

Abstract
Semisynthetic rifamycin derivatives such as rifampicin (Rif) are first line treatments for tuberculosis and other bacterial infections. Historically, synthetic modifications made to the C-3/C-4 region of the rifamycin naphthalene core, like those seen in Rif, have yielded the biggest improvements in pharmacological properties. However, modifications found in natural product rifamycin congeners occur at other positions in the structure. The kanglemycins (Kangs) are a family of rifamycin congeners with a unique collection of natural modifications including a dimethylsuccinic acid appended to their polyketide backbone. These modifications confer activity against the single most common clinically relevant Rif resistance (RifR) mutation in the antibiotic’s target, the bacterial RNA polymerase (RNAP). Here we evaluate the in vivo efficacy of Kang A, the parent compound in the Kang family, in a murine model of bacterial peritonitis/sepsis. We then set out to improve its potency by combining its natural tailoring modifications with semisynthetic derivatizations at either its acid moiety or in the C-3/C-4 region. A collection of C-3/C-4 benzoxazino Kang derivatives exhibit improved activity against wild-type bacteria, and acquire activity against the second most common clinically relevant RifR mutation. The semisynthetic analogue 3′-hydroxy-5′-[4-isobutyl-1-piperazinyl] benzoxazino Kang A (Kang KZ) protected mice against infection with either Rif sensitive MRSA or a highly virulent RifRStaphylococcus aureus strain in a neutropenic peritonitis/sepsis model and led to reduced bacterial burdens. The compounds generated in this study may represent promising candidates for treating RifR infections.
Keywords: rifampicin; rifamycin; kanglemycin, antibiotic resistance; Staphylococcus aureus; Mycobacterium tuberculosis
Rifamycin SV, a natural product produced by Amycolatopsis mediterranei, was first used for treating tuberculosis more than half a century ago. Since that time, numerous semisynthetic derivatives of rifamycin SV have been generated in an effort to improve its pharmacological properties.1 The most important of these, rifampicin (Rif), is a cornerstone in modern treatments for tuberculosis. Rif contains a methylpiperazine group appended to the C-3 position of the rifamycin naphthalene ring system. This modification overcame the limited oral bioavailability of rifamycin SV and also improved the potency of the compound against Mycobacterium tuberculosis.2 Although the development of Rif has led to dramatic improvements in the treatment of tuberculosis, resistance to the antibiotic (RifR) poses a significant challenge.3 RifR most commonly results from mutations in the bacterial RNA polymerase (RNAP), the target of the rifamycins, with substitutions at amino acids H451 and S456 accounting for the majority of mutations observed in clinical isolates of RifRM. tuberculosis.4 In addition to Rif, there are currently three other semisynthetic rifamycin analogues in clinical use: rifapentine, rifabutin, and rifaximin.1 Rifapentine exhibits a longer half-life than rifampicin and can therefore be used on a more intermittent dosing schedule. Rifabutin shows reduced cytochrome P450 (CYP) induction, which is a significant problem when rifampicin is used in patients receiving other chemotherapeutics. Rifaximin, the most recently approved analogue, is poorly absorbed via the oral route but is useful for treating infections of the gastrointestinal tract.
The large number of previously prepared semisynthetic rifamycins provide a wealth of structure–activity relationship data. Invariant among both natural rifamycin congeners and semisynthetic derivatives is a naphthalene core and a polyketide backbone. The vast majority of rifamycin derivatives that retain potent antibiotic activity, including all of those currently used in the clinic, are modified at either C-3 and/or C-4 of the naphthalene ring system (Figure 1a).1,2,5−11 Crystal structures of clinically used rifamycin derivatives in complex with RNAP reveal that modifications of the naphthalene core are tolerated because they extend toward an open space in the polymerase active site that is adjacent to the nucleotide binding pocket.12,13 Derivatives modified at either the C-11 or C-25 position have also been generated, although none of these are in clinical use.14,15 Modifications at most other chemically accessible positions in the rifamycin structure generally have detrimental effects on antibiotic activity. In particular, the free hydroxyl groups at C-1, C-8, C-21, and C-23 form critical hydrogen bonds with the polymerase and therefore cannot be easily modified without significantly affecting potency.1
Figure 1.

Structures of rifamycin SV and kanglemycin A. (a) Potential entry points for synthesis. The vast majority of previous synthetic modifications have been made at C-3 and/or C-4 of the rifamycin ring system. Evolution of rifamycin SV has resulted in the addition of features such as the K-acid, which are found in other regions of the molecule that have been largely inaccessible for synthesis. The K-acid provides a new entry point for generating novel semisynthetic derivatives. (b) Position of the K-acid relative to the nascent RNA transcript in the RNAP active site. The nucleotides were modeled into the crystal structure of the Mycobacterium smegmatis RNAP crystal structure in complex with Kang A (PDB ID: 6CCE) by superimposition with the RNAP transcription initiation complex from Thermus thermophilus (PDB ID: 4Q4Z).
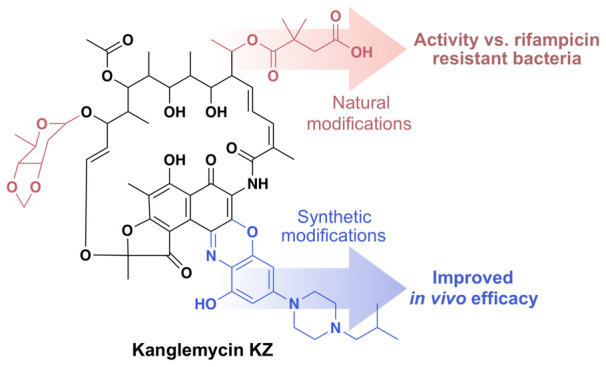
While the C-3 and C-4 positions represent the most common entry points for generating semisynthetic rifamycin derivatives, modifications seen in natural rifamycin congeners are not limited to these sites. For instance, members of the kanglemycin (Kang) family of rifamycin congeners possess a deoxysugar (K-sugar) at C-27 and a dimethylsuccinic acid (K-acid) moiety stemming from an ethyl branch at C-20 (Figure 1).16−18 Remarkably, these tailoring modifications confer activity against RNAP variants carrying some of the most prevalent RifR mutations, including the S456L mutation that represents the single most common RifR mutation in M. tuberculosis clinical isolates. Structural and mechanistic analyses suggest that the K-sugar may stabilize binding of the Kangs to RNAPs carrying the S456L mutation, while the K-acid provides a new mechanism of inhibition by blocking an earlier stage of transcript elongation than Rif.16,17
The activity of the Kangs against bacteria carrying the most common RifR mutation observed in M. tuberculosis clinical isolates suggests that they could have valuable applications in treating RifR infections.17 However, to the best of our knowledge their efficacy in vivo has not been reported previously. In the present study, we assessed the in vivo activity of Kang A, the parent compound in the Kang family, in a murine model of bacterial peritonitis/sepsis. Our initial evaluation of the compound revealed limited bioavailability and in vivo efficacy. With the aim of improving these properties, we generated a series of Kang derivatives using two different semisynthesis strategies. First, we explored the use of the K-acid moiety as a new entry point for synthesis. We found that while modification of the K-acid in some cases leads to improved potency against wild-type bacteria, these modifications come at the expense of activity against RifR strains. As an alternative strategy, we evaluated the effect of combining proven synthetic modifications of the C-3/C-4 region with the natural tailoring modifications found on the Kangs. We found that these combinations of natural and synthetic modifications offer benefits in terms of potency against susceptible and resistant strains and also provide significantly improved bioavailability and in vivo activity in a murine infection model.
Results and Discussion
In Vivo Efficacy of Kang A
The Kangs are natural products produced by the soil bacterium, Amycolatopsis vancoresmycina. Although several Kang congeners are produced by A. vancoresmycina, the major product is Kang A. Because of the comparative ease with which we could access Kang A we used it as our starting material in our synthesis work. For this study we began by establishing some basic pharmacokinetic properties of Kang A (Supporting Table S1). We were particularly interested in evaluating the in vivo bioavailability of the compound, as this was a significant limitation of previously studied natural product rifamycins.2 We found that the bioavailability of Kang A was below detectable levels following oral dosing, although the compound had some bioavailability (6.84%) when delivered by intraperitoneal (IP) injection. Using IP dosing, we tested the antibacterial activity of Kang A against methicillin-resistant S. aureus (MRSA) in a kidney infection model. Infected mice received IP injections of Kang A (15 mg/kg) or Rif (15 mg/kg) at 2, 4, and 8 h post infection. There was no overt morbidity or mortality observed for the different treatments at the end point of the study (24 h post infection). While Rif sterilized the kidneys of infected mice, treatment with Kang A resulted in a comparatively modest 1.8 log reduction in bacterial burdens in kidneys (Supporting Table S2).
Synthesis of K-Acid Derivatives
In an effort to improve the in vivo efficacy of Kang A we created a series of semisynthetic Kang A analogues. In our initial semisynthesis studies we explored modifications of the K-acid, which along with the K-sugar, is one of the main structural features that differentiate the Kangs from other rifamycins. The K-acid was an appealing initial synthetic handle for a number of reasons. First, it has not been previously explored as a synthesis entry point. In fact the general region of the ansa backbone from which the K-acid extends has been largely inaccessible for semisynthetic studies using other rifamycin congeners.1 Second, within the RNAP active site, the K-acid extends toward a large open pocket adjacent to the nucleotide binding site (Figure 1b).17 We predicted that this opening could accommodate semisynthetic modifications without impairing the antibiotic’s inhibition of the polymerase (by comparison, the K-sugar binds within a tighter pocket). Third, while the K-sugar is predicted to play a key role in allowing the Kangs to bind to RifR RNAP variants, the K-acid appears to serve an ancillary mechanistic role in inhibiting both mutant and wild-type forms of the polymerase,7 a function we thought might be therapeutically dispensable. Finally, we rationalized that the charged nature of the K-acid could reduce entry of Kang A into cells thereby limiting its bioavailability.
We generated our initial library of Kang A analogues by coupling a structurally diverse collection of primary and secondary amines to the K-acid. The individual amines in this collection fell into seven general structural classes: aliphatic amines, cyclic amines, aromatic amines, carboxylic acid amines, phosphate mimics, sugars, and cyclic amino acid analogues (tryptophan, tyrosine, phenylalanine, histidine) (Figure 2a; Supporting Figures S1–S7). The phosphate mimics were included in an effort to mimic the triphosphate portion of a nucleotide bound in the RNAP active site. Beyond the phosphate mimics, it was difficult to rationally design modifications based on the X-ray crystal structure of RNAP in complex with Kang A. The initial collection of amines was therefore intended to broadly sample a variety of distinct chemical classes. The product of each 0.4 mg scale amide coupling reaction was purified by HPLC and its identity was verified by LC/MS (Supporting Figures S1–S7). The concentration of each new analogue was determined based on UV absorbance (395 nm) and comparison to a standard curve generated with known quantities of Kang A.
Figure 2.

Synthesis and activity of Kang amides. A screen of more than 100 Kang amides identified 17 compounds with improved activity against wild-type (WT) S. aureus compared to Kang A. (a) Reaction used for the synthesis of Kang amides and summary of screening hits with improved activity against WT S. aureus. (b) MIC values (μg/mL) for Kang A and Rif against WT and RifR H481Y and S486L S. aureus strains. (c) Structural modifications and MIC values (μg/mL) of hits against WT and RifRS. aureus strains. Amines used for synthesis are shown. A subset of the semisynthetic derivatives, highlighted in red, was subjected to downstream analyses. The C5 and N29 amides were synthesized and screened using diastereomeric mixtures of 2-cyclopropylpyrrolidine and 2-methylpyrrolidine, respectively.
Activity Screening of K-Acid Derivatives
We generated more than 100 Kang amides over the course of two rounds of synthesis (Supporting Figures S1–S7). The antibacterial activity of each amide analogue was evaluated against Rif sensitive S. aureus as well as S. aureus strains carrying either an H481Y or an S486L RNAP mutation.19 These mutations correspond to the two most common RifR mutations found in M. tuberculosis clinical isolates (M. tuberculosis RNAP H451Y and S456L). While Kang A shows strong activity against the S486L mutant, it is not active against the H481Y variant (Figure 2b). Rif is inactive against both mutants. The best amide analogues showed a time-dependent improvement in inhibition of S. aureus growth, with the highest levels of inhibition relative to Kang A occurring after 12 h and less dramatic differences in growth inhibition occurring at later time points. The reason for the time dependent inhibition of these analogues remains to be determined. We therefore monitored the MICs for all amides at a 12 h time point to ensure detection of analogues that had an effect on even the early growth of S. aureus. In the first round of synthesis, amides with increased potency against wild-type S. aureus fell into three structural classes: aliphatic amides, cyclic amides, and aromatic amides (Figure 2a,c). The most potent compounds from the first round of synthesis from each of these structural classes were the amides synthesized from tert-butylamine (J5; MIC = 0.000061 μg/mL), benzylamine (J4; MIC = 0.000061 μg/mL), as well as cyclopropanemethylamine (B1; MIC = 0.00098 μg/mL) and 2-methylpyrrolidine (N29; MIC = 0.00098 μg/mL; Figure 2c). In general, modification of the K-acid resulted in at least a 16-fold reduction in activity against the S486L strain. Like Kang A, most were inactive against the H481Y strain, with the exception of the amide of F6, which weakly inhibited the growth of this strain (MIC = 64 μg/mL; Figure 2c).
In a second round of synthesis, we expanded the structural diversity around three of our most potent initial hits: amides J4, J5, and N29 (Supporting Figure S1–S3). In each case five to seven additional Kang A amides were synthesized using primary and secondary amines related to those that yielded these three hits. Amides with improved potency included the methoxy-containing aromatic amides, C13, F1, and F2, which showed modest 4-fold increases in activity relative to Kang A, and a fluorinated aromatic amine, N4, which ranked among the most potent compounds we generated against wild-type S. aureus (MIC = 0.000061 μg/mL; Figure 2c). We also identified a pyrrolidine-containing amide with a cyclopropyl moiety (C5) which had an MIC of 0.00024 μg/mL against wild-type S. aureus, representing a 4-fold improvement in activity compared to the structurally related N29 amide from our first round of screening (Figure 2c). Interestingly, the structure of C5 combines the pyrrolidine substructure of N29 with a second highly active moiety, the cyclopropyl functionality from B1. As we observed in our first round of screening, the amides generated in our second round had reduced activity against the S486L mutant and were inactive against the H481Y mutant.
In total we identified 17 amide modifications that showed increased potency compared to Kang A against wild-type S. aureus (Figure 2c). The activity of these compounds is consistent with our prediction that the open pocket in the RNAP active site adjacent to the Kang binding site is sufficiently large to accommodate modifications made to the K-acid. However, the results also reveal that the K-acid plays an important role in allowing the Kangs to inhibit the S486L RNAP variant and that any improvement in activity of the amides against Rif susceptible bacteria is likely to come at the cost of reduced activity against RifR strains. The fact that the addition of even minimal amide side chains (such as the C23 methylamide and D4 dimethylamide; Supporting Figure S1) resulted in reduced activity against the S486L mutant demonstrates that an intact carboxylic acid is likely to be evolutionarily optimized for strong inhibition of the RifR enzyme. In the RNAP-Kang A crystal structure, the K-acid forms a salt bridge with a nearby arginine.17 It is possible that this interaction, which is disrupted by replacing the acid with an amide, makes an important contribution to the activity of the antibiotic against the S486L RNAP variant.
Synthesis and Screening of C-3/C-4 Derivatives
Since an intact acid moiety appears to be essential for potent activity of Kang A against bacteria carrying the S486L RNAP variant, we considered other modification strategies that would not detrimentally affect the activity against RifR bacteria. The C-3 and C-4 positions of rifamycin have historically been the most fruitful sites for semisynthetic modifications.1 In addition to offering potential increases in potency, some modifications in this region of the structure also confer improved bioavailability,1,2 which we observed as a key limitation of Kang A in our initial pharmacological profiling study. Of particular interest to us were a series of benzoxazinorifamycin analogues, in which the C-3/C-4 region of the naphthoquinone is fused with a hydroxylated benzoxazino functionality.11,20 Although at present there are no benzoxazinorifamycins in clinical use, several were reported to have superior activity compared to Rif against wild-type bacteria and interestingly, some RifR strains.20−24 Among previously synthesized benzoxazorifamycins, a 3′-hydroxy-5′-aminobenzoxazino derivative containing an isobutyl piperazine side chain known as rifalazil has been the most studied.1
Using a two-step reaction11,14 that involves initially generating a 3-hydroxy-benzoxazino intermediate (Kint) of Kang A followed by functionalization of Kint with a secondary amine, we created a series of benzoxazino derivatives of Kang A (Figure 3a). We focused primarily on piperazines and other cyclic amine side chains, as similar modifications were found to confer potent antibiotic activity to other rifamycins (Figure 3b).11,20 We also tested an N-benzylmethylamine-containing side chain (Z4), which was meant to mimic the potent J4 benzylamide modification we identified in our amide screening studies. We prepared 13 benzoxazino derivatives at 1 mg scale and purified them by HPLC. The identity of the purified products was confirmed by LC/MS (Supporting Figure S8). All Kang benzoxazino derivatives were assayed for antibacterial activity against wild-type S. aureus and the H481Y and S486L mutant strains (Figure 3b). Several of the new compounds (C4z, KZ, Z6, and Z8) showed 4-fold improvements in activity compared to Kang A against the wild-type bacteria. Interestingly, the KZ compound has the same N-isobutyl piperazine-containing side chain found in rifalazil, while Z6 differs from KZ by only one carbon in its side chain. Compounds with bulkier side chains such as Z7, Z9, Z10, Z11, and N31z did not show improved activity. Unlike the amide analogues, some of the benzoxazino derivatives, in particular KZ and Z6, retained good activity against the S486L mutant (MIC = 1 μg/mL). These derivatives also acquired modest activity against the H481Y mutant, with MIC values of 16 μg/mL (Figure 3b). The H481Y mutation normally results in a particularly high level of resistance to many Rif analogues. The compounds produced in this study may therefore represent useful starting points for the generation of derivatives that are even more potent against this difficult to treat RifR mutation.
Figure 3.

Synthesis and activity of C-3/C-4 benzoxazino Kang derivatives. (a) Benzoxazino modification synthesis reaction. (b) Structural modifications and MIC values (μg/mL) of Kang benzoxazino derivatives against wild-type (WT) and RifRS. aureus strains. Amines used for synthesis are shown. The MIC values for the parent compound, Kang A, were 0.016 μg/mL, >64 μg/mL, and 0.25 μg/mL against the WT, H481Y, and S486L strains, respectively. A subset of the semisynthetic derivatives, highlighted in red, was subjected to downstream analyses. N29z was synthesized and screened as a diastereomeric mixture of 2-methylpyrrolidine.
Activity of Derivatives against Mycobacteria
Given the long history of the rifamycin family of antibiotics in the treatment of tuberculosis, we were interested in examining the activity of our new semisynthetic Kang analogues against M. tuberculosis. We tested the antibiotic activity of a subset of our most potent Kang amides (B1, C5, E4, F6, J4, and J5) and C-3/C-4 benzoxazino analogues (C4, KZ, N29z, Z6, Z8, and Z10) against wild-type and RifR strains of M. tuberculosis H37rv and in an in vitro assay against purified mycobacterial RNAP from M. smegmatis (Figure 4). The M. smegmatis enzyme exhibits a very high level of sequence identity with M. tuberculosis RNAP, including all amino acids that directly interact with Kang A/Rif. We found that the amides had similar activity to Kang A against the wild-type M. tuberculosis strain, with the C5 and F6 amides showing modest 4-fold improvements in activity compared to Kang A (Figure 4a). With the exception of the B1 and C5 amides, the amides had reduced activity against a RifR S456L strain (Figure 4a), providing further evidence that modification of the K-acid comes at the expense of activity against this common RifR mutant. Of the C-3/C-4 benzoxazino derivatives, KZ and N29z exhibited the most potent activity against the wild-type M. tuberculosis strain, with MIC90 values that were 8- and 4-fold lower than that of Kang A, respectively (Figure 4b). Against the RifR S456L strain, the C-3/C-4 derivatives showed between 4- and 16-fold improvements in activity relative to Kang A (Figure 4b). This strain was completely resistant to Rif at the highest concentration tested (25 μg/mL; Figure 4a).
Figure 4.

(a) Activity of a subset of the Kang amides against wild-type (WT) and S456L RifRM. tuberculosis. MIC90 (μg/mL) values are shown. (b) Activity of a subset of the C-3/C-4 benzoxazino Kang derivatives against WT and S456L RifRM. tuberculosis. MIC90 (μg/mL) values are shown. (c) In vitro transcription assay showing the activity of the subset of Kang amides against purified M. smegmatis RNAP. Compounds were evaluated at the concentrations indicated for their ability to inhibit the production of a radiolabeled transcript. (d) In vitro transcription assay showing the activity of the subset of C-3/C-4 benzoxazino Kang derivatives against purified M. smegmatis RNAP.
In vitro, the amides inhibited the purified mycobacterial RNAP with equal or slightly lower potency compared to Kang A, with transcription reduced at a concentration of 0.1 μM of antibiotic and strongly inhibited at 1 μM (Figure 4c). The C-3/C-4 derivatives all had reduced activity against the purified mycobacterial polymerase compared to Kang A (Figure 4d). These findings suggest that the enhanced antibiotic activity of the compounds observed during screening may be due to increased passage of the compounds into the bacterial cells rather than more potent inhibition of RNAP.
In Vivo Efficacy of Lead Structures
Lead compounds were selected from both the Kang amide and the C-3/C-4 benzoxazino derivative series for in vivo analysis (Figure 5a). From the K-acid derivatives, we chose the J4 amide, which showed extremely potent activity against wild-type S. aureus and could be easily produced in high yields from Kang A. From the C-3/C-4 derivatives, we selected KZ, which exhibited strong activity against wild-type S. aureus and also had promising activity against S. aureus strains carrying two common RifR mutations. As we predicted, preliminary pharmacokinetic analysis showed that capping the K-acid with the J4 amide or adding the C-3/C-4 benzoxazino modification of KZ led to marked improvements in bioavailability compared to Kang A (Figure 5b; Supporting Table S1).
Figure 5.

In vivo activity of top leads from semisynthesis in comparison to Kang A and Rif. (a) Structural modifications of lead compounds: the J4 Kang amide and the KZ benzoxazino analogue. (b) IP and PO bioavailability of J4 and KZ in comparison to Kang A. BLQ, below limit of quantification. (c) Efficacy of Kang A, J4, KZ, and Rif in treating MRSA in a neutropenic murine acute peritonitis/septicemia model. (d) Efficacy of KZ and Rif in treating infection with a highly virulent S. aureus strain with RifR S486L RNAP in the neutropenic murine acute peritonitis/septicemia model. For panels (c) and (d), infected mice received IP injections of drug (15 mg/kg) or vehicle (5% DMA plus 30% Captisol) at 2, 4, and 8 h post infection. The y-axis indicates bacterial burdens in kidneys at 24 h postinfection. Limit of detection (LOD) for burden quantification was calculated as 100 CFU/g of kidney. The results shown represent the average bacterial burden from six mice. Error bars indicate standard deviation. Asterisks indicate treatments that resulted in a statistically significant reduction in burden (P < 0.05) relative to the vehicle treated group. Insets, percent survival of mice at 24 h for each treatment.
The in vivo efficacies of J4 and KZ were evaluated with the same murine neutropenic peritonitis/sepsis model described earlier for Kang A using MRSA. As with Kang A, there was no overt morbidity or mortality observed upon treatment with J4 or KZ using 15 mg/kg IP doses. J4 caused a modest reduction in bacterial burdens in the kidneys of infected mice, while KZ was successful in sterilizing the kidneys (Figure 5c, Supporting Table S2). Given the good activity of the KZ compound against RifRS. aureus in our MIC assays, we were curious to see whether KZ would show in vivo efficacy in treating a RifR infection. We tested the ability of KZ to reduce bacterial burden and protect mice upon infection with a highly virulent S. aureus strain carrying the RifR S486L RNAP variant. While infection with the Rif sensitive MRSA strain tested earlier was not lethal to the mice over the 24 h course of the previous experiment, infection with the RifR strain resulted in the death of half of the vehicle treated mice. As expected, Rif failed to significantly reduce bacterial burdens in the kidneys of mice infected with the RifR strain, leading to the survival of only 17% of mice (Figure 5d, Supporting Table S3). In contrast, treatment with KZ caused a significant reduction in bacterial burdens (>1.8 log) and led to the survival of all mice tested over the course of the experiment. These results demonstrate that the KZ compound could be a valuable lead for the design of drugs for treating RifR infections.
Conclusions
The Kangs represent interesting scaffolds for the development of chemotherapeutics due to their activity against RifR bacteria. In this study, we aimed to improve the in vivo activity of Kang A by generating a series of semisynthetic derivatives. The distinctive structural features of the Kangs and in particular the K-acid moiety provided a facile entry point for generating amide derivatives. Modifications of this region of the antibiotic’s structure were not obvious or easily accessible to chemists using other rifamycins as starting materials. Our study therefore suggests that the identification of natural product congeners of other established drugs can present new avenues for semisynthetic modification. The fact that several of the amides generated in this study were active against S. aureus and in vitro against purified RNAP confirmed our prediction that the open pocket in the RNAP active site adjacent to the Kang binding site is sufficiently large to accept modifications built onto the K-acid. However, the reduced activity of the amide derivatives against the S. aureus S486L strain suggests that the K-acid plays a more important role in inhibiting this RNAP mutant than originally thought; it is likely that both the K-acid and K-sugar have been evolutionarily optimized for activity against the RifR enzyme. Moreover, the J4 amide, which was among the most potent amides against wild-type S. aureus cells during screening, did not prove efficacious in our in vivo analysis of the compound.
To date, the vast majority of semisynthetic rifamycin derivatives have been modified on the naphthalene ring system. All rifamycin analogues currently in clinical use have been modified at this part of their structures. The availability of this portion of the Kang A structure for synthetic derivatization allowed us to test the effect of combining a proven C-3/C-4 modification, which was known to yield improvements in activity against some RifR bacteria and which we also hypothesized could help increase the bioavailability of the compound, with the natural tailoring modifications of Kang A, which confer potent activity against the dominant S486L mutation. The result was a series of benzoxazino derivatives that showed broad spectrum activity against wild-type bacteria as well as two of the most common RifR strains. While the activity of the derivatives against the H481Y mutant was only moderate, these compounds may inspire the design of additional Kang analogues with more potent activity against this highly resistant mutant. The KZ benzoxazino derivative had the additional benefit of conferring increased in vivo bioavailability, which likely contributed to the success of this compound in treating the MRSA infected mice in our peritonitis/septicemia model. Importantly, we found the KZ compound was active in vivo against a highly virulent S. aureus strain carrying the RifR S486L RNAP mutation. The compounds generated in our study provide new leads for the development of drugs for treating RifR infections. Moreover, our results suggest that the Kangs and other natural product antibiotic congeners represent a valuable source of structural variations that can be paired with proven synthetic modifications to yield useful combinations of pharmacologically relevant properties.
Methods
Isolation of Kang A
Kang A was isolated from fermentations of Amycolatopsis vancoresmycina (NRRL B-24208). Five μL of a frozen glycerol spore stock of A. vancoresmycina was used to inoculate 50 mL of TSB media (Oxoid) in a 125 mL baffled flask. The culture was grown for 48 h with shaking at 30 °C and 200 rpm. 200 μL of the saturated culture was used to inoculate 72 × 50 mL R5A media (100 g/L sucrose, 0.25 g/L K2SO4, 10.12 g/L MgCl2·6H2O, 10 g/L glucose, 0.1 g/L casamino acids, 20.5 g/L MOPS, 5 g/L yeast extract, and 2 g/L NaOH) containing 1.5 g Diaion HP-20 resin (Sigma) and a 1” × 1” stainless steel metal mesh (for increased aeration) in 125 mL baffled flasks. The cultures were incubated at 30 °C with shaking at 200 rpm. After 10 days, HP-20 resin was removed from the cultures by filtration and washed with 2 × 500 mL water. Material bound to the resin was eluted using 2 × 500 mL methanol. The resulting crude extract was fractionated by flash chromatography (RediSep Rf, High Performance Gold 50 g HP C18 resin) using a linear gradient of 30–100% acetonitrile:water with 0.1% acetic acid over 30 min. A small portion of each fraction was analyzed by LC/MS on a Waters Acquity H-Class UPLC. Fractions containing Kang A were further purified by HPLC on a 10 mm × 150 mm C18 column (Waters) using an isocratic method of 42% acetonitrile with 0.1% formic acid at a flow rate of 2.5 mL/min. Purified Kang A was isolated with a yield of approximately 5 mg per liter of culture.
Synthesis of Kang Amides
For the synthesis of Kang amides, each reaction utilized a total of 0.4 mg (0.4 μmol) Kang A. A 0.2 M stock of Kang A was prepared in dimethylformamide (DMF). 0.4 M stocks (in DMF) were prepared for each of the following: 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), triethylamine (TEA), and each amine to be coupled to the Kang A acid. As the amines containing carboxylic acid, phosphonate and sulfonate moieties generally had poor solubility in DMF, solutions of these amines were instead prepared in water. Two μL of each reagent were transferred to a 1.5 mL Eppendorf tube in the order: Kang A, TEA, HATU, and amine. Reactions were performed at 25 °C. Reactions were allowed to proceed overnight with gentle agitation on a vortexer. The following day, reactions were diluted with 100 μL DMF and purified by HPLC with a 10 mm × 150 mm C18 column (Waters) and a linear gradient of 30–95% acetonitrile:water with 0.1% formic over 30 min at a flow rate of 3.5 mL/min. The identity of each purified Kang amide was verified by LC/MS and the purity was determined to be 95% or greater. The concentration of each purified analogue was evaluated using UV absorbance (395 nm) by comparison to a standard curve generated with known quantities of Kang A (Supporting Figure S9). Amides generated from the aromatic amines N34 and N35 had altered UV absorbances at 395 nm compared to Kang A and were instead produced in larger scale (∼1 mg) and quantified by mass. Amides selected for additional studies were resynthesized using multiple 0.4 mg reactions, purified as described above, and quantified by mass. The C5 and N29 amides were synthesized and screened using diastereomeric mixtures of 2-cyclopropylpyrrolidine and 2-methylpyrrolidine, respectively. The lead compound from the Kang amides, J4, was further characterized by analysis of its UV spectrum, high resolution mass and mass fragmentation, and by 1D and 2D NMR, including comparison of its NMR spectra to those of Kang A (Supporting Figures S10–S22, Table S4).
Antibiotic Assays against S. aureus
Minimum inhibitory concentration (MIC) assays were performed against wild-type S. aureus ATCC 12600 or H481Y or S486L RifR strains19 using a serial 1:4 dilution of each compound. A single colony of S. aureus was used to inoculate 15 mL of Luria–Bertani (LB) broth and grown overnight. The next day, 10 μL of the saturated overnight culture was diluted with 50 mL fresh LB. 80 μL of the diluted cells were aliquoted into each well of a 96-well plate. Compounds were resuspended in DMSO and a serial 1:4 dilution of each compound was prepared in a separate 96-well plate, so that upon transfer of 20 μL of each diluted compound to the plate containing the bacteria, the concentration of compound in the first well was 64 μg/mL. Each compound was tested in duplicate. Plates were sealed using a Breathe-Easy air permeable membrane and incubated at 30 °C with shaking at 200 rpm for 12 h. MIC values were reported as the lowest concentration of the compound that inhibited visible bacterial growth.
Synthesis of C-3/C-4 Derivatives
Benzoxazino derivatives of Kang A were generated in a two-step reaction, as reported previously for the synthesis of benzoxazinorifamycins.11,20 All reactions were performed at 25 °C. In the initial step, 1 mg (1 μmol) of Kang A was reacted with 2-aminoresoricinol hydrochloride (Sigma) in a 1:1 molar ratio in 20 μL of 1:1 toluene:tetrahydrofuran (THF) to afford the hydroxylated benzoxazino intermediate, Kint. The reaction was allowed to proceed for approximately 24 h. To the product of the first reaction was added 20 μL DMSO, 1 mg MnO2 (11.5 μmol) and a 2-fold molar excess (relative to the Kang A starting material) of one of the secondary amines shown in Figure 3. The second reaction was allowed to proceed with shaking for 24 h, at which point 500 μL of methanol was added to the reaction and insoluble materials were removed by centrifugation. The reaction products were purified by HPLC as described above for the Kang amides. A second round of HPLC purification for the benzoxazino derivatives utilized a gradient of 75–95% methanol:water with 0.1% formic acid over 30 min at a flow rate of 3.5 mL/min. The identity of each purified benzoxazino derivative was verified by LC/MS and purity was determined to be at least 95%. The compounds were screened for activity against S. aureus wild-type and H481Y and S486L RifR strains of S. aureus using the same protocol used for screening the Kang amides. Compounds selected for additional studies were resynthesized using multiple 1 mg reactions and purified as described above. Compound N29z was synthesized and screened as a diastereomeric mixture of 2-methylpyrrolidine. The lead compound from the Kang benzoxazino derivatives, KZ, was further characterized by analysis of its UV spectrum, high resolution mass and mass fragmentation, and by 1D and 2D NMR, including comparison of its NMR spectra to those of Kang A (Supporting Figures S10–S17 and S23–S27, Table S4).
Antibiotic Assays against M. tuberculosis
M. tuberculosis H37Rv was purchased from the ATCC (Manassas, Viriginia). The RifR S456L strain was obtained from the Kreiswirth laboratory (Center for Discovery and Innovation, Nutley, New Jersey). Bacteria were growth overnight in 7H9 broth (Becton, Dickinson and Company 271310), plus 0.2% glycerol, and 20% 5× albumin-dextrose complex (ADC). The 5× ADC solution was prepared using 25 g/L Bovine Serine Albumin, 10 g/L dextrose, and 4.2 g/L NaCl. For MIC90 assays, bacteria were grown for 7 days at 37 °C. Compounds were suspended in DMSO. A series of 2-fold dilutions of the compounds were prepared in the bacterial media and added to a 96-well round-bottom cell culture plate (Corning Incorporated, Costat 3799). Bacterial stocks were prepared by making a 1:100 dilution of the seven day old cultures and 100 μL of the stocks were added to the wells of the 96-well plate. Each well contained a total volume of 200 μL. Plates were incubated for 7 days at 37 °C at which point MIC90 values were determined by adding Alamar Blue to the 96-well plate and then reading the absorbance in each well at 570 nm. Assays were performed in duplicate or triplicate.
In Vitro Transcription Assays
Recombinantly produced RNAP was purified from M. smegmatis strain MGM6029 as previously described.17 Transcription assays were performed by mixing 50 nM RNAP holoenzyme in transcription buffer (10 mM Tris HCl, pH 7.9, 50 mM KCl, 10 mM, MgCl2, 1 mM DTT, 5 μg/mL bovine serum albumin (BSA) and 0.1 mM EDTA) with different concentrations of antibiotic in a total reaction volume of 20 μL. The RNAP/antibiotic mixtures were first incubated at 37 °C for 5 min to permit binding of the antibiotics to the polymerase. To form the RNAP open complex, 10 nM of AP3 promoter25 was added to each reaction and the tubes were incubated for 15 min at 37 °C. To initiate transcription, a nucleotide mixture (200 μM ATP, 200 μM CTP, 200 μM GTP, 50 μM unlabeled UTP and 1.25 μCi (0.3 μM) γ-P32–UTP) was added to each tube. Reactions were allowed to proceed for 15 min at 37 °C before being stopped by the addition of buffer containing 0.5× TBE, pH 8.3, 8 M urea, 30 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol. Reactions were then heated at 95 °C for 10 min and loaded onto a polyacrylamide gel (23% acrylamide/bis acrylamide (19:1), 6 M urea, and 1× TBE, pH 8.3). Gels were run for 1.5 h at 1000 V, before being exposed on a phosphoroimaging plate (GE Healthcare) for 12 h. Gels were imaged using a Typhoon 9400 Variable Imager (Amersham Biosciences).
In Vivo Mouse Pharmacokinetics
All animal experiments were approved by the Institutional Animal Care and Use Committee of Hackensack University Medical Center for Discovery and Innovation, and were conducted in compliance with their guidelines. Female CD-1 mice were weighed and received a single dose of Kang A, J4, or KZ via IV (5 mg/kg), PO (5 mg/kg), and IP (5 mg/kg) dosing routes. Compound was formulated as a solution in 5% DMA/95% (4% Cremophor in water). Sequential bleeds were collected at 0.017, 0.25, 1, 3, 5, and 7 h post dose via the tail snip method for IV dosing and 0.5, 1, 3, and 5 h post dose for PO and IP dosing. Blood (50 μL) was collected in capillary microvette EDTA blood tubes and kept on ice prior to centrifugation at 1500g for 5 min. The supernatant (plasma) was transferred into a 96-well plate and stored at −80 °C. LC-MS/MS analysis was performed on a Sciex Applied Biosystems Qtrap 6500+ triple-quadrupole mass spectrometer coupled to a Shimadzu Nexera 2 HPLC system to quantify each drug in plasma. Neat 1 mg/mL DMSO stocks of each compound were serial diluted in 50/50 acetonitrile/water to create standard curves and quality control spiking solutions. Standards and QCs were created by adding 10 μL of spiking solutions to 90 μL of drug free plasma (CD-1 K2EDTA Mouse, Bioreclamation IVT). Ten μL of control, standard, QC, or study sample were added to 100 μL of acetonitrile protein precipitation solvent containing internal standard (10 ng/mL Verpamil). Extracts were vortexed for 5 min and centrifuged at 4000 rpm for 5 min. 75 μL of supernatant were transferred for HPLC-MS/MS analysis and diluted with 75 μL of Milli-Q deionized water. Chromatography was performed on an Agilent Zorbax SB-C8 column (2.1 × 30 mm; particle size, 3.5 μm) using a reverse phase gradient. Milli-Q deionized water with 0.1% formic acid was used for the aqueous mobile phase and 0.1% formic acid in acetonitrile for the organic mobile phase. Multiple-reaction monitoring of precursor/product transitions in electrospray positive-ionization mode was used to quantify the analytes. The following MRM precursor/product transitions were used for Kang A (982.26/822.20), J4 (1071.35/911.20), KZ (1227.43/1067.30) and Verapamil (455.4/165.2). Data processing was performed using Analyst software (version 1.6.2; Applied Biosystems Sciex).
In Vivo Efficacy Studies
Kang A, J4, KZ and Rif were evaluated for their efficacy in treating MRSA in a neutropenic murine acute peritonitis/septicemia model. The drugs were prepared in the vehicle, consisting of 5% DMA and 30% Captisol in sterile water for injection. Female outbred Swiss Webster mice (∼6 weeks old) were housed in individually ventilated cages and maintained in accordance with American Association for Accreditation of Laboratory Care criteria. The animal study was approved by Hackensack Meridian Health’s Institutional Animal Care and Use Committee under protocol number 260. MRSA strain COL was acquired through the Kreiswirth Laboratory (Center for Discovery and Innovation, Nutley, New Jersey). This strain was originally obtained from the Tomasz Laboratory at the Rockefeller University. S. aureus ATCC 12600 carrying an S486L RNAP mutation was used to test the in vivo efficacy of KZ against a RifR strain. Bacterial strains were grown overnight in Mueller–Hinton Broth at 37 °C with shaking. The cultures were centrifuged, the supernatant was removed and the bacteria were gently washed once in sterile saline. The optical density at 600 nm was monitored in a spectrophotometer. The bacteria were resuspended in 5% mucin. The suspension provided a challenge inoculum of approximately 2.0 × 104 CFU per mouse in a volume of 0.5 mL. Inoculum counts were verified by viable counts on LB plates spread with dilutions of the inoculum and incubated at 37 °C for 24 h. A murine neutropenic peritonitis/sepsis model was used to assess the efficacy of the treatment.26 Mice were rendered neutropenic by receiving 150 mg/kg cyclophosphamide on day −4 and 100 mg/kg cyclophosphamide on day −1 prior to infection. Mice were manually restrained and inoculated with approximately 2.0 × 104 CFU of MRSA strain COL or the S. aureus ATCC 12600 strain with the S486L RNAP mutation in a volume of 0.5 mL in 5% hog mucin and 0.9% NaCl via IP injection. Treatment was initiated at 2 h post infection. Mice were given IP injections of vehicle (5% DMA plus 30% Captisol), or 15 mg/kg of Rif, Kang A, J4, or KZ at 2, 4, and 8 h post infection. Six mice were used for each treatment. Mice were observed twice daily for mortality and morbidity and possible signs of acute toxicity. Abnormal clinical signs were recorded if observed. At 24 h post infection, mice were humanely euthanized by CO2 narcosis. Kidneys were aseptically removed, homogenized and enumerated for bacterial burden by CFU counts by plating on MSA agar. All graphic data are expressed as columnar average data points by group and were statistically analyzed by analysis of variance (ANOVA) using computer Prism software (Prism 8; GraphPad Software, Inc., San Diego, CA). Burden difference between testing and control groups was assessed by post hoc analysis, using Holm–Sidak’s multiple comparison test. A P value of <0.05 is considered statistically significant.
Acknowledgments
We thank Elizabeth Campbell, Seth Darst, and Mirjana Lilic for help with in vitro transcription assays. We thank Enriko Dolgov, Nathaly Cabrera, and Rosa Hernandez for their assistance with in vivo efficacy studies. We thank Richard Ebright for providing RifR strains of S. aureus. This work was supported by the Bill and Melinda Gates Foundation grant OPP1117928 and by NIH grants 1U19AI142731 and S10-OD023524.
Glossary
Abbreviations
- Rif
Rifampicin
- Kang
Kanglemycin
- RifR
Rifampicin resistant
- RNAP
RNA polymerase
- IP
intraperitoneal
- MRSA
methicillin-resistant Staphylococcus aureus
- MIC
minimal inhibitory concentration.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00223.
Table S1: Pharmacokinetic properties of Kang A, J4, and KZ; Table S2: Comparison of bacterial burdens in mouse kidneys infected with MRSA strain COL following treatment with Kang A, J4, KZ, or Rif; Table S3: Comparison of bacterial burdens in mouse kidneys infected with S. aureus ATCC 12600 carrying an S486L RNAP mutation following treatment with KZ or Rif; Table S4: 1H and 13C chemical shifts of Kang A, J4, and KZ; Figure S1: Complete collection of aliphatic amines used in the synthesis of Kang amides; Figure S2: Complete collection of cyclic amines used in the synthesis of Kang amides; Figure S3: Complete collection of aromatic amines used in the synthesis of Kang amides; Figure S4: Complete collection of carboxylic acid amines used in the synthesis of Kang amides; Figure S5: Complete collection of phosphate mimic amines used in the synthesis of Kang amides; Figure S6: Complete collection of sugar amines used in the synthesis of Kang amides; Figure S7: Complete collection of Phe/Trp/Try/His analogue amines used in the synthesis of Kang amides; Figure S8: Complete collection of amines uses in the synthesis C-3/C-4 Kang derivatives; Figure S9: Calibration curve used to determine the concentration of Kang amides; Figure S10: UPLC traces and UV spectra of Kang A, J4, and KZ; Figure S11: Mass fragmentation analysis of Kang A, J4, and KZ; Figure S12: HRMS and NMR data used to verify the structures of J4 and KZ; Figure S13: 1H NMR spectrum of Kang A in CD2Cl2; Figure S14: 13C NMR spectrum of Kang A in CD2Cl2; Figure S15: HMQC spectrum of Kang A in CD2Cl2; Figure S16: HMBC spectrum of Kang A in CD2Cl2; Figure S17: COSY spectrum of Kang A in CD2Cl2; Figure S18: 1H NMR spectrum of J4 in CDCl3; Figure S19: 13C NMR spectrum of J4 in CDCl3; Figure S20: HSQC NMR spectrum of J4 in CDCl3; Figure S21: HMBC NMR spectrum of J4 in CDCl3; Figure S22: COSY NMR spectrum of J4 in CDCl3; Figure S23: 1H NMR spectrum of KZ in DMSO-d6; Figure S24: 13C NMR spectrum of KZ in DMSO-d6; Figure S25: HSQC NMR spectrum of KZ in DMSO-d6; Figure S26: HMBC NMR spectrum of KZ in DMSO-d6; Figure S27: COSY NMR spectrum of KZ in DMSO-d6 (PDF)
The authors declare the following competing financial interest(s): Sean F. Brady is the founder of Lodo Therapeutics.
Supplementary Material
References
- Aristoff P. A.; Garcia G. A.; Kirchhoff P. D.; Showalter H. D. (2010) Rifamycins--obstacles and opportunities. Tuberculosis (Oxford, U. K.) 90, 94–118. 10.1016/j.tube.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Sensi P. (1983) History of the development of rifampin. Clin. Infect. Dis. 5 (Suppl 3), S402–S406. 10.1093/clinids/5.Supplement_3.S402. [DOI] [PubMed] [Google Scholar]
- Zumla A.; George A.; Sharma V.; Herbert R. H. N.; Oxley A.; Oliver M. (2015) The WHO 2014 global tuberculosis report--further to go. Lancet Glob Health 3, e10–e12. 10.1016/S2214-109X(14)70361-4. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S.; Musser J. M. (1998) Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis 79, 3–29. 10.1054/tuld.1998.0002. [DOI] [PubMed] [Google Scholar]
- Arioli V.; Berti M.; Carniti G.; Randisi E.; Rossi E.; Scotti R. (1981) Antibacterial activity of DL 473, a new semisynthetic rifamycin derivative. J. Antibiot. 34, 1026–1032. 10.7164/antibiotics.34.1026. [DOI] [PubMed] [Google Scholar]
- Bellomo P.; Marchi E.; Mascellani G.; Brufani M. (1981) Synthesis and antibacterial activity of some esters, amides, and hydrazides of 3-carboxyrifamycin S. Relationship between structure and activity of ansamycins. J. Med. Chem. 24, 1310–1314. 10.1021/jm00143a010. [DOI] [PubMed] [Google Scholar]
- Bujnowski K.; Synoradzki L.; Darłak R.ła. C.; Zevaco T. A.; Dinjus E. (2016) Semi-synthetic zwitterionic rifamycins: a promising class of antibiotics; survey of their chemistry and biological activities. RSC Adv. 6, 114758–114772. 10.1039/C6RA22880A. [DOI] [Google Scholar]
- Marsili L.; Pasqualucci C. R.; Vigevani A.; Gioia B.; Schioppacassi G.; Oronzo G. (1981) New rifamycins modified at positions 3 and 4. Synthesis, structure and biological evaluation. J. Antibiot. 34, 1033–1038. 10.7164/antibiotics.34.1033. [DOI] [PubMed] [Google Scholar]
- Sanfilippo A.; Della Bruna C.; Marsili L.; Morvillo E.; Pasqualucci C. R.; Schioppacassi G.; Ungheri D. (1980) Biological activity of a new class of rifamycins. Spiro-piperidyl-rifamycins. J. Antibiot. 33, 1193–1198. 10.7164/antibiotics.33.1193. [DOI] [PubMed] [Google Scholar]
- Sensi P.; Maggi N.; Ballotta R.; Fürész S.; Pallanza R.; Arioli V. (1964) Rifamycins XXXV. Amides and Hydrazides of Rifamycin B.. J. Med. Chem. 7, 596–602. 10.1021/jm00335a005. [DOI] [PubMed] [Google Scholar]
- Yamane T.; Hashizume T.; Yamashita K.; Konishi E.; Hosoe K.; Hidaka T.; Watanabe K.; Kawaharada H.; Yamamoto T.; Kuze F. (1993) Synthesis and biological activity of 3′-hydroxy-5′-aminobenzoxazinorifamycin derivatives. Chem. Pharm. Bull. 41, 148–155. 10.1248/cpb.41.148. [DOI] [PubMed] [Google Scholar]
- Campbell E. A.; Korzheva N.; Mustaev A.; Murakami K.; Nair S.; Goldfarb A.; Darst S. A. (2001) Structural mechanism for rifampicin inhibiton of bacterial RNA polymerase. Cell 104, 901–912. 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Artsimovitch I.; Vassylyeva M. N.; Svetlov D.; Svetlov V.; Perederina A.; Igarashi N.; Matsugaki N.; Wakatsuki S.; Tahirov T. H.; Vassylyev D. G. (2005) Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell 122, 351–363. 10.1016/j.cell.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Li J.; Ma Z.; Chapo K.; Yan D.; Lynch A. S.; Ding C. Z. (2007) Preparation and in vitro anti-staphylococcal activity of novel 11-deoxy-11-hydroxyiminorifamycins. Bioorg. Med. Chem. Lett. 17, 5510–5513. 10.1016/j.bmcl.2007.08.048. [DOI] [PubMed] [Google Scholar]
- Wehrli W.; Zimmermann W.; Kump W.; Tosch W.; Vischer W.; Zak O. (1987) CGP 4832, a new semisynthetic rifamycin derivative highly active against some gram-negative bacteria. J. Antibiot. 40, 1733–1739. 10.7164/antibiotics.40.1733. [DOI] [PubMed] [Google Scholar]
- Mosaei H.; Molodtsov V.; Kepplinger B.; Harbottle J.; Moon C. W.; Jeeves R. E.; Ceccaroni L.; Shin Y.; Morton-Laing S.; Marrs E. C. L.; Wills C.; Clegg W.; Yuzenkova Y.; Perry J. D.; Bacon J.; Errington J.; Allenby N. E. E.; Hall M. J.; Murakami K. S.; Zenkin N. (2018) Mode of action of kanglemycin A, an ansamycin natural product that is active against rifampicin-resistant Mycobacterium tuberculosis. Mol. Cell 72, 263–274. 10.1016/j.molcel.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek J.; Lilic M.; Montiel D.; Milshteyn A.; Woodworth I.; Biggins J. B.; Ternei M. A.; Calle P. Y.; Danziger M.; Warrier T.; Saito K.; Braffman N.; Fay A.; Glickman M. S.; Darst S. A.; Campbell E. A.; Brady S. F. (2018) Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat. Commun. 9, 4147. 10.1038/s41467-018-06587-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N. J.; Fu Y.; Yan G. H.; Bao G. H.; Xu C. F.; He C. H. (1988) Isolation and structure of a new ansamycin antibiotic kanglemycin A from a Norcardia. J. Antibiot. 41, 264–267. 10.7164/antibiotics.41.264. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Degen D.; Ebright Y. W.; Ebright R. H. (2012) Frequency, spectrum, and nonzero fitness costs of resistance to myxopyronin in Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 6250–6255. 10.1128/AAC.01060-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H.; Tomioka H.; Sato K.; Emori M.; Yamane T.; Yamashita K.; Hosoe K.; Hidaka T. (1991) In vitro antimycobacterial activities of newly synthesized benzoxazinorifamycins. Antimicrob. Agents Chemother. 35, 542–547. 10.1128/AAC.35.3.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghazeh S. L.; Pan X.; Arain T.; Stover C. K.; Musser J. M.; Kreiswirth B. N. (1996) Comparative antimycobacterial activities of rifampin, rifapentine, and KRM-1648 against a collection of rifampin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Antimicrob. Agents Chemother. 40, 2655–2657. 10.1128/AAC.40.11.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M.; Suchland R. J.; Carswell J. A.; Van Duzer J.; Buxton D. K.; Brown K.; Rothstein D. M.; Stamm W. E. (2005) Activities of rifamycin derivatives against wild-type and rpoB mutants of Chlamydia trachomatis. Antimicrob. Agents Chemother. 49 (9), 3974–3976. 10.1128/AAC.49.9.3974-3976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. K.; Mullin S.; Osburne M. S.; van Duzer J.; Siedlecki J.; Yu X.; Kerstein K.; Cynamon M.; Rothstein D. M. (2006) In vitro activity of novel rifamycins against rifamycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 50, 827–834. 10.1128/AAC.50.3.827-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein D. M.; Suchland R. J.; Xia M.; Murphy C. K.; Stamm W. E. (2008) Rifalazil retains activity against rifampin-resistant mutants of Chlamydia pneumoniae. J. Antibiot. 61, 489–495. 10.1038/ja.2008.65. [DOI] [PubMed] [Google Scholar]
- Gonzalez-y-Merchand J. A.; Colston M. J.; Cox R. A. (1996) The rRNA operons of Mycobacterium smegmatis and Mycobacterium tuberculosis: comparison of promoter elements and of neighbouring upstream genes. Microbiology 142 (3), 667–674. 10.1099/13500872-142-3-667. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Wang H.; Labroli M.; Koseoglu S.; Zuck P.; Mayhood T.; Gill C.; Mann P.; Sher X.; Ha S.; Yang S. W.; Mandal M.; Yang C.; Liang L.; Tan Z.; Tawa P.; Hou Y.; Kuvelkar R.; DeVito K.; Wen X.; Xiao J.; Batchlett M.; Balibar C. J.; Liu J.; Xiao J.; Murgolo N.; Garlisi C. G.; Sheth P. R.; Flattery A.; Su J.; Tan C.; Roemer T. (2016) TarO-specific inhibitors of wall teichoic acid biosynthesis restore beta-lactam efficacy against methicillin-resistant staphylococci. Sci. Transl. Med. 8, 329ra32. 10.1126/scitranslmed.aad7364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.