Abstract
Die Kinderendokrinologie beschäftigt sich mit der Physiologie und Pathophysiologie der Synthese und Wirkung von Hormonen. Endokrine Störungen des Kindesalters können dabei primär oder sekundär im Rahmen anderer Grunderkrankungen auftreten. Viele dieser Erkrankungen sind chronischer Natur. In den letzten Jahren konnte durch die rasante Entwicklung der molekulargenetischen Diagnostik eine erhebliche Verbesserung des Verständnisses endokriner Störungen des Kindesalters erreicht werden. Gleichzeitig wurde durch technischen Fortschritt eine deutliche Verbesserung in der Versorgung von Kindern und Jugendlichen mit Diabetes mellitus erreicht. Im vorliegenden Kapitel wird ein aktueller Überblick über das Fachgebiet der Kinderendokrinologie und Diabetologie gegeben. Dieser umfasst u. a. Physiologie und Pathophysiologie von Wachstum, Pubertät, der Funktion von Schilddrüse, Nebenschilddrüse, des Kalzium-Phosphat-Stoffwechsels, der Nebenniere, der Hypothalamus-Hypophysen-Achse, der Gonaden sowie von Fehlbildungen der Genitalorgane. Ein ausführliches Kapitel behandelt die verschiedenen Formen des Diabetes mellitus im Kindes und Jugendalter.
Wachstum
Physiologie des Längenwachstums
Somatisches Längenwachstum ist ein komplexer Prozess der durch nichtendokrine und endokrine Einflussfaktoren beeinflusst wird. Es ist zu einem hohen Anteil genetisch determiniert. Aktuelle Untersuchungen zeigen, dass mehr als 700 Gene für die erreichte Erwachsenengröße relevant sind; trotz des Wissenszuwachses durch moderne genomweite Analysen können aber derzeit nur etwa 20% des genetischen Beitrags zum Körperwachstum erklärt werden. Viele der Effekte der bislang identifizierten Genvarianten werden durch Wachstumshormon (WH) und den insulinähnlichen Wachstumsfaktor-1 („insulin-like growth factor-1“, IGF-1) vermittelt. Auf tierexperimentellen Untersuchungen basierend wurde geschätzt, dass bei Fehlen von WH und IGF-1 weniger als 20 % des normalen Körperwachstums erreicht werden können.
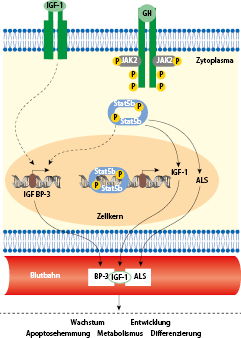
Pulsatil sezerniertes hypophysäres Wachstumshormon gelangt in die Zirkulation und bindet an den Wachstumshormonrezeptor (GHR). Hierdurch wird eine intrazelluläre Kaskade aktiviert und u. a. in der Leber die Synthese von IGF-1 induziert, das dann gebunden an das Protein „insulin-like growth factor binding protein 3“ (IGFBP-3) in einem ternären Komplex maßgeblich die endokrine Wirkung von Wachstumshormon vermittelt. Zusätzlich üben WH und IGF-1 auch lokal am Knochen wachstumsstimulierende Effekte aus (Abb. 26.1).

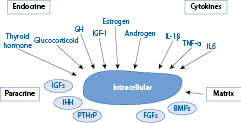
Anatomisch ist die sog. Wachstumsfuge des langen Röhrenknochens wichtigster Ort des Geschehens; sie integriert zirkulierende endokrine und nichtendokrine Signale, lokale parakrine Effekte mit inflammatorischen Signalen und Einflüssen der extrazellulären Matrix und vermittelt durch die Chondrozytenproliferation das Wachstum der langen Röhrenknochen (Abb. 26.2).

Die Regulation des Längenwachstums wird in Abhängigkeit der Lebensphase in der Kindheit durch unterschiedliche Einflussfaktoren dominiert: So unterliegt nach dem ICP-Modell von Karlberg (1989) das Wachstum im Kleinkindesalter (sog. Infancy-Komponente kindlichen Wachstums) überwiegend nutritiven Einflüssen und dem jeweiligen genetischen Hintergrund. In der anschließenden Childhood-Komponente mit deutlich abnehmender Wachstumsgeschwindigkeit scheint die Wachstumshormon-IGF-1-Achse eine dominante Rolle zu spielen. Anschließend kommt es in der Pubertätsphase unter dem Einfluss der Sexualsteroide zu einem geschlechtsunterschiedlichen Wachstumsschub, bevor das Längenwachstum zum Abschluss kommt.
Auxiologie
Meßmethoden
Bei der Beurteilung des kindlichen Wachstums ist eine longitudinale Erhebung von Körperlänge (Messung im Liegen, typischerweise in einer Meßschale) bzw. Körperhöhe (Messung im Stehen) mit einem geeigneten Messinstrument (Stadiometer) notwendig. Diese erfolgt in bestimmten Altersphasen standardisiert vorgegeben im Rahmen der kinderärztlichen Vorsorgeuntersuchungen.
Wachstumskurven und Bewertung von Wachstumsdaten
Die erhobenen Meßwerte werden nach Eintragen in geeignete populationsspezifische Perzentilkurven eingetragen. Während die weiterhin gebräuchlichen Wachstumskurven nach Brandt-Reinken auf einer longitudinalen Erhebung von Wachstumsdaten einer Population basieren sind die aktuelleren Kurven des Robert-Koch-Instituts (KIGGS-Studie; https://www.rki.de/DE/Content/Gesundheitsmonitoring/Gesundheitsberichterstattung/GBEDownloadsB/KiGGS_Referenzperzentile.pdf?__blob=publicationFile) oder die Kurven nach Kromeyer-Hauschild auf Querschnitterhebungen basierend. Verwendet werden sollten möglichst aktuelle Kurven, die auf Referenzdaten geeigneter Herkunft beruhen. Darüber hinaus existieren eine Reihe erkrankungsspezifischer Perzentilen. Deren Verwendung ist insbesondere dann sinnvoll, wenn das Längenwachstum betroffener Kinder sich deutlich vom Wachstum der nichtbetroffenen Referenzpopulation unterscheidet (z. B. krankheitsspezifische Wachstumskurven für Ullrich-Turner- oder Down-Syndrom).
Während die grafische Darstellung der altersbezogenen Körperhöhe bei der Einordnung der Körperhöhe im Verhältnis zur Referenzpopulation hilft und sich hierdurch ein Perzentilrang bestimmen lässt, ist für die Einschätzung, ob ein auffälliges und womöglich pathologisches Wachstum vorliegt die Bestimmung der Wachstumsgeschwindigkeit von größerer Bedeutung. Diese setzt zwei Messungen der Körperhöhe in einem Abstand von mindestens sechs (besser 12 Monaten) mit einem geeigneten Messinstrument voraus.
Darüber hinaus können Abweichungen der Längenentwicklung vom Wachstum der Referenzpopulation durch das Berechnen sog. „standard deviation scores“ (SDS) erfassen:
Dabei umfasst der Normalbereich sämtliche Körperhöhen zwischen -2 bis +2 SDS. Der SDS-Wert erlaubt eine Bewertung der Körperhöhe auch außerhalb der Verwendung von Perzentilen; longitudinal kann auch eine Veränderung der altersbezogenen SDS-Werte Hinweis auf eine Wachstumsstörung sein.
Körperproportionen
Insbesondere bei der Einschätzung, ob eine syndromale Wachstumsstörung vorliegt kommt der Bestimmung von Körperproportionen eine wichtige Rolle zu. Hierbei werden u. a. die Körperhöhe im Sitzen auf einem standardisierten Sitzhocker („Sitzhöhe“), das Verhältnis Oberlänge zu Unterlänge und die Armspannweite herangezogen. Die Bewertung erfolgt mittels Heranziehen geeigneter Referenzdaten.
Knochenreifung
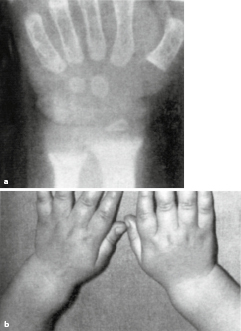
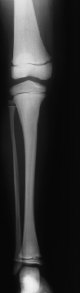
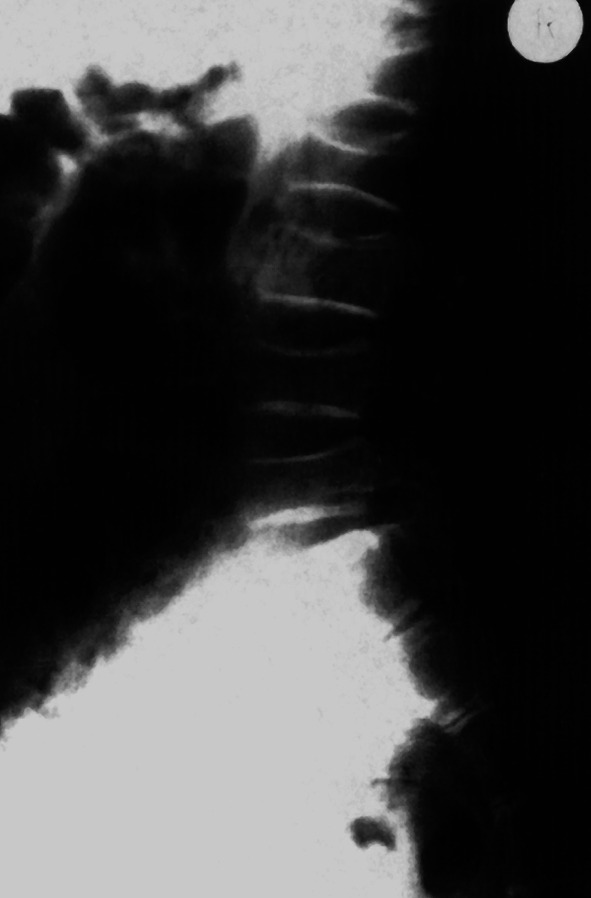
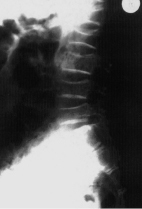
Ein weiterer wichtiger Parameter bei der Einschätzung kindlichen Längenwachstums ist die biologische Reife des Individuums; diese wird erhoben durch das sog. „Knochenalter“. Konfiguration und Größe der Knochen von Handwurzel und Fingern werden dabei entweder mit historischen Referenzaufnahmen verglichen (z. B. Knochenatlas von Greulich und Pyle) oder automatisiert analysiert (z. B. BoneExpert). Aus der Summe des jeweiligen Knochenreifungszustands wird ein „Knochenalter“ ermittelt, dieses mit dem chronologischen Alter in Beziehung gesetzt und hierdurch Hinweise auf das verbleibende Wachstumspotenzial gewonnen.
Endlängenprognose
Eine Abschätzung der Erwachsenengröße eines Kindes kann mit verschiedenen Prognosemodellen vorgenommen werden. Dabei ist zu berücksichtigen, dass diese Prognosen umso ungenauer sind, je jünger das Kind ist, da ggf. während der weiteren Entwicklung des Kindes noch wachstumsrelevante Einflüsse wie verfrühter oder verspäteter Pubertätsbeginn, psychische oder physische Belastungen mit dem Wachstumspotenzial interferieren können.
Genetische Zielgröße („target height“): Der körperhöhenrelevante genetische Hintergrund eines Kindes bildet sich in den Körperhöhen der biologischen Eltern ab: Die Zielgröße des Kindes nach Tanner ist die mittlere Körperhöhe beider Eltern, zu der bei Jungen 6,5 cm addiert und von der bei Mädchen 6,5 cm subtrahiert werden. Allerdings ist der Streubereich dieser Zielgröße mit ±8,5 cm erheblich.
Projizierte Endlänge: eine recht grobe Abschätzung der Erwachsenenlänge ist möglich unter Verwendung des präpubertären Körperlängen SDS-Werts. Bei Übereinstimmung von Lebensalter und biologischer Reife lässt sich bei Verwendung geeigneter Perzentilen die Erwachsenenlänge abschätzen (korrespondierender adulter Körperlängen-SDS-Wert).
Knochenalterbasierte Endlängenprognose: basierend auf Körperhöhenmessung und korrespondierendem Knochenalter lässt sich mit entsprechenden Tabellen (z. B. von Bayley/Pinneau) die Erwachsenengröße abschätzen. Auch hier muss berücksichtigt werden, dass Krankheit/Gesundheit, Pubertätsentwicklung und Körpergewicht/BMI Einfluss auf die Erwachsenengröße haben und damit die Genauigkeit der Endlängenprognose einschränken.
Varianten des Wachstums und Kleinwuchs
Wachstum ist ein dynamischer Prozess, der die Veränderungen der Körperhöhe über die Zeit beschreibt. Eine anhaltend unterdurchschnittliche Wachstumsgeschwindigkeit ist meist pathologisch und sollte zu einer erweiterten Diagnostik führen, selbst wenn die absolute Körpergröße noch im Normalbereich liegt. Umgekehrt kann ein nach der Säuglingsphase identifizierter Kleinwuchs fortbestehen und mit einer normalen Wachstumsgeschwindigkeit einhergehen, ohne dass eine pathologische Krankheitsursache vorliegt (z. B. familiärer Kleinwuchs).
Definition
Kinder, deren Körperhöhe oder -länge unterhalb des altersbezogenen 3. Perzentils liegt, sind kleinwüchsig. Dieses statistische Kriterium erfüllen bei Benutzung aktueller Referenzdaten 3% aller deutschen Kinder. Kleinwuchs kann bei Geburt vorliegen oder entsteht später durch zu langsames oder zu früh endendes Wachstum.
Differenzialdiagnose des Kleinwuchses und von Wachstumsstörungen
Die Differenzialdiagnose des Kleinwuchses ist breit; eine Auswahl relevanter Differenzialdiagnosen ist in Tab. 26.1 dargestellt (Abb. 26.3). Im klinischen Alltag sind die beiden Diagnosegruppen „familiärer Kleinwuchs“ und die „konstitutionelle Verzögerung von Wachstum und Pubertät“ die häufigsten Ursachen für eine Vorstellung in der Wachstumssprechstunde.
| Anamnese | Routinediagnostik | Spezielle Diagnostik |
|---|---|---|
| Messung der Körperhöhen der Eltern (und Geschwister); Berechnung der familiären Zielgrößenbereichs | Messung von Körperhöhe, Wachstumsgeschwindigkeit, Körperproportionen, Gewicht/BMI | Ggf. radiologische und/oder genetische Diagnostik bei V. a. ossären KW |
| Elterliche Pubertätsentwicklung (Menarche der Mutter, Pubertät des Vaters) | Erfassen der Pubertätsstadien nach Tanner, Bestimmung des Knochenalters (Röntgen linke Hand) | |
| IGF-I, IGFBP-3 (WH-Mangel); TSH, fT4 (Hypothyreose) |
Bei Hinweis auf WH-Mangel: STH-Stimulationstest und/oder STH-Spontansekretionsanalyse Ggf. MRT-ZNS (Hypophyse!) |
|
| Schwangerschaftsverlauf (Konsum teratogener Substanzen?), Geburtsparameter | Mutterpass einsehen, ggf. neuropädiatrische Mitbeurteilung | Sonographie, Echokardiographie |
| Familiäre Vorerkrankungen, Familienanamnese | Bei Mädchen mit unerklärtem Kleinwuchs: Karyotyp bestimmen | |
|
Relevante Vorerkrankungen oder Vortherapien? Ernährungsanamnese (Fütterstörungen?, Malnutrition?) |
Internistische Untersuchung; b. B. Bestimmung von: Differenzialblutbild, CRP, BSG, Ferritin, Eisen (Anämie, Infektion, Zöliakie, Mukoviszidose); GPT, GOT, γGT, AP, Albumin (Hepatopathie); Kreatinin, HN, Na, K, Ca, Ph, Blutgasanalyse, UrinStix (Nephropathie); IgA-anti-Endomysium, IgA-anti-Transglutaminase, Gesamt IgA (Zöliakie) | Erweiterte spezifische Diagnostik in Abhängigkeit Verdachtsdiagnose (z. B. spezielle Stoffwechseldiagnostik, genetische Diagnostik |

Konstitutionelle Verzögerung von Entwicklung und Pubertät
Hierbei handelt es sich um eine häufige Normvariante mit meist subnormaler Wachstumsgeschwindigkeit in der Kindheit und verzögertem Pubertätsbeginn. Obwohl eine hohe Familiarität besteht (anamnestisch oft späte Menarche der Mutter oder später Wachstumsschub des Vaters) ist der genetische Hintergrund weitestgehend unklar. Radiologisch findet sich eine verzögerte Knochenreifung. Die betroffenen „Spätentwickler“ erreichen in der Regel ohne hormonelle Intervention eine Erwachsenengröße innerhalb des familiären Zielgrößenbereichs.
Familiärer Kleinwuchs
Wie eingangs beschrieben übt eine Vielzahl verschiedener genetischer Varianten Einfluss auf unsere Körperwachstum aus, sodass die Erwachsenengröße letztlich das Integral von wachstumsattenuierenden und -fördernden Varianten darstellt; ein familiärer Kleinwuchs wird daher meist als Normalvariante angesehen. Allerdings sind die Grenzen zwischen niedrignormalem und pathologischem Wachstum fließend. In Einzelfällen können daher einer familiären Wachstumsstörung auch milde monogene Störungen u. a. des Knochenwachstums im Sinne eines ossären Kleinwuchses zugrunde liegen. Daher ist bei der Beurteilung familiärer Wachstumsstörungen die Beurteilung von Körperproportionen und potenziell assoziierter phänotypischer Merkmale von Relevanz für die Abgrenzung von pathologischem und physiologischem Wachstum.
Wichtige Differenzialdiagnosen des Kleinwuchses
Idiopathischer und familiärer Kleinwuchs
Konstitutionelle Verzögerung von Wachstum und Pubertät
Intrauteriner Kleinwuchs (Small for gestational age (SGA) ohne Aufholwachstum)
Chromosomale Störungen mit Aneuploidie (z. B. Ullrich-Turner-Syndrom, Down-Syndrom)
Syndromale Erkrankungen (z. B. Noonan-Syndrom, Silver-Russell-Syndrom, Prader-Willi-Syndrom, DiGeorge-Syndrom, velokardiofaziales Syndrom u. a.)
Skelettdysplasien (häufig mit disproportioniertem Kleinwuchs; z. B. Achondroplasie, Hypochondroplasie, spondyloepiphysäre Dysplasie, Dyschondrosteose etc.)
Malnutrition
- Organische Ursachen einer Wachstumsstörung
- Kardiale Ursachen
- Pulmonale Ursachen
- Lebererkrankungen
- Gastrointestinale Erkrankungen
- Renale Ursachen
- Chronische Anämien
- Muskuläre und neurologische Erkrankungen
- Chronisch entzündliche Erkrankungen
Endokrine Erkrankungen (z. B. Wachstumshormonmangel, Cushing-Syndrom, Hypothyreose, Leprechaunism, Diabetes mellitus, Laron-Syndrom und andere Störungen der WH-IGF-I-Achse)
- Metabolische Störungen
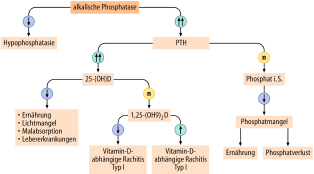
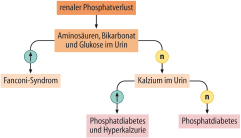
- Störungen des Kalzium-Phosphat-Metabolismus
- Störungen des Kohlenhydratmetabolismus
- Störungen des Lipidmetabolismus
- Störungen des Aminosäuren- und Proteinstoffwechsels
- Störungen des Knochenstoffwechsels
Psycho-soziale Ursachen (z. B. psychosoziale Deprivation, Anorexia nervosa, Depression u. a.)
- Iatrogene Ursachen
- Hochdosierte systemische bzw. lokale Glukokortikoidtherapie
- Schädel- und Ganzkörperbestrahlung
- Chemotherapie
Therapie
Therapeutisches Ziel bei Kindern und Jugendlichen mit Kleinwuchs ist eine Normalisierung des Längenwachstums und eine Vermeidung einer Infantilisierung Betroffener. Bei allen sekundären Kleinwuchsformen steht die Behandlung der Grunderkrankung im Vordergrund (z. B. glutenfreie Ernährung bei Zöliakie).
Obwohl die konstitutionelle Entwicklungsverzögerung und die Mehrzahl der Fälle eines familiären Kleinwuchses als Normvarianten angesehen werden, bei denen eine Endlänge im Bereich der familiären Zielgröße erreicht wird, so kann u. U. doch eine psychologisch/psychotherapeutische Unterstützung mit dem Ziel eines verbesserten Adaptationsprozesses hinsichtlich Kleinwuchs merkmalsbedingter Stressbewältigung hilfreich sein. Ergänzend sollte insbesondere bei den Kleinwuchsformen, die mit anderen assoziierten Problemen verknüpft sind oder bei denen das Risiko einer Alltagsbehinderung besteht eine Anbindung an eine der Selbsthilfegruppen angeraten werden (z. B. Bund kleinwüchsiger Menschen und ihre Familien „BKMF“, Ullrich-Turner-Syndrom-Vereinigung Deutschland, etc.).
Endokrine Therapien sind in Form einer STH-Therapie für folgende Indikationen verfügbar:
hypophysärer Wachstumshormonmangel (Substitutionstherapie),
vorgeburtliche Wachstumsverzögerung (SGA) ohne postnatales Aufholwachstum (supraphysiologisch),
Ullrich-Turner-Syndrom (supraphysiologisch),
Prader-Willi-Syndrom (supraphysiologisch),
SHOX-Defizienz (supraphysiologisch),
Kleinwuchs bei chronischer Niereninsuffizienz (supraphysiologisch).
Darüber hinaus ist für die seltenen Formen der Wachstumshormoninsensitivität eine Therapie mit rekombinantem IGF-1 möglich.
Bei verschiedenen Formen des ossären Kleinwuchses kann in Abhängigkeit der individuellen Befunde in Einzelfällen eine operative Verlängerung von insbesondere Ober- und Unterschenkel durchgeführt werden. Darüber hinaus sind derzeit medikamentöse Therapien der FGFR3-vermittelten Skelettdysplasien in klinischer Erprobung (Kap. 1).
Hochwuchs
Definition
Ein Hochwuchs liegt bei Kindern und Jugendlichen vor, deren Körperhöhe oder -länge oberhalb des altersbezogenen 97. Perzentils liegt. Dieses statistische Kriterium erfüllen bei Benutzung aktueller Referenzdaten 3% aller deutschen Kinder. Dabei sollte ein transienter Hochwuchs aufgrund einer temporären Entwicklungsbeschleunigung von anderen Ursachen abgegrenzt werden.

| Beispielhafte Hochwuchsvariante | Merkmale | |
|---|---|---|
| Transienter Hochwuchs |
- Pubertas und Pseudopubertas präcox - Hyperthyreose - Adiposogigantismus (alimentäre Adipositas) - Konstitutionelle Beschleunigung von Wachstum und Pubertät |
Beschleunigte Knochenreifung |
| Permanente Hochwuchsformen | - Familiärer Hochwuchs | Positive Familienanamnese; Größe im familiären Zielgrößenbereich |
| - Idiopathischer Hochwuchs | ||
| Hochwuchs bei numerischen Chromosomenanomalien |
- Klinefelter-Syndrom - 47,XYY-Syndrom - 47,XXX-Syndrom |
Assoziierte phänotypische Merkmale |
| Syndromaler Hochwuchs mit relevanter Dysproportionierung | - Marfan-Syndrom (Abb. 26.4) | Arachnodaktylie, Linsenluxation, kardiovaskuläre Komplikationen |
| - Homozysteinurie | Psychomotorische Retardierung | |
| Überwuchssyndrome |
- Wiedemann-Beckwith-Syndrom - Simpson-Golabi-Behmel-Syndrom - PTEN-Hamartoma-Syndrom - Proteus-Syndrom - Sotos-Syndrom |
Assoziierte phänotypische Merkmale, Tumorneigung |
| Hypophysärer Gigantismus |
- Isoliertes Hypophysenadenom - Mutation in GPR101 oder AIH |
Erhöhtes IGF-1, IGFBP-3, fehlende WH-Suppression nach Glukosebelastung, pathologisches MRT |
| Seltene Endokrinopathien |
- Defekte im Östrogenrezeptor - Aromatasemangel - Familiärer Glukokortikoidmangel |
Assoziierte phänotypische Merkmale |
Diagnostik
Neben Anamnese und Wachstumsverlauf (perzentilenflüchtiges Wachstum? Elterliche Zielgröße?), klinischer Untersuchung (Dysproportionierung? Pubertätsstatus? Syndromale Auffälligkeiten?) gehören die Bestimmung des Knochenalters (Reifungsbeschleunigung?) und von IGF-I und IGFBP-3 zur Basisdiagnostik. Diese wird ergänzt durch einen WH-Suppressionstest und ein zerebrales MRT bei Verdacht auf ein Wachstumshormon produzierendes Adenom der Hypophyse. Bei klinischen Hinweisen auf eine numerische Chromosomenanomalie sollte eine Karyotypisierung durchgeführt werden.
Therapie
Primär steht bei Kindern und Jugendlichen mit Hochwuchs die Therapie der zugrundeliegenden Ursache im Vordergrund. Eine GnRH-Agonisten-Therapie eines Kindes mit Pubertas präcox centralis und sekundärem Hochwuchs führt z. B. bei ausreichender Therapiedauer meist zu einer Normalisierung des Wachstums und einer Endlänge im familiären Zielgrößenbereich. Allerdings werden bei Patienten mit konstitutionellem, idiopathischen oder syndromalem Hochwuchs immer wieder Alltagsbelastungen wie orthopädische Probleme, Schwierigkeiten bei der Suche nach geeigneter Kleidung, Notwendigkeit angepassten Mobiliars und andere soziale Belastungen angeführt, sodass in Einzelfällen bei hochwüchsigen Kindern, insbesondere bei Mädchen, eine hochwuchsattenuierende Therapie aus psychosozialen Gründen notwendig werden kann. Die Bewertung einer überdurchschnittlichen Körpergröße ist interindividuell und interkulturell sehr unterschiedlich. Es herrscht Konsens, dass eine Behandlung nur in Fällen durchgeführt werden sollte, wenn die minimale Endgrößenprognose bei Jungen über 200 cm oder bei Mädchen über 185 cm liegt.
Die medikamentöse Therapie erfolgt durch eine supraphysiologische Verabreichung von Sexualsteroiden. Hierdurch wird ein früherer und rascherer Verlauf der Pubertät induziert und dadurch ein vorzeitiger Epiphysenfugenschluss erreicht. Allerdings gibt es Hinweise, dass eine wachstumsattenuierende Therapie zu einer reduzierten Fertilität behandelter Mädchen führen könnte. Eltern und Jugendliche sollten daher ausführlich über die Effektivität und Nebenwirkungen der Therapie aufgeklärt werden.
Chirurgisch steht die beidseitige Epiphysiodese als Therapiealternative zur medikamentösen Therapie zur Verfügung. Diese kann insbesondere bei Patienten mit dysproportioniertem Hochwuchs zu einer Normalisierung der Körperproportionen beitragen; größere Fallserien behandelter Patienten stehen allerdings noch nicht zur Verfügung.
Pubertät
Die Pubertät ist die Lebensphase die sich an die Kindheit anschließt und in der sich die Geschlechtsreife entwickelt. Sie ist charakterisiert durch ausgeprägte körperliche, kognitive, emotionale und psychosoziale Veränderungen. Sie wird reguliert durch eine komplexe Interaktion inhibierender und aktivierender Faktoren. Die Pubertät wird ausgelöst durch eine Re-Aktivierung der Hypothalamus-Hypophysen-Gonaden-Achse, die nach frühen Phasen der Aktivierung in der Mittschwangerschaft und postnatal in der sog. Minipubertät mit ca. 6 Monaten in eine Ruhephase übergeht. Obwohl sich unser Verständnis der Regulation dieses Netzwerks deutlich vergrößerte sind die Mechanismen der Pubertätsauslösung nicht vollständig bekannt.
Der Zeitpunkt des Pubertätsbeginns und des Pubertätsfortschritts variiert abhängig von familiärem Hintergrund, Ethnizität und von Umweltfaktoren; er unterlag in den letzten 150 Jahren einem deutlichen säkularen Trend, der sich aber zumindest in Deutschland in den letzten zwei Jahrzehnten deutlich abgeschwächt hat.
Pubertätsentwicklung
Bei der (Re)aktivierung des sog. Pubertätsgenerators, die den Pubertätsbeginn markiert, kommt es zu einer zunächst vorwiegend nächtlichen pulsatilen Freisetzung von GnRH im Hypothalamus, das über den portalen Gefäßplexus zur Hypophyse transportiert wird und dort zu einer Freisetzung der Gonadotropine LH und FSH führt (Details Abb. 26.5). Diese gelangen über die periphere Zirkulation zu den Gonaden und induzieren dort Synthese und Freisetzung von Sexualsteroiden (Testosteron, Östradiol, Progesteron), welche sowohl über eine stimulierende als auch inhibierende Rückkopplung die hypothalamisch/hypophysäre Freisetzung von GnRH und Gonadotropinen modulieren können.

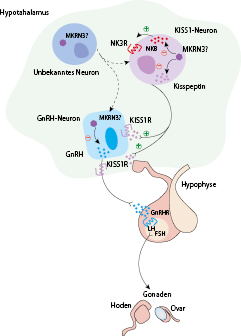
Unter dem zunehmenden Einfluss der Sexualsteroide kommt es beim Mädchen zu einer Vergrößerung der Brustdrüse und einer Größenzunahme des Uterus, während Testosteron beim Jungen zur Ausprägung der männlichen sekundären Geschlechtsmerkmale beiträgt.
Die äußeren Merkmale der Pubertätsentwicklung werden nach Marshall und Tanner in verschiedene Stadien eingeteilt (Abb. 26.6): Schambehaarung („Pubesbehaarung“, P1 bis P6; Tab. 26.3), Brustentwicklung (B1 bis B5; Tab. 26.4) und Genitalentwicklung (G1 bis G5; Tab. 26.5). Hierbei wird das Hodenvolumen mittels eines Orchiometers geschätzt, das Vergleichsovoide unterschiedlichen Volumens enthält.
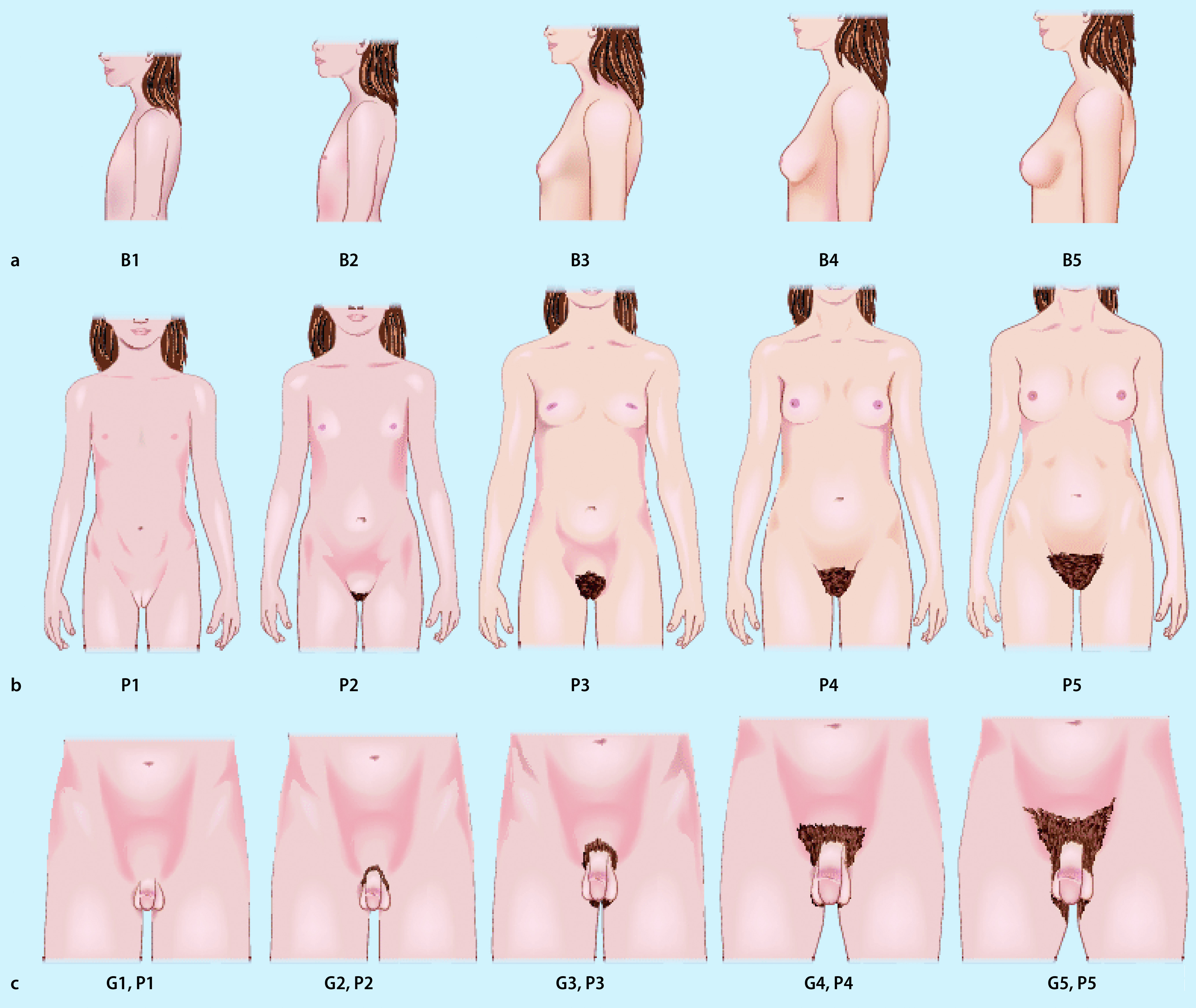
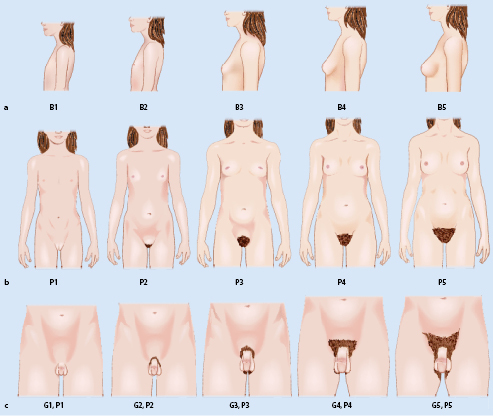
| PH1 | Präpuberal – keine Pubesbehaarung |
| PH2 | Spärliches Wachstum von langen, leicht pigmentierten, flaumigen Haaren, glatt oder gering gekräuselt. Sie erscheinen hauptsächlich an der Peniswurzel bzw. entlang der großen Labien |
| PH3 | Beträchtlich dunklere, kräftigere und stärker gekräuselte Haare. Behaarung geht über die Symphyse etwas hinaus |
| PH4 | Behaarung entspricht dem Erwachsenentyp, die Ausdehnung ist aber noch beträchtlich kleiner. Noch keine Ausbreitung auf die Innenseite der Oberschenkel |
| PH5 | In Dichte und Ausdehnung wie beim Erwachsenen, aber nach oben horizontal begrenzt (Dreieckform); Übergang auf Oberschenkel |
| PH6 | Bei 80% der Männer und 10% der Frauen kommt es zu weiterer Ausbreitung der Behaarung nach oben zum Nabel hin |
| B1 | Fehlende Brustentwicklung, keine palpable Drüse |
| B2 | Brustknospung. Brustdrüse und Warzenhof sind leicht erhaben |
| B3 | Brustdrüse ist stärker vergrößert als der Warzenhof. Die Form entspricht der einer erwachsenen Brust |
| B4 | Die Drüse im Warzenhofbereich hebt sich mit einer eigenen Kontur vom übrigen Anteil der Brust ab |
| B5 | Die Vorwölbung im Warzenhofbereich des Stadiums B 4 weicht in die abgerundete Kontur der erwachsenen Brust zurück |
| G1 | Infantil, Hodenvolumina <3 ml |
| G2 | Vergrößerung des Skrotums, Hodenvolumina 4–8 ml |
| G3 | Vergrößerung des Penis in die Länge, Vergrößerung von Testes und Skrotum |
| G4 | Penis wird dicker, Entwicklung der Glans, Skrotalhaut wird dunkler, Samenerguss |
| G5 | Genitalien ausgereift wie beim erwachsenen Mann, reife Spermien |
Der zeitliche Ablauf der Pubertät ist in Abb. 26.7 dargestellt.

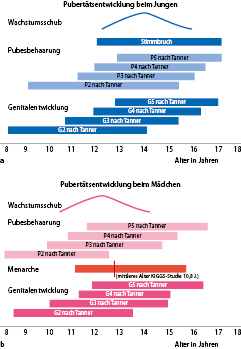
Pubertätsentwicklung bei Mädchen
Die Pubertät beim Mädchen beginnt mit der Entwicklung der Brustdrüsen (Thelarche). Diese tritt derzeit im Mittel mit 10,5 Jahren auf (Abb. 26.7b). Tab. 26.4 zeigt die weitere Entwicklung der Brustdrüse, wobei bei einigen Mädchen das Stadium B3 direkt in das Stadium B5 übergeht. Nahezu zeitgleich mit der Thelarche kommt es zur Entwicklung von Sekundärbehaarung (Pubarche). Diese steht initial v. a. unter dem Einfluss von adrenalen, weniger gonadalen Steroiden (Adrenarche); sie kann im Einzelfall auch der Thelarche vorangehen. Etwa zwei Jahre nach der Thelarche kommt es bei Mädchen zur ersten Regelblutung (Menarche). In dem zwischen 2003 und 2006 durchgeführten Kinder- und Jugendsurvey des Robert-Koch-Instituts (KIGGS) wurde ein Menarchemedian von 12,8 Jahren angeben. Das Menarchealter wird u. a. durch Ethnizitität, Sozialstatus und BMI moduliert. Nachdem vor dem Beginn der Pubertätsentwicklung die Wachstumsgeschwindigkeit deutlich abfällt, steigt diese mit Beginn der Pubertät infolge einer insbesondere östrogenstimulierten vermehrten Freisetzung von hypophysärem Wachstumshormon erneut an auf ein Maximum im Alter von ca. 12 Jahren (Pubertätswachstumsschub).
Pubertätsentwicklung bei Jungen
Beim Jungen ist das erste Pubertätsmerkmal die Vergrößerung des Hodenvolumens (>3 ml) in einem durchschnittlichen Alter von 12 Jahren. Im Anschluss daran kommt es zur Entwicklung von Sekundärbehaarung (Pubarche), die allerdings für viele Jungen das erste selbst bemerkte Pubertätsmerkmal darstellt. Durch die Zunahme der gonadalen Sexualsteroidsynthese kommt es im Verlauf zu einer Vergrößerung des Penis (G1–G5) und zu Bartwuchs. Erste Spermien im Morgenurin lassen sich im Mittel im Alter von 13,4 Jahren nachweisen (Spermarche). Im Gegensatz zu Mädchen tritt der Pubertätswachstumsschub beim Jungen deutlich später auf. Die maximale pubertäre Wachstumsgeschwindigkeit liegt im Median bei 14 Jahren. Oft letztes Pubertätsmerkmal ist die Mutation (Stimmbruch), die im KIGGS-Survey im Median von 15 Jahren berichtet wurde (bei großer Variabilität).
Normvarianten der Pubertätsentwicklung
Konstitutionelle Verzögerung von Wachstum und Pubertät
Abschn. 26.1
Prämature Thelarche
Eine isolierte vorzeitige Entwicklung der Brustdrüse ohne sonstige Pubertätszeichen wird als prämature Thelarche bezeichnet. Die Ätiologie der prämaturen Thelarche ist nicht bekannt. Sie tritt typischerweise innerhalb der ersten 2 Lebensjahre auf; die Brustentwicklung geht oft nicht über palpables subareoläres Brustdrüsengewebe hinaus (B2). Man findet keine Aktivierung der Hypothalamus-Hypophysen-Gonadenachse; das Knochenalter ist nicht akzeleriert. Ebenso finden sich keine Einflüsse auf Längenwachstum oder Uterusentwicklung.
Eine Therapie ist nicht erforderlich. Allerdings sollte eine longitudinale Supervision der Patientinnen angeregt werden, da die Abgrenzung zur Pubertas präcox im Einzelfall erst im Verlauf möglich wird.
Prämature Pubarche
Das isolierte vorzeitige Auftreten von Sekundärbehaarung wird als prämature Pubarche bezeichnet (Pubarche bei Mädchen vor dem 8., bei Jungen vor dem 9. Geburtstag).
Eine prämature Pubarche scheint bei Kindern gehäuft aufzutreten, die mit für das Gestationsalter zu niedrigen Geburtsmaßen zur Welt kamen (SGA). Bei diesen Kindern scheint die prämature Pubarche zusätzlich mit einem erhöhten Risiko für metabolische Folgeerkrankungen sowie dem Auftreten eines polyzystischen Ovarsyndroms (PCOS) assoziiert zu sein.
Die prämature Pubarche ist Folge einer prämaturen Aktivierung der Nebennierenrindenaktivität (Adrenarche); deren biochemisches Merkmal ist der laborchemische Nachweis erhöhter Konzentrationen von DHEA und DHEAS.
Klinisch können milde Zeichen einer Hyperandrogenämie wie unreine Haut oder verfrüht auftretender Schweißgeruch bemerkt werden. Es besteht oft kein oder nur eine geringe Akzeleration der Skelettentwicklung ohne Beschleunigung des Längenwachstums. Abzugrenzen sind Erkrankungen mit erhöhter adrenaler Androgenproduktion (z. B. nichtklassisches adrenogenitales Syndrom, androgenproduzierende Tumoren, etc.).
Eine Verlaufsbeobachtung über 6–12 Monate ist hilfreich, um eine Pubertas präcox sicher abgrenzen zu können.
Eine Therapie ist nicht erforderlich. Bei ehemaligen SGA-Kindern mit prämaturer Pubarche sollte eine Verlaufsbeobachtung hinsichtlich der genannten Risiken angeraten werden.
Pubertätsgynäkomastie
Mit Beginn der Sexualsteroidproduktion kommt es bei vielen Jungen (50–90%) zu einer transienten, meist beidseitigen Vergrößerung der Brustdrüse. Ursächlich ist vermutlich eine vermehrte Aromatisierung von Testosteron zu Östradiol. Klinisch lässt sich subareolär oft nur eine kleine Verhärtung tasten. Abzugrenzen ist eine Lipomastie bei adipösen Jugendlichen.
Differenzialdiagnostische Überlegungen sind nur bei atypischem Manifestationsalter oder zusätzlicher klinischer Symptomatik notwendig (z. B. auffällige Medikamentenanamnese; u. a. Neuroleptika, Spironolakton) oder Vorliegen zusätzlicher assoziierter phänotypischer Merkmale (z. B. Hinweise auf ein Klinefelter-Syndrom).
Eine Therapie ist meist nicht notwendig.
Vorzeitige Pubertätsentwicklung (Pubertas präcox)
Definition
Das vorzeitige Auftreten von Pubertätsmerkmalen in einem Alter von mehr als zwei Standardabweichungen vor dem durchschnittlichen Pubertätsbeginn wird als Pubertas präcox bezeichnet. Dies korrespondiert zum Auftreten erster Pubertätsmerkmale bei Mädchen vor dem 8. und bei Jungen vor dem 9. Geburtstag.
Die vorzeitigen Pubertätsstörungen können in gonadotropinabhängige (Pubertas präcox vera oder centralis) und in gonadotropinunabhängige Pubertätsstörungen (Pseudopubertas präcox) unterschieden werden. Während sich bei der Pubertas präcox vera die Pubertätsmerkmale zwar verfrüht, aber in der typischen Sequenz manifestieren treten Pubertätszeichen bei der Pseudopubertas präcox unabhängig vom normalen Pubertätsverlauf auf.
Pubertas präcox vera
Epidemiologie
Die Pubertas präcox vera (oder centralis) tritt mit einer Inzidenz von 1:5.000 bis 1:10.000 auf. Es besteht eine deutliche Mädchenwendigkeit (Mädchen sind 5- bis 10-mal häufiger betroffen).
Ätiologie/Pathogenese
In der Mehrzahl der Fälle ist die Pubertas präcox beim Mädchen idiopathisch. Bei einem Teil der Patienten konnten Mutationen in pubertätsrelevanten Genen gefunden werden (u. a. KISS1, KISS1R [syn. GPR54], MKRN3, DLK1). Seltene Ursachen für eine Pubertas präcox sind ZNS-Tumoren (insbesondere Hamartome), andere ZNS-Läsionen, eine vorangehende Schädelbestrahlung oder Endokrinopathien (z. B. eine lange unbehandelte Hypothyreose). Bei Jungen finden sich organische Ursachen einer Pubertas präcox vera zu einem deutlich höheren Anteil (40–90% bei Jungen gegenüber 10–30% bei Mädchen).
Klinik
Neben der vorzeitigen Pubertätsentwicklung findet sich ein beschleunigtes Längenwachstum als Ausdruck der sexualsteroidvermittelten Zunahme der Wachstumshormonsekretion. Allerdings kommt es durch die Beschleunigung der Knochenreifung, die sich radiologisch als Knochenalterakzeleration manifestiert, zu einem vorzeitigen Verschluss der Epiphysenfugen und damit zum Risiko einer adulten Kleinwüchsigkeit unterhalb des individuellen familiären Zielgrößenbereichs. Die frühe Ausbildung der Geschlechtsmerkmale kann in Abhängigkeit des Manifestationsalters und der individuellen Situation zu einer erheblichen psychosozialen Belastung führen.
Diagnose
Biochemisch ist eine Messung der basalen Gonadotropinkonzentration aufgrund deren pulsatiler Sekretion meist nicht weiterführend. Im GnRH-Test lassen sich aber bei Vorliegen einer zentralen Pubertas präcox die Gonadotropine auf pubertäre Werte mit einer Dominanz von LH gegenüber FSH stimulieren (LH/FSH-ratio >1). Typischerweise finden sich erhöhte Serumkonzentrationen für die Sexualsteroide Östradiol oder Testosteron.
Radiologisch zeigt die Kochenalterbestimmung ein akzeleriertes Knochenalter; sonographisch lassen sich beim Mädchen eine Maturierung des Uterus sowie Follikelzysten der Ovarien nachweisen. Zur Abklärung einer organischen Ursache sollte bei allen Jungen und bei Mädchen mit Manifestion vor dem 6. Geburtstag eine zerebrale Kernspintomographie erfolgen.
Therapie
Indikationen für eine medikamentöse Intervention sind das o. g. Risiko einer Reduktion der adulten Körpergröße sowie die große psychosoziale Belastung durch die verfrühte Pubertätsentwicklung der oft sehr jungen Kinder.
Aufgrund des variablen Verlaufs empfiehlt sich vor Einleitung einer Behandlung eine 3- bis 6-monatige Beobachtungsphase, da bei einem Bruchteil der Patienten nach verfrühtem Beginn der weitere Pubertätsprogress langsam verläuft. Schreitet die Pubertätsentwicklung rasch fort sollte eine medikamentöse Therapie mit GnRH-Agonisten eingeleitet werden. Diese führen nach einer initialen Stimulation („flare-up“) im weiteren Verlauf durch eine „down-Regulation“ hypophysärer GnRH-Rezeptoren infolge kontinuierlicher GnRH-Exposition und Wegfall der pulsatilen Hypophysenstimulation zu einem Abfall der Konzentrationen für Gonadotropine und Sexualsteroide. Hierdurch wird der weitere Pubertätsprogresss verhindert und bei rechtzeitigem Therapiebeginn die Erwachsenenlänge verbessert.
Pseudopubertas präcox
Definition
Unter einer Pseudopubertas präcox werden Erkrankungen subsummiert, die zu einer vorzeitigen Entwicklung von Pubertätsmerkmalen führen, ohne dass eine Aktivierung des GnRH-Pulsgenerators vorliegt.
Ursachen
Exogene Hormonexposition (akzidentelle Ingestion von Östrogenen/Adrogenen, Umweltexposition z. B. durch Phytoöstrogene),
Hormonproduzierende Tumore (z. B. HCG-produzierendes Pinealom, androgenproduzierender Nebennierentumor, gonadale Tumoren wie Ovarialtumor oder Leydigzelltumor),
Adrenale Androgensynthese infolge Enzymdefekt (insbesondere kongenitales adrenogenitales Syndrom; Abschn. 7.5.5),
GnRH-unabhängige gonadale Aktivität (Testotoxikose, autonome Ovarialzysten, McCune-Albright-Syndrom).
Diagnose
Klinisch zeigt sich in Abhängigkeit der Ätiologie eine isosexuelle oder heterosexuelle vorzeitige Pubertätsentwicklung, mit korrespondierend erhöhten Androgenen oder Östrogenen. Radiologisch findet sich wie bei der zentralen Pubertas präcox eine Beschleunigung der Knochenentwicklung. Im GnRH-Test zeigt sich aber nur eine präpubertäre oder sogar supprimierte Stimulierbarkeit der Gonadotropine LH und FSH.
Therapie
Da bei der Pseudopubertas präcox keine zentrale Aktivierung zugrunde liegt sind GnRH-Agonisten nicht wirksam. Bei Erkrankungen mit autonomer peripherer Östrogensynthese wie z. B. bei autonomen Ovarialzysten oder dem McCune-Albright-Syndrom kann ein Therapieversuch mit Aromataseinhibitoren oder selektiven Östrogenmodulatoren erwogen werden.
Verspätete Pubertätsentwicklung (Pubertas tarda)
Definition
Eine Pubertas tarda beim Mädchen liegt vor, wenn im Alter von 13,5 Jahren noch keine Pubertätsmerkmale vorliegen sowie wenn im Alter von 15 Jahren die Menarche noch nicht eingetreten ist.
Beim Jungen liegt eine Pubertas tarda vor, wenn im Alter von 15 Jahren noch keine Pubertätsmerkmale eingetreten sind.
Bei beiden Geschlechtern pathologisch ist ein zu langsamer Pubertätsprogress (wenn die Pubertätsdauer mehr als 5 Jahre beträgt) oder wenn es zu einem Pubertätsstillstand von mehr als 18 Monaten kommt.
Ätiologie
Die Ursachen einer verzögerten Pubertätsentwicklung sind vielfältig. Sie beinhalten u. a. schwere Allgemeinerkrankungen, Malnutrition und Essstörungen, aber auch extreme sportliche Anstrengungen wie bei Leistungssportlern. Unter den endokrinen Ursachen sind zentrale hypothalamisch/hypophysäre Störungen mit niedrigen Serumkonzentrationen von Gonadotropinen und Sexualsteroiden (hypogonadotroper Hypogonadismus) zu unterscheiden von peripheren Störungen der Gonadenfunktion mit niedrigen Sexualsteroiden, aber via feedback erhöhten Gonadotropinen (hypergonadotroper Hypogonadismus).
Normvarianten
Wichtigste Differenzialdiagnose einer Pubertas tarda mit der biochemischen Konstellation eines hypogonadotropen Hypogonadismus ist die konstitutionelle Verzögerung von Wachstum und Pubertät (Abschn. 26.1.3).
Hypogonadotroper Hypogonadismus
Definition
Hypothalamische oder hypophysäre Störungen, die zu einer Mindersekretion von Gonadotropinen und damit zu einer verminderten Sexualsteroidsynthese führen, bezeichnet man als hypogonadotropen Hypogonadismus. Die Differenzialdiagnose des hypogonadotropen Hypogonadismus ist in Tab. 26.6 dargestellt.
| Ursache | Erkrankung |
|---|---|
| Funktioneller hypothalamischer Hypogonadismus |
- Leistungssport (z. B. Turnerinnen) - Chronische Erkrankungen, z. B. zystische Fibrose, chronisch entzündliche Darmerkrankung, terminale Niereninsuffizienz) - Esstörungen wie Anorexia nervosa |
| Posttraumatisch/Postoperativ/Post radiationem | - Nach z. B. Hypophysentumoroperation |
| ZNS-Tumor; andere Systemerkrankungen |
- Kraniopharyngeom - Langerhans-Histiozytose - Germinom - Prolaktinom - Speichererkrankungen - Hypophysitis - Xanthogranulom |
| Endokrinopathien |
- Isolierter hypogonadotroper Hypogonadismus - Kallmann-Syndrom (Kombination von hypogonadotropem Hypogonadismus mit [partieller] Anosmie infolge Migrationsstörung der GnRH-Neurone) - Multiple Hypophysenvorderlappeninsuffizienz |
| Sonstige |
- Syndromale Erkrankungen (z. B. Prader-Willi-Syndrom) - ZNS-Fehlbildungen (z. B. septooptische Dysplasie) |
Diagnose
Basal niedrige Serumkonzentrationen von Gonadotropinen und Sexualsteroiden, im GnRH-Test fehlender Anstieg der Gonadotropinsekretion. Zum Ausschluss einer partiellen Anosmie bei V. a. Kallmann-Syndrom sollte eine standardisierte Riechprüfung durchgeführt werden. Ergänzend wird zum Ausschluss zentralnervöser Pathologien eine zerebrale MRT durchgeführt. Radiologisch findet sich meist ein retardiertes Knochenalter.
Therapie
Abschn. 7.1.4.3.
Hypergonadotroper Hypogonadismus
Definition
Primäre Störungen der Gonadenfunktion mit niedrigen Sexualsteroiden, aber via feedback erhöhten Gonadotropinen, werden als hypergonadotroper Hypogonadismus bezeichnet.
Die Ätiologie unterscheidet sich zwischen den Geschlechtern. Beim Mädchen stellt das Ullrich-Turner-Syndrom die wichtigste und häufigste Differenzialdiagnose dar (Inzidenz 1:2.500; Tab. 26.7). Sehr viel seltener sind andere Formen der Gonadendysgenesie oder -insuffizienz, z. B. als Spätfolge nach Chemotherapie, Radiatio, postoperativ oder im Rahmen einer polyglandulären Insuffizienz.
| Ullrich-Turner-Syndrom | Klinefelter-Syndrom | |
|---|---|---|
| Ätiologie | Numerische Chromosomenaberration (Karyotyp 45,X0, oder 46,XX,del oder 46,Xr(X) in> 5% analysierter Mitosen) | Numerische Chromosomen-aberration (Karyotyp 47,XXY oder höhergradige Polysomie) |
| Häufigkeit | 1:2500 | 1.800 bis 1:1000 |
| Klinik |
Variabel! - Regelhaft Kleinwuchs - Meist Gonadendysgenesie - Häufige Otitiden als Kleinkind - Haut- und Hautanhangsgebilde: (Nageldysplasien, zahlreiche Pigmentnävi) - Assoziierte Malformationen: bikuspide AO-Klappe, Aortenisthmusstenose, Hufeisenniere, faziale Dysmorphien, Pterygium colli, Cubitus valgus - Erhöhte Autoimmunität (z. B. erhöhte Inzidenz an Autoimmunthyreoiditis, T1DM, Zöliakie) - 90% durchschnittlicher IQ (gehäuft einzelne spezielle Einschränkungen wie bei räumlichem Sehen oder Aufmerksamkeit) |
Variabel! - Häufig (relativer) Hochwuchs - Variable Gonadeninsuffizienz mit postpubertär häufig kleinen Testes - Gynoider Habitus mit spärlicher Körperbehaarung und Gynäkomastie - Gehäuft Kryptorchismus, selten Mikropenis - Meist durchschnittliche Intelligenz - Gehäuftes Auftreten von Verhaltensauffälligkeiten und Sprachentwicklungs- oder -verarbeitungsstörungen sowie fraglich von Erkrankungen des Autismus-Spektrums |
| Diagnose | Chromosomenanalyse | Chromosomenanalyse |
| Therapie |
- Ggf. Therapie des Kleinwuchses mit rekombinantem Wachstumshormon - Sexualsteroidsubstitution - Therapie assoziierter Erkrankungen (z. B. kardiologische, HNO-ärztliche, reproduktionsmedizinische Mitbetreuung) |
- Sexualsteroidsubstitution - Therapie assoziierter Erkrankungen (z. B. logopädische oder psychologische Mitbetreuung) |
Beim Jungen stellt das Klinefelter-Syndrom eine häufige Ursache des primären Hypogonadismus dar (Inzidenz 1:1000, Tab. 26.7). Andere Ursachen sind erworbene Anorchien, z. B. posttraumatisch oder durch bilaterale Hodentorsion, nach Vanishing-testis-Syndrom, als Spätfolge nach Chemotherapie, nach Radiatio, postoperativ oder im Rahmen einer polyglandulären Insuffizienz.
Diagnose
Biochemisch finden sich erniedrigte Sexualsteroide, präpubertär oft noch normwertig niedrige Gonadotropinkonzentrationen, die aber während und nach der Pubertät deutlich über den Normalbereich hinaus ansteigen. Radiologisch findet sich ein retardiertes Knochenalter. Beim Ullrich-Turner- und Klinefelter-Syndrom erfolgt die Diagnosesicherung durch eine Karyotypisierung.
Therapie
Bei nachgewiesenem Hypogonadismus ist das therapeutische Ziel, durch Substitution von Sexualsteroiden einen möglichst normnahen Pubertätsverlauf zu erreichen.
Mädchen
Bei bereits bekanntem Hypogonadismus sollte ein möglichst zeitgerechter Start der Pubertätsinduktion angestrebt werden. Auch bei Mädchen mit Ullrich-Turner-Syndrom wird mittlerweile in der aktuell gültigen Leitlinie ein Beginn der Pubertätsinduktion zwischen 11 und 12 Jahren empfohlen, um durch eine normnahe Einleitung der Sexualsteroidsubstitution Risiken für die Knochengesundheit und psychosoziale Belastungen zu vermeiden. Die Substitution erfolgt dabei durch eine orale oder transdermale Östrogengabe in steigender Dosierung und wird im Verlauf in Abhängigkeit der Brust- und Uterusentwicklung durch eine zyklische Gestagengabe ergänzt.
Jungen
Die Behandlung des Hypogonadismus beim Jungen besteht aus einer topischen oder intramuskulären Testosterongabe in ansteigender Dosierung; eine orale Pubertätsinduktion und Androgenersatz sind durch den First-Pass-Effekt der Leber und die nicht selten hepatotoxische Androgenwirkung kein gängiges Therapieprinzip.
Allerdings wird durch eine isolierte Androgensubstition kein testikuläres Wachstum und keine Stimulation der Spermatogenese erreicht. Daher kann in Einzelfällen von sekundärem oder tertiären Hypogonadismus die Pubertätsinduktion durch eine kombinierte Gonadotropinsubstitution (FSH, HCG) erfolgen.
Hypothalamus-Hypophysen-Achse
Die Hypothalamus-Hypophysen-Achse integriert zahlreiche periphere endokrine Signale und zentralnervöse Einflüsse und übt über die Sekretion zahlreicher Hormone eine zentrale Rolle hinsichtlich Körperwachstum und Energiehaushalt, Salz-Wasser-Haushalt und Reproduktion aus.
Aus endokriner Perspektive kommen insbesondere den Kerngebieten von Nucleus arcuatus (ARC) sowie Nuclei para- und periventricularis (PVH) besondere Bedeutung zu, da dort verschiedene Releasinghormone gebildet werden, die über den portalen Kreislauf die hypophysäre Hormonsekretion beeinflussen. Zusätzlich sind die supraoptischen und periventrikulären Kerngebiete endokrin relevant, da deren Axone mit der Neurohypophyse verbunden sind und die Sekretion von antidiuretischem Hormon (ADH) und Oxytocin regulieren.
Anatomisch besteht die Hypophyse aus dem Hypophysenvorderlappen (HHL; Adenohypophyse mit pars anterior und intermedia) sowie dem Hypophysenhinterlappen (HHL; Abb. 26.8). In der Adenohypophyse werden 6 verschiedene Hormone gebildet und sezerniert:
Somatotropin (STH),
Prolaktin (PRL) ,
adrenokortikotrophes Hormon (ACTH),
thyreoideastimulierendes Hormon (TSH) und
die Gonadotropine (luteinisierendes Hormon, LH; follikelstimulierendes Hormon, FSH).

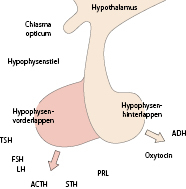
Diese gelangen über den Sinus petrosus in die periphere Zirkulation; während die Hormone des HHL direkt sezerniert werden.
Störungen der Hypothalamus-Hypophysen-Achse umfassen neben Fehlbildungssyndromen, angeborenen genetischen Störungen mit isolierten oder kombinierten HVL-Ausfällen, isoliertem Exzess einzelner Hormone oder Hormonexzess im Rahmen von syndromalen Erkrankungen zahlreiche erworbene Ursachen (z. B. paraneoplastisch, posttraumatisch, postoperativ, post radiatio).
Somatotropin (STH)
Wachstumshormonmangel
Physiologie
Abschn. 26.1.1.
Neben der wachstumsstimulierenden Wirkung übt STH auch Effekte u. a. auf den Metabolismus, die Muskel-Knochen-Einheit und das kardiovaskuläre System aus, sodass auch ein im Erwachsenenalter bestehender STH-Mangel behandlungsbedürftig sein kann.
Epidemiologie
Die Inzidenz des STH-Mangels wird mit 1:3.000 bis 1:10.000 geschätzt. Neben syndromalen Formen (z. B. bei einer septooptischen Dysplasie oder anderen Mittellinienfehlbildungen) kommen ursächlich neben Geburtstraumata (gehäuft nach Beckenendlage), Schädel-Hirn-Traumata, hypothalamisch/hypophysäre Tumoren und Folgen der Tumortherapie insbesondere genetische Ursachen des STH-Mangels in Frage (isoliert: Mutationen in GH1- oder GHRHR-Gen, kombiniert mit anderen Ausfällen als Folge von Mutationen in PIT-1, PROP1, LHX3, LHX4, andere).
Klinik
Neugeborene mit STH-Mangel kommen meist mit normalen Geburtsmaßen zur Welt, zeigen aber häufig Hypoglykämien und insbesondere bei kombiniertem Ausfall mit ACTH eine cholestatische Hepatopathie. Neben einem puppenartigen Aussehen können ggf. phänotypisch ein mittlerer Schneidezahn oder ein Mikropenis vorliegen. Nach der Säuglingsphase kommt es häufig infolge einer unterdurchschnittlichen Wachstumsgeschwindigkeit zu einem perzentilenflüchtigen Wachstum und zu einem Kleinwuchs unterhalb des familiären Zielgrößenbereichs.
Diagnose
Die Diagnose eines STH-Mangels im Kindesalter basiert wesentlich auf auxiologischen Kriterien. Eine normwertige Wachstumsgeschwindigkeit macht das Vorliegen eines STH-Mangels sehr unwahrscheinlich.
Aufgrund der pulsatilen Freisetzung ist eine isolierte Bestimmung von STH für die Diagnose des STH-Mangels nicht weiterführend (Ausnahme: Neugeborenes).
Ein wichtiger Mediator der hypophysären Wachstumshormonwirkung ist der insulinähnliche Wachstumsfaktor IGF-1. Dieser zirkuliert im Serum als Teil eines ternären Komplexes zusammen mit dem IGF-Bindungsprotein-3 (IGFBP-3) und der sog. säurelabilen Untereinheit (acid labile subunit; ALS). Bei Kindern mit STH-Mangel liegen meist erniedrigte Serumkonzentrationen für IGF-1 oder IGFBP-3 vor. Normwertige Konzentration für IGF-1 und IGFBP-3 machen einen STH-Mangel unwahrscheinlich. Umgekehrt gibt es aber neben einem STH-Mangel zahlreiche andere Ursachen für eine Erniedrigung der IGF-1-Serumkonzentration, wie Malnutrition, Adipositas oder chronische Entzündungen.
Radiologisch findet sich typischerweise bei Kindern mit einem STH-Mangel ein retardiertes Knochenalter.
Sind die auxiologischen, biochemischen und radiologischen Kriterien für die Verdachtsdiagnose eines STH-Mangels erfüllt, wird typischerweise eine Stimulation der STH-Sekretion durch pharmakologische Stimuli durchgeführt (z. B. Arginin-, Clonidin-, Glukagon- oder Insulin-Hypoglykämie-Test). Als auffällig wird eine maximale stimulierte STH-Konzentration <8 ng/ml gewertet. Alternativ wird in Einzelfällen eine Analyse der nächtlichen Spontansekretion von STH herangezogen. Zur differenzialdiagnostischen Abgrenzung der häufigen Differenzialdiagnose „konstitutionelle Verzögerung von Wachstum und Pubertät“ ist vor Durchführung der Stimulationstestung ein sog. „priming“ mit Sexualsteroiden notwendig bei ab einem Alter von 8 Jahren für Mädchen und ab 10 Jahren für Jungen, die zur Testdurchführung noch keine Pubertätsmerkmale aufweisen.
Wurde ein STH-Mangel diagnostiziert sollte zum Ausschluss einer Pathologie des ZNS eine zerebrale Kernspintomographie durchgeführt werden.
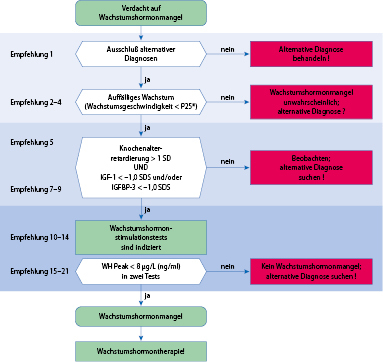
In der aktuell gültigen S2-Leitlinie der AWMF zur Diagnostik ist der in Abb. 26.9 hinterlegte Algorithmus vorgesehen, der die Bedeutung der auxiologischen Parameter bei der Diagnostik des STH-Mangels unterstreicht.

Therapie
Die Therapie des STH-Mangels besteht aus einer Substitution des defizienten STH in einer Dosierung von 25–30 µg/kgKG/d in Form von rekombinantem STH durch eine einmalig tägliche subkutane Injektion. Typischerweise kommt es hierdurch insbesondere zu Beginn der Therapie zu einem deutlichen Aufholwachstum; bei früher Diagnosestellung wird eine Erwachsenengröße im elterlichen Zielgrößenbereich erreicht.
Bei Patienten mit sog. absolutem STH-Mangel (max. stimuliertes STH <3 ng/ml; oft kombiniert mit Defizienz anderer HVL-Hormone) kann ggf. die Notwendigkeit einer lebenslangen Substitutionstherapie bestehen.
Wachstumshormonexzess
Ein Wachstumshormonexzess in Form eines hypophysären Gigantismus ist eine der Differenzialdiagnosen einer kindlichen Makrosomie bzw. eines Hochwuchses. Es handelt sich dabei um eine extrem seltene Diagnose im Kindesalter.
Laborchemisch finden sich bei diesen Patienten erhöhte Serumkonzentrationen an IGF-1 und IGFBP-3 sowie aufgrund der autonomen STH-Produktion eine fehlende Supprimierbarkeit von STH durch eine orale Glukosebelastung. Radiologisch findet sich u. U. ein hypophysäres Mikroadenom, das dann operativ entfernt werden sollte.
Thyreoideastimulierendes Hormon (TSH)
Zentrale Hypothyreose
Hypophysäres TSH wird unter dem Einfluss von hypophysärem Thyreotropin-Releasing-Hormon (TRH) freigesetzt, bindet an den TSH-Rezeptor der Schilddrüsenzelle und stimuliert dort die Schilddrüsenhormonbiosynthese. Bei Entwicklungs- oder Funktionsstörung der Schilddrüse kommt es infolge der ausbleibenden negativen Rückkopplung zu einer Stimulation der TSH-Sekretion, die man im Neugeborenenscreening auf eine angeborene Hypothyreose diagnostisch nutzt (Kap. 4).
Sehr viel seltener als primäre Hypothyreosen kommen sekundäre Hypothyreosen vor, entweder als isolierter oder als kombinierter Ausfall zusammen mit anderen Hypophysenhormonen.
Cave
Derzeit wird eine angeborene zentrale (sekundäre) Hypothyreose in Deutschland durch das Neugeborenenscreening nicht erkannt, da in der Mehrzahl nur TSH-Konzentrationen über einer Konzentration von 15 mU/l rückgemeldet werden.
Bei im Kindes- und Jugendalter erworbener zentraler Hypothyreose sollte eine zerebrale MRT zum Ausschluss einer zerebralen Raumforderung durchgeführt werden. Molekulargenetisch finden sich bei kombinierten HVL-Ausfällen Mutationen in Genabschnitten, die für die Hypophysen-/Hypothalamusentwicklung relevant sind (u. a. PIT1, PROP1, LHX3, u. a.) oder bei isolierter zentraler Hypothyreose Mutationen u. a. im IGSF-1 oder β-TSH-Gen.
TSH-Überproduktion
Eine autonome TSH-Überproduktion durch ein Adenom ist im Kindesalter eine Rarität.
Adrenokortikotrophes Hormon (ACTH)
ACTH-Mangel
Physiologie
Hypophysäres ACTH wird unter dem Einfluss von hypothalamischem CRH im HVL gebildet; CRH ist ein wichtiger Regulator der Stressantwort des menschlichen Organismus. ACTH entsteht aus der Proteolyse von Propiomelanocortin (POMC); es reguliert die adrenale Steroidogenese. Ein ACTH-Mangel kann isoliert (z. B. durch T-PIT-Mutation) oder in Kombination mit anderen Hormondefizienzen auftreten (z. B. i. R. von PROP1-Mutationen). Mutationen in POMC-Gen führen zum klinischen Bild eines ACTH-Mangels und einer durch einen MSH-Mangel induzierten Adipositas.
Klinisches Bild und Diagnose
Klinisch zeigen die betroffenen Patienten das Bild eines sekundären Hypokortisolismus mit zunehmender Abgeschlagenheit und Leistungsschwäche; zusätzlich können Hypoglykämien auftreten. Die mineralokortikoide Funktion der Nebenniere ist dabei nicht beeinträchtigt. Die Diagnosestellung erfolgt durch einen CRH-Test (in dem eine unzureichende ACTH- und Kortisolstimulation beobachtet wird) oder durch einen Metopirontest, bei dem durch Hemmung der adrenalen Kortisolsynthese via negative Rückkopplung beim Gesunden, aber nicht ACTH-Defizienten, eine deutlich ACTH-Stimulation erreicht wird.
Therapie
Die Therapie besteht aus Substitution der Glukokortikoiddefizienz in Form von Hydrokortison.
ACTH-Exzess (M. Cushing))
Klinisches Bild und Diagnose
Ein M. Cushing durch eine vermehrte hypophysäre ACTH-Sekretion ist eine im Kindesalter seltene Erkrankung. Im Gegensatz zu Erwachsenen gibt es häufiger oligosymptomatische Manifestationen des klassischen klinischen Bilds. Dieses besteht aus einer Wachstumsstörung und stammbetonten Adipositas, einem sog. Vollmondgesicht mit einem „Büffelnacken“, einer Muskelschwäche sowie dermatologischen Besonderheiten wie dem Vorliegen von Striae rubrae, einer vermehrten Körperbehaarung und einer oft dünnen und empfindlichen Haut. Hinzu können ein arterieller Hypertonus und eine pathologische Glukosetoleranz kommen. Nicht selten sind psychische Veränderungen assoziiert.
Diagnostisch sollte die Ausscheidung von Kortisol im Sammelurin, ein Kortisoltagesprofil (Mitternachtskortisol), Dexamethasonhemmtest und CRH-Test erfolgen. Radiologisch kann die zerebrale Kernspintomographie u. U. erst im Verlauf ein Mikroadenom der Hypophyse sichtbar machen. Ergänzend kommt in Einzelfällen ein Sinus-petrosus-sampling zum Einsatz.
Therapie
Therapie der Wahl ist die transphenoidale Entfernung des hypophysären Mikroadenoms. Ergänzend stehen verschiedene medikamentöse Optionen zur Hemmung der ACTH- und Kortikoidbiosynthese zur Verfügung.
Gonadotropine
Hypogonadotroper Hypogonadismus und zentrale Pubertas präcox: Abschn. 7.1.3 und Abschn. 7.1.4.
Prolaktin
Prolaktin wird im HVL unter verschiedenen stimulierenden und hemmenden hypothalamischen und peripheren Einflüssen gebildet (u. a. Stimulation durch TRH und PRH, Hemmung durch Somatostatin).
Ein Prolaktinmangel im Kindesalter ist meist Teil einer multiplen Hypophyseninsuffizienz in Kombination mit anderen Hormonausfällen (z. B. infolge Mutation in PIT-1).
Hyperprolaktinämien treten meist als Folge von Stresssituationen (z. B. postiktal) auf.
Prolaktinome sind im Kindes-und Jugendalter sehr seltene Erkrankungen, die sich beim Mädchen v. a. durch eine primäre oder sekundäre Amenorrhö, beim Jungen v. a. durch eine Gynäkomastie und Galaktorrhö manifestieren. Radiologisch finden sich Mikro- oder Makroadenome der Hypophyse. Therapeutisch kommen in erster Linie Dopaminagonisten zum Einsatz.
Antidiuretisches Hormon
Diabetes insipidus centralis
Physiologie
Das antidiuretische Hormon Arginin-Vasopressin (AVP; syn. ADH) wird in den Kerngebieten des Hypothalamus gebildet, axonal in die Neurohypophyse transportiert und dort sezerniert. Seine Freisetzung wird v. a. über die Serumosmolalität reguliert, sodass unter physiologischen Bedingungen eine Natriumkonzentration von 140 mmmol/l erhalten wird. Bei einer verminderten oder fehlenden Sekretion von AVP kommt es zu einem Wasserverlust mit der Folge einer Hyponatriämie, einem sog. Diabetes insipidus (DI).
Ätiologie und Pathogenese
Man unterscheidet die seltenen familiären Formen vom idiopathischen oder organischen D. insipidus. Die seltenen familiären Formen beinhalten Patienten mit Mutation im Neurophysin-II-Gen sowie syndromale Formen (z. B. DIDMOAD-Syndrom: Diabetes Insipidus, Diabetes Mellitus, Optikusatrophie, Deafness). Sehr viel häufiger sind die organischen Formen des DI. Diese beinhalten Fehlbildungssyndrome (z. B. im Rahmen einer septooptischen Dysplasie), Tumoren der Hypothalamus-/Hypophysenregion (Kraniopharyngeome, Germinome, u. a.), inflammatorische Störungen (Langerhanszell-Histiozytose, Infundibulohypophysitis, u. a.), iatroge Ursachen (u. a. postoperativ) oder Schädel-Hirn-Traumata.
Verbleibt die Ursache ungeklärt (idiopathischer DI) sollte eine längerdauernde Supervision mit Wiederholung der zerebralen MRT durchgeführt werden, da einige Ursachen des DI erst im Verlauf deutlich werden (z. B. Germinome).
Klinisches Bild
Patienten mit einem zentralen DI weisen eine im Verlauf z. T. sehr ausgeprägte Polydipsie und Polyurie auf (z. T >12 l/d). Oft ist der Beginn schleichend, sodass der genaue Beginn anamnestisch nicht mehr eindeutig eruiert werden kann. Ab dem Zeitpunkt, ab dem der Flüssigkeitsverlust nicht mehr ausreichend über die Polydispie gedeckt werden kann kommt es zu Dehydratation, z. T. mit Fieber und Gewichtsverlust, bei jüngeren Kindern ggf. eine Gedeihstörung.
Diagnose
Laborchemisch findet sich eine verminderte Urinosmolalität und eine Hypernatriämie. Die Diagnose erfolgt im standardisierten Durstversuch unter stationären Bedingungen. Im Durstversuch ist v. a. die Veränderung der Urinosmolalität diagnostisch relevant, die direkte ADH-Bestimmung ist meist nicht weiterführend (ggf. ergänzende Bestimmung von Copeptin).
Wichtigste Differenzialdioagnosen sind der nephrogene DI (Kap. 25) sowie die habituelle Polydipsie.
Ist die Diagnose zentraler DI gesichert muss eine (repetitive) Schnittbildgebung, meist eine zerebralen MRT, durchgeführt werden, ggf. ergänzt durch eine Bestimmung der HVL-Hormone und bei V. a. Germinom oder LCH ggf. eine Lumbalpunktion und Liquoranalytik.
Therapie
Diese besteht aus Ersatz des defizienten ADH in Form des synthetischen ADH-Analogons DDAVP, entweder in oraler, intranasaler, selten parenteraler Form; typischerweise reicht eine 2- bis 3-mal tägliche Substitution aus.
SIADH (Syndrom der inadäquaten ADH-Sekretion; syn. Schwartz-Bartter-Syndrom)
Als SIADH wird Elektrolytstörung bezeichnet, bei der es trotz Normonatriämie zu einer unangemessenen Sekretion von ADH kommt. In der Folge bildet sich eine „Wasserintoxikation“ mit Hyponatriämie aus. Die Ursachen sind vielfältig: es dominiert ein medikamenteninduziertes SIADH (verschiedene Chemotherapeutika wie Vincristin, Cyclophosphamid, verschiedene Antikonvulsiva, u. a.) neben zentralnervösen Störungen (ZNS-Tumor/-Trauma, inflammatorische Störungen wie Meningitis/Enzepahlitis). Die Symptome der Hypervolämie und Hyponatriämie beinhalten u. a. Kopfschmerzen, eine arterielle Hypertonie mit Übelkeit, Erbrechen oder Krampfanfällen. Da ein SIADH nicht selten im Rahmen komplexer Erkrankungen unter einer Polypharmakotherapie auftritt, kommen hier die Symptome der Grunderkrankung (und Therapie) hinzu. Die Differenzialdiagnose der Hyponatriämie ist schwierig. Wegweisend ist der Befund einer relativ zu hohen Urinsomolalität im Vergleich zur hypoosmolaren Serumosmolalität. Grundpfeiler der Therapie ist die Behandlung der Ursache (ggf. Ersatz des auslösenden Agens) in Kombination mit einer Flüssigkeitsrestriktion.
Schilddrüse
Hypothyreose
Definition
Unter einer Hypothyreose versteht man eine Störung der Schilddrüsenfunktion, die zu einer Minderversorgung des Organismus mit Schilddrüsenhormon führt. Eine Hypothyreose kann angeboren vorliegen oder später erworben werden.
Liegt die Funktionsstörung im Bereich der Schilddrüse selbst spricht man von einer primären Hypothyreose. Eine sekundäre Hypothyreose entsteht infolge eines hypophysären TSH-Mangels, eine tertiäre Hypothyreose infolge einer hypothalamischen TRH-Defizienz. Die angeborene Hypothyreose ist mit einer Inzidenz von 1:3000 die häufigste angeborene Störung des Endokriniums.
Ätiologie und Pathogenese
Die Differenzialdiagnose der angeborenen Hypothyreose ist in Tab. 26.8 dargestellt:
| Angeborene Hypothyreose (kein oder nur hypoplastisches SD-Gewebe in loco typico) | Angeborene Hypothyreose (normales oder vermehrtes SD-Gewebe in loco typico) |
|---|---|
| Athyreose | Pendred-Syndrom (Kombination mit Innenohrschwerhörigkeit durch Mutation im Pendrin-Gen (SLC6A4) |
| Hypoplastische Schilddrüse mit Hypothyreose | Thyreoglobulinsynthesedefekt (verdächtige Konstellation: Neugeborenes mit TSH-Erhöhung, Hypothyreose, nachweisbarer/vergrößerter Scihlddrüse und niedrigem oder nicht detektierbarem Thyreoglobulin) |
| Schilddrüsenektopie mit Hypothyreose | Jodtyrosin-Dejodase-Mangel (Mutation im DEHAL1-Gen; initial meist normale SD-Funktion [unauffälliges Neugeborenenscreening]; im Verlauf nach „Jodverarmung“ Entwicklung einer Hypothyreose |
Primäre angeborene Hypothyreose (CH)
Ätiologie
Die Differenzialdiagnose einer konnatalen primären Hypothyreose ist in Tab. 26.8 dargestellt; dabei dominieren anatomische Anomalien wie Athyreose und hypoplastische Schilddrüsen. Die Pathogenese der Hypothyreose ist nur partiell verstanden (u. a. Mutationen in verschiedenen transkriptionsfaktorkodierenden Genen wie PAX8). Sehr viel seltener finden sich Störungen der Schilddrüsenhormonbiosynthese (rechte Spalte Tab. 26.8).
Eine wichtige, aber seltene Form der transienten erworbenen Hypothyreose ist die durch transplazentaren Übergang von blockierenden TSH-Rezeptor-AK-vermittelte Hypothyreose bei Kindern von Müttern mit einer autoimmunologischen Schilddrüsenfunktionsstörung.
Zusätzlich kann eine neonatale Exposition gegenüber Jod (Kontrastmittel, Desinfektiva) über den Wolff-Chaikoff-Effekt die Schilddrüsenhormonbiosynthese blockieren und u. U. zu einer Monate anhaltenden, transienten Hypothyreose führen.
Klinisches Bild
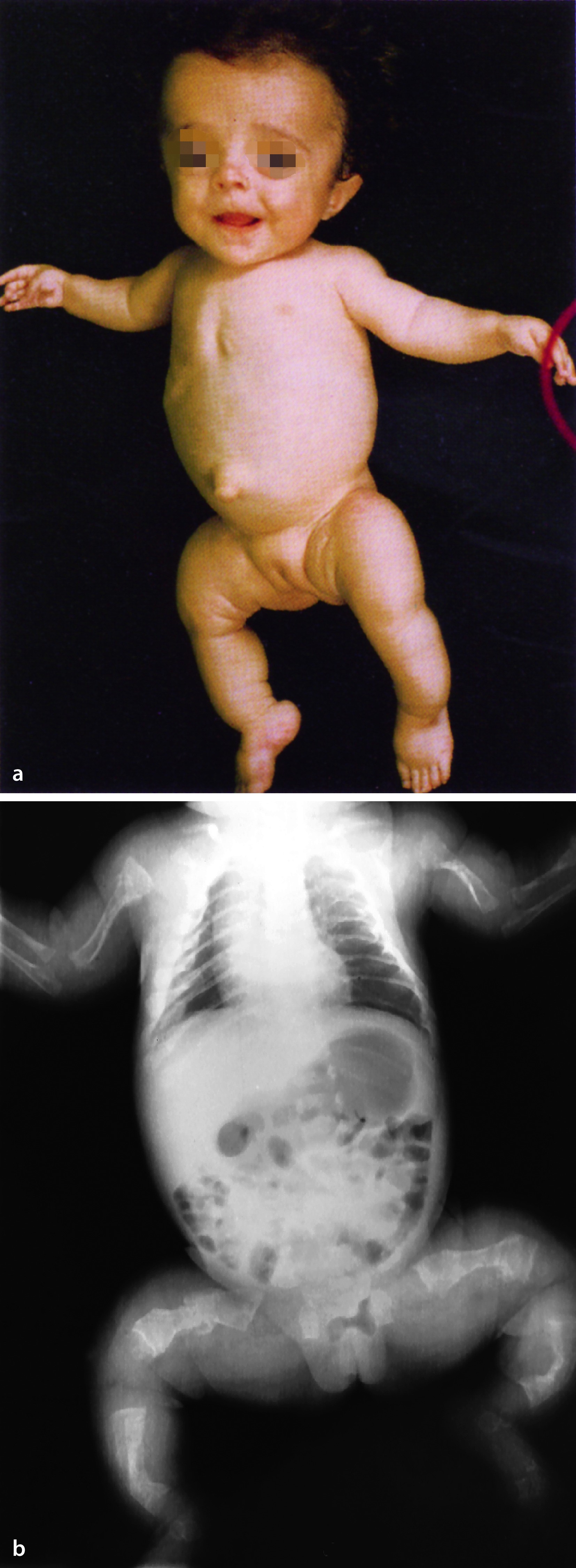
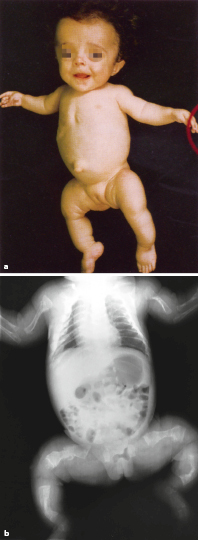
Zu den klinischen Symptomen der angeborenen Hypothyreose gehören (Abb. 26.10):
Ikterus prolongatus,
Hypothermie und Bradykardie,
Makroglossie,
Nabelhernie,
auffällige Hauttextur (ödematös, trocken, pastös),
Adynamie, Muskelhypotonie,
Makroglossie,
weit offene große Fontanelle, verzögerte Ossifikation anderer Knochenkerne.

Seit der Einführung des Neugeborenenscreening auf eine erhöhte TSH-Konzentration ist das Vollbild des Kretinismus selten geworden. Allerdings gibt es weiterhin Fälle von verpasstem oder falsch-negativem Neugeborenenscreening; insbesondere bei sehr unreifen Frühgeborenen mit einem Geburtsgewicht <1.500 g scheint es gehäuft zu einem verzögerten TSH-Anstieg zu kommen, sodass hier ggf. die Diagnose einer Hypothyreose durch ein erhöhtes Screening-TSH erst in einem Zweit- oder Drittscreening gestellt wird. Darüber hinaus wird in den meisten Screeningprogrammen eine zentrale Hypothyreose durch das isolierte TSH-Screening nicht erkannt (Abschn. 7.2.2).
Diagnose und Differentialdiagnose
Typischerweise basiert der Verdacht auf eine angeborene primäre Hypothyreose auf einem auffälligen Befund im Neugeborenenscreening (durchgeführt zwischen 36. und 72. Lebensstunde). Allerdings sichert ein im Screening gefundener TSH-Wert >15 mU/l noch nicht die Diagnose einer CH; hierfür muss der Nachweis gleichzeitig erniedrigter peripherer Schilddrüsenhormonkonzentrationen (T4, T3) erbracht werden. Bei einer TSH-Konzentration zwischen 15 und 50 mU/l wird zunächst eine Kontrolle des Screenings vorgenommen. Wurde im Neugeborenenscreening bereits eine TSH-Konzentration >50 mU/l gefunden, muss vor Beginn einer Substitutionstherapie noch eine Bestimmung der Serumkonzentration von TSH, Gesamt- oder freiem T4 erfolgen, um eine hypothyreote Stoffwechsellage zu bestätigen. Sind die peripheren Schilddrüsenhormone erniedrigt bei gleichzeitig erhöhter TSH-Konzentration sollte rasch eine Substitutionstherapie eingeleitet werden.
Neben der Bestimmung von Thyreoglobulin und Schilddrüsenautoantikörpern wird die Diagnostik der CH durch eine Sonographie der Schilddrüse ergänzt; hierbei finden sich in >80% anatomische Auffälligkeiten der Schilddrüse (Athyreose, hypoplastische oder ektope SD). Bei Früh- und Neugeborenen sollte ggf. auch eine Bestimmung der Jodausscheidung im Urin ergänzt werden, wenn eine peripartale Jodexposition nicht ausgeschlossen werden kann.
Therapie
Bei einer CH wird mit eine Substitutionstherapie durch Levothyroxin in einer initialen Dosierung zwischen 10–15 µg/kgKG/Tag begonnen; im weiteren Verlauf nimmt der gewichtsbezogene Substitutionsbedarf ab. Bei Behandlungsbeginn innerhalb der ersten zwei Lebenswochen kann erfreulicherweise von einer normalen kognitiven Entwicklung der betroffenen Kinder ausgegangen werden.
Nach dem zweiten Lebensjahr sollte ein standardisierter Auslassversuch durchgeführt werden. Bei nicht eindeutiger Diagnose einer CH oder V. a. eine transiente Hypothyreose (wie z. B. bei Nachweis von SD-Autoantikörpern) kann der Auslassversuch ggf. bereits früher in einem Alter von 6–12 Monaten diskutiert werden.
Hyperthyreose
Definition
Eine Hyperthyreose im Kindes- und Jugendalter ist entweder durch eine vermehrte Schilddrüsenhormonproduktion oder eine vermehrte Schilddrüsenhormonwirkung charakterisiert. Häufigste Ursache ist ein M. Basedow, andere Ursachen sind in Tab. 26.9 aufgelistet. Insgesamt besteht eine deutliche Mädchenwendigkeit.
| Altersgruppe | Ätiologie |
|---|---|
| Neugeborenes/Säuglingsalter | Transiente neonatale Hyperthyreose durch maternale, transplazentar übergegangene stimulierende TSH-Rezeptor-AK |
| Angeborene Hyperthyreose durch aktivierende Mutation im TSH-Rezeptor | |
| Syndromale Erkrankungen mit Hyperthyreose, z. B. bei aktivierender Mutation im Gsα-Protein (McCune-Albright-Syndrom) | |
| Kindes- und Jugendalter | M. Basedow |
| Initiale hyperthyreote Phase einer Autoimmunthyroiditis Hashimoto | |
| Autonomes Schilddrüsenadenom | |
| TSH-produzierendes HVL-Adenom | |
| Hyperthyreosis factitia (akzidentelle, iatrogene oder bewusste SD-Hormoneinnahme) |
Ätiologie und Pathogenese
Häufigste Ursache einer Hyperthyreose im Kindesalter ist der M. Basedow. Dieser wird durch die stimulierende Wirkung von TSH-Rezeptor-spezifischen Immunglobulinen (TSI oder TRAK) erklärt. Allerdings ist der alleinige biochemische Nachweis von TRAKs nicht immer mit dem klinischen Bild eines M. Basedow assoziiert, da zwischen blockierenden und aktivierenden TRAKs unterschieden werden muss. Ggf. können diese koexistent vorkommen, sodass ein Überwiegen stimulierender gegenüber inhibierenden TRAKs zum klinischen Bild einer Hyperthyreose führt. Das Auftreten eines M. Basedow assoziiert mit bestimmten HLA-Haplotypen (vermehrt HLA DR-3, -A1 und -B8). Zusätzlich wurde ein spezifischer Polymorphismus des CTL4A-Gens gehäuft bei Patienten mit einem M. Basedow gefunden.
Neben dem immunogen vermittelten M. Basedow kann eine hyperthyreote Stoffwechsellage auch in der Frühphase einer Autoimmunthyreoditis vorkommen; u. U. erlaubt das serologische Muster von TSH-Rezeptor-Auto-AK, anti-TPO- und anti-Thyreoglobulinantikörpern nicht direkt eine klare Zuordnung zu entweder einem M. Basedow oder einer Hashimoto-Autoimmunthyreoiditis.
Bei Vorliegen eines mütterlichen M. Basedow können TSH-Rezeptor-stimulierende mütterliche Autoantikörper transplazentar auf den Feten übergehen und beim Neugeborenen bis zur Elimination der TRAK-AK aus dem kindlichen Organismus eine transiente Hyperthyreose verursachen.
Sehr viel seltener sind nichtimmunogene Formen der Hyperthyreose (Tab. 26.9).
Klinisches Bild
Beim Neugeborenen mit transplazentar übergegangenen mütterlichen Antikörpern hängt der Manifestationszeitpunkt u. a. davon ab, ob die Mutter eine thyreostatische Therapie erhielt, da ggf. die transplazentare Passage der Thyreostatika den Zeitpunkt der Hyperthyreoseentwicklung beim Neugeborenen verzögern kann. Nach Elimination der Thyreostatika (typischerweise nach einigen Tagen) entwickeln diese Kinder Zeichen der Hyperthyreose.
Symptome einer Hyperthyreose im Kindesalter
- Neugeborene
- Oft niedriges Geburtsgewicht, Anamnese von IUGR
- Tachykardie, Tachyarrhythmie
- Vermehrte Irritabilität (DD Drogenentzugssyndrom)
- Bei thyreotoxischer Krise sepsisähnliches Krankheitsbild (Tachykardie, Herzinsuffizienz, Organomegalie, Polyglobulie, Leukozytopenie, Thrombozytopenie)
- Im Verlauf ggf. prämature Synostose von Schädelnähten
- Ältere Kinder
- Oft protrahierter Beginn mit unspezifischen Befunden (Gewichtsverlust, Abgeschlagenheit, vermehrte Reizbarkeit, Myopathie, Ruhetremor)
- Tachykardie, ggf. arterielle Hypertonie
- Vermehrtes Schwitzen
- Endokrine Orbitopathie, insbesondere Exophthalmus
- Nykturie, sekundäre Enuresis
- Knochenalterakzeleration
Diagnose und Differenzialdiagnose
Diagnostisch wegweisend sind der Nachweis der erhöhten Schilddrüsenhormone (T3, T4) sowie der meist supprimierten TSH-Konzentration. Ein serologischer Nachweis von TSH-Rezeptor-Antikörpern (TRAK) beim Neugeborenen macht eine Neugeborenenhyperthyreose durch transplazentaren Übergang maternaler AK wahrscheinlich. Beim älteren Kind ist der Nachweis von TRAK (ggf. in Kombination mit anti-TPO oder anti-Thyreoglobulin-AK) meist beweisend für die Diagnose eines M. Basedow.
Sonographisch finden sich eine inhomogene Schilddrüsenhormonstruktur bei häufig vergrößertem Schilddrüsenvolumen; die Dopplersonographie zeigt eine deutliche Hyperperfusion.
Eine szintigraphische Diagnostik ist im Kindes- und Jugendalter selten indiziert; sie kann ggf. bei fehlendem Antikörpernachweis bei der seltenen Differenzialdiagnose eines autonomen Adenoms differenzialdiagnostisch weiterhelfen.
Therapie und Prognose
Die hyperthyreote Stoffwechsellage bei Kindern mit M. Basedow wird thyreostatisch entweder mit Carbimazol oder Thiamazol behandelt; die früher gebräuchliche Therapie mit Propylthiouracil ist heute aufgrund des häufigen Auftretens schwerwiegender hepatischer Probleme obsolet. Oft ist zusätzlich in der Initialphase eine zusätzliche Behandlung mit β-Rezeptorenblockern notwendig.
Beim Neugeborenen mit einer schwerwiegenden Hyperthyreose wird neben einer thyreostatischen Therapie ggf. eine zusätzliche Behandlung mit Jodid in Form von Lugol-Lösung notwendig, um die Schilddrüsenhormonbiosynthese zu hemmen; ergänzend können Kortikosteroide mit dem Ziel einer verminderten Dejodierung zu T3 oder eine Plasmapherese zum Einsatz kommen.
Mittlerweile liegen mehrere Studien vor, die ein geringeres Relaps-Risiko nach verlängerter thyreostatischer Therapie berichten. Kann langfristig keine Remission erreicht werden (in Abhängigkeit der vorangehenden Dauer bei ca. 50–60% der Patienten) oder kommt es unter Therapie zu relevanten Nebenwirkungen kann eine definitive Therapie in Form einer operativen Thyreoidektomie oder einer Radiojodtherapie notwendig werden. Diese sollte in Zentren durchgeführt werden, die in der Behandlung von Kindern und Jugendlichen über hinreichende Erfahrung verfügen.
Autoimmunthyreoiditis
Definition
Die Autoimmunthyreoiditis Hashimoto beschreibt eine erworbene Schilddrüsenerkrankung, die durch eine immunogene Infiltration der Schilddrüse verursacht wird. Histologisch findet sich eine lymphozytäre Infiltration der Schilddrüse. Mädchen und Frauen sind häufiger als das männliche Geschlecht betroffen.
Ätiologie und Pathogenese
Trotz ihrer Häufigkeit ist die Ätiologie der Autoimmunthyreoiditis nicht vollständig verstanden. Es besteht eine gewisse genetische Prädisposition (gehäuft bei HLA-Typ DR4 oder 5). Außerdem scheint eine exogene Jodzufuhr bei Prädisposition ihre Entstehung zu begünstigen. Sie kann isoliert auftreten oder in Kombination mit anderen immunologischen Endokrinopathien (z. B. bei Patienten mit einem Diabetes mellitus Typ 1, bei Kindern mit einem Down- oder Ullrich-Turner-Syndrom oder im Rahmen eines sog. Schmidt-Syndroms, APS2).
Klinisches Bild
Entzündung und (transient oder konstant) hypothyreote Stoffwechsellage mit konsekutiver TSH-Erhöhung (Wachstumsreiz!) führen zu einer Vergrößerung des Schilddrüsenvolumens; dieses kann zu einer Dysphagie oder einem Kloßgefühl führen. Die Schilddrüsenfunktion kann (initial) hyper-, aber auch euthyreot oder hypothyreot sein. Assoziierte Beschwerden können Abgeschlagenheit, Gewichtszunahme, vermehrter Haarverlust, zervikale Lymphadenopathie oder Konzentrationsprobleme beinhalten.
Diagnose und Differenzialdiagnose
Die Diagnose wird durch den Nachweis von Schilddrüsenautoantikörpern (anti-TPO, anti- Thyreoglobulin) sowie ein auffälliges sonographisches Muster (irreguläre, inhomogen echoarme Schilddrüse) gestellt.
Therapie
Im Falle einer hypothyreoten Stoffwechsellage oder Vorliegen einer Struma bei Hashimoto-Thyreoiditis sollte eine Therapie mit Levothyroxin durchgeführt werden. Für eine antioxidative Therapie mit Selen, die im Erwachsenenalter z. T. mit einer Reduktion von Antikörpertitern oder Verbesserung der Schilddrüsenfunktion in Verbindung gebracht wurde, liegen bislang in der Kinderheilkunde keine supportiven Daten vor.
Struma
Definition
Eine Vergrößerung des Schilddrüsenvolumens über den alters- und geschlechtsspezifischen Normalbereich hinaus wird als Struma bezeichnet.
Ätiologie und Pathogenese
Während in der Vergangenheit ein Jodmangel die häufigste Ursache für eine Struma war, ist diese durch die verbesserte Jodversorgung in Deutschland deutlich seltener geworden. Neben einem Jodmangel können Nitrate, infektiöse Erkrankungen und Autoimmunthyreoiditiden zur Entstehung einer Struma führen. Bei konnatalen Strumen sollte ein Schilddrüsenhormonbiosynthesedefekt (u. a. Pendred-Syndrom, Thyreglobulinsynthesedefekt) ausgeschlossen werden.
Klinisches Bild
Die Vergrößerung des Schilddrüsenvolumens kann ggf. als isolierte Vergrößerung des Halsumfangs auffallen oder durch lokale Verdrängung zu einer Dysphagie, Heiserkeit sowie selten zu einer Schmerzsymptomatik führen. In Abhängigkeit davon, ob eine diffuse Struma oder eine Knotenstruma vorliegt kann ggf. eine lokale Verhärtung bzw. ein Schilddrüsenknoten tastbar sein.
Eingeteilt wird die Struma anhand der WHO-Klassifikation in Stadien:
0: keine Struma,
1a: nur palpatorisch erfassbare, nicht sichtbare Schilddrüsenvergrößerung,
1b: bei maximaler Reklination sichtbare Schilddrüse,
2: bei normaler Kopfhaltung sichtbare Schilddrüse,
3: stark vergrößerte Schilddrüse.
Diagnose
Neben der klinischen Erfassung wird eine Struma insbesondere durch die Sonographie erfasst und verlaufskontrolliert. Wichtige Aspekte hierbei sind neben der Volumetrie die Schilddrüsenstruktur sowie das Vorhandensein/Abwesenheit von Schilddrüsenknoten.
Therapie
Diese erfolgt in Abhängigkeit von der Ätiopathogenese der Struma. Bei Verdacht auf Jodmangel erfolgt eine Therapie mit Jodid (bei Säuglingen in einer Dosierung von 50–80 µg/d, bei Kleinkindern 100 µg/d und bei älteren Kindern in einer Dosis von 150–200 µg/d. Zurückhaltung hinsichtlich einer Jodidsubstitution sollte bei V. a. Hashimoto-Thyreoiditis geübt werden, die im Kindesalter hier evtl. der immunogene Prozess weiter stimuliert werden kann. Ebenso sollte bei Vorliegen einer Knotenstruma eine Autonomie ausgeschlossen werden.
Schilddrüsenknoten
Definition
Unter einem Schilddrüsenknoten versteht man eine klinisch palpable oder sonographisch nachweisbare bindegewebig abgegrenzte knotige Veränderung des Schilddrüsenparenchyms. Schilddrüsenknoten kommen in der Kindheit im Vergleich zum Erwachsenenalter deutlich seltener vor. Allerdings ist in den vergangenen Jahren eine zunehmende Detektion von Schilddrüsenknoten zu verzeichnen. Dies beruht u. a. auf der deutlich verbesserten Qualität der Sonographie sowie auf der Zunahme von Routineultraschalluntersuchungen im Rahmen der Vorsorgeuntersuchungen. Abzugrenzen sind sog. „Pseudoknoten ohne Kapsel“, wie sie z. B. im Rahmen einer Autoimmunthyreoiditis vorkommen. Hier finden sich meist sonographisch echoarme Regionen innerhalb von Schilddrüsengewebe normaler Echostruktur ohne umgebende bindegewebige Kapsel.
Ätiologie und Pathogenese
Die Ätiologie von Schilddrüsenknoten ist heterogen; diese umfasst u. a. regressive Veränderungen von Jodmangelstrumen, Schilddrüsenzysten, Adenome und im Kindesalter sehr viel seltener maligne Schilddrüsentumore. Anamnestisch relevant sind hierbei insbesondere eine vorangehende Bestrahlung der Schilddrüsenregion (onkologische Vorgeschichte?) sowie die Familienanamnese (z. B. bei MEN2a, PTEN-Hamartoma-Tumor-Syndrom, u. a.).
Klinisches Bild
Häufig handelt es sich um asymptomatische Zufallsbefunde im Rahmen einer Routinesonographie. Seltener imponieren palpable knotige Veränderungen mit lokalen Symptomen wie Dysphagie, Klossgefühl oder Heiserkeit. Nichtverschieblichkeit, derber Tastbefund oder Kombination mit einer zervikalen Lymphadenopathie können Hinweise auf eine Schilddrüsenneoplasie sein.
Diagnose und Differenzialdiagnose
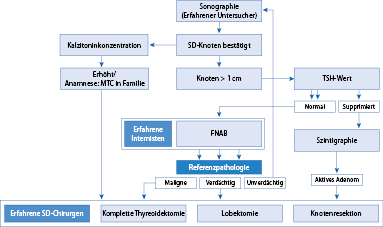
Die Diagnose eines Schilddrüsenknoten ist Domäne der Sonographie. In Abhängigkeit vom sonographischen Bild (Volumen des Knotens, homogene versus irreguläre Struktur, Perfusion, Verkalkung) kann ggf. zusätzliche Diagnostik indiziert sein (Abb. 26.11). Serologische Marker zur differenzialdiagnostischen Abgrenzung beinhalten eine Bestimmung der TSH- und Schilddrüsenhormonkonzentration (Hinweise auf autonomes Adenom?), Bestimmung der Schilddrüsenautoantikörper (Pseudoknoten bei Hashimoto-Thyreoiditis?), und eine Kalzitoninbestimmung (Ausschluss medulläres Schilddrüsenkarzinom). Hierbei ist allerdings zu berücksichtigen, dass der Nachweis von Schilddrüsenautoantikörpern ein Schilddrüsenmalignom nicht sicher ausschließt. In Abhängigkeit von Grunderkrankung und klinischem Befund wird in den häufigen Fällen kleiner Knoten eine sonographische Verlaufskontrolle vorgenommen. Bei Knoten >1 cm und/oder Wachstumstendenz und/oder auffälliger Struktur muss eine weiterführende Diagnostik erfolgen. Diese beinhaltet ggf. eine Feinnadelpunktion oder eine Schilddrüsenszintigraphie. Uneindeutige Feinnadelbiopsiebefunde oder der V. a. ein malignes Geschehen machen eine chirurgische Klärung erforderlich. Meist erfolgt hierzu eine Hemithyreoidektomie mit Schnellschnittbeurteilung. Bei Karzinomnachweis ist eine vollständige Thyreoidektomie erforderlich, ggf. ergänzt durch eine „neck dissection“ und eine Radiojodtherapie.

Therapie
Diese erfolgt in Abhängigkeit der jeweiligen Diagnose. Bei Verdacht auf Adenom oder Malignom sollte eine Vorstellung in einem erfahrenen chirurgischen Zentrum mit Expertise in der Behandlung von Kindern erfolgen. Im Falle eines operativen Vorgehens entscheiden die anamnestisch/klinische Befundkonstellation und der intraoperative Schnellschnitt über den Umfang des chirurgischen Vorgehens.
Nebenschilddrüse
Die vier Nebenschilddrüsen sind für die kalziumregulierte Freisetzung von Parathormon verantwortlich. Parathormon wird ähnlich wie Insulin aus einem Propeptid prozessiert und in den Epithelkörperchen gespeichert.
Dabei agiert Kalzium an der Nebenschilddrüse als Ligand für den „Calcium sensing Rezeptor, CaSR“. Niedrige Serumkalziumkonzentrationen führen via Aktivierung des CaSR zu einer Stimulation der Freisetzung von Parathormon. Zusätzlich stimulieren erhöhte Serumkonzentrationen von Phosphat die PTH-Freisetzung.
Zur Vermittlung seiner Wirkung bindet PTH an einen G-Protein-gekoppelten Rezeptor. Hierdurch wird u. a. den Tubuluszellen der Niere die 1,25-Hydroxilierung von Vitamin D3 stimuliert. Am Knochen führt PTH zu einer vermehrten Freisetzung von Kalzium.
Neben Parathormon wird in einer Reihe von Geweben ein PTH-ähnliches Peptid, PTH-related-Protein (PTHRP) gebildet. Diese kann zusätzlich paraneoplastisch gebildet werden und zu einer tumorinduzierten Hyperkalzämie führen.
Hypoparathyreoidismus
Definition
Unter der Diagnose Hypoparathyreoidismus fasst man Erkrankungen zusammen, die mit einer verminderten PTH-Sekretion einhergehen.
Ätiologie und Pathogenese
Die Ätiologie eines Hypoparathyreoidismus ist heterogen; man unterscheidet den primären Hypoparathyreoidismus von sekundären Formen.
Hypoparathyreoidismus im Kindesalter
-
A.Isoliert
- Familiär (autosomale und X-chromosomal; CaSR, CGM2, GNA11, PTH; SOX3, FHL1)
- Nichthereditär
-
B.Syndromal
- Mitochondropathien (u. a. MELAS- und Kearns-Sayre-Syndrom)
- LCHAD-Mangel
- DiGeorge-Syndrom (Mikrodeletionssyndrom 22q11.2; Catch22)
- APECED-Syndrom (AIRE-Mutation)
- Barakat-Syndrom (Nierendysplasie, Innenohrschwerhörigkeit; AD; GATA3-Mutation)
- Kenney-Caffey-Syndrom
- Sanjad-Sakati-Syndrom (Kleinwuchs, psychomotorische Retardierung, Dysmorphie; TBCE-Mutation)
-
C.Sekundär
- Postoperativ
- Speichererkrankungen (Hämosiderose, M. Wilson)
- Paraneoplastisch/nach Bestrahlung
- Hypomagnesämie
Klinisches Bild
Durch den Mangel an Parathormon kommt es in Abhängigkeit der nutritiven Kalziumzufuhr zu einer Hypokalzämie und Hyperphosphatämie. Dieses kann in milden Formen asymptomatisch oder mit moderaten Muskelschmerzen einhergehen; in schwereren Fällen führt die Hypokalzämie typischerweise zum klinischen Bild einer Tetanie mit hypokalzämischen Krampfanfällen. Eine chronische Hypokalzämie kann zu Veränderungen der Haut und Hautanhangsgebilde führen (trockene Haut, erhöhte Brüchigkeit von Nägeln, Nageldystrophie), Schmelzdefekten und intrazerebrale Verkalkungen (Basalganglien!). Durch die verminderte Kalziumrückresorption ist insbesondere nach Therapieeinleitung das Risiko für eine Nephrolithiasis erhöht.
Diagnose
Der laborchemische Befund einer Hypokalzämie, ggf. mit Hyperphosphatämie und niedriger Parathormonserumkonzentration ist wegweisend für die Diagnose eines Hypoparathyreoidismus. Ergänzend sollte die renale Kalziumausscheidung bestimmt werden. Bei Mutationen im CaSR bei einer autosomal dominanten Hypokalzämie (ADH) ist diese klassischerweise inadäquat hoch. Differenzialdiagnostisch sollte eine Hypomagnesämie ausgeschlossen werden.
Therapie
Ziel der Therapie des Hypoparathyreoidismus ist eine Anhebung der Serumkalziumspiegel in den unteren Normalbereich. Aufgrund von Sicherheitserwägungen sowie der Notwendigkeit der subkutanen Applikation wurde rekombinantes PTH bei Kindern bislang nur in Rahmen von klinischen Studien eingesetzt. Therapeutisch kommen Vitamin-D-Metabolite zum Einsatz; aufgrund der guten Steuerbarkeit insbesondere 1,25-(OH)2-Vitamin D3 (Kalzitriol) oder 1α-(OH)-Vitamin D3 (1α-Diol), in Kombination mit oraler Kalziumsubstitution.
Störungen des Parathormon PTH/PTH-related Peptid Signalwegs (iPPSD; Pseudohypoparathyreoidismus)
Definition
Unter einem Pseudohypoparathyreoidismus werden verschiedene Erkrankungen subsummiert, die funktionell einem Hypoparathyreoidismus mit der laborchemischen Konstellation von Hypokalzämie und Hyperphosphatämie entsprechen, bei denen aber nicht ein Mangel an Parathormon, sondern eine Störung der PTH-Wirkung zugrunde liegt. Auf Grundlage eines erweiterten klinischen, biochemischen und (epi)genetischen Verständnisses wurde vorgeschlagen diese früher als Pseudo- oder Pseudopseudohypoparathyreodismus bezeichneten Erkrankungen aktuell unter dem Begriff „Störungen des PTH/PTHrP-Signalweges“ zusammenzufassen.
Ätiologie und Pathogenese
Die Signalkette, die nach Bindung von PTH an den G-Protein-gekoppelten Rezeptor aktiviert wird ist in Abb. 26.12 dargestellt.
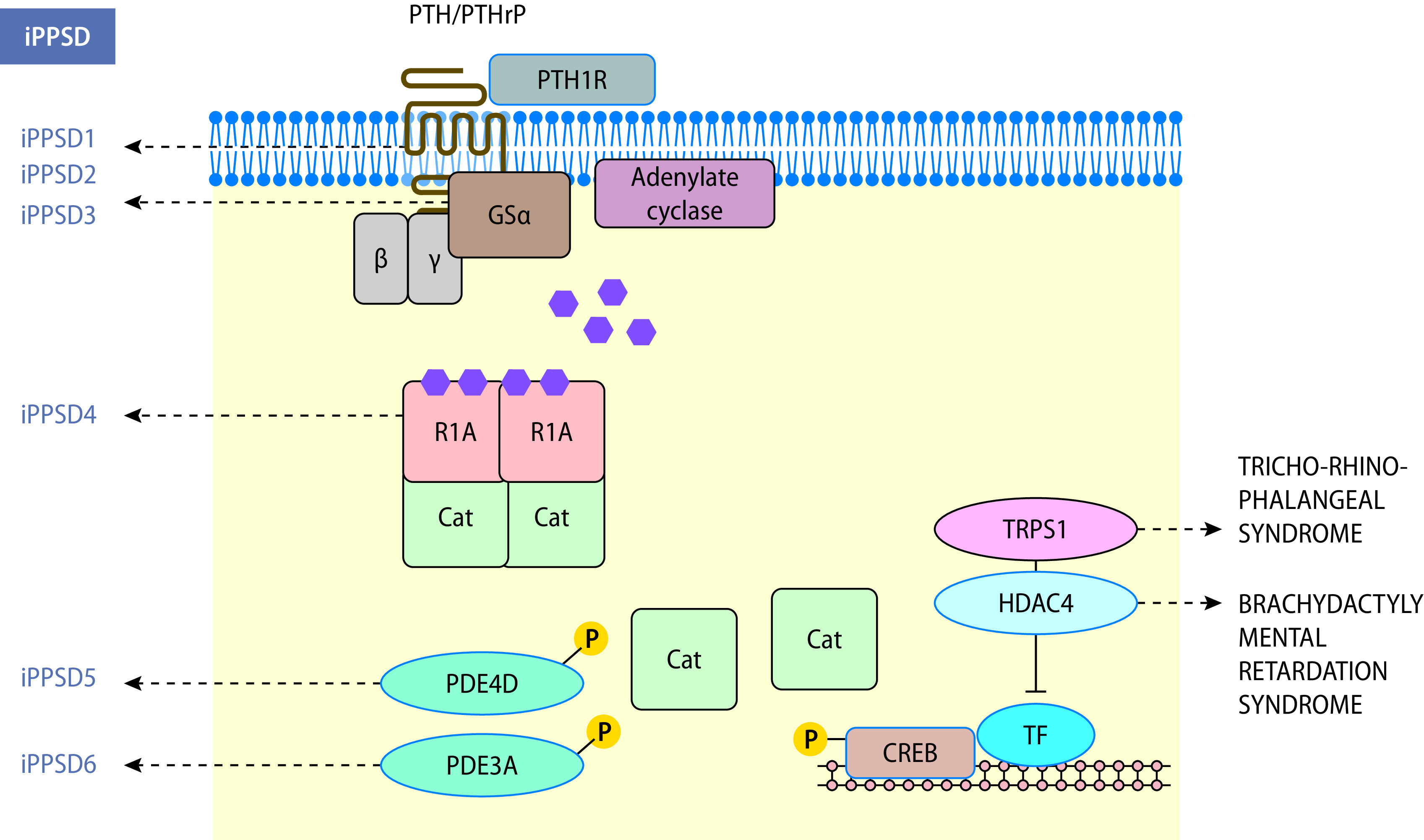
Mutationen im GNAS-Gen oder von nachgeschalteten Signalproteinen sind verantwortlich für funktionelle Störungen der PTH-Wirkung, die sich je nach zugrundeliegender Veränderung klinisch/biochemisch unterscheiden. Als molekulare Ursachen einer iPPSD sind bekannt inaktivierende Mutationen im PTH-Rezeptor, inaktivierende heterozygote Mutationen im GNAS-Lokus, Methylierungsveränderungen des GNAS-Lokus, verursacht u. a. durch Deletionen/Duplikationen oder eine paternale uniparentale Disomie von Chromosom 20q sowie heterozygote Mutationen im PRKAR1A-, PDE4D- oder PDE3A-Gen.
Mutationen im GNAS-Gen werden in familiären Fällen autosomal dominant vererbt. Allerdings liegen in der Mehrzahl der Fälle Neumutationen zugrunde. Alternatives splicing des GNAS-Lokus ist verantwortlich für zahlreiche unterschiedliche Transkripte. In der Mehrzahl der Gewebe unterliegt die Expression dieser Transkripte einem genomischen Imprinting, mit monoallelischer Expression des maternalen Allels.
Klinisches Bild
Das klinische Bild von Erkrankungen mit gestörter PTH-Wirkung umfasst die nach neuer Klassifikation Major-Kriterien sowie Minor-Kriterien (Tab. 26.10).
| Kriterien | Befund | Diagnostik | Differenzialdiagnosen (Auswahl) |
|---|---|---|---|
| Major-Kriterien | 1) PTH-Resistenz | Ca (ion.+Gesamt), PO4, PTH | - Normokalzämischer Hyperparathyreoidismus |
| 25-OH-Vitamin D, alkalische Phosphatase | - Niereninsuffizienz | ||
| Magnesium | - Ursachen des sekundären Hyperparathyreoidismus | ||
| Kreatinin | |||
| Urin-Ca, Urin-PO4 | |||
| Ggf. PTH-Infusionstest | |||
| 2) Heterotope Ossifikation |
- Fibrodysplasia ossificans progressiva - Posttraumatische Ossifikation |
||
| 3) Brachydaktylie | Klinische Untersuchung, Röntgen von Hand/Fuß |
- Ullrich-Turner-Syndrom - Tricho-rhino-pharyngeale Syndrome (TRPS I–III) - DeLange-Syndrom |
|
| Minor-Kriterien | TSH-Resistenz | TSH, T3/4, SD-Antikörper, Sonographie | TSH-R-Mutation |
| Andere Hormonresistenzen | IGF-1, Calcitonin, Gonadotropine | ||
| Psychomotorische Beeinträchtigung | MRT des ZNS, Entwicklungstestung | ||
| IUGR | Schwangerschafts- und Geburtsanamnese | ||
| Übergewicht | Perzentilen | ||
| Faziale Auffälligkeiten | Untersuchungsbefund |
Diagnose
Bei Vorliegen eines Major-Kriteriums (1) oder (2) oder Vorliegen von Major-Kriterium (3) und mindestens zwei Minorkriterien kann die Diagnose einer iPPSD gestellt werden (Tab. 26.10). Beachtenswert ist, dass sich einige Befunde wie Brachydaktylie oder Übergewicht erst im Verlauf entwickeln.
Therapie
In Analogie zum Hypoparathyreoidismus wird die Hypokalzämie mit Vitamin-D-Derivaten behandelt. Allerdings wird bei den Störungen des Parathormonsignalwegs eine Kalziumserumkonzentration im mittleren bis oberen Normalbereich angestrebt um die erhöhte PTH-Konzentration zu normalisieren. Hintergrund hierfür ist die Sorge, dass langfristig erhöhte PTH-Konzentrationen am Knochen zu einer Knochenresorption führen können (beachte: durch maternale oder paternale Expression der GNAS-Allele infolge genomischem imprinting ist die Expression und PTH-Resistenz gewebeabhängig verschieden!). Darüber hinaus ist die Hyperkalziurie im Gegensatz zum Hypoparathyreoidismus von geringerer Bedeutung, da die PTH-Funktion im distalen Tubulus erhalten ist. Im proximalen Tubulus hingegen besteht eine PTH-Resistenz. Da hierdurch keine effiziente 1,25-Hydroxilierung aus 25-OH-Vitamin D möglich ist, sollte möglichst Calcitriol zur Therapie verwendet werden.
Ergänzend sollten zusätzliche Endokrinopathien in Abhängigkeit der zugrundeliegenden Erkrankung behandelt werden, wie Substitution einer Hypothyreose bei TSH-Resistenz.
Primärer und sekundärer Hyperparathyreoidismus
Definition
Unter einem Hyperparathyreoidismus fasst man Zustände mit chronischer PTH-Mehrsekretion zusammen. Diese können entweder primär im Rahmen einer autonomen Mehrsekretion oder sekundär als Folge einer Hypokalzämie auftreten.
Ätiologie und Pathogenese
Der primäre Hyperparathyreoidismus ist im Kindesalter extrem selten; er kommt eher in der Adoleszenz als im jüngeren Kindesalter vor. Die Ursache ist weitestgehend unbekannt, der PHP kann im Rahmen einer Nebenschildrüsenhyperplasie, eines Adenoms oder Karzinoms auftreten. Familiäre Formen des Hyperparathyreoidismus kommen vor im Rahmen einer multiplen endokrinen Neoplasie Typ 1 oder 2a oder im Rahmen einer familiären hypokalziurischen Hyperkalziurie (FHH), inaktivierende, Letztere wird verursacht durch Mutation im Calcium-sensing-Rezeptor-Gen. Bei letzterer ist die PTH-Konzentration oft nicht in dem Ausmaß erhöht wie bei einem primären Hyperparathyreodismus, aber inadäquat hoch bezogen auf die Serumkalziumkonzentration. Biallelische CaSR-Mutationen können zum schweren neonatalen Hyperparathyreoidismus führen, einem Krankheitsbild, das durch Hyperkalzämie, Ateminsuffizienz und Knochendemineralisierung gekennzeichnet ist.
Der sekundäre Hyperparathyreoidismus entsteht meist auf dem Boden einer Hypokalzämie, z. B. im Rahmen von Vitamin-D-Mangel-Rachitiden oder im Rahmen einer Niereninsuffizienz mit verminderter Synthese von 1,25-OH-Vitamin-D3 (Kap. 25).
Klinisches Bild
Das klinische Bild ist durch die Hyperkalzämie geprägt. Es beinhaltet Inappetenz, ggf. Übelkeit und Erbrechen, Obstipation, psychische Veränderungen und bedingt durch die Hyperkalziurie eine Polydipsie und -urie mit dem konsekutiven Bild einer Nephrolithiasis. Am Knochen führt die langfristig erhöhte PTH-Exposition zu einer Demineralisierung mit reduzierter Knochendichte und Frakturen. Im Gegensatz zum Erwachsenenalter scheint der PHP im Kindesalter seltener asymptomatisch zu verlaufen.
Diagnose
Diagnostisch wegweisend ist die Konstellation von Hyperkalzämie bei gleichzeitig erhöhter Parathormonkonzentration. Sonographisch können Sonographie und Schnittbildgebung (MRT, CT) diagnostisch hilfreich sein. Häufig sind zusätzliche Untersuchungen (Szintigraphie, PET, andere) notwendig.
Therapie
Die Behandlung des primären Hyperparathyreoidimus basiert auf der chirurgischen Entfernung. Im Falle des Vorliegens eines isolierten Adenoms wird dieses extirpiert, bei einer Hyperplasie aller Nebenschilddrüsen erfolgt eine vollständige Parathyreoidektomie mit Autotransplantation eines Nebenschildrüsenanteils (üblicherweise im Bereich des Unterarms). Ergänzend sollte bei Kindern eine multiple endokrine Neoplasie (MEN) ausgeschlossen werden und bei positivem Mutationsnachweis eine Familienuntersuchung ergänzt werden.
Nebenniere
Anatomische Grundlagen und Physiologie
Die kindliche Nebenniere entwickelt sich aus mesodermalen (Nebennierenrinde) und ektodermalen Anteilen (Nebennierenmark). In der Fetalperiode verschiebt sich das Größenverhältnis der zunächst zweischichtigen Nebenniere und Niere massiv, von einer zunächst deutlich das Nierenvolumen überschreitenden fetalen Nebenniere hin zu einem verbleibenden kleinen kappenförmigen Nierenaufsatz beim Erwachsenen. Dieser Prozess beginnt bereits kurz vor der Geburt; ab dem 8. Schwangerschaftsmonat kommt es zu einer Involution der sog. fetalen Innenzone („Fetokortex“) und zu einer weiteren Differenzierung der dreischichtigen Außenzone zur dreischichtigen Nebennierenrinde mit Regionen die spezifische Enzymsysteme exprimieren:
Unter der Nebennierenkapsel liegt die Zona glomerulosa.
Als mittlere Schicht findet sich die Zona fasciculata.
Zum Nebennierenmark hin findet sich die Zona reticularis.
Die adrenale Steroidhormonsynthese verwendet als Baustein aller Steroide Cholesterin, sie ist dargestellt in Abb. 26.13. Unter dem Einfluss verschiedener Regulatoren wie ACTH und anderen werden unter Nutzung verschiedener Enzymsysteme Hormone mit grob schematisiert 3 Funktionsgruppen synthetisiert:
Die Mineralokortikoidsynthese findet vornehmlich in der Zona glomerulosa statt. Hierfür sind 3 Schlüsselenzyme notwendig: die 3β-Hydroxysteroid-Dehydrogenase Typ 2 (HSD3B2), die 21-Hydroxylase (CYP21A2), die Progesteron zu 11- Deoxykortikosteron umwandelt sowie die Aldosterosynthase (CYP11B2), die die letzten drei Schritte der Aldosteronsynthese katalysiert (11β-Hydroxylierung, 18-Hydroxylierung und 18-Methyloxidation). Physiologisch sind die Mineralokortikokoide Aldosteron und 11-Deoxykortikosteron für die tubuläre Natriumrückresorption und Kaliumexkretion von zentraler Bedeutung; ein Synthesedefizit kann rasch zu bedrohlichen Entgleisungen des Salz-Wasser-Haushalts führen.
Die Glukokortikoidsynthese findet in der Zona fasciculata unter dem Einfluss von ACTH statt. CYP17A1 katalysiert dort die 17α-Hydroxylierung von Pregnenolon und Progesteron. Zusammen mit den Enzymen der 3β-Hydroxysteroid-Dehydrogenase Typ 2 (HSD3B2), der 21-Hydroxylase (CYP21A2) und der 11-β-Hydroxylase wird dort aus Vorstufen Kortisol synthetisiert. Wichtige Vertreter der Glukokortikoide sind Kortisol, sein 11-Dehyrogenisierungsprodukt Kortison, Kortikosteron sowie 11-Desoxykortisol. Diese üben sowohl eine glukokortikoide wie auch mineralokortikoide Wirkung aus, wobei Kortison im Vergleich mit Kortisol eine ungleich geringere Wirksamkeit zeigt. Glukokortikoide stimulieren u. a. die Glukoneogenese aus Proteinen und sind daher in den meisten Geweben Proteinkatabol.
Die Sexualsteroidsynthese (Androgene, Östrogene) findet in der Zona reticularis statt. Die 17-Hydroxylase- und 17-20- Lyase-Kapazität der CYP17A1 katalysieren aus Pregnenolon und Progesteron die Synthese von DHEA und seiner sulfatierten Variante DHEAS. DHEAS ist das Schlüsselhormon bei der in der Vorpubertät auftretenden Adrenarche. Die 17β-Hydroxysteroid-Dehydrogenase Typ 5 (17βHSD5) ist notwendig für die Synthese der üblicherweise geringen adrenalen Testosteronsynthese. Aus Androstendion und Testosteron entstehen unter dem Einfluss der Aromatase die Östrogene Östron und Östradiol.

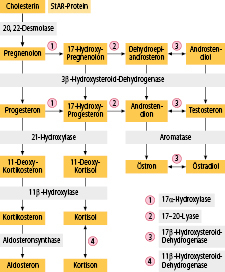
Zusammen mit den gonadalen Hormonen sind die Sexualsteroide für die Entwicklung der sekundären Geschlechtsmerkmale relevant.
Die Regulation der Glukokortikoidsynthese erfolgt überwiegend im Rahmen eines Regelkreises aus hypothalamisch gebildetem CRH, hypophysärem ACTH und unterliegt einer negativen Rückkopplung über zirkulierendes Kortisol. Demgegenüber wird die Mineralokortiokoidsynthese der Zona glomerulosa weitestgehend über das Renin-Angiotensin-System reguliert. Elektrolytverschiebungen und Veränderungen des Plasmavolumens modulieren im juxtaglomerulären Apparat der Niere die Reninsekretion. Hierüber wird über das Renin-Angiotensin-System u. a. die Mineralokortikoidsynthese beeinflusst.
Nebennierenunterfunktion
Ätiologie und Pathogenese
Störungen der Nebennierenfunktion können angeboren oder erworben auftreten. In Abhängigkeit der Lokalisation der Störung werden diese in primäre (adrenale), sekundäre (hypophysäre) oder tertiäre (hypothalamische) Funktionsstörungen eingeteilt. Je nach morphologischem Bild können diese als Nebenniereninsuffizienz mit Nebennieren-Hypoplasie oder mit Nebennierenhyperplasie auftreten.
Klassifikation der Störungen der Nebennierenfunktion
- Primäre NNR-Insuffizienz:
- Akute NNR-Insuffizienz (Addison-Krise)
- Chronische NNR-Insuffizienz (M. Addison)
- Kongenitale Nebennieren-Hypoplasie (u. a. Dax1-, SF1-, P450scc-Defekt, Triple-A-Syndrom)
- Familiäre Glukokortikoidinsuffizienz
- Familiäre Glukokortikoidresistenz
- Mineralokortikoidmangel
- Sekundäre NNR-Insuffizienz:
- Isolierter ACTH-Mangel (angeboren bei Tpit-Mutation, erworben)
- Panhypopituitarismus
- Tertiäre NNR-Insuffizienz:
- CRH-Mangel (hypothalamische Fehlbildung/Raumforderung)
- Iatrogen z. B. Glukokortikoidtherapie
Akute Nebenniereninsuffizienz (Addison-Krise)
Ätiologie
Die Ätiologie der akuten Nebenniereninsuffienz ist heterogen. Neonatal können beidseitige Nebennierenrindenblutungen im Rahmen von Geburtskomplikationen oder eine fulminante NNR-Insuffizienz im Rahmen eines Waterhouse-Friedrichsen-Syndroms ursächlich relevant sein (Kap. 23). Bei diesem Krankheitsbild kommt es durch bilaterale NNR-Blutungen im Rahmen eines septischen Krankheitsbilds zu einer Verbrauchskoagulopathie und akuter NNR-Insuffizienz. Weiterhin können andere Infektionen (Mykobakterien, HIV, andere), iatrogene Ursachen wie eine Steroidsynthese-inhibierende Medikation (u.a. Etomidate, Antimykotika) oder häufiger ein zu rasches Absetzen einer längerdauernden Glukokortikoidtherapie eine Addisonkrise verursachen (auch bei protrahierter Nutzung potenter inhalativer Steroide!).
Klinisches Bild
Im Vordergrund einer Addisonkrise steht der Salzverlust mit dem laborchemischen Befund einer Hyponatriämie, Hypochlorämie, Hyperkaliämie, Hypoglykämie sowie meist metabolischen Azidose. Häufig komplizieren Erbrechen und Durchfälle eine vorbestehende Apathie und Adynamie, ggf. Krampfanfälle i.R. von Hypoglykämie und Hyponaträmie. Es kann sich rasch ein lebensbedrohlicher Schockzustand mit Blutdruckabfall und Tachykardie entwickeln.
Diagnostik
Neben dem klinischen Bild sind die o.g. Laborveränderungen wegweisend; ergänzend sollte vor dem raschen Einleiten von Therapiemaßnahmen eine Blutprobe zur späteren Bestimmung endokriner Parameter abgenommen.
Therapie
Eine akute Addisonkrise bedarf einer unmittelbaren Substitution von Gluko- und Mineralokortikoiden, ergänzt durch eine parenterale Flüssigkeitszufuhr. Orientierend kann in den ersten 6 Lebensmonaten initial ein Hydrokortisonbolus von 25 mg, bei älteren Kindern von 50–100 mg gegeben werden, gefolgt von einer Hydrokortisondauerinfusion in einer Konzentration von 100–150 mg/m2 KOF/d. Eine isolierte Glukokortikoidapplikation wie z. B. von Prednison reicht nicht aus um die fehlende Mineralokortikoiddefizienz auszugleichen.
Der Flüssigkeitsersatz erfolgt in Abhängigkeit von Kreislaufsituation, Elektrolytkonzentration und Blutgasen meist in Form von isotonen Lösungen (ggf. 5–10%ige kohlehydrathaltige Vollelektrolytlösung).
Bei ausgeprägten Hyperkaliämien kann der Einsatz von Glukose-Insulin-Infusion, die Gabe von β-Sympathomimetika, Kalziumchlorid oder Natriumbikarbonat, von Ionenaustauschern bis hin zur Hämofiltration notwendig werden.
Chronische Nebenniereninsuffizienz (Morbus Addison)
Ätiologie
Unter dem Begriff eines M. Addison werden chronische Störungen der Nebennierenfunktion zusammengefasst.
Ursächliche Störungen bei M. Addison
Autoimmunadrenalitis: isoliert oder kombiniert mit anderen AI-Erkrankungen, z. B. bei Patienten mit Autoimmunpolyendokrinopathie Typ 1 (APECED-Syndrom; Mutation in AIRE-Gen) oder bei Autoimmunpolyendokrinopathie Typ 2 (Schmidt-Syndrom)
Adrenoleukodystrophie/Adrenomyeloneuropathie
Zellweger-Syndrom
M. Wolman
Infektionen (Tbc, Mykobakterien)
Medikamente (z. B. Adrenolytika wie o,p’-DDD)
Klinisches Bild
Meist erfolgt durch den protrahiert schleichenden Verlauf eine verspätete Diagnosestellung. Anamnestisch werden oft längerdauernder Leistungsabfall, Adynamie, Müdigkeit, Abfall schulischer Leistungen und eine Anorexie mit Gewichtsverlust berichtet. Der chronische Hypokortisolismus führt über eine negative Rückkopplung zu einer vermehrten CRH-vermittelten Induktion der POMC-Gen-Expression; hieraus resultiert eine Zunahme der ACTH/MSH-Synthese. Diese vermehrte MSH-Bildung ist die Ursache für eine bronzeartige Hautfarbe. Diese kann insbesondere an den Hautlinien der Handinnenflächen auftreten, im Kindesalter aber auch isoliert zu einer vermehrten Pigmentierung der Schleimhäute führen (u. a. von Mund und Lippen). Bei verspäteter Diagnose fällt oft ein Abknicken von Längen- und Gewichtsentwicklung sowie ggf. eine verzögerte Pubertätsentwicklung auf.
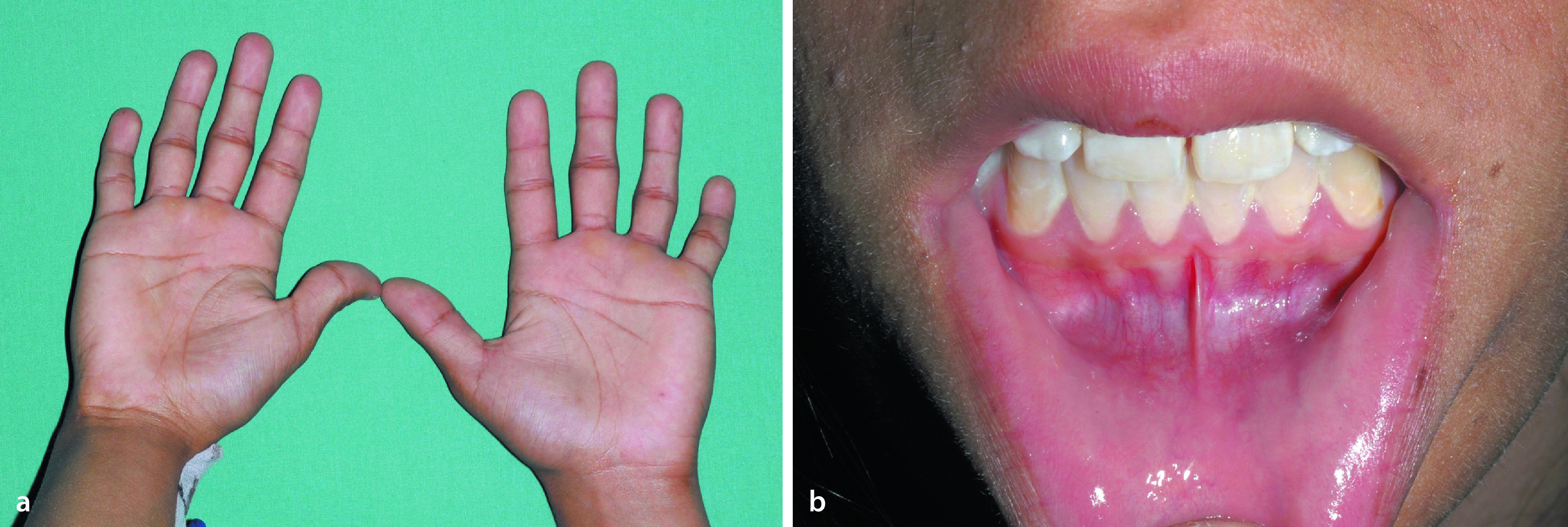
Diagnose
Neben klinischem Bild und Elektrolytveränderungen (Hyponatriämie, Hyperkaliämie, ggf. Hypoglykämie) kann eine Eosinophilie bestehen. Die Kortisolserumkonzentration und Ausscheidung kann erniedrigt oder niedrig-normal sein, bei allerdings deutlich erhöhter Plasma-ACTH-Konzentration. Im ACTH-Test findet sich eine nicht ausreichende Stimulation der Kortisolsekretion. Im Rahmen eines Salzverlustes fällt ergänzend eine erniedrigte Aldosteronkonzentration bei gleichzeitig erhöhter Plasma-Renin-Aktivität auf.
Therapie
Die Therapie des M. Addison besteht in einer lebenslangen Substitution von Gluko- und Mineralokortikoiden. Beim Kind und Jugendlichen ist Hydrokortison Mittel der Wahl; üblicherweise in einer Dosierung von 7–10 mg/m2 KOF/d. Dies wird ergänzt durch Mineralokortikoide (z. B. Fludrokortison) in einer Dosierung von 0,05–0,2 mg/d.
Beim Erwachsenen kommen mittlerweile retardierte Hydrokortisonderivate mit längerer Halbwertzeit zum Einsatz (z. B. Plenadren®). Im Kindesalter bestehen damit derzeit aber noch ungenügende Erfahrungen.
Von herausragender Bedeutung sind Patienten- und Elternschulung hinsichtlich einer Dosisanpassung bei interkurrenten Erkrankungen. In solchen Situationen muss die übliche Substitutionsdosis auf das 3- bis 5-fache erhöht oder ggf. eine Hydrokortisondauerzufuhr veranlasst werden.
Alle Patienten sollen einen entsprechenden Notfallausweis mit sich führen. Bei adäquater Substitutionstherapie ist die Prognose gut, auch wenn weiterhin durch krisenhafte Entgleisungen eine erhöhte Morbidität besteht.
Kongenitale Nebennierenhypoplasie
Durch den zunehmenden Einsatz molekulargenetischer Methoden konnten in den letzten Jahren zahlreiche seltene Formen der Nebenniereninsuffizienz mit Nebennierenhypoplasie aufgeklärt werden. Einige Formen sind in Tab. 26.11 dargestellt:
| Genetischer Defekt/Erkrankung | Erbgang | Klinisches Bild |
|---|---|---|
| DAX1-Defekt | X-chromosomal |
Primäre NNR-Insuffizienz (Säuglingsalter) Oft Maldeszensus testis Hypogonadismus mit Pubertas tarda |
| SF1-Defekt | AD |
Primäre NNR-Insuffizienz Untervirilisierung (bei Jungen; z. B. penoskrotale Hypospadie) |
| P450 scc-Defekt | AD/AR |
Frühgeburtlichkeit Primäre NNR-Insuffizienz DSD 46,XY |
| Familiäre Glukokortikoidinsuffizienz (= ACTH-Resistenz durch Mutation in MC2R) | AR |
Primäre NNR-Insuffizienz Ernährungsprobleme, Hyperpigmentierung |
| Triple-A-Syndrom (Mutation in AAAS-Gen) | AR |
Primäre NNR-Insuffizienz Alakrimie Achalasie |
Die Therapie erfolgt in Analogie zur Therapie des M. Addison, ggf. ergänzt durch erkrankungsspezifische Besonderheiten, insbesondere bei Vorliegen von Störungen der Geschlechtsdifferenzierung.
Adrenogenitales Syndrom
Definition
Die verschiedenen Formen der Nebennierenrindeninsuffizienz mit NNR-Hyperplasie werden im deutschen Sprachraum üblicherweise als adrenogenitales Syndrom bezeichnet. Im englischen Sprachraum sind diese unter dem Begriff „congenital adrenal hyperplasia“ subsummiert. In Abhängigkeit des zugrundeliegenden Enzymdefekts liegt eine verminderte Kortisolbiosynthese in Kombination mit meist vermehrter oder bei manchen Formen verminderter Androgensynthese vor. Durch den Hypokortisolismus kommt es im Rahmen der negativen Rückkopplung zu einer Stimulation der ACTH-Sekretion. Diese führt zu einer Stimulation der adrenalen Steroidsynthese oberhalb des jeweils zugrundeliegenden Enzymdefekts. Das hieraus resultierende klinische Bild ist Abb. 26.16 dargestellt, die biochemischen Veränderungen (Anstieg Metabolit vor Enzymdefekt, Mangel an Metabolit nach dem Defekt) sind aus Abb. 26.13 ableitbar.

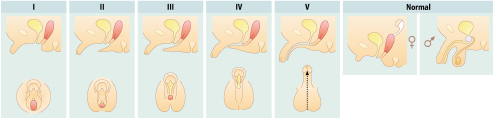
Adrenogenitales Syndrom durch 21-Hydroxylase-Mangel
Ätiologie, Pathogenese und Epidemiologie
Die weitaus häufigste Form eines adrenogenitalen Syndroms wird durch einen Defekt der 21-Hydroxylase (P450C21) verursacht. Die Inzidenz des 21-Hydroxylasemangels liegt bei 1:12.000, dies entspricht einer Heterozygotenfrequenz von 1:55. In Abhängigkeit der zugrundliegenden Mutation kann eine Restaktivität des Enzyms vorliegen, sodass klinisch verschiedene Formen unterschieden werden können (Abb. 26.15):
21-Hydroxylase-Mangel mit Salzverlust (SW; fehlende Kortisol- und Aldosteronbiosynthese, Hyperandrogenämie)
21-Hydroxylase-Mangel ohne Salzverlust, einfach virilisierend (SV; fehlende Kortisol-, aber ausreichende Aldosteronbiosynthese, Hyperandrogenämie)
nichtklassischer 21-Hydroxylase-Mangel („late onset“ = LO; milde eingeschränkte Kortisol- bei erhaltener Aldosteronbiosynthese, Hyperandrogenämie)

Das 21-Hydroxylase-Gen liegt auf Chromosom 6. Neben dem aktiven Gen (CYP21A2) liegt ein inaktives Pseudogen (CYP21A1) vor. Die enge Nachbarschaft und hohe Homologie von Gen und Pseudogen ist von Bedeutung für die Entstehung von Gendefekten durch crossing-over während der Meiose.
Klinisches Bild
Typischerweise fällt bei Mädchen nach Geburt eine unterschiedlich stark ausgeprägte Virilisierung des äußeren Genitale auf; diese wird nach Prader in verschiedene Schweregrade eingeteilt (Abb. 26.16). Dieses kann von einer leichten Klitorishypertrophie (Prader I) bis hin zu einem unauffälligen männlichen äußeren Genitale (Prader V) bei allerdings leerem Skrotum reichen. Das innere Genitale bei Mädchen ist immer unauffällig weiblich. Bei Jungen kann die Diagnose postpartal schwierig sein. Ggf. fällt bei betroffenen Knaben eine vermehrte Pigmentierung des Skrotums oder ein auffällig großes äußeres Genitale auf.
Seit Einführung des Neugeborenenscreenings auf erhöhtes 17-OH-Progesteron wird die Mehrzahl der Patienten im Rahmen des Screenings diagnostiziert. Zuvor kam es häufig in der 2. Lebenswoche zu vital bedrohlichen Salzverlustkrisen mit Gedeihstörung, Erbrechen, Exsikkose und dem laborchemischen Befund einer Hyponaträmie, Hypochlorämie, Hyperkaliämie und metabolischer Azidose. Ohne Therapie verläuft ein adrenogenitales Syndrom mit Salzverlust meist tödlich.
Patienten, die an einer milderen „Simple-virilizing“-Form des adrenogenitalen Syndroms leiden und bei denen eine Restaktivität der 21-Hydroxylase besteht fallen z. T. nicht im Neugeborenenscreening auf („Screeningversager“). Bei diesen fällt im weiteren Verlauf eine Pseudopubertas präcox auf. Bei betroffenen Mädchen kann sich dabei ggf. die Klitorishypertrophie aggravieren oder zusätzlich eine prämature Pubarche auftreten. Betroffene Jungen entwickeln häufig eine prämature Pubarche, ggf. kombiniert mit einer Genitalvergrößerung.
Durch die Hyperandrogenämie und die damit vermehrte hypophysäre Wachstumshormonsekretion sind betroffene Mädchen und Jungen oft im Verhältnis zur Elterngröße überdurchschnittlich groß. Allerdings kommt es gleichzeitig zu einer beschleunigten Knochenreifung mit der Konsequenz eines verfrühten Epiphysenfugenschlusses und dem Resultat einer niedrigen adulten Körpergröße.
Diagnose
Die Mehrzahl der Patienten wird durch das Neugeborenenscreening diagnostiziert (Analyt 17-Hydroxyprogesteron). Bei Frühgeborenen und kranken Neugeborenen werden oft erhöhte 17-OHP-Konzentrationen gemessen; daher ist die Verwendung gestationsalterspezifischer Referenzbereiche und ggf. die spätere Wiederholung des Screenings sinnvoll. Neben 17-OHP kommt der Bestimmung von 21-Desoxykortisol diagnostische Bedeutung zu.
Auffällige Screeningbefunde sollten durch venöse Bestimmung von 17-OHP kontrolliert werden; zusätzlich hat die Bestimmung von adrenalen Androgenen wie DHEAS, Androstendion und Testosteron diagnostische Relevanz.
Bei Patienten mit Salzverlust findet sich neben den Veränderungen der Serumelektrolyte meist eine erhöhte Plasmareninaktivität.
Die Urinanalytik mittels Gaschromatographie/Massenspektrometrie zeigt bei Patienten mit 21-Hydroxylasemangel ein charakteristisches Steroidprofil mit erhöhter Ausscheidung von Pregnantriol und Pregnantriolon. Diese wird sowohl in der Initialdiagnostik als auch zur Verlaufskontrolle der Therapieeinstellung genutzt.
Sonographisch kann bei Neugeborenen und jungen Säuglingen nicht selten eine Nebennierenhyperplasie gesehen werden; die Röntgenaufnahme der Hand zeigt bei älteren Kindern mit verzögerter Diagnosestellung oder unbefriedigender Stoffwechseleinstellung ein akzeleriertes Kochenalter.
Therapie
Therapeutisch steht die Substitution der defizienten adrenalen Steroide mit Hydrokortison und Fludrokortison im Vordergrund. Im Vergleich zur Nebenniereninsuffizienz ohne gleichzeitigen Androgenexzess ist eine etwas höhere Hydrokortisondosierung notwendig, damit nicht im Verlauf durch fortbestehenden Androgenexzess die Virilisierung fortschreitet. Die optimale Dosierung und Therapieeinstellung muss individuell erfolgen; als Richtdosis kann bei jungen Säuglingen mit rascher Veränderung der anthropometrischen Maße eine Hydrokortisondosierung von 10–15 mg/m2 KOF/d, bei älteren Kindern eine Dosierung in Höhe von 10–12 mg/m2 KOF/d dienen.
Die Substitution der defizienten Mineralokortikoide bei Patienten mit einem Salzverlust erfolgt in Form von Fludrokortison. Kleine Säuglinge benötigen eine vergleichsweise hohe Dosis (100–250 µg/m2 KOF/d), während ältere Kinder und Erwachsene oft mit 25–100 µg/m2 KOF/d gut eingestellt werden können.
Wie bei Patienten mit einem M. Addison sind Patienten- und Elternschulung hinsichtlich einer Dosisanpassung bei interkurrenten Erkrankungen von herausragender Bedeutung. In solchen Situationen muss die übliche Substitutionsdosis auf das 3- bis 5-fache erhöht oder ggf. eine Hydrokortisondauerzufuhr veranlasst werden.
Zusätzlich sollte eine Notfallmedikation (Hydrokortisonampullen) verordnet werden und ein Notfallausweis verfügbar sein.
Weiterhin kontrovers diskutiert werden Durchführung und Zeitpunkt geschlechtsangleichender genitalchirurgischer Eingriffe. Während verschiedene kinderchirurgisch/kinderurologische Experten eine frühe operative Korrektur im Säuglingsalter mit dem Argument einer geringeren Komplikationsrate sowie reduzierter Notwendigkeit von wiederholten vaginalen Bougierungen favorisieren wird von einigen Patientenverbänden das Abwarten der Einwilligungsfähigkeit der Patienten favorisiert.
Prognose
Die Prognose von Kindern mit einem adrenogenitalen Syndrom ist gut, vorausgesetzt es erfolgt eine adäquate Substitutionstherapie sowohl unter Alltagsbedingungen als auch im Rahmen interkurrenter krisenhafter Erkrankungen. Allerdings kann auch bei guter Therapieadhärenz, insbesondere bei verspäteter Diagnosestellung eine Einschränkung der Erwachsenengröße resultieren.
Eine Übertherapie mit Glukokortikoiden kann die Entstehung einer Adipositas, einer arteriellen Hypertonie und von metabolischen Komplikationen begünstigen.
Sowohl bei Männern wie Frauen mit einem adrenogenitalen Syndrom scheint die Fertilität etwas eingeschränkt zu sein. Ein kausaler Faktor hierfür scheint bei betroffenen Frauen in einer hyperandrogenämievermittelten Zyklusstörung zu liegen. Bei inadäquat eingestellten Jugendlichen und Männern wurden sog. testikuläre adrenale Resttumore (TART) mit einer eingeschränkten Fertilität assoziiert.
Pränatale Diagnostik und Therapie
Um eine pränatale Virilisierung von betroffenen weiblichen Feten zu verhindern ist eine pränatale Therapie möglich. Hierzu verwendet wird das plazentagängige Dexamethason. Die Therapie wird vor der 6. Schwangerschaftswoche begonnen und bis zum Ausschluss eines 21-Hydroxylasemangels oder Nachweis eines männlichen Karyotyps fortgeführt; bei weiblichen betroffenen Feten wird die Therapie bis zum Ende der Schwangerschaft fortgesetzt. Die pränatale Therapie ist aufgrund des ethischen Dilemmas einer Behandlung nicht betroffener Kinder und aufgrund von Daten zu potenziell negativen emotionalen und kognitiven Folgen sowie einer möglichen negativen fetalen Programmierung durch pränatale Glukokortikoidexposition umstritten und gilt daher als experimentelle Therapie.
Seltene Formen eines adrenogenitalen Syndroms
11β-Hydroxylase-Mangel
Der 11β-Hydroxylasemangel stellt mit etwa 5% nach dem 21-Hydroxylasemangel die zweithäufigste Form eines adrenogenitalen Syndroms dar. Ursächlich sind Mutationen im CYP11B1-Gen.
Klinisch findet sich bei Geburt bei betroffenen Mädchen meist eine deutliche Virilisierung. Allerdings entwickelt sich durch die mineralokortikoide Potenz von DOC (Metabolit vor dem Enzymblock) kein Salzverlust. Durch die erhöhte DOC-Konzentration entwickelt sich in den ersten Lebensjahren nicht selten eine schwer einstellbare arterielle Hypertonie. Infolge einer hohen klinischen Variabilität wird die Diagnose heute meist durch molekulargenetische Analyse des 11β-Hydroxylasegens gestellt.
Therapeutisch entspricht das Vorgehen dem des 21-Hydroxylasemangels.
3β-Hydroxysteroiddehydrogenase-Mangel
Pathogenetisch wurden Mutation im 3β-HSD-Typ-II-Gen gefunden. Klinisch kann das Genitale betroffener Mädchen unauffällig oder mild virilisiert auffallen, während bei betroffenen Jungen infolge der ebenfalls beeinträchtigten testikulären Androgenbiosynthese eine inkomplette Virilisierung (z. B. Hypospadie) vorliegen kann.
Defekte des StAR-Proteins (kongenitale Lipoidhyperplasie)
Das ACTH-abhängige StAR-Protein ist für den schnellen Cholesterintransport von der äußeren zur inneren Mitochondrienmembran und damit für die Umwandlung von Cholesterin zu Pregnenolon verantwortlich. Defekte im StAR-Gen führen zu einer intrazellulären Cholesterinakkumulation und der Abwesenheit fast aller Steroidhormone, sowohl in den Nebennieren wie den Gonaden.
Klinisch entsteht daher neben einer primären Nebenniereninsuffizienz bei betroffenen Jungen infolge der defizienten fetalen Androgenbiosynthese eine 46,XY-DSD. Betroffene Mädchen können eine leichte Klitorishypertrophie aufweisen. Bei manchen, aber nicht allen Patienten kann sonographisch eine adrenale Vergrößerung durch Cholesterinakkumulation gefunden werden.
Therapeutisch erfolgt eine Substitution der defizienten Steroidhormone.
Familiäre Glukokortikoidresistenz
Hierbei handelt es sich um eine seltene Form der NNR-Insuffizienz mit Nebennierenrindenhyperplasie. Sie wird verursacht durch Mutationen im Glukokortikoid-Rezeptor-Gen (NR3C1). Das klinische Bild ähnelt der familiären ACTH-Resistenz, ergänzt durch eine arterielle Hypertonie, einer hypokaliämischen Alkalose infolge ACTH-induzierter Mehrsekretion von Androgenen und Mineralokortikoiden.
Nebennierenrindenüberfunktion
Hyperkortisolismus
Definition, Ätiologie und Pathogenese
Unter einem Cushing-Syndrom werden alle Formen der Nebennierenüberfunktion zusammengefasst. Es ist mit einer Inzidenz von ca. 1:50.000 im Kindesalter selten. Man unterscheidet:
- ACTH-abhängige Störungen:
- ACTH-sezernierendes Hypophysenadenom (M. Cushing),
- CRH-Überproduktion (hypothalamischer Cushing),
- ektope ACTH-/CRH-Sekretion,
- ACTH-abhängige makronoduläre NNR-Hyperplasie.
- ACTH-unabhängige Störungen:
- adrenale Tumoren (Karzinome > Adenome),
- bilaterale mikronoduläre NNR-Hyperplasie,
- makronoduläre NNR-Hyperplasie (isoliert oder als Teil Carney-Komplex),
- McCune-Albright-Syndrom (GNAS1-Genmutation),
- iatrogen (Glukokortikoidtherapie).
Im Kindesalter stellt ein iatrogenes Cushing-Syndrom durch eine längerdauernde ACTH-Therapie (BNS-Anfälle) oder im Rahmen einer längeren immunsuppressiven Therapie weiterhin die häufigste Ursache für ein Cushing-Syndrom dar. Dem M. Cushing, also einer hypophysären ACTH-Mehrsekretion, liegt am häufigsten ein Mikroadenom der Hypophyse zugrunde; hierdurch kommt es zu einer bilateralen Nebennierenrindenhyperplasie.
Genetische Ursachen für einen M. Cushing kommen u. a. im Rahmen einer multiplen endokrinen Neoplasie Typ 1 (MEN-1) vor. Ein Cushing-Syndrom durch eine adrenale Hyperplasie kommt gehäuft vor bei Patienten einer MEN-1 und einem Carney-Komplex sowie im Rahmen adrenaler Karzinome bei Patienten mit Li-Fraumeni-Syndrom, MEN-1 oder selten bei Patienten mit einem Wiedemann-Beckwith-Syndrom.
Klinisches Bild
Im Kindesalter liegt bei betroffenen Patienten zunächst oft ein nichtcharakteristisches Bild vor, sodass die Diagnose oft erst verspätet nach Jahren gestellt wird.
Beim Vollbild eines Cushing-Syndroms weisen die Patienten eine stammbetonte Adipositas mit Entwicklung eines sog. „Büffelnackens“ auf. Oft liegen zusätzlich Striae rubrae in den Flanken vor. Das Gesicht ist rund („Vollmondgesicht“) mit typischerweise geröteten Wangen bei oft dünner und empfindlicher Hauttextur. Infolge des Glukokortikoidexzesses können eine arterielle Hypertonie und eine pathologische Glukosetoleranz vorliegen. Auxiologisch zeigt sich gerade im Langzeitverlauf eine Gewichtszunahme bei gleichzeitig schlechterem Intervallwachstum oder Wachstumsstillstand.
Begleitend findet sich vereinzelt eine erhöhte psychische Labilität bis hin zu psychiatrischen Krankheitsbildern oder dem Vorliegen einer Depression.
Bei älteren Kindern kann es zu einer verzögerten Pubertätsentwicklung, beim Mädchen zu einer primären oder sekundären Amenorrhö kommen.
Bei später Diagnose kann durch den bereits längerfristig bestehenden Glukokortikoidexzess eine Osteoporose bestehen, ggf. im Einzelfall mit Vorliegen von Wirbelkörperfrakturen.
Liegt dem Cushing-Syndrom eine adrenale Mehrproduktion von Glukokortikoiden und zusätzlich Androgenen oder Östrogenen zugrunde, kann ggf. ergänzend eine Pseudopubertas präcox vorliegen.
Diagnostik
Eine erweiterte Diagnostik wird bei entsprechendem klinischem Verdacht eingeleitet; dies wird allerdings in den letzten Jahren erschwert durch die rasante Zunahme von Kindern und Jugendlichen mit z. T. extremem Übergewicht und die dadurch z. T. erst spät eingeleiteten diagnostischen Maßnahmen.
Als Screeninguntersuchungen bei V. a. auf Vorliegen eines M. Cushing sind die u. g. Untersuchungen geeignet. Folgende Befunde können auf einen M. Cushing hinweisen:
Erhöhte Kortisol-Ausscheidung im Sammelurin,
Aufhebung der zirkadianen Rhythmik der Kortisolsekretion im Kortisol-Tagesprofil (fehlender Abfall des „Mitternachtskortisols“),
fehlende Suppression des Serumkortisols im niedrigdosierten Dexamethason-Kurztest.
Die erweiterte Diagnostik schließt den CRH-Test ein, der bei der Differenzierung von ACTH-abhängigem M. Cushing von Glukokortikoidexzessen anderer Genese hilfreich ist. So zeigen Patienten mit einem hypophysären M. Cushing nach CRH-Gabe einen zusätzlichen ACTH- und Kortisolanstieg, während dies bei ektoper ACTH-Produktion oder primär adrenalem Cushing-Syndrom nicht beobachtet wird. Zusätzlich zum Dexamethason-Kurztest existieren verschiedene Varianten des Dexamethasontest, für die unterschiedliche Sensitivitäten und Spezifitäten im Rahmen der Diagnostik des M. Cushing angegeben werden und die zur besseren Differenzierung der Genese des Glukokortikoidexzesses herangezogen werden können.
Neben der biochemischen Diagnostik ist die radiologische Darstellung von Hypothalamus und Hypophyse relevant; hier steht die Kernspintomographie mit Dünnschichtdarstellung der Hypophyse im Vordergrund (Abb. 26.17). Hier kann ein Mikroadenom der Hypophyse z. Z. erst im längeren Verlauf diskriminiert werden. Ergänzend kann in Einzelfällen ein Sinus petrosus sampling zur Lokalisationsdiagnostik notwendig werden. Darüber hinaus sollte je nach individuellem Befund eine Bildgebung der Nebennieren durch Sonographie, CT oder MRT erfolgen, ggf. ergänzt durch selektive Angiographie und Katheterisierung zur Lokalisationsdiagnostik.
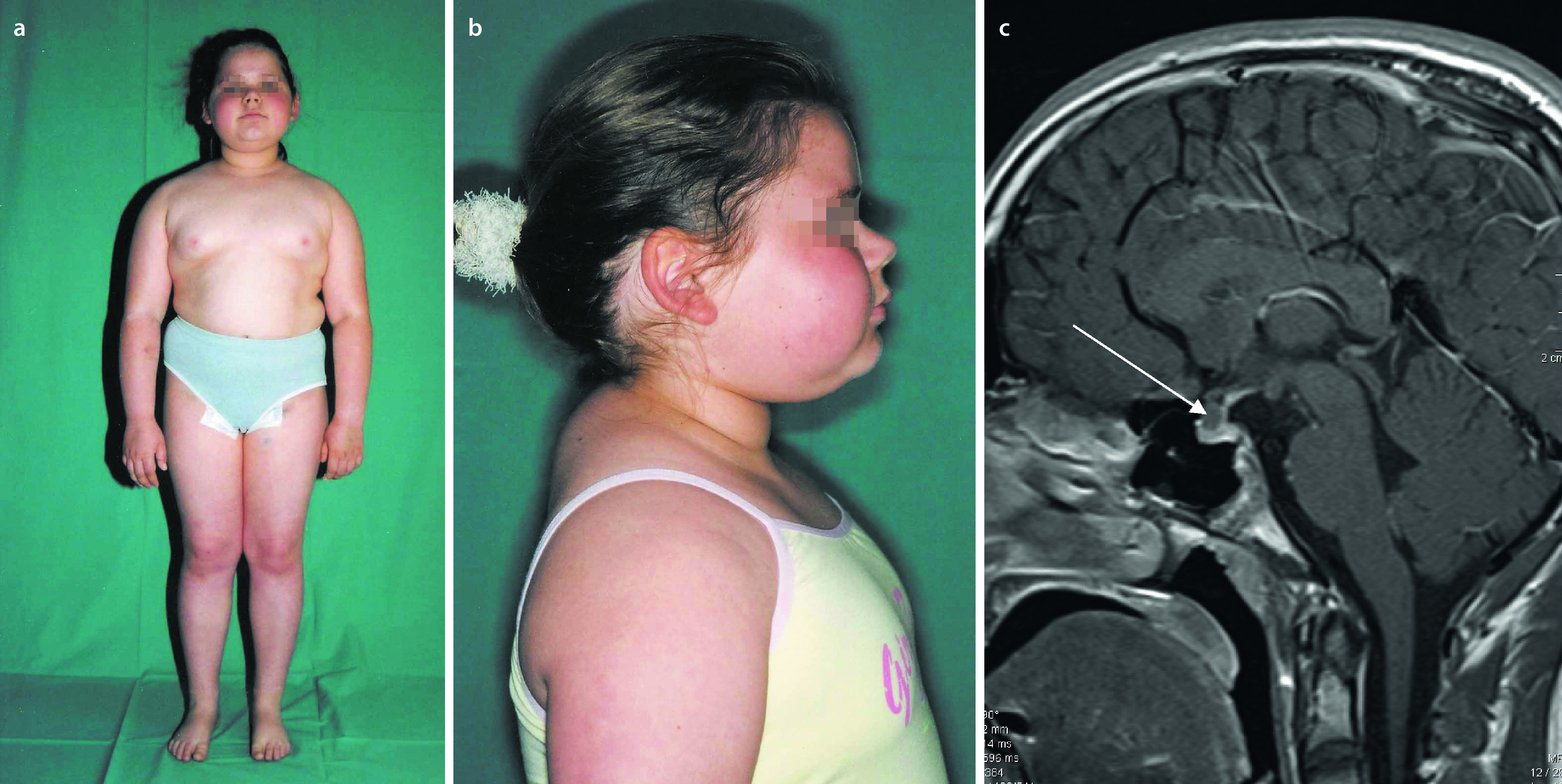
Therapie
Therapie der Wahl beim ACTH-abhängigen M. Cushing ist die transsphenoidale Resektion des Mikroadenoms in einem Zentrum mit diesbezüglich ausgewiesener hoher Kompetenz. Bei mehrfachen Rezidiven, erfolgloser Lokalisationsdiagnostik, erfolgloser medikamentöser Therapie und nicht ausreichender Hypophysenbestrahlung kann auch heute noch als ultima ratio eine bilaterale Adrenalektomie erfolgen.
Liegt ein Nebennierenadenom oder Karzinom dem Cushing-Syndrom zugrunde ist die unilaterale Adrenalektomie Therapie der Wahl.
Die medikamentöse Therapie ist die zweite Wahl bei Versagen einer chirurgischen Therapie oder nichtlokalisierbarer ektoper ACTH-Sekretion. Derzeit liegen drei pharmakologische Wirkgruppen vor:
Substanzen die direkt die hypophysäre ACTH-Sekretion beeinflussen,
die die adrenale Streroidbiosynthese inhibieren oder
Medikamente, die die Glukortikoidrezeptorwirkung antagonisieren.
Keines dieser Medikamente ist im Kindesalter zugelassen. Somatostatinanaloga wie Pasireotid sind bei ca. 30% der Erwachsenen mit M. Cushing effizient; bislang bestehen hier aber nur wenige Erfahrungen im Kindesalter. Sog. Adrenostatika wie Ketokonazol oder Mitotane weisen z. T. erhebliche unerwünschte Effekte auf und stehen daher eher im Hintergrund des therapeutischen Arsenals.
Mineralokortikoidexzess
Die Prävalenz der arteriellen Hypertonie im Kindesalter ist deutlich geringer als bei Erwachsenen. Allerdings findet sich bei Kindern mit arterieller Hypertonie eine höhere Wahrscheinlichkeit, dass die Hypertonie sekundär auf dem Boden einer Grunderkrankung basiert. Ein primärer Hyperaldosteronismus (Conn-Syndrom) ist allerdings im Kindesalter eine Rarität.
Ätiologie und Pathogenese
Ursachen für einen primären Hyperaldosteronismus umfassen aldosteronbildende Adenome, eine bilaterale Nebennierenhyperplasie sowie extrem selten Nebennieren-Karzinome.
Klinischer Befund
Bei den Patienten liegt eine häufig schwer einstellbare Hypertonie vor. Diese kann ergänzt sein durch Zephalgien und Schwindel, evtl. kombiniert mit Parästhesien und einer Polydipsie.
Diagnostik und Differentialdiagnose
Das Vorliegen einer Hypernaträmie in Kombination mit einer hypokalämischen Alkalose, eine erhöhte Aldosteronkonzentration im Plasma und Sammelurin sowie die supprimierte Plasmareninaktivität sind wegweisend.
Differenzialdiagnostisch ist der sekundäre hyperreninämische Hyperaldosteronismus bei renovaskulären Fehlbildungen oder hyperreninämischen Nierentumoren (z. B. Wilms-Tumor) sehr viel häufiger.
Darüber hinaus können folgende seltene Differenzialdiagnosen abgegrenzt werden:
Glukokortikoid-supprimierbarer Hyperaldosteronismus (GSH): bei Betroffenen lässt sich die arterielle Hypertonie und der Hyperaldosteronismus durch Dexamethasonapplikation normalisieren. Ursächlich ist ein „crossing over“ von Gensequenzen der beiden 11β-Hydroxylasegene, sodass das CYP11B2-Gen (Aldosteronsynthese) abnorm einer Regulation durch ACTH unterliegt und durch Glukokortikoide supprimiert werden kann.
Ein adrenogenitales Syndrom durch Mutation der 11β-Hydroxylase kann infolge der mineralkortikoiden Wirkung von DOC (Metabolit vor dem Enzymblock) zu einer arteriellen Hypertonie bei hyporeninämischem Hypoaldosteronimus führen.
Apparent mineralocorticoid excess (AME): Ursächlich hierfür ist eine Mutation in der 11β-Hydroxysteroid-Dehydrogenase Typ 2, sodass Kortisol nicht mehr zu Kortison inaktiviert wird und es zu einer protrahierten Stimulation des Mineralokortikoidrezeptors durch Glukokortikoide kommt. Der AME lässt sich biochemisch vom Conn-Syndrom differenzieren, da hier ein hyporeninämischer Hypoaldosteronismus vorliegt. Diagnostisch weiterführend ist die erhöhte Kortisol-Kortison-Ratio im Serum bzw. der abnorme Tetrahydrokortisol-Tetrahydrokortison-Quotient im Urin.
Eine sehr ähnliche klinisch/laborchemische Konstellation kann beim übermäßigen Konsum von Lakritze oder Carbenoxolon auftreten, da der Inhaltsstoff Glyzyrrhizinsäure die Aktivität der 11β-Hydroxysteroid-Dehydrogenase Typ 2 hemmt und es dadurch ebenfalls zu einem Pseudohyperaldosteronismus kommt.
Monogene tubuläre Defekte wie das Liddle-Syndrom (Mutation in epithelialem Natriumkanal, ENAC) oder das Geller-Syndrom (Mutation im Mineralokortiokoidrezeptor) können ebenfalls zu einer arteriellen Hypertonie mit hyporeninämischem Hypoaldosteronismus und metabolischer Alkalose führen.
Therapie
Bei Vorliegen eines isolierten Adenoms ist die unilaterale Adrenalektomie das gängige Therapieprinzip. Bei Patienten mit bilateraler Nebennierenhyperplasie oder Vorliegen eines Pseudohyperaldosteronismus kann eine kompetitive Hemmung des Mineralokortikoidrezeptors mittels Spironolacton versucht werden. Patienten mit GSH und AME sind mit niedrigen Glukokortikoiddosen effektiv behandelbar.
Gonaden
Hypogonadismus
Abschn. 7.2
Hodenhochstand
Kap. 25.2.4
Diabetes mellitus
Definition und Grundlagen
Der Begriff Diabetes mellitus beschreibt eine Stoffwechselstörung, die durch das Vorliegen einer Hyperglykämie infolge Insulinmangel oder Insulinresistenz oder eine Kombination beider Bedingungen gekennzeichnet ist.
Diagnosekriterien
Nach der aktuellen Klassifikation der WHO werden 3 Stadien des Diabetes unterschieden:
Stadium mit noch normaler Glukoseregulation und Normoglykämie,
Stadium mit gestörter Glukoseregulation und Hyperglykämie,
Stadium des Diabetes.
Der orale Glukosetoleranztest mit 1,75 g/kgKG bzw. max. 75 g Glukose wird diagnostisch zur Einordnung des Kohlehydratstoffwechsels herangezogen:
Eine Normoglykämie liegt vor bei einem Nüchternblutzucker von <100 mg/dl (<5,6 mmol/l) bzw. einem 2-h-Wert <140 mg/dl (<7,8 mmol/l).
Eine gestörte Glukoseregulation kann sich äußern als:
gestörte Nüchternglukose (impaired fasting glucose, IFG) mit einer Blutzuckerkonzentration zwischen 100–125 mg/dl (>5,6 und <6,9 mmol/l) bei einem normwertigen 2-h-Wert (<140 mg/dl bzw. <7,8 mmol/l)
oder als gestörte Glukosetoleranz (Impaired glucose tolerance, IGT) mit einem 2-h-Wert im OGT von 140–199 mg/dl (7,8–11,0 mmol/l).
Beide gelten als Risikogruppen für einen Diabetes mellitus.
Ein Diabetes mellitus liegt vor bei:
einer Nüchernglykämie >125 mg/dl (>6,9 mmol/l) oder
einem 2-h-Wert im OGT von >199 mg/dl (>11,0 mmol/l).
Zusätzlich kann der Diabetes mellitus anhand des HbA1c-Werts definiert werden: ein HbA1c unterhalb von 5,7% (<39 mmol/mol) schließt einen Diabetes mellitus aus, ein HbA1c >6,5% (47,54 mmol/mol) definiert einen Diabetes.
Die Diagnosekriterien sind in Abb. 26.19 dargestellt:

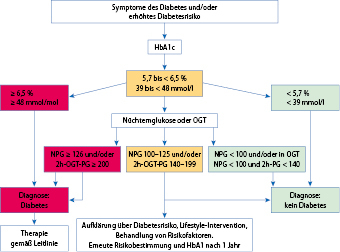
Epidemiologie
Während in der Gesamtheit aller Altersgruppen der Typ-2 Diabetes etwa 90% der Diabetesformen ausmacht ist im Kindes- und Jugendalter der Typ-1-Diabetes die dominierende Diabetesform. Er ist die häufigste Stoffwechselerkrankung im Kindes- und Jugendalter. Derzeit werden in Deutschland deshalb etwa 34.000 Kinder und Jugendliche in der Altersgruppe 0–19 Jahre behandelt. Auf Basis von lokalen Bevölkerungsregisterdaten ist mit einer jährlichen Neudiagnoserate von 3.200–3.700 Patienten mit Typ-1-Diabetes zu rechnen, wobei insbesondere bei den Patienten im Vorschulalter eine deutliche Zunahme der Inzidenz verzeichnet wurde. Jüngere Daten aus Baden-Württemberg ergaben eine Inzidenz von 19,4 Fällen/100.000 Einwohner für Kinder und Jugendliche in einem Alter unter 15 Jahren. Die Inzidenzrate scheint um 3–4% pro Jahr zu steigen. Es besteht ein deutliches Nord-Süd-Gefälle in der Inzidenz des Typ-1-DM mit hohen Neuerkrankungsraten in Skandinavien und niedrigeren in Mittelmeerländern (Ausnahme: Sardinien). Demgegenüber wird der Typ-2-Diabetes trotz der besorgniserregenden Veränderung der Gewichtsentwicklung von Kindern und Jugendlichen im Kindes- und Jugendalter derzeit noch ungleich weniger häufig diagnostiziert. Aktuelle Zahlen gehen von 800–900 Patienten mit einem Typ-2 DM in einem Alter zwischen 10 und 19 Jahren aus, bei allerdings vermutlich hoher Dunkelziffer.
Ätiologie
Ätiologische Klassifikation des Diabetes mellitus
-
I.Typ-1-Diabetes (β-Zellzerstörung, üblicherweise mit absolutem Insulinmangel)
- immunologisch bedingt
- idiopathisch
-
II.
Typ-2-Diabetes (von überwiegender Insulinresistenz mit relativem Insulinmangel bis hin zu Überwiegen eines Sekretionsdefekts mit Insulinresistenz)
-
III.Andere Diabetestypen mit bekannten Ursachen
-
A.Genetische Defekte der β-Zellfunktion:
- MODY 1 (Defekt in HNF-4α-Gen)
- MODY 2 (Defekt in Glukokinase-Gen)
- MODY 3 (Defekt in HNF-1α-Gen)
- MODY 4 (Defekt in HNF-1α-Gen)
- MODY 5 (Defekt in HNF-1αβ-Gen)
- MODY 6 (Defekt in Neuro-D1-Gen)
- MODY 7 (Defekt in KLF11-Gen)
- MODY 8 (Defekt in CEL-Gen)
- MODY 9 (Defekt in PAX4-Gen)
- MODY-like (Defekt in Insulin-Gen)
- Transienter neonataler Diabetes mellitus (TNDM)
- Permanenter neonataler Diabetes mellitus (PNDM)
- Mitochondrialer Diabetes
- Andere
-
B.Genetische Defekte der Insulinwirkung:
- Typ A-Insulinresistenz
- Leprechaunismus
- Rabson-Mendenhall-Syndrom
- Lipoatrophischer Diabetes
- Andere
-
C.Erkrankungen des exokrinen Pankreas:
- Pankreatitis
- Trauma/Pankreatektomie
- Neoplasie
- Mukoviszidose
- Hämochromatose
- Fibrokalkuläre Pankreatopathie
- Andere
-
D.Endokrinopathien:
- Akromegalie
- Cushing-Syndrom
- Glucagonom
- Phäochromozytom
- Hyperthyreose
- Somatostatinom
- Aldosteronom
- Andere
-
E.Medikamenten- oder toxininduziert:
- Pyrinuron, Pentamidin, Nikotinsäure, Glukokortikoide, Schilddrüsenhormone, Diazoxid, ß-Sympathomimetika, Thiazide, Dilantin, Interferon-γ, u. a.
-
F.Infektionen:
- Konnatale Röteln
- Zytomegalie
- Andere
-
G.Seltene Formen von immunologisch vermitteltem Diabetes:
- „Stiff man“-Syndrom
- Anti-Insulinrezeptor-Antikörper
- Andere
-
H.Andere genetische Syndrome, die mit Diabetes assoziiert sind:
- Down-Syndrom
- Klinefelter-Syndrom
- Ullrich-Turner-Syndrom
- Wolfram-Syndrom
- Friedreich-Ataxie
- Chorea Huntington
- Bardet-Biedl-Syndrom
- Myotone Dystrophie
- Porphyrie
- Prader-Willi-Syndrom
- Andere
-
A.
-
IV.
Gestationsdiabetes (Beginn oder Nachweis einer Glukoseintoleranz in der Schwangerschaft)
Hypoglykämie
Es gibt keine einheitlich evidenzbasierte Evidenz für eine Hypoglykämie bei Kindern mit einem Diabetes mellitus, da die relevanten biologischen Reaktionsmuster und Symptome bei sehr unterschiedlichen Blutzuckerkonzentrationen auftreten können. Auf Basis eines Expertenkonsensus wurde eine asymptomatische Hypoglykämie definiert als Blutzucker <65 mg/dl (<3,6 mmol/l) ohne gleichzeitiges Vorliegen von Symptomen der neuroendokrinen Gegenregulation. Erfahrungsgemäß kommt es aber unterhalb eines Blutzuckerschwellenwerts von 45 mg/dl zu kognitiven Beeinträchtigungen.
Asymptomatische und leichte Hypoglykämien treten bei Kindern mit Diabetes unter Insulintherapie nicht selten auf, das Auftreten schwerer Hypoglykämien sollte aber unbedingt vermieden werden. Häufige Ursachen einer Hypoglykämie beim Typ-1-Diabetiker sind Ernährungsfehler im Sinne verminderter Nahrungszufuhr nach Insulinjektion, vermehrte körperliche Belastung, z. B. im Rahmen sportlicher Betätigung oder Dosierungsfehler bei der Insulintherapie.
Symptome
Die klinischen Symptome einer Hypoglykämie sind entweder die Folge der Neuroglykopenie oder einer adrenergen Gegenreaktion. Die Symptomatik von Hypoglykämien beinhaltet u. a. Konzentrationsstörungen, Unruhe, Kopfschmerzen, Verwirrtheit, Aggressivität, Zittrigkeit, Kaltschweißigkeit, Hungergefühl oder Müdigkeit. Morgendliche Müdigkeit und Abgeschlagenheit, morgendliche Kopfschmerzen, Albträume oder nassgeschwitzte Bettwäsche können Hinweise auf eine nächtliche Hypoglykämie sein.
In Abhängigkeit der Stoffwechseleinstellung und Diabetesdauer variiert die Hypoglykämiewahrnehmung. So werden Symptome eines schnellen Blutzuckerabfalls bei durchschnittlich erhöhten Blutzuckerwerten bereits bei deutlich höheren Blutzuckern wahrgenommen als wenn niedrige Blutzucker oder Hypoglykämien häufig auftreten.
Hypoglykämien werden unterschieden in:
Leichte Hypoglykämien, die durch den Genuss von schnellresorbierbaren Kohlehydraten rasch ausgeglichen werden können und
schwere Hypoglykämien, die zu einer Bewusstseinseinschränkung bzw. einem Bewusstseinsverlust führen und bei denen der Betroffene auf Fremdhilfe angewiesen ist.
Therapie
Im Falle des Auftretens von Symptomen einer Hypoglykämie sollte der Betroffene unmittelbar nach Auftreten der Symptomatik rasch resorbierbare Kohlehydrate zu sich nehmen, um eine schwere Hypoglykämie mit Entwicklung einer Neuroglykopenie zu vermeiden. Dies kann u. a. durch Zufuhr von Traubenzucker (10–20 g), Apfelsaft (200 ml) oder Glukosegel erfolgen.
Bei schweren Hypoglykämien muss so rasch wie möglich Glukagon i.m. oder s.c. injiziert werden; alternativ kann eine Glukoseininjektion i.v. (0,2 g/kgKG) erfolgen (Cave: evtl. rekurrierende Hypoglykämien, daher sollte unbedingt nach Normalisierung der Bewusstseinslage eine orale Kohlehydratzufuhr erfolgen).
Alle Kinder und Jugendliche sollten ein Notfallset mit sich führen, das neben den für die Blutzuckermessung notwendigen Utensilien einen ausreichenden Vorrat an schnellen Kohlehydraten beinhaltet (z. B. Traubenzuckerplättchen).
Für Eltern und Sorgeberechtigte sollte ein Glukagon-Notfall-Kit verfügbar sein; hierfür müssen die Verantwortlichen in die Anwendung des Glukagons geschult sein!
Typ-1-Diabetes
Alle Kinder und Jugendliche sollten einen Notfallausweis mit sich führen, aus dem hervorgeht, dass sie an einem Typ-1-Diabetes leiden und wer im Notfall verständigt werden sollte.
Pathogenese
Neben den immunologisch vermittelten Formen des Typ-1-Diabetes-mellitus (T1DM) gibt es idiopathische, nichtimmunologisch vermittelte Formen des T1DM, auf die hier nicht weiter eingegangen wird.
Aus heutiger Sicht handelt es sich beim immunologisch vermittelten T1DM um eine Autoimmunerkrankung, bei der es durch eine gesteigerte zelluläre Immunität zu einer schubweisen Zerstörung der pankreatischen β-Zellen kommt. Die Auslösung des Krankheitsprozesses beruht dabei auf einer komplexen Interaktion von genetischen und Umweltfaktoren.
Unter den genetischen Faktoren spielen die Histokompatibilitätsantigene (HLA), die in die Präsentation von pankreatischen Antigenen an T-Zellen eingebunden sind, eine große Rolle. Hierbei kommt den Genen der HLA-Klasse II auf Chromosom 6p21.3 im Bereich der DR- und DQ-Region die größte Bedeutung zu.
Entsprechend der Bedeutung des HLA-Systems erhöht sich das Risiko an einem Typ-1-Diabetes zu erkranken, wenn Verwandte an einem Typ-1-Diabetes leiden (Tab. 26.14).
| Verwandschafts-Konstellation | Diabetesrisiko |
|---|---|
| Allgemeines Risiko in Gesamtbevölkerung <30 Jahre | 0,4% |
| Kind einer diabetischen Mutter | 4% |
| Kind eines diabetischen Vaters | 6% |
| Geschwister mit T1DM | 10% |
| Eineiige Zwillinge mit T1DM | 25–50% |
| Ein Geschwister und ein Elternteil | 25% |
| Ein Geschwister und beide Elternteile | 50% |
Der Autoimmunprozess im Pankreas führt zu einer „Insulinitis“ mit begleitendem Nachweis diabetesspezifischer Antikörper wie u. a. Inselzellantikörper (ICA), Antikörper gegen die Glutaminsäure-Decarboxylase (anti-GAD), gegen die Tyrosinphosphatase (anti-IA2) und gegen Insulin (IAA), die aber eher die Insulinitis widerspiegeln als dass sie kausal in den autoimmunen Krankheitsprozess eingebunden sind. Hierbei scheinen autoreaktive T-Lymphozyten im Zusammenspiel mit Effektor-T-Zellen und regulatorischen T-Zellen bei der Progression der Insulinitis eine wesentliche Rolle zu spielen.
Umweltfaktoren
Verschiedene Umweltfaktoren wurden wiederholt in Verbindung mit der Diabetesmanifestation in Verbindung gebracht:
So findet sich eine saisonale Häufung der Diabetesmanifestation im Herbst und Winter. Verschiedene Virusinfektionen, insbesondere Infektionen mit Enteroviren, wurden als pathogenetisch relevant diskutiert.
Auch die mikrobielle Besiedlung des Intestinums scheint phasenhaft in die Pathogenese der Diabetesentstehung eingebunden zu sein. So wurde angedeutet, dass eine intestinale Fehlbesiedlung Konsequenzen für die angeborenen Immunität bzw. Immuntoleranz mit sich bringt. Inwieweit eine Behandlung mit „günstigen“ Darmbakterien bei Risikopatienten eine Diabetesentstehung verhindern kann bleibt aber derzeit noch offen.
Frühe Zufuhr glutenhaltiger Nahrung in den ersten 3 Lebensmonaten scheint mit häufigerem Auftreten von Merkmalen der Inselzellautoimmunität einherzugehen. Gegenwärtig ist nicht eindeutig geklärt, inwieweit Stillen bzw. eine frühe Kuhmilchexposition das Risiko eines T1DM beeinflusst.
Der aus dem Zusammenspiel von Genetik und Umwelt verursachte Entzündungsprozess bedingt eine allmähliche Abnahme der Sekretionskapazität für Insulin, die lange inapparent verläuft (Abb. 26.20). Erst wenn die β-Zell-Masse und assoziierte Insulinsekretionskapazität unter einen kritischen Schwellenwert fällt (≈10% in Kindheit, 20% in Adoleszenz) kommt es zur Manifestation des Diabetes mellitus.

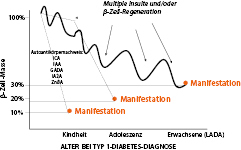
Durch den Insulinmangel bei gleichzeitig relativ gesehen verstärkter Glucagonwirkung kommt es zu einer reduzierten Glukoseverwertung sowie einer gesteigerten Glycogenolyse und Gluconeogenese. Zusätzlich führt der Insulinmangel zu einer verstärkten Lipolyse und verminderten Lipogenese. Dieses erhöhte Angebot freier Fettsäuren wird z. T. zu Ketonkörpern metabolisiert. Als Konsequenz resultiert eine Hyperglykämie und Hyperketonämie. Zusätzlich führt eine erhöhte osmotische Diurese infolge der Hyperglykämie zu ausgeprägten Flüssigkeits- und Elektrolytverlusten, die in Kombination mit der Hyperketonämie zur Komplikation einer diabetischen Ketoazidose führen können.
Klinisches Bild
Bei Manifestation bzw. Diagnosestellung werden meist typische anamnestische Informationen berichtet. Diese beinhalten u. a. das Auftreten einer Polyurie, Polydipsie, (sekundären) Enuresis oder eines Gewichtsverlusts. In Einzelfällen werden längerdauernde Entzündungen von Haut und Schleimhäuten (z. B. Soor-Kolpitis) berichtet. Zusätzlich sollte nach weiteren Diabeteserkrankten in der Familie gefragt werden.
Im Untersuchungsbefund ist das Ausmaß der Dehydratation relevant; es sollte eine Schätzung des Gewichtsverlusts während der vorangehenden Wochen abgeschätzt werden. Neben den Vitalparametern Herzfrequenz, Blutdruck, Temperatur sollte auf eine etwaige Bauchschmerzsymptomatik geachtet werden (Pseudappendizitis diabetica). Im Falle einer Ketoazidose kann eine Kussmaul-Atmung mit einem Fötor acetonämicus bestehen.
Diagnostik
Die Labordiagnostik bei Manifestation beinhaltet eine Blutzuckerbestimmung, eine Blutgasanalyse, einen Urinstatus mit Urinzucker- und Ketonkörperbestimmung, Elektrolyte inkl. Phosphat, renale Retentionsparameter, HbA1c sowie ein Differenzialblutbild.
Im weiteren Verlauf erfolgt die Bestimmung von Nüchterninsulin und C-Peptid, diabetesspezifischen Antikörpern (anti-GAD, ICA, IAA), Serumlipiden, Transaminasen sowie ein Screening auf assoziierte Erkrankungen wie Autoimmunthyreoiditis (TSH, fT4, SD-AK) oder Zöliakie (Transglutaminase-AK).
Der Verlauf des T1DM ist durch verschiedene Phasen charakterisiert, diese umfassen:
die (meist vor Diagnosestellung) bestehende Phase eines Prädiabetes mit noch ausreichender Insulinsekretionskapazität,
die Manifestation bzw. Diagnosestellung des Diabetes,
eine variabel stark ausgeprägte Remissionsphase („honeymoon“),
sowie die Phase der dauerhaften Insulinabhängigkeit.
Therapie
Neben der medizinischen Diagnostik und Therapie sind Inhalt und Kontext der Diagnosemitteilung „Diabetes mellitus“ von nachhaltiger Relevanz. Neben einer klaren Information zu praktischen Inhalten und Konsequenzen der Diagnose sollten mögliche Krankheitstheorien von Kind und Eltern dezidiert erfragt werden, um anhaltende Schuldgefühle auszuräumen bzw. zu vermeiden.
Die Therapie bei Kindern und Jugendlichen mit einem Diabetes mellitus muss sowohl die medizinischen Aspekte der Erkrankung als auch die psychosozialen Beeinträchtigungen einer spezifischen chronischen Erkrankung in Betracht ziehen. So dienen Einleitung, Schulung und Durchführung einer Diabetestherapie der Vermeidung akuter Stoffwechselentgleisungen als auch langfristig der Prävention diabetesbedingter Folgeerkrankungen. Gleichzeitig müssen aber altersübliche Alltagsanforderungen unter nun komplizierten Bedingungen bewältigt werden.
Ziel einer Diabetestherapie muss daher sein, dass die psychosoziale Entwicklung diabeteserkrankter Kinder durch den Diabetes mellitus und seine Therapie geringstmöglich beeinträchtigt und die Integration in Kindergarten, Schule sowie die spätere berufliche Ausbildung gewährleistet wird.
Um dies zu erreichen muss unter Berücksichtigung der individuellen Bedingungen und Ressourcen eine optimale Anleitung von Patient und Familie zu möglichst selbständig und selbstbestimmtem Umgang mit der Erkrankung erfolgen.
Mit dem Kind/Jugendlichen und dem jeweiligen familiären Umfeld werden individuelle Therapieziele formuliert, die eine weitgehende Normoglykämie bei gleichzeitiger Minimierung des Risikos von Hypoglykämien zum Ziel haben. Typischerweise wird dabei ein Nüchternblutzucker von 90–145 mg/dl bzw. ein postprandialer Butzucker von 90–180 mg/dl angestrebt; bei guter Stoffwechseleinstellung ist ein HbA1c-Wert <7,5% Therapieziel. Darüber hinaus ist die „time in range“, also der Zeitanteil bei dem der Blutzucker im angestrebten Bereich liegt relevant.
Erstbehandlung mit Ketoazidose
Trotz Fortschritten in Diagnostik und Therapie liegt doch etwa bei jedem 5. Patienten in Deutschland bei der Manifestation des Diabetes im Kindes- und Jugendalter eine Ketoazidose vor. Diese ist definiert durch einen erhöhten Blutzucker, einem pH-Wert <7,3 mit einem Plasmabikarbonatkonzentration <15 mol/l, begleitet von einer Ketonurie oder Ketonämie.
Drei Schweregrade der Ketoazidose werden unterschieden:
leicht (pH <7,3; Bikarbonat <15 mmol/l),
mittelschwer (pH <7,2; Bikarbonat <10 mmol/l) und
schwer (pH <7,1; Bikarbonat <5 mmol/l).
Klinisch steht bei Manifestation des T1DM mit Ketoazidose die Dehydratation mit trockener Haut und Schleimhäuten im Vordergrund. Ergänzend findet sich eine beschleunigte Atmung (Kussmaul-Atmung), die unter Umständen zur Fehldiagnose einer Pneumonie führen kann. Allerdings fällt üblicherweise der Acetongeruch der Atemluft als wichtiges diagnostisches Kriterium auf. Abdominelle Beschwerden im Rahmen der Ketose können zur Fehldiagnose einer abdominellen Problematik führen (Pseudoperitonitis diabetica). Glücklicherweise kommt es in weniger als 1% der Fälle zu der gefürchteten Komplikation eines Hirnödems; klinische Hinweise hierauf können Zephalgien und Schwindel, vermehrte Irritabilität, aber insbesondere sekundäre Störungen der Vigilanz und/oder Bradykardie mit Blutdruckanstieg nach Einleitung (inadäquat hochdosierter) Flüssigkeits- und Insulintherapie.
Als Differenzialdiagnose bei abdominellen Beschwerden in Kombination mit einer Dehydratation sollte an die Ketoazidose gedacht werden!
Therapeutisch ist bei Kindern mit Ketoazidose zunächst eine Flüssigkeitstherapie und Kreislaufstabilisierung indiziert. In Abhängigkeit vom Ausmaß der Kreislaufbeeinträchtigung kann initial eine Bolusgabe isotoner Lösung von 10–20 ml/kgKG über 1–2 h notwendig sein. Anschließend erfolgt ein Ausgleich des Flüssigkeitsdefizits über einen Zeitraum von 36–48 Stunden unter Verwendung plasmaisotoner Elektrolytlösungen (NaCl 0,9% oder Ringerlösung). Um das Risiko eines Hirnödems zu reduzieren, sollte die tägliche Infusionsmenge das 1,5- bis 2-fache des normalen Tagesbedarfs in Bezug auf Alter und Gewicht nicht übersteigen.
Azidosekorrektur und Beginn der Insulintherapie bedingen eine Verschiebung von Kalium von extra- nach intrazellulär, sodass hier ggf. ein entsprechender Ausgleich notwendig wird. Deshalb ist ein Kaliumersatz bei der Behandlung der diabetischen Ketoazidose erforderlich. Bei Ketoazidose und Hypokaliämie sollte die Kaliumsubstitution bereits im Rahmen der initialen Flüssigkeitstherapie erfolgen, bei Normokaliämie sollte diese mit der Insulintherapie einsetzen. Lediglich im Falle einer Hyperkaliämie sollte das Wiedereinsetzen der Diurese abgewartet werden bis dann die Kaliumsubstitution erfolgt. Aufgrund der genannten Verschiebungen unter Flüssigkeits- und Insulintherapie sollte die Kaliumkonzentration zu Beginn der Therapie engmaschig kontrolliert werden.
Darüber hinaus sollte die Natriumserumkonzentration überwacht werden, da das Absinken des Blutzuckers und der damit verbundene Abfall der Serumosmolalität zu einer Verschiebung freien Wassers und damit zu einem Anstieg der gemessenen Serumnatriumkonzentration führt. Gerade bei initial sehr hohen Blutzuckerwerten ist die Berechnung der korrigierten Natriumkonzentration mittels folgender Formel hilfreich:
Erfahrungsgemäß führt bereits die Einleitung der intravenösen Flüssigkeitszufuhr zu einer Absenkung des Blutzuckers, wenn auch die Azidose sich aufgrund der weiter fortschreitenden Ketogenese infolge fortbestehenden Insulinmangels meist nicht bessert.
Ein bis zwei Stunden nach Beginn der Rehydratation sollte daher zur weiteren Blutzuckerabsenkung und Azidosebehandlung eine intravenöse Insulintherapie in niedriger Dosis begonnen werden (0,1 IU/kgKG/h, bei sehr hohem Blutzucker oder kleinen Kindern 0,05 IU/kgKG/h oder weniger). Die Insulintherapie führt zu einer Absenkung des Blutzuckers um etwa 36–90 mg/dl/h (2–5 mmol/l/h).
Cave
Eine zu rasche Absenkung des Blutzuckers sollte vermieden werden!
Bei einer Blutzuckerkonzentration von <250 mg/dl sollte auf eine glukosehaltige Infusionslösung gewechselt werden (z. B. Ionosteril D5%), bei weiterem Abfall unter 200 mg/dl ggf. in höherer Glukosekonzentration. Dies ist notwendig, um einerseits das Risiko einer Hypoglykämie bei laufender Insulininfusion zu vermeiden, andererseits bei ansonsten notwendiger Reduktion der Insulinzufuhr nicht erneut die Ketogenese zu stimulieren.
Ein Azidoseausgleich sollte vermieden werden, da die Bikarbonatgabe aufgrund von Eektrolytverschiebungen das Hirnödemrisiko weiter erhöhen kann. Ausnahmen stellen evtl. die therapierefraktäre Kreislaufinsuffizienz bei schwerster Azidose (pH <6,9) oder eine lebensbedrohliche Hyperkaliämie dar; hier kann ggf. eine vorsichtige Bikarbonatgabe erwogen werden.
Zerebrale Krise und Hirnödem
Im Falle der glücklicherweise seltener gewordenen Komplikation einer zerebralen Krise kommt es typischerweise innerhalb der ersten 24 h nach Einleiten einer Flüssigkeits- und Insulintherapie zu zentralnervösen Symptomen. Diese beinhalten Kopfschmerzen, Erbrechen, Störungen der Vigilanz, im weiteren Verlauf Absinken der Herzfrequenz (>20 Schläge/min) und Blutdruckanstieg, abnormes Atemmuster, Störungen der Pupillomotorik, Hirnnervenparesen, Bewegungsstörungen mit Opisthotonus und Krampfanfälle.
Wichtig ist bei Verdacht auf Entwicklung einer zerebralen Krise die frühzeitige Therapie mit Mannitol einzuleiten (Mannitol 0,25–1,0 g/kgKG über 20 min). Bei Erfolglosigkeit der Mannittherapie kann ggf. ein Einsatz 3%iger hypertoner Kochsalzlösung i.v. (2,5–10 ml/kg über 10–15 min) erfolgen.
Hyperglykäm-hyperosmolares Koma (HHS)
Das HHS ist eine bei Kindern mit T1DM seltene Erkrankung, die aber mit hoher Morbidität und Mortalität verknüpft ist. Das HHS tritt häufiger bei Jugendlichen oder Erwachsenen mit Typ-2-Diabetes auf. Das HHS zeichnet sich durch die folgende Trias aus:
Hyperglykämie >600 mg/dl (33,3 mmol/l),
Hyperosmolarität >320 mosm/l und
maximal milder Azidose (ph >7,3, Serumbikarbonat >15 mmol/l) infolge fehlender oder milder Ketose.
Das HHS scheint häufiger bei Kindern afroamerikanischer Herkunft aufzutreten; es wird spekuliert dass pathogenetisch eine ineffektivere Ketogenese eine Rolle spielt.
Das HHS ist mit hoher Morbidität und Mortalität verbunden, Flüssigkeits- und Insulintherapie der oft drastisch erhöhten Blutzucker sollten daher auf der Intensivstation erfolgen!
Therapeutisch stehen bei Vorliegen eines HHS die Flüssigkeitstherapie und das langsame Absenken des oft massiv erhöhten Blutzuckers im Vordergrund. Ein sorgfältiges Monitoring zur Vermeidung von Komplikationen ist unbedingt erforderlich. Eine späte und niedrig dosierte Insulintherapie erscheint vorteilhaft.
Erstbehandlung ohne Ketoazidose
In der Mehrzahl der Fälle liegt bei der Manifestation keine Ketoazidose vor, sodass nicht zwingend eine Infusionstherapie und i.v.-Insulintherapie initiiert werden muss. Gleichwohl beginnen viele Zentren zunächst mit einer i.v.-Rehydratation und i.v.-Insulintherapie. Neben psychologischen Aspekten erleichtert dieses Vorgehen die Abschätzung des initialen Insulinbedarfs. Allerdings ist in den Fällen einer Manifestation ohne Azidose der direkte Beginn einer subkutanen Insulintherapie mittels ICT (ab Schulalter) oder CSII (Säuglinge oder Vorschulkinder) unter der Voraussetzung einer ausreichenden oralen Flüssigkeitszufuhr eine gangbare Alternative.
Dauerbehandlung
In der Dauertherapie von Kindern und Jugendlichen steht als medikamentöses Therapieprinzip die Insulintherapie im Vordergrund, da der immunologisch vermittelte T1DM mit der Folge eines absoluten Insulinmangels in der Pädiatrie klar im Vordergrund steht. Hand in Hand mit der Insulintherapie sind Kenntnisse zur Ernährung und Stoffwechselkontrolle sowie zum Umgang mit der chronischen Erkrankungen in besonderen Situationen (Erkrankung, Sport, Krisen, …) relevant. Diese Kenntnisse werden in wiederholten, altersadaptierten Einzel- oder Gruppenschulungen vermittelt mit dem Schulungsziel, den Patienten und seine Familie umfassend über die Erkrankung zu informieren und in die Lage zu versetzen, die chronische Erkrankung weitestgehend selbständig zu kontrollieren. Unumgänglich ist eine individualisierte psychosoziale Betreuung, um etwaige Krisen zu vermeiden oder rechtzeitig zu erkennen und ggf. zu intervenieren.
Insulintherapie
Die Insulintherapie von Kindern und Jugendlichen mit einem T1DM erfolgt heute in weit über 90% der Fälle als sog. intensivierte Insulintherapie, entweder mit Insulin-Pen, Insulinspritze oder mittels Insulinpumpentherapie (CSII). Die früher eingesetzte konventionelle Insulintherapie mit einem fest vorgegeben Insulin- und Mahlzeitenregime wird mittlerweile nur noch sehr selten eingesetzt. Bei der intensivierten Insulintherapie unterscheidet man zwei verschiedene Behandlungsmethoden, die sog. intensivierte konventionelle Insulintherapie (ICT) und die Behandlung mit einer Insulinpumpe.
Intensivierte konventionelle Insulintherapie (ICT)
Im Rahmen einer ICT erfolgt die Injektion eines Normalinsulins (Humaninsulin oder schnelles Insulinanalogon) vor jeder Mahlzeit (Prandialtherapie); das Verzögerungsinsulin wird 1- bis 3-mal täglich (meist morgens und abends, seltener auch mittags) verabreicht.
Bei der ICT können im Gegensatz zu der früher verwendeten konventionellen Insulintherapie Mahlzeiten flexibel eingenommen werden. Neben der erhöhten Flexibilität ermöglicht die ICT eine Verbesserung der Stoffwechseleinstellung, ist aber mit der Notwendigkeit vermehrter Blutzuckerkontrollen, einem etwas höheren Hypoglykämierisiko und höheren Kosten verknüpft.
Die Insulinapplikation erfolgt subkutan; um die Entwicklung einer Lipodystrophie mit dann ggf. schwer vorhersehbarem Resorptionsverhalten zu vermeiden, müssen Injektionsstellen regelmäßig gewechselt werden. In Abhängigkeit der Lokalisation der Injektionsstelle finden sich Unterschiede in der Absorptionsgeschwindigkeit:
schnell wirksames Insulin wird typischerweise in Gewebe mit schneller Resorption injiziert (z. B. periumbilikal),
während Basalinsuline in Gewebe mit langsamer Resorption (z. B. Gesäß oder Oberschenkel) injiziert werden.
Prandial wird kurzwirksames Insulin in Abhängigkeit von der Kohlehydratzufuhr verabreicht. Die Menge an Kohlehydraten einer Mahlzeit wird heute meist als sog. Kohlehydrateinheit (KE) angegeben; diesem entspricht eine Kohlehydratmenge von 10–12 g. Der Insulin-KE-Faktor liegt durchschnittlich zwischen 1–2 g/KE, er ist interindividuell unterschiedlich und abhängig von Alter, Größe, Gewicht, Geschlecht, Diabetesdauer und Nahrungszusammensetzung. Darüber hinaus ist die zirkadiane Rythmik der Insulinwirkung zu beachten: So werden morgens durchschnittlich 2 IU/KE oder mehr Insulin benötigt, während um die Mittagszeit die Hypoglykämieneigung mit der Insulinempfindlichkeit zunimmt und eher 1 IU/kgKG benötigt werden. Am frühen Abend steigt der Insulinbedarf wieder auf durchschnittlich 1,5–2 IU/kgKG an. Auch für die Gestaltung der Basisinsulintherapie bzw. Basalratengestaltung ist die zirkadiane Rhythmik von Relevanz. So steigt der Insulinbedarf zwischen 4 und 8 Uhr infolge vermehrter Sekretion kontrainsulinärer Hormone (wie z. B. STH oder Kortikoide) insbesondere während der Pubertät deutlich an. Besteht hier eine Insulinunterversorgung kommt es zu einer Morgenhyperglykämie („Dawn-Phänomen“).
Die Häufigkeit der Basalinsulinapplikation hängt u. a. von der Art des Basalinsulins (NPH-Insulin (langwirksames Analogon) und der Insulindosis ab. Der Basisinsulinbedarf liegt durchschnittlich bei etwa 0,2 IU/kgKG/d im Kleinkindalter und etwa bei 0,3 IU/kgKG/d im Kindesalter, kann aber während der Pubertät deutlich ansteigen.
Eine Übersicht (Auswahl) über die aktuell im Kindes- und Jugendalter zugelassenen Insuline ist in Tab. 26.15 dargestellt; Kombinationsinsulinpräparate werden im Kindesalter nur in Ausnahmen eingesetzt.
| Gruppe | Wirkeintritt (min) | Wirkmaximum (h) | Wirkdauer (h) | Präparat | Handelsname | Zugelassen ab Alter (J.) |
|---|---|---|---|---|---|---|
| Ultrakurz wirkende Insulinanaloga | 5 | 1(–2) | 3–4 | Faster acting insulin aspart | FIASP | Ab 18 Pädiatrische Zulassung angekündigt |
| Sehr kurz wirkende Insulinanaloga | 10 | 1–2 | 4 | Insulinglulisin | Apidra | Ab 6 |
| Insulin lispro | Humalog | Keine Altersbeschränkung | ||||
| Insulin lispro | Liprolog | Keine Altersbeschränkung | ||||
| Insulin aspart | Novorapid | Ab 2 | ||||
| Kurz wirkendes Normalinsulin | 20 | 2–4 | 8 | Humanes Normalinsulin | Insulin Insuman rapid Huminsulin normal Actrapid HM | Keine Altersbeschränkung |
| Mittellang wirkendes Basalinsulin | 30 | 4–6 | 14–20 | NPH-Insulin | Keine Altersbeschränkung | |
| Lang wirkende Basalinsulin-Analoga | 90 | - | 20 | Insulindetemir | Levemir | Ab 1 |
| 60 | - | 24 | Insulinglargin | Lantus | Ab 2 |
Insulinpumpentherapie
Rasante technische Weiterentwicklungen insbesondere in den letzten 15–20 Jahren führten zu einer dramatischen Zunahme des Anteils von diabetischen Kindern und Jugendlichen mit Insulinpumpentherapie. Aktuell werden etwas mehr als die Hälfte aller pädiatrischen Patienten mit einer Insulinpumpe behandelt. Im Kleinkind- und Vorschulalter wird in Deutschland fast jedes diabetische Kind aufgrund der deutlich besseren Steuerbarkeit mit einer CSII versorgt. Während sich in fast allen Altersgruppen bestimmte Vorteile für eine CSII finden lassen hat die ISPAD für einzelne Indikationen eine gesonderte Empfehlung zur CSII-Therapie empfohlen; diese beinhalten u. a.:
Säuglinge und Kinder im Vorschulalter,
Patienten mit ausgeprägtem Dawn-Phänomen,
schwere, rezidivierende und nächtliche Hypoglykämien,
Kinder mit ausgeprägter Nadelphobie.
Allerdings ist die Insulinpumpentherapie mit deutlich höheren Behandlungskosten verknüpft. Als weitere Einschränkung sollte erwähnt werden, dass der Erfolg einer CSII maßgeblich an die Therapiekompetenz und Adhärenz der Nutzer geknüpft ist. So ist die CSII nur erfolgreich, wenn Patient und Familie gut geschult weitestgehend selbständig das technische Werkzeug nutzen können und die erworbenen Kenntnisse tatsächlich nutzen.
Die derzeit in Deutschland zugelassenen Insulinpumpen werden außerhalb des Körpers angebracht; sie bestehen neben der elektronischen Hard- und Software aus Bildschirm, Insulinreservoir und Fördersystem, über die typischerweise über einen Katheter das Insulin in das Subkutangewebe transportiert wird. Abweichend hiervon erfreuen sich insbesondere im Jugendalter die sog. Patch-Pumpen zunehmender Beliebtheit, die direkt ohne Katheter auf der Haut angebracht wird.
Der Insulinbasisbedarf wird bei CSII-Therapie durch die fest programmierte, individuell ermittelte Basalrate gedeckt. Die prandiale Insulinabgabe erfolgt aktiv durch den Patienten als sog. Bolus, analog zum Insulin-KE-Faktor bei der ICT. Zusätzlich verfügen die meisten Insulinpumpen über Bolusberechnungsprogramme, die den Patienten bei der Berechnung unterstützen können.
Sensorunterstützte Pumpentherapie und closed loop
Mit der Einführung und raschen Weiterentwicklung von Glukosesensoren zur minimalinvasiven Messung von Glukose in Subkutis oder der interstitellen Flüssigkeit der Haut wurde die Grundlage für die sog. sensorunterstütze Pumpentherapie gelegt. Hierbei „kommunizieren“ Sensor und Insulinpumpe miteinander. Bei Unterschreiten einer definierten Glukosekonzentration oder zu raschem Abfall des gemessenen Gewebezuckers wird die Insulinabgabe gestoppt und ein Alarm ausgelöst. Bereits hierdurch konnte eine signifikante Reduktion von Hypoglykämien erreicht werden, ohne dass sich die Stoffwechseleinstellung der Patienten verschlechtert.
Im nächsten logischen Schritt kommt es zu einer weitgehenderen Steuerung der Insulinabgabe durch die vom Sensor übermittelten Messdaten, dem sog. Closed-loop-System. Ein erstes System hierzu ist in den USA bereits zugelassen (Medtronic MiniMed 670G®), nachdem gezeigt werden konnte, dass insbesondere nachts durch das closed loop eine bessere Stoffwechselkontrolle mit höherem Anteil von Glukosewerten im Zielbereich erreicht werden kann. Dieser technische Siegeszug führte zu einem neuen Bedarf an noch schneller resorbierbaren Insulinvarianten. Hier können die vermutlich in Bälde auch im Kindes- und Jugendalter zugelassenen ultrakurzwirksamen Insulinanaloga zu einer weiteren Verbesserung der Stoffwechselkontrolle beitragen.
Ernährung
Kinder und Jugendliche mit einem Diabetes mellitus benötigen keine andere Form der Ernährung als nichtdiabetische Kinder; es gelten die Empfehlungen einer optimierten Mischkost des Forschungsinstitutes für Kinderernährung. Die Ernährung sollte kohlehydratreich (>50%, bevorzugt komplexe KH) und fettkontrolliert sein (<35%).
Allerdings müssen Patienten und/oder ihre Familien in der Lage sein, den Kohlehydratanteil der Nahrung und deren Effekt auf die Blutglukose abzuschätzen. Während früher in Deutschland überwiegend der Begriff der „Broteinheit“ gebräuchlich war, der einem Kohlehydratäquivalent von 12 g D-Glukose entsprach wird heute eher eine Kohlehydrateinheit von 10–12 g Kohlehydraten als Maßeinheit herangezogen.
Als Faustregel für den Kalorienbedarf von Kindern gilt:
Kalorienbedarf (kcal/Tag) = Alter in Jahren × 100 +1.000
Zusätzlich sollten Kinder und Eltern darin geschult werden, dass vergleichbare Mengen an Kohlehydraten in Abhängigkeit von Beschaffenheit und Zubereitung, aber auch individuellen Besonderheiten einen unterschiedlichen Effekt auf den Blutzucker haben können (sog. glykämischer Index).
Diagnostik und Verlaufsbeurteilung
Blutglukose
Weiterhin wichtigster Parameter zur Beurteilung der akuten Stoffwechselsituation ist der typischerweise im Kapillarblut gemessene Blutzucker. Hier wird ein Nüchternblutzucker von 90–145 mg/dl bzw. ein postprandialer Blutzucker von 90–180 mg/dl angestrebt. Die Blutzuckerkonzentrationen sowie Kohlehydrat- und Insulinzufuhr und ggf. situative Besonderheiten (z. B. Sport, Infekt, …) sollten protokolliert werden.
Uringlukosemessung
Die Uringlukosemessung ist heutzutage infolge der geringen Sensitivität und den deutlich strengeren Therapiezielen, bei deren Einhaltung es nicht zu einem Überschreiten der tubulären Rückresorption kommt, klinisch bei der Therapiekontrolle nicht mehr relevant.
Ketonkörpermessung
Bei schlechter Stoffwechseleinstellung mit einem Mangel an Insulin und/oder einer mangelhaften Energiezufuhr (insbesondere von KH) können zu einer Hyperketonämie führen. Daher sollte in diesen Situationen eine Untersuchung von entweder Urin oder Blut auf Ketonkörper erfolgen (Abschn. 7.9, Ketoazidose).
HbA1c
Andauernde Exposition gegenüber einer erhöhten Blutzuckerkonzentration führt zu einem Anstieg glykierter Hämoglobine; HbA1c macht hierbei etwa 80% des Gesamt-HbA1 aus. Basierend auf einer mittleren Erythrozytenlebensdauer von 100–120 Tagen spiegelt der HbA1c-Wert die Stoffwechseleinstellung etwa der vorangehenden 6–8 Wochen wider. Therapieziel ist ein HbA1c-Wert <7,5% (<58 mmol/l).
Kontinuierliche Glukosemessung
Wie im Abschnitt Insulinpumpentherapie beschrieben, wurden im Bereich der kontinuierlichen Glukosemessung (CGM) erhebliche Fortschritte gemacht. Dies und die positive Entscheidung des IQWIG, die zu einer Kostenerstattung der CGM-Systeme durch die GKV geführt hat, veranlasste eine rasante Zunahme des Anteils von Patienten, die eine CGM nutzen. Dabei werden CGM-Systeme nicht nur in Verbindung mit Pumpen im Sinne einer sensorunterstützten Pumpentherapie eingesetzt, sondern auch ergänzend oder als Alternative zur blutigen Stoffwechselkontrolle.
Psychosoziale Faktoren
Die Diagnose T1DM bedingt, dass sich Kind und Familie neben alterstypischen Anforderungen zusätzlich mit zahlreichen Einschränkungen und Herausforderungen auseinandersetzen müssen. Diese beinhalten u. a., dass zur Vermeidung von akuten und chronischen Komplikationen lebenslang sowohl Monitoring als auch Therapie notwendig werden.
Dauerhafte Reflektion der Nahrungsmittelzufuhr, regelmäßig notwendige Stoffwechselkontrolle, hoher Zeit-, aber auch finanzieller Aufwand sind nur einzelne Aspekte die die Belastungen von Betroffenen und ihren Familien verdeutlichen. Gerade bei Heranwachsenden in der Pubertät wird diese Einschränkung der Unbeschwertheit und Verlust der Spontanität oft als sehr belastend empfunden.
Unumgänglich ist daher eine individualisierte psychosoziale Betreuung, um etwaige Krisen zu vermeiden oder rechtzeitig zu erkennen und ggf. zu intervenieren. Ein wesentlicher Pfeiler in der Vermeidung von Krisen sind wiederholte, altersadaptierte Einzel- oder Gruppenschulungen, um Patienten und deren Familien umfassend über die Erkrankung zu informieren und zu weitestgehender Selbständigkeit zu verhelfen (empowerment). In den meisten Zentren hat sich die Vermittlung von strukturierten Schulungskonzepten durch ein multiprofessionelles Team bewährt. Typischerweise besteht dieses aus pädiatrischen Endokrinologen/Diabetologen, Diabetesberatern, Ernährungsberatern, Kinderpsychologen und Sozialarbeitern. Letztere sind wichtig bei Information, Wissensvermittlung und Durchsetzung sozialrechtlicher Aspekte. Diese beinhalten u. a. Nachteilsausgleich mit einem Grad der Behinderung, aber auch Hilfestellung bei der Beantragung von familiären Unterstützungsmaßnahmen.
Folgeerkrankungen des T1DM
Wichtige Folgeerkrankungen/Langzeitkomplikationen des T1DM sind in Tab. 26.16 dargestellt. Dabei ist heute der Zusammenhang zwischen langfristiger Hyperglykämie und der diabetischen Mikroangiopathie unstrittig. Die diabetische Mikroangiopathie manifestiert sich insbesondere am Auge (diabetische Retinopathie), an der Niere (diabetische Nephropathie) sowie an den Nervenzellen (diabetische Neuropathie).
| Screeninguntersuchung auf | Screeningintervalle | Empfohlene Screeningmethode(n) | Interventionen |
|---|---|---|---|
| Retinopathie | Alle 1–2 Jahre ab dem 11. Lebensjahr oder ab 5 Jahren Diabetesdauer | Binokulare bimikroskopische Funduskopie in Mydriasis durch routinierten Augenarzt |
Verbesserung der glykämischen Kontrolle Lasertherapie |
| Nephropathie | Jährlich ab dem 11. Lebensjahr oder ab 5 Jahren Diabetesdauer |
Nachweis einer Mikroalbuminurie: – Konzentrationsmessung: 20–200 mg/l – Albumin-Exkretionsrate >20–<200 µg/min – Albumin-Kreatinin-Ratio |
Verbesserung der glykämischen Kontrolle ACE-Hemmer AT-I-Blocker Nikotinabstinenz |
| Neuropathie | Bei langfristig schlechter Stoffwechsellage jährlich ab dem 11. Lebensjahr oder ab 5 Jahren Diabetesdauer |
Anamnese Berührungsempfinden (Monofilament) Vibrationsempfinden (Stimmgabeltest) Eigenreflexe |
Verbesserung der glykämischen Kontrolle |
| Hypertonie | Alle 3 Monate, mindestens jährlich ab dem 11. Lebensjahr |
Ruhe-RR 24-h-RR bei mindestens 2-mal >95. Perzentile oder Mikroalbuminurie |
Lebensstilintervention (Bewegung, Salzrestriktion, Gewichtsreduktion, Reduktion Alkohol, Nikotin) Falls nicht erfolgreich: ACE-Hemmer |
| Hyperlipidämie | Innerhalb des ersten Jahres nach Diagnose, dann alle 2 Jahre, präpubertär alle 5 Jahre |
Bestimmung von – Gesamtcholesterin – HDL – LDL – Triglyzeride |
Diätetische Therapie Falls nicht erfolgreich: ab dem 8. Lebensjahr Statine |
Die diabetische Makroangiopathie beschreibt atherosklerotische Veränderungen der großen Gefäße, die mit großer Wahrscheinlichkeit ebenfalls bereits in der Kindheit initiiert wird.
Typ-2-Diabetes
Epidemiologie
Obwohl auch in Deutschland eine besorgniserregende Zunahme von Adipositas bei Kindern und Jugendlichen beobachtet wird, so werden derzeit doch noch ungleich weniger Fälle von Typ-2-Diabetes-mellitus (T2DM) im Kindes- und Jugendalter diagnostiziert. Aktuelle Zahlen gehen von 800–900 Patienten mit einem T2DM in einem Alter zwischen 10 und 19 Jahren aus, bei allerdings vermutlich hoher Dunkelziffer.
Pathogenese
Im Gegensatz zum Insulinmangel bei Patienten mit einem T1DM liegt bei der Glukoseverwertungsstörung des T2DM eine Insulinresistenz vor. Hierzu tragen u. a. verschiedene Adipokine und inflammatorische Zytokine bei. Ergänzend hierzu kommt es unter Beteiligung von erhöhten Konzentrationen zirkulierender freier Fettsäuren zu einer Beeinträchtigung der β-Zellfunktion.
Diagnose und klinisches Bild
Das klinische Bild des T2DM ist oft uncharakteristisch und schleichend. Das Vorliegen einer Ketoazidose bei Manifestation ist die Ausnahme.
Neben Übergewicht und Adipositas weisen fast alle Patienten eine Acanthosis nigricans auf. Dies beschreibt eine Hyperpigmentierung und Verdickung der Haut; insbesondere an Hals und Nacken, Achselhöhlen sowie den Intertriginärfalten.
Viele Patienten mit einem T2DM zeigen zusätzlich andere Marker des metabolischen Syndroms (arterielle Hypertonie, Dyslipidämie, polyzstisches Ovarsyndrom).
Therapie und Prognose
Im Vordergrund steht eine Modifikation von Faktoren des Lebensstils, analog zu den Therapieprogrammen bei pädiatrischen Patienten mit Adipositas. Wichtige Bausteine strukturierter Programme sind die Modifikation des Verhaltens (sowie des Ess- wie des Bewegungsverhaltens), eine Steigerung der körperlichen Bewegung und Verbesserung der Ernährung.
Bei Manifestation ist eine medikamentöse Therapie mit oralen Antidiabetika (insbesondere Metformin), sowie initial meist auch eine Insulintherapie notwendig. Andere Therapeutika wie Inhibitoren des Enzyms Dipeptidyl-Peptidase 4 (DPP-4) oder Agonisten des Rezeptors für Glukagon-like-Peptide-1 (GLP-1-Agonisten) sind derzeit in klinischer Erprobung.
Neben der Therapie von Adipositas und der diabetischen Stoffwechsellage muss ggf. eine Therapie der Komorbiditäten erfolgen (z. B. antihypertensive Therapie bei arterieller Hypertonie).
Zur Inzidenz von Folgeerkrankungen des T2DM im Kindes- und Jugendalter liegen bislang nur wenige Daten vor. Es scheint jedoch im Vergleich zu Patienten mit einem T1DM eher zu einer früheren Manifestation mikrovaskulärer Veränderungen wie der Retinopathie oder Nephropathie zu kommen.
Typ-3-Diabetes
Die Ätiologie des T3DM ist ausgesprochen heterogen; hier soll nur exemplarisch auf einige relevante Formen eingegangen werden.
MODY (maturation-onset diabetes of the young)
Die Gruppe der MODY-Diabetesformen fasst ätiopathogenetisch unterschiedliche autosomal-dominant vererbte Formen eines Diabetes mellitus zusammen.
Findet sich bei Kindern und Jugendlichen mit Diabetes mellitus betroffene Angehörige über 3 Generationen, ohne gleichzeitiges Vorliegen von Übergewicht oder autoimmune Genese sollte eine diagnostische Abklärung eines MODY erfolgen.
Oft wird ein MODY-Diabetes durch einen Erkrankungsbeginn vor dem 25. Lebensjahr, Fehlen einer Hyperketonämie bei Manifestation oder Stoffwechselentgleisung, autosomal dominanten Erbgang sowie Hinweisen auf einen primären β-Zelldefekt charakterisiert.
Derzeit sind 10 verschiedene MODY-Formen bekannt (Abschn. 7.9).
Klinisch ähnelt der MODY-Diabetes eher dem T2DM, mit oft langen uncharakteristischen Beschwerden oder asymptomatischem Verlauf. Nicht selten wird die Diagnose erst nach Diagnosestellung eines familiären Indexfalles gestellt.
Therapeutisch richtet sich die Behandlung nach dem zugrundeliegenden Defekt. Patienten mit MODY-3 lassen sich oft mit Gliniden oder Sulfonylharnstoffen behandeln.
Angeborener Hyperinsulinismus
Ätiologie und Pathogenese
Der kongenitale Hyperinsulinismus ist eine seltene Erkrankung. Ein angeborener Hyperinsulinismus im Kindesalter ist meist Teil einer genetisch determinierten Erkrankung, die zu einer übersteigerten Insulinsekretion führt. Noch seltener tritt im Kindesalter ein erworbener Hyperinsulinismus z. B. durch Insulinome im Rahmen einer MEN auf.
Meist führt der Hyperinsulinimus zu einer persistierenden Hypoglykämieneigung im frühen Kindesalter. Selten können in der Neugeborenenperiode transiente Formen vorkommen.
In den letzten Jahren wurden zahlreiche genetische Veränderungen identifiziert, die einem kongenitalen Hyperinsulinismus zugrundeliegen. Darüber hinaus kann ein Hyperinsulinismus Teil einer syndromalen Erkrankung wie des Wiedemann-Beckwith-Syndroms, Varianten von congenital disorders of glycosylation (CDG, insbes. Typ 1b) oder eines Costello-Syndroms sein.
Diagnostik und klinische Symptomatik
Durch den bereits pränatal bestehenden Hyperinsulinismus (Insulin ist ein potentes anaboles Hormon!) kann bereits vor Geburt eine Makrosomie auffallen. Viele Patienten zeigen in den ersten Lebenstagen eine Hypoglykämieneigung, nicht selten assoziiert mit Krampfanfällen im Rahmen einer Neuroglykopenie.
Schwere und rekurrierende Hypoglykämien sind ein Risiko für die kindliche Hirnentwicklung! Daher muss eine Hypoglykämie rasch erkannt und behandelt werden!
Ein wertvolles diagnostisches Kriterium ist der Kohlehydratbedarf, der zur Aufrechterhaltung eines normalen Blutzuckers notwendig ist. Werte über 10 mg/kgKG/min können auf einen kongenitalen Hyperinsulinismus hinweisen. Im Rahmen einer Hypoglykämie abgenommene Blutproben, die trotz Hypoglykämie nachweisbare Insulinkonzentrationen zeigen sind ebenfalls verdächtig auf einen Hyperinsulinismus.
Für das weitere therapeutische Vorgehen ist relevant, ob eine diffuse oder eine fokale Form des Hxperinsulinismus vorliegt. Zur Unterscheidung wird eine „[18F]Fluoro-L-DOPA-Positron-Emission-Tomography-Computed Tomography“ herangezogen; die Untersuchung sollte in diesbezüglich erfahrenen Zentren durchgeführt werden.
Therapie
In der akuten Hypoglykämie wird als Akuttherapie vor Allem eine hochdosierte Glukosezufuhr, ggf. ergänzt durch Glukagongabe. In der medikamentösen Dauertherapie stehen Diazoxid, Somatostatin oder Somatostatinanaloga sowie eine Ernährungstherapie zur Verfügung.
Fokale Krankheitsformen lassen sich durch eine Pankreasteilresektion meist heilen. Schlechter sind die Ergebnisse bei diffusem Hyperinsulinismus. Hier kann eine subtotale Pankreasresektion ggf. die Symptomatik stabilisieren oder abmildern. Allerdings droht bei 95%iger Resektion langfristig die Entwicklung eines Diabetes mellitus und/oder einer exokrinen Pankreasinsuffizienz.
Zyklusstörungen
Wegen Menstruationsstörungen suchen bis zu 75% aller Mädchen einen Arzt auf. In diesem Kapitel soll auf praxisrelevante Zyklusstörungen und deren Therapie eingegangen werden.
Das mittlere Alter, in dem die Menarche einsetzt, beträgt in Deutschland aktuell 12,2 Jahre. Die Menarche tritt ca. 2½ Jahre nach Beginn der Thelarche auf. Mädchen empfinden die Menstruation einerseits als Geschenk, welches ein Gefühl der Unversehrtheit und Intaktheit vermittelt, und sehen sie als Symbol von Fruchtbarkeit und weiblicher Gesundheit. Andererseits erleben Mädchen die Monatsblutungen auch als Hygienekrise, Einschränkung der sexuellen Attraktivität und Bewegungsfreiheit. Von einer normalen Zykluslänge spricht man, wenn die Blutungsabstände zwischen 25 und 33 Tage betragen.
Zyklusstörungen bei Jugendlichen unterscheiden sich nicht von denen im Erwachsenenalter. Die Blutungsstörungen werden eingeteilt in Amenorrhö (primär und sekundär), Regeltempostörungen (Oligomenorrhö und Polymenorrhö), Regeltypusstörungen (Hypermenorrhö und Hypomenorrhö), Zusatzblutungen, Dauerblutungen und die Dysmenorrhö (primär und sekundär). Lediglich die primäre Amenorrhö (Fehlbildung als Ursache: Abschn. 7.11) und die juvenile Dauerblutung (Abschn. 7.12.1) sind typische Zyklusstörungen des jugendlichen Mädchens.
In den ersten 2 Jahren nach der Menarche sind unregelmäßige Blutungen physiologisch, dahinter verbergen sich häufig anovulatorische Zyklen. Eine Diagnostik von Blutungsstörungen vor Ablauf von 3 Jahren nach Beginn der Menarche empfiehlt sich deshalb nur bei sich entwickelten Androgenisierungszeichen (Akne, Hirsutismus). In diesem Fall sollte auf eine Hyperandrogenämie abgeklärt werden.
Juvenile Dauerblutung
Leitsymptom
Unter einer juvenilen Dauerblutung versteht man eine zyklusunabhängige, anhaltende Blutung in der Adoleszenz, häufig tritt diese in den ersten beiden Jahren nach Eintritt der Menarche auf.
Eine Anovulation mit Follikelpersistenz ist die häufigste Ursache der juvenilen Dauerblutung, die dysfunktionelle juvenilen Dauerblutung. Ansonsten muss auch an eine Hämophilie gedacht werden. Im Falle einer anovulatorischen Dauerblutung kommt es zu einer Östrogendominanz mit Überproliferation des Endometriums und aufgrund der nicht vorhandenen Gestagenwirkung (ohne Ovulation keine Gestagenbildung) bleibt die sekretorische Transformation des Endometriums aus.
Die Mädchen entwickeln rasch eine Anämie und fallen durch Müdigkeit und Leistungsminderung auf.
Diagnostik
Anamnese (v. a. vorangegangene Blutungsneigung, Zyklusanamnese),
Schwangerschaftstest,
Inspektion äußeres Genital u. a auch zum Ausschluss von genitalen Verletzungen,
Sonographie abdominal (falls Geschlechtsverkehr erfolgt von vaginal): Endometriumsdicke!!!
- Blutentnahme:
- BB, Ferritin, Gerinnungsscreening bei V. a. Blutungsneigung,
- primär keine Hormonbasisdiagnostik notwendig.
Spekulumuntersuchung nur nötig, falls die sonstigen Untersuchungen keine aussagekräftige Ursache erbringen.
Therapie
Die Therapie sieht zum einen den Blutungsstopp und zum anderen die Behandlung der Anämie vor. Die Therapie der Dauerblutung ist primär abhängig von der Endometriumsdicke, somit ist die Ultraschalluntersuchung das wichtigste Diagnostikum.
Bei flachem Endometrium wird mit einer Estrogenmonotherapie (z. B. Estradiolvalerat 2 mg) für 10 Tage gefolgt von einer Kombinationstherapie (Östrogen-Gestagen-Kombination) von mindestens 12 Tagen therapiert. Bei hochaufgebautem Endometrium ist die Monotherapie mit Gestagenen (z. B. Dydrogesteron 10 mg oder Chlormadinonacetat 2 mg) indiziert. Sollte die Dauerblutung schon länger bestehen, so empfiehlt sich auch bei hochaufgebauten Endometrium die primäre Behandlung mit einer Kombinationstherapie (z. B. Estradioldvalerat + Dienogest). Bei bestehender Hämophilie muss eine kausale Therapie zusammen mit den Hämostaseologen festgelegt werden.
Kommt es auch hierunter nicht zum erwünschten Erfolg, kann zusätzlich Tranexamsäure gegeben werden.
In absoluten Ausnahmefällt kommt es trotz eingeleiteter hormoneller Therapie nicht zum Blutungsstopp, in diesem Fall muss eine Hysteroskopie mit Abrasio durchgeführt werden.
Eine weiterführende Therapie nach erfolgreicher Behandlung ist abhängig
vom Ausmaß der Anämie: besteht eine ausgeprägte Anämie würde sich die Weiterführung kombinierten Hormontherapie oder eines kombinierten Kontrazeptivums anbieten. Besteht keine ausgeprägte Anämie muss die initiale Therapie nicht fortgesetzt werden.
Von einer bestehender Hämophilie: bei bestehender Hämophilie bieten sich am ehesten kombinierte Kontrazeptiva an, die kontinuierlich (im Langzyklus) gegeben werden sollten.
Besteht keine ausgeprägte Anämie oder ist keine Hämophilie bekannt, so muss nicht zwingende eine fortbestehende Hormontherapie eingeleitet werden.
Die Therapie der Anämie wird mit den üblichen Eisenpräparaten primär oral bis zur Normalisierung des Hb-Werts durchgeführt.
Dysmenorrhö
Die Dysmenorrhö ist definiert als Unterbauchschmerzen während der Menstruation, die nicht selten kolikartig ablaufen. Zusätzlich kommt es häufig zu Übelkeit, Erbrechen, Diarrhö, Kopfschmerzen oder Migräneattacken und auch psychovegetativen Beschwerden wie Niedergeschlagenheit. Nicht selten sind die Beschwerden so massiv, dass die Betroffenen im Alltag eingeschränkt sind und somit in der Schule fehlen oder nicht am Sportunterricht teilnehmen können.
Die Dysmenorrhö ist die häufigste gynäkologische Erkrankung von Jugendlichen, aber lediglich 15% der betroffenen Mädchen konsultieren aufgrund der Schmerzen einen Arzt. Ein Großteil der betroffenen Jugendlichen wird erst durch die Anamnese, also das aktive danach fragen, diagnostiziert. Lediglich 1/3 der Mädchen mit Dysmenorrhö nutzen vor Erstdiagnose durch den Arzt ein Schmerzmittel. Es scheint in den Köpfen der Betroffenen, wie auch deren Mütter, die Meinung vor zu herrschen: Schmerzen, egal wie stark, gehören zur Regelblutung dazu.
Unterschieden wird die primäre Dysmenorrhö, ohne organische Ursachen und ab Menarche bestehend, von der sekundären Dysmenorrhö mit organischen Ursachen wie z. B. Myome oder Endometriose.
Die Inzidenz der Dysmenorrhö durch Endometriose scheint bei Jugendlichen wesentlich höher zu sein, als von uns Ärzten angenommen. Endometriose ist eine gutartige Erkrankung, bei der sich Gebärmutterschleimhaut (Endometrium) an den Eierstöcken, Eileitern, Darm, Blase und/oder dem Bauchfell ansiedelt. Als typisches Symptom kommt es zu Unterbauchschmerzen anfangs zyklisch während der Regelblutung. Langfristig leiden die Patientinnen aber auch an Unterbauchschmerzen außerhalb der Blutung. Daneben finden sich Schmerzen beim Wasserlassen, Stuhlgang und Geschlechtsverkehr. Die Erkrankung geht mit hohem Lebensqualitätsverlust einher und nicht selten können die Betroffenen nicht am Schul- und/oder Berufsleben teilnehmen. Einige Frauen werden erst aufgrund einer Sterilitätsproblematik diagnostiziert.
Leitsymptom
Symptome, die für eine Endometriose bei Jugendlichen sprechen, unterscheiden sich teilweise von denen bei Erwachsenen und sind:
nichtzyklische Unterbauchschmerzen,
schwere Dysmenorrhö,
Einfluss der Symptome auf das Fehlen in der Schule oder generell auf das Alltagsleben,
Nichtansprechen auf nichtsteroidale Antiphlogistika (NSAR) oder kombinierte orale Kontrazeptiva,
Dyschezie während Menstruation,
Darmkrämpfe,
Blasenschmerzen,
Depression und Angstzustände.
Diagnostik
Der Goldstandard für die Diagnostik ist die Laparoskopie mit Gewinnung einer Histologie, diese Tatsache erschwert die Diagnose. Per Palpation können bei ausgeprägter Endometriose Knötchen bzw. Verhärtungen im kleinen Becken getastet werden. Bei ovarieller Endometriose können per Ultraschall Endometriosezysten dargestellt werden.
Bei Mädchen mit Dysmenorrhö sollte eine gynäkologische Untersuchung mit Inspektion, ggf. Spekulum, Palpation und Ultraschall durchgeführt werden.
Therapie
Unabhängig ob von einer primären oder sekundären Dysmenorrhö aufgrund von Endometriose ausgegangen wird empfiehlt sich nachfolgendes Schema:
Bei milden Beschwerden sollte eine Einstellung oder auch Optimierung mit Schmerzmitteln v. a. NSAR (z. B. Ibuprofen) gewichtsadaptiert erfolgen. Wichtig ist es, diese Schmerzmittel an den Tagen der Beschwerden so früh wie möglich zu beginnen und an diesen Tagen kontinuierlich zu verabreichen.
Bei stärkeren Beschwerden oder ausbleibendem Erfolg mit Schmerzmitteln, ist die Gabe von kombinierten oralen Kontrazeptiva indiziert. Zur Therapie der Dysmenorrhö eignen sich besonders Präparate mit den Gestagenen Dienogest, Chlormadinonacetat oder Drospirenon, aber auch Präparate mit Levonorgestrel haben einen Einfluss auf die Dysmenorrhö. Natürlich sollte zuvor das Thromboserisiko abgeschätzt und hiernach verordnet werden. Die erste Kontrolle sollte nach 3 Monaten erfolgen. Bei Besserung ohne komplette Beschwerdefreiheit empfiehlt sich, frühestens nach 3, besser nach 6 Monaten, die kontinuierliche Gabe des Kombinationspräparats, um eine Blutung zu unterdrücken oder eine Umstellung auf ein anderes Gestagen.
Sollte es hiermit erneut zu keiner Besserung kommen, empfiehlt sich die Laparoskopie zur Diagnose einer Endometriose und gleichzeitiger Entfernung von Endometrioseherden. Postoperativ kann prophylaktisch ein kombiniertes Kontrazeptivum zur Prophylaxe indiziert sein.
Bei Anwendung dieses Schemas können unnötige operative Eingriffe verhindert werden. Nach einem Review 2013 hierzu konnte bei Mädchen, die bei therapierefraktären chronischen Unterbauchschmerzen laparoskopiert worden waren, bei 75% eine Endometriose gesehen und entfernt werden. Wurden Mädchen lediglich aufgrund einer Dysmenorrhö laparoskopiert, wurde nur noch bei 49% eine Endometriose diagnostiziert.
Contributor Information
J. Wölfle, Email: joachim.woelfle@ukb.uni-bonn.de
P. G. Oppelt, Email: patricia.oppelt@uk-erlangen.de
L. Wünsch, Email: lutz.wuensch@uksh.de
J. O. Semler, Email: joerg.semler@uk-koeln.de
E. Schönau, Email: eckhard.schoenau@uk-koeln.de
C. Petersen, Email: petersen.claus@mh-hannover.de