

Abstract
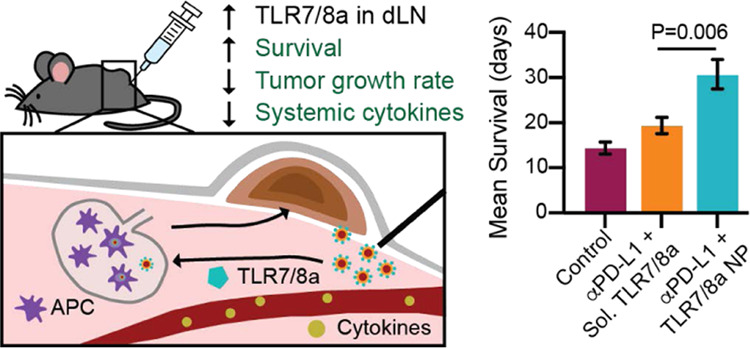
Cancer immunotherapy can be augmented with toll-like receptor agonist (TLRa) adjuvants, which interact with immune cells to elicit potent immune activation. Despite their potential, use of many TLRa compounds has been limited clinically due to their extreme potency and lack of pharmacokinetic control, causing systemic toxicity from unregulated systemic cytokine release. Herein, we overcome these shortcomings by generating poly(ethylene glycol)–poly(lactic acid) (PEG–PLA) nanoparticles (NPs) presenting potent TLR7/8a moieties on their surface. The NP platform allows precise control of TLR7/8a valency and resulting surface presentation through self-assembly using nanoprecipitation. We hypothesize that the pharmacokinetic profile of the NPs minimizes systemic toxicity, localizing TLR7/8a presentation to the tumor bed and tumor-draining lymph nodes. In conjunction with antiprogrammed death-ligand 1 (anti-PD-L1) checkpoint blockade, peritumoral injection of TLR7/8a NPs slows tumor growth, extends survival, and decreases systemic toxicity in comparison to the free TLR7/8a in a murine colon adenocarcinoma model. These NPs constitute a modular platform for controlling pharmacokinetics of immunostimulatory molecules, resulting in increased potency and decreased toxicity.
Introduction
Cancer immunotherapies based on immune checkpoint antibodies continue to gain interest for clinical development on account of their tremendous therapeutic potential. The most widely used cancer immunotherapies are antibodies that prevent the suppressive checkpoint interactions of CTLA4 with CD80/CD86 and PD1 with PD-L1. Though anti-CTLA4 and anti-PD1 or anti-PD-L1 therapies have shown great efficacy in some cancers, the overall response rates are highly variable on account of multifarious tumor cell evasion mechanisms.1 Supplementing PD-L1 checkpoint blocking antibodies with toll-like receptor agonist (TLRa) adjuvants, as well as other innate activators, such as stimulator of interferon gene (STING) agonists, has shown great promise toward overcoming resistance mechanisms responsible for low response rates.2−5 In particular, TLR7/8a, which are typically structural mimics of single-stranded RNA (ssRNA), can elicit extremely potent immune responses, and have been shown to synergize with immune checkpoint therapies.6 Unfortunately, the applicability of TLR7/8a compounds in cancer immunotherapy is currently limited to skin cancers, including metastatic cancers presenting on the skin, as systemic distribution of these compounds results in severe toxicity.7−9 Control over the pharmacokinetics, pharmacodynamics, and biodistribution of these compounds is crucial for their translation into the clinic, emphasizing the need for optimized drug delivery approaches.
TLR7/8a primarily activate pathways in innate immune cells, including dendritic cells (DCs) and macrophages, by mimicking ssRNA that are natural ligands for pathogen-associated molecular patterns (PAMPs). Activation of TLR7/8 boosts antigen presentation by DCs and macrophages through downstream signaling and cytokine production as part of the multifaceted adaptive immune response. Synergy with PD-L1 blockade results from co-administration of TLR7/8a compounds, whereby TLR7/8 activation aids DCs residing in the tumor and tumor-draining lymph nodes in priming naïve T-cells toward tumor antigens, resulting in tumoricidal behavior that can then be enhanced and prolonged by the addition of PD-L1 blockade.10,11 Immunosuppressive tumors experience low levels of T-cell priming, which typically renders PD-L1 checkpoint blockade ineffective. In contrast, the addition of stimulatory molecules like TLR7/8a can initiate innate immune cell activation and kick-start downstream T-cell responses.
As the TLR7/8 ligand binding site can be found on the endosomal lumen of DCs and macrophages, TLR7/8a compounds that are covalently bound to macromolecular constructs can activate TLR7/8 receptors without needing to be released.12 Moreover, the pharmacokinetic properties for such a construct would be strictly dictated by the nanoparticle (NP) properties. Numerous analogs of Resiquimod (R848), a popular TLR7/8a with high specificity and potency, have been developed that have functional handles (e.g., primary amines) that make them amenable to conjugation to macromolecular constructs. Multiple approaches to polymer conjugation of these TLR7/8a compounds have been effective in modulating the pharmacokinetics and pharmacodynamics of these analogues, which has proven to generate more potent adjuvants for therapeutic and prophylactic vaccines. For example, Lynn et al. found that the morphology of NPs formed from TLR7/8a-conjugated polymers had a significant impact on both induction of cytotoxic T cells and antibody production against a co-presented antigen.13,14 Nuhn et al. have likewise shown that pH-responsive polymeric micelles with conjugated TLR7/8a ligands are effective in eliciting a local immune response with low toxicity that synergizes with PD-L1 antibody antagonists.15,16
One primary benefit afforded by TLR7/8a conjugation to macromolecular constructs is the opportunity to drive more favorable tissue distribution and pharmacokinetics that give rise to enhanced therapeutic efficacy and lower toxicity. Altering the biodistribution and pharmacokinetics of TLR7/8a impacts which immune cells are exposed and become activated and, consequently, can lead to dramatically different pharmacodynamic properties. We hypothesized that conjugation of TLRa moieties to the exterior of poly(ethylene glycol)–poly(lactic acid) (PEG–PLA) NPs would provide an opportunity to not only control potency through TLRa valency on the NP, but also to control biodistribution, pharmacokinetics, and pharmacodynamics (Scheme 1). Core-shell NPs comprising PEG–PLA block copolymers are a modular platform that have been widely employed for drug delivery applications, and have been evaluated in numerous human clinical trials, and are considered highly biocompatible.17−21 Typically, drug cargo are encapsulated into the core of the NPs, while conjugation of targeting ligands to the exterior of the NPs has been employed for recognition and localization of the NPs for selective delivery to certain cells.22−25 Here, we show that TLRa moieties can be conjugated to the terminus of the PEG corona to directly present potent innate activators on the surface of the NPs. Physical mixing of functionalized PEG–PLA polymers and plain, unmodified PEG–PLA polymers at various ratios prior to nanoprecipitation provides a facile approach to controlling the density of conjugated TLRa moieties on the NP surface. Nanoprecipitation of the PEG–PLA block copolymers results in stable kinetically frozen micelle particles, as shown by their lack of a critical micelle concentration (CMC).26 In this work, we evaluate these TLR7/8a-PEG–PLA NPs in a murine model of colon adenocarcinoma (MC38) and demonstrate that they slow tumor growth, extend survival, and decrease systemic toxicity in comparison to the free TLR7/8a when used in combination with anti-PD-L1 checkpoint blockade. As such, we demonstrate that the PEG–PLA NPs constitute a promising and highly modular NP construct for improving the efficacy and safety of the potent TLRa ligands in augmenting cancer immunotherapy.
Scheme 1. TLR7/8a Delivery in a Subcutaneous Murine Tumor Model.
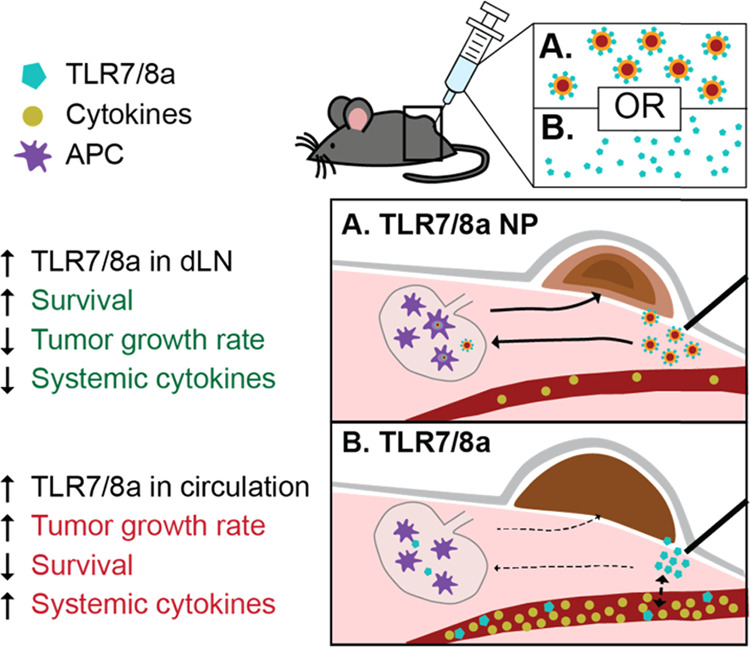
(A) Peritumoral TLR7/8a NP delivery results in lower levels of circulating cytokines (brown circles), enhanced drainage to lymph nodes, and potent activation of the TLR7/8 receptors in antigen presenting cells (APCs) (purple cells) like DCs and macrophages. The altered cytokine profile and enhanced drainage of TLR7/8a to lymph nodes significantly slows tumor growth, extends survival, and results in less systemic toxicity. (B) Peritumoral free TLR7/8a delivery results in increased levels of many circulating cytokines, nonspecific diffusion throughout the body, and likely less APC activation in lymph nodes. This delivery route leads to greater systemic toxicity and weaker antitumor effects.
Materials and Methods
Reagents
All chemicals were purchased through Sigma-Millipore, unless stated otherwise. Synthesis of alkynated mannose, alkynated TLR7/8a, and PEG–PLA block copolymers is described in the Supporting Information.
Materials Characterization
NMR was obtained using an Inova 300 MHz NMR spectrometer with a Varian Inova console using VNMRJ 4.2 A software. Number-average (Mn) and weight-average (Mw) molar mass and dispersity (Đ = Mw/Mn) of the polymers were obtained from gel permeation chromatography (GPC) carried out using a Dionex Ultimate 3000 instrument (including pump, autosampler, and column compartment) outfitted with an ERC Refractomax 520 refractometer. The columns were Jordi Resolve DVB 1000 Å, 5 μm, 30 cm × 7.8 mm, and a Mixed Bed Low, 5 μm, 30 cm × 7.8 mm, with a Jordi Resolve DVB Guard Column, 1000 Å, 5μ, 30 cm × 7.8 mm, 5 cm × 7.8 mm. Dimethylformamide (DMF) with 10 mM LiBr was used as the eluent at 1 mL/min at room temperature. Poly(ethylene glycol) was used to calibrate the GPC system. Analyte samples at 2 mg/mL were filtered through a nylon membrane with 0.2 μm pore size before injection (20 μL). Data was analyzed using Chromeleon GPC/SEC Software.
Nanoparticle Synthesis and Characterization
NPs were prepared as previously reported.27 A 1 mL solution of a combination of PEG–PLA, TLR7/8a-PEG–PLA, and Mannose-PEG–PLA (depending on the experiment, as shown in Table S2) in acetonitrile (50 mg/mL) was added dropwise to 10 mL of water under a high stir rate (600 rpm). NPs were purified by ultracentrifugation over a filter (molecular weight cutoff of 10 kDa; Millipore Amicon Ultra-15) followed by resuspension in phosphate buffered saline to a final concentration of 200 mg/mL. NPs were characterized by dynamic light scattering (DLS) to determine the NP diameters and ζ-potential for the NPs (Tables S3 and S4).
In Vitro RAW-Blue Reporter Assay
The RAW-Blue reporter cell line (InvivoGen, raw-sp) was used in this study. The cells were cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) supplemented with l-glutamine (2 mM), d-glucose (4.5 g/L), 10% heat inactivated fetal bovine serum (Atlanta Biologicals), and penicillin (100 U/mL)/streptomycin (100 μg) and zeocin (100 μg/mL; Invivogen). Serial dilutions of free TLR7/8a or one of the TLR7/8a NP formulations (20 μL) were added to a 96-well tissue culture-treated plate to final concentrations ranging from 0.08 to 10 μg/mL. About 10 000 cells were added to each well in 180 μL of media. The cells were cultured for 24 h at 37 °C in a CO2 incubator before following manufacturer instructions for SEAP quantification (absorbance at 655 nm). Fits were generated using the “log(agonist) vs response – Find EC50” in GraphPad Prism with the lower bound constrained to a constant value (0.22) for all fits.
Animal Studies
Eight–ten weeks old female C57BL/6 mice were obtained from Charles River and were cared for according to the Institutional Animal Care and Use guidelines. Animal studies were performed in accordance with the guidelines for the care and use of laboratory animals; all protocols were approved by the Stanford Institutional Animal Care and Use Committee.
In Vivo Serum IFNα Quantification
Mice were injected with buffer (200 μL) containing free TLR7/8a or one of the TLR7/8a NP formulations (25 μg of TLR7/8a dose). Mice were injected intraperitoneal (IP) since this administration route resulted in quantifiable cytokine levels across treatment groups. The serum was collected at the indicated times by tail vein blood collection and stored at −80 °C. The serum IFNα concentrations were determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (PBL Assay Science). Absorbance was measured at 450 nm in a Synergy H1 microplate reader (BioTek). Cytokine concentrations were calculated from the standard curves and represented as ng/mL.
MC38 Tumor Inoculation and Treatments
The MC38 colon carcinoma cell line was purchased from Kerafast and cultured using DMEM (Thermo Fisher Scientific) supplemented with l-glutamine (2 mM), 0.1 mM nonessential amino acids, 10% heat inactivated fetal bovine serum (Atlanta Biologicals), penicillin (100 U/mL)/streptomycin (100 μg), and 10 mM Hepes (Sigma-Aldrich). 5 × 105 MC38 cells suspended in 100 μL of phosphate-buffered saline (PBS) were injected subcutaneously (SC) on the right side of the back of C57BL/6 mice. Mice received IP injections on days 8, 10, 12, and 15 post inoculation with either PBS or 100 μg of rat monoclonal antimouse aPD-L1 antibody (clone 10F.9G2; Bio-X Cell). At the same time as the IP injections, mice were injected subcutaneously (SC) with 50 μL of free TLR7/8a or high-valency TLR7/8a NPs. The free TLR7/8a treatment also included PEG–PLA NPs without any conjugated TLR7/8a to account for any effects of the polymer NPs themselves. Tumor growth was monitored by measuring the tumors with digital calipers (Mitutoyo Digimatic Caliper) 3 days a week. Tumor area was calculated from a length and width measurement (area = length x width). Mice were euthanized when tumor area exceeded 150 mm2. On day 50, the tumor-free survivor was rechallenged by SC injection on the right flank with 5 × 105 MC38 cells and tumor area monitoring was continued.
Liver Toxicity Assessment
The mice were treated SC with 50 μL of either free TLR7/8a or high-valency TLR7/8a NPs (n = 3 for each group) 4 times. Injections occurred on days 0, 2, 4, and 7. On day 8, mice were sacrificed and blood was collected by cardiac puncture. Whole blood samples were submitted to the Animal Diagnostic Laboratory in the Stanford Department of Comparative Medicine Veterinary Service Center for the quantification of alanine aminotransferase (ALT) and asparate aminotransferase (AST).
Statistical Analysis
Statistical analysis in Figures 2−4 and 5 was done using GraphPad prism software. Data in Figure 2A,B were fit using a log(agonist) vs response fit constrained to 0.22 (average of the unconstrained minimum values) for the minimum response value. The EC50 and maximum response values were extrapolated from the fits and reported in Figure 2C. Mean values in Figure 2D,E were compared by ordinary one-way analysis of variance (ANOVA) with multiple comparisons to the control group (free TLR7/8a). In Figure 4C–E, mean fluorescence intensity (MFI) values were analyzed by t test. Statistical analysis of the MFI values shown on the heatmap in Figure 4B is shown in Table S4. A t test was run to compare NP treatment to free TLR7/8a treatment for each individual cytokine. Additional corrections were done to take into account error from multiple comparisons in the Luminex assay including a false-discovery rate (FDR) two-stage step-up method of Benjamini, Krieger, and Yekutieli correction and multiple comparisons with the Bonferroni method. In Figure 4F, mean % change in mass values on day 7 (final day of treatment) was analyzed by t test with multiple comparisons to the control group. *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant.
Figure 2.

In vitro and in vivo activity of TLR7/8a NPs. (a, b) Activity graphs across a range of TLR7/8a concentrations (0.08–10 μg/mL) delivered on NPs at different densities, on NPs with or without mannose, or as free TLR7/8a to the RAW-Blue murine macrophage reporter cell line (Invivogen). The absorbance at 655 nm is an output for TLR activation in this cell line. (c) EC50 values (using a log(agonist) vs response) and maximum absorbance values for each activation curve. (d) ELISA analysis of IFNα in serum of C57BL/6 mice 3–18 h after intraperitoneal administration of NP or free TLR7/8a treatments (n = 3). (e) Area-under-the-curve (AUC) of IFNα in serum from 3 to 18 h (n = 3). (d, e) Data depict mean ± standard error of the mean (SEM); values were analyzed by ordinary one-way ANOVA with multiple comparisons to the control group (*p < 0.05, **p < 0.01).
Figure 4.

Evaluation of the toxicity of combination immunotherapies. (a) Timeline for murine colon adenocarcinoma (MC38) inoculation and serum collection. Blood for Luminex analysis was collected 2 h after the first treatment. (b) Heatmap depiction of mean MFI cytokine levels in mouse serum as determined by Luminex (n = 3). Cytokines are listed in descending order based on average fold change (Δ = Average Free TLR7/8a MFI/Average TLR7/8 NP MFI). p-values are presented in Table S4. (c) Levels of select cytokines that are critical for the anticancer response (n = 3). (d) Levels of select proinflammatory cytokines in serum (n = 3). (e) Levels of select chemokines in serum (n = 3). (c–e) Data depict mean ± SEM; p-values were analyzed by t test. (f) Change in mouse body mass over the course of treatment (n = 8). Young, healthy mice show a weight gain of about 5% of their body mouse each week.39 Data depict mean ± SEM; p-values were analyzed by ordinary one-way ANOVA at day 7 with multiple comparisons to the control group. For all data, *p < 0.5, **p < 0.01, ***p < 0.001.
MC38 Tumor Growth and Survival Statistical Analysis
The mice were assigned randomly to 4 treatment groups (i) no treatment, (ii) aPD-L1, (iii) aPD-L1 + free TLR7/8a, and (iv) aPD-L1 + TLR7/8a NPs. For statistical analysis, the tumor area required additional transformation using the natural logarithm to meet the assumptions of homoscedasticity. Analysis was performed in JMP Pro 14. To test if tumor growth differed between treatments, we used a restricted maximum likelihood (REML) mixed model. The mouse was included as a random effect subject. The interaction between treatment and time tested whether treatment altered tumor growth over time. Post-hoc pair-wise comparisons were done between treatment groups and a Bonferroni correction was used to adjust for multiple comparisons (α = 0.008). p-values for pair-wise comparisons (α = 0.008): aPD-L1 vs control, p = 0.0001; TLR7/8a vs control, p = 0.0001; NP TLR7/8a vs control, p < 0.0001; aPD-L1 vs free TLR7/8a, p = 0.7263; NP TLR7/8a vs aPD-L1, p < 0.0001; NP TLR7/8a vs free TLR7/8a, p < 0.0001. Survival statistical analysis was performed in SAS Version 9.4. To test if survival differed between treatments, we used a maximum likelihood parametric regression with censored data. Least-squared means were used to compare survival time between individual treatments and Tukey–Kramer post-hoc tests were used to correct for multiple comparison.
Body Mass Measurements
Mouse body mass was monitored every other day for the first 10 days following the start of treatment using a digital kitchen scale with 0.1 g resolution.
Luminex
Two hours after the first treatment, blood samples were collected by tail vein bleeds. Blood was collected in serum centrifuge tubes (Sarstedt), incubated at RT for 30–60 min, and was spun at 10 000 RCF for 5 min. The serum was collected and kept frozen at −80 °C until use for Luminex analysis. The Luminex assay was performed by the Human Immune Monitoring Center at Stanford University. Mouse 38-plex Procarta kits were purchased from eBiosciences/Affymetrix/Thermo Fisher, Santa Clara, California, and used according to the manufacturer’s recommendations with modifications as described. Briefly, the beads were added to a 96-well plate and washed in a Biotek ELx405 washer. The samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature for 1 h followed by overnight incubation at 4 °C with shaking. Cold (4 °C) and room-temperature incubation steps were performed on an orbital shaker at 500–600 rpm. Following the overnight incubation, the plates were washed in a Biotek ELx405 washer and then biotinylated detection antibody was added for 75 min at room temperature with shaking. The plate was washed as above, and streptavidin-PE was added. After incubation for 30 min at room temperature, a wash was performed as above and reading buffer was added to the wells. Each sample was measured in duplicate. The plates were read using a Luminex 200 with a lower bound of 50 beads per sample per cytokine. Custom Assay Chex control beads were purchased from Radix Biosolutions, Georgetown, Texas, and are added to all wells.
Results and Discussion
As the basis of a modular PEG–PLA NP platform, we first synthesized azide-terminated PEG–PLA from N3-PEG-OH. The block copolymers were subsequently functionalized with alkyne derivatives of TLR7/8a or mannose (Figures 1A, S1, and S2). A series of NPs with varying surface densities of the TLR7/8a ligand and mannose were formed from physical mixtures of modified and unmodified PEG–PLA by nanoprecipitation according to the standard protocols (Figure 1B).17−21 This approach allows for NPs to be prepared with consistent sizes and surface charge by ζ-potential, regardless of the identity or density of the molecule(s) attached to the NP surface (Tables S2 and S3). Further, as the PLA polymers comprising the core are biodegradable and the 5 kDa PEG polymers of the corona are sufficiently small to allow for renal clearance, the constituents of these constructs can be readily eliminated from the body to prevent bioaccumulation after treatment.
Figure 1.
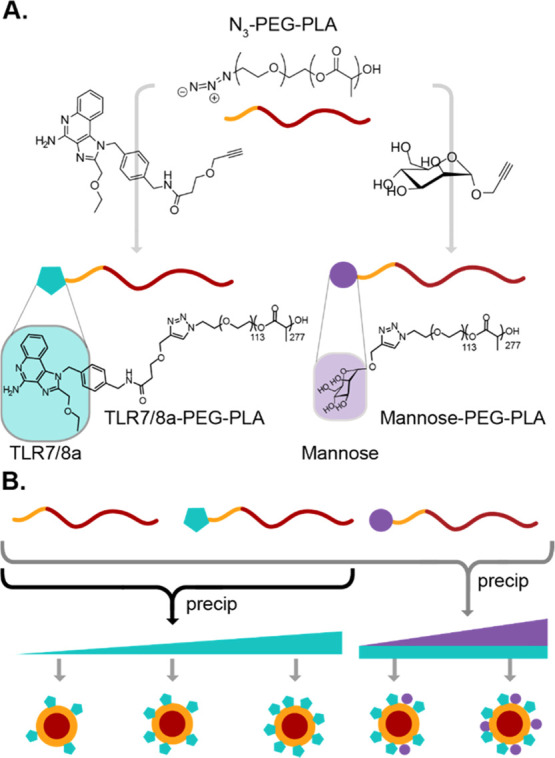
Preparation of TLR7/8a NPs. (a) N3-PEG–PLA block copolymers can be modified by alkyne-azide “click” chemistry with either a mannose analog or a potent TLR7/8a. (b) Physical mixing of modified PEG–PLA with different termini at various ratios with unmodified PEG–PLA enables simple manufacturing of the nanoparticles by nanoprecipitation with control of surface presentation of the conjugated moieties.
In vitro studies were conducted to evaluate the impact of TLR7/8a and mannose valency on the NPs and their potency in innate immune cell activation. We hypothesized that an optimal density of TLR7/8a ligand would arise, exhibiting the greatest potency. NPs of 30 nm have been shown to clear by lymphatic drainage, and we target NPs of this size for this study.28−32 Moreover, recent studies of TLR7/8a-functional macromolecular constructs indicate that mannose functionalization can increase recognition and internalization by mannose-binding C-type lectins.14,33 We therefore hypothesized that introduction of mannose to the NP surface alongside TLR7/8a may improve NP uptake and therefore receptor activation.14,33 We used RAW-Blue transgenic mouse macrophage reporter cells (Invivogen) to evaluate the potency of interferon regulatory factor (IRF) activation by TLR7/8a-tethered NPs. In these assays, the cells were incubated with TLR7/8a at a range of concentrations (0.08–10 μg/mL) either in free form or tethered to PEG–PLA NPs (TLR7/8a NPs) at different densities to generate concentration-dependent activation curves. The density of TLR7/8a on NPs influenced the observed EC50 values and maximum values of the activation curves (Figure 2A,C), which are indicators of TLRa potency. A lower EC50 is optimal because it indicates that a lower TLR7/8a concentration is needed to reach the half-maximum activation. TLR7/8a presented at a medium or low density on the surface of the NPs (medium/low valency) resulted in EC50 values that were between 3 and 4-fold greater than EC50 for the free TLR7/8a curve (Figure 2A,C). Unlike the NPs with low TLR7/8a valency, which had a very low maximum activation, the NPs with medium and high TLR7/8a valency had similar maximum activation values to that of free TLR7/8a (Figure 2A,C).
In these assays, we also evaluated medium-valency TLR7/8a-functional NPs bearing either low or high mannose valency in the same RAW-Blue macrophage reporter cell line assay to quantify TLR activation. We found that the presence of mannose increased activation, decreasing EC50 values by approximately 2-fold, regardless of the mannose density. Yet, both mannose densities led to modest decreases in maximum activation compared to the free TLR7/8a and the medium-valency TLR7/8a NPs without mannose (Figure 2B,C).
These results demonstrate that tethering TLR7/8a to the surface of the PEG–PLA NPs slightly decreases the potency of the molecule in vitro and co-presentation of mannose on the NP surface does not significantly alter the potency of the construct. This may be due to the mannose only being present as a monosaccharide, and more complex presentation is necessary to promote lectin-mediated opsonization.34 In the case of TLR7/8a-functional PEG–PLA NPs, the difference in potency and maximum activation across various TLR7/8a densities may be due to the pattern of TLR7/8a presentation, which can lead to receptor clustering that is necessary for the downstream responses observed.35 Previously reported TLR7/8a delivery systems exhibit similar results whereby tethering of a TLR7/8a to a macromolecular construct lead to decreased potency in cell-based assays when compared to the free TLR7/8a, even when these constructs exhibited greater potency in vivo.15 It is important to consider that in vitro cell assays do not take into account biodistribution and pharmacokinetics, and TLRa molecules may act upon a broad array of immune cells in different tissues within an organism. For this reason, we determined the potency of TLR7/8a NPs over time in vivo to evaluate the impact of TLR7/8a density on the time frame of innate immune cell activation.
In these assays, serum IFNα concentration was quantified at several time points following intraperitoneal (IP) administration of TLR7/8a NPs or free TLR7/8a in C57BL/6 mice. IFNα is a critical cytokine produced in response to TLR7/8 activation that is known to contribute to productive antitumor responses.36 Free TLR7/8a treatment led to an early spike (3 h) in serum IFNα levels followed by a rapid decrease (Figure 2D). In contrast, all NP-based treatments led to peak serum IFNα levels at 6 h, and these levels remained elevated through 9 h (Figure 2D). The high-valency TLR7/8a NP treatment generated significantly higher IFNα levels at 6 and 9 h post administration compared to the free TLR7/8a treatment. Indeed, the high-valency TLR7/8a NPs exhibited a significantly higher area-under-the-curve (AUC) of serum IFNα levels over the 18 h period than all other NP treatments evaluated (Figure 2D,E). These in vivo experiments showed that TLR7/8a NP treatment prolonged IFNα serum concentrations following a single administration compared to free TLR7/8a treatment. We expect the higher potency in vivo of the high-valency NPs is the result of differences in immune cell composition and the in vivo processing of the NPs. Based on these results, the high-valency TLR7/8a NP was identified as the most promising adjuvant candidate to augment checkpoint blockade therapy in the MC38 model using an antibody against PD-L1 (aPD-L1). The effectiveness of cancer immunotherapy depends on activation of tumor-specific cytotoxic T lymphocytes (CTLs).37 The immune checkpoint blockade antibody aPD-L1 blocks an interaction between tumor cells and CTLs that inhibits CTL activation, therefore improving tumor killing.38 Unfortunately, response rates of only ∼20% have been reported for this antibody treatment clinically, likely due to an insufficient number of activated CTLs reaching the tumor.38 Potent yet nontoxic TLR7/8 activation in the tumor and tumor-draining lymph node has the potential to promote CTL recruitment and activation to the tumor, potentially synergizing with aPD-L1 therapy and ultimately resulting in more effective and safe immunotherapy than either component alone.
In these studies, the MC38 cells were injected subcutaneously (SC) on the right flank of the C57BL/6 mice. A series of four doses of each treatment began once tumors were measurable—on day 8 following tumor cell inoculation (Figure 3A). We chose to start treatment once all tumors were measurable and treated with a relatively low dose of aPD-L1 since treating a visible tumor and administering therapies with low toxicity are important for translation. Mouse mass and tumor area were monitored until the tumors reached euthanasia criteria at 150 mm2 (Figure 3A). We evaluated four treatment groups: (i) intraperitoneal (IP) phosphate-buffered saline (PBS) injections with peritumoral (PT) unmodified PEG–PLA NP (no TLR7/8a), (ii) IP aPD-L1 with PT unmodified PEG–PLA NP (no TLR7/8a), (iii) IP aPD-L1 with PT free TLR7/8a, and (iv) IP aPD-L1 with PT high-valency TLR7/8a NP. Control mouse tumors treated with only saline and unmodified PEG–PLA NPs grew consistently and quickly (Figure 3B). Mice that received aPD-L1 treatment alone (i.e., alongside unmodified PEG–PLA NPs) exhibited slightly more varied patterns of tumor growth (Figure 3C). Mice in the cohort that received aPD-L1 treatment and free TLR7/8a experienced relatively slowed tumor growth, but ultimately all mice succumbed to the tumors (Figure 3D).
Figure 3.

Treatment of murine colon adenocarcinoma (MC38). (a) Timeline for MC38 inoculation and treatment. The mice (C57BL/6) were inoculated with half a million MC38 cells SC on the right flank. Measurement and treatments began 8 days later when the tumors were palpable. Treatments were given 4 times over 8 days. Mice were euthanized when tumors reached 150 mm2. (b–e) Tumor growth curves over time for individual mice that received PBS injections as a control (b), IP aPD-L1 treatment (c), IP aPD-L1 and peritumoral (PT) free TLR7/8a treatment (d), and IP aPD-L1 and PT NP TLR7/8a treatment (e) (n = 8). (f) Average tumor growth for each treatment group for the first 10 days following the start of treatment (n = 8). (g) Survival curves showing percent survival over the duration of the study of all treatment groups. (h) Mean survival for each treatment group. (f) Data depict mean ± standard error of the mean (SEM); the values were analyzed by ordinary one-way ANOVA at day 10 with multiple comparisons to the control group (*p < 0.05, **p < 0.01). (h) Survival means are shown as inverse link transformed least squares mean ± SE. Tukey–Kramer post-hoc tests were used to correct for multiple comparisons.
In contrast, the mice that received aPD-L1 treatment and TLR7/8a NPs exhibited tumor growth curves that varied quite substantially, with some tumors shrinking before growing out, others growing out at a similar rate to those treated with free TLR7/8a, and some completely controlling the tumors (Figure 3E,G). This was the only treatment group where some mouse tumors receded completely during the study (n = 2) and one of these mice was ultimately cured (Figure 3E). The cured mouse was rechallenged with the MC38 cells 50 days after the start of the initial treatments and did not regrow a tumor, suggesting that immune memory was generated against the cancer during the initial round of treatment. The primary limitation of TLR7/8a therapy is extreme systemic toxicities.40 In this work, we sought to both enhance the efficacy and decrease the toxicity of TLR7/8a treatments by tethering the TLR7/8a molecules to PEG–PLA NPs. As agonism of TLR7/8 causes high levels of many cytokines, particularly proinflammatory cytokines resulting in systemic immune activation and flu-like symptoms,12 we hypothesized that presentation of TLR7/8a on the surface of PEG–PLA NPs would restrict immune activity to the local tumor environment and the tumor-draining lymph nodes, limiting systemic exposure. Since the TLR7/8a molecule on its own is small, it can rapidly enter systemic circulation, whereas the TLR7/8a NPs are in a size regime, approximately 30 nm, that has been shown to drain passively to lymph nodes.28−32,41 In these assays, we assessed toxicity by conducting Luminex analysis of cytokines in mouse serum 2 h after the initial treatment and by measuring mouse body mass over the course of treatment (Figure 4A).
Cytokines are key players in the anticancer immune response that act by triggering cell differentiation, inhibiting growth, and attracting specific leukocytes to an area of inflammation, among other functions. Type I IFNs, for example, are responsible for priming of tumor-specific CD8 T cells and attracting natural killer (NK) cells and other leukocytes to the tumor site by promoting production of CXCL9 and CXCL1.42 Unfortunately, high levels of systemic cytokines, including those that are useful and necessary in the proper abundance at the appropriate location, can cause harmful and dysregulated immune responses in distal parts of the body.
With Luminex analysis, we quantified a large panel of cytokines in serum collected 2 h after the first treatment injection (Figure 4A,B). We chose this timepoint because we were interested in differences in the acute response to TLR7/8a when delivered in the free form versus the NP form. We chose to assess toxicity in tumor-bearing mice since the presence of tumors can impact TLR7/8a drainage, but we also wanted to test for toxicity before tumor burden between groups diverged too much so that the background was consistent.
Luminex analysis showed lower overall systemic cytokine responses in the mice that received the TLR7/8a NP treatment as compared to the free TLR7/8a treatment (Figure 4B). Select cytokines known to be implicated in potent anticancer responses but which are also extremely toxic at high concentrations were selected prior to running the analysis and were plotted separately as bar graphs (Figure 4C–E). High serum concentrations of the activating cytokines IFNα and IL-12 have been observed in a number of preclinical and clinical trials to be associated with autoimmune effects and flu-like symptoms.43 In these assays, treatment with TLR7/8a NPs resulted in lower levels of serum IFNα and IL-12 than treatment with the free TLR7/8a (Figure 4C). Likewise, high levels of proinflammatory cytokines such as TNFα and IL17A in the serum, a common side effect of TLR7/8a treatment,44 are linked with general inflammation as well as sepsis and lupus.45,46 The mice receiving the TLR7/8a NP treatment had significantly lower systemic levels of both of these cytokines than those receiving the free TLR7/8 treatment (Figure 4D). Moreover, while chemokines are critical for antitumor responses since they can attract various leukocytes to the site of the tumor,42 chemokines such as IP10 or CCL2 at high concentrations in serum unfortunately lead to systemic sclerosis.47 Again, we observed that serum concentrations of both IP10 and CCL2 were significantly lower in mice receiving TLR7/8a NP treatment as compared to the free TLR7/8a treatment (Figure 4E). Although cytokines in Figure 4B are based on the fold change between group averages, it is important to note that there are not clear functional cutoffs for serum cytokine levels. A slight change in one cytokine may contribute more to overall toxicity or efficacy than a very large change in another that appears much more significant. Overall, TLR7/8a NPs exhibited significantly reduced systemic levels of many cytokines that are known to contribute to toxic effects of TLR7/8a, likely on account of the altered biodistribution and pharmacokinetics afforded by conjugation of the TLR7/8a compound to the PEG–PLA NP construct.
On the final day of treatment, the mice receiving the TLR7/8a NP treatment had a significantly higher average body mass than all other groups (Figure 4F). Severe toxicity and/or illness typically lead to a decrease in body mass in mice. The mice used in these experiments were 8-weeks-old at the start of treatment and as such would be expected to gain about 5% body mass each week.39 The sham-treated control mice did not gain as much mass as expected, suggesting that the cancer alone is quite unhealthy for the mice (Figure 4F). Moreover, aPD-L1 treatment alone and augmented with free TLR7/8a treatment led to the similar poor thriving of the mice. In contrast, the low toxicity of the TLR7/8a NP treatment observed through assessment of systemic cytokines was corroborated by this aggregate measure, whereby this treatment enabled mice to gain the expected percentage of their body mass throughout the treatment period (Figure 4F). As an additional assessment of systemic toxicity, serum levels of the liver enzymes alanine aminotransferase (ALT) and asparate aminotransferase (AST) were determined. Enzyme levels in mice that received 4 SC doses of either free TLR7/8a or TLR7/8a NPs were compared to the levels of the untreated control mice. Treatment with TLR7/8a NPs led to similarly low levels of both AST and ALT as was seen in the control mice, while treatment with free TLR7/8a led to higher and more varied levels of AST and ALT (Figure S4).
Conclusions
Potent TLR7/8 agonists presented on the surface of the PEG–PLA NPs were shown to retain their agonism in vitro, with mannose-functionalized particles showing a negligible increase in potency. In vivo, the NPs presenting TLR7/8 agonists led to prolonged and elevated levels of type I IFN compared to the free TLR7/8a. In a murine cancer model, these TLR7/8a NPs were shown to potently synergize with PD-L1 checkpoint blockade to slow tumor growth and extend survival while reducing systemic cytokine release and decreasing toxicity. Overall, this study reports a simple, modular approach to preparation of the PEG–PLA nanoparticles bearing potent TLR7/8 agonists that can overcome the current toxicity limitations of the TLR7/8a compounds and generate a viable complement to PD-L1 checkpoint therapy with enhanced safety and efficacy.
Acknowledgments
The authors would like to thank members of the Appel Lab for their useful discussion and advice throughout this project. The authors would also like to thank Juliana Idoyaga for helpful discussions regarding this work.
Glossary
Abbreviations Used
- CCR2
CC chemokine receptor 2
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
- TLC
thin layer chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00812.
Composition of the nanoparticles tested (Table S1); nanoparticle size and ζ-potential (Tables S2 and S3); raw Luminex data (Table S4) (PDF)
Author Contributions
# A.A.A.S. and E.C.G. contributed equally. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
A.A.A.S. was funded by grant NNF18OC0030896 from the Novo Nordisk Foundation and the Stanford Bio-X Program. E.C.G. was funded by the NIH-funded Cell and Molecular Biology Training Program (T32 GM007276). This research was financially supported by the Center for Human Systems Immunology with Bill and Melinda Gates Foundation (OPP1113682) as well as the American Cancer Society (RSG-18-133-01).
The authors declare the following competing financial interest(s): A.A.A.S, E.C.G., G.A.R., and E.A.A. are listed as authors on a provisional patent application describing the technology reported in this manuscript.
Supplementary Material
References
- Ren D.; Hua Y.; Yu B.; Ye X.; He Z.; Li C.; Wang J.; Mo Y.; Wei X.; Chen Y.; Zhou Y.; Liao Q.; Wang H.; Xiang B.; Zhou M.; Li X.; Li G.; Li Y.; Zeng Z.; Xiong W. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19 10.1186/s12943-020-1144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins S. R.; Vasilakos J. P.; Deschler K.; Grigsby I.; Gillis P.; John J.; Elder M. J.; Swales J.; Timosenko E.; Cooper Z.; Dovedi S. J.; Leishman A. J.; Luheshi N.; Elvecrog J.; Tilahun A.; Goodwin R.; Herbst R.; Tomai M. A.; Wilkinson R. W. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. 10.1186/s40425-019-0724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. A.; Minn A. J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Su L.; Morin M. D.; Jones B. T.; Mifune Y.; Shi H.; Wang K.-w.; Zhan X.; Liu A.; Wang J.; Li X.; Tang M.; Ludwig S.; Hildebrand S.; Zhou K.; Siegwart D. J.; Moresco E. M. Y.; Zhang H.; Boger D. L.; Beutler B. Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti–PD-L1 to eliminate melanoma in mice. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E8698–E8706. 10.1073/pnas.1809232115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.; Wang H.; Wang Z.; Cai H.; Lu Z.; Li Y.; Du M.; Huang G.; Wang C.; Chen X.; Porembka M. R.; Lea J.; Frankel A. E.; Fu Y.-X.; Chen Z. J.; Gao J. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017, 12, 648–654. 10.1038/nnano.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Kaneko F.; Yao S. Y.; Ahmadi A.; Zhang S. S.; Hosoya T.; Kaneda M. M.; Varner J. A.; Pu M.; Messer K. S.; Guiducci C.; Coffman R. L.; Kitaura K.; Matsutani T.; Suzuki R.; Carson D. A.; Hayashi T.; Cohen E. E. W. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397 10.1172/jci.insight.93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits E. L. J. M.; Ponsaerts P.; Berneman Z. N.; Van Tendeloo V. F. I. The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy. Oncologist 2008, 13, 859–875. 10.1634/theoncologist.2008-0097. [DOI] [PubMed] [Google Scholar]
- Pandit A. S.; Geiger E. J.; Ariyan S.; Narayan D.; Choi J. N. Using topical imiquimod for the management of positive in situ margins after melanoma resection. Cancer Med. 2015, 4, 507–512. 10.1002/cam4.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S.; Kozhaya L.; Martiniuk F.; Meng T.-C.; Chiriboga L.; Liebes L.; Hochman T.; Shuman N.; Axelrod D.; Speyer J.; Novik Y.; Tiersten A.; Goldberg J. D.; Formenti S. C.; Bhardwaj N.; Unutmaz D.; Demaria S. Topical TLR7 Agonist Imiquimod Can Induce Immune-Mediated Rejection of Skin Metastases in Patients with Breast Cancer. Clin. Cancer Res. 2012, 18, 6748–6757. 10.1158/1078-0432.CCR-12-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcmann M.; Drobits B.; Sibilia M. How imiquimod licenses plasmacytoid dendritic cells to kill tumors. Oncoimmunology 2012, 1, 1661–1663. 10.4161/onci.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspord C.; Tramcourt L.; Leloup C.; Molens J.-P.; Leccia M.-T.; Charles J.; Plumas J. Imiquimod Inhibits Melanoma Development by Promoting pDC Cytotoxic Functions and Impeding Tumor Vascularization. J. Invest. Dermatol. 2014, 134, 2551–2561. 10.1038/jid.2014.194. [DOI] [PubMed] [Google Scholar]
- Dowling D. J. Recent Advances in the Discovery and Delivery of TLR7/8 Agonists as Vaccine Adjuvants. ImmunoHorizons 2018, 2, 185–197. 10.4049/immunohorizons.1700063. [DOI] [PubMed] [Google Scholar]
- Lynn G. M.; Laga R.; Darrah P. A.; Ishizuka A. S.; Balaci A. J.; Dulcey A. E.; Pechar M.; Pola R.; Gerner M. Y.; Yamamoto A.; Buechler C. R.; Quinn K. M.; Smelkinson M. G.; Vanek O.; Cawood R.; Hills T.; Vasalatiy O.; Kastenmuller K.; Francica J. R.; Stutts L.; Tom J. K.; Ryu K. A.; Esser-Kahn A. P.; Etrych T.; Fisher K. D.; Seymour L. W.; Seder R. A. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 2015, 33, 1201–1210. 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn G. M.; Chytil P.; Francica J. R.; Lagova A.; Kueberuwa G.; Ishizuka A. S.; Zaidi N.; Ramirez-Valdez R. A.; Blobel N. J.; Baharom F.; Leal J.; Wang A. Q.; Gerner M. Y.; Etrych T.; Ulbrich K.; Seymour L. W.; Seder R. A.; Laga R. Impact of Polymer-TLR-7/8 Agonist (Adjuvant) Morphology on the Potency and Mechanism of CD8 T Cell Induction. Biomacromolecules 2019, 20, 854–870. 10.1021/acs.biomac.8b01473. [DOI] [PubMed] [Google Scholar]
- Nuhn L.; Van Hoecke L.; Deswarte K.; Schepens B.; Li Y.; Lambrecht B. N.; De Koker S.; David S. A.; Saelens X.; De Geest B. G. Potent anti-viral vaccine adjuvant based on pH-degradable nanogels with covalently linked small molecule imidazoquinoline TLR7/8 agonist. Biomaterials 2018, 178, 643–651. 10.1016/j.biomaterials.2018.03.026. [DOI] [PubMed] [Google Scholar]
- Nuhn L.; De Koker S.; Van Lint S.; Zhong Z.; Catani J. P.; Combes F.; Deswarte K.; Li Y.; Lambrecht B. N.; Lienenklaus S.; Sanders N. N.; David S. A.; Tavernier J.; De Geest B. G. Nanoparticle-Conjugate TLR7/8 Agonist Localized Immunotherapy Provokes Safe Antitumoral Responses. Adv. Mater. 2018, 30, e1803397 10.1002/adma.201803397. [DOI] [PubMed] [Google Scholar]
- Shi J. J.; Xiao Z. Y.; Kamaly N.; Farokhzad O. C. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc. Chem. Res. 2011, 44, 1123–1134. 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- Valencia P. M.; Hanewich-Hollatz M. H.; Gao W. W.; Karim F.; Langer R.; Karnik R.; Farokhzad O. C. Effects of ligands with different water solubilities on self-assembly and properties of targeted nanoparticles. Biomaterials 2011, 32, 6226–6233. 10.1016/j.biomaterials.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf N.; Bielenberg D. R.; Kolishetti N.; Muus C.; Banyard J.; Farokhzad O. C.; Lippard S. J. alpha(V)beta(3) Integrin-Targeted PLGA-PEG Nanoparticles for Enhanced Anti-tumor Efficacy of a Pt(IV) Prodrug. ACS Nano 2012, 6, 4530–4539. 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone T. C.; Kulak N.; Pridgen E. M.; Farokhzad O. C.; Langer R.; Lippard S. J. Nanoparticle Encapsulation of Mitaplatin and the Effect Thereof on In Vivo Properties. ACS Nano 2013, 7, 5675–5683. 10.1021/nn401905g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N.; Grenier P.; Mahmoudi M.; Lima E. M.; Appel E. A.; Dormont F.; Lim J.-M.; Karnik R.; Langer R.; Farokhzad O. C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777 10.1038/s41467-017-00600-w. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1-8.
- Cheng J.; Teply B. A.; Sherifi I.; Sung J.; Luther G.; Gu F. X.; Levy-Nissenbaum E.; Radovic-Moreno A. F.; Langer R.; Farokhzad O. C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.; Kolishetti N.; Lippard S. J.; Farokhzad O. C. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1850–1855. 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieszawska A. J.; Kim Y.; Gianella A.; van Rooy I.; Priem B.; Labarre M. P.; Ozcan C.; Cormode D. P.; Petrov A.; Langer R.; Farokhzad O. C.; Fayad Z. A.; Mulder W. J. M. Synthesis of Polymer-Lipid Nanoparticles for Image-Guided Delivery of Dual Modality Therapy. Bioconjugate Chem. 2013, 24, 1429–1434. 10.1021/bc400166j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. Y.; Xie K.; Zhang X.-Q.; Pridgen E. M.; Park G. Y.; Cui D. S.; Shi J. J.; Wu J.; Kantoff P. W.; Lippard S. J.; Langer R.; Walker G. C.; Farokhzad O. C. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18638–18643. 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almoustafa H. A.; Alshawsh M. A.; Chik Z. Technical aspects of preparing PEG-PLGA nanoparticles as carrier for chemotherapeutic agents by nanoprecipitation method. Int. J. Pharm. 2017, 533, 275–284. 10.1016/j.ijpharm.2017.09.054. [DOI] [PubMed] [Google Scholar]
- Appel E. A.; Tibbitt M. W.; Webber M. J.; Mattix B. A.; Veiseh O.; Langer R. Self-assembled hydrogels utilizing polymer-nanoparticle interactions. Nat. Commun. 2015, 6, 6295 10.1038/ncomms7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Melia M. J.; Lund A. W.; Thomas S. N. The Biophysics of Lymphatic Transport: Engineering Tools and Immunological Consequences. iScience 2019, 22, 28–43. 10.1016/j.isci.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palao-Suay R.; Martín-Saavedra F. M.; Rosa Aguilar M.; Escudero-Duch C.; Martín-Saldaña S.; Parra-Ruiz F. J.; Rohner N. A.; Thomas S. N.; Vilaboa N.; San Román J. Photothermal and photodynamic activity of polymeric nanoparticles based on α-tocopheryl succinate-RAFT block copolymers conjugated to IR-780. Acta Biomater. 2017, 57, 70–84. 10.1016/j.actbio.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S. T.; van der Vlies A. J.; Simeoni E.; Angeli V.; Randolph G. J.; O’Neil C. P.; Lee L. K.; Swartz M. A.; Hubbell J. A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- Schudel A.; Chapman A. P.; Yau M.-K.; Higginson C. J.; Francis D. M.; Manspeaker M. P.; Avecilla A. R. C.; Rohner N. A.; Finn M. G.; Thomas S. N. Programmable multistage drug delivery to lymph nodes. Nat Nanotechnol. 2020, 15, 491–499. 10.1038/s41565-020-0679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schudel A.; Sestito L. F.; Thomas S. N. Winner of the society for biomaterials young investigator award for the annual meeting of the society for biomaterials, April 11–14, 2018, Atlanta, GA: S-nitrosated poly(propylene sulfide) nanoparticles for enhanced nitric oxide delivery to lymphatic tissues. J. Biomed. Mater. Res., Part A 2018, 106, 1463–1475. 10.1002/jbm.a.36348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. S.; Hirosue S.; Raczy M. M.; Bonilla-Ramirez L.; Jeanbart L.; Wang R.; Kwissa M.; Franetich J.-F.; Broggi M. A. S.; Diaceri G.; Quaglia-Thermes X.; Mazier D.; Swartz M. A.; Hubbell J. A. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 2019, 18, 175–185. 10.1038/s41563-018-0256-5. [DOI] [PubMed] [Google Scholar]
- Adams E. W.; Ratner D. M.; Seeberger P. H.; Hacohen N. Carbohydrate-mediated targeting of antigen to dendritic cells leads to enhanced presentation of antigen to T cells. ChemBioChem 2008, 9, 294–303. 10.1002/cbic.200700310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme K. W.; Compton T. Innate Sensing of Viruses by Toll-Like Receptors. J. Virol. 2004, 78, 7867–7873. 10.1128/JVI.78.15.7867-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L.; Galluzzi L.; Kepp O.; Smyth M. J.; Kroemer G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- Sharma P.; Hu-Lieskovan S.; Wargo J. A.; Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W.; Wolchok J. D.; Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Body Weight Information for C57BL/6J. https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/body-weight-chart-000664. (accessed 17 Jul, 2020).
- Kobold S.; Wiedemann G.; Rothenfusser S.; Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy 2014, 6, 1085–1095. 10.2217/imt.14.75. [DOI] [PubMed] [Google Scholar]
- Foroozandeh P.; Aziz A. A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339 10.1186/s11671-018-2728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O.; Cornen S.; Daëron M.; Morel Y.; Medzhitov R.; Vivier E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. 10.1038/s41586-019-1593-5. [DOI] [PubMed] [Google Scholar]
- Lasek W.; Zagożdżon R.; Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy?. Cancer Immunol. Immunother. 2014, 63, 419–435. 10.1007/s00262-014-1523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeratna R. D.; Makinen S. R.; McCluskie M. J.; Davis H. L. TLR agonists as vaccine adjuvants: comparison of CpG ODN and Resiquimod (R-848). Vaccine 2005, 23, 5263–5270. 10.1016/j.vaccine.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Debandt M.; Vittecoq O.; Descamps V.; Le Loët X.; Meyer O. Anti-TNF-α-induced systemic lupus syndrome. Clin. Rheumatol. 2003, 22, 56–61. 10.1007/s10067-002-0654-5. [DOI] [PubMed] [Google Scholar]
- Nalbandian A.; Crispín J. C.; Tsokos G. C. Interleukin-17 and systemic lupus erythematosus: current concepts. Clin. Exp. Immunol. 2009, 157, 209–215. 10.1111/j.1365-2249.2009.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado A. The Th1 chemokine IP-10 in Systemic sclerosis. Clin. Ter. 2014, 165, e436–e441. 10.7417/ct.2014.1789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.