

Abstract
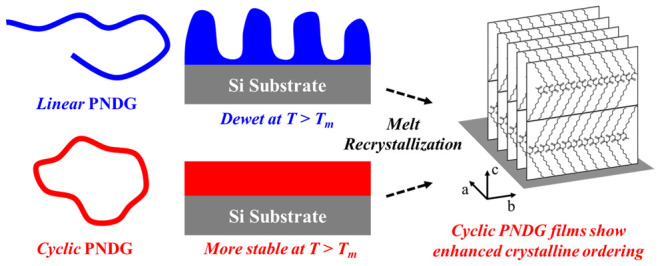
We investigated the effect of cyclic chain topology on the molecular ordering and thermal stability of comb-shaped polypeptoid thin films on silicon (Si) substrates. Cyclic and linear poly(N-decylglycine) (PNDG) bearing long n-decyl side chains were synthesized by ring-opening polymerization of N-decylglycine-derived N-carboxyanhydrides. When the spin-coated thin films were subjected to thermal annealing at temperatures above the melting temperature (T > Tm), the cyclic PNDG films exhibited significantly enhanced stability against melt-induced dewetting than the linear counterparts (l-PNDG). When recrystallized at temperatures below the crystallization temperature (T < Tc), the homogeneous c-PNDG films exhibit enhanced crystalline ordering relative to the macroscopically dewetted l-PNDG films. Both cyclic and linear PNDG molecules adopt cis-amide conformations in the crystalline film, which transition into trans-amide conformations upon melting. A top-down solvent leaching treatment of both l/c-PNDG films revealed the formation of an irreversibly physisorbed monolayer with similar thickness (ca. 3 nm) on the Si substrate. The physisorbed monolayers are more disordered relative to the respective thicker crystalline films for both cyclic and linear PNDGs. Upon heating above Tm, the adsorbed c-PNDG chains adopt trans-amide backbone conformation identical with the free c-PNDG molecules in the molten film. By contrast, the backbone conformations of l-PNDG chains in the adsorbed layers are notably different from those of the free chains in the molten film. We postulate that the conformational disparity between the chains in the physically adsorbed layers versus the free chains in the molten film is an important factor to account for the difference in the thermal stability of PNDG thin films. These findings highlight the use of cyclic chain topology to suppress the melt-induced dewetting in polymer thin films.
Introduction
Macromolecules with cyclic topology have attracted growing interests because of their unique structural and physical properties relative to the linear analogues. Because of the absence of chain ends, cyclic polymers exhibit unusual relaxation behaviors and diffusive motions that deviate from the classic reptation theory of linear polymers in concentrated systems.1−8 The closed contour shape of cyclic polymers also gives rise to more compact coil-like conformations that are often described as crumpled globules4 or “lattice animal-like” structures with folded loops in a self-similar manner.5,9 It has also been shown that the cyclic topology has a significant impact on the self-organization or melt recrystallization of polymers, which can lead to a change in melting point, nucleation rate, crystalline growth kinetics, and crystallinity, depending on the molecular weight (Mn) and polymer types.10−14
Recent studies on topological effects of cyclic polymers have also been focused on thin film systems where polymer chains are confined to nanometer length scales between interfaces. For example, Foster and co-workers reported a retarded surface fluctuation for cyclic polystyrene molten films relative to the linear counterparts within a low molecular weight regime.15,16 Torkelson and co-workers have reported a diminished free surface effect on the glass transition temperature (Tg) of cyclic PS chains, which led to a much smaller Tg reduction at the free surface region as compared to the linear PS films.17 Several studies have also been focused on the influence of cyclic topology on dewetting or instability of thin polymer films prepared on solid substrates, which often presents a challenge in the design of thin film devices (e.g., conformal coatings, organic photovoltaics, antifouling coatings, and biosensors). Albert and co-workers have shown that cyclic poly(ε-caprolactone) (Mn = 6 kg/mol) thin films have enhanced stability against melt-induced dewetting relative to the linear analogues.18 Shi, Jiang, and co-workers investigated the film instability of linear and cyclic polystyrene (PS) film on silanized Si substrate and showed that cyclic PS (Mn = 20 kg/mol) films dewetted more slowly than the linear analogues.19
When the polymers are confined at nanometer scales, interfaces are known to have significant impacts on the structure and properties of thin polymer films. Within the past decade, the research literature regarding interfacial effects have emphasized a key point: The formation of an irreversible adsorbed layer at the polymer–substrate interface from the melt plays a vital role in determining the structure, chain dynamics, and thermal stability of substrate-supported thin polymer films.20−31 Depending on the polymer characteristics and thermal processing history, polymer chains that directly interact with the substrate surface via physisorption can possess different chain conformation, adsorbed amount, surface coverage, and density on the substrate.20,21,24,26−28,30,32 Recent studies have revealed a strong correlation between autophobic dewetting of thin polymer films and the nanoarchitectures/conformations of adsorbed chains at the interface, where the conformational difference between the adsorbed chains and free chains plays a significant role.25,29−31 Interestingly, by using the top-down solvent leaching method (also known as Guiselin’s approach),33 it has been found that cyclic PS melt forms thicker irreversibly adsorbed layers on solid substrates than the linear PS melt, which can dramatically alter the interfacial effect on chain dynamics and dewetting behaviors of polymer thin films.16
Polypeptoids are structural mimics of polypeptides and have recently attracted much attention due to their potential for biotechnological applications (e.g., antifouling coatings, drug/gene delivery, and biosensor).34−39 They are biodegradable and minimally cytotoxic toward various human cell lines.39 In addition, polypeptoids exhibit excellent thermal processability, enhanced protease stability, and good solubility in common organic solvents due to the absence of hydrogen bonding and stereogenic centers along the backbone, in sharp contrast to polypeptides.34−39 Recent synthetic developments in the organo-mediated ring-opening polymerization (ROP) of N-substituted glycine-derived N-carboxyanhydride have enabled access to well-defined linear and cyclic polypeptoids with tunable side-chain chemistry.40−42 It has been found that both linear and cyclic polypeptoids with relatively long n-alkyl side chains (4 ≤ S ≤ 14, where S is the number of carbon atoms in the linear n-alkyl group) exhibit two phase transitions with increasing temperature, i.e., a crystalline phase to a “sanidic” liquid crystalline (LC) mesophase transition and the LC mesophase-to-isotropic melt transition.42,43 In the crystalline phase, the polypeptoids chain adopt a board-like structure where the backbone is fully extended in an all cis-amide conformation and is approximately coplanar with the n-alkyl side chains.43,44 The intermolecular interactions between n-alkyl side chains likely arise from van der Waals interactions and packing constraints in the crystalline lattice, whereas the interactions between all cis-amide backbones may involve additional contributions, such as CH···O hydrogen bonding or amide dipolar interactions.43−45 The all-cis-amide backbone conformation, which is more compact and possesses a higher degree of ordering than the all-trans-amide conformation, allows for more favorable intra- and intermolecular interactions during self-assembly processes.44,45 While significant advances have been made toward understanding of the structure and assembly properties of comb-shaped polypeptoids in bulk and in solution, the understanding of how these polypeptoids behave under nanoconfinement or near interfaces is limited. Because of the unique combination of biophysiochemical properties of polypeptoids, understanding the fundamental structure–property relationships of polypeptoid thin films is important toward rational design of thin-film-based biotechnological devices, such as antifouling coatings, biosensors, and bioelectronics.
In this study, we investigated the effect of cyclic topology on the structural ordering and thermal stability of thin polymer films based on comb-shaped crystallizable polypeptoid bearing long n-alkyl side chains (i.e., poly(N-decylglycine)). Both l-PNDG and c-PNDG spin-coated films were found to be highly crystalline with board-shaped molecules stacked side-by-side normal to the silicon substrate. Interestingly, upon prolonged thermal annealing at T > Tm, l-PNDG films exhibited a significant extent of dewetting relative to the analogous c-PNDG films. A top-down solvent leaching treatment of the l/c-PNDG films revealed the presence of irreversibly and physically adsorbed PNDG monolayers at the polymer–solid interface. While the thicknesses of the monolayers are nearly identical, cyclic PNDG adsorbed chains show enhanced molecular ordering relative to the linear adsorbed chains. We postulate that the difference in the interfacial structure of adsorbed chains is one important factor that contributes to the notable discrepancy in the stability of cyclic and linear PNDG thin films against melt-induced dewetting. Cyclic topology can be used to suppress the melt-induced dewetting of comb-shaped polypeptoid films over appreciable time scales while simultaneous promoting crystalline ordering of the film.
Results and Discussion
Structural Elucidation of Linear and Cyclic PNDG Thin Films before and after Melt-Induced Dewetting
Linear and cyclic PNDG thin films (l-PNDG: DPn = 52, Mn = 10.4 kg/mol, PDI = 1.14; c-PNDG: DPn = 50, Mn = 10.2 kg/mol, PDI = 1.11 (Table S1 and Figures S1–S3)) with different thicknesses were spin-coated onto HF-treated Si substrates (hereafter denoted as Si substrates) and then subjected to thermal annealing at T = 200 °C for prolonged time under vacuum. (Note: the thermal annealing temperature is significantly higher than the isotropic melting transition temperature of the bulk polymers (Tm ≈ 174 °C for l-PNDG and Tm ≈ 169 °C for c-PNDG).42) Figure 1 shows representative optical microscopic (OM) images of 48 and 111 nm thick thin films of linear and cyclic PNDG (hereafter denoted as l/c-PNDG films) on Si substrates after annealed at T = 200 °C for 15 h. As seen from the figure, all the l-PNDG thin films broke up into islands or droplet-based patterns, which is a clear indication of dewetting. It should be noted that the original as-cast films were homogeneous, and no sign of dewetting was observed (Figure S5), which suggests that the dewetting process occurred during the thermal annealing at T ≫ Tm. By contrast, c-PNDG thin films with the same thicknesses remain relatively homogeneous without any notable formation of islands or droplets, indicating enhanced film stability under identical thermal annealing conditions. The differences in the thermal stability and homogeneity of the thermally annealed l/c-PNDG films were further revealed by X-ray reflectivity (XRR) analysis. Figure 2 shows the XRR curves plotted as a function of the scattering vector (qz) in the direction perpendicular to the surface. The XRR curve for the thermally annealed c-PNDG film exhibit well-defined Kiessig fringes with a periodicity of Δqz = 0.013 Å–1, in stark contrast to that of the l-PNDG film where the Kiessig fringes are notably absent. In addition, a sharp Bragg reflection at qz = 0.258 Å–1 corresponding to a d-spacing of 2.47 nm (via d = 2π/q) was observed for both thermally annealed linear and cyclic PNDG films. This spacing is consistent with the distance between adjacent backbones of PNDG polymers that are separated by the long n-decyl side chains along the out-of-plane direction in the crystalline state (Figure S6).42−44,46−49 The XRR data for the annealed c-PNDG film with qz ≤ 0.22 Å–1 were fitted by using a two-layer model with a Si substrate and a polypeptoid layer. The best fit (the black solid line) to the XRR profile of the c-PNDG film was calculated based on the electron density profile above the Si substrate, as shown in the inset of Figure 2. It should be noted that the electron density (ρe) of the PNDG film is estimated to be 0.32 e·Å–3 based on the bulk density of PNDG (0.95 g/cm3)48,49 and was fixed during the fitting. The best fit to the data yields a thickness of 48.1 ± 0.1 nm and a surface root-mean-square (RMS) roughness of 0.85 ± 0.05 nm for the c-PNDG film. As a result, we conclude that the thermally annealed c-PNDG film is relatively homogeneous and retains the bulk density on the Si substrate.
Figure 1.
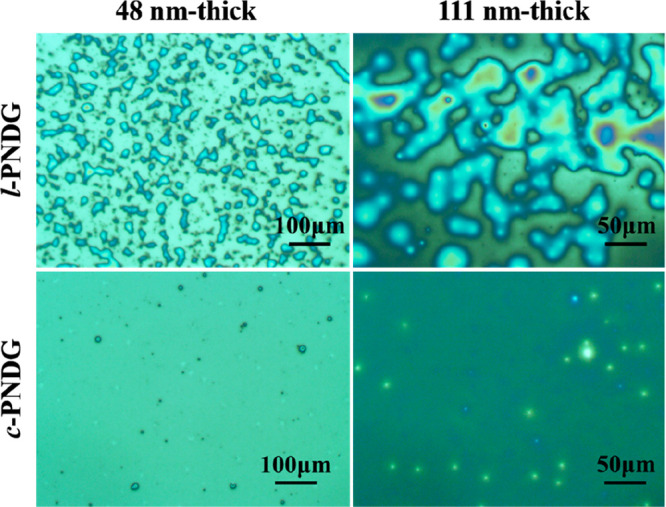
(a) Representative optical microscopic (OM) images of the 48 nm thick and 111 nm thick l-PNDG and c-PNDG films prepared on Si substrates after thermally annealed at 200 °C for 15 h.
Figure 2.
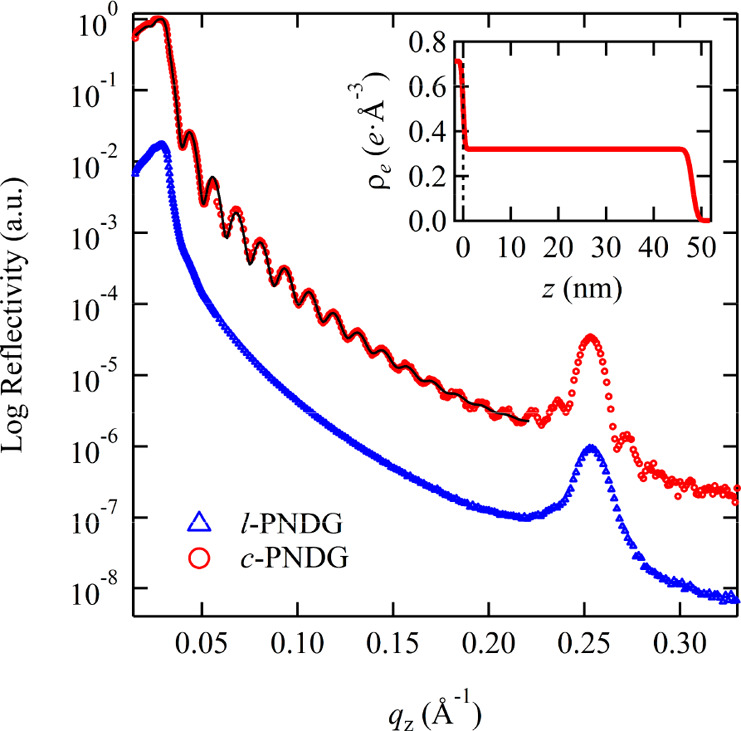
XRR curves of the 48 nm thick c-PNDG (red circles) and l-PNDG (blue triangles) films prepared on Si substrates after thermally annealed at 200 °C for 15 h. The solid black line corresponds to the best-fits of the electron density (ρe) profile against the distance (z) from the Si surface shown in the inset.
In situ GIWAXD was used to investigate the structure and phase transition of l/c-PNDG thin films on the Si substrate. Figure 3 shows the representative two-dimensional (2D) GIWAXD profiles of the 48 nm thick l/c-PNDG thin films measured before, during, and after thermal annealing at T = 200 °C. (Note: the 2D GIWAXD patterns before and after the thermal annealing were collected at T = 25 °C.) As seen in Figure 3a,d, prior to thermal annealing, both the as-cast linear and cyclic PNDG films show arc-shaped primary (001), secondary (002), and tertiary (003) peaks at q = 0.254, 0.508, and 0.762 Å–1 in the out-of-plane (qz) direction, respectively (Figure 4a). The primary (001) reflection gives a d-spacing of 2.47 nm, which corresponds to the distance between adjacent PNDG backbones separated by the long n-decyl side chains along the crystallographic c-axis in the crystalline state.42−44,46−49 Meanwhile, a broad ring-like diffraction is observed near q = 1.4 Å–1 with a notably higher intensity in the in-plane (qxy) than the out-of-plane (qz) direction for both l/c-PNDG as-cast films (Figure 4a,b). This diffraction corresponds to the face-to-face packing of PNDG backbones, i.e., the (100) packing with a d-spacing of 0.45 nm along the crystallographic a-axis.44 These crystal dimensions along a- and c-axes suggest that the l/c-PNDG molecules in the as-cast films have adopted the board-like conformations with a fully extended cis-amide backbone (Figure 5a) being nearly coplanar with the n-decyl side chains, similar to those reported for the comb-shaped polypeptoids in the bulk crystalline state.44 The board-like l/c-PNDG molecules are preferentially stacked side-by-side in the substrate-normal direction and face-to-face in the substrate-parallel direction, which is the so-called “edge-on” crystalline orientation (Figure 5b). The predominant edge-on crystalline orientation is likely attributed to the preferential segregation of long n-decyl side chains at both the free surface and polymer–solid interface to lower the interfacial energy.
Figure 3.

2D GIWAXD images of the 48 nm thick l-PNDG (top row) and c-PNDG (bottom row) films prepared on Si before, during, and after the thermal annealing process. The out-of-plane (qz) and in-plane (qxy) directions are indicated by arrows.
Figure 4.

(a, b) One dimensional (1D) GIWAXD profiles of the as-cast l/c-PNDG films (48 nm in thickness) along (a) qz and (b) qxy directions measured at T = 25 °C before annealing. (c) 1D GIWAXD profiles of the l/c-PNDG films measured at T = 200 °C during annealing, which were obtained by azimuthally integrating of the corresponding 2D images (Figure 3b,e) from 0 to 90° azimuthal angle with respect to the qxy-axis. The corresponding 1D GIWAXD along the qz direction is shown in the inset. (d, e) 1D GIWAXD profiles of the annealed l/c-PNDG thin films along the (d) qz and (e) qxy directions after being cooled to T = 25 °C. Note that c-PNDG and l-PNDG are shown in red and blue dots, respectively.
Figure 5.

(a) Schematics showing the all trans- and cis-amide backbone conformations of l/c-PNDG (R = n-decyl group). (b) Schematic illustration of the change in molecular packing of l/c-PNDG films upon heating/cooling. Possible chain folding and cyclic topology were omitted for clarity.
To further elucidate the effect of chain topology on the molecular ordering of PNDGs in the films, the full width at half-maximum (β) of the (001) reflection in the qz direction obtained by fitting to a Gaussian function was corrected for instrumental resolution and used to estimate the crystalline size (or the structural coherence, L) of l/c-PNDG crystals from the Scherrer equation L = (0.9λ)/(β cos θ), where 0.9 is the Scherrer constant, θ is the scattering angle, and λ is the X-ray wavelength. The L001 values were found to be 11.3 and 9.4 nm for the c-PNDG and l-PNDG as-cast films, respectively, indicating an enhanced molecular ordering of cyclic PNDG relative to the linear PNDG molecules in the substrate-normal direction within the 48 nm thick films prior to any thermal treatment. Interestingly, the broad (100) peak in the qxy direction is less intense for the c-PNDG as-cast film relative to the l-PNDG film under the same X-ray exposure time, suggesting a somewhat diminished ordering for the face-to-face stacking of the board-like PNDG molecules in the substrate-parallel direction for the former film than the latter.
Upon heating the samples at T = 200 °C, which is far above the bulk melting temperatures of l/c-PNDGs,42 only two broad isotropic ring-like reflections located at q = 0.33 Å–1 and q = 1.29 Å–1 are discernible in the GIWAXD profiles (Figures3b,e and 4c), corresponding to d-spacings of 1.90 and 0.49 nm, respectively. The azimuthally isotropic scattering patterns indicate the lack of preferred molecular orientation inside the molten films. In addition, the azimuthally integrated intensities of the ring-like reflections measured in situ at T = 200 °C during thermal annealing are much weaker than those measured at T = 25 °C (i.e., both before and after annealing) (Figure 4). The two d-spacings obtained at T = 200 °C clearly deviate from the (001) and (100) d-spacings of the crystalline PNDGs with the cis-amide backbone conformation. Interestingly, they are consistent with the PNDG molecules adopting a board-shape conformation with a trans-amide backbone (Figure 5a) being nearly coplanar with the n-decyl side chains, as predicted by molecular dynamics simulations.44 Hence, the melting of l/c-PNDG films correspond to a structural transition from a crystalline packing of board-shape molecules with cis-amide backbone conformations to randomly oriented board-shape molecules with trans-amide backbone conformation (Figure 5b). The absence of the higher order diffraction peaks indicates the lack of long-range molecular ordering of the board-shape molecules at T > Tm. It is interesting that the linear and cyclic PNDGs maintain a board-shape conformation in both the crystalline and molten state. This is presumably due to the strong and unique backbone–backbone interaction present in polypeptoid molecules.
The recrystallization of l/c-PNDG thin films was triggered upon cooling from the isotropic melt, evidenced by the emergence of sharp diffraction peaks in the GIWAXS profiles (Figures 3c,f and 4d,e). Separate in situ temperature-dependent GIWAXD measurements (Figure S7) have shown that the crystallization temperatures (Tc) of l/c-PNDG thin films are nearly identical (Tc ≅ 140 °C), which is consistent with the crystallization temperatures of bulk l/c-PNDGs determined by DSC analysis.42 At room temperature, a series of well-defined diffraction arcs corresponding to the side-by-side packing of board-like PNDG molecules (00l) were observed in the qz direction for both linear and cyclic PNDG films (Figures 3c,f and 4d), whereas the (100) reflection corresponding to the face-to-face packing of PNDG molecules appeared as streaks in the qxy direction (Figures 3c,f and 4e). The (001) and (100) d-spacings of the recrystallized l/c-PNDG films obtained by GIWAXD were determined to be d001 = 2.45 nm and d100 = 0.45 nm, respectively, indicating the l/c-PNDGs have readopted the cis-amide backbone conformation on the Si substrate upon cooling below Tc (Figure 5b). The recrystallized l/c-PNDG films have clearly restored “edge-on” crystalline orientation with significantly enhanced anisotropic molecular ordering relative to their respective as-cast films. In addition to the streak-like (100) peak located at qxy = 1.4 Å–1, a broad off-axis reflection near q = 1.4 Å–1 is also discernible at ∼30° off the qxy-axis, which is attributed to the overlapped (101) and (102) reflections in the nonorthogonal crystalline lattice (Figure S6).44 The appearance of the off-axis reflection is consistent with enhanced molecular ordering in the recrystallized PDNG films relative to the as-cast films. It should be noted that unlike shorter PNDG segments or oligomers (i.e., DPn ≤ 20), which often show distinct higher order (10l) reflections,43,44,50 the (10l) peaks in both 52-mer l-PNDG and 50-mer c-PNDG films are poorly resolved. Similar findings have also been reported in relatively high molecular weight PNDG homopolymers and block copolymers.42,50 We attribute this to the chain folding of longer PNDG molecules in the crystalline state, resulting in diminished long-range ordering along the crystallographic a-axis relative to the oligomeric counterparts.
The structural coherence L001 in the qz direction has been calculated via the Scherrer equation (Figure 4d). The L001 of the recrystallized c-PNDG film (16.8 nm) was found to be larger than that of the recrystallized l-PNDG film (15.9 nm). Consistently, the broad (100) peak in the qxy direction was also found to be much more intense for the recrystallized c-PNDG film than the linear counterpart (Figure 4e). In addition, the structural coherence in both substrate-normal and substrate-parallel directions is significantly higher in recrystallized films than their respective as-cast films. These combined results indicate that recrystallization under the same supercooling condition has resulted in the formation of linear and cyclic PNDG films with enhanced anisotropic molecular ordering, in spite of the macroscopic dewetting for the linear PNDG film; the molecular ordering in the recrystallized cyclic PNDG film is more long ranged relative to the linear counterpart in both the substrate-normal and substrate-parallel directions.
It is clear that with same film thickness, thermal annealing condition, and nearly identical chain length (or molecular weight) cyclic PNDG films are more stable against dewetting than the linear counterparts. Meanwhile, the difference in the chain topology did not alter the preferred molecular conformation and orientation of PNDGs in the crystalline or molten films.
Structural Elucidation of Physisorbed l/c-PNDG Layers at the Substrate–Polymer Interface
Thermal annealing of the spin-coated polymer films (including both amorphous and semicrystalline polymers) at T > Tg or Tm is known to facilitate the formation of irreversibly adsorbed polymer nanolayers on solid surfaces via physisorption where noncovalent interaction is involved.20−25,27,28,32,51−53 AFM analysis of the dewetted region of the l-PNDG film on the Si (Figure S8) revealed that the dewetted region is not “empty” but covered by a thin “wetting polymer layer” on the Si substrate. This suggests the occurrence of autophobic dewetting, where a layer of polymers covalently or noncovalently attached on a solid substrate “thermodynamically repels” the nongrafted but otherwise identical molecules.54−57 Several previous studies have shown that the formation of an irreversible adsorbed layer and its local structural evolution at the interface are responsible for the thermal stability (or wettability) of supported thin polymer films.19,25,29,31,58 The unfavorable entropic interaction between the free chains and the strongly adsorbed chains (also known as the “flattened chains”) is believed to be the driving force for the occurrence of autophobic dewetting,25,29,56,59 resulting in no interpenetration of chains at the free polymer/adsorbed polymer interface.25,53 By contrast, when the adsorbed chains are loosely attached and allow interpenetration with free chains at the interface, they then act as “connectors” that can effectively promote interfacial adhesion and prevent dewetting of the polymer film from the substrate.25,31,53,60,61
Inspired by these earlier reports, we further investigated the structure of adsorbed PNDG polymers at the substrate interface using a top-down solvent leaching method previously developed by Guiselin.33 After being preannealed at T = 200 °C for 15 h, both the dewetted l-PNDG and the relatively stable c-PNDG films were subjected to solvent leaching with chloroform, a good solvent for both l/c-PNDGs, to remove the unadsorbed chains and unveil the interfacially adsorbed chains. The detailed solvent leaching process is summarized in the Supporting Information. Figure 6a shows the XRR profiles of the l-PNDG and c-PNDG residual layers after leaching with chloroform to remove the unadsorbed chains. The XRR curves for both l-PNDG and c-PNDG residual layers showed well-defined interference fringes with the first two minima located at almost the same position near qz = 0.10 Å–1 and qz = 0.32 Å–1, indicating the presence of thin layers with similar thickness on the Si substrates. The lengths of the first oscillation periods, i.e., Δqz ≅ 0.22 Å–1, give an approximated thickness of ∼2.9 nm via d = 2π/Δqz. However, we found that the Δqz values of the other oscillation periods in the XRR curve are different, indicating the presence of nonuniform electron density along the surface-normal direction in the residual layer, which will be further discussed (vide infra, Figures 6c and 8).
Figure 6.
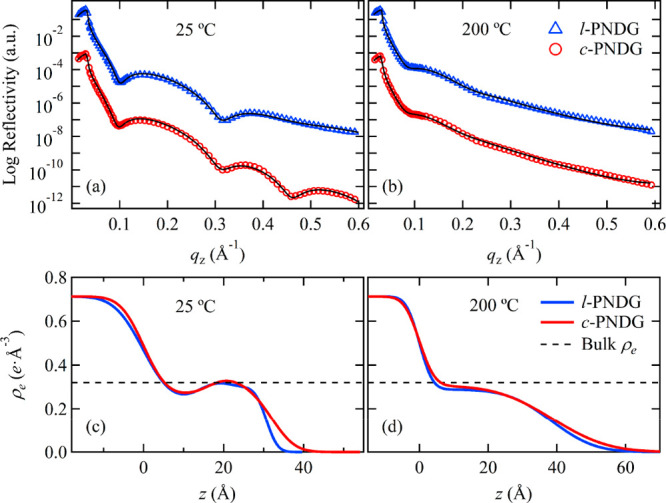
(a, b) XRR curves of the c-PNDG (red circles) and l-PNDG (blue triangles) residual layers (i.e., irreversibly adsorbed layers) measured at (a) 25 °C and (b) 200 °C in situ, respectively. The solid black line corresponds to the best fits to the data based on the electron density (ρe) profile against the distance (z) from the Si surface shown in (c, d). The dotted line in (b) corresponds to the ρe value of bulk PNDG with a density of 0.95 g/cm3.
Figure 8.
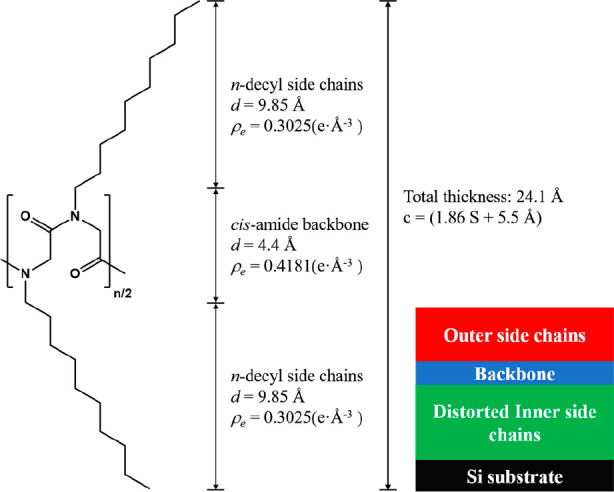
Schematic representation of the molecular structure of l/c-PNDG with a cis-amide backbone conformation. The theoretical dimensions and electron densities of the backbone and side-chain regions are illustrated. The four-layer model used to fit the XRR curves of the adsorbed monolayers measured at 25 °C is shown in the inset.
To determine the chemical composition of these residual layers on Si, we performed X-ray photoelectron spectroscopy (XPS) measurements, and the results are summarized in Figure S9. In the XPS scans, the prominent peak with a binding energy of 284.7 eV is identifiable with the C 1s peak typically found in the aliphatic carbons in the n-decyl side chains (as well as possible hydrocarbon contaminations from the sample).62 An additional shoulder peak corresponding to the carbons of the amide (C(=O)N) groups in the PNDG backbone with a binding energy near 288.1 is also discernible.62,63 In addition, a strong N 1s peak with a binding energy of 400.0 eV that corresponds to the nitrogen atoms of the amide group in the PNDG backbone was also observed in the spectra of the l-PNDG and c-PNDG residual layers,62 which was absent in the spectra of the bare Si substrates. Hence, XPS data provide compelling evidence that the residual layers are composed of adsorbed PNDG molecules, which cannot be removed from the substrate during the chloroform leaching.
Figure 7 shows the GIWAXD results of the nanometer thick l-PNDG and c-PNDG residual layers, that is, irreversibly adsorbed layers. At 25 °C, both l-PNDG and c-PNDG adsorbed layers did not exhibit any signals associated with the (001), (002), and (003) diffractions along qxy (Figure 7e) or other directions (Figure S10), indicating the absence of ordered side-by-side packing of PNDG molecules in the adsorbed layers, in sharp contrast to their respective thin films. The only discernible peak is the streak-like (100) reflections located near qxy = 1.4 Å–1 for both l-PNDG and c-PNDG adsorbed layers, as shown in the horizontal line cuts of the 2D GIWAXD patterns along the qxy direction (Figure 7e). This indicates the adsorbed l/c-PNDGs still adopt a board-shape molecular geometry with a cis-amide conformation and maintain a face-to-face molecular packing in the substrate-parallel direction (i.e., an edge-on orientation), which is identical with the crystalline packing observed in their respective thin film. With the same X-ray exposure time, the (100) peak for the c-PNDG adsorbed layer is slightly sharper relative to that of the l-PNDG adsorbed layer (Figure 7a,b,e), suggesting a more ordered face-to-face stacking of the adsorbed chains in the substrate-parallel direction for the former than the latter. As the l/c-PNDG adsorbed layers are only of ca. 3 nm thick, we conclude that the adsorbed layers are composed of a single molecular layer of l/c-PNDGs with an “edge-on” monolayer lamellae on the Si surface.
Figure 7.

(a–d) 2D GIWAXD images of the (a, c) c-PNDG and (b, d) l-PNDG adsorbed layers on a Si substrate measured at 25 and 200 °C. (e, f) Line-cut GIWAXD profiles of the c-PNDG (red dots) and l-PNDG (blue dots) adsorbed layers along the qxy direction measured at (e) 25 and (f) 200 °C. Note that the 1D GIWAXD intensity data have been shifted vertically for clarity. The (100) peak position is indicated by the red arrows shown in (a) and (b). The red dashed line in (c) is to guide the eye. In (c), the reflection at q = 0.33 Å–1 is indicated by the black arrows.
On the basis of the structural ordering and orientation of the l/c-PNDG adsorbed chains, we adopt a multislab model for the electron density profile (Figure 8) to fit the XRR curves of the adsorbed layers (Figure 6a) measured at 25 °C. Given that the cis-amide backbone and n-decyl side chains possess different electron densities (ρe), a four-layer model that composed of a bottom Si substrate and three layers on top: a middle layer of cis-amide backbone units sandwiched by two layers of n-decyl side chains, was used to fit the data (Figure 8). The details of the four-layer model fitting are described in the Supporting Information. In brief, the free parameters used in the best fits of the XRR data (Figure 6a) are the ρe and thickness of the inner n-decyl layer (h1) and the interfacial roughness at the multiple interfaces, i.e., silicon/inner n-decyl layer interface (σSi), inner n-decyl layer/cis-amide backbone interface (σ1), cis-amide backbone/outer n-decyl layer interface (σ2), and outer n-decyl layer/air interface (σ3). The parameters obtained from the best fits to the XRR data are tabulated in Table 1. We found the total thicknesses of the l-PNDG (h1 + h2 + h3 = 30.7 ± 1.0 Å) and c-PNDG adsorbed layers (32.3 ± 1.0 Å) to be comparable with only a 1.6 Å difference. Based on the ρe profiles shown in Figure 6c, the ρe values of the inner region for l-PNDG and c-PNDG adsorbed layers were about 12% lower than the theoretical ρe of the n-decyl side chains with the cis-amide polypeptoid backbone.44 This indicates that the inner n-decyl chains in both l-PNDG and c-PNDG adsorbed layers are less compact relative to those in the crystalline bulk or the outer n-decyl chains in the monolayers. There is also a clear difference in the interfacial roughness between the l-PNDG and c-PNDG adsorbed layers (Figure 6c): the c-PNDG adsorbed layer has slightly lower roughness at the silicon/inner n-decyl layer interface (σSi = 4.6 ± 0.5 Å), but nearly 2 times higher roughness at the free surface (σ3 = 4.5 ± 0.5 Å) than the l-PNDG counterpart (σSi = 5.7 ± 0.6 Å, σ3 = 2.3 ± 0.2 Å). This finding is in good agreement with the AFM results (Figure 9). On the basis of AFM imaging analysis, we found that the surface RMS roughness of the c-PNDG adsorbed layer (∼4.5 Å) is much larger than that of the l-PNDG counterpart (∼2.9 Å), in good agreement with the XRR results. Interconnected fibrillar nanostructures for both l-PNDG and c-PNDG adsorbed layers are also discernible from the corresponding phase images, which are likely attributed to the molecular packing of adsorbed chains at the interface. The nanofibrils in c-PNDG adsorbed layer are thicker and longer, suggesting higher molecular ordering of c-PNDG adsorbed chains along in-plane direction than linear ones, which is consistent with the GIWAXD results.
Table 1. XRR Fitting Parametersa for l/c-PNDG Adsorbed Monolayers at 25 and 200 °C.
| T (°C) | polymer | σSi (Å) | ρe,1 (e Å–3) | h1 (Å) | σ1 (Å) | ρe,2 (e Å–3) | h2 (Å) | σ2 (Å) | ρe,3 (e Å–3) | h3 (Å) | σ3 (Å) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | l-PNDG | 5.7 | 0.22 | 16.4 | 5.9 | 0.42 | 4.4 | 3.5 | 0.30 | 9.9 | 2.3 |
| c-PNDG | 4.6 | 0.26 | 18.0 | 4.6 | 0.42 | 4.4 | 3.8 | 0.30 | 9.9 | 4.5 | |
| 200 | l-PNDG | 3.0 | 0.29 | 38.4 | 9.9 | ||||||
| c-PNDG | 3.5 | 0.31 | 38.7 | 12.8 |
σSi: silicon/inner n-decyl layer interface (σSi). ρe,1, ρe,2, and ρe,3 are the electron densities of the first, second, and third layers from the Si surface, respectively. h1, h2, and h3 are the thicknesses of the first, second, and third layers from the Si surface, respectively. σ1, σ2, and σ3 are the surface RMS roughnesses of the first, second, and third layers from the Si surface, respectively. Note that ρe,2, ρe,3, h2, and h3 were fixed to the theoretical dimension and electron density of the backbone and side chains derived from the previous report.44 Note that the error values of h1, h2, and h3 are ±1.0 Å. The error values of σSi, σ1, σ2, and σ3 are ∼10% of the σ values. The error values of ρe,1, ρe,2, and ρe,3 are ∼10% of the ρe values.
Figure 9.
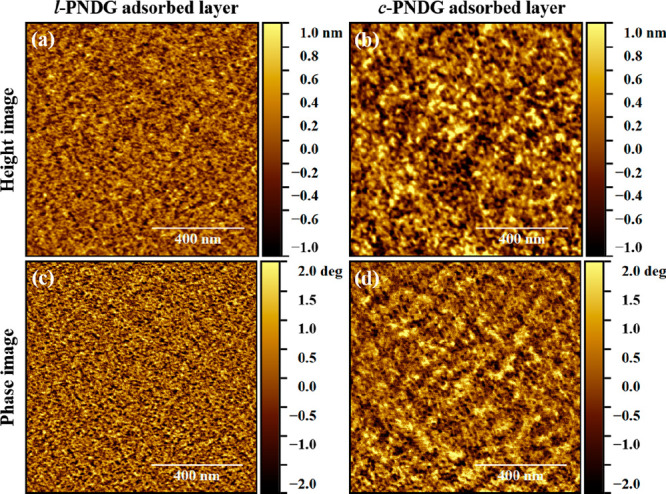
Representative AFM height images of the (a) l-PNDG and (b) c-PNDG adsorbed layers. The corresponding phase images are shown in (c) and (d), respectively. The scales of height and phase images are ±1 nm and ±2°, respectively. The scan size of all images is 1 μm × 1 μm.
Interestingly, when the monolayer is heated at T > Tm, the in situ GIWAXD result for the c-PNDG adsorbed monolayer exhibits two ring-like reflections at q = 0.33 and 1.29 Å–1 at 200 °C (Figure 7c,f and Figure S10), indicating the adsorbed chains of c-PNDG adopt the extended trans-amide backbone conformations with the board-like molecules randomly oriented inside the monolayer, similar to the conformational state observed for the 48 nm thick molten films. By contrast, for the l-PNDG adsorbed monolayer at 200 °C, only a ring-like reflection located at q ∼ 1.58 Å–1 is discernible, which corresponds to a d-spacing of 4.0 Å. This d-spacing does not match any theoretical molecular dimension of PNDG in extended chain conformations (trans or cis-amide), indicating the l-PNDG adsorbed chains may be trapped in an out-of-equilibrium state with a distorted chain conformation. We postulate that the difference in the molecular ordering in the molten state of the adsorbed layers is attributed to the topological difference of the polymers: cyclic topology reduces the conformational freedom of the polymer chains, thus favoring more ordered backbone conformation and molecular packing at the solid–polymer interface. Upon irreversible adsorption, it is likely that the cyclic chains adopt mutually threaded “lattice animal-like” structures with folded loops in two dimensions at the solid interface,5,9 thereby maximizing the contacts between the n-decyl side chains and the Si surface and maintaining certain compactness in the substrate-parallel direction.
Upon heating to 200 °C, drastic changes in the XRR curves of both adsorbed l/c-PNDG monolayers were observed (Figure 6b). The substantial smearing of the interference fringes of the XRR curves is attributed to the significant roughening of the monolayers along with an apparent loss of ρe contrast between PNDG backbone and side chains. Hence, the XRR curves at 200 °C were fitted by using a two-layer model with a Si substrate and a single polypeptoid layer. The electron density profiles obtained from the best fits to the data are shown in Figure 6d. The fitting parameters are tabulated in Table 1. Both cyclic and linear PNDG adsorbed layers were found to exhibit a similar ∼34% increase in the overall thickness along with a significant increase in the surface roughness upon heating to 200 °C. Meanwhile, the overall ρe of the adsorbed layers is reduced. These drastic changes in the electron density profiles near the Si substrate are attributed to the phase transitions of adsorbed chains upon heating at T > Tm. As mentioned, the 2D GIWAXD results of both l-PNDG and c-PNDG adsorbed layers at 200 °C (Figure 7) have revealed isotropic diffraction patterns, indicating the absence of any preferential molecular orientation on the substrate surface. These combined results suggest that upon heating to 200 °C (above Tm) the l-PNDG and c-PNDG adsorbed monolayers transition from an ordered (crystalline) state to a disordered molten (amorphous) state, which is accompanied by an increase in thickness (or free volume) and decrease in average density.
It should be mentioned that 15 h annealing at 200 °C is sufficient to reach equilibrium of chain adsorption for both l/c-PNDGs on the Si substrate. By using the top-down solvent leaching method, we conducted independent adsorption studies of l/c-PNDGs on Si substrates by determining the growth of residual adsorbed layers as a function of preannealing time (5 min–50 h) at 200 °C. The thickness of the l/c-PNDG adsorbed layers was found to increase rapidly at the early stage of adsorption and reached a plateau thicknesses after 2–4 h of annealing at 200 °C (Figure S11). There is no significant difference in the adsorption kinetics between cyclic and linear chains, and the overall trend of adsorption process is similar to the findings on other polymer melt systems.20,22,23,32,51
Molecular Origin for the PNDG Film Stability against Melt-Induced Dewetting
At temperatures above Tm, l-PNDG films undergo autophobic dewetting to a much greater extent than the c-PNDG films, resulting in the formation of macroscopic droplets and islands for the linear PNDG film. What remains unclear is why the molecular topology (cyclic vs linear) has such a significant impact on the relative stability of comb-shaped polypeptoid thin films against melt-induced dewetting. From the liquid contact angle measurement (Figure S12), it was found that both adsorbed l/c-PNDG monolayers (ca. 3 nm in thickness) exhibit the same surface tensions as those in 48 nm thick films (Figure S4), indicating that the difference in the autophobicity between linear and cyclic PNDG adsorbed chains is not due to enthalpic contributions (or macroscopic interfacial tension differences).
A recent study by Wang et al. investigated the film instability of linear and cyclic polystyrene (PS) film on silanized Si substrate and showed that cyclic PS (Mn = 20 kg/mol) films dewetted more slowly than the linear analogues.19 It was found that the irreversibly adsorbed layers formed by cyclic PS were more than 30% thicker than those formed by linear chains, which was believed to be the key for the enhanced thermal stability of cyclic PS films.19 By using Si as substrate, Foster et al. showed that the adsorbed layer is 70% thicker for cyclic PS chains than for the linear analogues.16 An early theoretical study of a weakly attractive polymer–substrate system has also predicted a greater amount of adsorption of cyclic chains to the substrate than linear chains.64 Because the adsorption process of a polymer chain toward a solid surface is enthalpically driven and entropically unfavored, adsorption of cyclic polymers is expected to cause less configurational entropy penalty due to their topological constraints than that of the linear polymers,64−66 while the enthalpic gains for both cyclic and linear chains are the same due to identical segmental structure. Consequently, if the cyclic chain topology can confer a greater extent of polymer adsorption on the substrate resulting in thicker adsorbed layers, it may amplify the substrate effect (or the impact of adsorbed chains) on the molecular motion or the surface fluctuation of a polymer film via long-range perturbation,15,16,20,21,27,67−69 thereby suppressing the dewetting.
In contrast to these early reports, we have found the thickness and electron density profiles of the adsorbed layers for cyclic and linear PNDG films to be nearly identical. As a result, we postulate that the relative thermal stability of cyclic and linear PNDG films mainly arises from the structural disparity of their adsorbed layers and not from their thicknesses/total adsorbed amounts on the substrate. At 25 °C, the c-PNDG adsorbed monolayer shows higher surface roughness and better in-plane molecular ordering than the l-PNDG monolayer. Upon heating at T = 200 °C above the melting temperature (Tm), both cyclic and linear PNDG adsorbed layers become thicker and less dense as they lose orientational ordering. Interestingly, c-PNDG adsorbed chains at 200 °C adopt the same board-shape molecular geometry with the trans-amide backbone conformation as the free c-PNDG chains in the molten film. By contrast, the conformation of l-PNDG adsorbed chains at 200 °C are more disordered and notably different from the board-shape conformation of the free l-PNDG chains with the trans-amide backbone in the molten film. As a result, the disordered l-PNDG adsorbed chains “thermodynamically repel” the neighboring unadsorbed chains to minimize the unfavorable contacts and limit interpenetration of unadsorbed chains at the free polymer/adsorbed polymer interface, eventually resulting in autophobic dewetting of the l-PNDG film. We reason that the conformational disparity between the linear PNDG molecules in the adsorbed layer and the molten film provides the thermodynamic driving force for the autophobic dewetting for the linear PNDG films. In the case of c-PNDG film, the continuity in the chain conformation from the adsorbed monolayer to the neighboring polymer melt plays an active role in stabilizing the interface and suppressing the autophobic dewetting.
One aspect that remains unclear is how the c-PNDG free chains in the film interact with the adsorbed chains under the cyclic topological constraints and whether there is any interdiffusion at the interface. According to previous neutron reflectivity studies on PS and PEO systems, the adsorbed chains show interdiffusion with the chemically identical free chains in the melt up to a certain extent only if the chains are loosely attached at the interface.31,53 For strongly adsorbed chains with a flattened conformation, no interdiffusion has been observed at the free polymer-adsorbed polymer interface.31,53 Considering the lack of chain ends and the presence of long n-decyl side chains in polypeptoids, we speculate that the interdiffusion between free chains and adsorbed chains of comb-shaped c-PNDG would be more restricted relative to conventional coil-like polymers without bulky side chains.
In addition to structural factors, we shall discuss the effect of polymer mobility which may also play a role in determining the film stability and dewetting process. Because of the unusual conformational properties conferred by the absence of free chain ends, the molecular motion of a cyclic polymer often behaves differently from its linear analogue.8 Apart from the topological effect on the dynamical properties of bulk polymer melts, the molecular motion or the mobility gradient of a confined polymer film can be strongly influenced by the nanoscale structure and dynamics of adsorbed chains at the polymer–solid interface.15,16,20,21,27,67−69 Using X-ray photon correlation spectroscopy, Foster et al. observed slower surface fluctuations of cyclic PS as compared to linear PS analogues, which were attributed to the bulk Tg differences and the interplay between the free surface effect and immobilized adsorbed layer effect.15,16 Torkelson et al. have reported a weaker perturbation of the free surface of cyclic PS thin film relative to the linear PS counterpart by Tg measurement of thin films with varying thickness.17 The slower surface fluctuations of cyclic PS have been linked to the irreversibly adsorbed of chains at the substrate interface, where a 70% thicker adsorbed layer was found for cyclic PS chains as compared to linear analogues.16 If the c-PNDG melts in a confined thin film geometry have slower molecular mobility relative to that of l-PNDG, we may expect a suppression in the autophobic dewetting process. A full investigation on dynamical properties of l/c-PNDG films will be the subject of future experiments.
Conclusions
We investigated the thermal properties and structural ordering of comb-shaped polypeptoid thin films with different chain topologies (linear vs cyclic) on a Si substrate. When thermally annealed at T > Tm, both l-PNDG and c-PNDG chains adopt a board-like geometry with mainly trans-amide backbone conformation and are randomly oriented inside the film. When cooled below the crystallization temperature (T < Tc), the l/c-PNDGs maintained a board-shape geometry with predominantly cis-amide backbone conformation and pack into highly ordered lamellar structure with the crystallographic c-axis preferentially aligned in the substrate-normal direction. Cyclic PNDG films were found to exhibit significantly enhanced stability against melt-induced dewetting and improved crystalline ordering relative to the linear PNDG films which ruptured into macroscopic droplets during the thermal annealing process.
Investigation of the interfacial structure at the polymer–Si interface has revealed the formation of physically adsorbed monolayer at the Si surface for both l/c-PNDG films. While both l/c-PNDG physisorbed chains exhibited a face-to-face molecular packing in the direction parallel to the Si surface at room temperature (T < Tc), the c-PNDG monolayer show thicker fibrillar structures and higher surface roughness than the linear counterpart. When annealed at T > Tm, c-PNDG adsorbed chains can still undergo a cis-to-trans amide backbone conformational transition similar to that observed in the bulk film. This ensures a conformational continuity between the adsorbed chains and free chains in the bulk molten film, thus inhibiting the dewetting of c-PNDG films. By contrast, the conformation of l-PNDG adsorbed chains was found to differ from that of the free chains in the molten film. The conformational difference results in unfavorable interaction between the chemically identical molecules in the adsorbed layer and bulk film, thus contributing to pronounced melt-induced dewetting in l-PNDG film. This study highlights the use of cyclic chain topology to suppress the melt-induced dewetting and enhance molecular ordering of crystallizable polymer thin films. The formation of comb-shaped polypeptoid monolayers that irreversibly physisorbed at the solid surface with a preferred edge-on lamellae orientation may also serve as a simple and versatile approach for a broad spectrum of surface modification/functionalization purposes, such as surface sensing and antifouling coatings.
Acknowledgments
The authors thank Robin McCarley for providing access to the contact angle goniometer as well as Jayne C. Garno and Neepa M. K. Kuruppu Arachchige for providing access to and training of the AFM instrument. The polymer synthesis and characterization of polymer thin films were supported by the National Science Foundation (CHE 1609447). The Synchrotron X-ray scattering experiments were supported by the U.S. Department of Energy (DOE) under EPSCoR Grant DE-SC0012432 with additional support from the Louisiana Board of Regents. N.J. also acknowledges partial financial support from the Fundamental Research Funds for the Central Universities (Grant FRF-TP-17-001A1). This research used the Complex Materials Scattering (CMS/11-BM) beamline, operated by the National Synchrotron Light Source II at Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-SC0012704.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.0c01205.
Experimental section, NMR spectra for monomer and l/c-PNDG samples, MALDI-TOF MS spectra of the l/c-PNDG samples, static contact angle results for l/c-PNDG thin films and adsorbed layers, optical microscopy images for l/c-PNDG as-cast films, general crystalline packing and unit cell dimensions of comb-shaped polypeptoids, additional in situ high-temperature GIWAXD results, additional AFM results, XPS spectra for the adsorbed layers, additional 1D GIWAXD plots for the adsorbed layers, adsorption study of l/c-PNDGs on Si substrates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- De Gennes P.-G.Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, 1979. [Google Scholar]
- Doi M.; Edwards S. F.. The Theory of Polymer Dynamics; Oxford Science: Oxford, 1986. [Google Scholar]
- Cates M. E.; Deutsch J. M. Conjectures on the statistics of ring polymers. J. Phys. (Paris) 1986, 47 (12), 2121–2128. 10.1051/jphys:0198600470120212100. [DOI] [Google Scholar]
- Grosberg A. Y.; Nechaev S. K.; Shakhnovich E. I. The role of topological constraints in the kinetics of collapse of macromolecules. J. Phys. (Paris) 1988, 49 (12), 2095–2100. 10.1051/jphys:0198800490120209500. [DOI] [Google Scholar]
- Kapnistos M.; Lang M.; Vlassopoulos D.; Pyckhout-Hintzen W.; Richter D.; Cho D.; Chang T.; Rubinstein M. Unexpected power-law stress relaxation of entangled ring polymers. Nat. Mater. 2008, 7 (12), 997–1002. 10.1038/nmat2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halverson J. D.; Lee W. B.; Grest G. S.; Grosberg A. Y.; Kremer K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. I. Statics. J. Chem. Phys. 2011, 134 (20), 204904. 10.1063/1.3587137. [DOI] [PubMed] [Google Scholar]
- Halverson J. D.; Lee W. B.; Grest G. S.; Grosberg A. Y.; Kremer K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. II. Dynamics. J. Chem. Phys. 2011, 134 (20), 204905. 10.1063/1.3587138. [DOI] [PubMed] [Google Scholar]
- Richter D.; Gooßen S.; Wischnewski A. Celebrating soft matter’s 10th anniversary: Topology matters: Structure and dynamics of ring polymers. Soft Matter 2015, 11 (44), 8535–8549. 10.1039/C5SM01994J. [DOI] [PubMed] [Google Scholar]
- Rubinstein M. Dynamics of ring polymers in the presence of fixed obstacles. Phys. Rev. Lett. 1986, 57 (24), 3023–3026. 10.1103/PhysRevLett.57.3023. [DOI] [PubMed] [Google Scholar]
- Pérez-Camargo R. A.; Mugica A.; Zubitur M.; Müller A. J.. Crystallization of cyclic polymers. In Polymer Crystallization I: From Chain Microstructure to Processing; Auriemma F., Alfonso G. C., de Rosa C., Eds.; Springer International Publishing: Cham, 2017; pp 93–132. [Google Scholar]
- Zaldua N.; Liénard R.; Josse T.; Zubitur M.; Mugica A.; Iturrospe A.; Arbe A.; De Winter J.; Coulembier O.; Müller A. J. Influence of Chain Topology (Cyclic versus Linear) on the Nucleation and Isothermal Crystallization of Poly(L-lactide) and Poly(D-lactide). Macromolecules 2018, 51 (5), 1718–1732. 10.1021/acs.macromol.7b02638. [DOI] [Google Scholar]
- Zardalidis G.; Mars J.; Allgaier J.; Mezger M.; Richter D.; Floudas G. Influence of chain topology on polymer crystallization: Poly(ethylene oxide) (PEO) rings vs. Linear chains. Soft Matter 2016, 12 (39), 8124–8134. 10.1039/C6SM01622G. [DOI] [PubMed] [Google Scholar]
- Shin E. J.; Jeong W.; Brown H. A.; Koo B. J.; Hedrick J. L.; Waymouth R. M. Crystallization of cyclic polymers: Synthesis and crystallization behavior of high molecular weight cyclic poly(ε-caprolactone)s. Macromolecules 2011, 44 (8), 2773–2779. 10.1021/ma102970m. [DOI] [Google Scholar]
- Schäler K.; Ostas E.; Schröter K.; Thurn-Albrecht T.; Binder W. H.; Saalwächter K. Influence of chain topology on polymer dynamics and crystallization. Investigation of linear and cyclic poly(ε-caprolactone)s by 1H solid-state NMR methods. Macromolecules 2011, 44 (8), 2743–2754. 10.1021/ma102838c. [DOI] [Google Scholar]
- Wang S.-F.; Jiang Z.; Narayanan S.; Foster M. D. Dynamics of surface fluctuations on macrocyclic melts. Macromolecules 2012, 45 (15), 6210–6219. 10.1021/ma2028215. [DOI] [Google Scholar]
- He Q.; Narayanan S.; Wu D. T.; Foster M. D. Confinement effects with molten thin cyclic polystyrene films. ACS Macro Lett. 2016, 5 (9), 999–1003. 10.1021/acsmacrolett.6b00497. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Elupula R.; Grayson S. M.; Torkelson J. M. Major impact of cyclic chain topology on the Tg-confinement effect of supported thin films of polystyrene. Macromolecules 2016, 49 (1), 257–268. 10.1021/acs.macromol.5b02474. [DOI] [Google Scholar]
- Kelly G. M.; Haque F. M.; Grayson S. M.; Albert J. N. L. Suppression of melt-induced dewetting in cyclic poly(ε-caprolactone) thin films. Macromolecules 2017, 50 (24), 9852–9856. 10.1021/acs.macromol.7b02200. [DOI] [Google Scholar]
- Wang L.; Xu L.; Liu B.; Shi T.; Jiang S.; An L. The influence of polymer architectures on the dewetting behavior of thin polymer films: From linear chains to ring chains. Soft Matter 2017, 13 (17), 3091–3098. 10.1039/C7SM00379J. [DOI] [PubMed] [Google Scholar]
- Fujii Y.; Yang Z. H.; Leach J.; Atarashi H.; Tanaka K.; Tsui O. K. C. Affinity of polystyrene films to hydrogen-passivated silicon and its relevance to the t-g of the films. Macromolecules 2009, 42 (19), 7418–7422. 10.1021/ma901851w. [DOI] [Google Scholar]
- Napolitano S.; Wubbenhorst M. The lifetime of the deviations from bulk behaviour in polymers confined at the nanoscale. Nat. Commun. 2011, 2, 260–267. 10.1038/ncomms1259. [DOI] [Google Scholar]
- Gin P.; Jiang N.; Liang C.; Taniguchi T.; Akgun B.; Satija S. K.; Endoh M. K.; Koga T. Revealed architectures of adsorbed polymer chains at solid-polymer melt interfaces. Phys. Rev. Lett. 2012, 109 (26), 265501. 10.1103/PhysRevLett.109.265501. [DOI] [PubMed] [Google Scholar]
- Jiang N.; Shang J.; Di X.; Endoh M. K.; Koga T. Formation mechanism of high-density, flattened polymer nanolayers adsorbed on planar solids. Macromolecules 2014, 47 (8), 2682–2689. 10.1021/ma5003485. [DOI] [Google Scholar]
- Asada M.; Jiang N.; Sendogdular L.; Sokolov J.; Endoh M. K.; Koga T.; Fukuto M.; Yang L.; Akgun B.; Dimitriou M.; Satija S. Melt crystallization/dewetting of ultrathin PEO films via carbon dioxide annealing: The effects of polymer adsorbed layers. Soft Matter 2014, 10 (34), 6392–6403. 10.1039/C4SM00683F. [DOI] [PubMed] [Google Scholar]
- Jiang N.; Wang J.; Di X.; Cheung J.; Zeng W.; Endoh M. K.; Koga T.; Satija S. K. Nanoscale adsorbed structures as a robust approach for tailoring polymer film stability. Soft Matter 2016, 12 (6), 1801–1809. 10.1039/C5SM02435H. [DOI] [PubMed] [Google Scholar]
- Burroughs M. J.; Napolitano S.; Cangialosi D.; Priestley R. D. Direct measurement of glass transition temperature in exposed and buried adsorbed polymer nanolayers. Macromolecules 2016, 49 (12), 4647–4655. 10.1021/acs.macromol.6b00400. [DOI] [Google Scholar]
- Panagopoulou A.; Napolitano S. Irreversible adsorption governs the equilibration of thin polymer films. Phys. Rev. Lett. 2017, 119 (9), 097801 10.1103/PhysRevLett.119.097801. [DOI] [PubMed] [Google Scholar]
- Xu J.; Liu Z.; Lan Y.; Zuo B.; Wang X.; Yang J.; Zhang W.; Hu W. Mobility gradient of poly(ethylene terephthalate) chains near a substrate scaled by the thickness of the adsorbed layer. Macromolecules 2017, 50 (17), 6804–6812. 10.1021/acs.macromol.7b00922. [DOI] [Google Scholar]
- Li X.; Lu X. Evolution of irreversibly adsorbed layer promotes dewetting of polystyrene film on sapphire. Macromolecules 2018, 51 (17), 6653–6660. 10.1021/acs.macromol.8b01141. [DOI] [Google Scholar]
- Zuo B.; Zhou H.; Davis M. J. B.; Wang X.; Priestley R. D. Effect of local chain conformation in adsorbed nanolayers on confined polymer molecular mobility. Phys. Rev. Lett. 2019, 122 (21), 217801. 10.1103/PhysRevLett.122.217801. [DOI] [PubMed] [Google Scholar]
- Barkley D. A.; Jiang N.; Sen M.; Endoh M. K.; Rudick J. G.; Koga T.; Zhang Y.; Gang O.; Yuan G.; Satija S. K.; Kawaguchi D.; Tanaka K.; Karim A. Chain conformation near the buried interface in nanoparticle-stabilized polymer thin films. Macromolecules 2017, 50 (19), 7657–7665. 10.1021/acs.macromol.7b01187. [DOI] [Google Scholar]
- Durning C. J.; O’Shaughnessy B.; Sawhney U.; Nguyen D.; Majewski J.; Smith G. S. Adsorption of poly(methyl methacrylate) melts on quartz. Macromolecules 1999, 32 (20), 6772–6781. 10.1021/ma981785k. [DOI] [Google Scholar]
- Guiselin O. Irreversible adsorption of a concentrated polymer solution. Europhys. Lett. 1992, 17 (3), 225–230. 10.1209/0295-5075/17/3/007. [DOI] [Google Scholar]
- Kirshenbaum K.; Barron A. E.; Goldsmith R. A.; Armand P.; Bradley E. K.; Truong K. T. V.; Dill K. A.; Cohen F. E.; Zuckermann R. N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (8), 4303–4308. 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Lahasky S. H.; Guo L.; Lee C.-U.; Lavan M. Polypeptoid materials: Current status and future perspectives. Macromolecules 2012, 45 (15), 5833–5841. 10.1021/ma202319g. [DOI] [Google Scholar]
- Sun J.; Zuckermann R. N. Peptoid polymers: A highly designable bioinspired material. ACS Nano 2013, 7 (6), 4715–4732. 10.1021/nn4015714. [DOI] [PubMed] [Google Scholar]
- Gangloff N.; Ulbricht J.; Lorson T.; Schlaad H.; Luxenhofer R. Peptoids and polypeptoids at the frontier of supra- and macromolecular engineering. Chem. Rev. 2016, 116 (4), 1753–1802. 10.1021/acs.chemrev.5b00201. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Simpson J. M.; Xu X.; He H.; Zhang D.; Yin L. Cationic polypeptoids with optimized molecular characteristics toward efficient nonviral gene delivery. ACS Appl. Mater. Interfaces 2017, 9 (28), 23476–23486. 10.1021/acsami.7b06031. [DOI] [PubMed] [Google Scholar]
- Chan B. A.; Xuan S.; Li A.; Simpson J. M.; Sternhagen G. L.; Yu T.; Darvish O. A.; Jiang N.; Zhang D. Polypeptoid polymers: Synthesis, characterization, and properties. Biopolymers 2018, 109 (1), e23070. 10.1002/bip.23070. [DOI] [PubMed] [Google Scholar]
- Guo L.; Zhang D. Cyclic Poly(α-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxylanhydrides. J. Am. Chem. Soc. 2009, 131 (50), 18072–18074. 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]
- Guo L.; Lahasky S. H.; Ghale K.; Zhang D. N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydrides toward Poly(α-peptoid)s: Kinetic, Mechanism, and Architectural Control. J. Am. Chem. Soc. 2012, 134 (22), 9163–9171. 10.1021/ja210842b. [DOI] [PubMed] [Google Scholar]
- Lee C.-U.; Li A.; Ghale K.; Zhang D. Crystallization and Melting Behaviors of Cyclic and Linear Polypeptoids with Alkyl Side Chains. Macromolecules 2013, 46 (20), 8213–8223. 10.1021/ma401067f. [DOI] [Google Scholar]
- Greer D. R.; Stolberg M. A.; Xuan S.; Jiang X.; Balsara N. P.; Zuckermann R. N. Liquid-Crystalline Phase Behavior in Polypeptoid Diblock Copolymers. Macromolecules 2018, 51 (23), 9519–9525. 10.1021/acs.macromol.8b01952. [DOI] [Google Scholar]
- Greer D. R.; Stolberg M. A.; Kundu J.; Spencer R. K.; Pascal T.; Prendergast D.; Balsara N. P.; Zuckermann R. N. Universal Relationship between Molecular Structure and Crystal Structure in Peptoid Polymers and Prevalence of the cis Backbone Conformation. J. Am. Chem. Soc. 2018, 140 (2), 827–833. 10.1021/jacs.7b11891. [DOI] [PubMed] [Google Scholar]
- Edison J. R.; Spencer R. K.; Butterfoss G. L.; Hudson B. C.; Hochbaum A. I.; Paravastu A. K.; Zuckermann R. N.; Whitelam S. Conformations of peptoids in nanosheets result from the interplay of backbone energetics and intermolecular interactions. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (22), 5647–5651. 10.1073/pnas.1800397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-U.; Smart T. P.; Guo L.; Epps T. H.; Zhang D. Synthesis and Characterization of Amphiphilic Cyclic Diblock Copolypeptoids from N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydride. Macromolecules 2011, 44 (24), 9574–9585. 10.1021/ma2020936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-U.; Lu L.; Chen J.; Garno J. C.; Zhang D. Crystallization-Driven Thermoreversible Gelation of Coil-Crystalline Cyclic and Linear Diblock Copolypeptoids. ACS Macro Lett. 2013, 2 (5), 436–440. 10.1021/mz300667n. [DOI] [PubMed] [Google Scholar]
- Sun J.; Teran A. A.; Liao X. X.; Balsara N. P.; Zuckermann R. N. Crystallization in sequence-defined peptoid diblock copolymers induced by microphase separation. J. Am. Chem. Soc. 2014, 136 (5), 2070–2077. 10.1021/ja412123y. [DOI] [PubMed] [Google Scholar]
- Sun J.; Jiang X.; Lund R.; Downing K. H.; Balsara N. P.; Zuckermann R. N. Self-assembly of crystalline nanotubes from monodisperse amphiphilic diblock copolypeptoid tiles. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (15), 3954–3959. 10.1073/pnas.1517169113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N.; Yu T.; Darvish O. A.; Qian S.; Mkam Tsengam I. K.; John V.; Zhang D. Crystallization-driven self-assembly of coil–comb-shaped polypeptoid block copolymers: Solution morphology and self-assembly pathways. Macromolecules 2019, 52 (22), 8867–8877. 10.1021/acs.macromol.9b01546. [DOI] [Google Scholar]
- Housmans C.; Sferrazza M.; Napolitano S. Kinetics of irreversible chain adsorption. Macromolecules 2014, 47 (10), 3390–3393. 10.1021/ma500506r. [DOI] [Google Scholar]
- Bal J. K.; Beuvier T.; Unni A. B.; Chavez Panduro E. A.; Vignaud G.; Delorme N.; Chebil M. S.; Grohens Y.; Gibaud A. Stability of polymer ultrathin films (<7 nm) made by a top-down approach. ACS Nano 2015, 9 (8), 8184–8193. 10.1021/acsnano.5b02381. [DOI] [PubMed] [Google Scholar]
- Jiang N.; Sen M.; Zeng W.; Chen Z.; Cheung J. M.; Morimitsu Y.; Endoh M. K.; Koga T.; Fukuto M.; Yuan G.; Satija S. K.; Carrillo J.-M. Y.; Sumpter B. G. Structure-induced switching of interpolymer adhesion at a solid-polymer melt interface. Soft Matter 2018, 14 (7), 1108–1119. 10.1039/C7SM02279D. [DOI] [PubMed] [Google Scholar]
- Hare E. F.; Zisman W. A. Autophobic liquids and the properties of their adsorbed films. J. Phys. Chem. 1955, 59 (4), 335–340. 10.1021/j150526a014. [DOI] [Google Scholar]
- Shull K. R. Wetting autophobicity of polymer melts. Faraday Discuss. 1994, 98 (0), 203–217. 10.1039/fd9949800203. [DOI] [Google Scholar]
- Reiter G.; Auroy P.; Auvray L. Instabilities of thin polymer films on layers of chemically identical grafted molecules. Macromolecules 1996, 29 (6), 2150–2157. 10.1021/ma950297z. [DOI] [Google Scholar]
- Ferreira P. G.; Ajdari A.; Leibler L. Scaling law for entropic effects at interfaces between grafted layers and polymer melts. Macromolecules 1998, 31 (12), 3994–4003. 10.1021/ma9712460. [DOI] [Google Scholar]
- Wang L.-N.; Zhang H.-H.; Xu L.; Liu B.-Y.; Shi T.-F.; Jiang S.-C.; An L.-J. Dewetting kinetics of thin polymer films with different architectures: Effect of polymer adsorption. Chin. J. Polym. Sci. 2018, 36 (8), 984–990. 10.1007/s10118-018-2111-1. [DOI] [Google Scholar]
- Reiter G.; Khanna R. Negative excess interfacial entropy between free and end-grafted chemically identical polymers. Phys. Rev. Lett. 2000, 85 (26), 5599–5602. 10.1103/PhysRevLett.85.5599. [DOI] [PubMed] [Google Scholar]
- Raphael E.; De Gennes P. G. Rubber adhesion with connector molecules. J. Phys. Chem. 1992, 96, 4002–4007. 10.1021/j100189a018. [DOI] [Google Scholar]
- Reiter G.; Schultz J.; Auroy P.; Auvray L. Improving adhesion via connector polymers to stabilize non-wetting liquid films. Europhys. Lett. 1996, 33 (1), 29–34. 10.1209/epl/i1996-00299-3. [DOI] [Google Scholar]
- Briggs D.Surface Analysis of Polymers by XPS and Static SIMS; Cambridge University Press: Cambridge, 1998. [Google Scholar]
- Statz A. R.; Meagher R. J.; Barron A. E.; Messersmith P. B. New peptidomimetic polymers for antifouling surfaces. J. Am. Chem. Soc. 2005, 127 (22), 7972–7973. 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- Van Lent B.; Scheutjens J.; Cosgrove T. Self-consistent field theory for the adsorption of ring polymers from solution. Macromolecules 1987, 20 (2), 366–370. 10.1021/ma00168a024. [DOI] [Google Scholar]
- Chen Z.; Escobedo F. A. Influence of polymer architecture and polymer-wall interaction on the adsorption of polymers into a slit-pore. Phys. Rev. E 2004, 69 (2), 021802 10.1103/PhysRevE.69.021802. [DOI] [PubMed] [Google Scholar]
- Ye S.; Tang Q.; Yang J.; Zhang K.; Zhao J. Interfacial diffusion of a single cyclic polymer chain. Soft Matter 2016, 12 (47), 9520–9526. 10.1039/C6SM02103D. [DOI] [PubMed] [Google Scholar]
- Koga T.; Jiang N.; Gin P.; Endoh M. K.; Narayanan S.; Lurio L. B.; Sinha S. K. Impact of an irreversibly adsorbed layer on local viscosity of nanoconfined polymer melts. Phys. Rev. Lett. 2011, 107 (22), 225901. 10.1103/PhysRevLett.107.225901. [DOI] [PubMed] [Google Scholar]
- Perez-de-Eulate N. G.; Sferrazza M.; Cangialosi D.; Napolitano S. Irreversible adsorption erases the free surface effect on the Tg of supported films of poly(4-tert-butylstyrene). ACS Macro Lett. 2017, 6 (4), 354–358. 10.1021/acsmacrolett.7b00129. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Rafailovich M. H.; Sokolov J.; Strzhemechny Y.; Schwarz S. A.; Sauer B. B.; Rubinstein M. Long-range effects on polymer diffusion induced by a bounding interface. Phys. Rev. Lett. 1997, 79 (2), 241–244. 10.1103/PhysRevLett.79.241. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.