

Abstract
Clinical data have provided evidence that schistosomiasis can promote hepatocellular carcinogenesis. c‐Jun and STAT3 are critical regulators of liver cancer development and progression. The aim of the present study was to investigate the hepatocellular activation of c‐Jun and STAT3 by Schistosoma mansoni infection. Expression and function of c‐Jun and STAT3 as well as proliferation and DNA repair were analyzed by western blotting, electrophoretic mobility‐shift assay, and immunohistochemistry in liver of S. mansoni–infected hamsters, Huh7 cells, primary hepatocytes, and human liver biopsies. Hepatocellular activation of c‐Jun was demonstrated by nuclear translocation of c‐Jun, enhanced phosphorylation (Ser73), and AP‐1/DNA‐binding in response to S. mansoni infection. Nuclear c‐Jun staining pattern around lodged eggs without ambient immune reaction, and directionally from granuloma to the central veins, suggested that substances released from schistosome eggs were responsible for the observed effects. In addition, hepatocytes with c‐Jun activation show cell activation and DNA double‐strand breaks. These findings from the hamster model were confirmed by analyses of human biopsies from patients with schistosomiasis. Cell culture experiments finally demonstrated that activation of c‐Jun and STAT3 as well as DNA repair were induced by an extract from schistosome eggs (soluble egg antigens) and culture supernatants of live schistosome egg (egg‐conditioned medium), and in particular by IPSE/alpha‐1, the major component secreted by live schistosome eggs. The permanent activation of hepatocellular carcinoma–associated proto‐oncogenes such as c‐Jun and associated transcription factors including STAT3 by substances released from tissue‐trapped schistosome eggs may be important factors contributing to the development of liver cancer in S. mansoni–infected patients. Therefore, identification and therapeutic targeting of the underlying pathways is a useful strategy to prevent schistosomiasis‐associated carcinogenesis.
Abbreviations
- EMSA
electrophoretic mobility‐shift assay HBV, hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- JNK
c‐Jun N‐terminal kinase
- MCM2
Minichromosome Maintenance Complex Component 2
- PCNA
proliferating cell nuclear antigen
- SEA
soluble egg antigens
- UV
ultraviolet
Schistosomiasis is one of the most important parasitic infections worldwide that is caused by trematodes of the genus Schistosoma. Three major species of schistosomes infect humans in 78 countries: Schistosoma mansoni, S. haematobium, and S. japonicum. The disease causes severe clinical symptoms as well as socioeconomic problems and more than 200,000 deaths per year.1 According to the World Health Organization (WHO), at least 206.5 million people required preventive treatment in 2018 (WHO 2018, Schistosomiasis – fact sheet). Schistosomiasis is increasingly imported into temperate climates by immigrants from and travellers to endemic areas.2, 3 A recent epidemiological case study investigating an outbreak of urogenital schistosomiasis in Corsica (France) demonstrated the potential risk of schistosomiasis to spread into novel areas.4
During infection with S. mansoni, the paired adult worms, which lodge in mesenterial veins, produce approximately 300 eggs per day per couple. Half of the eggs migrate through the intestinal tissue toward the gut lumen, from where they are expelled. The other half, however, are carried away with the blood stream into the liver, where they get trapped. In both the gut and the liver tissue, the schistosome eggs induce an inflammatory reaction with granuloma formation.5 All evidence suggests that schistosome eggs, and not adult worms, induce the morbidity caused by schistosome infections.5 Eggs of S. mansoni trapped in the liver sinusoids continuously secrete hepatotoxic and immunologically active antigens such as IPSE/alpha‐1 (IL‐4 inducing principle from S. mansoni eggs) and omega‐1.6, 7 Recently, it has been demonstrated that IPSE/alpha‐1 representing a pathogen‐secreted and host nucleus–infiltrating protein (infiltrin) might act as a transcription factor controlling the host–parasite interaction at the molecular level.8 Moreover, egg‐secreted immunologically active antigens are involved in recruitment of inflammatory and immune cells, leading to the formation of periovular granulomas and eventually chronic fibrosis in infected individuals.9 In S. mansoni infections, the fibrotic process takes 5‐15 years and leads in turn to progressive occlusion of the portal veins, portal hypertension, splenomegaly, collateral venous circulation, portocaval shunting, and gastrointestinal varices.5
Among the five human‐pathogenic species of schistosomes, S. mansoni and S. japonicum are linked to hepatocellular carcinoma (HCC).10 Clinical studies and animal experiments have suggested that schistosomiasis promotes the development of HCC but also colorectal cancer, prostate cancer, and giant follicular lymphomas.11, 12 Furthermore, schistosomiasis likely potentiates hepatic injury when coinciding with hepatitis B virus (HBV) and hepatitis C virus (HCV) infections.10 S. mansoni infection in a mouse model for HCC accelerated hepatic dysplastic changes and promoted earlier onset of cancer formation with a more aggressive nature.13 Currently, it is discussed whether the aggravated carcinogenesis in the case of HBV‐ or HCV‐schistosome coinfection results from intensified liver lesions or an impairment of the immune system.10 Carcinogenic mechanisms have been described for blood and liver flukes, including chronic inflammation, metabolic oxidative stress induced by parasite‐derived products, and host‐tissue damage during parasite development, along with the active wound healing, immunosuppression, and a reduced immunosurveillance, which could initiate or promote hepatocellular carcinogenesis.14, 15 However, definite biomolecular risk factors linking S. mansoni infection to HCC are still unknown.
HCC is the second leading cause of cancer mortality worldwide, with 745,000 deaths annually.7 HCC predominantly develops in the context of liver cirrhosis following chronic liver damage due to exposure to liver carcinogens or infection with HBV or HCV.8 Chronic liver injury is frequently accompanied by inflammation driving the compensatory proliferation of surviving hepatocytes into cirrhosis.8 c‐Jun and STAT3 are proto‐oncogenes or transcription factors whose activation promotes the development and progression of dysplasia during HCC tumorigenesis.11, 14 In 2001, a clinical study initially suggested that the coordinated expression of c‐fos and c‐jun in HCC may reflect the coordinated tumor cell cycle of progression and proliferation.16 c‐Jun is a major regulator of hepatocyte survival17 and hepatocyte proliferation during regeneration.18 In vivo studies demonstrated the importance of c‐Jun for liver cancer initiation and survival by suppression of apoptosis.19, 20 In transgenic mice, the HCV core protein potentiates chemically induced HCC through c‐Jun and STAT3 activation, which in turn enhances cell proliferation, suppresses apoptosis, and impairs oxidative DNA damage repair. This finally leads to hepatocellular transformation.21 STAT3 is a member of the signal transducer and activator of transcription (STAT) family, which is inactive in nonstimulated cells, but is rapidly activated by various stimuli like cytokines and growth factors.22 The feedback‐controlled STAT3 activity ensures transient STAT3 activation in normal cells, whereas STAT3 is often found to be constitutively activated in liver cancer cells.23 STAT3 signaling contributes to tumor cell proliferation, invasion, and survival in HCC.24 STAT3 activation also functions as a potent immune checkpoint for multiple antitumor immune responses.25 STAT3‐positive HCCs are more aggressive,24 and STAT3 is a marker for poor prognosis in liver cancer.26 The aim of this study was to examine whether S. mansoni infection is associated with the activation of c‐Jun and STAT3, which are vitally important for the development of HCC.
Materials and Methods
Human Material
Pseudonymized human liver samples were kindly provided by Dr. Janssens and Dr. Van Eyken from Ziekenhuis Oost‐Limburg and the tissue bank of the German Center for Infection Research after approval by the ethics committee (project ID: 0064). According to the ethics vote, informed consent was not required for our retrospective analyses of archived tissues.
Animal Model
For maintaining the S. mansoni life cycle, Biomphalaria glabrata snails were used as intermediate hosts and Syrian hamsters (Mesocricetus auratus) as final hosts. The strain of S. mansoni originated from a Liberian isolate obtained from Bayer AG (Monheim, Germany). Bisex (= mixed sex) and single‐sex (= unisexual) worm populations were generated by polymiracidial and monomiracidial intermediate‐host infections, respectively.27 Hamsters of both sexes were analyzed by immunohistochemistry. Only female hamsters were analyzed for semiquantitative assays and electrophoretic mobility‐shift assay (EMSA).
All animal experiments have been done in accordance with the European Convention for the Protection of Vertebrate Animals used for experimental and other scientific purposes (ETS No 123; revised Appendix A) and have been approved by the Regional Council (Regierungspräsidium) Giessen with approval number V54‐19 c 20/15 c GI 18/10 Nr. A1/2014.
Soluable Egg Antigens Isolation and Egg‐Conditioned Medium Production
S. mansoni eggs were prepared from livers of bisex infected hamsters at day 46 post infection, and soluble egg antigens (SEA) was isolated from such eggs as described earlier with minor modifications.27 SEA protein concentration was determined colorimetrically employing the Advanced Protein Assay (Cytoskeleton, Inc., Denver, CO) according to the manufacturer's instructions.
Egg‐conditioned medium (ECM) was prepared by incubation of liver eggs in nonsupplemented M199 medium (Gibco, Thermo Fisher Scientific, Darmstadt, Germany) in a humidified incubator with 5% CO2 at 37°C for 6 days. Medium was collected and cleared by centrifugation at 12,000g. The supernatant was aliquoted, frozen in liquid nitrogen, and stored at –80°C for further use.
Purification of IPSE/ALPHA‐1
Natural IPSE (nIPSE)/alpha‐1 was purified from SEA by a two‐step chromatography procedure. The first step was a cation exchange chromatography described in detail by Schramm et al.,7 followed by an affinity chromatography via binding to a monoclonal anti‐IPSE/alpha‐1 antibody coupled to N‐hydroxysuccinimide‐activated sepharose. Bound IPSE/alpha‐1 was eluted from the column by 0.1 M glycin/HCl, pH 2.8, and immediately neutralized to pH 7.5 with 1 M Tris solution. Fractions were controlled by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), silver staining, and western blot for content and purity of IPSE/alpha‐1, dialyzed against phosphate‐buffered saline, pH 7.5, concentrated to a final suitable concentration, and stored in aliquots at −80°C.
Cell Culture Experiments
Huh7 cells at 80% confluency were stimulated in vitro with SEA, ECM, or IPSE as indicated in the figures or legends. Cells from one well of a 24‐well plate were lysed in 60 µL Laemmli‐buffer for western blot analysis.
Human primary hepatocytes were plated and cultured according to the manufacturer’s protocols (Cellartis Enhanced hiPS‐HEP v2 Kits; Takara, Göteborg, Sweden). Following the initial plating and culture protocol, the medium was changed to William´s Medium E, and the cells were treated with the indicated doses of SEA and IPSE for the recommended time.
Phospho‐Kinase Proteome Profiler Array
Lysates were prepared according to the manufacturer´s protocol from primary human hepatocytes cultured to ~80% confluency in one well of a 6‐well plate. The proteome profiler arrays were carried out as indicated in the manual (R&D Biosystems, Abington, United Kingdom).
Immunohistochemistry/Immunofluorescence
Detection of c‐Jun (Cell Signaling Technology, Leiden, the Netherlands), Ki67 (Abcam, Cambridge, United Kingdom), Minichromosome Maintenance Complex Component 2 (MCM2; CST), proliferating cell nuclear antigen (PCNA; CST), Ubiquityl‐PCNA (Lys164) (CST), γ‐H2AX (CST), and hepatocyte nuclear factor 4 alpha (HNF4α; CST) was performed as described.28, 29 Appropriately concentrated antibodies were used for isotype control in order to avoid unspecific staining.
Western Blot Analysis
Protein samples were prepared from total liver lysates and boiled in Laemmli‐buffer for 5 minutes, chilled on ice, and subjected to 10% SDS‐PAGE and transferred to polyvinylidene difluoride membranes. Visualization of proteins was performed by horseradish peroxidase (HRP)–linked antibodies and the ECL Chemiluminescence Detection Kit (SuperSignal West Pico Chemiluminescent Substrate; ThermoFisher Scientific) according to the manufacturer’s protocols. Semiquantitative analysis of obtained signals was done with the BioDoc Analyze 2.1 imaging system (Biometra, Göttingen, Germany).
Preparation of Nuclear Protein Extracts
Nuclear lysates were prepared from the hamster livers as per the instruction given in NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Dreieich, Germany). The nuclear extracts were stored at ‐80°C until further use.
Electrophoretic Mobility‐Shift Assay
The double‐stranded activator protein 1 (AP‐1) or STAT3 consensus oligonucleotides were labeled with biotin (Seqlab, Goettingen, Germany). Nuclear extracts (3 µL) were mixed with 17 µL of binding buffer (10 mM Tris‐HCl [pH 7.5], 50 mM NaCl, 1 mM EDTA, 1 mM DDT, 5% glycerol) containing 0.5 ng of end‐labeled probe. For competition studies, the binding reactions were performed in the presence of 100‐fold excess of unlabeled AP‐1 or STAT3 consensus oligonucleotides. After incubation for 20 minutes at room temperature, the complexes were separated on a native 5% polyacrylamide gel and transferred to a nylon membrane (Carl Roth, Karlsruhe, Germany). The DNA was crosslinked for 15 minutes by ultraviolet (UV) light to the membrane in a transilluminator, and visualization of proteins was performed by HRP‐linked streptavidin and the ECL Chemiluminescence Detection Kit according to the manufacturer’s protocol.
Statistical Analysis
Statistical analysis was performed with SPSS V. 22.0 software (SPSS Inc., IBM Corp., Armonk, NY). One‐way analysis of variance test was applied in order to define differences between expression and phosphorylation levels in western blotting. Significant differences are pointed out (*P < 0.05).
Results
S. mansoni Eggs Lodge in the Liver of Infected Syrian Hamsters and Induce Granuloma Formation
S. mansoni eggs enter the liver through the portal vein and get trapped in sinusoids around portal tracts (Fig. 1A). Sinusoidal‐trapped eggs recruited inflammatory and immune cells, leading to the formation of periovular granulomas (Fig. 1B,C). Thus, the hamster model for S. mansoni infection used in the current study demonstrated typical clinical characteristics comparable to liver damage in infected humans.
Figure 1.
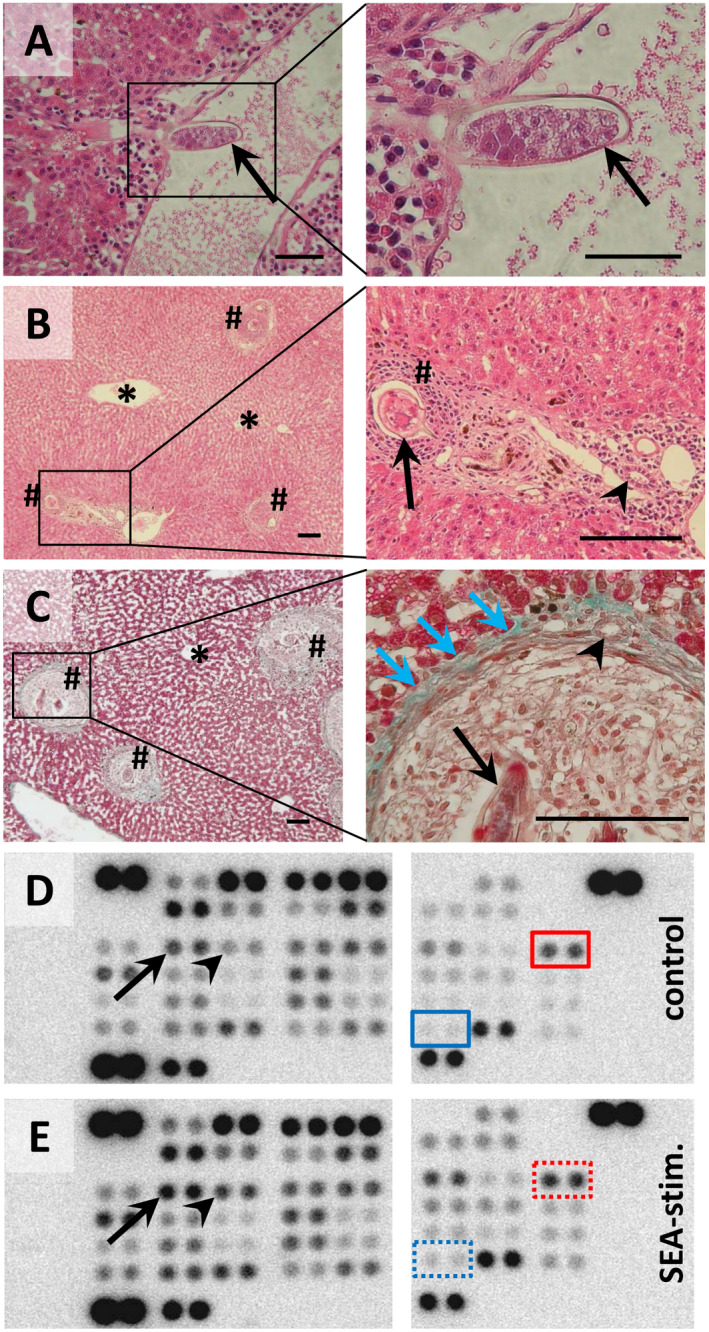
Liver‐trapped eggs of S. mansoni during lodging and granuloma formation in Syrian hamsters. (A) S. mansoni egg (arrow) entering the liver parenchyma through a branch of the portal vein. (B,C) Eggs were trapped in the liver around the portal tracts. At 6 weeks post infection, the eggs were surrounded by a dense population of immune cells, followed by a band of fibrovascular tissue (blue arrows), leading to the formation of a mature granuloma (hash). The parenchyma around the central vein (star) was free from granuloma. Formalin‐fixed, paraffin‐embedded liver sections stained with hematoxylin and eosin (A,B) or Masson Goldner staining kit (C). Arrowheads indicate bile ducts. Magnifications are ×200 (A left panel), ×400 (A,C right panels), ×50 (B,C left panels), and ×100 (B right panel). Space bars are 50 µm (A) and 100 µm (B,C). (D,E) Phospho‐kinase Proteome Profiler arrays of control‐ (D) and SEA‐stimulated (E) human hepatocytes (hiPS‐Hep). The Human Phospho‐Kinase Array is a rapid, sensitive, and economical tool to simultaneously detect the relative levels of phosphorylation of 43 kinase phosphorylation sites. The Human Phospho‐Kinase Array is divided into two parts (left and right panels of each figure) and each kinase is analyzed in duplicate. The red boxes indicate activated c‐Jun, and the blue boxes indicate activated STAT3 (dotted boxes: enhanced activation by SEA stimulation). Please note that other factors that have been described in the context of hepatic carcinogenesis were activated by SEA stimulation, e.g., CREB (arrow) or HSP27 (arrowhead).
SEA‐Activated c‐Jun and STAT3 in Primary Human Hepatocytes
Currently, there are only a few details available about the role of hepatocytes during S. mansoni egg–induced liver injury. Therefore, we investigated whether S. mansoni‐egg antigens directly interact with hepatocytes to stimulate the activation of distinct intracellular signaling pathways. To this end, a phospho‐kinase array approach was performed, demonstrating that c‐Jun and STAT3, two nuclear factors that are known to be essential for hepatocellular carcinogenesis, were activated by stimulation of primary human hepatocytes with SEA (Fig. 1D,E).
c‐Jun is Activated in Hepatocytes Adjacent to S. mansoni Eggs
Immunohistochemistry demonstrated nuclear translocation of c‐Jun (red arrows) in hepatocytes around granuloma (hash, border: dashed line) in the livers of S. mansoni‐infected hamsters (Fig. 2A,B).
Figure 2.
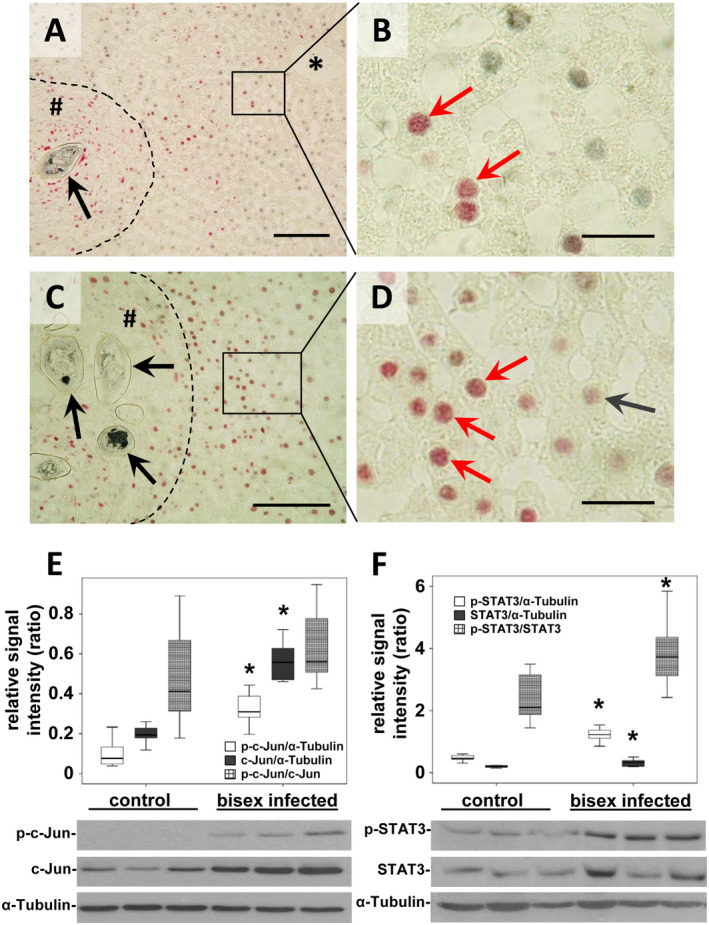
Activation of c‐Jun in hepatocytes around S. mansoni egg–induced granuloma. (A‐D) Coimmunostaining (red arrows) for c‐Jun (red) and HNF4α (gray) depicted the nuclear translocation of c‐Jun in hepatocytes joining granuloma and central veins (*). Note the directional hepatocellular activation pattern of c‐Jun from granuloma to central vein (A). Enhanced nuclear translocation of c‐Jun (red arrows) next to granuloma around egg‐conglomerates (C,D) *central vein, # granuloma, dashed line granuloma border, black arrow S. mansoni egg. Magnification ×200 (A), ×1,000 (B,D), ×400 (C), bars 100 µm (A,C), 20 µm (B,D). (E) Semiquantitative western blot analysis and subsequent assessment of optical density revealed enhanced expression and activation of c‐Jun (p‐Ser73) in S. mansoni (bisex infected, ♀+♂)–infected hamsters in comparison with single‐sex infected hamsters (no egg production) that served as controls. Note that the ratio of p‐c‐Jun/c‐Jun increased only by trend. (F) Western blot analysis demonstrated induced expression and activation of STAT3 (p‐Ser705) in S. mansoni (♀+♂)–infected hamsters in comparison with controls. n = 9/group for statistical analysis; representative blots are depicted, *P < 0.05.
Nuclear translocation of c‐Jun appeared more prominent in hepatocytes adjoining granuloma and in directional pattern between granuloma and central veins (Fig. 2A). The costaining of c‐Jun in combination with HNF4α identified hepatocytes as the major hepatic cell population showing c‐Jun activation (Fig. 2B). Moreover, a potentiated nuclear translocation of c‐Jun was observed in hepatocytes around granuloma bearing multiple eggs (Fig. 2C,D). Furthermore, the activation of c‐Jun was also demonstrated in hepatocytes around initially settled eggs without surrounding immune cells (Supporting Fig. S1). The globally enhanced expression and phosphorylation of c‐Jun and STAT3 was demonstrated by semiquantitative western blot analysis (Fig. 2E,F). Expression and phosphorylation patterns of c‐Jun and STAT3 were normalized to tubulin. The ratio of the phosphorylated forms in relation to the nonphosphorylated forms is depicted in Fig. 2E,F, and statistical data are summarized in Table 1.
Table 1.
Relative Expression and Phosphorylation of Nuclear Factors and Proliferation Markers
| Control | Bisex infected | P Value | |
|---|---|---|---|
| p‐c‐Jun/α‐tubulin | 0.10 (0.05‐0.15) | 0.40 (0.21‐0.58) | 0.002 |
| c‐Jun/α‐tubulin | 0.20 (0.16‐0.23) | 0.56 (0.49‐0.64) | <0.001 |
| p‐c‐Jun/ c‐Jun | 0.49 (0.30‐0.68) | 0.69 (0.45‐0.93) | 0.153 |
| p‐STAT3/α‐tubulin | 0.48 (0.38‐0.59) | 1.21 (1.05‐1.37) | <0.001 |
| STAT3/α‐tubulin | 0.21 (0.16‐0.26) | 0.33 (0.24‐0.41) | 0.015 |
| p‐STAT3/STAT3 | 2.40 (1.81‐2.98) | 4.04 (2.93‐5.15) | 0.008 |
| MCM2/α‐tubulin | 0.24 (0.11‐0.38) | 1.23 (0.89‐1.58) | <0.001 |
| PCNA/α‐tubulin | 0.37 (0.22‐0.51) | 0.96 (0.64‐1.27) | 0.001 |
Mean (95% confidence interval) of the indicated ratios of densitometrically assessed signals of western blots is depicted (n = 9 per group).
Abbreviations: MCM2, Minichromosome Maintenance Complex Component 2; PCNA, proliferating cell nuclear antigen.
EMSAs were performed to verify that the infection with S. mansoni exerted stimulatory effects on the binding of nuclear extracts to an AP‐1 consensus oligonucleotide that binds the c‐Jun homodimer and the Jun/Fos heterodimer.30 S. mansoni infection increased the binding of nuclear extracts to AP‐1 consensus oligonucleotides when compared with single‐sex infected controls (Fig. 3A). The specificity of DNA–protein complex formation was confirmed by competition experiments with 100‐fold molar excess of unlabeled AP‐1 oligonucleotide (Fig. 3A). After demonstrating that STAT3 is significantly activated in S. mansoni‐infected hamsters (Fig. 2F), we confirmed the DNA‐binding ability of nuclear pSTAT3 in EMSA using nuclear pSTAT3 and labelled STAT3 consensus oligonucleotide (Fig. 3A). To demonstrate the specificity of the binding activity, an unlabeled STAT3 binding‐site DNA probe was added to the reaction in 100‐times molar excess.
Figure 3.
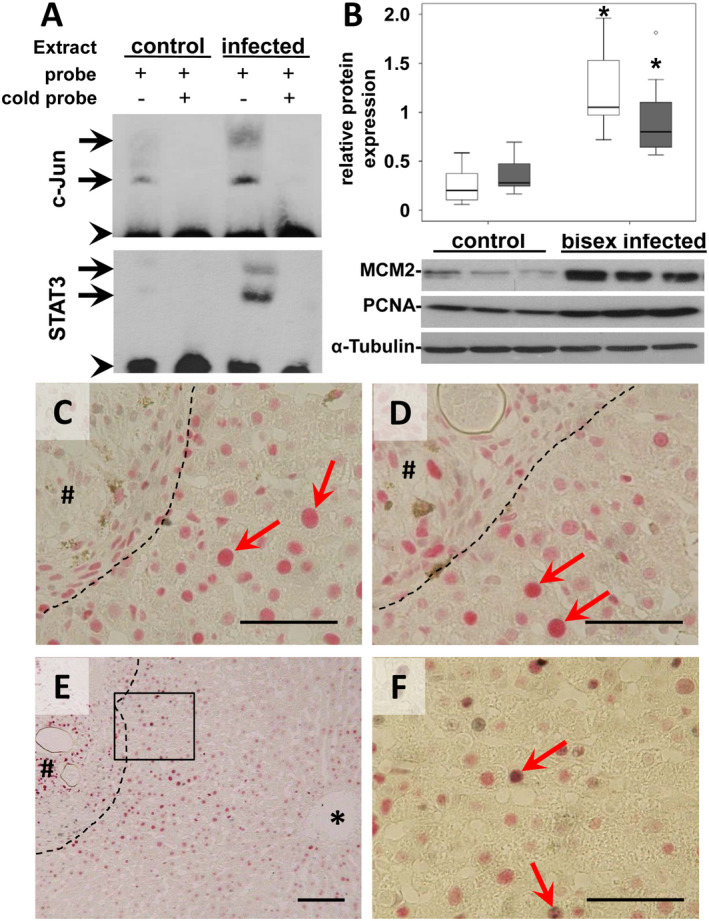
AP‐1/DNA binding and induction of proliferation markers. (A) The upper EMSA assay demonstrated the enhanced AP1/DNA binding in two different complexes (arrows) using liver extracts of S. mansoni–infected hamsters in comparison with single‐sex infected controls. The preparation of nuclear extracts and the binding to the labelled consensus oligonucleotide were performed in the absence of a competitor or in the presence of 100‐fold excess of the unlabeled AP‐1 consensus oligonucleotide (cold probe). Binding of STAT3 to biotin‐labeled DNA probes is shown in the lower EMSA blot. To compete with the binding, an unlabeled STAT3 binding‐site DNA probe was added to the reaction in 100‐times molar excess. The STAT3‐DNA complexes are marked by arrows. Arrowhead indicates free probe. (B) Western blot analysis and subsequent assessment of optical density of the signals depicted enhanced expression of the MCM2 and the PCNA in S. mansoni–infected hamsters. MCM2 (white bars) PCNA (gray bars), n = 9/group for statistical analysis; representative blots are depicted, *P < 0.05. (C,D) Immunohistological nuclear staining (red arrows) of MCM2 (C, red) and PCNA (D, red) was shown in hepatocytes closely located around granuloma. Magnification ×400, bars 50 µm. (E,F) Immunohistological costaining of c‐Jun (red) and Ki67 (gray) demonstrated the activation of the cell cycle in hepatocytes with prominently activated c‐Jun (red arrows). Magnification ×100 (C), ×400 (D), bars 100 µm (C), 50 µm (D). *central vein, # granuloma, dashed line granuloma border.
The AP‐1 transcription factor c‐Jun and STAT3 are well‐characterized key regulators of hepatocyte proliferation.18 Therefore, we next analyzed cellular proliferation factors MCM2, PCNA, and Ki67 that also have been described as prognostic molecular markers in the context of HCC.31 The western blot analysis revealed significantly enhanced hepatic expression of MCM2 and PCNA in S. mansoni‐infected hamsters (Fig. 3B; Table 1). The relative expression of MCM2 and PCNA was normalized to tubulin loading controls. In addition, enhanced nuclear staining of MCM2 and PCNA suggested initiation of DNA replication in hepatocytes around granuloma (Fig. 3C,D). Furthermore, proliferation marker Ki67 was shown to colocalize with c‐Jun in the same nuclei of hepatocytes around granuloma induced by S. mansoni eggs (Fig. 3E,F). Similar to the c‐Jun staining pattern, Ki67 was also stained in the nuclei of hepatocytes in directional pattern between granuloma and central veins (Fig. 3E). In addition, costaining of the DNA replication marker PCNA and the cell cycle inhibitor p27 demonstrated their colocalization in the same nuclei of hepatocytes around granuloma (Supporting Fig. S2).
SEA, ECM, and IPSE Activate Proto‐Oncogene c‐Jun and STAT3 in Primary Human Hepatocytes and Huh7 Cells
The c‐Jun staining pattern raised the hypothesis that the observed activation of c‐Jun is caused by substances released from the eggs and transported with the blood flow from portal tracts toward central veins (Figs. 2A and 3E). In order to demonstrate egg‐dependent activation of c‐Jun, primary human hepatocytes (hiPS‐Hep) and the human HCC cell line Huh7 were stimulated with SEA in vitro. The time‐ and concentration‐dependent activation of c‐Jun was demonstrated by western blot and immunofluorescence (Fig. 4A,B). A maximum of c‐Jun activation was observed 4 hours after addition of SEA to the culture medium, and c‐Jun expression and phosphorylation was still more prominent after 24 hours in comparison with cells that were treated with control medium (Fig. 4A). The activation of c‐Jun in Huh7 cells by SEA was confirmed by enhanced expression and phosphorylation as well as nuclear translocation (Fig. 4A,B; Supporting Fig. S3). Also, S. mansoni ECM stimulated c‐Jun and STAT3 activation in Huh7 cells (Fig. 4C), strongly indicating that products that are secreted from the eggs are responsible for the activation of c‐Jun. The SEA‐induced expression and activation of c‐Jun was inhibited by a potent, cell‐permeable, and selective inhibitor of c‐Jun N‐terminal kinase (JNK) (Fig. 4D).
Figure 4.
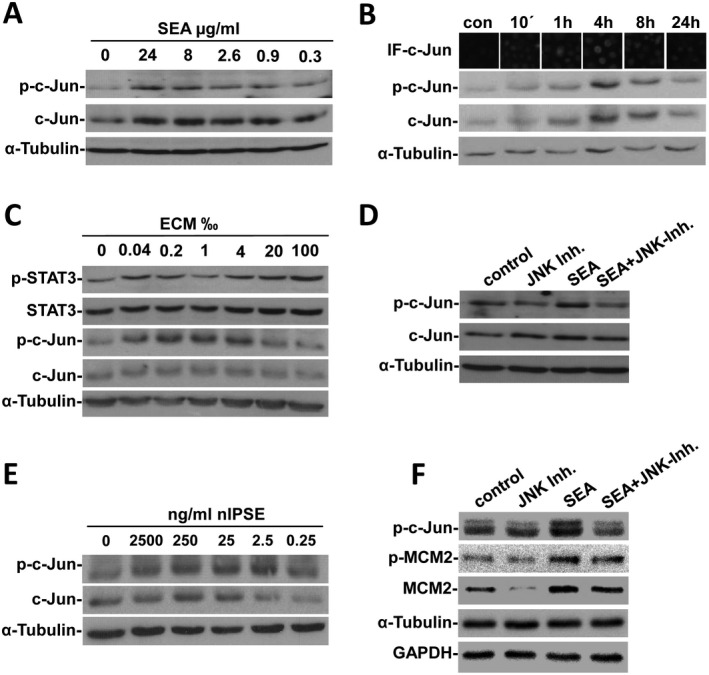
SEA, ECM, and IPSE can activate c‐Jun and STAT3 as well as markers for DNA replication and cell cycle inhibition in vitro. (A) Concentration‐dependent expression and activation of c‐Jun by SEA was shown by western blot. (B) IF and western blot analysis of c‐Jun and p‐c‐Jun demonstrated activation of c‐Jun by SEA in Huh7 cells with maximal stimulation after 4 hours. (C) Stimulation with ECM induced a concentration‐dependent expression and phosphorylation of c‐Jun and STAT3 in Huh7 cells. (D) The SEA‐activated expression and phosphorylation of c‐Jun was inhibited by a specific inhibitor for JNK in Huh7 cells. (E) Western blot analysis for c‐Jun as well as for their phosphorylated forms showed a concentration‐dependent activation of both factors nIPSE in Huh7 cells. (F) The SEA‐induced activation of c‐Jun, MCM2, and cleavage of caspase 3 were prevented by inhibition of JNK in primary hiPS‐Hep. For control and for zero‐concentration in titration experiments, appropriate buffers of SEA, ECM, or IPSE samples were applied. The in vitro activation of c‐Jun and STAT3 fluctuated with different charges of SEA and ECM but also with different passages of Huh7 exhibiting different basal activation of the transcription factors. Nevertheless, in vitro assays were repeated at least 2‐fold, and representative blots and stainings with clear activation patterns were depicted. Abbreviations: con, control; IF, immunofluorescence.
In order to demonstrate the activation of c‐Jun in Huh7 cells by purified nIPSE/alpha‐1, we stimulated freshly seeded cells in vitro, employing titration of IPSE/alpha‐1. Interestingly, expression was maximal by stimulation with 250 ng/mL nIPSE/alpha‐1, whereas phosphorylation of c‐Jun was highest at lower concentrations of 2.5 ng/mL nIPSE/alpha‐1 (Fig. 4E). These results were also reproduced with recombinant IPSE/alpha‐1 produced from human embryonic kidney cells (data not shown). Similar assays for the stimulation of JNK, c‐Jun, and MCM2 by soluble factors of S. mansoni eggs were also performed in primary hepatocytes (representative western blots are depicted in Supporting Fig. S4). The SEA‐stimulated activation of c‐Jun and MCM2 was abrogated by inhibition of JNK, the proximate upstream activator of c‐Jun (Fig. 4F).
DNA Double‐Strand Breaks Occurred in c‐Jun+ Hepatocytes
Genomic instability, DNA damage, and the induction of DNA repair pathways are common hallmarks of most human cancers.32 It has been demonstrated before that the activation of the JNK pathway might contribute to DNA damage in liver cells including increased phosphorylation of related proteins such as H2AX,33 indicating DNA double‐strand breaks.34 We demonstrate the S. mansoni egg–regulated activation of hepatocellular cancer–associated factors c‐Jun and STAT3 as well as proliferation markers in the hamster model. Furthermore, DNA replication marker PCNA and the cell cycle inhibitor p27 appear in the same nuclei of hepatocytes around granuloma. Simultaneous DNA replication and cell cycle inhibition clearly indicated replication stress, which can cause DNA damage. These results suggested that the hepatocytes in our model might be damaged by replication stress.35 Therefore, we aimed to analyze markers for DNA damage, which is a hallmark of replication stress.35 The phosphorylation of H2AX is the first step in DNA repair, and nuclear foci of phosphorylated H2AX represent DNA double‐strand breaks in a 1:1 manner.34 We visualized the phosphorylation pattern of H2AX (γ‐H2AX‐S139) by immunohistochemical staining in livers of hamsters infected with S. mansoni. Immunohistological costaining of c‐Jun (gray) in combination with γ‐H2AX (red) demonstrated the phosphorylation of H2AX in hepatocyte nuclei with prominently activated c‐Jun (Fig. 5A,B, red arrows). Typical staining patterns of nuclear foci of γ‐H2AX (red) were visible in γ‐H2AX‐stained liver slices without any nuclear costaining (Fig. 5C,D, red arrows). As γ‐H2AX has been shown to be involved in apoptosis,36 terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining was performed. In contrast to the multitude of γ‐H2AX‐positive hepatocyte nuclei around granuloma, apoptotic hepatocytes were rarely found in TUNEL‐stained livers of S. mansoni–infected hamsters (Fig. 5E, green arrows). The typical γ‐H2AX ring showing early apoptosis37 was not observed in any of the γ‐H2AX‐positive hepatocytes. As we could show that PCNA is upregulated in S. mansoni–infected hamsters, we aimed to analyze whether PCNA was ubiquitinated. Indeed, PCNA was ubiquitinated in bisex infected hamsters, indicating translesion DNA synthesis.38
Figure 5.
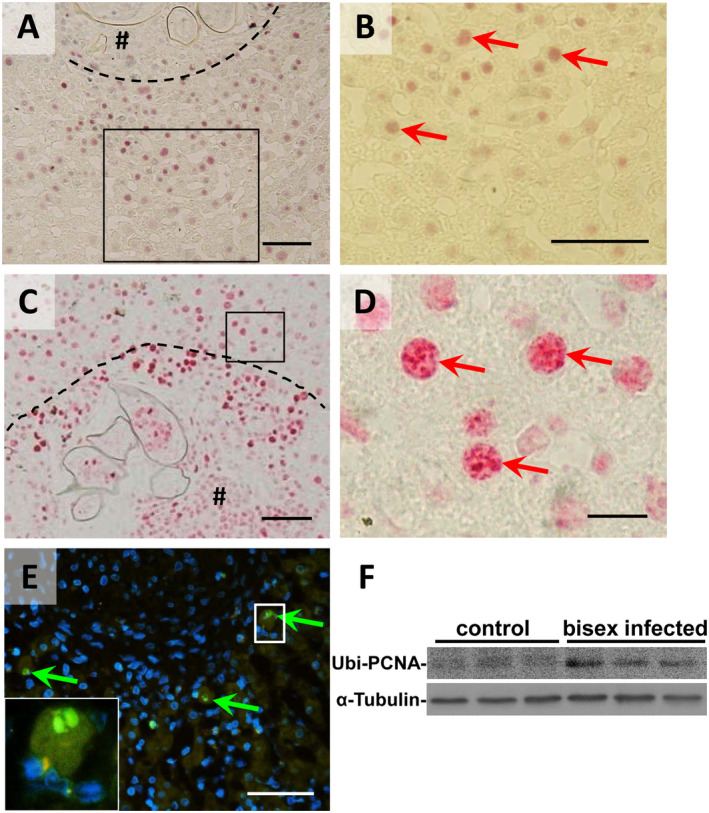
DNA double‐strand breaks in c‐Jun+ hepatocytes. (A,B) Immunohistochemical costaining of c‐Jun (gray) in combination with γ‐H2AX (red) demonstrated the phosphorylation of H2AX in hepatocyte nuclei with prominently activated c‐Jun (red arrows). (C,D) Immunohistochemical staining of γ‐H2AX (red) without any nuclear costaining visualized nuclear foci containing thousands of molecules at DNA damage sites (red arrows). (E) Apoptotic hepatocytes were rarely found in TUNEL‐stained livers of S. mansoni–infected hamsters (green arrows). Magnification ×100 (A,C), ×200 (E), ×400 (B), and ×1,000 (D). Bars 50 µm (A‐C,E) and 10 µm (D). # granuloma, dashed line granuloma border. (F) Western blot analysis of Ubiquityl‐PCNA (Lys164).
Hepatocellular Activation of c‐Jun and H2AX in Patients with Schistosomiasis
In order to obtain a model‐independent proof of the most important results from hamster and cell cultures, c‐Jun, Ki67, and γ‐H2AX were stained on a liver slice of a male Egyptian patient aged 38 years and diagnosed with schistosomiasis. No other infections were diagnosed in the patient. Pathological assessment of a puncture biopsy demonstrated a residual state of the disease with partly dystrophic and calcified eggs (Fig. 6A,B). Interestingly, nuclei of hepatocytes in direct vicinity of granuloma were positively stained for c‐Jun (Fig. 6C,D), γ‐H2AX (Fig. 6E,F), and Ki67 (Supporting Fig. S5) even in this late stage of the disease and long time after primary infection and hepatic lodging of the eggs. The same findings were validated in a recently published case of a 26‐year‐old patient from Eritrea who was diagnosed with hepatosplenic schistosomiasis by liver biopsy39 (representative c‐Jun immunostainings are depicted in Supporting Fig. S6).
Figure 6.
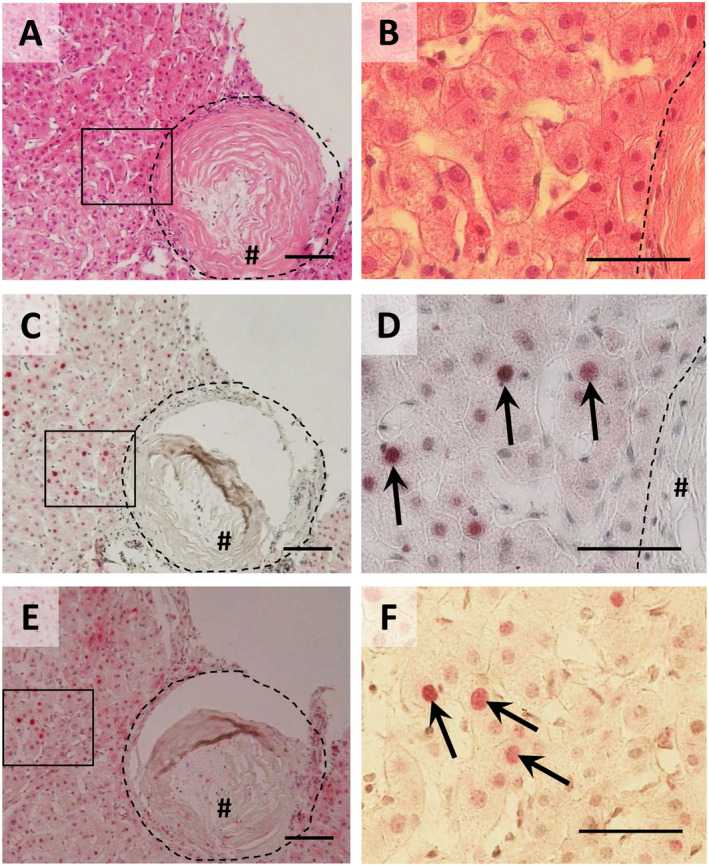
Hepatocellular activation of c‐Jun and H2AX in human schistosomiasis (A,B) hematoxylin and eosin staining demonstrated a residual state of the disease with partly dystrophic and calcified eggs. (C,D) Immunohistochemical staining of c‐Jun (red) in combination with hematoxylin nuclear staining points out nuclear translocation of c‐Jun (arrows) in hepatocytes around the granuloma (hash). (E,F) Immunohistochemical staining of γ‐H2AX (red) demonstrated DNA double strand breaks (arrows) in hepatocytes around granuloma (hash). Magnification ×100 (A,C,E) and ×400 (B,D,F). Bars 100 µm (A,C,E) and 50 µm (B,D,F). # granuloma, dashed line granuloma border.
Discussion
A large epidemiologic study of schistosomiasis including 89,180 individuals showed the prevalence of mainly two Schistosoma species in Egypt, S. mansoni and S. haematobium. S. haematobium, the predominant etiologic agent for urinary schistosomiasis, is carcinogenic to humans, inducing squamous cell carcinoma of the urinary bladder and bladder cancer.40 S. haematobium–associated bladder cancer has been associated with mutations in hras 41 and kras 42 genes and dysregulated expression of Bcl‐2 and tumor protein p53.43 Case reports have described associations of S. mansoni with prostatic adenocarcinoma, colorectal cancer, and bladder cancer.44, 45, 46 Experiments in mice demonstrated that infection with S. mansoni can accelerate hepatic dysplastic changes in the presence of other risk factors, inducing earlier cancer appearance with a more aggressive nature.13 Clinical studies demonstrated that the coinfection of HBV and HCV in combination with S. mansoni aggravate the clinical course of hepatitis but also of hepatocellular carcinogenesis.10, 47, 48 Schistosomiasis‐induced Th2 polarization has been discussed as one mechanism in promoting viral persistence; it might be responsible for increased viral load in the case of coinfection and thus might cause a higher risk of HCC.48 The altered expression of p53 in patients with S. mansoni colitis‐related colorectal cancer suggested that schistosome infections may induce carcinogenesis by targeting oncogenes.49 Furthermore, it was recently supposed that cancer induction by S. mansoni infection could result from somatic mutations in oncogenes and in the regulation of immune responses leading to activation of several host signaling cancer pathways.15 Nevertheless, the permanent activation of c‐Jun and STAT3 as critical regulators in liver cancerogenesis and progression in human HCC has not yet been described in the context of S. mansoni infection.11, 14
In the current study, we demonstrated by phospho‐kinase‐proteome profiler array that the proto‐oncogene c‐Jun is among the molecules activated in primary human hepatocytes stimulated with SEA. Furthermore, we have shown that c‐Jun is activated in hepatocytes around S. mansoni eggs and that they are permanently activated in hepatocytes around mature granuloma in infected hamsters. Moreover, perpetual activation of c‐Jun was even demonstrated in the active and residual state of the disease with partly dystrophic and calcified eggs in biopsies of patients with schistosomiasis. The activation of c‐Jun and STAT3, the directional activation pattern of c‐Jun, the correlation between quantity of eggs per granuloma and surrounding c‐Jun activation, and the nuclear c‐Jun translocation next to freshly settled eggs not surrounded by granuloma supported the hypothesis that c‐Jun activation can be caused by components released from the eggs. Additionally, the activation of c‐Jun and STAT3 was shown in vitro after stimulation with ECM and IPSE derived from S. mansoni eggs. Taken together, these results evidently demonstrated that stimulating factors are secreted by the eggs and, furthermore, that IPSE is at least one of the egg‐secreted components inducing c‐Jun activation in hepatocytes. The clinical relevance of these results was proven in human biopsies of patients with schistosomiasis. A similar effect with regard to the activation of proto‐oncogenes in our model has recently been described for myeloid‐derived suppressor cells.50 The authors suggested that a S. japonicum infection could promote the expansion of myeloid‐derived suppressor cells by SEA‐dependent activation of the JAK/STAT3 pathway.50 Thus, egg‐secreted antigen‐dependent perpetual activation of the JNK and STAT3 pathway described in the current study might be a general molecular mechanism associated with carcinogenesis in Schistosoma, or generally in parasite infections that are associated with oncogene activation.15 Indeed, this hypothesis is strengthened by the fact that we also observed hepatocellular c‐Jun activation around granuloma in a patient infected with S. haematobium (data not shown). In our study, we have also provided evidence that the HCC‐promoting downstream processes of the JNK and STAT3 signal cascades such as proliferation and replicative stress with DNA double‐strand breaks are induced in the hamster model as well as in the human biopsy sample. Furthermore, ubiquitylated PCNA indicated active translesion DNA synthesis, which is likely associated with DNA‐replication stress.38 These findings open a new route for subsequent studies analyzing the schistosome egg–induced transformation of hepatocytes.
In conclusion, the HCC‐associated transcription factors c‐Jun and STAT3 are permanently induced in hepatocytes of acute and chronic S. mansoni egg–infiltrated liver. Also, downstream processes such as proliferation and DNA double‐strand breaks are perpetually activated in c‐Jun‐positive hepatocytes around liver‐trapped eggs. Thus, our data suggest an aspect of how intrahepatically trapped S. mansoni eggs could directly influence hepatocarcinogenesis.
Supporting information
Acknowledgment
This research was supported by GILEAD Förderprogramm Infektiologie. We are grateful to the DZIF biobank and Dr. Janssens and Dr. Van Eyken from Ziekenhuis Oost‐Limburg, as well as to Dr. D. Tappe from BNITM for providing human samples. We thank Heike Müller and Dagmar Leder, as well as Christina Scheld, Georgette Stovall, and Bianca Kulik for excellent technical assistance.
See Editorial on Page 376
Supported by grants from GILEAD (support program Infectiology 2017), the Behring‐Röntgen Stiftung (#60‐0002), the Deutsche Forschungsgemeinschaft (RO957/10), and the University Hospital Giessen and Marburg (UKGM).
Potential conflict of interest: Elke Roeb advises Gilead, Pfizer, and Falk.
References
Author names in bold designate co‐first authorship.
- 1. Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet 2014;383:2253‐2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hatz CFR. Schistosomiasis: An underestimated problem in industrialized countries? J Travel Med 2005;12:1‐2. [DOI] [PubMed] [Google Scholar]
- 3. Lingscheid T, Kurth F, Clerinx J, Marocco S, Trevino B, Schunk M, et al. Schistosomiasis in European travelers and migrants: analysis of 14 years TropNet surveillance data. Am J Trop Med Hyg 2017;97:567‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boissier J, Grech‐Angelini S, Webster BL, Allienne JF, Huyse T, Mas‐Coma S, et al. Outbreak of urogenital schistosomiasis in Corsica (France): An epidemiological case study. Lancet Infect Dis 2016;16:971‐979. [DOI] [PubMed] [Google Scholar]
- 5. Olveda DU, Olveda RM, McManus DP, Cai P, Chau TN, Lam AK, et al. The chronic enteropathogenic disease schistosomiasis. Int J Infect Dis 2014;28:193‐203. [DOI] [PubMed] [Google Scholar]
- 6. Abdulla MH, Lim KC, McKerrow JH, Caffrey CR. Proteomic identification of IPSE/alpha‐1 as a major hepatotoxin secreted by Schistosoma mansoni eggs. PLoS Negl Trop Dis 2011;5:e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schramm G, Falcone FH, Gronow A, Haisch K, Mamat U, Doenhoff MJ, et al. Molecular characterization of an interleukin‐4‐inducing factor from Schistosoma mansoni eggs. J Biol Chem 2003;278:18384‐18392. [DOI] [PubMed] [Google Scholar]
- 8. Pennington LF, Alouffi A, Mbanefo EC, Ray D, Heery DM, Jardetzky TS, et al. H‐IPSE is a pathogen‐secreted host nucleus infiltrating protein (infiltrin) expressed exclusively by the Schistosoma haematobium egg stage. Infect Immun 2017. Sep 18. 10.1128/IAI.00301-17. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chuah C, Jones MK, Burke ML, McManus DP, Gobert GN. Cellular and chemokine‐mediated regulation in schistosome‐induced hepatic pathology. Trends Parasitol 2014;30:141‐150. [DOI] [PubMed] [Google Scholar]
- 10. Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology 2004;127:5‐16. [DOI] [PubMed] [Google Scholar]
- 11. El‐Tonsy MM, Hussein HM, Helal T, Tawfik RA, Koriem KM, Hussein HM. Schistosoma mansoni infection: is it a risk factor for development of hepatocellular carcinoma? Acta Trop 2013;128:542‐547. [DOI] [PubMed] [Google Scholar]
- 12. Palumbo E. Association between schistosomiasis and cancer. Infect Dis Clin Pract 2007;15:145‐148. [Google Scholar]
- 13. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557‐2576. [DOI] [PubMed] [Google Scholar]
- 14. Toda KS, Kikuchi L, Chagas AL, Tanigawa RY, Paranagua‐Vezozzo DC, Pfiffer T, et al. Hepatocellular carcinoma related to Schistosoma mansoni infection: case series and literature review. J Clin Transl Hepatol 2015;3:260‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Tong H, Brindley PJ, Meyer CG, Velavan TP. Parasite infection, carcinogenesis and human malignancy. EBioMedicine 2017;15:12‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuen MF, Wu PC, Lai VC, Lau JY, Lai CL. Expression of c‐Myc, c‐Fos, and c‐jun in hepatocellular carcinoma. Cancer. 2001;91:106‐112. [DOI] [PubMed] [Google Scholar]
- 17. Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE, et al. The transcription factor c‐Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 2012;55:408‐418. [DOI] [PubMed] [Google Scholar]
- 18. Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, et al. c‐Jun/AP‐1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev 2006;20:2306‐2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eferl R, Ricci R, Kenner L, Zenz R, David J‐P, Rath M, et al. Liver tumor development. c‐Jun antagonizes the proapoptotic activity of p53. Cell 2003;112:181‐192. [DOI] [PubMed] [Google Scholar]
- 20. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation is controlled by AP‐1 through SIRT6‐dependent inhibition of survivin. Nat Cell Biol 2012;14:1203‐1211. [DOI] [PubMed] [Google Scholar]
- 21. Machida K, Tsukamoto H, Liu JC, Han YP, Govindarajan S, Lai MM, et al. c‐Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide‐dependent impairment of oxidative DNA repair. Hepatology 2010;52:480‐492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL‐6 family of cytokine receptors. Oncogene 2000;19:2548‐2556. [DOI] [PubMed] [Google Scholar]
- 23. Liu T, Ma H, Shi W, Duan J, Wang Y, Zhang C, et al. Inhibition of STAT3 signaling pathway by ursolic acid suppresses growth of hepatocellular carcinoma. Int J Oncol 2017;51:555‐562. [DOI] [PubMed] [Google Scholar]
- 24. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF‐kappaB inhibits tumor promotion and progression by preventing oxidative stress‐driven STAT3 activation. Cancer Cell 2010;17:286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat Rev Cancer 2014;14:736‐746. [DOI] [PubMed] [Google Scholar]
- 26. Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006;130:1117‐1128. [DOI] [PubMed] [Google Scholar]
- 27. Grevelding CG. Genomic instability in Schistosoma mansoni. Mol Biochem Parasitol 1999;101:207‐216. [DOI] [PubMed] [Google Scholar]
- 28. Roderfeld M, Rath T, Pasupuleti S, Zimmermann M, Neumann C, Churin Y, et al. Bone marrow transplantation improves hepatic fibrosis in Abcb4‐/‐ mice via Th1 response and matrix metalloproteinase activity. Gut 2012;61:907‐916. [DOI] [PubMed] [Google Scholar]
- 29. Roderfeld M, Rath T, Voswinckel R, Dierkes C, Dietrich H, Zahner D, et al. Bone marrow transplantation demonstrates medullar origin of CD34+ fibrocytes and ameliorates hepatic fibrosis in Abcb4‐/‐ mice. Hepatology 2010;51:267‐276. [DOI] [PubMed] [Google Scholar]
- 30. Singh S, Aggarwal BB. Activation of transcription factor NF‐kappa B is suppressed by curcumin (diferuloylmethane) corrected. J Biol Chem 1995;270:24995‐25000. [DOI] [PubMed] [Google Scholar]
- 31. Qin LX, Tang ZY. The prognostic molecular markers in hepatocellular carcinoma. World J Gastroenterol 2002;8:385‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khanna KK, Jackson SP. DNA double‐strand breaks: Signaling, repair and the cancer connection. Nat Genetics. 2001;27:247‐254. [DOI] [PubMed] [Google Scholar]
- 33. Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase‐3. Mol Cell 2006;23:121‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuo LJ, Yang LX. Gamma‐H2AX ‐ a novel biomarker for DNA double‐strand breaks. Vivo 2008;22:305‐309. [PubMed] [Google Scholar]
- 35. Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine‐induced stalled replication forks and their collapse upon S‐phase checkpoint abrogation. Mol Cancer Ther 2007;6:1239‐1248. [DOI] [PubMed] [Google Scholar]
- 36. Rogakou EP, Nieves‐Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 2000;275:9390‐9395. [DOI] [PubMed] [Google Scholar]
- 37. Solier S, Sordet O, Kohn KW, Pommier Y. Death receptor‐induced activation of the Chk2‐ and histone H2AX‐associated DNA damage response pathways. Mol Cell Biol 2009;29:68‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mailand N, Gibbs‐Seymour I, Bekker‐Jensen S. Regulation of PCNA‐protein interactions for genome stability. Nat Rev Mol Cell Biol 2013;14:269‐282. [DOI] [PubMed] [Google Scholar]
- 39. Thijs L, Messiaen P, van der Hilst J, Madoe V, Melis C, van Eyken P, et al. Hepatic schistosomiasis with massive splenomegaly: A case report and literature review. Acta Gastroenterol Belg 2018;81:93‐96. [PubMed] [Google Scholar]
- 40. Yosry A. Schistosomiasis and neoplasia. Contrib Microbiol 2006;13:81‐100. [DOI] [PubMed] [Google Scholar]
- 41. Czerniak B, Deitch D, Simmons H, Etkind P, Herz F, Koss LG. Ha‐ras gene codon 12 mutation and DNA ploidy in urinary bladder carcinoma. Br J Cancer 1990;62:762‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Botelho MC, Veiga I, Oliveira PA, Lopes C, Teixeira M, da Costa JM, et al. Carcinogenic ability of Schistosoma haematobium possibly through oncogenic mutation of KRAS gene. Adv Cancer Res Treat 2013;2013:pii: 876585. [PMC free article] [PubMed] [Google Scholar]
- 43. Chaudhary KS, Lu QL, Abel PD, Khandan‐Nia N, Shoma AM, el Baz M, et al. Expression of bcl‐2 and p53 oncoproteins in schistosomiasis‐associated transitional and squamous cell carcinoma of urinary bladder. Br J Urol 1997;79:78‐84. [DOI] [PubMed] [Google Scholar]
- 44. Basílio‐de‐Oliveira CA, Aquino A, Simon EF, Eyer‐Silva WA. Concomitant prostatic schistosomiasis and adenocarcinoma: case report and review. Braz J Infect Dis 2002;6:45‐49. [DOI] [PubMed] [Google Scholar]
- 45. Salim OEH, Hamid HK, Mekki SO, Suleiman SH, Ibrahim SZ. Colorectal carcinoma associated with schistosomiasis: a possible causal relationship. World J Surg Oncol 2010;8:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiremit MC, Cakir A, Arslan F, Ormeci T, Erkurt B, Albayrak S. The bladder carcinoma secondary to Schistosoma mansoni infection: a case report with review of the literature. Int J Surg Case Rep 2015;13:76‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abruzzi A, Fried B, Alikhan SB. Coinfection of Schistosoma species with hepatitis B or hepatitis C viruses. Adv Parasitol 2016;91:111‐231. [DOI] [PubMed] [Google Scholar]
- 48. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 49. Madbouly KM, Senagore AJ, Mukerjee A, Hussien AM, Shehata MA, Navine P, et al. Colorectal cancer in a population with endemic Schistosoma mansoni: is this an at‐risk population? Int J Colorectal Dis 2007;22:175‐181. [DOI] [PubMed] [Google Scholar]
- 50. Burke ML, Jones MK, Gobert GN, Li YS, Ellis MK, McManus DP. Immunopathogenesis of human schistosomiasis. Parasite Immunol 2009;31:163‐176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials