

Abstract
CRISPR-Cas9 is a widely employed genome-editing tool with functionality reliant on the ability of the Cas9 endonuclease to introduce site-specific breaks in double-stranded DNA. In this system, an intriguing allosteric communication has been suggested to control its DNA cleavage activity through flexibility of the catalytic HNH domain. Here, solution NMR experiments and a novel Gaussian accelerated Molecular Dynamics (GaMD) simulation method are used to capture the structural and dynamic determinants of allosteric signaling within the HNH domain. We reveal the existence of a millisecond timescale dynamic pathway that spans HNH from the region interfacing the adjacent RuvC nuclease and propagates up to the DNA recognition lobe in full-length CRISPR-Cas9. These findings reveal a potential route of signal transduction within the CRISPR-Cas9 HNH nuclease, advancing our understanding of the allosteric pathway of activation. Further, considering the role of allosteric signaling in the specificity of CRISPR-Cas9, this work poses the mechanistic basis for novel engineering efforts aimed at improving its genome editing capability.
Keywords: CRISPR-Cas9, protein dynamics, allostery, NMR, MD simulations
Graphical Abstract
Introduction
The CRISPR-Cas9 enzyme machine has exciting applications in genome editing and numerous investigations have sought to harness its mechanism for therapeutic bioengineering.1-2 Cas9 is an RNA-guided DNA endonuclease, which generates double-stranded breaks in DNA by first recognizing its protospacer-adjacent motif (PAM) sequence and then cleaving the two DNA strands via the HNH and RuvC nuclease domains.3 Structural studies of Cas9 have employed crystallographic4-6 and cryo-EM7-8 techniques, revealing several well-defined subdomains, including the catalytic domains, a recognition (REC) lobe and a PAM interacting (PI) region (Figure 1A). In parallel, Förster Resonance Energy Transfer (FRET) techniques provided insight into the large-scale conformational changes that occur during nucleic acid processing.9-11 These and other biophysical studies have been invaluable to our current understanding of Cas9 function.12-13 Building on this experimental information, computational investigations have been carried out to describe the conformational and dynamic requirements underlying Cas9 mechanistic action. All-atom Molecular Dynamics (MD) simulations have described the conformational activation of the Cas9 protein toward the binding and enzymatic processing of nucleic acids.14-16 These investigations also revealed the ability of Cas9 to propagate the DNA binding signal across the HNH and RuvC nuclease domains for concerted cleavage of double-stranded DNA.17 Notably, biochemical experiments and MD simulations have jointly indicated a dynamically driven allosteric signal throughout Cas9, where the intrinsic flexibility of the catalytic HNH domain regulates the conformational activation of both nucleases, therefore controlling the DNA cleavage activity 9,17 Detailed knowledge of this allosteric mechanism and of the conformational control exerted by HNH is essential for understanding Cas9 function and for engineering efforts aimed at improving the specificity of this system through modulation of its allosteric signaling.18 In this respect, an in-depth investigation necessitates the use of experimental techniques such as solution nuclear magnetic resonance (NMR) to quantify the motional timescales critical to this allosteric crosstalk. NMR can readily detect subtle conformational fluctuations at the molecular level, with precise information about the local dynamics on picosecond (ps) to nanosecond (ns) timescales (i.e. the so-called fast dynamics), as well as those occurring over microseconds (μs) to milliseconds (ms) (i.e. slow dynamics), both of which contribute to allosteric signaling.19-22 The power of solution NMR is magnified when coupled to MD simulations23-24 that capture protein fluctuations and conformations on the same timescales of NMR experiments, offering an interpretation at the atomic scale while also describing the subtle changes that characterize protein allostery.25-27
Figure 1. NMR Spectrum of HNH.

(a) Architecture of the Cas9 protein (PDB code: 4UN3),5 highlighting its protein domains as follows: HNH (green), RuvC (blue), PAM interacting region (PI, gold) and recognition lobe (REC, gray). In the close-up view, a model of the HNH structure determined from NMR data (green) is overlaid with that of HNH from the full-length Cas9 (gray). (b) 1H-15N HSQC NMR spectrum of the HNH nuclease domain from S. pyogenes Cas9 (the inset reports two peaks out of range). (c) Consensus chemical shift index (CSi), indicating the predicted secondary structure for the HNH construct based on the NMR chemical shifts (black bars from 0 to 1 indicate α-helix, while bars from 0 to −1 indicate β-sheet, see the Methods section) compared to that of HNH from the full-length Cas9 (shown on top of the graph as sequence, with α-helical and β-sheet regions indicated as tubes and arrows).
Here, we probe the structural and dynamic determinants of allosteric signaling in the Cas9 HNH nuclease by means of solution NMR and all-atom MD simulations. A novel construct of the HNH nuclease domain from S. pyogenes Cas9 has been engineered that maintains the fold and properties of the wild-type (WT, i.e. full-length) Cas9 protein and allows the characterization of its multi-timescale conformational dynamics by solution NMR spectroscopy and MD simulations. To comprehensively access the slow timescale dynamics of the system at the atomic scale, we performed accelerated MD (aMD). aMD is an enhanced sampling methodology that applies a boost potential to the simulation, thereby accelerating transitions between low-energy states. The method has previously been shown to access slow dynamical motions in biomolecules, in excellent agreement with NMR experiments.28-30 However, the use of aMD for large biomolecular systems, such as CRISPR-Cas9, can suffer from high statistical noise, which hampers the characterization of the correct statistical ensemble.31-32 To overcome this limitation, a novel Gaussian accelerated MD (GaMD) method33 has been proposed, which uses harmonic functions to construct a boost potential that is adaptively added to the simulation (see the Methods section). GaMD has been applied to large biomolecular complexes, successfully describing the long timescale dynamics of CRISPR-Cas934-36 and G-protein coupled receptors.37-38 Hence, while overcoming the limitations of the early aMD methodology, GaMD still holds the method capability to describe slow dynamical motions, which are relevant for allostery,19 and to provide an atomic-level comparison with NMR experiments.28-30 As a result of our experimental and theoretical approach, we identify a dynamic pathway that connects HNH and RuvC through contiguous ms timescale motions, while also highlighting its propagation to the REC lobe to enable the information transfer for concerted cleavage of DNA. Structure-based prediction of the NMR chemical shifts further reveal the agreement between experiments and computations, indicating that the structural/dynamic features derived from GaMD simulations represent the experimental results well at the molecular level. Overall, the integrated approach employed in this study enabled access to the intrinsic conformational fluctuations of the Cas9 HNH nuclease, which are essential for allosteric signaling in CRISPR-Cas9. Our combined NMR and theoretical approach paves the way for the complete mapping of allosteric signaling and determination of its role in the enzymatic function and specificity of Cas9.
Results
Structural features of the HNH nuclease
To determine the structural features of the isolated HNH domain, we employed solution NMR and X-ray crystallography. First, the structure of the HNH domain (Figure 1A) was derived from the assigned 1H-15N HSQC NMR spectrum (Figure 1B) and backbone assignments.39 Backbone assignments were uploaded to the CS23D server in order to predict the HNH structure from composite NMR chemical shift indices.40 Figure 1A shows a model of the HNH structure determined from NMR data (close-up view, green) overlaid with that of HNH from the full-length Cas9 (gray). The predicted structure is remarkably similar to that of full-length Cas9 (PDB code: 4UN3)5 displaying Cα root-mean-squared-deviation RMSD = 0.688 Å. The NMR model also highlights small helical turns in regions of poor electron density in the full-length Cas9 structure, as well as an extension of the C-terminal α-helix.
The secondary structure of this construct determined from Cα and Cβ chemical shift indices is in good agreement with that of the HNH domain from the full-length Cas9 (Figure 1C), indicating that the engineered protein is a good representation of this fold in solution. Circular dichroism (CD) spectroscopy is consistent with a predominantly α-helical protein (Figure S1), in agreement with the X-ray structure of the full-length Cas9.5 Interestingly, the thermal stability of this engineered construct is 12 °C higher than that of full-length Cas9, despite identical amino acid sequences, and is soluble to concentrations exceeding 1 mM.
The similarity of our construct to that of HNH from the full-length Cas9 supports the reliability of the predicted structure. A further confirmation is provided by the X-ray crystal structure of the HNH construct solved at 1.3 Å resolution (Figure 2). This X-ray crystal structure aligns well to that of the full-length Cas9 (PDB code 4UN3)41 and the predicted NMR structure, with a Cα RMSD values of 0.549 Å and 0.479 Å, respectively, with the most significant difference due to a crystal contact in the experimental lattice pushing the N-terminal helix inward (Figure 2, inset top).
Figure 2. X-ray structure of HNH.

The X-ray structure of the isolated HNH domain (PDB code: 6O56, green), solved at 1.30 Å resolution, is overlaid with the X-ray structure of the HNH domain from the full-length S. pyogenes Cas9 (PDB code: 4UN3, gray).5
The overall fold of HNH from full-length Cas9 is therefore well-maintained in the isolated domain. The residues L791–E802 and T858–S872 form two flexible loop regions, as suggested by NMR. An α-helix is introduced in residues Q794–E798 and an additional solvent exposed loop comprised of residues T858–S872 forms a small α-helix at D861–R864 (Figure 2, inset bottom), also observed in the structural model from the NMR chemical shifts. Lastly, a small extension of the C-terminal α-helix is also confirmed in this novel X-ray structure.
Experimental conformational dynamics of the HNH nuclease
In order to experimentally probe timescales relevant to allostery, we first analyzed the dynamics of HNH with the method of Bracken and coworkers.42 Here, potential sites of ps–ns and μs–ms flexibility have been identified through the analysis of the R1R2 product. Motions on each of these timescales contribute to allostery; ps-ns motions have been associated with allosteric activation as effector molecules induce favorable changes in configurational entropy,43-44 leading to a population shift from inactive-to-active states, while μs–ms processes are often commensurate with the rates of catalytic reactions.45-46 With respect to the individual longitudinal and transverse relaxation rates, the R1R2 product attenuates the contribution of motional anisotropy and more clearly illuminates sites of chemical exchange. The R1R2 values for each residue in HNH (Figure 3A and Table S1) highlight several locations of ps–ns and μs–ms flexibility. Twenty residues display R1R2 values above 1.5σ of the 10% trimmed mean, due to the significant influence of Rex related to μs–ms motion. Measured Rex parameters are consistent with this interpretation (Figures 3A, S2, S3 and Table S1). A lower number of residues (i.e., 13) fall below 1.5σ of the mean, suggesting potential influence of ps–ns dynamics at these sites, with the mean R1R2 value corresponding to an average order parameter (S2) value of 0.85, where . Steady-state 1H-[15N] NOE were also measured and the order parameter (S2) was determined for assigned residues in HNH with RELAX.47 Regions of ps–ns flexibility (i.e. high configurational entropy) are observed in residues 822–843 and 890–904. Consistent with these data, in the X-ray structure of full-length Cas9 residues 822–843 are exposed toward the solvent, while residues 890–904 comprise flexible loop regions.5 In order to expand on this analysis, we quantified the conformational exchange parameters associated with millisecond dynamics of the HNH nuclease by Carr–Purcell–Meiboom–Gill (CPMG) relaxation dispersion experiments (Figure 3B and Table S2). Residues displaying slow timescale (ms) dynamics correspond to K782–E786, 1788, K789, G792, Q794–E798, Y815–N818, R820, E827–D829, H840, V841, S851, D853, K855, E873 and L900 in the full-length Cas9. Many of these sites are also indicated by the R1R2 analysis. Rates of conformational exchange (kex) at these sites range from 800 – 2900 s−1 with an average kex = 1761 ± 414. Interestingly, kex values determined from CPMG experiments show an approximate bimodal distribution, with a large number of residues showing 1000 ≤ kex ≤ 2000 s−1 and fewer residues with 2000 ≤ kex ≤ 3000 s−1 (Figure S4). Regions with larger kex values are primarily confined to the periphery and N-terminus of HNH. Residues with smaller kex values comprise the majority of the HNH-REC interface and central dynamic core.
Figure 3. HNH Dynamics Measured by NMR.

(a) Plots of R1R2, Rex, and the order parameter (S2, determined from model-free analysis of T1, T2, 1H-[15N]-NOE measurements) for the HNH nuclease. The R1R2 parameters were measured at 600 (black) and 850 (red) MHz. The red dashed line on the Rex plot denotes the 1.5σ from the 10% trimmed mean of the data. The Rex (right, top) and S2 (right, bottom) values are mapped onto the HNH structure and colored according to the adjacent legends. (b) Selected CPMG relaxation dispersion curves collected at 600 (solid lines) and 850 (dashed lines) MHz.
Allosteric signaling pathway
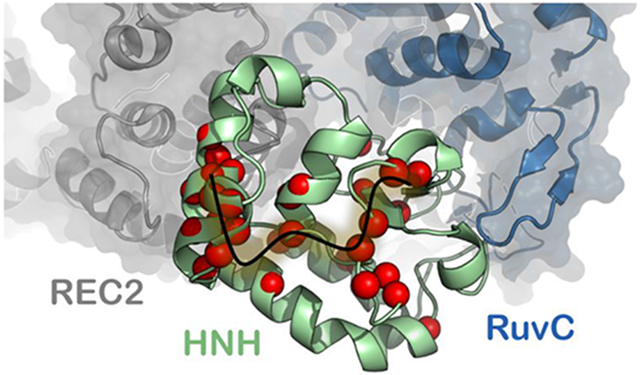
The ms dynamics observed in CPMG relaxation dispersion experiments are of particular interest for the identification of the Cas9 signaling pathways, due to the established importance of slow dynamical motions in allosteric regulation.19 Residues displaying slow timescale (ms) dynamics (Table S2) form clusters in three regions of HNH (Figure 4, red spheres), two of which are the interface with the region REC2 of the recognition lobe and with RuvC (i.e., the HNH–REC2 and HNH–RuvC interfaces), while the third region is located in the core of HNH. This well-defined subset of flexible residues within HNH therefore bridges the RuvC and REC2 interfaces, forming a contiguous dynamic pathway within the isolated HNH domain. This pathway of flexible residues connecting HNH–RuvC and HNH–REC2 agrees well with the available experimental evidence that indicate the existence of allosteric communication within CRISPR-Cas9. Previous experimental studies revealed that the nuclease activity of the spatially distant HNH and RuvC domains is coupled, facilitating concerted cleavage of the DNA strands.9 In parallel, MD simulations have shown that HNH and RuvC display highly coupled dynamical motions.17 This prior work employed Community Network Analysis48 to structure the Cas9 correlations, revealing a strong communication channel between HNH and RuvC. Taken together, experiments and simulations have indicated a tight dynamic “cross-talk” between HNH and RuvC, which underlie their coupled nuclease function. The REC2 region has been recently suggested to be involved in the activation of HNH through an allosteric regulation that also implicates the REC3 region.41 The authors have shown that, upon binding a complementary RNA:DNA structure prone to undergo DNA cleavage, the REC3 region modulates the motions of the neighboring REC2, which in turn contacts HNH and sterically regulates its access to the scissile phosphate. MD simulations of the fully activated CRISPR-Cas9 complex revealed that highly coupled motions between REC2, REC3 and HNH are critical for the activation of the catalytic domain toward cleavage, supporting the existence of an allosteric signal.49
Figure 4. Allosteric signaling across HNH.

Flexible residues in the HNH construct (green) measured by CPMG relaxation dispersion NMR (red spheres) and the theoretical allosteric pathway (orange, transparent) optimizing the overall correlation between HNH residues at the RuvC (blue) and REC2 (gray) interfaces. A significant number of the sites identified by NMR are within the top 10 optimal pathways calculated from MD trajectories, suggesting that the experimentally measured dynamic pathway spanning the HNH domain correlates well with the computationally derived pathway for optimal information transfer.
A recent experimental study has further suggested that REC2 is critical in regulating the rearrangements of the DNA for double strand cleavage via the HNH and RuvC nuclease domains.50 Taken together, these findings strongly support the outcomes of the NMR experiments reported here, suggesting that the dynamic pathway spanning the isolated HNH domain is responsible for the information transfer between RuvC and REC2.
To gain insight into the allosteric signaling pathway within full-length Cas9, the complete CRISPR-Cas9 system in complex with the nucleic acids5 was subjected to extensive computational analyses that are suitable for the detection of allosteric effects.46,51-53 We combined correlation analyses and network models derived from graph theory to determine the most relevant pathways across HNH communicating RuvC with REC2. The computed pathways are composed by residue–to–residue steps that optimize the overall correlation (i.e., the momentum transport) between amino acids 789/794 and 841/858 (belonging to HNH but adjacent to RuvC and REC2, respectively). This yields an estimation of the principal channels of communication between RuvC and REC2. Interestingly, the pathway that maximizes the dynamic transmission between RuvC and REC2 through HNH (Figure 4, orange spheres) agrees remarkably well with the pathway experimentally identified in the HNH construct via CPMG relaxation dispersion (Figure 4, red spheres). Residues belonging to the computational pathway are G792*, Q794*, K797*, E798*, Y812, L813, Y814, L816*, Q817*, N818*, G819, R820*, D825, 1830, V838*, D839, H840*, 1841*, V842, P843†, Q844†, N854, K855*, V856, L857, T858, R859†, S860†, D861†,K862†; where the asterisk indicates that they also show slow dynamics in the HNH construct (as experimentally identified via CPMG relaxation dispersion and R1R2 (+1.5σ), Tables S1-S2) and the dagger indicates residues unassigned by NMR. Importantly, while our experimental approach is restricted to the slow dynamic residues, the computation of the optimal pathways considers motions in every possible timescale within the simulated window. Therefore, differences between the two approaches may originate from the fast-dynamic component present in the simulated pathways. On the other hand, several differences between the relaxation dispersion network and computationally derived network are due to missing NMR assignments for these residues (daggers).
These missing assignments are, in some cases, due to exchange broadening of the NMR signal, indicative of a highly flexible site. Also of note are the many sites of NMR-detected ms motions directly adjacent to the MD optimized pathway. Examples from the MD (NMR) pathways are 1) Y812 – Y814 (Y815); 2) D825, 1830 (E827, L828, D829); 3) D839 (V838, H840); and 4) N854, V856, L857, T858 (D853, K855). This consensus between the dynamic pathways experimentally observed in the HNH construct by NMR and in full-length Cas9 by MD indicates that the REC2–HNH–RuvC communication channel is conserved in the full-length enzyme.
To establish the functional role of the allosteric pathway, we performed mutational studies. We introduced alanine mutations into all residues belonging to the computational pathway that also show ms dynamics in CPMG relaxation dispersion studies (G792, Q794, K797, E798, L816, Q817, N818, R820, V838, H840, 1841, K855). We then computed the communication pathways between amino acids 789/794 and 841/858, analogously to the WT system (Figure 4). As a result, the communication pathway is disrupted, loosening the communication in the core of HNH (Figure S5A). We also mutated all residues that experimentally show ms dynamics (Table S2), confirming the disruption of the allosteric pathway (Figure S5B). Previous biochemical studies of systematic alanine scanning of the positively charged residues in the HNH domain provide further insight into how the disruption of the allosteric pathway can affects Cas9 function.54 In particular, alanine mutation of two residues, K855 and K810, modulate off-target cleavage of semi-complementary DNAs. Notably, K855 is shown here to be a key element of the allosteric pathway, as identified by both computations and CPMG relaxation dispersion (Figure 4). This indicates that by substituting this key allosteric residue, the selectivity of CRISPR-Cas9 is affected and highlights the potential for modulation of allosteric communication to aid in the design of improved CRISPR-Cas9 systems. This hypothesis, which is also supported by previous experiments41 warrants further investigation by NMR and molecular simulations and is currently underway in our laboratories.
Conformational dynamics of HNH in full-length Cas9
In order to compare the conformational dynamics of this novel HNH construct with those of the full-length Cas9, and to further interpret the outcomes of solution NMR experiments, we performed all-atom MD simulations of the isolated HNH domain (starting from the model structure predicted by NMR, as well as from our newly determined X-ray structure) and of the X-ray structure of the full-length Cas9.5 To access the slower timescale dynamics of the systems, we performed accelerated MD simulations, using a Gaussian accelerated MD (GaMD) method,33 which has been shown to describe the μs and ms dynamics of CRISPR-Cas9 quite well.14, 35-36 Indeed, while classical MD can detect fast stochastic motions responsible for spin relaxation, more sophisticated methods that enhance the sampling of the configurational space are required to access the slower motion quantified by solution NMR. Accelerated MD is a biased-potential method,55 which adds a boost potential to the potential energy surface (PES), effectively decreasing the energy barriers separating low-energy states, thus accelerating the occurrence of slower dynamic events. As shown by several independent reports, the method accurately reproduces the slow dynamics captured by solution NMR in biomolecular systems,28-30 therefore providing comparison with the experimental results reported here.
The simulated trajectories have been analyzed to compare the conformational dynamics of HNH in its isolated form and in full-length Cas9. We employed Principal Component Analysis (PCA), an established method to extract the dominant collective motions from the fluctuations of a simulated ensemble. By performing PCA, the dynamics of HNH along the first principal mode of motion – usually referred as “essential dynamics”56 – reveals remarkable similarities in the full-length Cas9 and in the isolated form, indicating that the conformational dynamics of HNH in full-length Cas9 are preserved in the isolated domain (Figures S6-S7). Interestingly, the residues of HNH that experimentally display ms dynamics (i.e., as captured from the CPMG relaxation dispersion and R1R2 (+1.5σ) measurements) are characterized by short amplitude motions in both the isolated form of HNH and when embedded in the full-length Cas9. Notably, these residues form a contiguous dynamic pathway, which is also found through network analysis (Figure 4). This indicates that the allosteric pathway identified through CPMG relaxation dispersion and network analysis is aligned to the Principal Mode of motion derived from the simulated ensemble.
Analysis of the root mean square fluctuations (RMSF) of individual Cα atoms further shows that the residues with slow timescale motions (experimentally identified via NMR) display small fluctuations in the simulations of both the isolated HNH and in the full-length Cas9 (Figure S8). This indicates that short amplitude motions and small fluctuations are conserved in the regions that form a continuous ms dynamic pathway connecting REC2–HNH–RuvC (Figure 4). In this respect, it is important to note that short amplitude motions, as well as small fluctuations, do not directly correspond to slow time scale dynamical motions. However, the consensus observed in both the HNH construct and within the full-length Cas9 indicates similar intrinsic dynamics along the pathway connecting REC2–HNH–RuvC, which has been experimentally derived via NMR (Figure 4). Inspection of the conformational ensemble accessed during the simulations reveals that the isolated HNH domain resembles the ensemble of the full-length system overall, with a remarkable similarity in terms of short amplitude motions and low fluctuations for the residues within the REC2–HNH–RuvC pathway (Figure S9). Overall, the analysis of the conformational dynamics shows that the HNH construct maintains the fold observed in full-length Cas9 in both the NMR structure based on the 1H-15N-HSQC data and the X-ray structure of the isolated HNH domain. This supports the connection between conformational dynamics captured via solution NMR and those of HNH inside full-length Cas9.
Simulated ensemble and NMR experiments
To gain insight into how well the structural and dynamical features captured by GaMD simulations represent the NMR experiments at the molecular level, the simulated trajectories were used to compute NMR chemical shifts with the SHIFTX2 code.57 We detected excellent sequence-specific agreement between predicted and experimental Cα, Cβ, and CO chemical shifts for the isolated HNH domain (Figure 5). The experimental distributions of backbone amide groups also display remarkable agreement between experimental and simulated values (Figure S10). This is a strong indication that the GaMD ensemble properly represents the NMR experiments at a molecular level. Another important aspect of these simulations is the similarity of HNH in full-length Cas9 and its isolated form. Figure 5 shows that these forms of HNH display very similar Cα, Cβ, and CO chemical shifts as well, indicating that HNH presents similar spectral trends when it is isolated or in full-length Cas9. This is therefore a further indication that the structural dynamics of the HNH construct predicted by NMR are comparable to those of HNH in full-length Cas9, supporting the comparison performed here. Importantly, the observed agreement between the computed and experimental spectra is also observed in the simulation replicas (Figure S10). Finally, to account for fluctuations in the Cα, Cβ, and CO chemical shifts during the simulated ensemble, we computed carbon chemical shifts over a set of static models derived from the GaMD trajectories (details are reported in the SI). We observe low fluctuations in the carbon chemical shifts within the simulated ensemble (Figure S11), retaining also excellent agreement with experimental measurements (Figure S12)
Figure 5.

Experimental vs. simulated carbon chemical shifts. Experimental and simulated NMR chemical shifts of 13Cα (top), 13Cβ (center), and 13CO (bottom) plotted for each residue in the HNH domain. Data in each plot compare the experimental chemical shifts (green circles) with those calculated from simulations of the isolated HNH domain (blue diamonds) and of the full-length Cas9 complex (black circles). All simulated spectra were computed as described in Methods utilizing GaMD trajectories.
Discussion
The power of the CRISPR-Cas9 system is its ability to perform targeted genome editing in vivo with high efficiency and increasingly improved specificity.41, 54, 58-59 In this system, an intriguing allosteric communication has been suggested to propagate the DNA binding signal across the HNH and RuvC nuclease domains to facilitate their concerted cleavage of the two DNA strands.9, 17 In this process, the intrinsic flexibility of the catalytic HNH domain would regulate the information transfer, exerting conformational control. Here, solution NMR experiments are used to capture the intrinsic motions responsible for the allosteric signaling across the HNH domain. We use a novel engineered construct of HNH that maintains the conformation and flexibility of full-length Cas9 to reveal the existence of a ms timescale dynamic pathway. This network spans the HNH domain from the region interfacing the RuvC domain and propagates up to the REC lobe at the level of the REC2 region (Figure 4). In-depth analysis of the allosteric signaling within the full-length Cas9 has been performed, by employing theoretical approaches that are suited for the detection of allosteric effects.46, 51-53 Indeed, the dynamic pathway experimentally observed in the HNH construct is conserved in the full-length Cas9 (Figure 4), confirming the existence of a communication channel between REC2–HNH–RuvC. This continuous pathway confirms the direct communication between the two catalytic domains, originally identified by the experimental work of Sternberg9 and supported by MD simulations,17 and also discloses their connection to the REC2 region. In this respect, single molecule FRET experiments have indicated that REC2 is critical for the activation of HNH through an allosteric mechanism that also involves the REC3 region.41 Accordingly, in the fully activated complex, the REC3 region would modulate the motions of the neighboring REC2, which in turn contacts HNH and regulates its access to the scissile phosphate. By doing so, the REC region would act as a “sensor” for the formation of a RNA:DNA structure prone to DNA cleavage, transferring the DNA binding information to the catalytic HNH domain in an allosteric manner. A tight dynamical interplay between REC2–REC3 and HNH has also been detected via MD simulations of the fully activated CRISPR-Cas9 complex, revealing that highly coupled motions of these regions are at the basis of the activation of HNH for DNA cleavage.49 A recent important contribution further suggested that REC2 regulates the rearrangements of the DNA to attain double strand cleavages via the HNH and RuvC nucleases.50 Altogether, these experimental outcomes strongly support the finding of a continuous dynamic pathway spanning HNH from RuvC to REC2, and suggest its functional role for the allosteric transmission. To further investigate the motions associated with allosteric signaling, the conformational dynamics of the HNH domain was investigated by means of accelerated MD simulations, which can probe long-timescale μs and ms motions in remarkable agreement with NMR experiments.28-30 Analysis of these conformational dynamics indicates that the HNH construct maintains the overall fold observed in full-length Cas9, and indicates conserved short amplitude motions and low fluctuations in the regions that form a continuous ms dynamic pathway connecting REC2–HNH–RuvC. Taken together, these computational outcomes suggest that the intrinsic conformational dynamics experimentally identified in the HNH construct reasonably resemble the dynamics of HNH in the full complex, supporting the connection between the two systems. Finally, mixed machine learning and structure-based prediction of the NMR chemical shifts from the simulated trajectories have also revealed the agreement between experiments and computations, indicating that the structural/dynamic features derived via GaMD simulations represent the experimental results at the atomic and molecular level.
Overall, by combining solution NMR experiments and MD simulations, we identified the dynamic pathway for information transfer across the catalytic HNH domain of the CRISPR-Cas9 system. This pathway, which spans HNH from the RuvC nuclease interface up to the REC2 region in the full-length Cas9, is suggested to be critical for allosteric transmission, propagating the DNA binding signal across the recognition lobe and the nuclease domains (HNH and RuvC) for concerted cleavage of the two DNA strands. This study also represents the first step toward a complete mapping of the allosteric pathway in Cas9 through solution NMR experiments. In this respect, despite modern experimental practices such as perdeuteration,60 transverse relaxation-optimized spectroscopy (TROSY),61 sparse isotopic labeling,62 and 15N-detection,63 the complete characterization of the slow dynamical motions responsible for the allosteric signaling has remained challenging, due to the size of the polypeptide chain of the Cas9 protein (~ 160 kDa). Future investigations – reliant upon ongoing experiments and computations in our research groups – will include the investigation of the information transfer between HNH and RuvC and the allosteric role of their flexible interconnecting loops.9, 17 Further, our joint NMR/MD investigations are being employed to understand the role of the recognition region within the allosteric activation. This is of key importance, since mutations within the REC lobe – at distal sites with respect to HNH – can control the activation of HNH and the specificity of the enzyme toward on-target DNA sequences.41, 54, 58-59 As such, by providing fundamental understanding of the intrinsic allosteric signaling within the catalytic HNH domain, the present study poses the basis for the complete mapping of the allosteric pathway in Cas9 and its role in the on-target specificity, helping engineering efforts aimed at improving the genome editing capability of the Cas9 enzyme.
Materials and Methods
Protein Expression and Purification.
The HNH domain of S. pyogenes Cas9 (residues 775-908) was engineered into a pET15b vector with an N-terminal His6-tag and expressed in Rosetta(DE3) cells in M9 minimal medium containing MEM vitamins, MgSO4 and CaCl2. Cells were induced with 0.5 mM IPTG after reaching an OD600 of 0.8 – 1.0 and grown for 16 – 18 hours at 22 °C post induction. The cells were harvested by centrifugation, resuspended in a buffer containing 20 mM HEPES, 500 mM KCl, and 5 mM imidazole at pH 8.0, lysed by ultrasonication and purified on a Ni-NTA column. NMR samples were dialyzed into a buffer containing 20 mM HEPES, 80 mM KCl, 1 mM DTT and 7.5% (v/v) D2O at pH 7.4.
X-ray Crystallography.
Following TEV cleavage of the His6-tag, HNH was subsequently purified by HiPrep 16/60 Sephacryl 100 S-100 HR gel filtration chromatography. Crystals were obtained with sitting drop vapor diffusion at room temperature with 48 mg/mL HNH 1:1 with the Molecular Dimensions Morpheus I Screen condition E4 (0.1 M mixture of [imidazole and MES] pH 6.5, 25% (v/v) mixture of [2-methyl-2,4-pentanediol, PEG1000, and PEG3350], and 0.3 M mixture of [diethylene glycol, triethylene glycol, tetraethylene glycol, and pentaethlyene glycol]). Diffraction data were collected on a Rigaku MicroMax-003i sealed tube X-ray generator with a Saturn 944 HG CCD detector and processed and scaled using XDS64 and Aimless in the CCP4 program suite.65 The HNH domain from full-length S. pyogenes Cas9 was used for molecular replacement (PDB: 4UN3)5 with Phaser in the PHENIX software package.66 Iterative rounds of manual building in Coot67 and refinement in PHENIX yielded the final HNH domain structure.
NMR Spectroscopy.
NMR spin relaxation experiments were carried out at 600 and 850 MHz on Bruker Avance NEO and Avance III HD spectrometers, respectively. All NMR spectra were processed with NMRPipe 68 and analyzed in SPARKY.69 Backbone chemical shift data was uploaded to the CS23D server for secondary structure calculations, and a fragment library including the structure of full-length Cas9 (PDB: 4UN5) was used. Carr-Purcell-Meiboom-Gill (CPMG) NMR experiments were adapted from the report of Palmer and coworkers,70 and performed at 25 °C with a constant relaxation period of 40 ms, a 2.0 second recycle delay, and τcp points of 0.555, 0.625, 0.714, 0.833, 1.0, 1.25, 1.5, 1.667, 2.5, 5, 10, and 20 ms. Relaxation dispersion profiles were generated by plotting R2 vs. 1/τcp and exchange parameters were obtained from fits of these data carried out with in-house scripts and in RELAX under the R2eff, NoRex, Tollinger (TSMFK01), and Carver-Richards (CR72 and CR72-Full) models.47, 71 Two-field relaxation dispersion data were fit simultaneously and uncertainty values were obtained from replicate spectra (see the Supporting Information, SI). Longitudinal and transverse relaxation rates were measured with relaxation times of 0(x2), 40, 80, 120, 160(x2), 200, 240, 280(x2), 320, 360, and 400 ms for T1 and 4.18, 8.36(x2), 12.54, 16.72, 20.9(x2), 25.08(x2), 29.26, 33.44, 37.62, and 41.8 ms for T2. Peak intensities were quantified in Sparky and the resulting decay profiles were analyzed in Mathematica with errors determined from the fitted parameters. Steady-state 1H-[15N] NOE were measured with a 6 second relaxation delay followed by a 3 second saturation (delay) for the saturated (unsaturated) experiments. All relaxation experiments were carried out in a temperature-compensated interleaved manner. Model-free analysis using the Lipari-Szabo formalism was carried out on dual-field NMR data in RELAX with fully automated protocols.47
Computational Structural Models.
MD simulations were performed on several model systems of the CRISPR-Cas9 complex and isolated HNH domain. MD simulations of the CRISPR-Cas9 complex were based on the X-ray structure of the full-length wild-type (WT) Cas9 protein, solved at 2.58 Å resolution (PDB code: 4UN3).5 This system comprises a total of 1368 amino acids, combined with RNA and DNA (details are in the SI). This nucleoprotein complex was used to simulate the CRISPR-Cas9 complex as WT, and with alanine mutations. Two mutated model systems were built by substituting alanine at key residues within the allosteric pathway identified in this work. The first mutant system substitutes alanine at residues belonging to the computational pathway that also show slow dynamics via CPMG relaxation dispersion (G792, Q794, K797, E798, L816, Q817, N818, R820, V838, H840, 1841, K855; Figure 4), while a second mutant system introduces alanine mutations at all residues that experimentally show ms dynamics (Table S2). MD simulations of the isolated HNH domain were based on both the NMR model derived from the 1H-15N-HSQC data (Figure 1A, close-up view) and the X-ray structure of HNH solved in this work at 1.3 Å resolution (Figure 2). Both systems are composed of residues 775–908 and align well to the HNH domain in the X-ray structure of the WT CRISPR-Cas9 (details are in the SI).5 All model systems were embedded in explicit water, adding Na+ counter-ions to neutralize the total charge. A total of ~220,000 atoms and a box size of ~145 x 110 x 147 Å3 has been reached for the CRISPR-Cas9 complex; while ~25,000 atoms and a box size of ~72 x 62 x 60 Å3 characterize the isolated HNH domain from both NMR and X-ray.
Molecular Dynamics (MD) Simulations.
The above-mentioned model systems were equilibrated through conventional MD. We employed the Amber ff12SB force field, which includes the ff99bsc0 corrections for DNA72 and the ff99bscO+χOL3 corrections for RNA.73-74 This force field model has been shown to properly describe the conformational dynamics of CRISPR-Cas9 during extensive MD simulations, performed using both classical17, 35, 75 and accelerated MD.34, 36 It has also been employed to perform multi–μ s continuous MD simulations,76 preserving the overall fold of the structure and capturing critical conformational changes. The TIP3P model was employed for waters.77 Hydrogen atoms were added assuming standard bond lengths and constrained to their equilibrium position with the SHAKE algorithm. Temperature control (300 K) was performed via Langevin dynamics,78 with a collision frequency γ = 1. Pressure control was accomplished by coupling the system to a Berendsen barostat,79 at a reference pressure of 1 atm and a relaxation time of 2 ps. All simulations have been carried out through a well-established protocol described in the SI. MD simulations were carried out in the NVT ensemble, collecting ~100 ns for each system (for a total of ~500 ns of production runs). These well-equilibrated systems have been used as the starting point for Gaussian accelerated MD (GaMD, details below). All simulations were performed with the GPU version of AMBER 16.80
Gaussian Accelerated MD Simulations (GaMD).
Accelerated MD (aMD) is an enhanced sampling method that adds a boost potential to the Potential Energy Surface (PES), effectively decreasing the energy barriers and accelerating transitions between low-energy states.55 The method extends the capability of MD simulations over long timescales, capturing slow μs and ms motions in excellent comparability with solution NMR experiments.28-30 Here, we applied a novel and robust aMD method, namely a Gaussian aMD (GaMD),33 which uses harmonic functions to construct a boost potential that is adaptively added to the PES, enabling unconstrained enhanced sampling and simultaneous reweighting of the canonical ensemble.
Considering a system with N atoms at positions , when the system potential is lower than a threshold energy E, the energy surface is modified by a boost potential as:
| [1] |
| [2] |
where k is the harmonic force constant. The two adjustable parameters E and k are automatically determined by applying the following three criteria. First, for any two arbitrary potential values and found on the original energy surface, if , ΔV should be a monotonic function that does not change the relative order of the biased potential values, i.e. . Second, if , the potential difference observed on the smoothed energy surface should be smaller than that of the original, i.e. . By combining the first two criteria with Eqn [1] and [2]:
| [3] |
where Vmin and Vmax are the system minimum and maximum potential energies. To ensure that Eqn. [4] is valid, k must satisfy . By defining , then 0 < k ≤1. Lastly, the standard deviation of ΔV must be narrow enough to ensure accurate reweighting using cumulant expansion to the second order: σΔV = k(E – Vavg)σV ≤ σ0, where Vavg and σV are the average and standard deviation of the system potential energies, σΔV is the standard deviation of ΔV and σ0 as a user-specified upper limit (e.g., 10 kBT) for accurate reweighting. When E is set to the lower bound, E = Vmin, according to Eqn. [4], k0 can be calculated as:
| [4] |
Alternatively, when the threshold energy E is set to its upper bound , k0 is:
| [5] |
if is calculated between 0 and 1. Otherwise, k0 is calculated using Eqn. [4], instead of being set to 1 directly as described in the original paper.33 GaMD yields a canonical average of an ensemble by reweighting each point in the configuration space on the modified potential by the strength of the Boltzmann factor of the bias energy, exp [βΔV(rt(i))] at that particular point.
Based on extensive tests on the CRISPR-Cas9 system,14, 36, 81 the system threshold energy is E = Vmax for all GaMD simulations. The boost potential was applied in a dual-boost scheme, in which two acceleration potentials are applied simultaneously to the system: (i) the torsional terms only and (ii) across the entire potential. A timestep of 2 fs was used. The maximum, minimum, average, and standard deviation values of the system potential (Vmax, Vmin, Vavg and σV) were obtained from an initial ~12 ns NPT simulation with no boost potential. GaMD simulations were applied to the CRISPR-Cas9 complex and our HNH domain construct. Each GaMD simulation proceeded with a ~50 ns run, in which the boost potential was updated every 1.6 ns, thus reaching equilibrium. Finally, ~400 ns of GaMD simulations were carried out in the NVT ensemble for each system in two replicas. Considering that we have simulated three model systems of the CRISPR-Cas9 complex (one system as WT and two systems including alanine mutations, as described above) and two model systems of the isolated HNH domain (as derived from NMR and X-ray crystallography), a total of ~2.4 μs of GaMD for the CRISPR-Cas9 complex and ~1.6 μs of GaMD for the isolated HNH domain were completed. Notably, the simulation length of ~400 ns per replica has shown to exhaustively explore the conformational space of the CRISPR-Cas9 system.14, 81
Determination of the Allosteric Pathways across the HNH domain.
The allosteric pathway for information transfer has been investigated by employing correlation analysis and graph theory.46, 51-53 First, the generalized correlations (GCij), which capture non-collinear correlations between pairs of residues i and j, are computed (details are in the SI).82 In a second phase, the GCij are used as a metric to build a dynamical network model of the protein.53 In this model, the protein amino acids residues constitute the nodes of the dynamical network graph, connected by edges (residue pair connection). Edge lengths, i.e., the internode distances in the graph, are defined using the GCij coefficients according to:
| [6] |
In the present work, two nodes have been considered connected if any heavy atom of the two residues is within 5 Å of each other (i.e., distance cutoff) for at least the 70 % of the simulation time (i.e., frame cutoff). This leads to the definition of a set of elements wij of the graph. In the third phase of the protocol, the optimal pathways for the information transfer between two nodes (i.e., two amino acids) are defined using the Dijkstra algorithm, which finds the roads, composed by internode connections, that minimize the total distance (and therefore maximize the correlation) between amino acids. In the present study, this protocol was applied on the trajectories of the full-length Cas9 simulated for ~400 ns of GaMD simulations and averaged over two replicas. The Dijkstra algorithm was applied between the amino acids 789/794 and 841/858, which belong to HNH and are located at the interface with RuvC and REC2, respectively. As a result, the routes that maximize the correlation between amino acids 789/794 and 841/858 are identified, providing residue–to–residue pathways that optimize the correlations (i.e., the momentum transport). With the optimal motion transmission pathway, the following 50 sub-optimal information channels where computed and accumulated and plotted on the 3D structure (Figure 4), to account for the contribution of the most likely sub-optimal pathways. To gain further insights into the REC2–HNH–RuvC signal transfer, this analysis was extended to the catalytic residues of the RuvC domain. Specifically, the pathways that maximize the dynamic correlation were computed between the amino acids of HNH located at the interface with REC2 (841/858) and the RuvC catalytic residues (S15, H983, D986, D10 and E762). Finally, the analysis of the signaling pathways across HNH was also performed on two CRISPR-Cas9 systems that introduce alanine mutations of the key residues forming the allosteric pathway identified in this work (G792, Q794, K797, E798, L816, Q817, N818, R820, V838, H840, I841, K855; Figure 4), and that experimentally display slow dynamics (Table S2).
Supplementary Material
Acknowledgments
We thank Prof. Martin Jinek for the WT Cas9 plasmid and Prof. Patrick Loria for helpful discussion of spin relaxation. We thank Prof. Luca Mollica for useful discussions. This work was supported by start-up funds from Brown University and funds from the COBRE Center for Computational Biology of Human Disease (P20GM109035) to GPL. This material is also based upon work supported by the National Science Foundation under Grant No. CHE-1905374, awarded to GP. Computer time for MD has been awarded by XSEDE via grant TG-MCB160059 (to GP).
Footnotes
Supporting Information
Experimental procedures, circular dichroism spectra, NMR relaxation parameters, principal component analysis, MD simulations, and simulated NMR chemical shift distributions are available free-of-charge at pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Hsu PD; Lander ES; Zhang F, Development and Aplications of CRISPR–Cas9 for Genome Engineering. Cell 2014, 1576, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna JA; Charpentier E, Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346 (6213), 1258096. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M; Chylinski K; Fonfara I; Hauer M; Doudna JA; Charpentier E, A Programmable Dual–RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M; Jiang F; Taylor DW; Sternberg SH; Kaya E; Ma E; Anders C; Hauer M; Zhou K; Lin S; Kaplan M; Iavarone AT; Charpentier E; Nogales E; Doudna JA, Structures of Cas9 Endonucleases Reveal RNA-mediated Conformational Activation. Science 2014, 343, 12479971–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anders C; Niewoehner O; Duerst A; Jinek M, Structural Basis of PAM-dependent Target DNA Recognition by the Cas9 Endonuclease. Nature 2014, 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimasu H; Ran FA; Hsu PD; Konemann S; Shehata SI; Dohmae N; Ishitani R; Zhang F; Nureki O, Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang FG; Taylor DW; Chen JS; Kornfeld JE; Zhou KH; Thompson AJ; Nogales E; Doudna JA, Structures of a CRISPR-Cas9 R–loop Complex Primed for DNA Cleavage. Science 2016, 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huai G; Li G; Yao R; Zhang Y; Cao M; Kong L; Jia C; Yuan H; Chen H; Lu D; Huang Q, Structural Insights into DNA Cleavage Activation of CRISPR–Cas9 System. Nat. Commun 2017, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sternberg SH; LaFrance B; Kaplan M; Doudna JA, Conformational Control of DNA Target Cleavage by CRISPR–Cas9. Nature 2015, 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagdas YS; Chen JS; Sternberg SH; Doudna JA, A Conformational Checkpoint between DNA Binding and Cleavage by CRISPR–Cas9. Sci. Adv 2017, 3, eaao002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osuka S; Isomura K; Kajimoto S; Komori T; Nishimasu H; Shima T; Nureki O; Uemura S, Real–time observation of flexible domain movements in Cas9. EMBO J doi: 10.1101/122069 2018, 37 (10), e96941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raper AT; Stephenson AA; Suo Z, Functional Insights Revealed by the Kinetic Mechanism of CRISPR/Cas9. J Am Chem Soc 2018, 140, 2971–2984. [DOI] [PubMed] [Google Scholar]
- 13.Shibata M; Nishimasu H; Kodera N; Hirano S; Ando T; Uchihashi T; Nureki O, Real–space and real–time dynamics of CRISPR–Cas9 visualized by high-speed atomic force microscopy. Nat Commiun 2017, 8 (1), 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palermo G; Miao Y; Walker RC; Jinek M; McCammon JA, CRISPR–Cas9 Conformational Activation as Elucidated from Enhanced Molecular Simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuo Z; Liu J, Structure and Dynamics of Cas9 HNH Domain Catalytic State. Sci Rep 2017, 7 (1), 17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palermo G; Miao Y; Walker RC; Jinek M; McCammon JA, Striking Plasticity of CRISPR–Cas9 and Key Role of Non–target DNA, as Revealed by Molecular Simulations. ACS Cent. Sci 2016, 2, 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palermo G; Ricci CG; Fernando A; Basak R; Jinek M; Rivalta I; Batista VS; McCammon JA, Protospacer Adjacent Motif–Induced Allostery Activates CRISPR–Cas9. J. Am. Chem. Soc 2017, 139, 16028–16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JS; Doudna JA, The Chemistry of Cas9 and its CRISPR Colleagues. Nat. Rev. Chem 2017, 1, 78. [Google Scholar]
- 19.Kern D; Zuiderweg ER, The role of dynamics in allosteric regulation. Curr Opin Struct Biol 2003, 13 (6), 748–57. [DOI] [PubMed] [Google Scholar]
- 20.Capdevila DA; Braymer JJ; Edmonds KA; Wu H; Giedroc DP, Entropy redistribution controls allostery in a metalloregulatory protein. Proc Natl Acad Sci U S A 2017, 114 (17), 4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wand AJ, Bringing disorder and dynamics in protein allostery into focus. Proc Natl Acad Sci U S A 2017, 114 (17), 4278–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzeng SR; Kalodimos CG, Protein Activity Regulation by Conformational Entropy. Nature 2012, 488, 236. [DOI] [PubMed] [Google Scholar]
- 23.Lisi GP; Loria JP, Solution NMR Spectroscopy for the Study of Enzyme Allostery. Chem. Rev 2016, 116 (11), 6323–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nussinov R, Introduction to Protein Ensembles and Allostery. Chem Rev 2016, 116, 6263–6266. [DOI] [PubMed] [Google Scholar]
- 25.Wodak SJ; Paci E; Dokholyan NV; Berezovsky IN; Horovitz A; Li J; Hilser VJ; Bahar I; Karanicolas J; Stock G; Hamm P; Stote RH; Eberhardt J; Chebaro J; Dejaegere A; Cecchini M; Changeux JP; Bolhuis PJ; Vreede J; Faccioli P; Orioli S; Ravasio R; Yan L; Brito C; Wyart M; Gkeka P; Rivalta I; Palermo G; McCammon JA; Panecka–Hofman J; Wade RC; Di Pizio A; Niv MY; Nussinov R; Tsai CJ; Jang H; Padhorny D; Kozakov D; McLeish T, Allostery in its many disguises: from theory to applications. Structure 2019, 27 (4), 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo J; Zhou HX, Protein Allostery and Conformational Dynamics. Chem Rev 2016, 116 (11), 6503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dokholyan NV, Controlling Allosteric Networks in Proteins. Chem Rev 2016, 116 (11), 6463–87. [DOI] [PubMed] [Google Scholar]
- 28.Markwick PRL; Bouvignies G; Blackledge M, Exploring Multiple Timescale Motions in Protein GB3 using Accelerated Molecular Dynamics and NMR Spectroscopy. J. Am. Chem. Soc 2007, 129, 4724–4730. [DOI] [PubMed] [Google Scholar]
- 29.Mukrasch MD; Markwick P; Biernat J; von Bergen M; Bernado P; Griesinger C; Mandelkow E; Zweckstetter M; Blackledge M, Highly Populated Turn Conformations in Natively Unfolded Tau Protein Identified from Residual Dipolar Couplings and Molecular Simulation. J. Am. Chem. Soc 2007, 129, 5235–5243. [DOI] [PubMed] [Google Scholar]
- 30.Salmon L; Pierce L; Grimm A; Ortega R, J.-L.; Mollica L; Jensen MR; van Nuland N; Markwick P; McCammon JA; Blackledge M, Multi–timescale Conformational Dynamics of the SH3 Domain of CD2–Associarws Protein using NMR Spectroscopy and Accelerated Molecular Dynamics. Angew. Chem. Int. Ed 2012, 51, 6103–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dedmon MM; Lindorff-Larsen K; Christodoulou J; Vendruscolo M; Dobson CM, Mapping long–range interactions in alpha–synuclein using spin–label NMR and ensemble molecular dynamics simulations. J Am Chem Soc 2005, 127 (2), 476–7. [DOI] [PubMed] [Google Scholar]
- 32.Best RB; Vendruscolo M, Determination of protein structures consistent with NMR order parameters. J Am Chem Soc 2004, 126 (26), 8090–1. [DOI] [PubMed] [Google Scholar]
- 33.Miao Y; Feher VA; McCammon JA, Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theor. Comput 2015, 11, 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palermo G; Miao Y; Walker RC; Jinek M; McCammon JA, CRISPR–Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc Natl Acad Sci UJ S A 2017, 114 (28), 7260–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palermo G, Structure and Dynamics of the CRISPR–Cas9 Catalytic Complex. J Chem Inf Model 2019, 59 (5), 2394–2406. [DOI] [PubMed] [Google Scholar]
- 36.Ricci CG; Chen JS; Miao Y; Jinek M; Doudna JA; McCammon JA; Palermo G, Deciphering Off–Target Effects in CRISPR–Cas9 through Accelerated Molecular Dynamics. ACS Cent. Sci 2019, 5, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao YL; McCammon JA, Graded activation and free energy landscapes of a muscarinic G–protein–coupled receptor. P Natl Acad Sci USA 2016, 113 (43), 12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miao YL; McCammon JA, Mechanism of the G–protein mimetic nanobody binding to a muscarinic G–protein–coupled receptor. P Natl Acad Sci USA 2018, 115 (12), 3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belato HB; East KW; Lisi GP, (1)H, (13)C, (15)N backbone and side chain resonance assignment of the HNH nuclease from Streptococcus pyogenes CRISPR–Cas9. Biomiol Nmr Assign 2019, 13 (2), 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wishart DS; Arndt D; Berjanskii M; Tang P; Zhou J; Lin G, CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucl. Acids Res. 2008, 36, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JS; Dagdas YS; Kleinstiver BP; Welch MM; Harrington LB; Sternberg SH; Joung JK; Yildiz A; Doudna JA, Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 2017, 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kneller JM; Bracken C, An Effective Method for the Discrimination of Motional Anisotropy and Chemical Exchange. J. Am. Chem. Soc 2002, 124, 1852–1853. [DOI] [PubMed] [Google Scholar]
- 43.Frederick KK; Marlow MS; Valentine KG; Wand AJ, Conformational entropy in molecular recognition by proteins. Nature 2007, 448 (7151), 325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tzeng SR; Kalodimos CG, Dynamic Activation of an Allosteric Regulatory Protein. Nature 2009, 462, 368. [DOI] [PubMed] [Google Scholar]
- 45.Whittier SK; Hengge AC; Loria JP, Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 2013, 341 (6148), 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lisi GP; East KW; Batista VS; Loria JP, Altering the allosteric pathway in IGPS suppresses millisecond motions and catalytic activity. Proc Natl Acad Sci U S A 2017, 114 (17), E3414–E3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bieri M; d’Auvergne EJ; Gooley PR, relaxGUI: A New Software for Fast and Simple NMR Relaxation Data Analysis and Calculation of ps–ns and us Motion of Proteins. J. Biomol. NMR 2011, 50, 147–155. [DOI] [PubMed] [Google Scholar]
- 48.Sethi A; Eargle J; Black AA; Luthey–Schulten Z, Dynamical networks in tRNA:protein complexes. Proc Natl Acad Sci U S A 2009, 106 (16), 6620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palermo G; Chen JS; Ricci CG; Rivalta I; Jinek M; Batista VS; Doudna JA; McCammon JA, Key Role of the REC Lobe during CRISPR–Cas9 Activation by ‘Sensing’, ‘Regulating’, and ‘Locking’ the Catalytic HNH Domain. Q. Rev. Biophys 2018, 51, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sung K; Park J; Kim J; Lee NK; Kim SK, Target Specificity of Cas9 Nuclease via DNA Rearrangement Regulated by the REC2 Domain. J Am Chem Soc 2018, 140, 7778–7781. [DOI] [PubMed] [Google Scholar]
- 51.Rivalta I; Sultan MM; Lee NS; Manley GA; Loria JP; Batista VS, Allosteric pathways in imidazole glycerol phosphate synthase. Proceedings of the National Academy of Sciences of the United States of America 2012, 109 (22), E1428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negre CFA; Hendrickson H; Rhitankar Pal R; Rivalta I; Ho J; Batista VS, Eigenvector Centrality Distribution for Characterization of Protein Allosteric Pathways. P Natl Acad Sci JSA 2018, 115, 12201–12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sethi A; Eargle J; Black AA; Luthey-Schulten Z, Dynamical networks in tRNA: protein complexes. P Natl Acad Sci JSA 2009, 106 (16), 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slaymaker IM; Gao L; Zetsche B; Scott DA; Yan WX; Zhang F, Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351 (6268), 84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hamelberg D; Mongan J; McCammon JA, Accelerated Molecular Dynamics: A Promising and Efficient Simulation Method for Biomolecules. J. Chem. Phys 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- 56.Amadei A; Linssen ABM; Berendsen HJC, Essential Dynamics of Proteins. Proteins 1993, 17 (4), 412–425. [DOI] [PubMed] [Google Scholar]
- 57.Han B; Liu Y; Ginzinger SW; Wishart DS, SHIFTX2: significantly improved protein chemical shift prediction. J Biomiol NMR 2001, 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kleinstiver BP; Pattanayak V; Prew MS; Tsai SQ; Nguyen NT; Zheng ZL; Joung JK, High–fidelity CRISPR–Cas9 nucleases with no detectable genome–wide off–target effects. Nature 2016, 529 (7587), 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Casini A; Olivieri M; Petris G; Montagna C; Reginato G; Maule G; Lorenzin F; Prandi D; Romanel A; Demichelis F; Inga A; Cereseto A, A highly specific spCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol 2018, 36, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venters RA; Farmer BTI; Fierke CA; Spicer LD, Characterizing the Use of Perdeuteration in NMR Studies of Large Proteins: 13C, 15N and 1H Assignments of Human Carbonic Anhydrase II. J. Mol. Biol 1996, 264, 1101–1106. [DOI] [PubMed] [Google Scholar]
- 61.Pervushin K; Riek R; Wider G; Wuthrich K, Attenuated T2 Relaxation by Mutual Cancellation of Dipole–Dipole Coupling and Chemical Shift Anisotropy Indicates an Avenue to NMR Structures of Very Large Biological Macromolecules in Solution. Proc. Natl. Acad. Sci. JSA 1997, 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tugarinov V; Kanelis V; Kay LE, Isotope Labeling Strategies for the Study of High–Molecular–Weight Proteins by Solution NMR Spectroscopy. Nat. Protoc 2006, 1 (2), 749–754. [DOI] [PubMed] [Google Scholar]
- 63.Takeuchi K; Arthanari H; Imai M; Wagner G; Shimada I, Nitrogen–detected TROSY Yields Comparable Sensitivity to Proton–Detected TROSY for Non-deuterated, Large Proteins under Physiological Salt Conditions. J. Biomiol. NMR 2016, 64, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kabsch W, XDS. Acta Cryst. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 Suite and its Current Developments. Acta Cryst. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams PD; Afonine PV; Bunkoczi G ; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse–Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: A Comprehensive Python-based System for Macromolecular Structure Solution. Acta Cryst. Sect. D 2010, (66), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emsley P, Lohkamp B, Scott WG, Cowtan KD; , Reatures and Development of Coot. . Acta Cryst. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- 69.Goddard TD; Kneller DG, SPARKY 3. 2008, University of California, San Francisco. [Google Scholar]
- 70.Loria JP; Rance M; Palmer AG 3rd, A Relaxation–Compensated Carr–Purcell–Meiboom–Gill Sequence for Characterizing Chemical Exchange by NMR Spectroscopy. J. Am. Chem. Soc 1999, 121 (10), 2331–2332. [Google Scholar]
- 71.Morin S; Linnet T; Lescanne M; Schanda P; Thompson GS; Tollinger M; Teilum K; Gagne S; Marion D; Griesinger C; Blackledge M; d’Auvergne EJ, relax: The Analysis of Biomolecular Kinetics and Thermodynamics using NMR Relaxation Dispersion Data. Bioinformatics 2014, 30 (15), 2219–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez A; Marchan I; Svozil D; Sponer J; Cheatham TE r.; Laughton, C. A.; Orozco, M., Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of Alpha/Gamma Conformers. Biophys. J. 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banas P; Hollas D; Zgarbova M; Jurecka P; Orozco M; Cheatham T. E. r. ; Sponer J; Otyepka M, Performance of Molecular Mechanics Force Fields for RNA Simulations: Stability of UUCG and GNRA Hairpins. J. Chemi. Theor. Comput 2010, 6, 3836–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zgarbova M; Otyepka M; Sponer J; Mladek A; Banas P; Cheatham TE; Jurecka P, Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. Journal of Chemical Theory and Computation 2011, 7 (9), 2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palermo G; Miao Y; Walker RC; Jinek M; McCammon JA, Striking Plasticity of CRISPR–Cas9 and Key Role of Non–target DNA, as Revealed by Molecular Simulations. ACS Cent Sci 2016, 2, 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Palermo G; Chen JS; Ricci CG; Rivalta I; Jinek M; Batista VS; Doudna JA; McCammon JA, Key role of the REC lobe during CRISPR–Cas9 activation by “sensing”, “regulating” and “locking” the catalytic HNH domain. Quarterly Review of Biophysics 2018, 51, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79 (2), 926–935. [Google Scholar]
- 78.Turq P; Lantelme F; Friedman HL, Brownian Dynamics: Its Applications to Ionic Solutions. J. Chem. Phys 1977, 66, 3039. [Google Scholar]
- 79.Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR, Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81, 3684. [Google Scholar]
- 80.Case DA; Betz RM; Botello–Smith W; Cerutti DS; Cheatham I,TE; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Homeyer N; Izadi S; Janowski P; Kaus J; Kovalenko A; Lee TS; LeGrand S; Li P; Lin C; Luchko T; Luo R; Madej B; Mermelstein D; Merz KM; Monard G; Nguyen H; Nguyen HT; Omelyan I; Onufriev A; Roe DR; Roitberg A; Sagui C; Simmerling CL; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Xiao L; M. YD; A. KP, AMBER 2016. University of California, San Francisco: 2016. [Google Scholar]
- 81.Palermo G, Structure and Dynamics of the CRISPR–Cas9 catalytic complex. J. Chem. Inf. Model 2019. [DOI] [PubMed] [Google Scholar]
- 82.Lange OF; Grubmuller H, Generalized correlation for biomolecular dynamics. Proteins–Structure Function and Bioinformatics 2006, 62 (4), 1053–1061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.