affinities are increased as a result of the activation of these one‐component transcription regulators; for example, in OmpR by N‐terminal phosphorylation. Consequently, the activated monomers bind to DNA, causing a conformational change, which, in turn, increases the affinity for a second monomer to form symmetrical or asymmetrical dimers (Rhee et al., 2008).
The strongest evidence demonstrating ToxR dimerization was derived from OmpR structural studies. The dimerization may be linked to the cytoplasmic domain in which the w‐HTH motif is located. As is known in w‐HTH protein family members, dimerization via such motifs occurs due to the close localization of the monomers after their binding to the DNA operator sequences and the subsequent interaction of the N‐terminal winged helix (Littlefield and Nelson, 1999). A recent study sheds light on such mechanisms for ToxR (Morgan et al., 2019). Therein, alanine‐scanning mutagenesis was performed to characterize the w‐HTH domain. Exchange mutants that lost their transcription factor activities but retained their DNA binding and possible interaction capabilities were analyzed. As a result, all characterized ToxR mutants which were identified to be transcriptionally inactive have also lost their ability to bind to DNA, including ompU and toxT operators. Although the w‐HTH region might be involved in activating transcription mechanisms, ToxR dimerization or other PPIs were not observed.
An interesting study highlighting the DNA‐dependent PPIs of CadC, a ToxR family member, in E. coli was recently reported by Brameyer et al. (2019). Such studies revealed the importance of the spatiotemporal localization and correlating transcriptional activity of CadC due to its low abundance (100 molecules per cell). They showed that activating stimuli (low pH and lysine availability) forced homodimerization and operator binding that, in turn, led to a detectable cluster formation of fluorescence labeled CadC proteins. The removal of these stimuli instantly dissolved such clusters. The authors, thereby, concluded a diffusion‐and‐capture mechanism that organizes membrane‐integrated receptors in response to DNA‐binding. Similar results have been also observed for TcpP in V. cholerae by single‐molecule tracking (Haas et al., 2014), where both, the toxT promoter and ToxR, were shown to play crucial roles in TcpP motility. TcpP motility is divided into fast, slow and immobile motion behaviors. From these, it was concluded that ToxR recruits TcpP to its toxT promoter using a modified hand‐holding mechanism after removing nucleoid‐associated proteins (NAPs) such as H‐NS.
The dimerization of ToxR and its PPIs with other proteins, its co‐activator ToxS for instance, has long been of interest. Using the λ phage reporter system in E. coli, it was demonstrated that ToxR is capable of forming dimers and that ToxS seems to play a role in enhancing ToxR dimerization. In this system, the N‐terminal DNA‐binding domain of λ repressor protein C1, which lacks a C‐terminal dimerization domain, was fused to the N‐terminal cytosolic part of ToxR to assess the ability of ToxR to dimerize (Dziejman and Mekalanos, 1994). The data demonstrated that the periplasmic domain of ToxR is important for dimerization, suggesting an out‐to‐inside dimerization model facilitated by ToxS. However, the latter findings were partially rejected (Dziejman et al., 1999). ToxR‐ToxS and ToxR‐ToxR PPIs were also verified using cross‐linker studies (Ottemann and Mekalanos, 1996). In these studies, ToxR homodimers were observed if ToxR was overexpressed; ToxR‐ToxS heterodimers were detected even under conditions of low expression. Moreover, in vitro analysis using purified periplasmic domains of ToxS and ToxR led to the identification of ToxR‐ToxS PPIs by utilizing NMR and reciprocal pull‐down assays (Midgett et al., 2017).
In V. cholerae, ToxRS has emerged as a key regulatory complex involved in virulence gene regulation. The transmembrane spanning domains of ToxRS offer unique possibilities for perceiving and transducing signals into transcriptional regulation programes. Some activating conditions and substances were identified as bile salts, alkaline pH, and nutrient availability (Matson et al., 2007; Childers and Klose, 2007; Peterson and Gellings, 2018). Despite its important role for virulence and environmental adaption, the exact mechanism of ToxR signal transduction and transcription factor activation remains to be characterized. Many studies, summarized above and recently published by Morgan et al. (2019), showed evidence for ToxR dimerization. However, no detailed information about the interaction interface and orientation is available. Insights about the complexity of the ToxR family protein activation have been derived from an analysis of cysteine‐based intra‐ and intermolecular disulfide bond formations in the periplasm. Some examples include bile salt (taurocholate)‐induced intermolecular disulfide bond formation and activation in TcpP (Yang et al., 2013) or cysteine‐dependent, intermolecular heterodimeric interactions of TcpP and ToxR under anaerobic conditions and subsequent virulence gene activation via toxT transcription (Fan et al., 2014). Finally, the cysteine residues in ToxR are associated with its transcriptional activity through intramolecular disulfide bond formation. Moreover, they also provide a signal for proteolysis once they appear in their reduced form (Ottemann and Mekalanos, 1996; Fengler et al., 2012; Lembke et al., 2018).
In summary, information on the interplay between ToxRS molecules remains fragmented and incomplete. In this study, we focus on ToxR PPIs and its known interaction factors. We found that ToxR‐ToxR PPIs were enhanced in the presence of ToxR operator binding sites, ToxS and bile. Additionally, ToxR‐ToxS PPIs were detected using an adenylate‐cyclase‐based bacterial two‐hybrid system in E. coli. Finally, we extend our previous model by showing that the intermolecular disulfide bond formation of ToxR periplasmic domains is DsbA/DsbC‐dependent in V. cholerae, and that formation of this homodimer is associated with enhanced transcriptional activity.
2. RESULTS
2.1. ToxS, DNA operator binding sites, and bile enhance ToxR PPIs
Transcription regulators containing w‐HTH domains rarely act by themselves but form dimers to induce specific cellular responses (Littlefield and Nelson, 1999). More than 30 years ago, it was postulated that ToxR either acts as a homodimer (Miller et al., 1987; Dziejman and Mekalanos, 1994; Ottemann and Mekalanos, 1996) or in cooperation with other proteins (DiRita and Mekalanos, 1991; Krukonis et al., 2000). However, the molecular mechanism behind ToxR PPIs and its activity is still poorly understood. To dissect the roles of ToxS, DNA operator binding sites, and environmental stimuli such as bile, in ToxR PPIs, a bacterial cAMP‐based two‐hybrid system (BACTH) (Karimova et al., 1998) was used in E. coli W3110 ∆cyaA. The BACTH system is accessible to membrane proteins and is based on the reconstitution of the T25 and T18 domains of the adenylate cyclase CyaA from Bordetella pertussis, resulting in cAMP synthesis. In our experiments, the N‐termini of potentially interacting proteins were fused to the C‐termini of the two CyaA fragments because of their predetermined orientation in the inner membrane (Figure 1a,b). The respective fusion proteins were tested separately for the expression in E. coli XL1‐Blue, DH5α λpir and BL21 (DE3) (Figure S1). For the investigation of putative ToxR‐ToxR and ToxR‐ToxS PPIs, E. coli strain W3110 ∆cyaA was co‐transformed with the combinations of pUT18C and pKT25 derivatives carrying translational fusions of T18‐ToxR/T25‐ToxR; T18‐ToxRW76R/T25‐ToxRW76R, co‐expressed with or without ToxS, respectively, and T25‐ToxR/T18‐ToxS‐FLAG.
FIGURE 1.
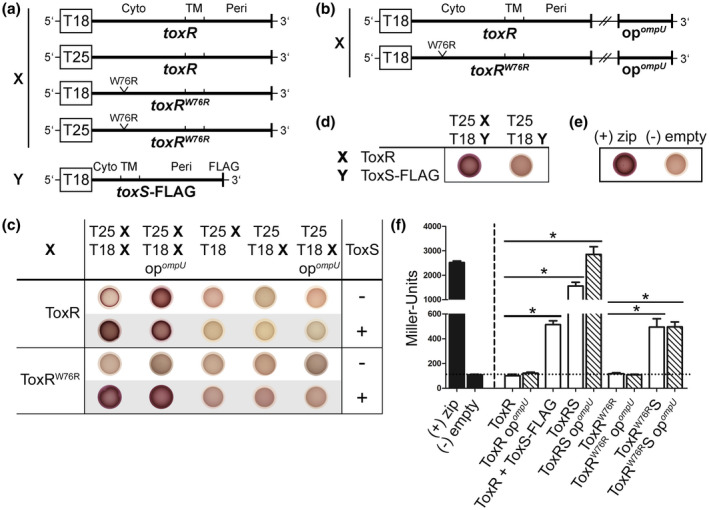
ToxS and ompU operator binding sites are key players in ToxR PPIs in E. coli. PPIs of the indicated ToxR and ToxS‐FLAG variants were tested in E. coli W3110 ∆cyaA using a bacterial cAMP‐based two‐hybrid system (BACTH), which is based on the functional complementation of the adenylate cyclase CyaA (Karimova et al., 1998). Strains are N‐terminal ToxR, ToxRW76R and ToxS‐FLAG translational fusions linked to the C‐termini of the B. pertussis CyaA T18 or T25 domains with or without V. cholerae ompU operator fragments (opompU), and co‐expressed ToxS (a, b). The leucine zipper of the yeast GCN4 protein (zip) was used as a positive complementation control (+), while the empty plasmids pKT25 and pUT18C served as negative controls (‐) (e, f). For the drop test (c, d, e), strains were grown in LB overnight and subsequently transferred to a single MacConkey maltose indicator plate to reveal the CyaA+ phenotype (red colonies indicate the utilization of maltose as a C‐source). PPIs are shown between the indicated ToxR or ToxRW76R translational fusions (designated as X) in the presence or absence of co‐expressed ToxS and ompU operator binding sites (opompU) (c). Panel (d) displays PPIs between ToxR and ToxS‐FLAG (designated as Y). Panel (f) shows quantifications of functional complementation between the indicated ToxR, ToxRW76R and ToxS‐FLAG hybrid proteins (white bars) in dependence of ompU operator binding sites (opompU, lined white bars) by measuring β‐galactosidase activities. The cells were grown in LB supplemented with 0.05 mM IPTG to the stationary phase. Strains in which ToxR and ToxRW76R PPIs were measured in the presence of co‐expressed ToxS were labeled with ToxRS or ToxRW76RS. Strains in which interactions between ToxR and ToxS‐FLAG were analyzed were labeled with ToxR + ToxS‐FLAG. The positive and negative controls are represented by black bars. The values are means of three biological replicates, each with technical triplicates with error bars, which represent the standard deviation. Interactions are reported as Miller Units. The asterisks indicate significantly different means with p < .05 for the respective columns, each tested against E. coli W3110 ∆cyaA pKT25‐ToxR pUT18C‐ToxR or pKT25‐ToxRW76R put18C‐ToxRW76R using one‐way ANOVA test with Bonferroni post hoc analysis [Colour figure can be viewed at wileyonlinelibrary.com]
Next, protein interactions were tested by spotting the resulting E. coli strains on MacConkey maltose agar plates (Figure 1c,d,e) and by measuring β‐galactosidase activities (Figure 1f). Positive interactions that generated an elevated adenylate cyclase activity were detected as red colonies on MacConkey maltose agar plates or through the increased expression of the lacZ reporter. We chose a cut‐off value of 100 Miller Units (Figure 1f), predetermined by the negative control, as indicative of a false positive interaction between the fusion proteins. This approach demonstrated that the co‐expression of ToxS with ToxR or the DNA‐binding‐deficient mutant ToxRW76R, resulted in a red colony phenotype and significantly increased lacZ expression levels compared to strains without ToxS (Figure 1c,f). Strains expressing ToxR or ToxRW76R alone displayed a white colony phenotype and Miller Units below or equal to the cut‐off level. Thus, we found that ToxR‐ToxR PPIs were enhanced in the presence of its operon partner ToxS.
Since ToxS was able to mediate ToxR‐ToxR PPIs, we were also interested in the interaction of ToxR with ToxS. Here, we were able to confirm ToxR‐ToxS‐FLAG PPIs using BACTH (Figure 1d,f), which, in turn, emphasizes the results of earlier studies by Midgett et al. (2017).
ToxR is a transcriptional regulator located in the inner membrane and binds to its operator binding sites after activation. Therefore, we also addressed the question of whether the ompU operator binding sites, also termed ToxR boxes, capture ToxR molecules to result in ToxR‐ToxR PPIs. Based on the direct repeat nature of ToxR‐binding sites in the ompU promoter region (5′‐TNAAA‐N5‐TNAAT‐3ʹ), located from −51 to −37 relative to the transcription start site (Goss et al., 2013), we suggest a cooperative binding of two ToxR molecules. To test our hypothesis, the V. cholerae ompU operator fragment (opompU) (Morgan et al., 2011) was cloned into pUT18C to provide ToxR with its natural DNA‐binding‐sites in E. coli (Figure 1b). Interestingly, the red colony phenotype (Figure 1c) indicated that the presence of the ompU operator binding sites triggered ToxR‐ToxR PPIs independently of ToxS. However, this could not be confirmed by the β‐galactosidase assay (Figure 1f). This implied that the MacConkey maltose agar plates exhibit a higher sensitivity for the evaluation of PPIs, which remains to be elucidated. Nevertheless, the ToxR‐ToxR PPIs were significantly increased in the strains that co‐expressed ToxS and provided ompU operators compared to those strains without ompU operators. In contrast, the ompU operators showed no effect on the ToxRW76R DNA‐binding‐deficient mutant with or without ToxS (Figure 1c,f). Thereby, we emphasize our above findings that ToxR‐boxes play a major role in ToxR‐ToxR PPIs, especially in the presence of ToxS.
During infection, bacterial pathogens of the small intestine are surrounded by adverse conditions, including bile salts that circulate between the intestine and the liver of vertebrates (Hofmann et al., 2010). Therefore, we tested whether incubation with the bile salt sodium deoxycholate (DC) has an impact on ToxR‐ToxR PPIs (Figure 2). Our results demonstrated that if toxS was co‐expressed, the addition of 0.1% DC increased PPIs between ToxR molecules. This indicates that DC represents a trigger factor that facilitates ToxR PPIs in dependence of ToxS. In contrast, the leucine zipper positive and negative controls did not respond to bile.
FIGURE 2.
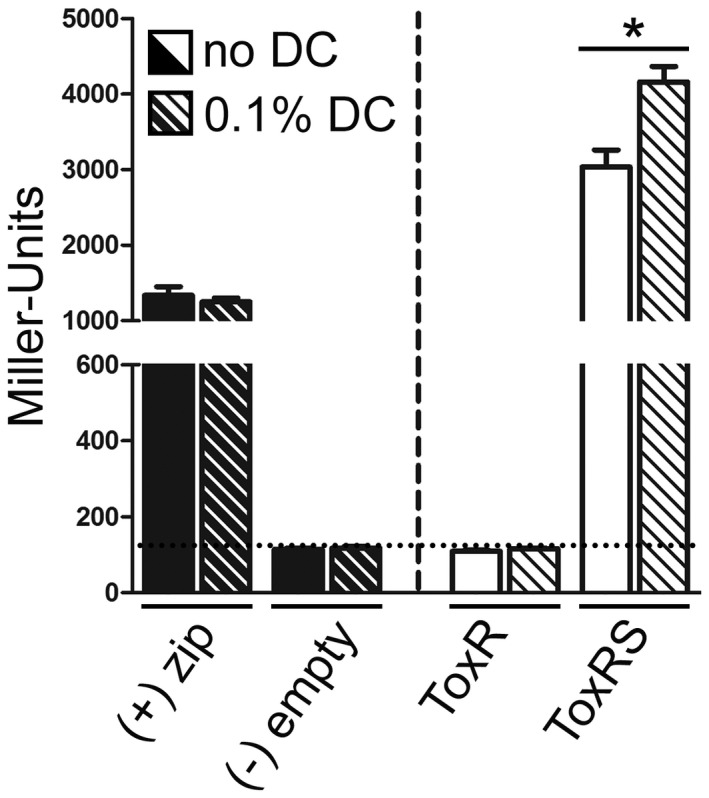
Bile salts (DC) stimulate ToxR PPIs in a ToxS dependent manner. PPIs of ToxR with or without co‐expressed ToxS were tested in dependence of bile salts in E. coli W3110 ∆cyaA using BACTH (Karimova et al., 1998). See experimental procedures for details. The leucine zipper of the yeast GCN4 protein (zip) was used as a positive complementation control (+), while the empty plasmids pKT25 and pUT18C served as negative controls (‐). Cells were grown in LB supplemented with 0.05 mM IPTG to the stationary phase in the absence or presence of 0.1% DC (sodium deoxycholate). The functional complementation of the controls (black bars) and ToxR with or without ToxS (white bars) was quantified by measuring β‐galactosidase activities. DC treated samples are indicated by lined bars. The values are means of three biological replicates, each with technical triplicates with error bars, which represent the standard deviation. The controls contain five values each. Interactions are reported as Miller Units. The asterisks indicate significantly different means with p < .05 using Student's t‐test
Taken together, we show that the membrane‐bound transcription regulator ToxR exhibited dynamic interaction states. In E. coli, the ToxR‐ToxR PPIs of the cytoplasmic domains were mediated by ToxS. These ToxR interactions were further enhanced by ompU operators provided on a plasmid or bile added into the growth media. Our results may indicate a hierarchical order in the generation of a functional ToxRS complex. The first step involves contact with ToxS, leading to significantly increased ToxR‐ToxR PPIs. Next, we observed that ToxR‐ToxR PPIs can further be stimulated in the presence of bile, but only if ToxS was present. Finally, ToxR operators capture preliminary formed ToxRS complexes leading to the highest ToxR‐ToxR PPI values measured.
2.2. ToxR transcription factor activity correlates with the formation of homodimers
Based on the abovementioned observations, we focused on PPIs taking place in the periplasmic domain of ToxR, namely by characterising disulfide bond formations and their influence on dimerization and activity. We recently demonstrated that the two cysteine residues in the periplasmic domain are responsible for the maintenance of ToxR stability and activity (Lembke et al., 2018). To find a connection between ToxR activity and homodimer formation, we focused on ToxR cysteine residues. To this end, native toxR or cysteine mutants (C236S, C293S or CC, the latter is an exchange of both cysteine residues with serine) were cloned into pFLAG‐MACTM under tac promoter expression control. As previously mentioned, we also cloned the operon partner gene toxS into the plasmids. When introduced into V. cholerae ΔtoxRS, the monomeric, dimeric, and oligomeric forms of FLAG‐tagged ToxR derivatives were analyzed by SDS‐PAGE and immunoblotting under non‐reducing conditions (Figure 3, see respective loading control in Figure S2). There, the disulfide bond‐dependent homodimerization and oligomerization of FLAG‐ToxR was decreased in the presence of ToxS, suggesting that ToxS competes for interactions with ToxR molecules for disulfide bond formation in the periplasm. The FLAG‐ToxRCC mutants lacking both cysteines showed a complete loss of the ability to form homodimers, which was independent of ToxS. Therefore, intermolecular disulfide bonds were responsible for the observed PPIs. To note, a proteolytic FLAG‐ToxRCC degradation fragment was observed when ToxS was co‐expressed. However, the ability of ToxR to form homodimers was restored in the FLAG‐ToxRC236S and FLAG‐ToxRC293S single cysteine mutants. Strikingly, high levels of homodimers were observed for the FLAG‐ToxRC293S mutant, indicating that the altered thiol redox state of Cys293 favored such dimer formations (Figure 3). These data also demonstrated that the periplasmic cysteine residues were close enough to form intermolecular disulfide bonds to yield homodimers.
FIGURE 3.
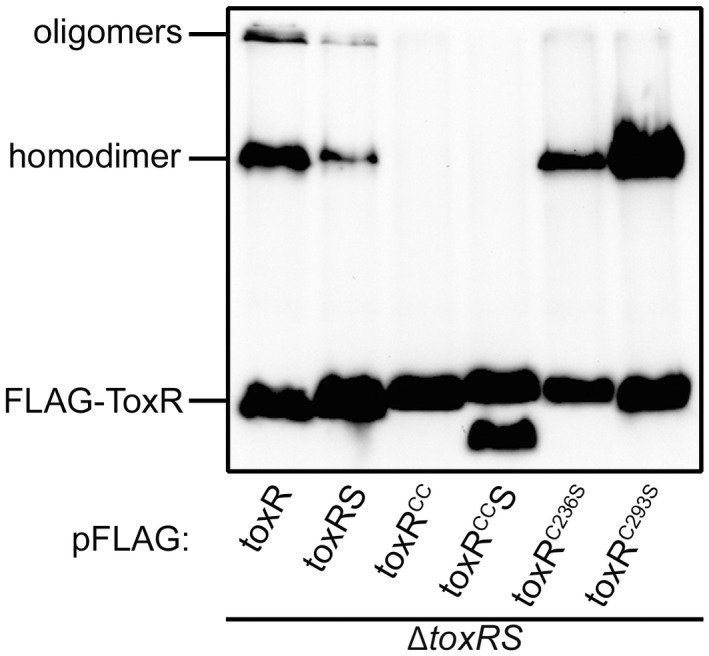
Cysteine dependent homodimer formation in ToxR. V. cholerae ΔtoxRS strains carrying toxR derivatives with or without its operon partner toxS on pFLAG‐MACTM were grown in LB. Samples were taken after 2 hr induction with 0.05 mM IPTG in the mid‐log phase and analyzed by immunoblotting using anti‐FLAG antibodies. Immunoblots were carried out under standard non‐reducing Laemmli buffer conditions
To determine the correlation between homodimer formation and ToxR activity, we monitored the PhoA activities and OmpU/T protein levels in parallel using strains with chromosomal ompU::phoA and ompT::phoA fusions. The ompU and ompT expression levels provide an excellent readout for ToxR activity, as they are inversely regulated by ToxR (Crawford et al., 1998; Li et al., 2000). We previously reported that the ToxRCC cysteine mutant is a target for regulated intramembrane proteolysis (RIP) (Lembke et al., 2018). We now show that RIP not only affected the ToxRCC mutant but also the single cysteine mutants when grown in M9 maltose minimal medium (Figure S3). Therefore, this experiment was carried out in a ΔdegP background in the mid‐log phase to ensure similar ToxR protein levels for ToxRWT and the cysteine mutants to allow a comparison of PhoA activities between different proteolysis prone toxR mutants (Figure S4a,b).
When grown in M9 maltose minimal medium, the strain expressing ToxRWT exhibited a more pronounced ompT expression compared to ompU (Figure 4a,b). As expected, the ΔtoxR control showed neither activated ompU transcription nor ompT repression. For comparison, chromosomal toxR cysteine mutants, constructed by exchanging one or both cysteines to serines (C236S, C293S or CC), were also analyzed. There, as expected, the toxRCC strain displayed significant regulatory deficiencies for ompU and ompT expression when compared to toxRWT, as we have previously shown (Lembke et al., 2018). Thus, this indicates a possible link between disulfide bond formation and ToxR activity. In contrast, the ToxRC236S mutant was able to activate ompU beyond the strain expressing ToxRWT, although simultaneous ompT repression seemed to be less evident. In particular, the ToxRC293S replacement mutant strongly activated ompU and repressed ompT significantly beyond the strain expressing ToxRWT. To be mentioned, this happened despite growing the strains under nutrient‐limiting conditions that do not favor ToxR activation. In addition, OmpU and OmpT protein expression patterns, which were detected by immunoblot analysis from the same cultures (Figure 4a,b), showed similar results to the PhoA activity measurements. To note, all the characterized strains featured a chromosomally toxS+ background.
FIGURE 4.
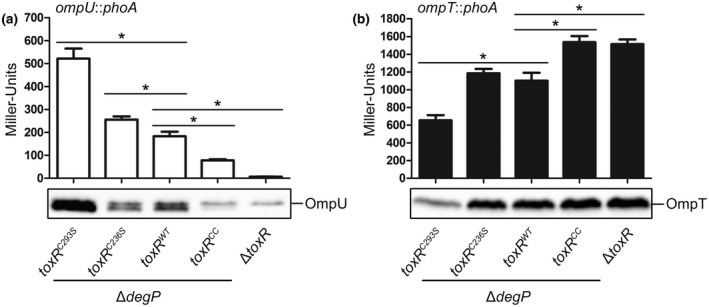
Transcription factor activity of toxR cysteine mutants. Shown are reporter gene activities of alkaline phosphatase PhoA (Miller Units) linked as operon fusions to either (a) ompU (white bars) or (b) ompT (black bars) in V. cholerae ΔdegP strains harboring various chromosomal cysteine mutations in toxR. Simultaneously, immunoblot analysis was performed under standard reducing Laemmli buffer conditions to detect OmpU or OmpT, respectively. Cells were grown in M9 maltose minimal medium and samples were taken in the mid‐log phase. V. cholerae ΔtoxR served as a negative control. The mean values with standard deviation are shown (n = 6). The asterisks indicate significantly different means with p < .05 for the respective columns each tested against ΔdegP toxRWT using one‐way ANOVA test, followed by Dunnett's post hoc test for multiple comparisons. To note, all the characterized strains featured a chromosomally toxS+ background
Taken together, these results indicate that ToxR cysteine residues contribute to the transcriptional activity of ToxR, presumably because they are required for intra‐ and intermolecular disulfide bond formation. Moreover, we conclude that cysteine‐dependent transcription factor activity correlates with the formation of homodimers, supporting the early view by Miller et al. (1987).
2.3. DNA‐binding triggers ToxR homodimer formation
Our results thus far suggest that ToxR homodimerization strongly correlates with its activation, ultimately resulting in the transcriptional regulation of genes such as ompU and ompT. These findings raised the question of which factors or conditions influence the ToxR‐ToxR PPIs. The DNA‐binding domains of the OmpR family proteins generally facilitate dimer formation once they are in contact with direct repeat DNA sequences (Yoshida et al., 2006). We, thus, investigated the effect of operator binding on ToxR dimerization in more detail in V. cholerae to expand the data derived from our BACTH analysis in E. coli.
The WT or single cysteine replacement mutants were grown in M9 maltose minimal medium to express the reduced (ToxRred) and oxidized (ToxRoxy) monomeric and dimeric forms of chromosomally encoded toxR. These were detected by SDS‐PAGE and immunoblotting under non‐reducing conditions (Figure 5a). To note, the following experiments have been carried out in a degP+ background to avoid distortions leading to artificial toxR expression patterns under non‐reducing conditions. Compared to the WT control, only the reduced monomeric form of ToxR was detected in the toxR single cysteine mutants, as they were unable to form intramolecular disulfide bonds (Ottemann and Mekalanos, 1996; Lembke et al., 2018). Notably, ToxR homodimers could only be detected in the toxRC293S mutant but not in the WT or the toxRC236S mutant in the various growth conditions tested (LB, M9 glucose or maltose minimal medium with and without NRES, AKI or media with bile salt supplementation) (data not shown). Since the addition of reducing agents in Laemmli buffer (β‐mercaptoethanol) (Laemmli, 1970) dissolved ToxRC293S homodimers, we deduced that these homodimers were formed by intermolecular disulfide bonds (compare Figure 5a with Figure S3). To determine the impact of DNA binding on ToxR‐ToxR PPIs, a W76R point mutation (according to the amino acid position as annotated by Heidelberg et al. (2000)) in the w‐HTH domain of ToxRC293S was introduced (resulting in ToxRC293SW76R). This amino acid substitution was first described by Morgan et al. (2011) as a mutation that is detrimental for DNA binding and activation of the ompU and toxT promoters. As presented in Figure 5a, the removal of operator binding abilities in ToxRC293SW76R consequently resulted in undetectable homodimer formation. This indicates that ToxR‐boxes may serve as an anchor point for PPIs between ToxR molecules, for example, homodimers. Since the toxR expression levels varied between toxRC293S and toxRC293SW76R strains, a more precise quantification analysis was performed to verify the impact of ToxR‐boxes on ToxR homodimerization. Shown in Figure 5b are the results of densitometric analysis (Figure S5). Data were calculated as absolute values of intensity per lane and sample and expressed as a percentage of the sum of both (homodimer and monomer) intensities. As a result, we saw a higher proportion of monomer relative to dimer formation for ToxRC293SW76R compared to ToxRC293S. These results indicate that loss of dimer formation correlates with the inability to bind to ToxR‐boxes. In summary, these results reveal a capture mechanism that organizes ToxR in the presence of operator sites to form cysteine‐dependent homodimers.
FIGURE 5.
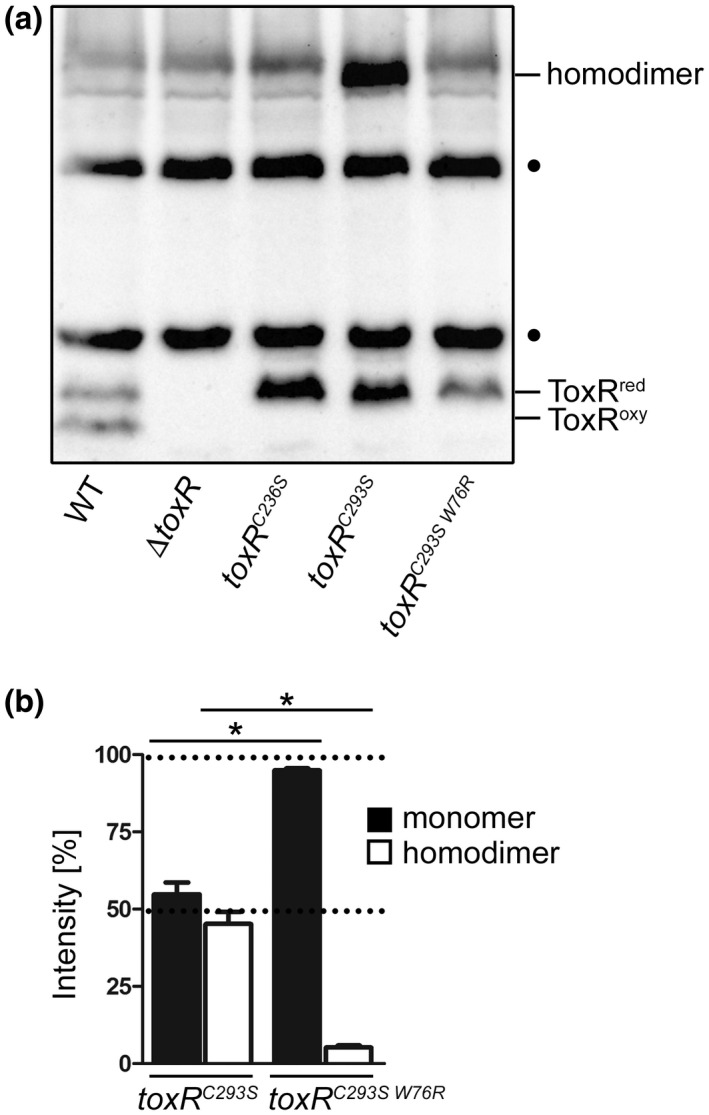
DNA binding triggers ToxR homodimer formation. (a) Shown is ToxR immunoblot analysis of V. cholerae WT, ΔtoxR, toxRC236S, toxRC293S, and toxRC293SW76R grown in M9 maltose minimal medium until the mid‐log phase was reached. Immunoblotting was performed under standard non‐reducing Laemmli buffer conditions using anti‐ToxR antibodies. (•) Represents nonspecific cross‐reacting background bands. (b) The column bar graph displays the protein band intensities of ToxR monomers (black bars) and homodimers (white bars) in V. cholerae toxRC293S compared to toxRC293SW76R as a result of densitometric analysis carried out under non‐reducing Laemmli buffer conditions (see representative immunoblot Figure S5a). Here, ToxR protein band intensities were measured per strain (both intensities add up to 100%) using Image Lab Software (BIO‐RAD). The mean values with standard deviation are shown (n = 6). The asterisks indicate significantly different means between toxRC293S and toxRC293SW76R monomers and homodimers with p < .05, respectively, using Student's t‐test. To note, all the characterized strains featured a chromosomally toxS+ background
2.4. DsbA and DsbC coordinate intra‐ and intermolecular disulfide bond formation in ToxR
Disulfide bonds are formed by the oxidation of two cysteine residues in close proximity, for example, 2.5 Å (Overington et al., 1992). This reaction can proceed spontaneously or with the help of enzymatic catalysts. Many secretory proteins, such as cholera toxin (Tomasi et al., 1979), undergo oxidative folding, in which they acquire intra‐ or intermolecular disulfide bonds to form higher‐order quaternary structures. The periplasmic space of Gram‐negative bacteria contains multiple disulfide bond‐forming enzymes, for example, DsbABCD, which catalyze the formation and isomerization of disulfide bonds. Since disulfide bond formation plays an essential role in ToxR activity and its homodimerization, Dsb proteins were studied in greater detail.
Cells were grown in M9 maltose minimal medium harboring mutations in the thiol‐disulfide oxidoreductase dsbA or the disulfide bond isomerase dsbC (Missiakas et al., 1995; Kadokura et al., 2003). SDS‐PAGE and immunoblotting were performed under non‐reducing conditions to expose the redox state of ToxR (Figure 6a,d), as well as its activation state, by monitoring OmpU (Figure 6b) and OmpT (Figure 6c) protein levels. For loading controls, see supplemental data (Figure S6). Furthermore, using densitometric analyses (Figure S7), we quantified the synthesis of OmpU/T. To note is that the ratio observed for ToxRred/oxy (see Figures 5 and 6) can be variable, depending on culture conditions and sample handling. Therefore, comparisons between different mutants always require the usage of the same culture media and growth conditions, best applied along with the same series of the experiment.
FIGURE 6.
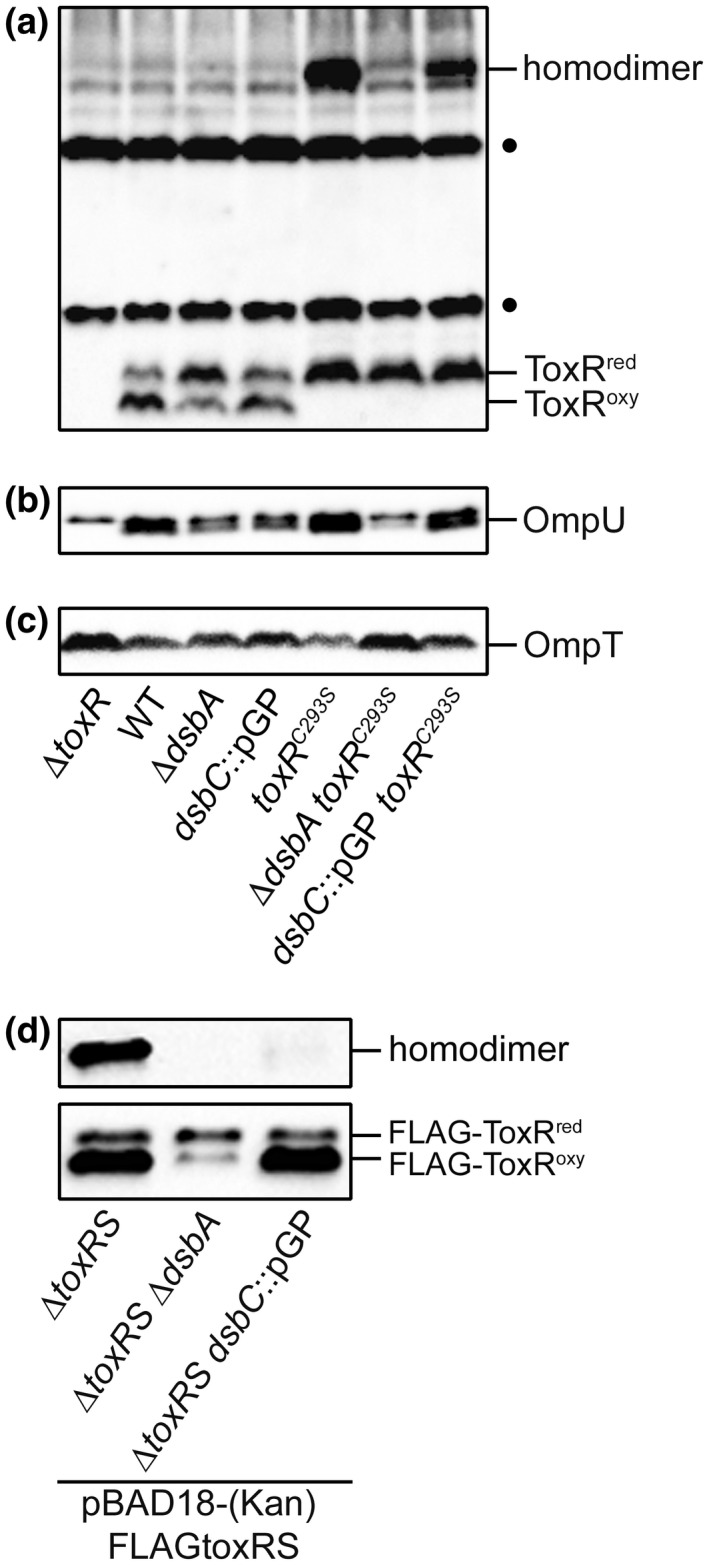
Dsb proteins influence ToxR inter‐ and intramolecular disulfide bond formation and its activity. V. cholerae strains harboring dsbA, dsbC and/or toxR mutations were grown in M9 maltose minimal medium until the mid‐log phase was reached. For ΔtoxRS strains carrying FLAGtoxRS on pBAD18‐Kan or pBAD18, samples were taken after 2 hr of induction with 0.1% arabinose when the cells reached the mid‐log phase. Immunoblotting was performed under standard non‐reducing Laemmli buffer conditions using anti‐ToxR (a) or anti‐FLAG antibodies (d); or reducing conditions using anti‐OmpU (b) or anti‐OmpT antibodies (c). (•) Represents nonspecific cross‐reacting background bands. It is to note, that Kang‐gel staining was performed to provide similar protein levels of all samples shown (Figure S6a,b). To note, all the characterized strains featured a chromosomally toxS+ background, except for the ΔtoxRS strain used
As was observed for chromosomal ToxRWT (Figure 6a–c), mutations in dsbA or dsbC significantly decreased OmpU but had no effect on OmpT protein levels (Figure S7) compared to the WT. This demonstrates a loss of ToxR activity due to the absence of Dsb proteins, especially for the ompU transcription activation. The overexpression of FLAG‐ToxRS revealed that the decreased activity of chromosomal ToxRWT in dsbA and dsbC mutants (Figure 6b,c) correlates with decreased homodimer formation (Figure 6d). It is to note that homodimer formation was not observed if ToxRS was expressed from chromosomally encoded loci but was readily detected if toxRS were overexpressed by the pBAD expression system (compare Figure 6a,d). Interestingly, there were no observable changes in the redox state of monomeric ToxRWT in the dsbC mutant compared to the WT (Figure 6a), indicating no interference in the redox equilibrium of the monomeric form. However, intramolecular disulfide bond formation in ToxRWT was disturbed in a dsbA mutant strain, as was shown previously (Lembke et al., 2018). In the study, the amount of monomeric ToxRred was higher than that of ToxRoxy. As an extension of our previous model, we suggest that DsbA introduces intramolecular disulfide bonds into newly translated ToxR polypeptides (Lembke et al., 2018). Only the monomeric, oxidized ToxR molecule (ToxRoxy) represents a substrate for the isomerase DsbC, which achieves the native disulfide proteome of the cell (Missiakas et al., 1995; Kadokura et al., 2003).
To decipher disulfide bond formations in toxRC293S, mutations in dsbA and dsbC were also introduced here. In particular, the toxRC293S mutant was able to activate ompU and repress ompT transcription beyond ToxRWT levels (Figure 4a,b) when grown in the M9 maltose minimal medium. In contrast to ToxRWT, ToxRC293S only possessed the option to form intermolecular disulfide bonds. The serine substitution of one of the two cysteine residues did not enable intramolecular disulfide bond formation but instead resulted in the monomeric ToxRred and the homodimeric form (Figure 6a). Changes in the redox status of ToxRC293S in the dsb mutants were, therefore, only detectable in the homodimers. As shown in the dsbA mutant, homodimer formation was abolished (Figure 6a) and the decreased activity of ToxRC293S became apparent for OmpU and OmpT expression (Figures 6b,c, S7). The insertion in dsbC had less impact on both porin expression levels, presumably because intramolecular disulfide bonds, which serve as DsbC substrates, cannot be formed in ToxRC293S.
Taken together, these results allowed us to confirm that the ToxR cysteine residues are critical for its activation state. Furthermore, we postulate that the transcriptional activity of ToxR correlates with the formation or interplay of cysteine‐dependent homodimers and that DsbA and DsbC contribute to this specific ToxR folding.
3. DISCUSSION
Only a minority of the one‐component systems are directly integrated into cytoplasmic membranes. Because more than 80% of the signal transduction pathways involve the binding of DNA, an arrangement of membrane‐bound signal transducers may place major constraints on their ability to interact with DNA (Ulrich et al., 2005; Jung et al., 2018). ToxR is one such membrane‐bound one‐component signal transducer that is required for V. cholerae’s lifestyle switch between the host and the environment. The dimerization of transcription factors often leads to enhanced DNA‐binding specificity and affinity—characteristics that mitigate the constraints on ToxR–DNA interactions (Littlefield and Nelson, 1999). In this study, we addressed how ToxR may overcome the difficulties that it experiences as a membrane‐bound transcriptional regulator by forming dynamic PPIs that depend on DNA operators, co‐activator ToxS, ToxR‐cysteine residues, Dsb mediated activities and ToxR activating stimuli (e.g., bile, DC). Therefore, we particularly focused on housekeeping genes (OmpU/T), since the cysteine residues seem to play an important role in their regulation (Fengler et al., 2012; Lembke et al., 2018).
The presence of direct repeat DNA sequences in operators, similar to OmpR operators in E. coli (Yoshida et al., 2006), recognized by ToxR (ToxR‐boxes) argues for the binding of ToxR dimers (Goss et al., 2013). Here, we demonstrate that ToxR DNA binding enhances ToxR‐ToxR PPIs and dimer formation. For example, we show in V. cholerae that the number of disulfide‐linked homodimers of a chromosomal ToxRC293S variant was significantly decreased once the protein was unable to bind its operators after the introduction of a W76R mutation in its w‐HTH domain (Morgan et al., 2011). These results were further supported by a bacterial cAMP‐based two‐hybrid system (BACTH) in E. coli. There, the presence of plasmid‐encoded V. cholerae ompU operator binding sites enhanced ToxR‐ToxR PPIs when ToxS was co‐expressed. To note, in the absence of co‐activator ToxS, the efficiency of interactions between these ToxR fusion proteins was not particularly strong. In comparison, the ToxRW76R operator‐binding‐deficient mutant displayed no enhancement of PPIs in the presence of the ompU operator‐binding sites. At this point, we propose that ToxR DNA operators may serve as an anchor point for the subsequent formation of ToxR‐ToxR PPIs and these interactions are further enhanced in the presence of ToxS (Figure 7). Brameyer et al. recently described a similar mechanism in E. coli where the ToxR‐like membrane‐bound transcriptional regulator CadC formed PPIs when external stresses activated the receptor which ultimately resulted in DNA binding (Brameyer et al., 2019). Owing to their membrane‐anchoring, ToxR‐like transcription regulators are limited in their spatial dynamics. However, the formation of homodimers may support these regulators to tether DNA close to the cytoplasmic membrane.
FIGURE 7.
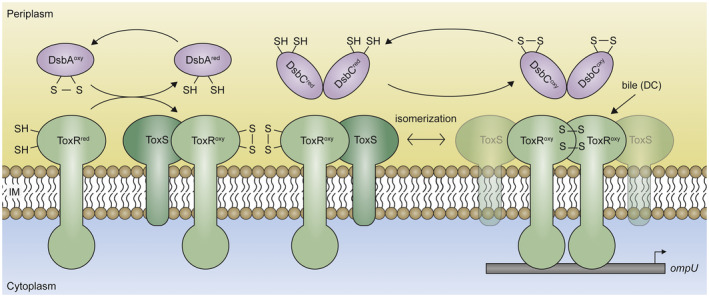
Activation mode and interaction patterns of ToxRS. Integral membrane proteins such as ToxRS are inserted into the cytoplasmic membrane co‐translationally, whereby ToxR exposes its reduced cysteine residues to the periplasm. These cysteines are then oxidized by DsbA to form intramolecular disulfide bonds, which represent the proteolytically stable form of ToxR. Increased ToxR‐ToxR PPIs can be detected if ToxS and ToxR‐boxes (e.g., ompU promoter) are available. Such an interaction is further strengthened in the presence of bile (sodium deoxycholate). As a model, we suggest that while ToxR molecules are bound to their operator sequences (ToxR‐boxes), temporarily formed ToxR dimers may exist due to intermolecular disulfide bond formation depending on DsbC or other mechanisms [Colour figure can be viewed at wileyonlinelibrary.com]
The model of a dynamic ToxR monomer and dimer formation derived from earlier studies suggests that the inactive form of ToxR is a monomer and the active one is a dimer. This was shown using ToxR‐PhoA fusions proteins or cross‐linking techniques, respectively (Miller et al., 1987; Ottemann and Mekalanos, 1996). However, an inconclusive picture regarding the ToxR localization and dimerization status still exists. The exact conditions necessary for the regulation of ompU/T are not known—both, cytosolic and soluble or membrane‐bound forms of ToxR were found to be sufficient for ompU/T regulation and TcpP controlled toxT regulation, independently of its periplasmic domain (Crawford et al., 2007). Moreover, other studies indicate that a cytosolic, soluble ToxR or ToxR periplasmic truncations only promote ctxAB gene expression in E. coli when fused to dimerization domains (e.g., Leu‐zipper). However, such constructs failed to show activity in V. cholerae (Dziejman et al., 1999). Furthermore, it was shown that PPIs also take place between TcpP and ToxR and that such interactions involve cysteine‐dependent disulfide bond formation in their periplasmic domains and anoxic growth conditions (Fan et al., 2014). Our data add to the current knowledge by showing that periplasmic cysteine residues interconnect ToxR molecules by disulfide bond formations via Dsb‐enzymes which plays a major role in ToxR transcriptional activity. The replacement of both cysteine residues with serine decreases ToxR activity. This was best demonstrated by monitoring ompU and ompT transcription in various cysteine mutants. Although the ToxR single cysteine mutants did not have the potential to form intramolecular disulfide bonds, these mutants were found to form homodimers. Interestingly, data obtained using Cys293 or Cys236 mutants demonstrated that periplasmic ToxR domains must come into immediate contact with each other in order to form intermolecular disulfide bonds. In addition, such close contact of the periplasm localized cysteine residues was highly dependent on the ability of ToxR to bind its operators, for example, ompU operators, as if there was signaling from the inside to the outside. Furthermore, both single cysteine ToxR mutants were able to activate ompU transcription beyond native ToxR levels in minimal medium. It should be emphasized, however, that the sole ability of ToxR homodimer formation does not automatically correlate with similar transcription factor strength of ToxR. This is particularly evident in the ToxRC293S mutant, which displayed a higher transcription activity than the ToxRC236S mutant or ToxRWT. Apart from steric limitations, the cysteine residues in ToxR may be able to assemble into tertiary or quaternary structures. For example, monomeric ToxR could be found in a reduced or oxidized state by the formation of intramolecular disulfide bonds. Furthermore, ToxR could build intermolecular disulfide bonds by a C236‐C236S, C293‐C293 or C236‐C293 linkage or oligomers, which would be connected in a chain‐like conformation.
Cysteine‐dependent homodimerization seemingly provokes ToxR activation, but equally important is the formation of intramolecular disulfide bonds, which are needed to stabilize ToxR molecules (Lembke et al., 2018). Although disulfide bonds are covalent linkages, they can be isomerized enzymatically by the correction system DsbCD (Figure 7). If ToxR, once activated and locked by intermolecular disulfide bonds, cannot be deactivated anymore, it may not be able to respond to changing environmental signals. This would cause severe problems in the stress response or energy homeostasis of the cell. Therefore, we examined the process of intra‐ and intermolecular disulfide bond formation in ToxR and were able to demonstrate that the Dsb system in the periplasm introduces and controls the correct arrangement of disulfide bonds in ToxR. We were able to confirm that DsbA is the primary electron donor for ToxR and ToxRC293S cysteine linkages (Lembke et al., 2018), assuming that their cysteine residues are in very close proximity (Landeta et al., 2018). DsbA possesses one of the highest redox potential values (−120 mV) among many known thiol‐disulfide oxidoreductases. This leads to rapid disulfide bond pairings, but these do not necessarily occur between the correct combinations of cysteines (Wunderlich et al., 1975; Grauschopf et al., 1995). Therefore, DsbA and DsbB must cooperate with the disulfide bond isomerization system DsbC and DsbD, to achieve the native proteome through the correction of false disulfide bonds (Kadokura et al., 2003). We found that DsbC is responsible for the formation of intermolecular disulfide bonds in ToxR homodimers. Furthermore, the lack of DsbA or DsbC, affected the regulation potency of native ToxR, best observed for ompU expression. This observation is in line with the decreased homodimer formation evidenced in overexpression studies. Unfortunately, we were unable to detect homodimers from native chromosomal expressed toxR. Nonetheless, we propose the following scenario (Figure 7). During de novo protein biosynthesis, the insertion of ToxR into the membrane exposes its thiol groups in the periplasm, which are, in turn, oxidized by the DsbAB system to form intramolecular disulfide bonds. DsbC then catalyses the exchange of ToxR disulfide bonds formed by DsbA under conditions of dimer formation that favor ToxR transcriptional activity and temporary homodimer conformations. It is tempting to speculate that ToxR homodimers are only transiently linked by an intermolecular disulfide bond under activating conditions, thus native ToxR homodimers may not be the abundant forms and therefore hard to detect. However, as soon as V. cholerae experiences less‐activating conditions, DsbC would conceivably dissolve this cysteine bridge, and ToxR would switch back to its monomeric form stabilized by intramolecular disulfide linkages. Further studies will be carried out to decipher the mechanisms of this redox switch.
Cysteines are important to the structure of proteins—they provide proteins with greater stability and allow them to better respond to environmental cues. However, cysteines can also cause incorrect folding. The operon partner of ToxR, ToxS, appears to stabilize ToxR in a conformation that is optimized for transcriptional activation (DiRita and Mekalanos, 1991; Ottemann and Mekalanos, 1996; Dziejman et al., 1999; Midgett et al., 2017; Lembke et al., 2018). Our results confirm the observations by Midgett et al. by showing ToxR–ToxS physical interactions using BACTH in E. coli (Midgett et al., 2017). We show that the interaction of ToxR with ToxS was significantly increased compared to ToxR–ToxR interactions in the absence of ToxS. Furthermore, we show that ToxS significantly increases ToxR–ToxR PPIs probably through the interaction of ToxR with ToxS itself. This was independent of the DNA‐binding capacity of ToxR, as demonstrated using a DNA‐binding‐deficient ToxRW76R mutant. Noteworthy, when ToxR is able to bind to its ToxR boxes (e.g., V. cholerae ompU operators) and ToxS is present, the maximum ToxR‐ToxR PPI was observed. To mention, disulfide‐linked homodimer formations decreased when toxS was co‐expressed in V. cholerae. However, our BACTH data in E. coli showed that ToxS concurrently enhances ToxR‐ToxR PPIs. It may be speculated that ToxS mitigates ToxR homodimer formation to diminish premature disulfide bond formations and favor specific ToxR interactions that may convert into an optimized transcriptional active complex that exhibits an ideal conformation for operator binding (Figure 7). Furthermore, ToxS may keep inter‐molecular disulfide bonds labile and therefore counteracted the DsbC isomerase action, which ultimately leads to the switching back and forth between inter‐ and intramolecular disulfide bonds. However, we need to interpret such data carefully, since overexpression of proteins may cause artificial effects. Further studies are needed to evaluate this issue.
Finally, we unraveled that DC was able to further enhance ToxR‐ToxR PPIs when toxS was co‐expressed. These observations extend the results published by Midgett et al., who reported that chenodeoxycholate interacts with the purified periplasmic domain of ToxR which then leads to enhanced interactions between ToxR and ToxS (Midgett et al., 2017). This indicates that DC may facilitate ToxR cooperativity to further stabilize or support the interactions within the ToxRS complex (Figure 7). To decipher this mechanism, future studies are needed to solve the protein structure of the ToxRS complex co‐crystallized with DC to identify possible conformational (e.g., homo‐, heterodimerization or sub‐domain) changes.
In this report, we focused on the molecular mechanism of the ToxR activation process mainly restricted to the ompU and ompT promoters, which are known to respond to bile. Subsequently, OmpU then confers the bacteria to bile resistance, an important physiological adaptation process, during the course of colonization in humans (Provenzano et al., 2000; Provenzano and Klose, 2000). Our obtained results are in accordance with previous observations that show that dimerization and other PPIs occur between the ToxR and ToxR‐ToxS molecules. This study extends the current view by showing that such ToxR PPIs are dynamic in response to ToxR‐boxes, cysteine disulfide bond formations, ToxS and the presence of bile (DC) (Figure 7). Since the binding ability for ompU‐operators increases ToxR PPIs in the presence of ToxS and the periplasmic domain plays a major role here, shown by the toxRC293S mutant, a signal path that leads from the inside to the outside seems very likely for ToxR. Still, it would be intriguing to speculate that environmental factors would initiate ToxR PPIs in an outside‐to‐inside direction. Here, the first hint is derived from bile (DC), representing an extracellular signal molecule. We show that it enhances the PPIs of ToxR in a similar manner in the presence of ToxS. In light of our results, we propose that sequential activation requirements such as that of ToxR may initially only form labile ToxR‐ToxR contacts. Such a preliminary complex then associates and gets stabilized by ToxS. If bile is present, more tightly bound ToxRS complexes are formed. Their binding to DNA operator binding sites then increases ToxR‐ToxR PPIs to the maximum. We hypothesize that the stability of ToxR complexes is further increased by the formation of transient intermolecular disulfide bonds during DNA binding, which is catalyzed by DsbA/DsbC, while PPIs between ToxR and ToxS may be reduced (Figure 7). Most of our data were derived from in vitro experiments; further in vivo studies will be carried out to determine the biological relevance of our findings. Regarding the mechanism of ToxR dimerization, some interesting questions still remain to be answered: what is the strength of the interaction between ToxR molecules and how does this change during the interplay with environmental factors; when exactly do ToxS‐ToxR complexes arise and when do they dissolve; does the binding of ToxR to its DNA operator sites result in conformational changes? Further comprehensive analyses of the mechanisms of action are needed to clarify these questions.
4. EXPERIMENTAL PROCEDURES
4.1. Strains, plasmids, and culture conditions
All bacterial strains and plasmids used in this study are listed in Table 1. Here, V. cholerae O1 El Tor Inaba P27459‐S was used as the wild‐type (WT) strain (Pearson et al., 1993). The E. coli strains XL1‐Blue, DH5α λpir, BL21 (DE3), and SM10 λpir were used for cloning, plasmid propagation, and conjugation (Kolter et al., 1978; Hanahan, 1983; Miller and Mekalanos, 1988), (New England Biolabs). Unless indicated otherwise, bacteria were routinely grown with aeration in lysogeny broth (LB), M9 maltose minimal medium or MacConkey maltose agar plates or in the respective liquid medium at 180 rpm at 37°C. When appropriate, supplements were added at the following final concentrations: streptomycin (Sm; 100 μg/ml), ampicillin (Ap; 50 or 100 μg/ml), chloramphenicol (Cm; 2 μg/ml), kanamycin (Km; 50 μg/ml), l‐arabinose (0.1%), sucrose (10%), maltose (0.2% or 1%), glucose (0.2%), isopropyl ß‐d‐1‐thiogalactopyranoside (IPTG; 0.05 or 0.5 mM), and sodium‐deoxycholate (DC; 0.1%).
TABLE 1.
Strains and plasmids used in this study
| Strains/Plasmids | Descriptions | References |
|---|---|---|
| E. coli strains | ||
| DH5αλpir | F‐ Δ(lacZYA‐argF)U169 recA1 endA1 hsdR17 supE44 thi‐1 gyrA96 relA1 λ::pir | Hanahan, (1983) |
| SM10λpir | thi thr leu tonA lacY supE recA::RPA‐2‐Te::Mu λpirR6K, Kmr | Miller and Mekalanos (1988) |
| XL1‐Blue | F ‐ ::Tn10 proA+B+ lacq Δ(lacZ)M151 recA1 endA1 gyrA46 (Nalr) thi hsdR17 (rK−mK+) supE44 relA1 lac | Bullock et al. (1987) |
| BL21 (DE3) | fhuA2 [lon] ompTgal (λ DE3) [dcm] ∆hsdS λ DE3=λsBamHIo ∆EcoRI‐B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 | NEB |
| W3110 ΔcyaA | F‐ λ‐ rpoS(Am) rph‐1 Inv(rrnD‐rrnE) ΔcyaA::scar | Herbst et al. (2018) |
| V. cholerae strains | ||
| WT | P27459‐S, O1 Inaba, El Tor, clinical isolate, Bangladesh 1976, spontaneous Smr | Pearson et al. (1993) |
| ΔtoxR | P27459‐S with deletion in toxR, Smr | Fengler et al. (2012) |
| ΔtoxRS | P27459‐S with deletion in toxR and toxS, Smr | Fengler et al. (2012) |
| toxRCC | P27459‐S with toxR replaced by FLAG‐toxRC236SC293S, Smr | Fengler et al. (2012) |
| toxRC236S | P27459‐S with toxR replaced by FLAG‐toxRC236S, Smr | This study |
| toxRC293S | P27459‐S with toxR replaced by FLAG‐toxRC293S, Smr | This study |
| toxRC293W76RS | P27459‐S with toxR replaced by FLAG‐toxRC293SW76R, Smr | This study |
| ΔdsbA | P27459‐S with dsbA replaced by km cassette, Smr, Kmr | Fengler et al. (2012) |
| dsbC::pGP | P27459‐with dsbC inserted by pGP704, Smr, Apr | Fengler et al., (2012) |
| ΔdsbA toxRC293S | P27459‐S ΔdsbA with toxR replaced by FLAG‐toxRC293S, Smr , Kmr | This study |
| dsbC::pGP toxRC293S | P27459‐S dsbC::pGP with toxR replaced by FLAG‐toxRC293S, Smr , Apr | This study |
| ΔdegP | P27459‐S with degP replaced by cat cassette, Smr, Cmr | This study |
| ΔdegP ompU::phoA | P27459‐S ΔdegP with insertion of pGP704phoA downstream of ompU, Smr , Cmr, Apr | This study |
| ΔtoxR ompU::phoA | P27459‐S with deletion in toxR and insertion of pGP704phoA downstream of ompU, Smr , Cmr, Apr | This study |
| ΔdegP toxRCC ompU::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC236SC293S and insertion of pGP704phoA downstream of ompU, Smr, Cmr, Apr | This study |
| ΔdegP toxRC236S ompU::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC236S and insertion of pGP704phoA downstream of ompU, Smr , Cmr, Apr | This study |
| ΔdegP toxRC293S ompU::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC293S and insertion of pGP704phoA downstream of ompU, Smr , Cmr, Apr | This study |
| ΔdegP ompT::phoA | P27459‐S ΔdegP with insertion of pGP704phoA downstream of ompT, Smr , Cmr, Apr | This study |
| ΔtoxR ompT::phoA | P27459‐S with deletion in toxR and insertion of pGP704phoA downstream of ompT, Smr , Cmr, Apr | This study |
| ΔdegP toxRCC ompT::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC236SC293S and insertion of pGP704phoA downstream of ompT, Smr , Cmr, Apr | This study |
| ΔdegP toxRC236S ompT::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC236S and insertion of pGP704phoA downstream of ompT, Smr , Cmr, Apr | This study |
| ΔdegP toxRC293S ompT::phoA | P27459‐S ΔdegP with toxR replaced by FLAG‐toxRC293S and insertion of pGP704phoA downstream of ompT, Smr , Cmr, Apr | This study |
| Plasmids | ||
| pKEK229 | OriR6K, mobRP4, sacB, Apr | Correa et al. (2000) |
| pCVD442 | OriR6K, mobRP4, sacB, Apr | Donnenberg and Kaper (1991) |
| pGP704 | OriR6K, mobRP4, Apr | Miller and Mekalanos (1988) |
| pBAD18‐Kan | Expression vector, oriColE1, arabinose Inducible, Kmr | Guzman et al. (1995) |
| pBAD18 | Expression vector, oriColE1, arabinose Inducible, Apr | Guzman et al. (1995) |
| pACYC184 | Cloning vector, orip15A, Tetr, Cmr | Rose (1988) |
| pFLAG‐MACTM | Expression vector with N‐terminal FLAG‐Tag, IPTG inducible, Apr | Sigma‐Aldrich |
| pKT25 | Expression vector, encodes for the first 224 AA of CyaA (T25 fragment, B. pertussis), C‐terminal heterologous protein fusion, IPTG inducible, Kmr | Karimova et al. (1998) |
| pUT18C | Expression vector, encodes for AA 225 to 399 of CyaA (T18 fragment, B. pertussis), C‐terminal heterologous protein fusion, IPTG inducible, Apr | Karimova et al. (1998) |
| pKEK229dsbA::km | pKEK229 carrying up and down fragments, Apr, Kmr | Fengler et al. (2012) |
| pCVD442degP::cat | pCVD442 carrying up and down fragment of degP flanking a cat cassette, Apr, Cmr | Vorkapic et al. (2019) |
| pCVD442toxR | pCVD442 carrying up and down fragment of toxR, Apr | Fengler et al. (2012) |
| pCVD442FLAGtoxRCC | pCVD442 carrying up and down fragment of FLAGtoxRC236C293S, Apr | Fengler et al. (2012) |
| pCVD442FLAGtoxRC236S | pCVD442 carrying up and down fragment of FLAGtoxRC236S, Apr | This study |
| pCVD442FLAGtoxRC293S | pCVD442 carrying up and down fragment of FLAGtoxRC293S, Apr | This study |
| pCVD442FLAGtoxRC293SW76R | pCVD442 carrying up and down fragment of FLAGtoxRC293SW76R, Apr | This study |
| pFLAGtoxR | toxR of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pFLAGtoxRS | toxR and toxS of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pFLAGtoxRCC | toxRC236SC293S point mutant of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pFLAGtoxRCCtoxS | toxRC236SC293S point mutant and toxS of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pFLAGtoxRC236S | toxRC236S point mutant of P27495‐S in pFLAG‐MACTM, Apr | This study |
| pFLAGtoxRC293S | toxRC293S point mutant of P27495‐S in pFLAG‐MACTM, Apr | This study |
| pFLAGtoxRW76RS | toxRW76RS of point mutant of P27495‐S in pFLAG‐MACTM, Apr | This study |
| pBAD18‐KanFLAGtoxRS | FLAG‐toxRS of P27495‐S in pBAD18‐Kan, Kmr | This study |
| pBAD18‐FLAGtoxRS | FLAG‐toxRS of P27495‐S in pBAD18, Apr | This study |
| pGP704dsbC | pGP704 carrying internal fragment of dsbC, Apr | Fengler et al. (2012) |
| pGP704phoAompU | pGP704phoA with ompU gene fragment, Apr | Lembke et al. (2018) |
| pGP704phoAompT | pGP704phoA with ompT gene fragment, Apr | Lembke et al. (2018) |
| pKT25‐zip | BACTH positive control, leucine zipper of GCN4 (yeast) fused to the T25 fragment, Kmr | Karimova et al. (1998) |
| pUT18C‐zip | BACTH positive control, leucine zipper of GCN4 (yeast) fused to the T18 fragment, Apr | Karimova et al. (1998) |
| pKT25toxR | toxR of P27495‐S in pKT25, Kmr | This study |
| pKT25toxRS | toxR and toxS of P27495‐S in pKT25, Kmr | This study |
| pKT25toxRW76R | toxRW76R of P27495‐S in pKT25, Kmr | This study |
| pKT25toxRW76RS | toxRW76R and toxS of P27495‐S in pKT25, Kmr | This study |
| pUT18CtoxR | toxR of P27495‐S in pUT18C, Apr | This study |
| pUT18CtoxRS | toxR and toxS of P27495‐S in pUT18C, Apr | This study |
| pUT18CtoxRW76R | toxRW76R of P27495‐S in pUT18C, Apr | This study |
| pUT18CtoxRW76RS | toxRW76R and toxS of P27495‐S in pUT18C, Apr | This study |
| pUT18CtoxSFLAG | toxS‐FLAG of P27495‐S in pUT18C, Apr | This study |
| pUT18CopompU | ompU operators O123 of P27495‐S in pUT18C, Apr | This study |
| pUT18CopompU toxR | toxR of P27495‐S in pUT18CopompU, Apr | This study |
| pUT18CopompU toxRS | toxR and toxS of P27495‐S in pUT18CopompU, Apr | This study |
| pUT18CopompU toxRW76R | toxRW76R of P27495‐S in pUT18CopompU, Apr | This study |
| pUT18CopompU toxRW76RS | toxRW76R and toxS of P27495‐S in pUT18CopompU, Apr | This study |
4.2. Strain and plasmid constructions
The primer (Thermo Fisher Scientific) used in this study for amplification as well as sequencing are listed in Table 2. PCR products and vectors were digested with the respective restriction endonucleases, ligated with T4 DNA ligase (NEB) and sequenced for validation (LGC Genomics) (data not shown).
TABLE 2.
Oligonucleotidesa (5ʹ‐3ʹ) used in this study
| Oligonucleotides (5′‐3′) used in this study | |
|---|---|
| BACTH | |
| Pst_pKT25_ToxR_fwd_BACTH | ATTCTGCAGTCGGATTAGGACACAACTC |
| Pst_pUT18C_ToxR_fwd_BACTH | ATTCTGCAGTTTCGGATTAGGACACAACT |
| XbaI_ToxR_rev_BACTH | ATTTCTAGACTACTCACACACTTTGATGG |
| ToxS_XbaI‐fwd | TAATCTAGAGATGCAAAATAGACACATCGCC |
| ToxS‐FLAG_EcoRI‐rev | TATGAATTCTGAAAATCTTCTCTCACTCGA |
| XbaI_ToxS_rev | ATTTCTAGATTAAGAATTACTGAACAGTACG |
| BamHI_ompUO123_fwd | ATTGGATCCTCCTAAATCGGGTCGGGT |
| KpnI_ompUO123_rev | AAAGGTACCATTGGTCATTGTTGTGTTCA |
| toxRC236S and toxRC293S substitution | |
| HindIII_toxR_5ʹ_FLAG | AATAAGCTTATGTTCGGATTAGGACACAACTCA |
| KpnI_toxR_3ʹ_FLAG | AATGGTACCCTACTCACACACTTTGATGGCAT |
| KpnI_toxR293S_3ʹ_FLAG | AATGGTACCCTACTCAGACACTTTGATGGCATCGTTA |
| toxRC236S_5ʹ | GGCTACCGTCAATCGAACTGAGCGTTAAAAAATACAATGA |
| toxRC236S_3ʹ | TCATTGTATTTTTTAACGCTCAGTTCGATTGACGGTAGCC |
| SacI_toxRS_1 | TTTGAGCTCATTTGGAAATCACATCGCGCAAAC |
| XbaI_toxRS_4 | TTTTCTAGAATGACGTTTCCCCGCGGTGAG |
| c_FLAGtoxR_3ʹ_F1 | TGTCATCGTCGTCCTTGTAGTCCATCTAATGTCCCAGTATCTCCCTGT |
| c_FLAGtoxR_5ʹ_F2 | GGGACAGGGAGATACTGGGACATTAGATGGACTACAAGGACGACGATGA |
| c_FLAGtoxR_3ʹ_F2 | CTACTCACACACTTTGATGGCAT |
| c_FLAGtoxRC293S_3ʹ_F2 | CTACTCAGACACTTTGATGGCAT |
| c_FLAGtoxR_5ʹ_F3 | AACCAGTTAACGCTGAATTACATTC |
| c_FLAGtoxRC293S_5ʹ_F3 | GTTGCTAACCCTAACGATGCCATCAAAGTGTCTGAG |
| toxRC293SW76R substitution | |
| F1_ToxRC293S‐W76R_XbaI_fwd | TTATCTAGAATCCGCCACGATGAAAGCCGA |
| F1_SOE_ToxRC293S‐W76R_rev | TTGCTCTCGCCGAACAAAGTCATGCAAATCATTGCGAGA |
| F2_SOE_ToxRC293S‐W76R_fwd | GACTTTGTTCGGCGAGAGCAAGGTTTTGAAGTCGATGAT |
| F2_ToxRC293S‐W76R_SacI_rev | TAAGAGCTCCAGACCGCAGCATCCAATTGC |
| pBAD18‐KanFLAGtoxRS, pBAD18‐FLAGtoxRS, pFLAGtoxRW76RS | |
| fwd_SacI_pFlagMAC_ShineD | TTAGAGCTCATAACAATTTCACACAGGAGA |
| FLAGtoxR_fw_KpnI | ATAGGTACCATGTTCGGATTAGGACACAACTCA |
| BglII_toxRS_3ʹFLAG | TTAAGATCTTTAAGAATTACTGAACAGTACGGT |
| Sequencing | |
| phoA‐seq‐rev | GCTCACCAACTGATAACCAC |
| SacIDsbA1 | TTTGAGCTCCAAGAAGAGATCCCGATCGTC |
| Kan_cassette_rv | TTAGAAAAACTCATCGAGCA |
| PhoA3ʹ 180 rev | GCTAAGAGAATCACGCAGA |
| pGP704_CVD_rv|15 | GATGTAACGCACTGAGAAG |
| pBAD_fwd | CCATAGCATTTTTATCCATAAG |
Restriction sites are underlined. Bold letters indicate codons changed to obtain desired amino acid mutations.
Suicide plasmids generating chromosomal deletions, amino acid substitutions or phoA fusions were achieved via PCR or SOE‐PCR (splicing by overlap extension) (Horton et al., 1989). Creation of deletion mutants was performed by cloning two DNA fragments of approximately 800 bp representing upstream and downstream of the target gene into pCVD442 (Donnenberg and Kaper, 1991). ToxR amino acid substitutions C236S and C293S were constructed using c_FLAGtoxR_5ʹ_F2, c_FLAGtoxR_3ʹ_F2, c_FLAGtoxRC293S_3ʹ_F2, SacI_toxRS_1, c_FLAGtoxR_3ʹ_F1, XbaI_toxRS_4, c_FLAGtoxR_5ʹ_F3 and c_FLAGtoxRC293S_5ʹ_F3, respectively. Fragments were amplified from pFLAGtoxRC236S, pFLAGtoxRC293S or chromosomal WT DNA to create pCVD442FLAGtoxRC236S and pCVD442FLAGtoxRC293S, respectively. Chromosomal DNA of V. cholerae P27459‐S ΔtoxR::FLAGtoxRC293S served as a template to generate the suicide plasmid pCVD442FLAGtoxRC236SW76R. The W76R point mutation (Morgan et al., 2011) in toxR was generated by SOE PCR utilizing primers listed in Table 2, subitem toxRC293SW76R substitution. The resulting plasmids were isolated from E. coli DH5α λpir, transformed into SM10 λpir and subsequently introduced into V. cholerae derivatives by conjugation (Donnenberg and Kaper, 1991). Transconjugants were selected on LB plates containing streptomycin and ampicillin. Sucrose counter‐selection and further selection steps were performed as described previously (Donnenberg and Kaper, 1991).
For the construction of the expression plasmid pFLAGtoxRC293S template DNA of the WT was used together with the primer pair HindIII_toxR_5ʹ_FLAG and KpnI_toxR293S_3ʹ_FLAG with the latter containing a point mutation within the DNA sequence that changed Cys293 to Ser293. The C236S point mutation in pFLAGtoxRC236S was generated by SOE PCR, using pFLAGtoxR as a template together with primers HindIII_toxR_5ʹ_FLAG, toxRC236S_3ʹ, toxRC236S_5ʹ, and KpnI_toxR_3ʹ_FLAG. Primers fwd_SacI_pFlagMAC_ShineD and XbaI_ToxS_rev were used to amplify PCR fragments derived from pFLAGtoxRS to construct pBAD18‐KanFLAGtoxRS and pBAD18FLAGtoxRS. The toxR, toxRS, toxRW76R, toxRW76RS, toxS‐FLAG, and ompU O123 operator fragments in pKT25 or pUT18C for the BACTH system were amplified by PCR using the BACTH primers listed in Table 2. The coding regions originate from WT DNA or pFLAGtoxRW76RS plasmid DNA which itself was generated by SOE PCR using primers FLAGtoxR_fw_KpnI, BglII_toxRS_3ʹFLAG, F1_SOE_ToxRC293S‐W76R_rev, and F2_SOE_ToxRC293S‐W76R_fwd. Subsequently, the pFLAG‐MACTM, pKT25, pUT18C (IPTG inducible) and pBAD (arabinose inducible) plasmids were electroporated into DH5α λpir, XL1‐Blue or BL21 (DE3) and monitored for expression before being introduced into E. coli W3110 ΔcyaA or V. cholerae derivatives.
4.3. Generation of cell extracts and immunoblot analysis
To verify protein expression in V. cholerae and E. coli, immunoblotting was performed. Whole cell lysates (WCL) were taken from cultures grown in LB overnight which were used to inoculate fresh LB or M9 maltose minimal medium to an OD600 of 0.1. Cells were grown at 37°C and 180 rpm until the mid‐log phase (OD600 = 0.4–0.6) was reached and subsequently collected before or after induction with IPTG (0.05–0.5 mM) or arabinose (0.1%) for 2 hr. Cells were resuspended in Laemmli buffer (Laemmli, 1970) with or without β‐mercaptoethanol, corresponding to reducing and non‐reducing conditions, respectively. The overall protein contents were analyzed to contain similar protein levels as described previously (Lembke et al., 2018). Following transfer on a AmershamTM ProtranTM 0.45‐µm nitrocellulose membrane (GE Healthcare Life Sciences), the membranes were blotted for OmpU, OmpT, ToxR or FLAG‐tagged proteins respectively (mouse anti‐OmpU and anti‐OmpT 1:3,000 (Salem et al., 2015), rabbit anti‐ToxR 1:1,000 (Fan et al., 2014), mouse anti‐FLAG M2 Peroxidase (HRP) 1:2,000 (Sigma)). The washing steps were performed as described previously (Lembke et al., 2018). Peroxidase secondary antibodies (horseradish peroxidase‐conjugated goat anti‐rabbit 1:10,000 or goat anti‐mouse 1:7,500 Dianova GmbH) were used for detection using ECL solution (Clarity™ Western ECL Blotting Substrates, BIO‐RAD) prior to visualization of the reactive protein bands using a Molecular Imager ChemiDocTM XRS System (BIO‐RAD). Quantification of ToxR protein band intensities was performed using Image Lab Software (BIO‐RAD). One immunoblot used for this analysis is shown (Figure S5).
4.4. Bacterial two‐hybrid analysis (BACTH)
For monitoring of protein–protein interactions in vivo, the bacterial adenylate cyclase‐based two‐hybrid (BACTH) system was performed as described in Karimova et al. (1998). V. cholerae ToxR and ToxRS derivatives or ToxS‐FLAG were fused to the 3ʹ end of the adenylate cyclase T25 or T18 fragment from Bordetella pertussis (CyaA), respectively. Additionally, V. cholerae ompU operators (opompU) were cloned on pUT18C. Prior to the interaction studies, plasmid functionality was tested in E. coli XL1‐Blue, Dh5αλpir or BL21 (DE3) by expression and sequencing. Oligonucleotide primers for cloning onto pKT25 and pUT18C plasmids are listed in Table 2. Positive complementary pUT18CtoxSFLAG, pKT25toxR and pUT18CtoxR plasmids (and its derivatives) were co‐transformed in E. coli K‐12 strain W3110 ∆cyaA (Herbst et al., 2018), which lacks endogenous adenylate cyclase activity. The transformants were selected on MacConkey agar (Becton Dickinson) plates supplemented with 50 µg/ml of ampicillin, 50 µg/ml of kanamycin, and 1% maltose for 24 hr at 30°C to reveal the CyaA+ phenotype (red colonies indicate maltose fermentation). For a clear presentation of PPIs, cells were grown at 37°C in LB medium overnight using selection for kanamycin and ampicillin. Subsequently, 5 µl of overnight culture was transferred directly onto a single MacConkey indicator plate to be compared and incubated for 24 hr at 30°C. As a positive complementation control, the leucine zipper of the yeast GCN4 protein was used (pKT25‐zip and pUT18C‐zip) whereas the empty pKT25 and pUT18C plasmids served as negative controls.
4.5. β‐Galactosidase and alkaline phosphatase assays
To determine transcriptional activity of chromosomal ToxR, pGP704phoAompU and pGP704phoAompT were introduced into V. cholerae derivatives by conjugation. Strains were inoculated from selective LB overnight cultures to an OD600 of 0.1 and grown in fresh selective M9 maltose minimal medium at 37°C and 180 rpm until the mid‐log phase (OD600 = 0.4–0.6) was reached. For the quantitative analysis of ToxR‐ToxR or ToxR‐ToxS‐FLAG protein–protein interactions in vivo, the BACTH system was used in E. coli W3110 ∆cyaA. The method is based on the positive regulation of β‐galactosidase expression by cAMP/CAP that will be produced upon functional complementation of the chimeric adenylate cyclase. There cultures were inoculated from selective LB overnight cultures to an OD600 of 0.1 and grown in fresh selective LB medium supplemented with 0.05 mM IPTG at 37°C and 180 rpm to the stationary phase in the absence or presence of 0.1% DC. For each assay 1–2 ml of culture was harvested by centrifugation respectively. Enzymatic activities for LacZ and PhoA were measured as described previously (Taylor et al., 1987; Miller, 1992) with at least three biological replicates each with technical triplicates.
4.6. Statistical analysis
The statistics in the respective experiments were carried out using GraphPad Prism 6.
Supporting information
Supplementary Material
Lembke M, Höfler T, Walter A‐N, et al. Host stimuli and operator binding sites controlling protein interactions between virulence master regulator ToxR and ToxS in Vibrio cholerae . Mol Microbiol. 2020;114:262–278. 10.1111/mmi.14510
REFERENCES
- Bischof, L.F. , Haurat, M.F. and Albers, S.V. (2019) Two membrane‐bound transcription factors regulate expression of various type‐IV‐pili surface structures in Sulfolobus acidocaldarius . PeerJ, 7, e6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameyer, S. , Rosch, T.C. , El Andari, J. , Hoyer, E. , Schwarz, J. , Graumann, P.L. , et al (2019) DNA‐binding directs the localization of a membrane‐integrated receptor of the ToxR family. Communications Biology, 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock, W.O. , Fernandez, J.M. and Short, J.M. (1987) XL1‐Blue—a high‐efficiency plasmid transforming recA Escherichia coli strain with β‐galactosidase selection. BioTechniques, 5, 376–379. [Google Scholar]
- Childers, B.M. and Klose, K.E. (2007) Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiology, 2, 335–344. [DOI] [PubMed] [Google Scholar]
- Correa, N.E. , Lauriano, C.M. , McGee, R. and Klose, K.E. (2000) Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Molecular Microbiology, 35, 743–755. [DOI] [PubMed] [Google Scholar]
- Crawford, J.A. , Kaper, J.B. and DiRita, V.J. (1998) Analysis of ToxR‐dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae . Molecular Microbiology, 29, 235–246. [DOI] [PubMed] [Google Scholar]
- Crawford, J. A. , Krukonis, E. S. , & DiRita, J. J. (2007). Membrane localisation of the ToxR winged‐helix domain is regulated for TcpP‐mediated virulence gene activation in Vibrio cholerae . Molecular Microbiology, 47, 1459–1473. [DOI] [PubMed] [Google Scholar]
- Dalia, A.B. , Lazinski, D.W. and Camilli, A. (2014) Identification of a membrane‐bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae . mBio, 5, e01028‐01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRita, V.J. and Mekalanos, J.J. (1991) Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell, 64, 29–37. [DOI] [PubMed] [Google Scholar]
- Donnenberg, M.S. and Kaper, J.B. (1991) Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive‐selection suicide vector. Infection and Immunity, 59, 4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziejman, M. , Kolmar, H. , Fritz, H.J. and Mekalanos, J.J. (1999) ToxR co‐operative interactions are not modulated by environmental conditions or periplasmic domain conformation. Molecular Microbiology, 31, 305–317. [DOI] [PubMed] [Google Scholar]
- Dziejman, M. and Mekalanos, J.J. (1994) Analysis of membrane protein interaction: ToxR can dimerize the amino terminus of phage lambda repressor. Molecular Microbiology, 13, 485–494. [DOI] [PubMed] [Google Scholar]
- Fan, F. , Liu, Z. , Jabeen, N. , Birdwell, L.D. , Zhu, J. and Kan, B. (2014) Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen‐limiting conditions. Infection and Immunity, 82, 1676–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengler, V.H. , Boritsch, E.C. , Tutz, S. , Seper, A. , Ebner, H. , Roier, S. , et al (2012) Disulfide bond formation and ToxR activity in Vibrio cholerae . PLoS ONE, 7, e47756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss, T.J. , Morgan, S.J. , French, E.L. and Krukonis, E.S. (2013) ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infection and Immunity, 81, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauschopf, U. , Winther, J.R. , Korber, P. , Zander, T. , Dallinger, P. and Bardwell, J.C. (1995) Why is DsbA such an oxidizing disulfide catalyst? Cell, 83, 947–955. [DOI] [PubMed] [Google Scholar]
- Guzman, L.‐M. , Beblin, D. , Carson, M.J. and Beckwith, J. (1995) Tight regulation, modulation, and high‐level expression by vectors containing the arabinose pBAD promotor. Journal of Bacteriology, 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.L. , Matson, J.S. , DiRita, V.J. and Biteen, J.S. (2014) Single‐molecule tracking in live Vibrio cholerae reveals that ToxR recruits the membrane‐bound virulence regulator TcpP to the toxT promoter. Molecular Microbiology. Available at: 10.1111/mmi.12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. (1983) Studies on transformation of Escherichia coli with plasmids. Journal of Molecular Biology, 166, 557–580. [DOI] [PubMed] [Google Scholar]
- Hase, C.C. and Mekalanos, J.J. (1998) TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae . Proceedings of the National Academy of Sciences of the United States of America, 95, 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg, J.F. , Eisen, J.A. , Nelson, W.C. , Clayton, R.A. , Gwinn, M.L. , Dodson, R.J. , et al (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae . Nature, 406, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, S. , Lorkowski, M. , Sarenko, O. , Nguyen, T.K.L. , Jaenicke, T. and Hengge, R. (2018) Transmembrane redox control and proteolysis of PdeC, a novel type of c‐di‐GMP phosphodiesterase. EMBO Journal, 37, e97825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, A.F. , Hagey, L.R. and Krasowski, M.D. (2010) Bile salts of vertebrates: structural variation and possible evolutionary significance. Journal of Lipid Research, 51, 226–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, R.M. , Hunt, H.D. , Ho, S.N. , Pullen, J.K. and Pease, L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Jung, K. , Fabiani, F. , Hoyer, E. and Lassak, J. (2018) Bacterial transmembrane signalling systems and their engineering for biosensing. Open Biology, 8, 180023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura, H. , Katzen, F. and Beckwith, J. (2003) Protein disulfide bond formation in prokaryotes. Annual Review of Biochemistry, 72, 111–135. [DOI] [PubMed] [Google Scholar]
- Karimova, G. , Pidoux, J. , Ullmann, A. and Ladant, D. (1998) A bacterial two‐hybrid system based on a reconstituted signal transduction pathway. Proceedings of the National Academy of Sciences of the United States of America, 95, 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney, L.J. (2002) Structure/function relationships in OmpR and other winged‐helix transcription factors. Current Opinion in Microbiology, 5, 135–141. [DOI] [PubMed] [Google Scholar]
- Kolter, R. , Inuzuka, M. and Helinski, D.R. (1978) Trans‐complementation‐dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell, 15, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Krukonis, E.S. , Yu, R.R. and Dirita, V.J. (2000) The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane‐localized transcriptional activators on a single promoter. Molecular Microbiology, 38, 67–84. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Landeta, C. , Boyd, D. and Beckwith, J. (2018) Disulfide bond formation in prokaryotes. Nature Microbiology, 3, 270–280. [DOI] [PubMed] [Google Scholar]
- Lembke, M. , Pennetzdorfer, N. , Tutz, S. , Koller, M. , Vorkapic, D. , Zhu, J. , et al (2018) Proteolysis of ToxR is controlled by cysteine‐thiol redox state and bile salts in Vibrio cholerae . Molecular Microbiology, 110, 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.C. , Crawford, J.A. , DiRita, V.J. and Kaper, J.B. (2000) Molecular cloning and transcriptional regulation of ompT, a ToxR‐repressed gene in Vibrio cholerae . Molecular Microbiology, 35, 189–203. [DOI] [PubMed] [Google Scholar]
- Littlefield, O. and Nelson, H.C. (1999) A new use for the ‘wing’ of the ‘winged’ helix‐turn‐helix motif in the HSF‐DNA cocrystal. Natural Structural Biology, 6, 464–470. [DOI] [PubMed] [Google Scholar]
- Maris, A.E. , Walthers, D. , Mattison, K. , Byers, N. and Kenney, L.J. (2005) The response regulator OmpR oligomerizes via beta‐sheets to form head‐to‐head dimers. Journal of Molecular Biology, 350, 843–856. [DOI] [PubMed] [Google Scholar]
- Martinez‐Hackert, E. and Stock, A.M. (1997) Structural relationships in the OmpR family of winged‐helix transcription factors. Journal of Molecular Biology, 269, 301–312. [DOI] [PubMed] [Google Scholar]
- Matson, J.S. , Withey, J.H. and DiRita, V.J. (2007) Regulatory networks controlling Vibrio cholerae virulence gene expression. Infection and Immunity, 75, 5542–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett, C.R. , Almagro‐Moreno, S. , Pellegrini, M. , Taylor, R.K. , Skorupski, K. and Kull, F.J. (2017) Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Molecular Microbiology, 105, 258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.H. (1992) A Short Course in Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Miller, V.L. , DiRita, V.J. and Mekalanos, J.J. (1989) Identification of toxS, a regulatory gene whose product enhances toxR‐mediated activation of the cholera toxin promoter. Journal of Bacteriology, 171, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, V.L. and Mekalanos, J.J. (1988) A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR . Journal of Bacteriology, 170, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, V.L. , Taylor, R.K. and Mekalanos, J.J. (1987) Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell, 48, 271–279. [DOI] [PubMed] [Google Scholar]
- Missiakas, D. , Schwager, F. and Raina, S. (1995) Identification and characterization of a new disulfide isomerase‐like protein (DsbD) in Escherichia coli . EMBO Journal, 14, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, S.J. , Felek, S. , Gadwal, S. , Koropatkin, N.M. , Perry, J.W. , Bryson, A.B. , et al (2011) The two faces of ToxR: activator of ompU, co‐regulator of toxT in Vibrio cholerae . Molecular Microbiology, 81, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, S.J. , French, E.L. , Plecha, S.C. and Krukonis, E.S. (2019) The wing of the ToxR winged helix‐turn‐helix domain is required for DNA binding and activation of toxT and ompU . PLoS ONE, 14, e0221936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann, K.M. and Mekalanos, J.J. (1996) The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. Journal of Bacteriology, 178, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington, J. , Donnelly, D. , Johnson, M.S. , Šali, A. and Blundell, T.L. (1992) Environment‐specific amino acid substitution tables: tertiary templates and prediction of protein folds. Protein Science, 1, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, G.D. , Woods, A. , Chiang, S.L. and Mekalanos, J.J. (1993) CTX genetic element encodes a site‐specific recombination system and an intestinal colonization factor. Proceedings of the National Academy of Sciences of the United States of America, 90, 3750–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, K.M. and Gellings, P.S. (2018) Multiple intraintestinal signals coordinate the regulation of Vibrio cholerae virulence determinants. Pathogens and Disease, 76 Available at: 10.1093/femspd/ftx1126. [DOI] [PubMed] [Google Scholar]
- Provenzano, D. and Klose, K.E. (2000) Altered expression of the ToxR‐regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proceedings of the National Academy of Sciences of the United States of America, 97, 10220–10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, D. , Schuhmacher, D.A. , Barker, J.L. and Klose, K.E. (2000) The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infection and Immunity, 68, 1491–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, J.E. , Sheng, W. , Morgan, L.K. , Nolet, R. , Liao, X. and Kenney, L.J. (2008) Amino acids important for DNA recognition by the response regulator OmpR. Journal of Biological Chemistry, 283, 8664–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, R.E. (1988) The nucleotide sequence of pACYC184. Nucleic Acids Research, 16, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem, W. , Leitner, D.R. , Zingl, F.G. , Schratter, G. , Prassl, R. , Goessler, W. , et al (2015) Antibacterial activity of silver and zinc nanoparticles against Vibrio cholerae and enterotoxic Escherichia coli . International Journal of Medical Microbiology, 305, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer, S. , Egan, S. , Larsen, M.R. , Bartlett, D.H. and Kjelleberg, S. (2006) Unravelling the role of the ToxR‐like transcriptional regulator WmpR in the marine antifouling bacterium Pseudoalteromonas tunicata . Microbiology, 152, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Taylor, R.K. , Miller, V.L. , Furlong, D.B. and Mekalanos, J.J. (1987) Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proceedings of the National Academy of Sciences of the United States of America, 84, 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsch, L. , Koller, C. , Donhofer, A. and Jung, K. (2011) Detection and function of an intramolecular disulfide bond in the pH‐responsive CadC of Escherichia coli . BMC Microbiology, 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi, M. , Battistini, A. , Araco, A. , Roda, L.G. and D'Agnolo, G. (1979) The role of the reactive disulfide bond in the interaction of cholera‐toxin functional regions. European Journal of Biochemistry, 93, 621–627. [DOI] [PubMed] [Google Scholar]
- Ulrich, L.E. , Koonin, E.V. and Zhulin, I.B. (2005) One‐component systems dominate signal transduction in prokaryotes. Trends in Microbiology, 13, 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorkapic, D. , Mitterer, F. , Pressler, K. , Leitner, D.R. , Anonsen, J.H. , Liesinger, L. , et al (2019) A broad spectrum protein glycosylation system influences type II protein secertion and associated phenotypes in Vibrio cholerae . Frontiers in Microbiology. Available at: 10.3389/fmicb.2019.02780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich, F. , Ronai, A. , Speth, V. , Seelig, J. and Blume, A. (1975) Thermotropic lipid clustering in tetrahymena membranes. Biochemistry, 14, 3730–3735. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Liu, Z. , Hughes, C. , Stern, A.M. , Wang, H. , Zhong, Z. , et al (2013) Bile salt‐induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proceedings of the National Academy of Sciences of the United States of America, 110, 2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. and Isberg, R.R. (1997) Transcriptional regulation of the Yersinia pseudotuberculosis pH6 antigen adhesin by two envelope‐associated components. Molecular Microbiology, 24, 499–510. [DOI] [PubMed] [Google Scholar]
- Yoshida, T. , Qin, L. , Egger, L.A. and Inouye, M. (2006) Transcription regulation of ompF and ompC by a single transcription factor, OmpR. Journal of Biological Chemistry, 281, 17114–17123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material