

Abstract
Background and Aims
Progressive familial intrahepatic cholestasis (PFIC) 6 has been associated with missense but not biallelic nonsense or frameshift mutations in MYO5B, encoding the motor protein myosin Vb (myoVb). This genotype‐phenotype correlation and the mechanism through which MYO5B mutations give rise to PFIC are not understood. The aim of this study was to determine whether the loss of myoVb or expression of patient‐specific myoVb mutants can be causally related to defects in canalicular protein localization and, if so, through which mechanism.
Approach and Results
We demonstrate that the cholestasis‐associated substitution of the proline at amino acid position 600 in the myoVb protein to a leucine (P660L) caused the intracellular accumulation of bile canalicular proteins in vesicular compartments. Remarkably, the knockout of MYO5B in vitro and in vivo produced no canalicular localization defects. In contrast, the expression of myoVb mutants consisting of only the tail domain phenocopied the effects of the Myo5b‐P660L mutation. Using additional myoVb and rab11a mutants, we demonstrate that motor domain‐deficient myoVb inhibited the formation of specialized apical recycling endosomes and that its disrupting effect on the localization of canalicular proteins was dependent on its interaction with active rab11a and occurred at the trans‐Golgi Network/recycling endosome interface.
Conclusions
Our results reveal a mechanism through which MYO5B motor domain mutations can cause the mislocalization of canalicular proteins in hepatocytes which, unexpectedly, does not involve myoVb loss‐of‐function but, as we propose, a rab11a‐mediated gain‐of‐toxic function. The results explain why biallelic MYO5B mutations that affect the motor domain but not those that eliminate myoVb expression are associated with PFIC6.
Abbreviations
- ANO
anoctamin
- ATP8B1
adenosine triphosphatase phospholipid transporting 8b1
- BC
bile canaliculi
- CRISPR
clustered regularly interspaced short palindromic repeat
- EGFP
enhanced green fluorescent protein
- HEK
human embryonic kidney
- hiHep
human‐induced hepatocyte
- KO
knockout
- Mrp
multidrug resistance‐associated protein
- MVID
microvillus inclusion disease
- Myc
myelocytomatosis
- myoVb
myosin Vb
- PFIC
progressive familial intrahepatic cholestasis
- SDS
sodium dodecyl sulfate
- TGN
trans‐Golgi Network
- TPN
total parenteral nutrition
Hepatocytes are polarized epithelial cells with basolateral/sinusoidal plasma membrane domains that is orientated to the blood circulation and apical/canalicular plasma membranes that form the bile canaliculi (BC) through which bile is safely moved out of the liver. Tight junctions separate the sinusoidal and canalicular domains and prevent the mixing of bile and blood. Defects in the polarized distribution or function of cell surface proteins can cause severe liver diseases.1 Of these, progressive familial intrahepatic cholestasis (PFIC) is characterized by the inability of hepatocytes to secrete bile into the canaliculi, resulting in the buildup of bile components and liver failure. PFIC can be caused by mutations in different genes,2 including adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1); PFIC1), ATP Binding Cassette Subfamily B Member 11 (ABCB11) (PFIC2), ATP Binding Cassette Subfamily B Member 4 (ABCB4) (PFIC3), tight junction protein 2 (TJP2) (PFIC4), and nuclear receptor subfamily 1, group H, member 4 (NR1H4; PFIC5). ATP8B1, ABCB11, and ABCB4 encode canalicular membrane transporters. Mutations in these proteins affect their expression, canalicular localization, or function and consequently impair bile salt secretion (ABCB11/bile salt export pump [BSEP]) or phospholipid dynamics in the canalicular membrane (ATP8B1, multidrug resistance protein 3). NR1H4 encodes the farnesoid X receptor, a transcription factor that regulates the expression of ABCB11/BSEP. TJP2 encodes the tight junction protein zona occludens‐2, and mutations in these presumably lead to the leaking of bile out of the canaliculi.
Recently, mutations in the MYO5B gene were reported in a group of patients with PFIC who presented elevated bilirubin and bile acid levels with normal gamma‐glutamyl transpeptidase (GGT) levels and did not have mutations in any of the other PFIC genes.3, 4 Unique MYO5B mutations were associated with each affected family. MYO5B encodes the actin‐filament–based motor protein myosin Vb (myoVb). MyoVb binds selected small guanosine triphosphatase (GTPase) rab proteins, including the trans‐Golgi Network (TGN)‐associated and/or recycling endosome‐associated rab8 and rab11a, and has been implicated in apical plasma membrane protein trafficking. Mutations in MYO5B can also cause microvillus inclusion disease (MVID),5, 6, 7, 8 a congenital enteropathy characterized by intractable diarrhea and malabsorption and, at the cellular level, the mislocalization of apical brush border proteins. Notably, many—but not all—patients with MVID also develop cholestasis, leading to liver failure.9
How MYO5B mutations may lead to PFIC is not known. Given the effect of MYO5B mutations on the apical localization of brush border proteins in enterocytes in MVID, it is possible that myoVb is similarly needed for the correct localization of BC proteins in hepatocytes and can cause cholestasis when mutated. In vitro studies in the hepatic WIF‐B9 cell line, in which the ectopic expression of a rat myoVb tail fragment impaired canalicular protein trafficking,10 may support this hypothesis. In situ studies, however, showing no immunohistochemical abnormalities of canalicular transporters in liver biopsies of some patients with MYO5B mutations presenting severe cholestasis,3, 11 challenge this hypothesis. Furthermore, although missense, nonsense, and frameshift MYO5B mutations all have been associated with MVID (reviewed in van der Velde et al.7), biallelic nonsense and frameshift mutations predicted to eliminate myoVb expression are noticeably absent in patients with non‐MVID cholestasis.3, 12 Thus, not all pathogenic MYO5B mutations may lead to PFIC and/or canalicular protein localization defects.
Notably, causality between patient MYO5B mutations and the mislocalization of BC proteins in hepatocytes has not been experimentally addressed. The need to decisively determine whether a causal relationship exists between patient‐specific MYO5B mutations and canalicular protein localization defects is particularly relevant for PFIC presenting in patients with MVID. Indeed, because patients with MVID receive lifelong total parenteral nutrition (TPN), which itself may induce cholestasis and liver failure,13, 14 it is difficult to determine whether liver symptoms in these patients are MYO5B mutation or TPN induced.
The aim of this study was to address the causal relationship between patient MYO5B mutations and canalicular protein mislocalization and to clarify the PFIC disease mechanism in these patients. We demonstrate that myoVb is dispensable for the correct localization of BC proteins yet can cause cholestasis‐associated defects in their localization when mutated through an unexpected mechanism involving the small GTPase rab11a.
Materials and Methods
Cell Culture
HepG2 cells (American Type Culture Collection HB8065) were maintained in high‐glucose Dulbecco’s modified Eagle’s medium with 10% heat‐inactivated fetal bovine serum, 2 mM l‐glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere. For experiments, cells were plated on poly‐L‐lysine–coated coverslips and used 3 days later. Human embryonic stem cell line 9 (HUES9) cells were maintained on vitronectin in E8 (Thermo Fisher). Cells were passaged every 4‐5 days with 1% RevitaCell Supplement added to the cells overnight on day of passage. Differentiation of HUES9 cells to hepatocytes was as described.15
Viral Transduction
Lentiviral particles were produced using a second‐generation system based on pCMVdR8.1 and pVSV‐G. Human embryonic kidney (HEK) 293T cells in the amount of 1 × 106 were transferred to poly‐L‐lysine–coated 9 cm2 plates in 1.3 mL culture medium. Lentiviral vector in the amount of 1,200 ng, 1,000 ng pCMVdR8.1, and 400 ng pVSV‐G were mixed with 7.8 μL FuGENE HD in 200 μL Opti‐MEM and added to suspension HEK293T. After overnight incubation, medium was refreshed, and after 48 hours, viral particles were harvested and filtered (0.45 μm polyvinylidene fluoride [PVDF] membrane filter). One day after plating, cells were incubated with viral particles for 16 hours (supplemented with 8 μg/mL polybrene). Antibiotics (2.5 μg/mL puromycin, 4 μg/mL blasticidin) were added 24 hours after viral incubation.
Clustered Regularly Interspaced Short Palindromic Repeat Knockout
A lentiviral clustered regularly interspaced short palindromic repeat (CRISPR) construct targeting Exon3 of MYO5B was generated using the plentiCRISPR‐V2 vector (Addgene #52961) following provided protocols (guide RNA target sequence: tcttacggaatccagatatc). Cells were transduced and selected with puromycin as described. Cells were plated on poly‐L‐lysine coating at 18 cells/cm2 with untreated cells/cm2 as feeder layer. After 4 days, cells were selected with puromycin (2.5 μg/mL) to kill feeder cells, and remaining colonies were isolated as separate lines. To deplete MyoVb in HUES9 cells, the cells were incubated with MYO5B‐targeting lentiCRISPR viral supernatant for 5 hours (in E8 medium supplemented with 8 μg/mL polybrene). After 48 hours, cells were selected with puromycin (1 μg/mL). Selected cells were then plated at 28 cells/cm2, and the resulting colonies were mechanically passaged after 3 weeks. Clones were checked for myoVb knockout (KO) by means of a western blot.
Plasmids
Full‐length human myoVb‐coding sequence was amplified from HepG2 complementary DNA (cDNA) through PCR, including a myelocytomatosis (myc)‐encoding ‘5 overhang in the forward primer sequence. Amplified myc‐myoVb was inserted into pENTR1a vectors. All described myoVb mutants were generated by modification of this construct using the Q5 Site‐Directed Mutagenesis Kit (New England BioLabs) with primers designed in the NEBaseChanger tool. MyoVb and thereof derived mutant constructs were transferred to lentiviral vectors for mammalian expression through Gateway cloning using LR Clonase II (Thermo Scientific) as per manufacturer’s instruction. Full‐length myc‐myoVb constructs were transferred to pLenti‐cytomegalovirus (CMV)‐Blast‐DEST (706‐1; Addgene #17451), and myc‐myoVb tail domain constructs to pLenti‐CMV‐Puro‐DEST (w118‐1; Addgene #17452). Enhanced green fluorescent protein (EGFP)‐tagged wild type (WT) rab11a (rab11aWT) and the EGFP‐tagged dominant negative mutant of rab11a in which the serine at position 25 is subsitituted with an asparagine (rab11aS25N)16 were gifts from R.E. Pagano (Mayo Clinic).
Western Blotting
Cells were resuspended in radio immunoprecipitation assay buffer (150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris pH 8.0) containing protease inhibitors. Lysates were mixed with sample buffer (2% SDS, 5% β‐mercaptoethanol, 0.125M Tris–HCl, pH 6.8, 40% glycerol, 0.01% bromophenol blue) and incubated at 70°C for 10 minutes. Samples were resolved with SDS‐polyacrylamide gel electrophoresis and electro‐transferred onto PVDF membranes. Membranes were blocked with Odyssey‐blocking buffer and incubated overnight at 4°C with primary antibodies (Supporting Table S1). After incubation with secondary antibodies, immunoblots were scanned with the Odyssey (LI‐COR Biosciences). Relative quantification was performed using the Odyssey software.
Microscopy
Immunolabeling and fluorescence microscopy were performed essentially as described.3, 8 Antibodies used are listed in Supporting Table S1. Fluorescent multiplex immunohistochemistry of paraffin‐embedded liver tissue of a reported patient with PFIC63 and an anonymous donor with cholangitis as control was performed with standard procedure of Tyramine Signal Amplification. Fluorescent images were analyzed using a combination of ImageJ and Adobe Photoshop. For electron microscopy, cells were fixed by adding dropwise an equal volume fixative (2% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer). After 10 minutes, this mixture was replaced by pure fixative at room temperature for 30 minutes. After fixation in 1% osmium tetroxide/1.5% potassium ferrocyanide (4°C; 30 minutes), cells were dehydrated using ethanol and embedded in EPON epoxy resin. Then, 60‐nm sections were cut and contrasted using 2% uranyl acetate followed by Reynolds lead citrate. Images were captured with a Zeiss Supra 55 in scanning and transmission electron microscopy mode at 26 KV.
Real‐Time Quantitative PCR
RNA was harvested using trizol reagent (Sigma). RNA was reverse transcribed in the presence of oligo(dT)12‐18 (Invitrogen) and deoxyribonucleotide triphosphate (Invitrogen) with Moloney murine leukemia virus reverse transcriptase (Invitrogen). Gene expression levels were measured by real‐time quantitative RT‐PCR using ABsolute aPCR SYBR Green Master Mix (Westburg) in a Step‐One Plus Real‐Time PCR machine (Applied Biosystems), and the resulting data were analyzed using the LinRegPCR method. The primers used to mutate MYO5B cDNA were as follows:
Forward ‐ACCAGCTGCCGTTCTTACGGA‐.
Reverse ‐TGCGTTGTACATCAATTGGG‐.
Statistics
For all phenotype quantifications, statistical significance of differences between triplicate experiments was determined using Student t test (two‐tailed, unpaired, equal variance). Replicates represent quantifications of >200 cells. P values: *P < 0.05, **P < 0.01, ***P < 0.001.
Results
MyoVb Deficiency Does Not Disrupt Canalicular Protein Localization
Displacement of BC transporters to the cytoplasm of hepatocytes has been shown in liver biopsies of patients with MVID presenting with cholestasis and homozygous missense mutations (c.1979C>T/p.P660L)17 and a patient with non‐MVID PFIC with homozygous missense mutations (c.796T>C/p.C266R)3 in the MYO5B gene. Here, we demonstrate the mislocalization of BC transporters to intracellular compartments in hepatocytes of an additional patient with non‐MVID PFIC6 carrying a missense MYO5B mutation (c.437C>T/p.S158F3; Supporting Fig. S1), thereby expanding the number of patients with PFIC in which missense MYO5B mutations correlate with a cytoplasmic displacement of BC transporters. Intriguingly, in patients with cholestatic MVID with nonsense MYO5B mutations, a normal localization of BC transporters was reported,11 suggesting that loss of myoVb expression is not sufficient to induce BC transporter mislocalization. To address the requirement of MYO5B expression for the localization of BC transporters in hepatocytes, we examined the in vivo distribution of the ATP‐binding cassette sub‐family C member 2 (ABCC2)/multidrug resistance‐associated protein (Mrp) 2 and the structural BC protein radixin in the liver of whole‐body Myo5b KO mice.18 We observed their exclusive localization at BC, indistinguishable from wild‐type control mouse livers (Fig. 1A,B). Human HepG2 cells (HepG2Par) develop apical‐basolateral polarity and BC lumens between adjacent cells.19 In agreement with the observations in Myo5b KO mouse hepatocytes, HepG2 cells in which endogenous myoVb had been knocked out by CRISPR/CRISPR‐associated protein 9 (Cas9; Fig. 1C; Supporting Fig. S2A; HepG2KO cells) showed no defect in the canalicular localization of ABCC2/MRP2, which was indistinguishable from that in HepG2Par cells (Fig. 1D). Similar results were obtained with another canalicular protein, anoctamin (ANO)‐6 (Supporting Fig. S2B,C). We also generated a myoVb‐deficient HUES9 human pluripotent stem cell line through CRISPR/Cas9‐mediated gene KO (HUES9KO; Fig. 1E) and differentiated these to polarized hepatocyte‐like cells (human‐induced hepatocyte [hiHep]). These hiHeps form in vivo‐like multicellular BC (Fig. 1F).15 Similar to the results in HepG2KO cells, hiHeps derived from HUES9KO cells formed multicellular BC to which ABCC2/MRP2 exclusively localized, similar to HUES9Par‐derived hiHeps (Fig. 1F).
Figure 1.
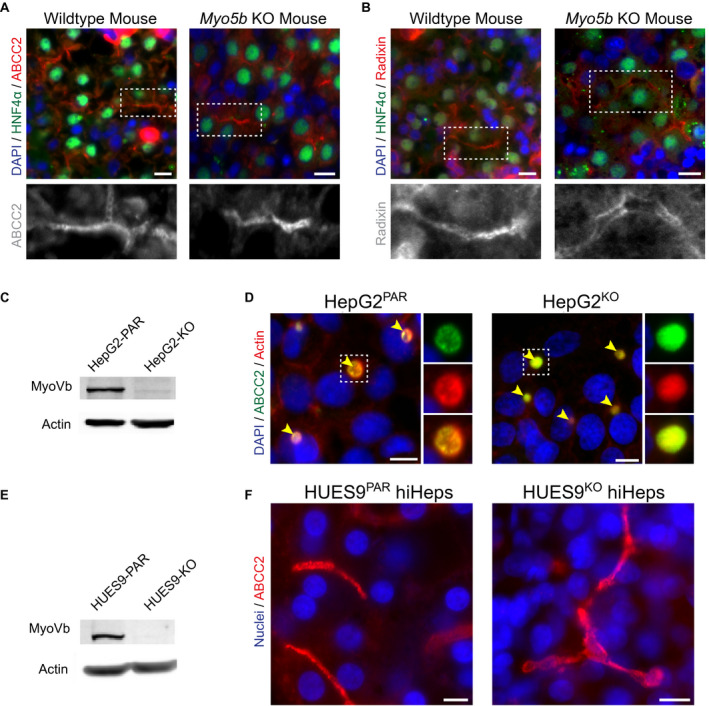
MyoVb deficiency does not disrupt canalicular protein localization. (A,B) Immunofluorescent staining of ABCC2 and radixin reveals their canalicular localization in both wild‐type and Myo5b KO mouse liver sections. HNF4α costaining marks hepatocytes. (C) KO of MYO5B in HepG2KO cells (treated with MYO5B‐targeting pLentiCRISPR V2) was confirmed by western blot (compared with parental line, HepG2PAR). (D) In HepG2 cells, localization of ABCC2 and F‐actin is unaffected by MYO5B KO (HepG2KO) compared with HepG2PAR control. Yellow arrowheads indicate BCs. (E) Western blot for myoVb in HUES9KO cells confirmed MYO5B KO (compared with parental line, HUES9Par). (F) HiHeps generated from HUES9KO cells exhibit BC formation comparable with HUES9Par‐derived hiHeps, with exclusive canalicular labeling of ABCC2. Scale bars: 10 μm.
These data show that missense myoVb mutations can correlate with a cytoplasmic displacement of BC transporters, whereas myoVb as such is dispensable for the correct localization of BC transporters at the canalicular membrane.
MVID‐Associated myoVb‐P660L Mutation Causes Intracellular Accumulation of Canalicular Proteins
Causality between patient MYO5B mutations and the mislocalization of BC proteins in hepatocytes has not been experimentally addressed. Investigation of such causality is particularly relevant for PFIC presenting in patients with MVID, as their long‐term parenteral nutrition obscures the etiology of hepatic dysfunction. We therefore focused on the founding homozygous c.1979C>T mutation in the MYO5B gene of Navajo patients with MVID.6 These patients display liver disease, and liver biopsies from these patients have been shown to display cytoplasmic displacement of canalicular bile acid transporters and signs of perturbed polarity.17
To determine whether this MYO5B mutation, leading to a P660L substitution in the myoVb protein, could be causally linked to canalicular protein localization defects, a full‐length human myc‐tagged MYO5B gene with the c.1979C>T mutation was constructed through site‐directed mutagenesis, and either the myc‐tagged wild‐type myoVb or myc‐myoVb‐P660L protein was expressed in HepG2KO cells. The expression of myoVb‐P660L in HepG2KO cells caused the intracellular accumulation of the BC proteins ABCC2/MRP2 and ANO6, when compared with HepG2KO cells expressing the wild‐type MYO5B gene (Fig. 2A‐C). This was accompanied by a reduction in the amount of BC (Supporting Fig. S2D). Notably, HepG2Par cells that expressed myoVb‐P660L showed a less severe phenotype (more BCs with subapical localization of the mutant protein and less intracellular accumulation of the mutant protein and canalicular proteins) when compared with HepG2KO cells that expressed myoVb‐P660L (Supporting Fig. S2E‐H, cf. Fig. 2). These data demonstrate that in the absence of wild‐type myoVb, mutant myoVb‐P660L caused the intracellular accumulation of BC‐resident proteins and provide evidence that this mutation is causally linked to the hepatic canalicular defects as observed in liver biopsies of the patients. Moreover, given the absence of a localization defect in myoVb‐depleted cells, the results also indicate that the disruptive effect of myoVb‐P660L on the localization of canalicular proteins in hepatocytes cannot be explained by the mere loss of myoVb function.
Figure 2.
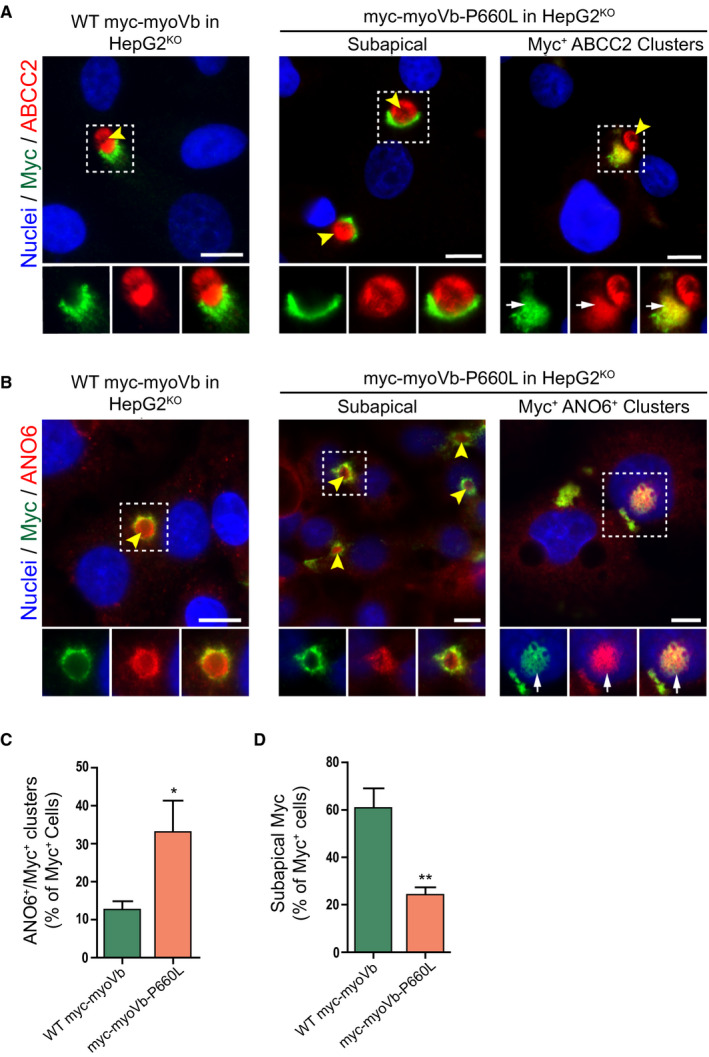
The MVID‐associated myoVb‐P660L mutation causes the intracellular accumulation of canalicular proteins. (A,B) Immunofluorescent microscopy images of myc‐tagged myoVb proteins, ABCC2 and ANO6, in HepG2KO expressing myc‐myoVb and myc‐myoVb‐P660L. HepG2KO cells expressing myoVb‐P660L show intracellular colocalization of myc and ABCC2 (white arrows). Yellow arrowheads indicate BCs. Scale bars: 10 μm. (C) Quantification of the percentage of myc‐positive cells that show intracellular clusters/accumulations of myc localized with ANO6. (D) Quantification of the percentage of myc‐positive cells that show subapical localization of myc‐tagged myoVb proteins.
Tail Domain of myoVb is Sufficient to Cause Intracellular Accumulation of Vesicles and BC Proteins
Because the mutated motor domain but not the absence of the myoVb protein produced a disease phenotype, we hypothesized that regions that were distal to the motor domain (IQ domains, coiled‐coil domains, and/or the globular tail domain) may have been instrumental to the disruptive effects on the localization of canalicular proteins. Therefore, we generated a human myoVb mutant that lacked the motor domain, IQ domains, and part of the coiled‐coil domain (Fig. 3A), which is similar to conventionally used dominant‐negative myoVb tail domain constructs (hereafter referred to as myoVb/Δ1‐1195). Similar to myoVb‐P660L, the expression of myoVb/Δ1‐1195 in HepG2KO cells resulted in the intracellular accumulation of ABCC2/MRP2 and ANO6 and a reduction in the number of BCs (Supporting Fig. S3A‐E). Notably, the effect of myoVb/Δ1‐1195 was more severe when compared with myoVb‐P660L and was also observed when expressed in HepG2Par cells (Fig. 3B‐E, Supporting Fig. S4A). Also, the canalicular protein dipeptidyl peptidase IV accumulated inside the cells (Supporting Fig. S4B,C). The myoVb/Δ1‐1195 mutant itself colocalized with the intracellular clusters (Fig. 3B). Electron microscopy of HepG2 cells expressing the myoVb mutant revealed the presence of large clusters of vesicles, which were not observed in control HepG2 cells (Fig. 3F). Finally, the expression of myoVb/Δ1‐1195 in pluripotent HUESPar stem cell–derived hepatocytes also caused the intracellular accumulation of ABCC2/MRP2 and ANO6 (Fig. S4D), which indicated that the effects caused by this mutant are not specific for the HepG2 cell line. Together, these results indicate that, in contrast to the loss of myoVb, the expression of the tail domain of myoVb mimicked the myoVb‐P660L–induced intracellular accumulation of BC proteins.
Figure 3.
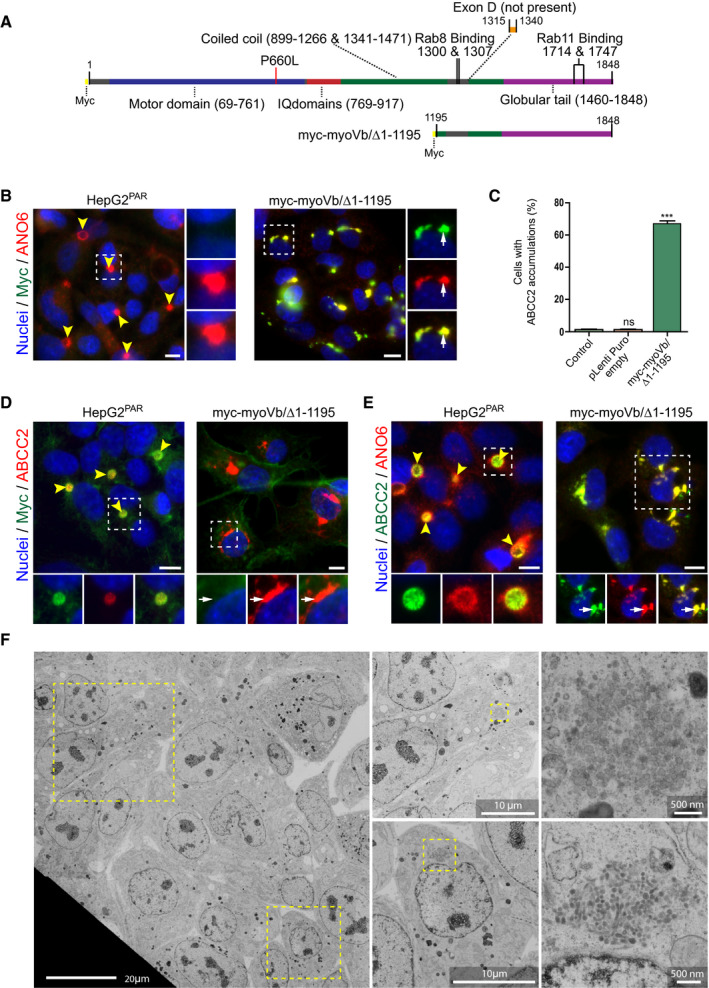
The tail domain of myoVb is sufficient to cause the intracellular accumulation of vesicles and BC proteins. (A) Schematic depiction of myoVb constructs. (B) Labeling of ANO6 and myc in HepG2Par cells expressing myc‐myoVb/Δ1‐1195 (white arrows indicate intracellular colocalization of both markers). (C) Quantification of the percentage of HepG2Par cells showing accumulation of ABCC2 (as shown in Fig. 4C) on expression of myc‐myoVb/Δ1‐1195 compared with untreated cells and cells transduced with an empty pLenti‐Puro construct (control). (D,E) Labeling of ABCC2 and F‐actin or ANO6 in HepG2Par cells expressing myc‐myoVb/Δ1‐1195 compared with untreated control. White arrows indicate intracellular accumulation of ABCC2 (and ANO6 in Fig. D). Scale bars: 10 μm unless labeled otherwise. Yellow arrowheads indicate BC. (F) Electron microscopy images of HepG2Par cells expressing myc‐myoVb/Δ1‐1195. Cells displayed large collections of vesicles (enlarged areas) that were not observed in control cells.
Motorless myoVb Induces Accumulation of Apical and Basolateral Proteins in Clustered Compartments with Mixed Recycling Endosome and TGN Identity
The intracellular clusters of BC proteins and the appearance of clusters of vesicles in myoVb mutant‐expressing cells suggested that these clusters represented intracellular organelles. To determine the identity of the mutant myoVb‐induced ABCC2/MRP2‐ and ANO6‐containing intracellular clusters, we performed immunofluorescence microscopy in cells colabeled with markers for different organelles. Proteins that make up BC microvilli, such as F‐actin, the ABCC2/MRP2‐ and F‐actin–binding protein radixin, or other phosphorylated proteins of the ezrin‐radixin‐moesi (ERM) family, did not colocalize with the BC protein–containing intracellular cluster in cells expressing myoVb/Δ1‐1195 (Fig. 4A, Supporting Fig. S5A), indicating that these clusters did not represent microvillus inclusions, which represent a hallmark of enterocytes in patients with MVID.20
Figure 4.
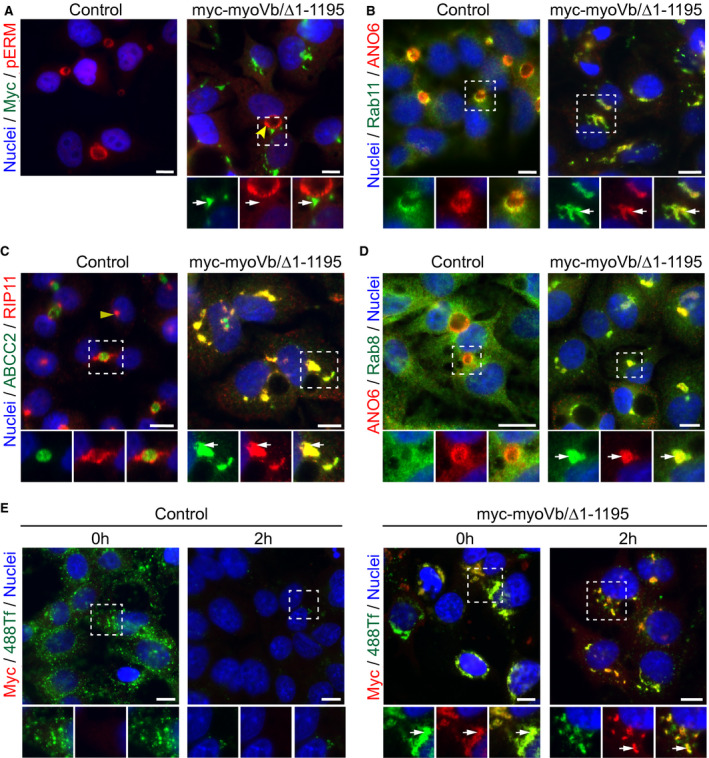
Motorless myoVb induces the accumulation of apical and basolateral proteins in clustered compartments that display recycling endosome identity. (A) Phospho‐proteins of the ezrin‐radixin‐moesi (ERM) family are not present in myc‐myoVb/Δ1‐1195 clusters (white arrows). Yellow arrowheads indicate BC. (B‐D) Labeling of rip11, rab11a, and rab8 with ANO6 or ABCC2 in HepG2 expressing myc‐myoVb/Δ1‐1195 (white arrows) compared with control. White arrows indicate colocalization of endosomal proteins with BC‐resident protein. The yellow arrowhead indicates juxta‐nuclear staining of rip11 in nonpolarized control cells. (E) Labeling of myc in control and myc‐myoVb/Δ1‐1195 expressing HepG2, fixed after 30 minutes incubation (t = 0 hours) with fluorescently labeled transferrin (388Tf) and after a 2‐hour chase period. 488‐Transferrin localized with myc‐myoVb/Δ1‐1195 clusters (white arrows) at t = 0 hours and at t = 2 hours. Scale bars: 10 μm.
By contrast, intracellular clusters containing BC proteins colocalized with the apical recycling endosome markers rab11a and its interacting protein rab11a‐family of interacting proteins number 5/rab‐interacting protein (rip11; Fig. 4B,C). This subcellular distribution of rab11a and its rip11 was markedly distinct from their exclusive subapical distribution in HepG2 cells that did not express the mutant protein. However, the subcellular distribution of rab11a and rip11 in HepG2KO cells and HepG2Par cells was indistinguishable, supporting the idea that myoVb expression is dispensable for canalicular polarity (Supporting Fig. S5B,C). Also, the recycling endosome‐associated rab8, which in control HepG2 cells showed a relatively dispersed cytoplasmic staining pattern, colocalized with the clusters in cells expressing the myoVb mutants (Fig. 4D). Lysosomal‐associated membrane protein 1, a marker of late endosomes/lysosomes, did not colocalize with the canalicular protein–containing clusters, and its subcellular distribution pattern was not visibly altered (Supporting Fig. S5D). In addition to BC proteins, the sinusoidal transferrin receptor and its ligand transferrin, which upon its endocytosis is recycled to the sinusoidal surface through recycling endosomes, was found to colocalize with the BC protein–containing clusters (Fig. 4E, Supporting Fig. S5E). Moreover, when fluorescently labeled transferrin was allowed to be endocytosed in control HepG2 cells or HepG2 cells expressing mutant myoVb, its subsequent recycling to the cell surface was inhibited in cells expressing the myoVb mutant, as evidenced by its persistent accumulation in the ANO6‐ and transferrin receptor‐containing clusters (Fig. 4E).
Although the cis‐Golgi protein giantin did not colocalize with the BC protein–containing clusters (Fig. 5A), we found that three markers of the TGN partly colocalized with the BC protein–containing clusters (Fig. 5B‐D). These included TGN46 and golgin‐97. With the exception of AP1y, which, in addition to its typical TGN‐like distribution pattern, was also observed in the subapical region of control HepG2 cells, TGN46 and golgin‐97 did not show a subapical localization in control HepG2 cells (Fig. 5B‐D). Together, these results indicate that the BC protein–containing clusters in cells expressing the myoVb mutant represented trafficking‐incompetent compartments with a mixed apical recycling endosome and TGN identity.
Figure 5.
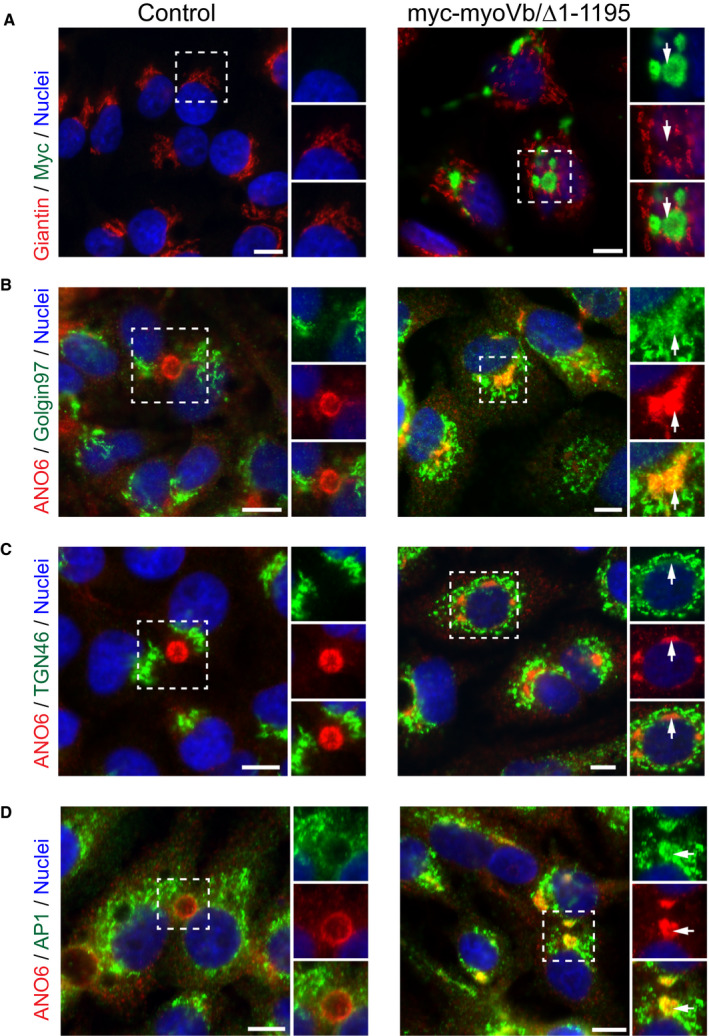
Motorless myoVb induces the accumulation of apical and basolateral proteins in clustered compartments that also display TGN identity. (A) Giantin labeling in HepG2 cells expressing myc‐myoVb/Δ1‐1195 compared with control. White arrows indicate lack of colocalization. (B,C) Golgin‐97 and TGN46 showed colocalization with intracellular cluster of ANO6 in HepG2 expressing myc‐myoVb/Δ1‐1195 (white arrows). (D) AP1y localized with ANO6 in intracellular clusters in HepG2 expressing myc‐myoVb/Δ1‐1195 (white arrows). Scale bars: 10 μm.
Disrupting Effect of Motorless myoVb on Canalicular Protein Localization Requires Active rab11a
We hypothesized that the myoVb tail domain induced canalicular defects through rab8 or rab11a function rather than competition with endogenous myoVb. To test whether the observed defects are mediated through rab8 or rab11a, we generated a myoVb mutant, which comprised only the globular tail domain (the last 383 amino acids) and did not contain the binding site for rab8 in ExonC/Exon30 (referred to as myoVb/Δ1‐1460; Fig. 6A). Like myoVb/Δ1‐1195, myoVb/Δ1‐1460 mutant led to the intracellular accumulation of BC proteins and a reduction in the number of BCs, albeit to a lesser extent than the myoVb/Δ1‐1195 mutant, suggesting that rab8 binding may contribute but is not essential to induce the effect (Fig. 6B,C, Supporting Fig. S5A). Indeed, substitution of glycine at position 1300 in myoVb/Δ1‐1195 to a leucine, a mutation known to abolish the interaction between myoVb and rab8,21 partially ameliorated its disrupting effects to the levels seen with the myoVb/Δ1‐1460 mutant (Fig. 6B,D). By contrast, when tyrosine at position 1714 in myoVb/Δ1‐1460 was mutated to glutamic acid (Y1714E; Fig. 6E‐G), a mutation that abolishes myoVb binding to rab11a,21 the intracellular accumulation of the canalicular proteins was completely abolished. Similarly, the introduction of this Y1714E mutation in the patient myoVb‐P660L mutant reduced the intracellular accumulation of the canalicular proteins (Fig. 7A‐C). Notably, the introduction of Y1714E in myoVb/Δ1‐1460 or myoVb‐P660L did not lead to an increase in the number of BC.
Figure 6.
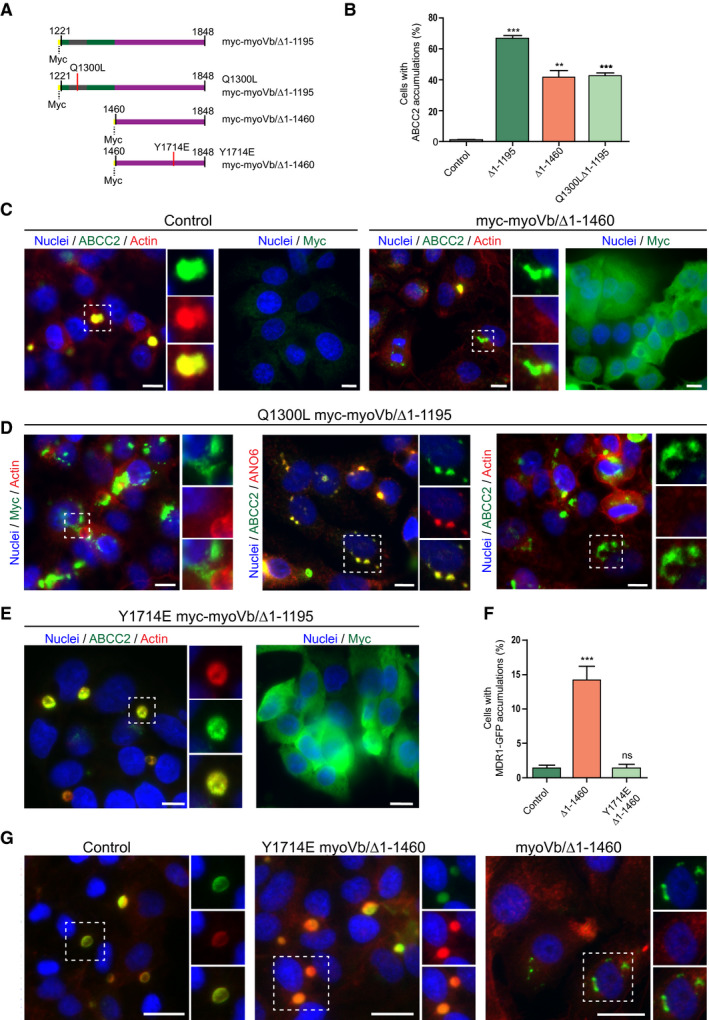
The role of rab8 and rab11a binding sites in the disrupting effect of motorless myoVb on canalicular protein localization. (A) Schematic depiction of the amino acid sequences of myoVb mutants. (B) Quantification of the percentage of HepG2 cells showing intracellular accumulation of ABCC2 on expression of myoVb tail domain mutants. (C) HepG2 cells expressing myc‐myoVb/Δ1‐1460 showed intracellular ABCC2 accumulation (white arrows). Myc labeling showed myc‐myoVb/Δ1‐1460 localized diffusely in the cytoplasm. (D) Labeling of ABCC2, F‐actin, and myc in HepG2 expressing myc‐myoVb/Δ1‐1195‐Q1300L and untreated control. White arrows indicate intracellular ABCC2 accumulation. (E) In HepG2 cells expressing myc‐myoVb/Δ1‐1460‐Y1714E ABCC2 localized at the BC with F‐actin (yellow arrowheads). Labeling for myc confirmed expression of the construct. (F) Quantification of the percentage of cells with intracellular multidrug resistance protein (MDR) 1‐GFP accumulations (depicted in Fig. 8G), on expression of myc‐myoVb/Δ1‐1460 or its Y1714E mutant variant. (G) HepG2 cells expressing myc‐myoVb/Δ1‐1460 showed intracellular accumulation of the coexpressed BC marker MDR1‐GFP (white arrows) but not when the Y1714E mutation was introduced in myc‐myoVb/Δ1‐1460.
Figure 7.
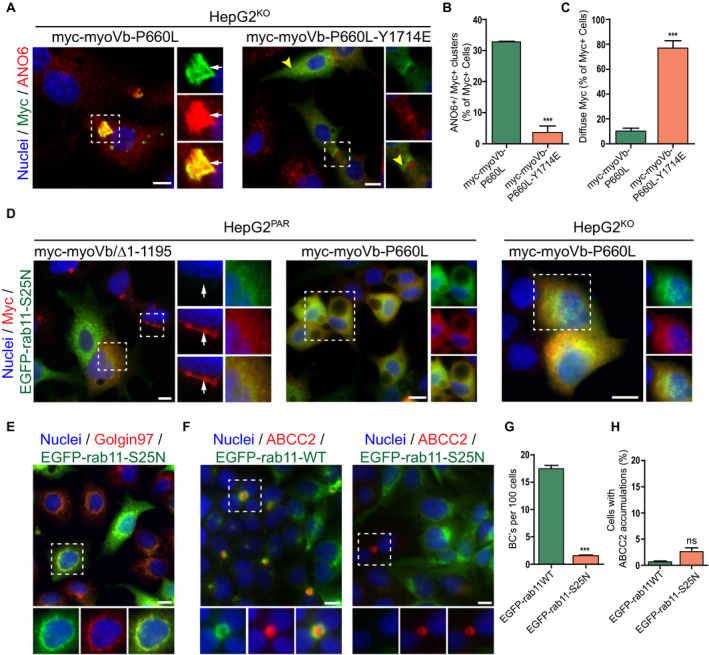
The disrupting effect of motorless myoVb on canalicular protein localization requires active rab11a. (A) ANO6 and myc labeling in HepG2KO expressing myc‐myoVb‐P660L or myc‐myoVb‐P660L‐Y1714E. Myc‐myoVb‐P660L frequently accumulated intracellularly with ANO6 (white arrows), whereas myc‐myoVb‐P660L‐Y1714E appeared diffuse in the cytoplasm or subapical (yellow arrowheads). (B) Quantification of the percentage of myc‐positive cells that show intracellular clusters/accumulations of myc localized with ANO6 in HepG2KO cells expressing myc‐myoVb‐P660L or myc‐myoVb‐P660L‐Y1714E. (C) Quantification of the percentage of myc‐positive cells that show subapical localization of myc in HepG2KO cells expressing myc‐myoVb‐P660L or myc‐myoVb‐P660L/Y1714E. (D) HepG2 coexpressing EGFP‐rab11aS25N and myc‐myoVb/Δ1‐1195 in HepG2Par cells (left), EGFP‐rab11aS25N and myc‐myoVb‐P660L in HepG2Par cells (middle), and EGFP‐rab11aS25N and myc‐myoVb‐P660L in HepG2KO HepG2 (right). In all three conditions, the presence of EGFP‐rab11aS25N prevented the clustering of the respective myoVb mutants. (E) EGFP‐rab11aS25N colocalized with golgin‐97 in HepG2 cells. (F) ABCC2 labeling in HepG2 overexpressing wild‐type EGFP‐rab11a or EGFP‐rab11aS25N. In EGFP‐rab11aS25N transduced cells, BC formation was only seen in cells with no or very low expression of the construct (right side, box). (G) Quantification of BC formation (expressed as BC/100 cells) in HepG2 cells expressing wild‐type EGFP‐rab11a or EGFP‐rab11aS25N. (H) Quantification of the percentage of HepG2 cells showing accumulation of ABCC2 on expression of wild‐type EGFP‐rab11a or EGFP‐rab11aS25N.
Moreover, the myoVb/Δ1‐1195 mutant, as well as the patient myoVb‐P660L mutant, failed to cause the intracellular clustering of ABCC2/MRP2 when expressed in HepG2 cells that also expressed the EGFP‐tagged mutant rab11aS25N (Fig. 7D), which is expected to shift the equilibrium of rab11a toward the guanosine diphosphate/nucleotide‐free state and exert dominant‐negative effects on endogenous rab11a by occupying the endogenous guanine nucleotide exchange factors (GEFs). Consistent with reports in other cells22 and the previously reported location of two rab11 GEFS, rab11‐interacting protein‐1, and Crag at the TGN, EGFP‐rab11aS25N colocalized with the TGN in HepG2 cells (Fig. 7E). Although the expression of EGFP‐tagged mutant rab11a‐S25N in HepG2 cells, as such, inhibited polarity development, it did not cause the intracellular clustering of BC proteins (Fig. 7F‐H). Cells expressing wild‐type EGFP‐rab11a showed normal BC formation and a subapical distribution of the EGFP‐rab11a, similar to wild‐type cells (Fig. 7F‐G). Thus, the intracellular clustering of BC proteins in cells expressing myoVb‐P660L or the myoVb tail domain is not phenocopied by loss of rab11a function. Together, we conclude that the interaction of the myoVb‐P660L or myoVb/Δ1‐1195 mutant with active rab11a is required for the myoVb‐P660L‐ or myoVb/Δ1‐1195–induced intracellular accumulation of BC proteins, but not inhibition of polarity development, and that loss of rab11a function inhibited polarity development independent of myoVb.
MVID‐Associated Nonsense MYO5B Mutations Producing Truncated myoVb Mutants do not Disrupt Hepatocyte polarity and Canalicular Protein Localization
We reasoned that if the disrupting effect of motor‐deficient myoVb mutants on the localization of canalicular proteins required their interaction with rab11a through the distal C‐terminal binding Y1714 residue, nonsense MYO5B mutations that cause a premature translation termination codon and the resultant synthesis of truncated myoVb proteins should not lead to polarity defects. We therefore generated different MYO5B nonsense mutations previously reported in patients with MVID and listed in the MVID registry (www.mvid-central.org), myoVb‐R363X (c.1087C>T), myoVb‐R1016X (c.5382C>T), and myoVb‐R1795X (c.5383C>T; Fig. 8A), and expressed these in HepG2KO cells. Note that in contrast to the myoVb‐R363X and myoVb‐R1016X mutants the myoVb‐R1795X mutant contains the rab11a‐binding site. Western blot analyses confirmed that these mutants led to the expression of truncated myoVb proteins at their predicted molecular weights (Fig. 8B). Fluorescence microscopy showed that the mutants failed to cause the intracellular accumulation of ABCC2/MRP2 (Fig. 8C). These results demonstrate that nonsense MVID–associated MYO5B mutations and the expression of resultant truncated myoVb mutants that lack the globular tail domain do not cause defects in BC formation and canalicular protein localization, and they support our observations that the C‐terminal region and a conserved interaction with rab11a is required for myoVb mutants to disrupt the localization of canalicular proteins.
Figure 8.
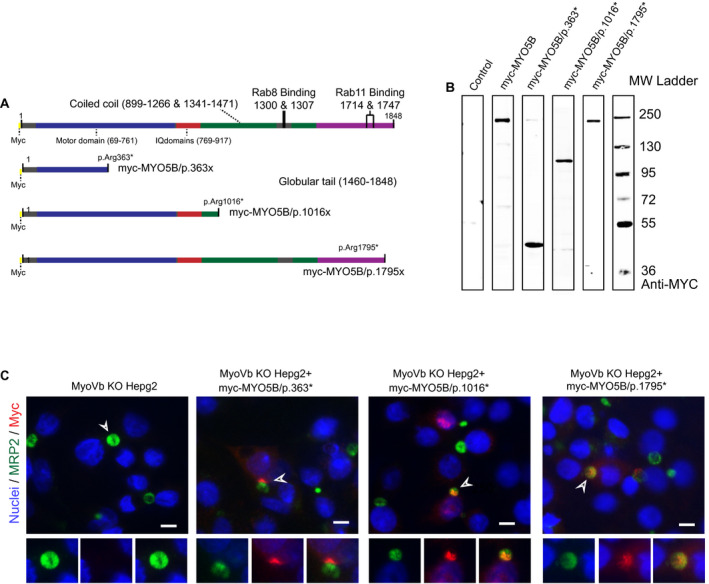
MVID‐associated nonsense MYO5B mutations producing truncated myoVb mutants do not disrupt hepatocyte polarity and canalicular protein localization. (A) Schematic depiction of the amino acid sequences of nonsense myoVb mutants. (B) Western blot showing the truncated myoVb mutant proteins expressed in HEK293 cells. (C) Immunofluorescence microscopy images showing the subcellular distribution of the truncated myoVb mutant proteins and the canalicular protein ABCC2. Scale bars: 10 μm.
Discussion
Determination of causality between patient MYO5B mutations and mislocalization of BC proteins in patients with cholestasis is essential to understand the etiology of the disease. In this study, we demonstrated that the founding Navajo myoVb‐P660L mutant6 when expressed in hepatic HepG2 cells caused the aberrant localization of canalicular proteins as well as the sinusoidal transferrin receptor to intracellular clusters. These data are in agreement with observations in hepatocytes in liver biopsies of patients with this mutation and therefore show that the liver symptoms and hepatocyte defects observed in Navajo patients with MVID17 are likely a direct consequence of their MYO5B mutation rather than a sole consequence of intestinal failure–induced or TPN‐induced liver damage. The causality between myoVb‐P660L and the mislocalization of canalicular transporters suggest similar mechanisms for PFIC6 in patients carrying other missense MYO5B mutations. Although different missense mutations may affect the myoVb protein differently,7, 12 the clear mislocalization of canalicular proteins to intracellular compartments as shown in this study for another PFIC6 patient with a different missense MYO5B mutation supports this.
Insight into the mechanism through which (mutant) myoVb (de)regulates hepatocyte function is necessary to understand the pathogenesis of the disease. We demonstrated that the loss of myoVb in human or mouse hepatocytes, as well as the expression of patients’ nonsense MYO5B mutations, did not cause the aberrant localization of canalicular proteins, indicating that myoVb function as such is not required for the correct localization of BC proteins. Instead, the hepatocyte polarity phenotype as observed in myoVb‐P660L–expressing cells was faithfully phenocopied by the expression of myoVb mutants that lacked the entire motor domain and consisted of only the tail domains of the myoVb protein.
These results indicated that the effects of myoVb‐P660L on the localization of canalicular proteins could not be explained by a mere loss of myoVb motor function. Indeed, additional mutagenesis experiments showed that the disrupting effect of myoVb motor domain mutants on the localization of canalicular proteins was critically dependent on their ability to interact with active rab11a. Furthermore, the absence of intracellular clusters of BC proteins on inhibition of rab11a activation indicated that the mechanism through which myoVb mutants exerted their effects on the distribution of canalicular proteins involved active rather than inhibited rab11a function.
A recent study demonstrated that the globular tail domain of myoVb induced the clustering of rab11a‐decorated lipid vesicles (liposomes) in a chemically defined in vitro reconstitution system by stimulating homotypic rab11‐rab11 interactions.23 Although this effect has thus far not been demonstrated in living cells, it would fit with the myoVb‐Y1714–dependent and rab11a‐dependent clustering of rab11a and associated cargo and the appearance of clusters of vesicles that we observed in cells expressing myoVb‐P660L or only the myoVb tail domain. Conceivably, the C‐terminal tail domain of mutant myoVb, on its displacement from its normal subapical location, in this way induced the ectopic clustering of TGN‐derived and/or recycling endosome–derived transport vesicles through rab11a and thereby perturbed the correct distribution of BC proteins.
The results from this study are relevant for understanding the unexplained genotype‐genotype correlations that have been reported for PFIC6. Indeed, PFIC in patients without MVID has been associated with only biallelic missense mutations and, in contrast to the enteropathy in MVID, has not been associated with biallelic MYO5B mutations that are predicted to result in the loss of myoVb protein expression, such as nonsense or frameshift mutations.3, 12 In agreement with these clinical findings, we found that the expression of truncated myoVb resulting from MVID‐associated nonsense MYO5B mutations did not cause a canalicular protein localization defect. Together with the findings that myoVb as such is not required for the correct localization of canalicular proteins in vitro and in vivo, and that myoVb mutants required active rab11a for their disruptive effect on canalicular protein localization, this study thus provides a direct and simple explanation for this genotype‐phenotype correlation in patients with non‐MVID PFIC6. It may also lead us to speculate that intrahepatic cholestasis in patients with MVID9, 14 is less likely to be caused by their MYO5B mutations when these involve nonsense mutations than when these involve missense mutations. In support of this, a patient with MVID was reported with only nonsense MYO5B mutations and presented with cholestasis with normal GGT levels for 9 months, but liver biopsies showed normal canalicular protein localization. Cholestasis in this patient later spontaneously resolved.11
The results of this study are relevant for exploring new treatment strategies, for example, for those patients with PFIC6 who are nonresponsive to routine treatment.3, 9 Our results suggest that the specific inhibition of the interaction between mutant myoVb and rab11a in the patients’ hepatocytes may ameliorate the harmful effects of the myoVb mutant on canalicular protein localization and thereby the PFIC in patients. This study thus paves the way for the discovery of small molecule inhibitors of this interaction and the exploration of their potential beneficial effects.
Finally, the ectopic expression of the globular tail domain of myoVb has been widely used to implicate the involvement of myoVb in intracellular trafficking of a variety of proteins in a variety of cell types.10, 24, 25, 26, 27 The results from our study, demonstrating that the effects of the myoVb tail domain are not necessarily mimicked by the loss of myoVb expression or loss of rab11a activity, yet depend on their interaction with active rab11a, suggest that the need to recheck the interpretation of some of the studies using this myoVb mutant is warranted.
Author Contributions
Conceptualization, A.W.O., Q.L., D.A., and S.C.D.IJ.; Methodology, A.W.O., Q.L., F.C., C.L., K.K., N.H., J.W., D.A., and S.C.D.IJ.; Investigation, A.W.O., Q.L., Y.Q, F.C., C.L., K.K., J.D., and N.H.; Writing Original Draft, A.W.O. ad S.C.D.IJ.,; Writing Review & Editing, A.W.O., Q.L., Y.Q., F.C., C.L., K.K., J.D., N.H., J.W., D.A., and S.C.D.IJ.,; Funding Acquisition, S.C.D.IJ. and J.W.; Resources, A.W.O., F.C., D.A., Y.Q., ; Supervision, S.C.D.IJ., D.A., and J.W.
Supporting information
Acknowledgment
We thank R. Prekeris for his kind gift of anti‐rip11 antibodies.
Supported by grants from the Nederlandse vereniging voor Gastroenterologie (to S. I. J.) and the Natural Science Foundation of China, Nos. 81873543 and 81570468 (to J. S. W.).
Potential conflict of interest: Nothing to report.
References
- 1. Gissen P, Arias IM. Structural and functional hepatocyte polarity and liver disease. J Hepatol 2015;63:1023‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis 2018;22:657‐669. [DOI] [PubMed] [Google Scholar]
- 3. Qiu YL, Gong JY, Feng JY, Wang RX, Han J, Liu T, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ‐glutamyltransferase cholestasis. Hepatology 2017;65:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gonzales E, Taylor SA, Davit‐Spraul A, Thébaut A, Thomassin N, Guettier C, et al. MYO5B mutations cause cholestasis with normal serum gamma‐glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017;65:164‐173. [DOI] [PubMed] [Google Scholar]
- 5. Müller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz‐Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet 2008;40:1163‐1165. [DOI] [PubMed] [Google Scholar]
- 6. Erickson RP, Larson‐Thomé K, Valenzuela RK, Whitaker SE, Shub MD. Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A 2008;146A:3117‐3119. [DOI] [PubMed] [Google Scholar]
- 7. van der Velde KJ, Dhekne HS, Swertz MA, Sirigu S, Ropars V, Vinke PC, et al. An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Hum Mutat 2013;34:1597‐1605. [DOI] [PubMed] [Google Scholar]
- 8. Dhekne HS, Hsiao NH, Roelofs P, Kumari M, Slim CL, Rings EH, et al. Myosin Vb and Rab11a regulate phosphorylation of ezrin in enterocytes. J Cell Sci 2014;127:1007‐1017. [DOI] [PubMed] [Google Scholar]
- 9. Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatol Baltim Md 2014;60:301‐310. [DOI] [PubMed] [Google Scholar]
- 10. Wakabayashi Y, Dutt P, Lippincott‐Schwartz J, Arias IM. Rab11a and myosin Vb are required for bile canalicular formation in WIF‐B9 cells. Proc Natl Acad Sci USA 2005;102:15087‐15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernández Caamaño B, Quiles Blanco MJ, Fernández Tomé L, Burgos Lizáldez E, Sarría Osés J, Molina Arias M, et al. Intestinal failure and transplantation in microvillous inclusion disease. [in Spanish] An Pediatr (Barc) 2015;83:160‐165. [DOI] [PubMed] [Google Scholar]
- 12. Dhekne HS, Pylypenko O, Overeem AW, Zibouche M, Ferreira RJ, van der Velde KJ, et al. MYO5B, STX3, and STXBP2 mutations reveal a common disease mechanism that unifies a subset of congenital diarrheal disorders: a mutation update. Hum Mutat 2018;39:333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitra A, Ahn J. Liver disease in patients on total parenteral nutrition. Clin Liver Dis 2017;21:687‐695. [DOI] [PubMed] [Google Scholar]
- 14. Anez‐Bustillos L, Dao DT, Potemkin AK, Perez‐Atayde AR, Raphael BP, Carey AN, et al. An intravenous fish oil‐based lipid emulsion successfully treats intractable pruritus and cholestasis in a patient with microvillous inclusion disease. Hepatology 2019;69:1353‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Overeem AW, Klappe K, Parisi S, Klöters‐Planchy P, Mataković L, du Teil Espina M, et al. Pluripotent stem cell‐derived bile canaliculi‐forming hepatocytes to study genetic liver diseases involving hepatocyte polarity. J Hepatol 2019;71:344‐356. [DOI] [PubMed] [Google Scholar]
- 16. Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, et al. Rab proteins mediate Golgi transport of caveola‐internalized glycosphingolipids and correct lipid trafficking in Niemann‐Pick C cells. J Clin Invest 2002;109:1541‐1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schlegel C, Weis VG, Knowles BC, Lapierre LA, Martin MG, Dickman P, et al. Apical membrane alterations in non‐intestinal organs in microvillus inclusion disease. Dig Dis Sci 2018;63:356‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cartón‐García F, Overeem AW, Nieto R, Bazzocco S, Dopeso H, Macaya I, et al. Myo5b knockout mice as a model of microvillus inclusion disease. Sci Rep 2015;5:12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohgaki R, Matsushita M, Kanazawa H, Ogihara S, Hoekstra D, van Ijzendoorn SC. The Na+/H+ exchanger NHE6 in the endosomal recycling system is involved in the development of apical bile canalicular surface domains in HepG2 cells. Mol Biol Cell 2010;21:1293‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davidson GP, Cutz E, Hamilton JR, Gall DG. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology 1978;75:783‐790. [PubMed] [Google Scholar]
- 21. Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase‐Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci USA 2011;108:2789‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen W, Feng Y, Chen D, Wandinger‐Ness A. Rab11 is required for trans‐Golgi network‐to‐plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol Biol Cell 1998;9:3241‐3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inoshita M, Mima J. Human Rab small GTPase‐ and class V myosin‐mediated membrane tethering in a chemically defined reconstitution system. J Biol Chem 2017;292:18500‐18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gupta A, Schell MJ, Bhattacharjee A, Lutsenko S, Hubbard AL. Myosin Vb mediates Cu+ export in polarized hepatocytes. J Cell Sci 2016;129:1179‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brock SC, Goldenring JR, Crowe JE. Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc Natl Acad Sci USA 2003;100:15143‐15148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fan GH, Lapierre LA, Goldenring JR, Sai J, Richmond A. Rab11‐family interacting protein 2 and myosin Vb are required for CXCR2 recycling and receptor‐mediated chemotaxis. Mol Biol Cell 2004;15:2456‐2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swiatecka‐Urban A, Talebian L, Kanno E, Moreau‐Marquis S, Coutermarsh B, Hansen K, et al. Myosin Vb is required for trafficking of the cystic fibrosis transmembrane conductance regulator in Rab11a‐specific apical recycling endosomes in polarized human airway epithelial cells. J Biol Chem 2007;282:23725‐23736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials