

Abstract
Centrosomes catalyze microtubule formation for mitotic spindle assembly1. Centrosomes duplicate once per cell cycle in a process controlled the kinase PLK42,3. Following chemical PLK4 inhibition, cell division in the absence of centrosome duplication generates centrosome-less cells that exhibit delayed, acentrosomal spindle assembly4. Whether PLK4 inhibitors can be leveraged for cancer treatment is not yet clear. Here, we show that acentrosomal spindle assembly following PLK4 inhibition depends on levels of the centrosomal ubiquitin ligase TRIM37. Low TRIM37 accelerates acentrosomal spindle assembly and improves proliferation following PLK4 inhibition, whereas high TRIM37 inhibits acentrosomal spindle assembly, leading to mitotic failure and cessation of proliferation. The Chr17q region containing the TRIM37 gene is frequently amplified in neuroblastoma and in breast cancer5–8, which renders these cancer types highly sensitive to PLK4 inhibition. TRIM37 inactivation improves acentrosomal mitosis because TRIM37 prevents PLK4 self-assembly into centrosome-independent condensates that serve as ectopic microtubule-organizing centers. By contrast, elevated TRIM37 expression inhibits acentrosomal spindle assembly via a distinct mechanism that involves degradation of the centrosomal component CEP192. Thus, TRIM37 is a critical determinant of mitotic vulnerability to PLK4 inhibition. Linkage of TRIM37 to prevalent cancer-associated genomic changes, including 17q gain in neuroblastoma and 17q23 amplification in breast cancer, may offer an opportunity to use PLK4 inhibition to trigger selective mitotic failure and provide new avenues to treatments for these cancers.
MAIN
Cells entering mitosis have two centrosomes that catalyze microtubule generation for assembly of the mitotic spindle1. Each centrosome has a centriole at its core that recruits a proteinaceous matrix called the pericentriolar material that nucleates and anchors microtubules9. Centrioles duplicate in a cell cycle-coupled process controlled by the Polo family kinase PLK42,3. To explore the utility of PLK4 inhibition in cancer, we developed the selective and cellularly active PLK4 inhibitor centrinone4,10. In the presence of centrinone, continued cell division without centriole duplication generates centrosome-less cells4. Cells lacking centrosomes remain capable of forming a bipolar spindle; however, spindle assembly and chromosome alignment are delayed and error-prone4,11–14. Following centrinone treatment of non-transformed human RPE1 cells, chromosome segregation fails in ~10% of cells, leading to eventual growth arrest13.
TRIM37 controls response to centrinone
In a genome-wide screen for genes whose inactivation enables sustained proliferation of centrinone-treated RPE1 cells, we identified the ubiquitin ligase TRIM3713. TRIM37 loss did not alter the duration of mitosis in cells with centrosomes (DMSO) but rescued delayed spindle assembly and chromosome segregation failure in cells lacking centrosomes (centrinone; Fig. 1a,b, Extended Data Fig. 1a–e; Video S1;13). To determine if elevating TRIM37 levels had the opposite effect, we conditionally overexpressed TRIM37 (Extended Data Fig. 1a–c). An ~4-fold increase in TRIM37 did not affect mitotic timing in cells with centrosomes but significantly increased mitotic duration and chromosome segregation failure in centrinone-treated cells (Fig. 1a,b; Extended Data Fig. 1d,e; Video S1). Analysis of 4 additional clones with varying elevation of TRIM37 indicated that the magnitude of the mitotic defects in centrinone-treated cells was proportional to the amount of TRIM37 (Extended Data Fig. 1c,f). Thus, the extent of mitotic challenge imposed by centrosome loss due to PLK4 inhibition depends on TRIM37 in a bi-directional fashion: TRIM37 loss improves outcomes whereas TRIM37 elevation makes them significantly worse.
Figure 1. TRIM37 levels determine mitotic outcomes and cancer-specific sensitivity to PLK4 inhibition.
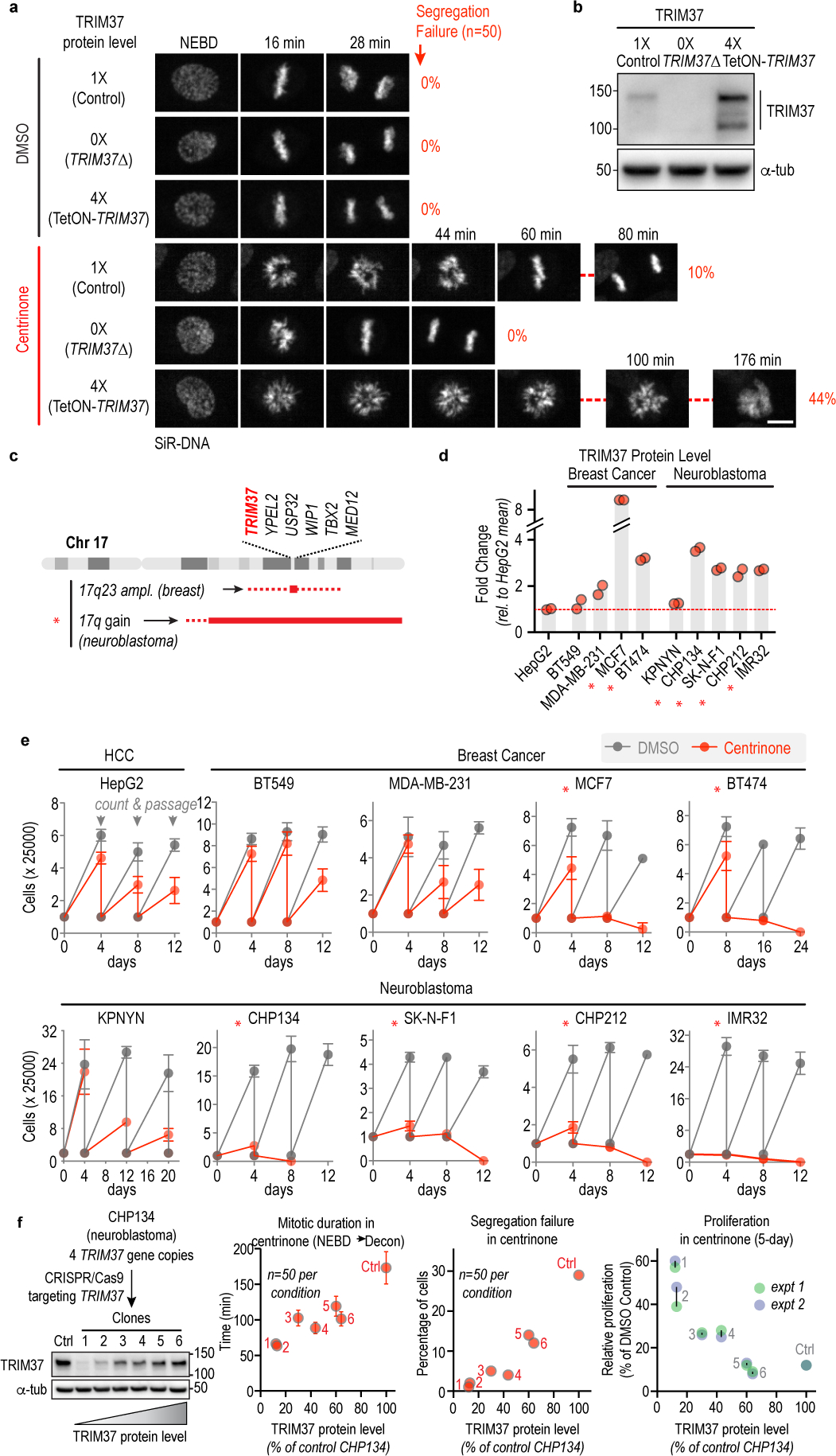
(a) Still images from timelapse sequences showing chromosomes in RPE1 cells with normal (1X), no (0X, TRIM37Δ) or 4-fold increased (4X) TRIM37 protein levels after treatment with DMSO or centrinone. Scale bar, 10 μm. Rates of chromosome segregation failure for the same conditions are also indicated. (b) Immunoblot shows TRIM37 in the 3 analyzed lines; α-tubulin serves as a loading control. (c) Schematic of the chromosome 17q region containing TRIM37 that is amplified in specific cancer contexts. (d) Graph shows TRIM37 protein level, measured by semi-quantitative immunoblotting, for the indicated breast cancer and neuroblastoma cell lines. (e) Passaging-based proliferation analysis for the indicated cell lines treated with DMSO (grey) or centrinone (red). (f) (left) Immunoblot of CHP134 clones in which CRISPR/Cas9-based inactivation of 1 or more of the 4 TRIM37 gene copies was used to vary TRIM37 protein levels. α-tubulin serves as a loading control. (right) Graphs plot mitotic duration, chromosome segregation failure frequency, and proliferation in centrinone as a function of TRIM37 protein level in the engineered CHP134 cell lines. Error bars in mitotic duration are 95% CI. For gel source data see Supplementary Figure 1.
TRIM37 elevation in specific cancers
The TRIM37 locus is at the border of 17q22 and 17q23, a chromosomal region amplified in a number of cancers, most prominently ~50–60% of neuroblastomas and ~10% of breast cancers5–8 (Fig. 1c). Consistent with the prevalence of 17q amplification in neuroblastomas6, TRIM37 mRNA is significantly higher in neuroblastoma, compared to other pediatric cancers (Extended Data Fig. 1g;15). As expected from the tumor expression data, cell lines derived from neuroblastomas and a subset of breast cancers also exhibited high TRIM37 expression (Extended Data Fig. 1h,i;16). To assess if elevated TRIM37 expression in cancers confers enhanced sensitivity to PLK4 inhibition, we analyzed two breast cancer (BT474 and MCF7) and four neuroblastoma (CHP134, SK-N-F1, CHP212 and IMR32) cell lines with amplification of TRIM37; four cancer cell lines that lack TRIM37 amplification – derived from neuroblastoma (KPNYN), breast cancer (BT549 and MDA-MB-231) and hepatic cancer (HepG2) – served as controls (Extended Data Fig. 1j). Immunoblotting confirmed elevation of TRIM37 protein in cell lines with TRIM37 amplification (Fig. 1d; Extended Data Fig. 2a–c). Passaging-based proliferation analysis revealed that non-amplified cancer cell lines behaved similarly to the >20 previously characterized cancer cell lines4, in that they continued to proliferate in centrinone, albeit at a reduced rate due to increased mitotic errors (Fig. 1e; Extended Data Fig. 2b; centrosome depletion was confirmed in these cell lines; Extended Data Fig. 2d4). By contrast, the six cancer cell lines with elevated TRIM37 failed to proliferate in centrinone, suggesting synthetic lethality with PLK4 inhibition (Fig. 1e).
To address the causal relationship between cancer-specific elevation of TRIM37 and sensitivity to PLK4 inhibition, we employed CHP134 neuroblastoma cells, which exhibit high sensitivity to centrinone (Extended Data Fig. 2e–g; Video S2). As CHP134 cells have 4 genes encoding TRIM37, variable targeting of the distinct gene copies enabled generation of a 6-clone “allelic series”, with TRIM37 protein levels ranging from ~10% to 70% of that in parental CHP134 cells (Fig. 1f). Live imaging revealed a striking correlation between the amount of TRIM37 and the severity of the mitotic defects following PLK4 inhibition (Fig. 1f; Extended Data Fig. 2h). As these data would predict, proliferation in centrinone was inversely correlated with TRIM37 protein levels (Fig. 1f; Extended Data Fig. 2h). Thus, in the context of a TRIM37-amplified cancer cell line, TRIM37 levels dictate sensitivity to PLK4 inhibition.
TRIM37 inhibits PLK4 condensation
We next asked why reducing TRIM37 levels improves acentrosomal mitosis whereas increasing TRIM37 levels makes it prone to fail. Surprisingly, the results indicate that the effects of decreasing versus increasing TRIM37 protein levels are mechanistically distinct. In RPE1 and CHP134 cells with reduced levels of TRIM37, PLK4 was found both at centrosomes and frequently in a single large condensate distinct from the centrosome13 (Fig. 2a,d; Extended Data Fig. 3a); condensate formation was not a consequence of increased PLK4 abundance (Fig. 2b; Extended Data Fig. 3b). Localization of 12 centrosomal proteins, including two (CEP192 and CEP152) that interact with PLK42, indicated that only PLK4 was in the ectopic condensate (Extended Data Fig. 3c,d). The PLK4-containing condensate in TRIM37Δ cells did not nucleate microtubules during interphase (not shown); however, as cells progressed into mitosis, ~25% of condensates acquired additional centrosomal components and nucleated microtubules (Extended Data Fig. 3e). These ectopic microtubule-generating centers clustered with one of the two centrosome-based spindle poles, resulting in bipolar division (not shown). In centrinone-treated TRIM37Δ cells, instead of a single large condensate, PLK4 was present in an array of smaller condensates that contained a larger subset of centrosomal components (Fig. 2c,d; Extended Data Fig. 3d;13). These small condensates functioned as robust microtubule-organizing centers in both interphase and mitosis13 (Fig. 2e, Extended Data Fig. 3f). Although why condensates in interphase centrinone-treated TRIM37Δ cells recruit multiple centrosomal components is unclear, their ability to nucleate microtubules explains why reducing TRIM37 levels improves mitosis in centrinone (Fig. 1a,b,f; Extended Data 1d,e).
Figure 2. TRIM37 prevents the formation of PLK4 condensate-based ectopic microtubule-organizing centers.
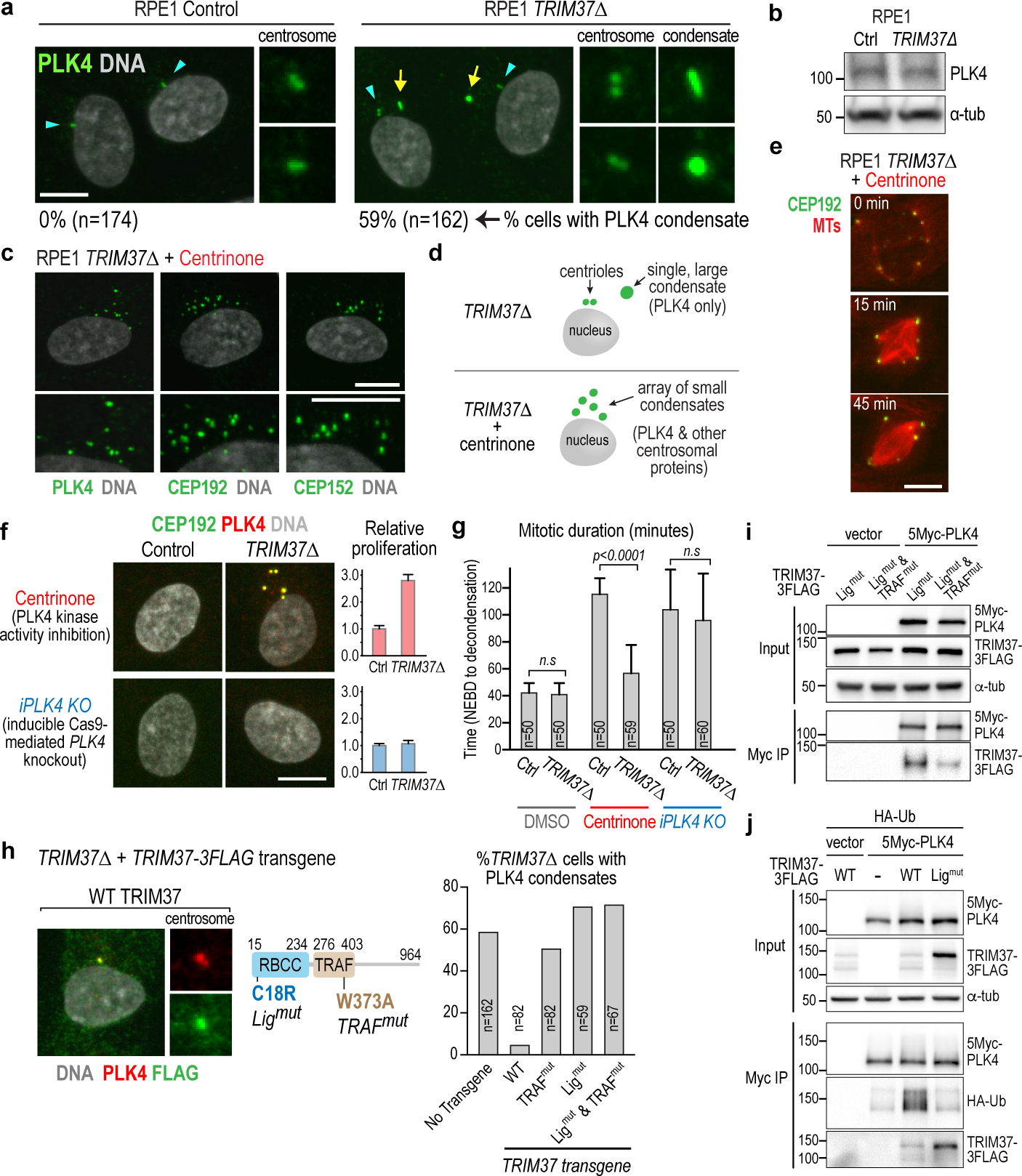
(a) Immunofluorescence images localizing PLK4 in interphase RPE1 cells. PLK4 localizes to centrioles (cyan arrowheads) and to a single ectopic condensate (yellow arrows) in TRIM37Δ cells. (b) Immunoblot showing PLK4 protein levels are not altered in TRIM37Δ cells. (c) Immunofluorescence images of TRIM37Δ cells that lack centrioles due to treatment with centrinone showing centrosome components in an array of small condensates. (d) Schematic highlighting differences between the single large condensate in TRIM37Δ cells with centrioles (top) and the small condensates in cells that lack centrioles due to treatment with centrinone (bottom). (e) Images of centrinone-treated TRIM37Δ cells with in situ mNG-tagged CEP192 and transgene-expressed red fluorescent microtubule-binding domain. Times are minutes after NEBD. (f) Control or TRIM37Δ RPE1 cells with in situ-tagged CEP192 treated with centrinone to inhibit PLK4 activity (top) or after inducibly knocking out PLK4 (bottom). Immunofluorescence (left) and plots of relative proliferation (right) show that foci formation and improved proliferation of centrinone-treated TRIM37Δ cells require PLK4 protein. Error bars are SD (n=3). (g) Mitotic duration analysis of the conditions in (f). Error bars are 95% CI. (h) (left) Images showing localization to the centrosome of FLAG-tagged WT TRIM37 expressed in TRIM37Δ RPE1 cells. (middle) schematic summarizing the ligase and TRAF domain TRIM37 mutations. (right) Graph plotting percent of cells with condensates after expression in TRIM37Δ cells of FLAG-tagged WT or mutant TRIM37 proteins. (i) Interaction analysis of TRIM37 variants with PLK4 following co-expression and PLK4 immunoprecipitation. Low expression of wild-type TRIM37 prompted use of ligase-mutant TRIM37 in this analysis. (j) TRIM37 ligase activity-dependent ubiquitination of PLK4, observed following co-expression. α-tubulin serves as a loading control for the input in (i) and (j). Scale bars are 10 μm. For gel source data see Supplementary Figure 1.
The above data suggest that TRIM37 prevents the self-assembly of PLK4 to form condensates that recruit other centrosomal components and function as ectopic microtubule-nucleating centers. This model predicts a requirement for catalytically-inhibited PLK4 to form the microtubule-generating condensates. To test this, we inducibly knocked out PLK4 in control and TRIM37Δ cells (iPLK4 KO; Extended Data Fig. 3g). Similar to centrinone treatment, induced PLK4 knockout resulted in centrosome loss. However, in the absence of PLK4, TRIM37Δ did not cause formation of ectopic assemblies containing centrosomal proteins, nor improve acentrosomal division (Fig. 2f,g; Extended Data Fig. 3h). Thus, in TRIM37Δ cells, catalytically-inhibited PLK4 scaffolds formation of condensates that improve acentrosomal division by acting as ectopic microtubule-generating centers. Under low ionic strength conditions, purified PLK4 self-assembles into spherical condensates17–19. Our observations suggest that in cells this intrinsic property of PLK4 is held in check by TRIM37.
TRIM37 ubiquitinates PLK4
TRIM37 is a TRIpartite Motif ubiquitin ligase that has been localized to peroxisomes20 but for which no centrosomal localization has been reported. In TRIM37Δ cells, epitope-tagged wildtype TRIM37 localized to centrosomes and prevented PLK4 condensate formation (Fig. 2h; Extended Data Fig. 4a,c). TRIM37 has an RBCC (RING, B-box, coiled-coil) ubiquitin ligase domain and a TRAF domain predicted to mediate a protein-protein interaction21. Point mutations engineered to disrupt ligase activity22 or TRAF domain-ligand interaction23,24 prevented suppression of condensate formation in TRIM37Δ cells (Fig. 2h; Extended Data Fig. 4a–c). Ligase-inactive TRIM37 expression was elevated (Extended Data Fig. 4a), suggesting autoregulation by ligase activity. Notably, ligase-inactive TRIM37 localized to PLK4 condensates and centrosomes, in a TRAF domain-dependent fashion (Extended Data Fig. 4c). As ligase-inactive mutants can act as substrate traps, the presence of ligase-inactive TRIM37 in condensates that contain PLK4 but no other centrosomal components suggested a direct TRIM37-PLK4 interaction. Supporting this idea, ligase-inactive TRIM37 associated with PLK4 when co-expressed in human cells, and this interaction was reduced in a TRAF domain mutant (Fig. 2i; Extended Data Fig. 4d). Co-expression of tagged ubiquitin revealed TRIM37-dependent ubiquitination of PLK4 (Fig. 2j), without apparent reduction of PLK4 levels, consistent with regulation of PLK4 self-assembly and not stability (Fig. 2a,b). While high TRIM37 expression in MCF7 cells has been reported to modulate gene expression by monoubiquitinating histone H2A22, there was no reduction in monoubiquitinated H2A in TRIM37Δ RPE1 cells (Extended Data Fig. 4e), and transcript levels of PLK4 and other centrosomal components were not altered by changes in TRIM37 expression (Extended Data Fig. 4f–h). Thus, TRIM37 acts to prevent PLK4 self-assembly rather than control its expression.
TRIM37 and acentrosomal mitotic foci
We next focused on understanding why elevated TRIM37 leads to mitotic failure in PLK4-inhibited cells (Fig. 1a,b,f; Extended Data 1d,e). As inducible PLK4 knockout and centrinone treatment exhibit similar magnitude mitotic defects (Fig. 2f,g), sensitivity to PLK4 inhibition caused by elevated TRIM37 expression cannot be due to TRIM37 limiting formation of PLK4-scaffolded ectopic microtubule-organizing centers; if it were, then a PLK4 knockout should exhibit more severe mitotic defects than centrinone treatment. To address how elevated TRIM37 expression enhances sensitivity to PLK4 inhibition, we monitored cells with an in situ-tagged fluorescent fusion of the pericentriolar material protein CEP192. While no concentration of centrosomal proteins was detected during interphase in centrinone-treated cells4, following nuclear envelope breakdown (NEBD), CEP192 and a collection of centrosomal proteins gradually coalesced to form foci positioned at the spindle poles (Fig. 3a; Extended Data Fig. 5a–c). Formation of these foci was observed with the same timing and frequency after induced PLK4 knockout (Fig. 3b,d; Extended Data Fig. 5d,f). In TRIM37-overexpressing RPE1 cells treated with centrinone, foci containing CEP192 and other centrosomal components failed to form (Fig. 3c,d; Extended Data Fig. 5e,f). In centrinone-treated CHP134 neuroblastoma cells with TRIM37 amplification, no acentrosomal mitotic foci were observed unless TRIM37 levels were reduced (Fig. 3e). Notably, presence of these acentrosomal mitotic foci correlated with bipolar spindle formation and chromosome segregation (Fig. 1a,b; Extended Data Fig. 5f). Thus, elevated TRIM37 inhibits the formation of mitotic pericentriolar material foci that occurs coincident with acentrosomal spindle assembly.
Figure 3. Acentrosomal spindle assembly and pericentriolar material coalescence is suppressed by elevated TRIM37 in PLK4-inhibited cells.
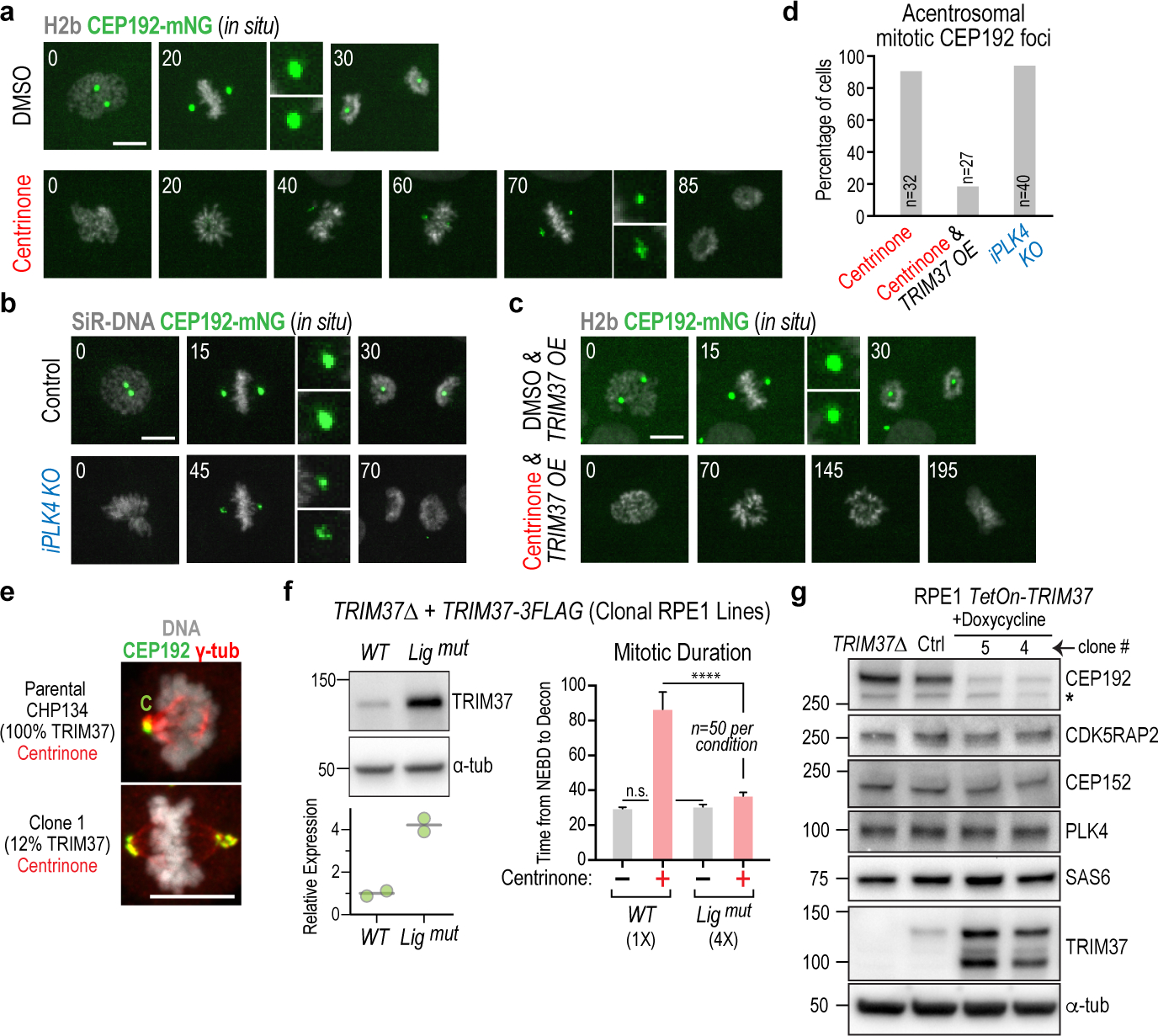
(a) Images of mitosis in DMSO or centrinone-treated RPE1 cells expressing in situ mNG-tagged CEP192. Acentrosomal spindle assembly in centrinone-treated cells is accompanied by CEP192 coalescence into foci at the spindle poles. Times are minutes after NEBD. (b) Images of mitosis in control and PLK4 knockout cells expressing in situ mNG-tagged CEP192, showing that formation of foci at the spindle poles does not require PLK4 protein. Times are minutes after NEBD. (c) Images of mitosis in TRIM37-overexpressing cells with in situ mNG-tagged CEP192 following treatment with DMSO or centrinone. Elevated TRIM37 expression suppresses coalescence of pericentriolar material components and frequently results in cells exiting mitosis without segregating their chromosomes. Times are minutes after NEBD. (d) Frequency of CEP192 coalescence to form mitotic foci for the indicated conditions. (e) Images of centrinone-treated mitotic CHP134 neuroblastoma parental cells or a clone with reduced TRIM37 expression (Fig. 1f). Acentrosomal foci are only at spindle poles following TRIM37 reduction in this TRIM37-amplified cell line. C: centrosome. (f) Ligase activity is required for elevated TRIM37 to increase sensitivity to PLK4 inhibition. (left) WT or ligase-inactive TRIM37 were expressed in TRIM37Δ RPE1 cells and clonal lines isolated; WT TRIM37 was expressed at a level comparable to endogenous TRIM37 (not shown); ligase-inactive TRIM37 was expressed at a ~4-fold higher level. (right) 4-fold overexpression of ligase-inactive TRIM37 suppresses, rather than enhances, mitotic defects in centrinone. (g) Effect of elevated TRIM37 expression on the indicated centrosomal components. CEP192 levels declined significantly whereas other tested components were not significantly affected. Asterisk marks a background band. α-tubulin serves as a loading control in (f) and (g). Scale bars are 10 μm. For gel source data see Supplementary Figure 1.
TRIM37 ligase regulates CEP192 stability
To define the molecular mechanism by which elevated TRIM37 enhances sensitivity to PLK4 inhibition, we first assessed if TRIM37 ligase activity was important. ~4-fold overexpression of ligase-dead TRIM37 failed to enhance mitotic defects following centrinone treatment (Fig. 3f), in contrast to comparable overexpression of wild type TRIM37 (Fig 1a,b). In fact, ligase-dead TRIM37 improved mitotic outcomes in centrinone because it phenocopied loss of TRIM37 in terms of promoting formation of PLK4 condensates that serve as ectopic microtubule-generating centers (Extended Data Fig. 4c). Thus, ligase activity is required for elevated TRIM37 to render mitosis sensitive to PLK4 inhibition. Immunoblotting of a panel of centrosome components in control, TRIM37Δ and TRIM37-overexpressing cells revealed that CEP192 levels were significantly decreased when TRIM37 was overexpressed, whereas levels of other analyzed components were unchanged (Fig. 3g). Notably, TRIM37 overexpression did not significantly affect the transcriptome (Extended Data Fig. 5g). The effect of TRIM37 on CEP192 protein levels was enhanced following centrinone treatment (Extended Data Fig. 5h), suggesting that centrosomes protect CEP192 from TRIM37-dependent degradation. This protection could be direct, mediated by CEP192 localization to centrosomes, or indirect, resulting from prolonged mitosis in absence of centrosomes. Centrosome-dependent protection of CEP192 likely explains why mitosis in cells with centrosomes is not affected by increased TRIM37 levels (Fig. 1a,b; Extended Data Fig. 1d,e).
As elevated TRIM37 reduces CEP192 protein levels and selectively disrupts acentrosomal mitosis, we next tested if reducing CEP192 levels by a different means also disrupted acentrosomal but not centrosomal mitosis. As CEP192 is essential, it is not possible to delete the gene encoding it and assess centrinone sensitivity. Instead, we partially inhibited CEP192 using a short-term conditional knockout that was well-tolerated in mock-treated cells (Fig. 4a; Extended Data Fig. 6a). Partial CEP192 inhibition selectively disrupted mitosis in centrinone-treated cells (Fig. 4a), analogous to TRIM37 overexpression (Fig. 1a,b; Extended Data Fig. 1d,e). A similar result was observed using an shRNA in a CHP134 clonal line with reduced TRIM37 expression (Extended Data Fig. 6b). These functional data indicate that elevated TRIM37 activity confers enhanced sensitivity to PLK4 inhibition by reducing CEP192 levels. Consistent with this model, co-expression of TRIM37 in HEK293 cells reduced CEP192 but not CEP152 levels in a ligase activity-dependent manner (Fig. 4b) and CEP192 co-immunoprecipitated with ligase-inactive TRIM37 (Fig. 4c). CEP192 is a multifunctional scaffold that binds PLK4 to control centriole duplication25–27 and to the mitotic kinases PLK1 and Aurora A to control pericentriolar material assembly28. Co-expression of CEP192 fragments with TRIM37 indicated that a ~450 amino acid C-terminal region of CEP192, distinct from previously characterized CEP192 interaction regions, bound robustly to TRIM37, was ubiquitinated in a TRIM37-dependent manner, and was required for TRIM37-dependent degradation (Fig. 4d; Extended Data Fig. 6c). These data indicate that CEP192, like PLK4, is a direct TRIM37 target. However, in contrast to PLK4, CEP192 protein levels are controlled by TRIM37 ligase activity, especially when centrosomes are absent. Thus, elevated TRIM37 confers sensitivity to PLK4 inhibition by causing a reduction in CEP192 levels.
Figure 4. Elevated TRIM37 leads to a reduction in CEP192 that confers enhanced sensitivity to PLK4 inhibition.
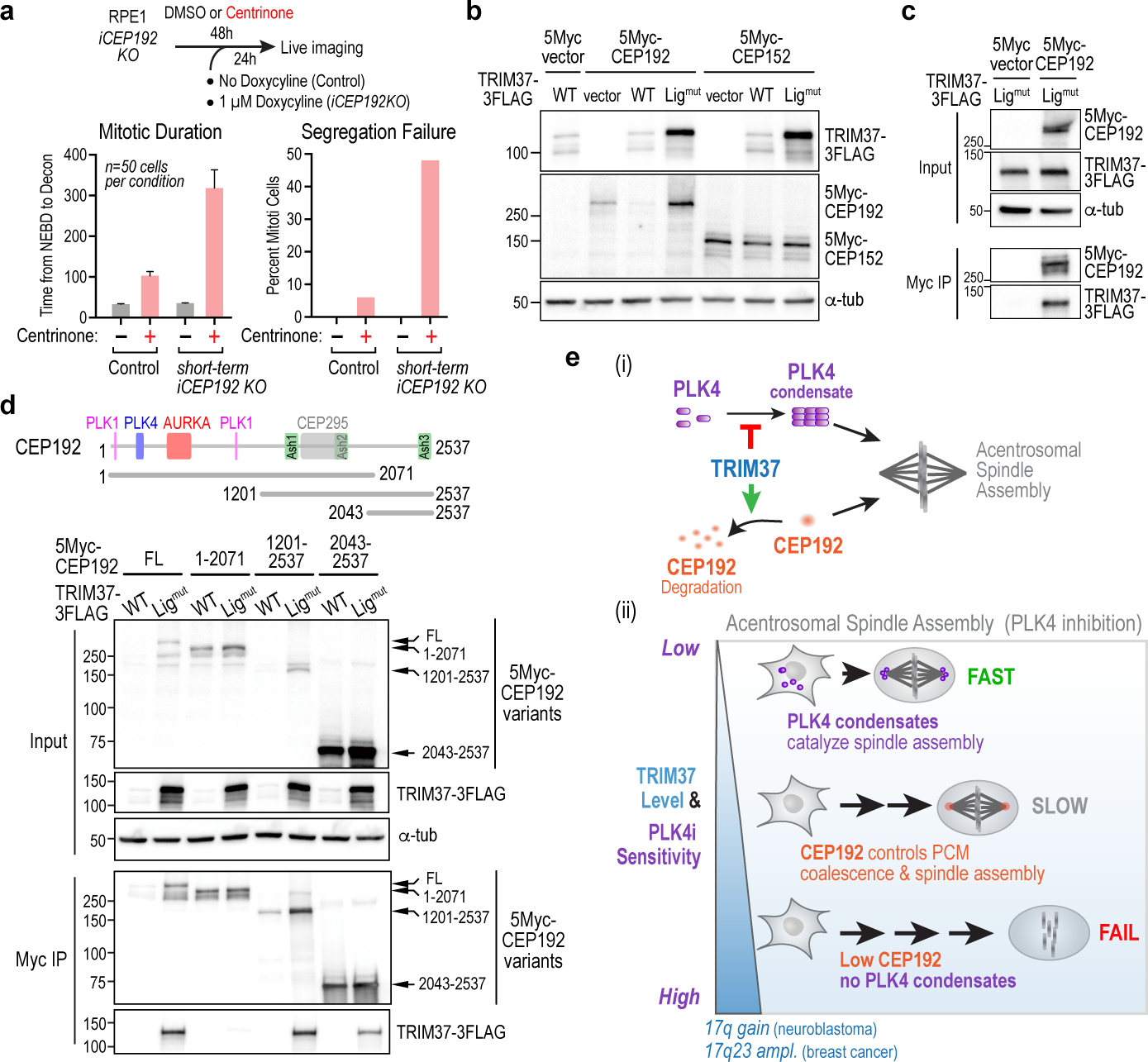
(a) (top) Approach for partial CEP192 inhibition using a short-term inducible knockout. (bottom) Graphs plot mitotic duration and percent segregation failure. Short-term inducible CEP192 knockout does not affect mitosis in DMSO-treated cells but significantly enhances mitotic defects in centrinone-treated cells. (b) Co-expression with WT or ligase-inactive TRIM37 shows that CEP192, but not CEP152, protein levels are controlled by TRIM37 ligase activity. α-tubulin serves as a loading control. (c) Interaction analysis showing that co-expressed ligase-inactive TRIM37 associates with CEP192. α-tubulin serves as an input loading control. (d) Schematic highlights key interaction sites in CEP192. Input blot shows effects of co-expressed TRIM37 (wildtype or ligase-inactive) on stability of CEP192 fragments; immunoprecipitation blot assesses association with ligase-mutant TRIM37. α-tubulin serves as an input loading control. Note that when CEP192 is unable to interact with TRIM37 due to deletion of its C- terminus (1–2071), levels are not affected by TRIM37 WT co-expression. (e) Model depicting how TRIM37 exerts bi-directional control over acentrosomal mitosis following PLK4 inhibition. (i) Two ligase activity-dependent functions of TRIM37 are to prevent PLK4 self-assembly into condensates that nucleate microtubules and to target CEP192 for degradation. (ii) When TRIM37 levels are low, PLK4 forms condensates that catalyze robust acentrosomal spindle assembly. When TRIM37 levels are normal, TRIM37 prevents PLK4 from forming condensates; after mitotic entry, foci containing pericentriolar material components coalesce concomitant with slow acentrosomal spindle assembly. When TRIM37 levels are high, CEP192 levels are reduced and there are no PLK4 condensates—consequently, acentrosomal spindle assembly fails. Amplification of the genomic region containing TRIM37 in neuroblastoma and a subset of breast cancers highlights the potential for synthetic lethality with PLK4 inhibition in specific cancer contexts. For gel source data see Supplementary Figure 1.
Xenograft sensitivity to PLK4 inhibition
While centrinone is highly selective for PLK4 and effective in culture, its pharmacokinetic profile has precluded use in tumor models (not shown). We therefore used inducible shRNA to test sensitivity of CHP134 xenografts to PLK4 inhibition. Induction of a PLK4 shRNA mimicked centrinone treatment, causing a reduction in centrosome number and rapid loss of CHP134 viability (Extended Data Fig. 7a–d). Xenograft tumors were generated using two CHP134 PLK4-shRNA clones in nude mice and the feed switched to induce shRNA expression (Extended Data Fig. 7e). PLK4 shRNA induction suppressed tumor growth for both clones (Extended Data Fig. 7f). To assess if the magnitude of the reduction of CHP134 xenograft tumor growth was influenced by the amount of TRIM37 present, we analyzed tumor formation by CHP134 parental cells and two derived clonal lines with reduced expression of TRIM37 (Fig. 1f; clones 1 & 2). Low TRIM37-expressing CHP134 clones exhibited poorer tumor growth than parental CHP134 cells (not shown); this result was reminiscent of prior work on TRIM37 as an oncoprotein in breast cancer22. As tumor growth was influenced by TRIM37 levels, it was not feasible to analyze the effect of different levels of TRIM37 expression. Nevertheless, the xenograft tumor experiments highlight the potential for PLK4 inhibition as a therapeutic strategy in neuroblastoma, and potentially also in other TRIM37-elevated cancers.
Conclusion
In conclusion, we show that the centrosomal ubiquitin ligase TRIM37 functions as a rheostat that controls cell division in the presence of chemical PLK4 inhibition (Fig. 4e). The molecular mechanisms by which low versus high TRIM37 expression influence mitosis in PLK4-inhibited cells are surprisingly distinct (Fig. 4e). Neuroblastoma and breast cancer cells with genomic amplification of TRIM37 are highly sensitive to PLK4 inhibition; an independent effort reached a similar conclusion for 17q23-amplified breast cancers29. In these, as well as in other cancer types with amplification of the TRIM37 locus, PLK4 inhibition offers a new approach for selectively triggering mitotic failure. The potential for PLK4 inhibition as a therapeutic strategy is particularly compelling in the case of neuroblastoma, which is the most common extra-cranial pediatric cancer and accounts for ~13% of pediatric cancer deaths30,31. About half of neuroblastoma is high-risk31,32 and nearly 80% of the high-risk cases have gain of 17q6. The current mortality for high-risk neuroblastoma is ~50% and survivors suffer treatment-related morbidity33. The results reported here should stimulate development of new highly selective PLK4 inhibitors with improved drug properties for testing in preclinical and clinical studies.
METHODS
Antibodies
Antibodies against CEP192 (1–211 aa; used at 0.5 μg/ml for immunofluorescence and immunoblotting), SAS-6 (501–657 aa; used at 0.5 μg/ml for immunofluorescence), and PLK4 (814–970 aa; used 1 μg/ml for immunofluorescence) were previously described4. The following antibodies were purchased from commercial sources, with their working concentrations indicated in parentheses: anti-TRIM37 (1:2,000 for immunoblotting; A301–174A; Bethyl Laboratories, Inc.); anti-PLK4 (1:500 for immunoblotting; clone 6H5, MABC544; Merck Millipore); anti-CEP152 (1:1,000 for immunofluorescence; ab183911; Abcam); anti-CEP152 (1:1,000 for immunoblotting; A302–479A-T; Bethyl Laboratories); anti-CDK5RAP2 (1:1,000 for immunofluorescence; ab86340; Abcam); anti-CDK5RAP2; (1:1,000 for immunoblotting; A300–554A-T; Bethyl Laboratories); anti–γ-tubulin (1:1,000 for immunofluorescence; GTU-88; Sigma-Aldrich); anti-pericentrin (1 μg/ml for immunofluorescence; ab4448; Abcam), anti-CPAP (1:400 for immunofluorescence; 11517–1-AP; Proteintech); anti-CCDC14 (1:100 for immunofluorescence; PA5–31759; Thermo Fisher Scientific); anti-CEP63 (1:100 for immunofluorescence; 06–1292; Merck-Millipore); anti-KIAA0753 (1:500 for immunofluorescence; HPA023494; Sigma-Aldrich); anti-PCM-1 (1:400 for immunofluorescence; #5259; Cell Signaling Technology); anti-CEP135 (1:500 for immunofluorescence; ab75005; Abcam); anti-α-tubulin (1:5000 for immunoblotting; DM1A; Sigma-Aldrich); anti-FLAG (1:1,000 for immunoblotting; F1804; Sigma-Aldrich); anti-Myc (1:5,000 for immunoblotting; monoclonal antibody 9E10, M4439; Sigma-Aldrich); anti-HA (1:500 for immunoblotting; monoclonal antibody 16B12; BioLegend). Secondary antibodies were purchased from Jackson ImmunoResearch and GE Healthcare.
Cell lines
All cell lines used in this study are described in Extended Data Table 1. RPE1 (hTERT RPE-1), CHP212, IMR32, SK-N-F1, BT474, BT549, MCF7, MDA-MB-231, and HepG2 were obtained from the American Type Culture Collection (ATCC). The CHP134 cell line was obtained from Sigma-Aldrich (ECACC general collection). KPNYN was a gift from Peter Zage (UC San Diego). Cell lines obtained from ATCC and Sigma were cultured as recommended. Each growth medium was supplemented with 100 IU/ml penicillin and 100 μg/ml streptomycin. All cell lines except FreeStyle 293-F cells were maintained at 37°C and 5% CO2. FreeStyle 293-F cells were maintained at 37°C and 8% CO2 in air on an orbital shaker platform rotating at 125 rpm. To inhibit PLK4 and deplete centrosomes, cells were treated with centrinone for the indicated amount of time4. Centrinone was diluted from a 1 mM stock; all treatments were at a final concentration of 150 nM centrinone and 0.015 % DMSO, control treatments were 0.15% DMSO.
The RPE1 TRIM37Δ cell line has been described13. TRIM37Δ and USP28Δ knockout in the RPE1 CEP192-mNeonGreen background were generated as described13. In brief, double-stranded oligonucleotides for specific guide RNAs targeting USP28 (TGAGCGTTTAGTTTCTGCAG) or TRIM37 (CTCCCCAAAGTGCACACTGA) were cloned into PX459 [a gift from Feng Zhang (Addgene plasmid # 48139; http://n2t.net/addgene:48139; RRID:Addgene_48139)34. RPE1 cells were plated in 10 cm plates at 500,000 cells/plate the day before transfection. Cells were transfected with plasmid using Lipofectamine 3000 according to the manufacturer’s instructions (ThermoFisher). Two days after transfection, 100 nM centrinone was added. After 10 days, centrinone-resistant RPE1 cells were plated at a density that supported direct picking of clones from 10 cm plates. One week after re-plating, multiple colonies were observed at a density that supported direct picking of clones. Gene knockout was determined by genotyping of the sequence surrounding the CRISPR cut site35 and/or by immunoblotting.
CHP134 cell lines with variable amount of TRIM37 expression were generated using CRISPR/Cas9. CHP134 cells were plated in 6 well dishes at 200,000 cells/well the day before transfection. On the day of transfection, a synthetic crRNA targeting exon 5 of TRIM37 (CTCCCCAAAGTGCACACTGA) was hybridized with synthetic tracrRNA, assembled with Cas9 protein and synthetic tracrRNA into RNPs, and then transfected into CHP134 using Lipofectamine RNAiMAX according to the manufacturer’s instructions (Thermo Fisher Scientific). To obtain clonal lines, single cells were plated into 96-well plates and expanded. Targeting of TRIM37 was determined by genotyping of the region surrounding the CRISPR cut site35 and immunoblotting.
RPE1 CEP192-mNeonGreen cells were generated using CRISPR/Cas9 in combination with rAAV-mediated delivery of the repair construct as previously described36. The gRNA was designed to cut close to the stop codon of CEP192 (CGACTAATTGGTGAAGCTCT) and cloned into PX45934. The repair construct was cloned into pSEPT and contains the left and right flanking region of the gRNA target site (960 bp and 672 bp); the mNeonGreen coding sequence, for C-terminal fusion to CEP192, and the neomycin resistance gene aminoglycoside phosphotransferase from Tn5 was cloned between the left and right homology arms. The expression of the neomycin resistance gene is linked to endogenous CEP192-NeonGreen expression through a P2A sequence.
The following transgenes were stably integrated into the genome using lentiviral constructs (see Extended Data Table 2): H2B-mRFP (EF1alpha promoter); mRuby2-MAP4-MBD (EF1alpha promoter; neomycin resistance gene); TRIM37-C18R-mNeonGreen (hPGK promoter; blasticidin resistance gene) and TRIM37–3FLAG (WT, C18R, W373A and C18R W373A; UbC promoter; neomycin resistance gene). Cell lines with inducible overexpression of TRIM37 were generated by sequential lentiviral integration of TetOn3G (inducible transactivator protein; hPGK promoter) and TRIM37 (doxycycline inducible TRE3GS promoter). Cell lines for inducible knockout of PLK4 or CEP192 were generated by sequential lentiviral integration of Cas9 (Edit-R Inducible Lentiviral Cas9; Dharmacon) and a PLK4 or CEP192 gRNA expressing plasmid, which is based on the lentiGuide-Puro plasmid37. The PLK4 gRNA (TCATATTACGAGTCAGTAGG) targets exon 5 in the kinase domain coding region. The CEP192 gRNA (AGGGAGTGTCCGAGTGCCCG) targets exon 19. The lentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid # 52963; http://n2t.net/addgene:52963; RRID:Addgene_52963). TRIM37 and Cas9 expression was induced with 1 μg/ml doxycycline. Viral particles were generated by transfecting the lentiviral construct into HEK-293T cells using Lenti-X Packaging Single Shots (Clontech). 48 hours after transfection, virus-containing culture supernatant was harvested and added to the growth medium of cells in combination with 2.5–8 μg/ml polybrene (EMD Millipore). Populations of each cell line were selected by FACS or antibiotics (Blasticidin, 5 μg/ml; Neomycin, 400 μg/ml; Puromycin, 10 μg/ml for RPE1). Single clones were isolated in 96 well plates. The lentiviral vector expressing CEP192 shRNA was purchased from Dharmacon (GAGGCATCAGTTAATACTGAT). The PLK4-shRNA (sequence: CAGTATAAGTGGTAGTTTA) was expressed from an integrated lentiviral vector and expressed from a doxycycline inducible promoter (construct name: V3SH11252–225330936 piSMART hEF1a/TurboGFP inducible). Single clones were selected for tumor xenograft experiments.
Immunofluorescence and quantification of centrosomal signals
For immunofluorescence, 10,000 cells per well were seeded into 96-well plates one day before fixation. Cells were fixed in 100 μl ice-cold methanol for 7 minutes at −20 °C. Cells were washed twice with washing buffer (PBS containing 0.1% Triton X-100) and blocked with blocking buffer (PBS containing 2% BSA, 0.1% Triton X-100 and 0.1% NaN3) overnight. After blocking, cells were incubated for 1–2 hours with primary antibody in fresh blocking buffer (concentrations as indicated above). Cells were washed three times with washing buffer, prior to 1-hour incubation with the secondary antibody and DNA staining Hoechst 33342 dye. Finally, cells were washed three times with washing buffer prior to inspection. Images were acquired on a CV7000 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40X (0.95 NA) or a 60X (water, 1.2 NA) U-PlanApo objective and a 2560×2160 pixel sCMOS camera (Andor). Image acquisition was performed using CV7000 software.
Live cell imaging
Live cell imaging was performed on the CQ1 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40X 0.95 NA U-PlanApo objective and a 2560×2160 pixel sCMOS camera (Andor) at 37°C and 5% CO2. Image acquisition and data analysis were performed using CQ1 software and ImageJ, respectively.
Cells were seeded into 96-well polystyrene plates at 6,000 – 10,000 cells/well, 24 hours before imaging unless indicated otherwise. Imaging conditions varied according to the experimental setup. For imaging H2B-RFP or SiR-DNA 5 × 2 μm z-sections in the RFP or FarRed channel (25% power, 150 ms) were acquired in each field at 4- or 5-minute intervals for 6–24 hours. The DNA marker SiR-DNA was added 2 h prior to imaging at a working concentration of 0.5 μM. For imaging CEP192-NeonGreen and/or mRuby-MAP4-MBP 8 × 1.2 μm z-sections in the GFP and/or RFP channel (50% power, 150 ms) were acquired in each field at 4- to 15-minute intervals for 6–12 hours. DMSO or centrinone treatment was conducted for 3 cell cycles prior to start of imaging, unless noted otherwise; cell cycle duration for RPE1 and neuroblastoma cell lines was measured by live imaging of each cell line and quantifying time from NEBD of a mother cell to NEBD of its daughters.
Proliferation and viability assays
For the passaging assays, cells were seeded into 6 well plates in triplicate at 25,000 cells/well and treated with 150 nM centrinone or DMSO. At 96-hour or 192-hour intervals, cells were harvested, counted and re-plated at 25,000 cells/well. Cell counting was performed using a TC20 automated cell counter (Bio-Rad).
For ATPlite viability assays, 550–750 cells were seeded into 96 well culture plates (Corning 3603) in 180 μl and incubated overnight at 37°C and 5% CO2. The following day 20 μl media containing 1.25 μM centrinone (or equal volume with DMSO as control) was added to each well to obtain a final concentration of 125 nM. After 5 days of incubation at 37°C and 5% CO2, 100 μl ATPlite (Perkin Elmer) was added prior to luminescence measurement with a Tecan Infinite M1000 Pro Multilabel Microplate Reader.
Immunoblotting
For immunoblotting, cells were cultured in 10 cm plates, harvested at 50–80% confluence and lysed by sonication in RIPA buffer + protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Cell extracts were stored at −80°C until use. Before use, extract concentrations were normalized based on a Protein Assay (Bio-Rad). For every sample, 20–30 μg protein/lane was run on Mini-PROTEAN gels (Bio-Rad), and transferred to PVDF membranes using a TransBlot Turbo system (Bio-Rad). Blocking and antibody incubations were performed in TBS-T + 5% nonfat dry milk. Detection was performed using HRP-conjugated secondary antibodies (GE Healthcare) with WesternBright Sirius (Advansta) or SuperSignal West Femto (Thermo Fisher Scientific) substrates. Membranes were imaged on a ChemiDoc MP system (Bio-Rad).
Protein expression in FreeStyle 293-F cells and immunoprecipitation
For co-expression and immunoprecipitation assays, FLAG-tagged TRIM37 and Myc-tagged PLK4, CEP192, or CEP152 (see Extended Data Table 2) were expressed in different combinations in FreeStyle 293-F cells (Thermo Fisher Scientific). cDNA constructs for CEP192 and CEP152 transient expression25 were gifts from Kyung S. Lee (National Cancer Institute). The empty 5Myc plasmid, which was used as negative control, is a derivative of CS2P (Addgene #17095) and is designed for expression of C-terminally Myc-tagged proteins. Cell transfection was performed using FreeStyle MAX Reagent and OptiPRO SFM according to the manufacturer’s guidelines (Thermo Fisher Scientific). 20 ml of cells at 1 × 106 cells/ml were transfected with a total of 25 μg DNA constructs. 43–48 hours after transfection, cells were harvested and washed with PBS. The cells were resuspended in lysis buffer (20 mM Tris/HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM EGTA, 1 mM dithiothreitol, 2 mM MgCl2 and EDTA-free protease inhibitor cocktail (Roche)) and lysed in an ice-cold sonicating water bath for 5 minutes. After 15-minute centrifugation at 15,000 x g and 4 °C, whole cell lysates were incubated with Pierce Anti-c-Myc magnetic beads (Thermo Fisher Scientific) for 2 hours at 4 °C. The beads were washed five times with lysis buffer and resuspended in SDS sample buffer. For immunoblotting, equal volumes of samples were run on Mini-PROTEAN gels (Bio-Rad) and transferred to PVDF membranes using a TransBlot Turbo system (Bio-Rad). Blocking and antibody incubations were performed in TBS-T plus 5% nonfat dry milk or in TBS-T plus 5% BSA. Immunoblotting was performed as described above.
Detection of protein ubiquitination
To detect ubiquitination of PLK4 and CEP192 by TRIM37, DNA constructs of Myc-tagged PLK4 or CEP192 along with HA-tagged ubiquitin and FLAG-tagged TRIM37 were expressed in FreeStyle 293-F cells for 48 hours. HA-ubiquitin was a gift from Edward Yeh (Addgene plasmid # 18712; http://n2t.net/addgene:18712 ; RRID:Addgene_18712)38. The cells were lysed in 20 mM Tris/HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM EGTA, 1 mM dithiothreitol, 2 mM MgCl2, EDTA-free protease inhibitor cocktail (Roche) and 5 mM N-ethylmaleimide. Immunoprecipitation and immunoblotting were performed as described above.
Statistical analysis
p-values are from t-tests conducted using Prism v8 (GraphPad). In Fig. 2g, 3f, Extended data Figs. 1d, 1g, 2g, 4e and 6b unpaired t-tests assuming equal standard deviation were performed. For Extended Data Fig. 7f, unpaired t-tests with Welch’s correction, which does not assume equal standard deviations, were performed at each time point; only the significantly different (p<0.05) timepoints are marked. p-values are labeled as follows: p>0.05 (n.s), p<0.05 (*), p<0.01 (**), p<0.001(***) and p<0.0001 (****).
RNA-Seq Analysis
RNA was purified from three independent samples for each analyzed cell line using RNeasy Plus Mini Kit. RNA library synthesis and sequencing were performed by the Genomics Center of the Institute for Genomic Medicine at UC San Diego. Samples were sequenced on a HiSeq4000 platform (SR75). The sequencing data was aligned to the human genome UCSC hg19 with STAR aligner39. Differential expression of genes was determined using DESeq240. Genes with low counts (<10 reads average for the triplicates) or outliers (highly skewed value in one of the triplicates) were excluded from the analysis. Results were visualized using Prism v8 and InteractiVenn41.
CHP134 xenograft tumor analysis
Tumor xenografts were initiated by inoculation of CHP134 human neuroblastoma cells stably transduced with an inducible PLK4 shRNA. Two independent clones (ODCL108 & ODCL109) were used to generate xenograft tumors in 6-week old BALB/c nude female mice. Cells (1 × 107/animal) were suspended in 1:1 phosphate-buffered saline:Matrigel (Corning); 100 μL volume of the cell suspension was injected into the subcutaneous right flank. For the inducible PLK4 shRNA, ODCL108 animals were randomized (2 groups with 9 mice each) and switched to irradiated control or doxycycline-containing (625 mg/kg) feed when tumors reached an average of about 150 mm3; ODCL109 animals exhibited slower tumor growth and were randomized (2 groups with 9 mice each) and switched when tumors reached an average of about 100 mm3. Tumor size was calculated by standard caliper measurement, using volume = (width2 × length)/2. All procedures related to animal handling, care and treatment followed guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of BioDuro, San Diego, following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). BioDuro’s limit on conventional mouse xenograft tumor size is 2000 mm3. One P7 clone control animal exceeded this limit on the final study day prior to all P7 animals being sacrificed.
Extended Data
Extended Data Figure 1. Effect of varying TRIM37 levels on sensitivity to PLK4 inhibition and TRIM37 expression profile in patient-derived tumors and cancer cell lines.
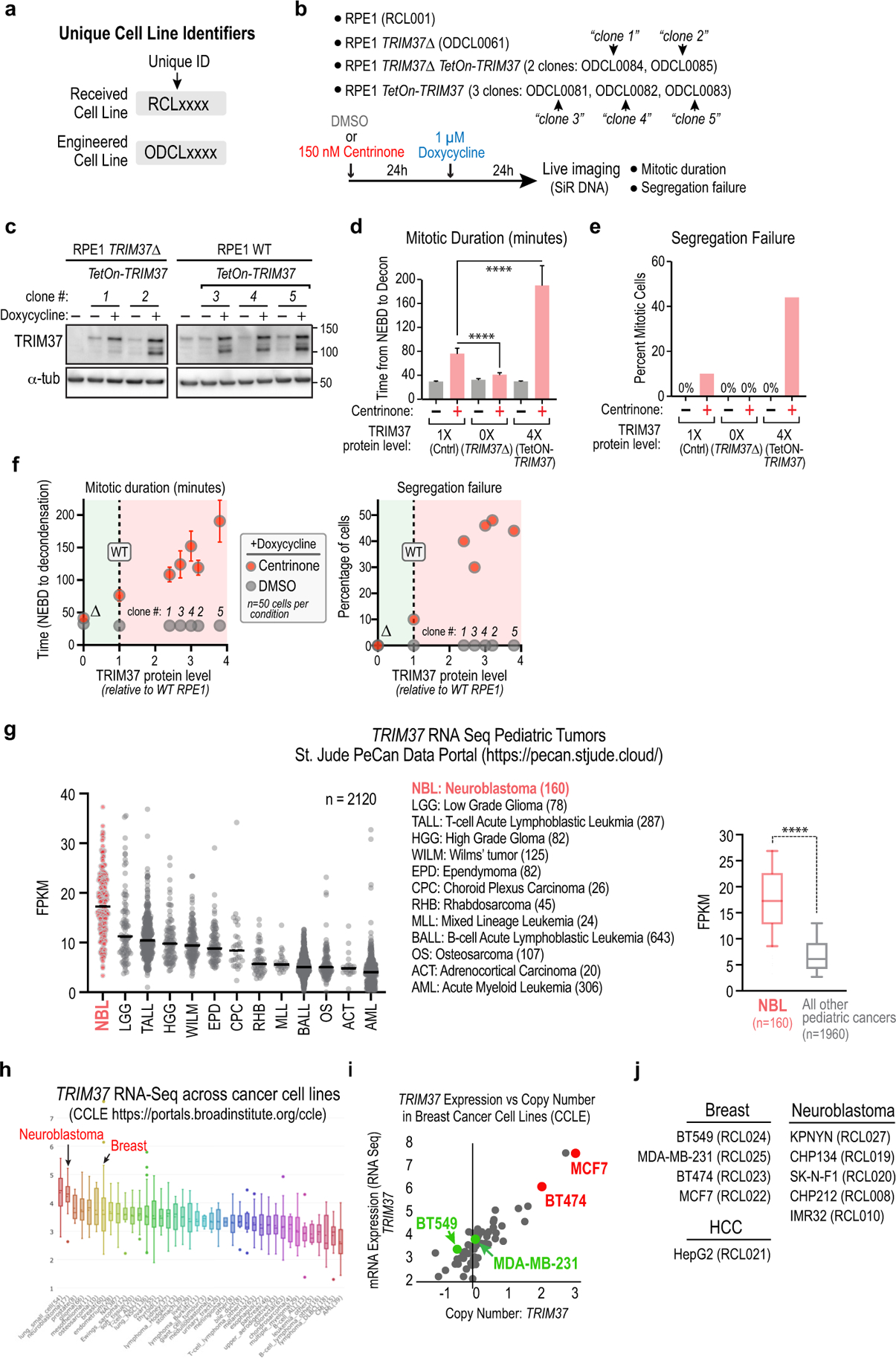
(a) Unique cell line identifiers are used to describe the cell lines in each experiment. The RCL prefix refers to cell lines received from an external source, such as the American Type Culture Collection (ATCC). The ODCL prefix refers to cell lines engineered from received cell lines. (b) Cell line code and experimental protocol for the analysis of mitotic duration and chromosome segregation failure. Clone 5 is the Tet-ON TRIM37 cell line shown in Fig. 1a,b, which overexpresses TRIM37 ~4-fold relative to parental RPE1 cells. (c) Immunoblots of the RPE1 cell lines described in (b); transgene-encoded TetON-TRIM37 expression was induced for 24h. (d,e) Graphs plotting mitotic duration (d) and the frequency of chromosome segregation failure (e) following treatment with DMSO (−) versus centrinone (+) for the 3 analyzed cell lines shown in Fig. 1a,b; n= 50 for each condition. Error bars are 95% CI. (f) Live imaging-based analysis was used to measure mitotic duration and segregation failure for the cell lines described in (b); values are plotted versus TRIM37 protein level measured by semi-quantitative western blotting. Each cell line was treated with DMSO (grey) or centrinone (red) and doxycycline before live imaging – experimental scheme is shown in panel (b). 50 cells were analyzed per condition. Error bars are 95% CI. In DMSO, the analyzed cell lines exhibited normal mitotic duration and segregation fidelity regardless of TRIM37 protein level. By contrast, in centrinone, loss of TRIM37 reduced mitotic duration and segregation failure percentage (green shading) whereas increased TRIM37 protein levels led to a proportional increase in mitotic duration and segregation failure rate (red shading). (g) (left) Graph plotting TRIM37 mRNA level in 2120 pediatric tumors, representing 13 different cancer types (data is from the St. Jude PeCan Data Portal15). All pediatric cancer types with >10 tumors analyzed are shown. Values for individual tumors (dots) and the median value (black lines) are plotted. The 3-letter codes to the right of the graph describe the 13 pediatric cancer types. Neuroblastoma tumors exhibit the highest TRIM37 expression. (right) Box-and-whiskers plot, comparing TRIM37 mRNA levels in neuroblastoma tumors to that all other pediatric cancer type tumors. The range represents 10–90th percentile of the data; the p-value shown is from an unpaired t-test. (h) Graph plotting TRIM37 mRNA levels across cancer cell lines described in the Cancer Cell Line Encyclopedia (CCLE16; https://portals.broadinstitute.org/ccle). (i) mRNA expression versus copy number from CCLE data for breast cancer cell lines. Two cell lines with high TRIM37 copy number and expression (MCF7 and BT474; red), as well as two cell lines with normal copy number and expression (MDA-MB-231 and BT549; green) are marked. (j) List of breast cancer and neuroblastoma cell lines used for analysis in Fig. 1d–e. HepG2 is a hepatocellular carcinoma-derived cell line with similar TRIM37 expression to control RPE1 cells. For gel source data see Supplementary Figure 1.
Extended Data Figure 2. Analysis of TRIM37 protein levels and centrinone efficacy in different cancer cell lines and comparison of mitosis in RPE1 and CHP134 neuroblastoma cells following centrinone treatment.
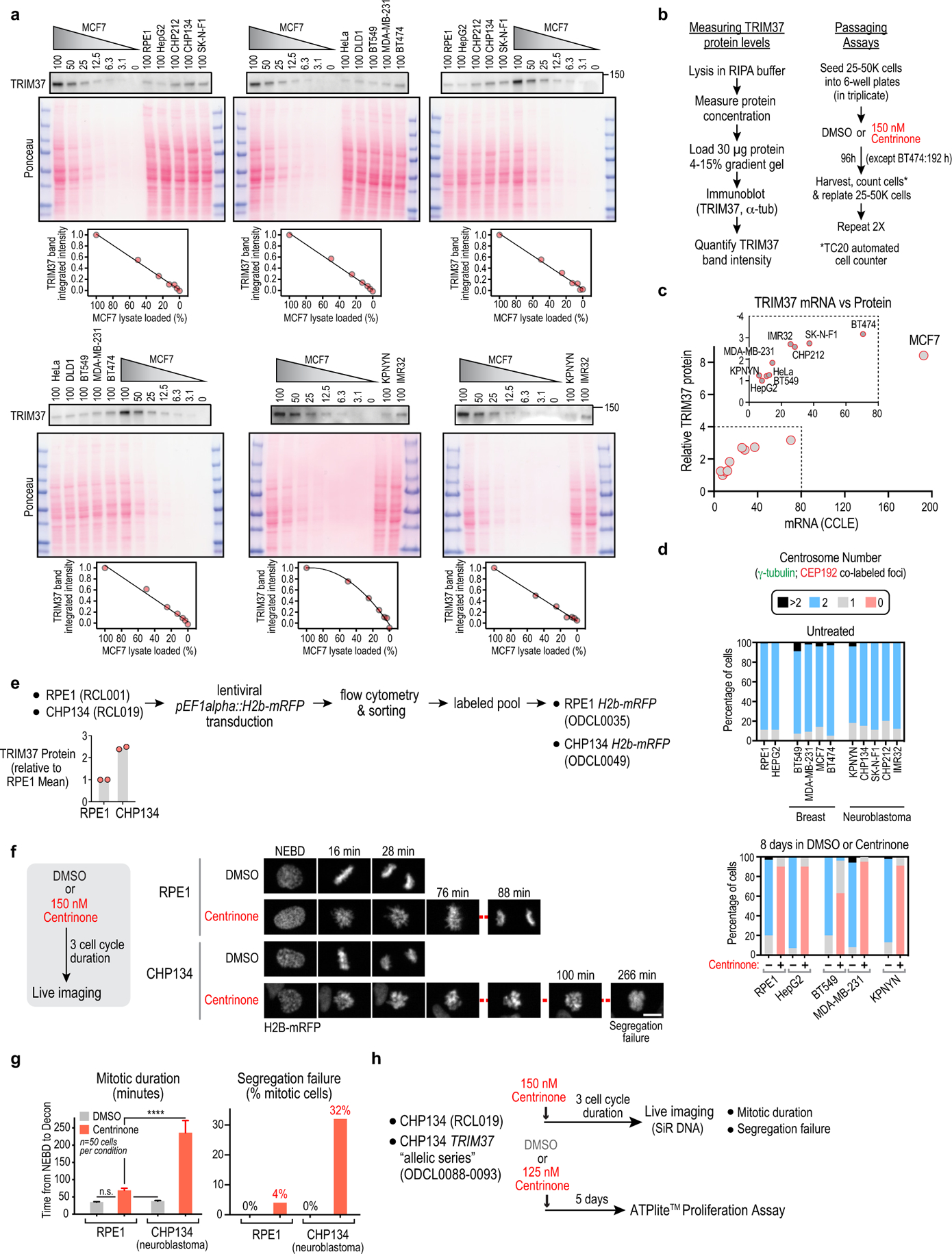
(a) Immunoblots used to quantify TRIM37 protein level across different cell lines. TRIM37 immunoblots are shown above the corresponding Ponceau-stained blots. MCF7 cells have the highest TRIM37 transcript levels and copy number in the CCLE (Extended Data Fig. 1i). Serial dilutions of MCF7 extract were loaded next to extracts from other cell lines on each blot and TRIM37 band intensities across a serial dilution of MCF7 cell extract were used to generate a standard curve (graphs below each blot); measured intensities for other cell line extracts were converted into relative expression values using the standard curve. TRIM37 protein level in HepG2 cells was set to 1 and values measured for other cell lines were plotted relative to the HepG2 level in Fig 1d. (b) Schematics of protocols used to measure TRIM37 protein levels (left) and conduct passaging-based proliferation analysis of cancer cell lines (right). (c) Comparison of TRIM37 mRNA and protein levels across cancer cell lines. mRNA levels are from the CCLE and were transformed from a logarithmic (base 2) to a linear scale. Protein levels are the mean value of two measurements conducted as in (a) and are plotted relative to the amount of TRIM37 in HepG2, a non-amplified cancer cell line. Inset graph excludes MCF7, which shows exceptionally high TRIM37 mRNA and protein levels. (d) Quantification of centrosome number in the indicated cell lines and conditions (n = 100 for each condition). Centrosomes were defined as co-localized foci of CEP192 and γ-tubulin in fixed interphase cells. In the absence of any treatments, there is mild centrosome amplification in the breast cancer cell lines and in one neuroblastoma cell line. Following 8-day centrinone treatment, a significant proportion of the cells from cell lines with relatively low sensitivity to centrinone lacked centrosomes. (e) Schematic of method used to generate a pool of cells expressing H2b-mRFP for each of the indicated cell lines. TRIM37 protein level relative to RPE1, measured by semi-quantitative immunoblotting. (f) Images are stills from timelapse sequences of H2b-RFP expressing mitotic RPE1 and CHP134 cells. Both cell lines exhibit rapid mitosis (~30 minutes) with no segregation failure in DMSO. Following centrinone treatment, CHP134 cells exhibited significantly more delayed mitosis and higher rates of segregation failure compared to RPE1 cells. Scale bar is 10 μm. (g) Quantification of mitotic duration and segregation failure comparing RPE1 and CHP134 cells. (h) Schematic of protocols used to analyze mitotic duration, segregation failure and viability of the CHP134-derived cell line panel with different levels of TRIM37 protein.
Extended Data Figure 3. Analysis of TRIM37Δ cells, rescue with TRIM37 variants and generation of inducible PLK4 knockout.
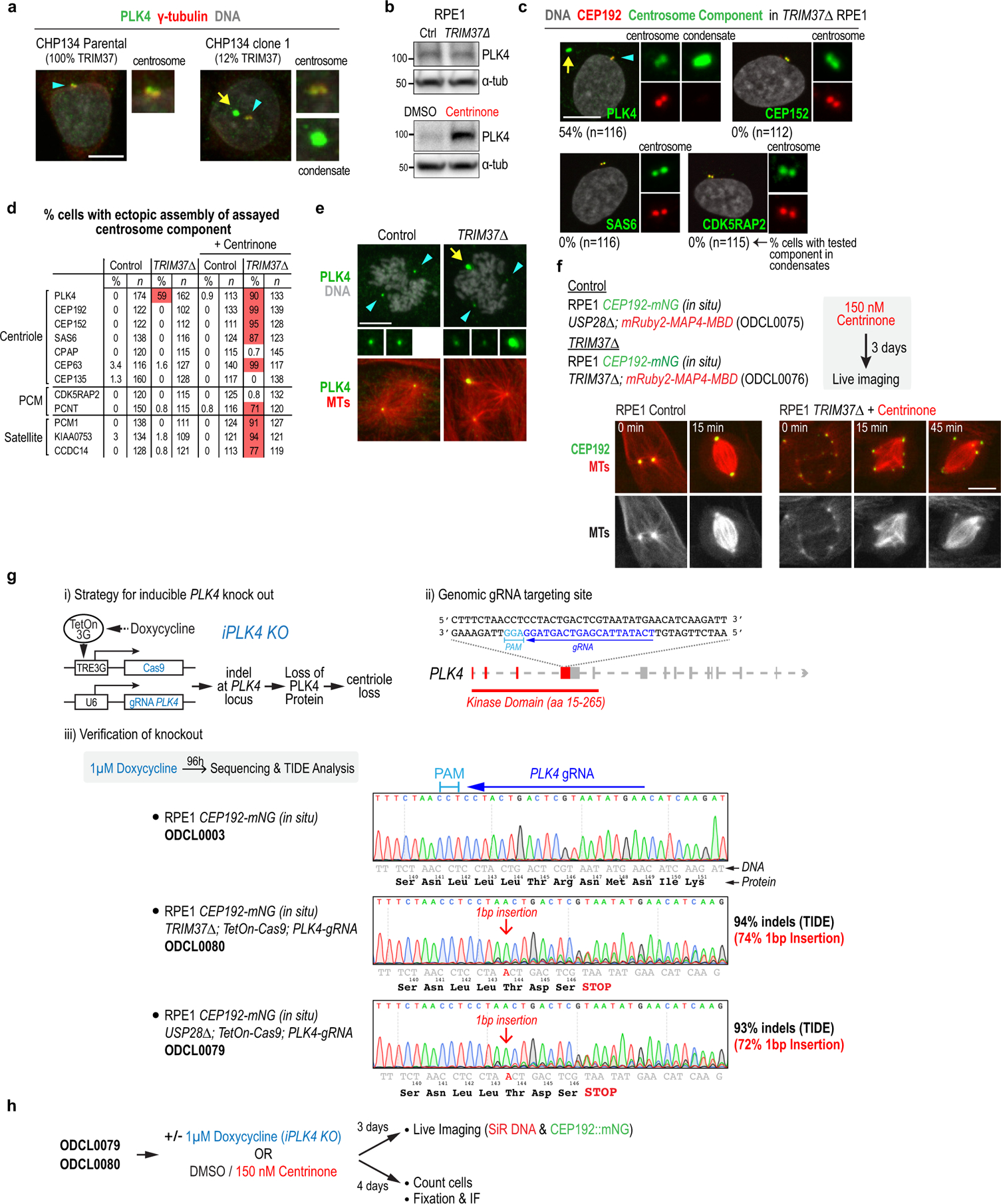
(a) PLK4 condensate formation in CHP134 neuroblastoma cells with reduced TRIM37 expression. Parental CHP134 cells, which have 4 copies of the TRIM37 gene, were compared to clone 1 (Fig. 1f; ~12% TRIM37 expression relative to parental CHP134). PLK4 condensates were observed in 23% of the clone 1 cells with reduced TRIM37 expression but in none of the parental cells (n=100 for each). (b) Immunoblots in RPE1 cells comparing the effect on PLK4 protein levels of TRIM37 deletion (top) versus inhibition of PLK4 kinase activity using centrinone (bottom). PLK4 protein levels were elevated ~ 7-fold by following kinase activity inhibition (7.4 ± 1.1 mean±SD, n=3) confirming that the detected band corresponds to PLK4. The TRIM37Δ blot is the same as in Fig. 2b. (c) Immunofluorescence images of the indicated centrosomal components in TRIM37Δ cells. Scale bar, 10 μm. (d) Summary of immunofluorescence analysis in interphase cells. (e) Immunofluorescence image showing microtubule organization by a PLK4 condensate in a mitotic TRIM37Δ cell. Scale bar, 10 μm. (f) (top) Schematic of the protocol used to conduct live imaging of CEP192 and microtubules. (bottom) Images of control and centrinone-treated TRIM37Δ cells with in situ mNG-tagged CEP192 and a transgene that expresses a red fluorescent microtubule-binding domain. Times in minutes after NEBD are noted on each panel. Scale bar, 10 μm. The merged TRIM37Δ images are the same as those shown in Fig. 2e. (g) Description and validation of inducible PLK4 knockout engineered in TRIM37Δ and control (USP28Δ) cells. USP28Δ cells were used as the control because inactivation of USP28 prevents p53 activation and G1 arrest that is observed as a consequence of delayed mitosis following centrosome loss in RPE1 cells13. Note that USP28Δ has no effect on the mitotic consequences of centrosome loss13 and enables comparison with TRIM37Δ cells, which prevent p53 activation following centrinone treatment by accelerating mitosis in the absence of centrosomes. The gRNA sequence used to target PLK4 exon 5 is depicted and the efficacy of the inducible knockout in both cell lines was validated by inducing Cas9 expression using doxycycline for 4 days, followed by sequencing and TIDE analysis35. Sequence traces show high frequency of indels, with a 1 bp insertion being the most common outcome. (h) Schematic of protocol used to compare centrinone treatment to inducible PLK4 knockout in Fig. 2f,g. For gel source data see Supplementary Figure 1.
Extended Data Figure 4. Generation of TRIM37 variants and analysis of the effect of TRIM37 loss on H2a ubiquitination and transcription.
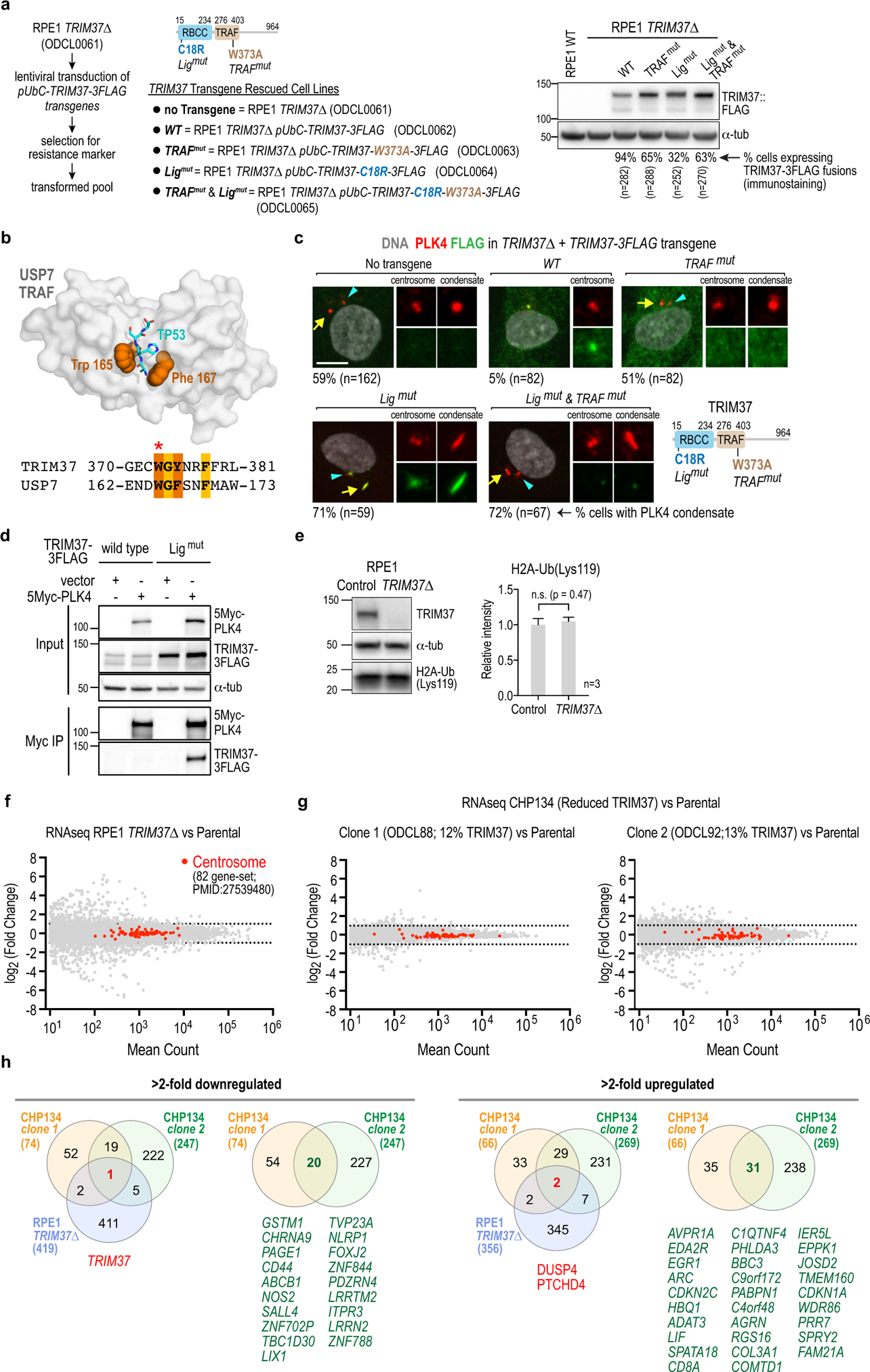
(a) (left) Method used to generate cell lines for testing rescue with transgenes encoding wildtype and mutant variants of TRIM37. Schematic of TRIM37 shows the point mutants engineered in the ligase and TRAF domains. (right) Blot shows expression of transgene-encoded TRIM37 variants in the pools selected for marker resistance; percentage of cells expressing the indicated fusions is shown below the blot. (b) Structural homology-based strategy used to engineer the TRIM37 TRAF domain mutant. Diagram illustrates the USP7 TRAF domain (gray surface) bound to a p53 peptide (cyan stick) with key binding residues W165 and F167 in orange spheres (PDB:3MQR). Sequences below show similarity between TRIM37 and USP7 in the peptide binding pocket; the conserved tryptophan (W165 in USP7; W373 in TRIM37) was mutated to alanine to generate the TRIM37 TRAF mutant. (c) Images illustrating the effect of expressing WT TRIM37 or engineered variants disrupting ligase activity or TRAF domain interactions in TRIM37Δ cells. (d) Interaction analysis employing co-expression of TRIM37 and PLK4 followed by PLK4 immunoprecipitation. Wild-type (WT) TRIM37 is expressed at significantly lower levels than ligase-mutant (C18R) TRIM37, suggesting that TRIM37 autoregulates its own stability. The low expression of WT TRIM37 led us to use ligase-mutant TRIM37 for the interaction analysis shown in Fig. 2i. (e) (left) Immunoblot of H2A-Ub(Lys119), comparing control and TRIM37Δ RPE1 cells. α-tubulin serves as a loading control. (right) Quantification of band intensities indicates that TRIM37 does not reduce ubiquitination of Lys119 in histone H2A. (f) RNA-Seq analysis comparing parental and TRIM37Δ RPE1 cells. A previously defined set of 82 genes encoding centrosomal components43 is marked in red to highlight lack of change in their mRNA levels. (g) RNA-Seq analysis comparing parental CHP134 cells to two clones with significantly lower expression of TRIM37. The two clones shown are clone 1 and clone 2 in Fig. 1f. The centrosome 82-gene set is highlighted in red. (h) Lists of >2-fold downregulated or upregulated genes. Each of the 3 test lines (RPE1 TRIM37Δ, CHP134 clone 1, CHP134 clone 2) was compared to the parental line in order to identify genes with statistically significant >2-fold changes. Cross-comparison of all 3 test lines and of the 2 CHP134 clones is summarized in the Venn diagrams; gene names for shared differentially expressed genes are shown below each Venn diagram. For gel source data see Supplementary Figure 1.
Extended Data Figure 5. Effect of TRIM37 overexpression on coalescence of pericentriolar material and acentrosomal mitosis.
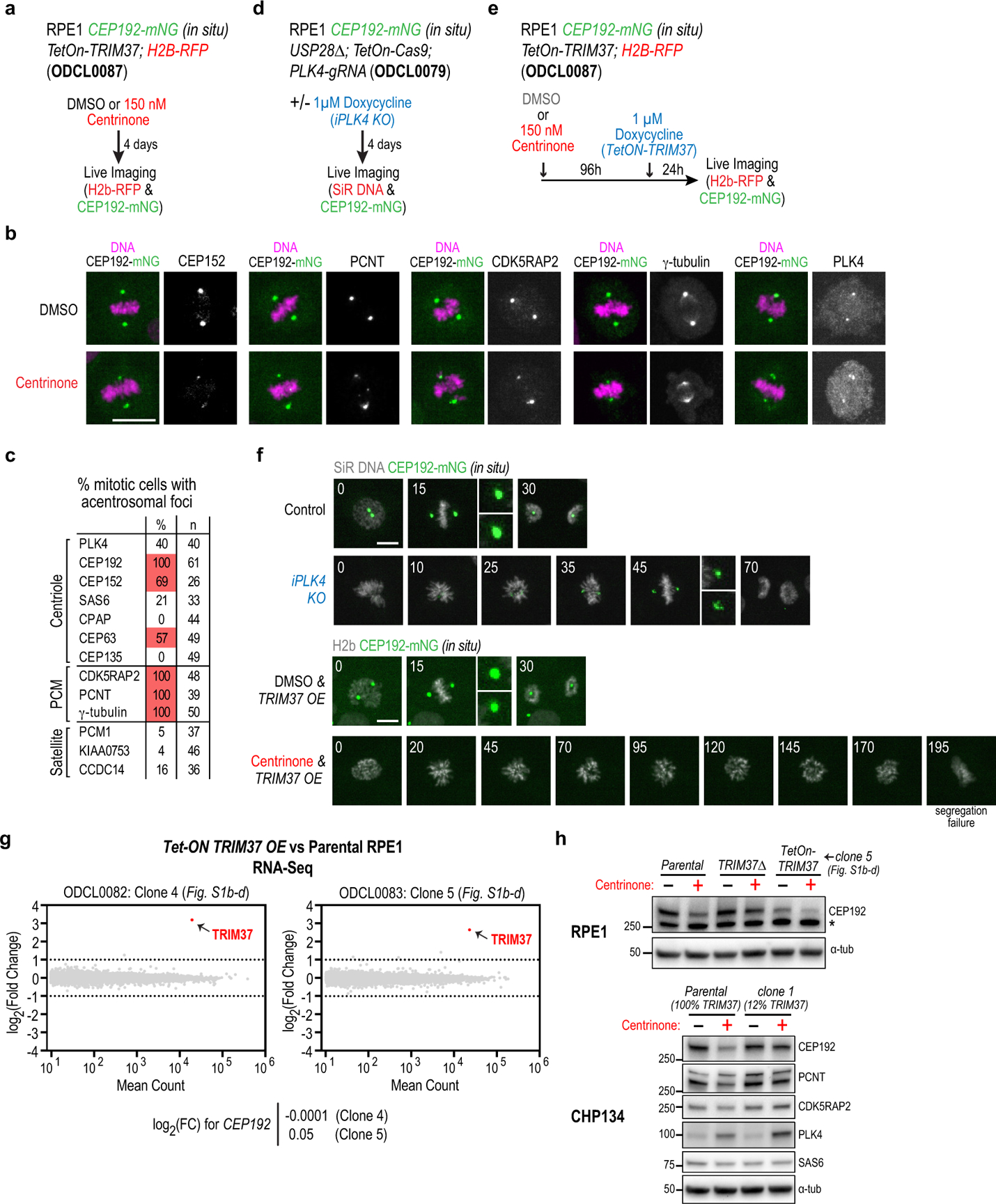
(a) Schematic of protocol used to analyze the effect of centrinone treatment on CEP192 mitotic dynamics by live imaging, shown in Fig. 3a. (b) Immunofluorescence images of centrosome components in DMSO versus centrinone-treated mitotic RPE1 cells. Scale bar, 10 μm. (c) Summary of immunofluorescence analysis showing which centrosome components were detected in the foci at the poles of acentrosomal spindles in centrinone-treated cells. (d) Schematic of protocol used to inducibly knock out PLK4 and monitor CEP192 dynamics in mitosis by live imaging, shown in Fig. 3b. (e) Schematic of protocol used to overexpress TRIM37 and monitor CEP192 dynamics in mitosis by live imaging, shown in Fig. 3c. Note that this is the same cell line used for the analysis shown in Fig. 3a (no dox induction); the analysis in these two conditions was conducted in parallel. (f) Additional panels from the timelapse image sequences shown in Fig. 3b and 3c for the inducible PLK4 knockout and TRIM37 overexpression, respectively. Times in minutes after NEBD are noted on each panel. (g) RNA-Seq analysis comparing two clones overexpressing TRIM37 to parental RPE1 cells. Elevated TRIM37 transcript levels are evident in both clones. No significant changes in the global transcriptome were otherwise observed. (h) Evidence that centrosomes protect CEP192 from TRIM37-dependent degradation. (top) Immunoblots of the indicated RPE1 cell lines with and without centrinone treatment. In cells overexpressing TRIM37, centrinone treatment further reduces CEP192 levels; by contrast, in TRIM37Δ cells, centrinone treatment only modestly affected CEP192 levels. Note that RNA-Seq analysis indicated no significant change in CEP192 transcript levels between cell lines with varying levels of TRIM37. (bottom) Immunoblots of CHP134 parental cells and a derived clone with ~12% expression of TRIM37. Centrinone strongly reduced CEP192 in the parental cells but not in the clone with reduced TRIM37 expression. α-tubulin serves as a loading control in both blots. For gel source data see Supplementary Figure 1.
Extended Data Figure 6. Evidence that CEP192 is the target of TRIM37 that accounts for enhanced sensitivity to PLK4 inhibition.
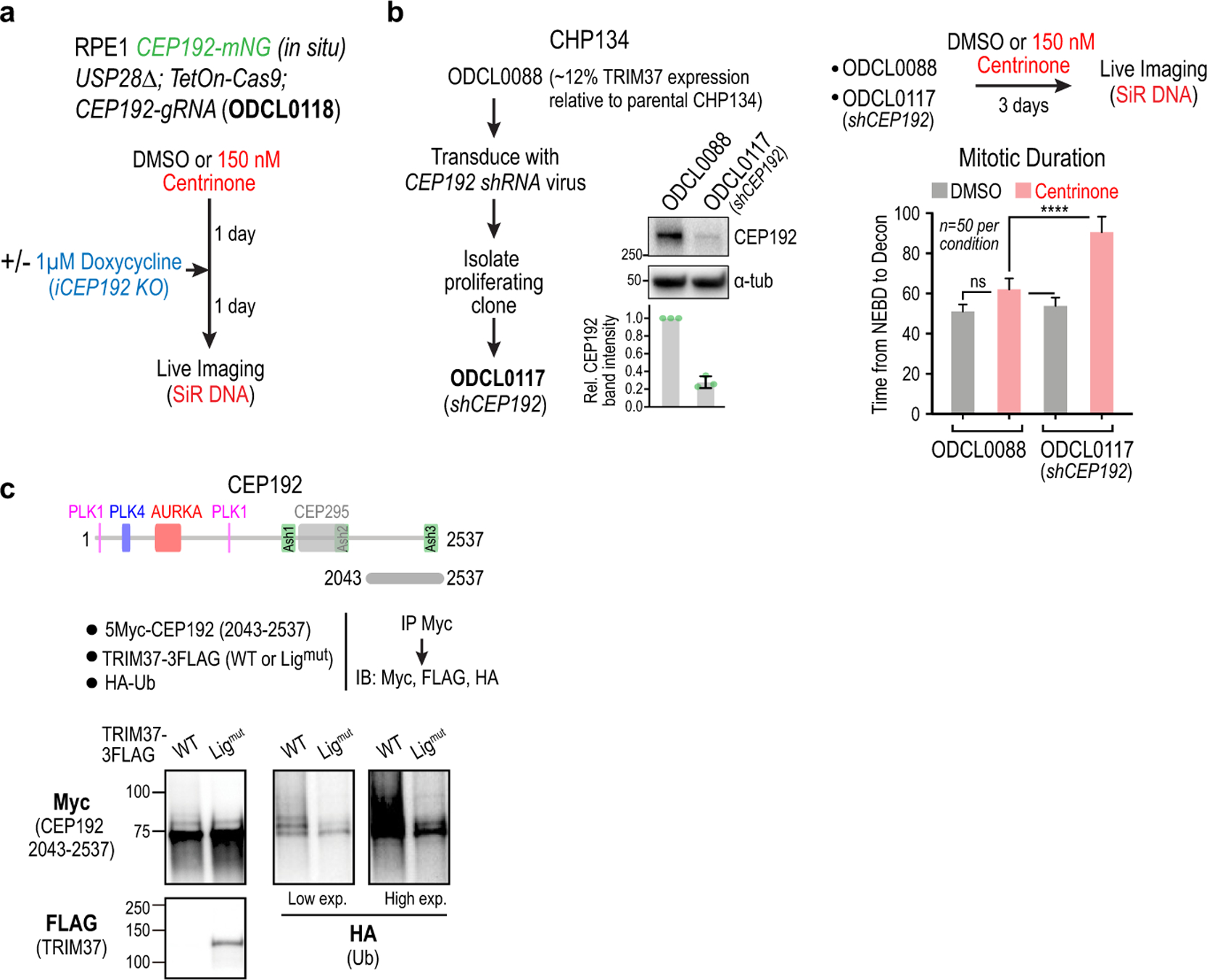
(a) Schematic for partial CEP192 inhibition using a short-term inducible knockout, followed by live imaging of mitosis. (b) Evidence in CHP134 cells that CEP192 is a functionally significant target of TRIM37. A CHP134 clonal cell line with reduced TRIM37 expression (~12% relative to parental CHP134 cells) was stably transduced with a CEP192 shRNA that reduced expression by ~75% (immunoblot and quantification below). Live imaging of mitosis showed that while reduction of CEP192 levels had no significant effect on the duration of mitosis in DMSO-treated cells, it significantly extended mitotic duration in centrinone-treated cells. (c) Evidence that the C-terminus of CEP192 is ubiquitinated in a TRIM37-dependent manner. The experiment shown in Fig. 4d included co-transfection of HA-tagged ubiquitin. Shown here is the HA-ubiquitin blot (together with FLAG and Myc blots) of the immunoprecipitated C-terminal fragment that binds TRIM37. Ubiquitination of this fragment was enhanced in the presence of WT relative to ligase-mutant TRIM37. The FLAG blot shown is the same as in Fig. 4d; the Myc blot is a different exposure of the blot shown in Fig. 4d. The other CEP192 fragments are not shown because their stability was affected by co-expression with WT but not ligase-mutant TRIM37, which makes comparisons of ubiquitination profiles difficult. For gel source data see Supplementary Figure 1.
Extended Data Figure 7. Inducible PLK4 shRNA-based CHP134 xenograft tumor analysis.
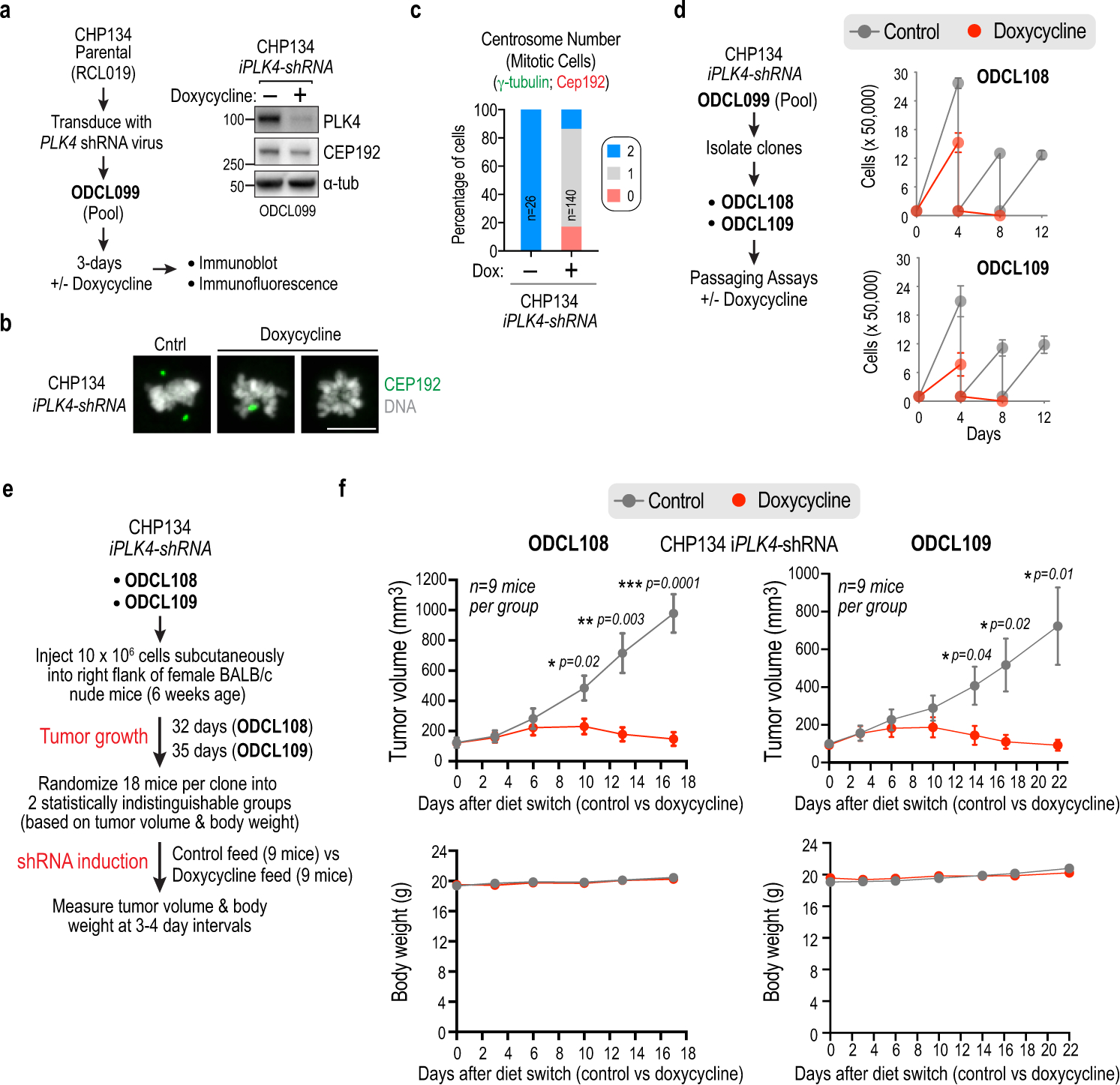
(a) (left) Schematic illustrating the generation and characterization of a CHP134 pool with stably integrated inducible PLK4 (iPLK4) shRNA. Following viral transduction, 3 day-induction of the shRNA with doxycycline, immunoblotting and immunofluorescence was used to assess PLK4 depletion and centrosome loss. (right) Immunoblot shows depletion of PLK4 as well as reduction of CEP192, as is also observed with centrinone; this reduction was dependent on high TRIM37 expression in CHP134 cells, as it was not observed following induction of PLK4 shRNA in a CHP134-derived line with ~10% TRIM37 expression (not shown). (b) Immunofluorescence images showing loss of centrosomes, detected using CEP192, following induction of PLK4 shRNA. Scale bar, 10 μm. (c) Quantification of centrosome number after 3-day induction of PLK4 shRNA. Longer induction was associated with extensive lethality, as is also observed with centrinone treatment of CHP134 cells. (d) (left) Schematic of isolation of CHP134 clones with stably integrated iPLK4 shRNA from the pool described in (a-c). (right) Results of passaging-based analysis showing that both clones exhibited rapid cessation of proliferation following induction of the shRNA. (e) Schematic of the workflow for tumor xenograft analysis with the two CHP134 iPLK4-shRNA clones. Tumors were generated in female BALB/c nude mice, and the shRNA was induced by switching to a doxycycline-containing diet. Tumor volume and body weight were measured over time after induction. (f) Time course of tumor growth in BALB/c nude mice for the two CHP134 PLK4 shRNA clonal lines following induction of shRNA (Doxycyline) versus no induction (Control). Error bars are SEM. Statistical significance was evaluated using unpaired t-tests. For gel source data see Supplementary Figure 1.
Extended Data Table 1.
Human cell lines used in this study.
| Parental cell lines | ||||
|---|---|---|---|---|
| OD cell line code | Name | Source | Clonal or Polyclonal | Catalog # |
| RCL001 | hTERT RPE-1 | ATCC | n/a | CRL-4000 |
| RCL008 | CHP212 | ATCC | n/a | CRL-2273 |
| RCL010 | IMR32 | ATCC | n/a | CCL-127 |
| RCL019 | CHP134 | Sigma | n/a | 06122002 |
| RCL020 | SK-N-F1 | ATCC | n/a | CRL-2142 |
| RCL021 | HepG2 | ATCC | n/a | HB-8065 |
| RCL022 | MCF7 | ATCC | n/a | HTB-22 |
| RCL023 | BT-474 | ATCC | n/a | HTB-20 |
| RCL024 | BT-549 | ATCC | n/a | HTB-122 |
| RCL025 | MDA-MB-231 | ATCC | n/a | HTB-26 |
| RCL026 | Freestyle 293-F | Thermo Fisher Scientific | n/a | R79007 |
| RCL027 | KPNYN | Gift from Peter Zage (UCSD) | n/a |
| Engineered cell lines | ||||
|---|---|---|---|---|
| OD cell line code | Parental line | Modification(s) | Clonal or Polyclonal | Reference |
| ODCL0002 | hTERT RPE-1 | USP28Δ | Clonal | Meitinger et al. 2016 |
| ODCL0003 | hTERT RPE-1 | CEP192-mNeonGreen | Clonal | This study |
| ODCL0035 | hTERT RPE-1 | EF-1αpro-H2B-mRFP | Polyclonal | Meitinger et al. 2016 |
| ODCL0036 | CHP212 | EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0049 | CHP134 | EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0060 | SK-N-F1 | EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0061 | hTERT RPE-1 | TRIM37Δ | Clonal | Meitinger et al. 2016 |
| ODCL0062 | ODCL0061 | TRIM37Δ UbCpro-TRIM37–3FLAG | Polyclonal | This study |
| ODCL0063 | ODCL0061 | TRIM37Δ UbCpro-TRIM37-W373A-3FLAG | Polyclonal | This study |
| ODCL0064 | ODCL0061 | TRIM37Δ UbCpro-TRIM37-C18R-3FLAG | Polyclonal | This study |
| ODCL0065 | ODCL0061 | TRIM37Δ UbCpro-TRIM37-C18R-W373A-3FLAG | Polyclonal | This study |
| ODCL0068 | ODCL0061 | TRIM37Δ PGKpro-TRIM37-C18R-mNeonGreen | Polyclonal | This study |
| ODCL0070 | ODCL0068 | TRIM37Δ PGKpro-TRIM37-C18R-mNeonGreen | Polyclonal | This study |
| EF-1αpro-mRuby2-MAP4-MBD | ||||
| ODCL0071 | ODCL0003 | CEP192-mNeonGreen; USP28Δ | Clonal | This study |
| ODCL0072 | ODCL0071 | CEP192-mNeonGreen; USP28Δ; EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0073 | ODCL0003 | CEP192-mNeonGreen; TRIM37Δ | Clonal | This study |
| ODCL0074 | ODCL0073 | CEP192-mNeonGreen; TRIM37Δ; EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0075 | ODCL0071 | CEP192-mNeonGreen; USP28Δ; EF-1αpro-mRuby2-MAP4-MBD | Polyclonal | This study |
| ODCL0076 | ODCL0073 | CEP192-mNeonGreen; TRIM37Δ; EF-1αpro-mRuby2-MAP4-MBD | Polyclonal | This study |
| ODCL0077 | ODCL0071 | CEP192-mNeonGreen; USP28Δ; TRE3Gpro-Cas9 | Clonal | This study |
| ODCL0078 | ODCL0073 | CEP192-mNeonGreen; TRIM37Δ; TRE3Gpro-Cas9 | Clonal | This study |
| ODCL0079 | ODCL0077 | CEP192-mNeonGreen; USP28Δ; TRE3Gpro-Cas9; U6pro-gRNA-PLK4 | Polyclonal | This study |
| ODCL0080 | ODCL0078 | CEP192-mNeonGreen; TRIM37Δ; TRE3Gpro-Cas9; U6pro-PLK4-gRNA | Polyclonal | This study |
| ODCL0081 | RCL001 | TRE3GSpro-TRIM37 | Clone 3; ED Fig. 1b–f | This study |
| ODCL0082 | RCL001 | TRE3GSpro-TRIM37 | Clone 4; Fig. 3g, ED Fig. 1b–f | This study |
| ODCL0083 | RCL001 | TRE3GSpro-TRIM37 | Clone 5; Fig. 1a–b, 3g, ED Fig. 1b–f | This study |
| ODCL0084 | ODCL0061 | TRIM37Δ; TRE3GSpro-TRIM37 | Clone 1; ED Fig. 1b–f | This study |
| ODCL0085 | ODCL0061 | TRIM37Δ; TRE3GSpro-TRIM37 | Clone 2; ED Fig. 1b–f | This study |
| ODCL0086 | ODCL0003 | CEP192-mNeonGreen; TRE3GSpro-TRIM37 | Clonal | This study |
| ODCL0087 | ODCL0086 | CEP192-mNeonGreen; TRE3GSpro-TRIM37; EF-1αpro-H2B-mRFP | Polyclonal | This study |
| ODCL0088 | CHP134 | TRIM37+/−/−/− (17bp del; 7bp del; 1bp ins) | Clone 1; Fig. 1f, 3f, ED Fig. 2h | This study |
| ODCL0089 | CHP134 | TRIM37+/+/+/− (374bp del) | Clone 6; Fig. 1f, ED Fig. 2h | This study |
| ODCL0090 | CHP134 | TRIM37+/−/−/− (15bp del; 1bp ins; 7bp ins) | Clone 4; Fig. 1f, ED Fig. 2h | This study |
| ODCL0091 | CHP134 | TRIM37+/+/+/− (2bp del) | Clone 5; Fig. 1f, ED Fig. 2f | This study |
| ODCL0092 | CHP134 | TRIM37+/−/−/− (1bp del; 1bp del; 17bp ins) | Clone 2; Fig. 1f, ED Fig. 2h | This study |
| ODCL0093 | CHP134 | TRIM37+/+/+/− (3 copies wildtype; mutation unclear) | Clone 3; Fig. 1f, ED Fig. 2h | This study |
| ODCL0099 | CHP134 | hEF1a/TurboGFP inducible PLK4-shRNA | Polyclonal | This study |
| ODCL0108 | ODCL0099 | hEF1a/TurboGFP inducible PLK4-shRNA | Clone 1; ED Fig 7d–f | This study |
| ODCL0109 | ODCL0099 | hEF1a/TurboGFP inducible PLK4-shRNA | Clone 2; ED Fig 7d–f | This study |
| ODCL0117 | ODCL0088 | TRIM37+/−/−/− (clone 1); CEP192-shRNA | Clonal | This study |
| ODCL0118 | hTERT RPE-1 | CEP192-mNeonGreen; USP28Δ; | Polyclonal | This study |
| TRE3Gpro-Cas9; U6pro-gRNA-CEP192 | ||||
| ODCL0119 | ODCL0061 | TRIM37A; UBCpro-TRIM37 | Clonal | This study |
| ODCL0121 | ODCL0061 | TRIM37Δ; UBCpro-TRIM37-C18R | Clonal | This study |
pro = Promoter; del = deletion; ins = insertion.
Extended Data Table 2.
Plasmids used in this study.
| OD Plasmid code | Description | Purpose | Selection | Reference |
|---|---|---|---|---|
| pOD3789 | CMVpro-5MYC-PLK4 | Transient transfection | Ampicillin | This study |
| pOD3790 | CMVpro-TRIM37–3FLAG | Transient transfection | Ampicillin | This study |
| pOD3791 | CMVpro-TRIM37-C18R-3FLAG | Transient transfection | Ampicillin | This study |
| pOD3792 | CMVpro-TRIM37-C18R-W373A-3FLAG | Transient transfection | Ampicillin | This study |
| pOD3793 | CMVpro-5MYC-CEP152 | Transient transfection | Ampicillin | This study |
| pOD3794 | CMVpro-5MYC-CEP192 | Transient transfection | Ampicillin | This study |
| pOD3795 | EF-1αpro-H2B-mRFP | Lentiviral integration | Ampicillin | Meitinger et al. 2016 |
| pOD3796 | EF-1αpro-mRuby-hMAP4-MBP; SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3797 | UbCpro-TRIM37–3FLAG SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3798 | UbCpro-TRIM37-C18R-3FLAG; SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3799 | UbCpro-TRIM37-C18R-W373A-3FLAG; SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3800 | UbCpro-TRIM37-C18R-W373A-3FLAG; SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3801 | PGKpro-TRIM37-C18R-NeonGreen-P2A-BSD | Lentiviral integration | Ampicillin | This study |
| pOD3802 | U6pro-TRIM37gRNA (PX459) | Transient transfection | Ampicillin | Meitinger et al. 2016 |
| pOD3803 | U6pro-USP28-gRNA (PX459) | Transient transfection | Ampicillin | Meitinger et al. 2016 |
| pOD3804 | U6pro-PLK4-gRNA (lentiGuide-Puro) | Lentiviral integration | Ampicillin | This study |
| pOD3805 | TRE3GSpro-TRIM37 SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3806 | PGKpro-Tet-On-3G-P2A-BSD | Lentiviral integration | Ampicillin | This study |
| pOD3807 | UbCpro-TRIM37 SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3808 | UbCpro-TRIM37-C18R SV40pro-NeoR | Lentiviral integration | Ampicillin | This study |
| pOD3809 | CMVpro-5xMYC-Cep192 | Transient transfection | Ampicillin | This study |
| pOD3810 | CMVpro-5xMYC-Cep192 aa 1–2071 | Transient transfection | Ampicillin | This study |
| pOD3811 | CMVpro-5xMYC-Cep192 aa 1201–2537 | Transient transfection | Ampicillin | This study |
| pOD3812 | CMVpro-5xMYC-Cep192 aa 2043–2537 | Transient transfection | Ampicillin | This study |
pro = Promoter
Supplementary Material
ACKNOWLEDGEMENTS
We dedicate this manuscript to the memory of Dr. Tim Gahman, our valued colleague and friend, with whom we developed centrinone. This work was supported by an NIH grant to K.O. (GM074207) and by funds from the Hilton Ludwig Cancer Prevention Initiative to A.K.S. F.M. was supported by the German Science Foundation (ME 4713/1-1). M.O. and S.W. were supported by the Japan Society for the Promotion of Science. R.L.D, J.V.A, D.J., A.K.S., A.D., and K.O. received salary and other support from the Ludwig Institute for Cancer Research. We thank Peter Zage for the KPNYN cells.
Footnotes
DATA AVAILABILITY
The RNA-seq data in Extended Data Fig. 4f,g and Fig. 5g have been deposited in NCBI’s Gene Expression Omnibus42 and are accessible through GEO Series accession number (GSE148263, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148263). Source Data for Extended Data Fig. 7e,f are provided with the paper. Other data or materials are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
K.O., A.D., A.K.S., R.L.D., and F.M. are inventors on a pending patent application from the Ludwig Institute for Cancer Research Ltd, application number PCT/US2018/026720. “TRIM37 levels predict sensitivity to PLK4 inhibition in cancer cells”.
REFERENCES
- 1.Petry S Mechanisms of Mitotic Spindle Assembly. Annu Rev Biochem 85, 659–683, doi: 10.1146/annurev-biochem-060815-014528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nigg EA & Holland AJ Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19, 297–312, doi: 10.1038/nrm.2017.127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zitouni S, Nabais C, Jana SC, Guerrero A & Bettencourt-Dias M Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 15, 433–452, doi: 10.1038/nrm3819 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Wong YL et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160, doi: 10.1126/science.aaa5111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulavin DV et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 31, 210–215, doi: 10.1038/ng894 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Ho N et al. Delineation of the frequency and boundary of chromosomal copy number variations in paediatric neuroblastoma. Cell Cycle 17, 749–758, doi: 10.1080/15384101.2017.1421875 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet 31, 133–134, doi: 10.1038/ng888 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Liu Y et al. Targeting 17q23 amplicon to overcome the resistance to anti-HER2 therapy in HER2+ breast cancer. Nat Commun 9, 4718, doi: 10.1038/s41467-018-07264-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodruff JB, Wueseke O & Hyman AA Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci 369, doi: 10.1098/rstb.2013.0459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oegema K, Davis RL, Lara-Gonzalez P, Desai A & Shiau AK CFI-400945 is not a selective cellular PLK4 inhibitor. Proc Natl Acad Sci U S A 115, E10808–E10809, doi: 10.1073/pnas.1813310115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bazzi H & Anderson KV Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci U S A 111, E1491–1500, doi: 10.1073/pnas.1400568111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khodjakov A & Rieder CL Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol 153, 237–242, doi: 10.1083/jcb.153.1.237 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meitinger F et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214, 155–166, doi: 10.1083/jcb.201604081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sir JH et al. Loss of centrioles causes chromosomal instability in vertebrate somatic cells. J Cell Biol 203, 747–756, doi: 10.1083/jcb.201309038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376, doi: 10.1038/nature25795 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607, doi: 10.1038/nature11003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montenegro Gouveia S et al. PLK4 is a microtubule-associated protein that self-assembles promoting de novo MTOC formation. J Cell Sci 132, doi: 10.1242/jcs.219501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JE et al. Phase separation of Polo-like kinase 4 by autoactivation and clustering drives centriole biogenesis. Nat Commun 10, 4959, doi: 10.1038/s41467-019-12619-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto S & Kitagawa D Self-organization of Plk4 regulates symmetry breaking in centriole duplication. Nat Commun 10, 1810, doi: 10.1038/s41467-019-09847-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brigant B, Metzinger-Le Meuth V, Rochette J & Metzinger L TRIMming down to TRIM37: Relevance to Inflammation, Cardiovascular Disorders, and Cancer in MULIBREY Nanism. Int J Mol Sci 20, doi: 10.3390/ijms20010067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park HH Structure of TRAF Family: Current Understanding of Receptor Recognition. Front Immunol 9, 1999, doi: 10.3389/fimmu.2018.01999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatnagar S et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 516, 116–120, doi: 10.1038/nature13955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu M et al. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol 4, e27, doi: 10.1371/journal.pbio.0040027 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheng Y et al. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol 13, 285–291, doi: 10.1038/nsmb1067 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Kim TS et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci U S A 110, E4849–4857, doi: 10.1073/pnas.1319656110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SY et al. Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat Struct Mol Biol 21, 696–703, doi: 10.1038/nsmb.2846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD & Nigg EA Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci 126, 3223–3233, doi: 10.1242/jcs.129502 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Joukov V & De Nicolo A Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal 11, doi: 10.1126/scisignal.aar4195 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Yeow ZY et al. Exploiting TRIM37-driven centrosome dysfunction to eliminate 17q23-amplified breast cancer cells. Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung NK & Dyer MA Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 13, 397–411, doi: 10.1038/nrc3526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis CU & Shohet JM Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med 66, 49–63, doi: 10.1146/annurev-med-011514-023121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maris JM Recent advances in neuroblastoma. N Engl J Med 362, 2202–2211, doi: 10.1056/NEJMra0804577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oeffinger KC et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 355, 1572–1582, doi: 10.1056/NEJMsa060185 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168, doi: 10.1093/nar/gku936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaulich M & Dowdy SF Combining CRISPR/Cas9 and rAAV Templates for Efficient Gene Editing. Nucleic Acid Ther 25, 287–296, doi: 10.1089/nat.2015.0545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784, doi: 10.1038/nmeth.3047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamitani T, Kito K, Nguyen HP & Yeh ET Characterization of NEDD8, a developmentally downregulated ubiquitin-like protein. J Biol Chem 272, 28557–28562, doi: 10.1074/jbc.272.45.28557 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S & Huber W Differential expression analysis for sequence count data. Genome Biol 11, R106, doi: 10.1186/gb-2010-11-10-r106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heberle H, Meirelles GV, da Silva FR, Telles GP & Minghim R InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16, 169, doi: 10.1186/s12859-015-0611-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar R, Domrachev M & Lash AE Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210, doi: 10.1093/nar/30.1.207 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bauer M, Cubizolles F, Schmidt A & Nigg EA Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. EMBO J 35, 2152–2166, doi: 10.15252/embj.201694462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.