

Abstract
The 90-kDa heat shock protein (Hsp90) is a molecular chaperone that ensures cellular proteostasis by maintaining the folding, stabilization, activation, and degradation of over 400 client proteins. Hsp90 is not only critical for routine protein maintenance in healthy cells, but also during states of cellular stress, such as cancer and neurodegenerative diseases. Due to its ability to affect phosphorylation of numerous client proteins, inhibition of Hsp90 has been an attractive anticancer approach since the early 1990’s, when researchers identified a druggable target on the amino terminus of Hsp90 for a variety of cancers. Since then, 17 Hsp90 inhibitors that target the chaperone’s N-terminal domain, have entered clinical trials. None, however, have been approved thus far by the FDA as a cancer monotherapy. In these trials, a major limitation observed with Hsp90 inhibition at the N-terminal domain was dose-limiting toxicities and relatively poor pharmacokinetic profiles. Despite this, preclinical and clinical research continues to show that Hsp90 inhibitors effectively target cancer cell death and decrease tumor progression supporting the rationale for the development of novel Hsp90 inhibitors. Here, we present an in-depth overview of the Hsp90 inhibitors used in clinical trials. Finally, we present current shifts in the field related to targeting the carboxy-terminal domain of Hsp90 as well as to the development of isoform-selective inhibitors as a means to bypass the pitfalls of current Hsp90 inhibitors and improve clinical trial outcomes.
Keywords: Cancer, chaperones, geldanamycin, Grp94, Hsp90, novobiocin, TAS-116, TRAP1
1. INTRODUCTION
A vital characteristic of all eukaryotic cells is the requirement for proper molecular chaperone function. These chaperone proteins serve as part of a cell’s “quality control system” to ensure proteostasis. Specifically, chaperones maintain the folding, stability, activation, and degradation of intracellular proteins, ultimately contributing to overall cellular homeostasis [1]. Proteins that depend upon and/or interact with these molecular chaperones are called clients. Chaperone function is not only important for routine client maintenance, but also in response to cellular stress. In certain disease states, like cancer and neurodegenerative diseases, proteins are often misfolded, leading to aggregation, which increases the cellular dependence upon chaperone function [2]. In light of this, there is a significant amount of interest to design and develop novel therapeutics that target molecular chaperones, specifically the heat shock protein (Hsp) family.
The Hsp family of molecular chaperones was first observed in Drosophila melanogaster by Ritossa in the early 1960s as a result of a heat-induced change to chromosome appearance [3]. In the D. melanogaster cells, certain regions of chromosomes gained “puff sites” where there was an increase of RNA synthesis and subsequent changes to the pattern of protein expression [4–6]. Interestingly, a small subset of these proteins accounted for the majority of proteins with increased expression and was accordingly named heat shock proteins [7]. Since their discovery and characterization, expression levels of these chaperones are not only increased in response to heat, but other environmental stressors as well, such as inflammation, hypoxia, infection, and/or nutrient deprivation. This increase in Hsp expression is the result of the heat shock response (HSR) that is mediated by the transcription factor heat shock factor-1 (HSF-1) binding to its transcriptional element, the heat shock element (HSE) [8]. The Hsp family consists of a multitude of chaperones named according to molecular weight. In regard to novel therapeutic agents, inhibitors that disrupt the 90-kDa Hsp (Hsp90) function are at the forefront of development and exhibit high potential for clinical use in humans and more specifically, the treatment of cancer.
Hsp90 accounts for 1–2% of total protein concentration in unstressed cells, whereas chaperone expression levels increase to ~4–6% in response to cellular stress as a means to handle the increased demand for client protein stabilization [1, 9]. Regardless of cellular state, Hsp90 clientele includes a variety of proteins involved in several cellular processes and pathways, such as protein kinases, cell cycle regulators, transcription factors, and steroid hormone receptors, to name a few [10]. Interestingly, in the stressed state, which is common to malignancies, Hsp90 serves as a chaperone to numerous proteins that maintain the ten hallmarks of cancer [11, 12]. Taken together, these points highlight the opportunity that Hsp90 chaperone inhibition simultaneously targets multiple oncogenic pathways and proteins.
In the early 1990s, Whitesell and colleagues demonstrated Hsp90’s crucial role in oncogenic transformation by serendipitously inhibiting chaperone function with a benzoquinone ansamycin, geldanamycin (GDA), which initiated the concept of chaperone inhibition [13]. Prior to this finding, it was mechanistically unclear how oncogenic gene products, like tyrosine kinases and more specifically v-src, promoted the transformation of healthy cells into malignant ones [14]. GDA was presumed to be a tyrosine kinase inhibitor until Whitesell and colleagues showed that it bound to Hsp90 in a stable and specific manner. This binding not only disrupted the formation of the Hsp90-src heteroprotein complex, but it also inhibited transformation, highlighting the dependence of the cell on Hsp90 during oncogenesis [15]. Since this pivotal finding, Hsp90’s function and role in oncogenesis have been more well-defined, leading to the development of several compounds that target and inhibit the chaperone. Of these inhibitors, a handful have entered clinical trials for the treatment of cancer, but none yet have been approved for use as anti-neoplastic agents [16].
Although the need for Hsp90’s function has mostly been recognized in cancer, the molecular chaperone also plays an integral role in the pathogenesis of neurodegenerative diseases characterized by protein aggregation, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [17]. Interestingly, the proteins most implicated in protein aggregation in AD and PD are Hsp90 clients – β-amyloid (Aβ) peptide and tau and α-synuclein, respectively. For example, two hallmark indicators of AD are extracellular plaque deposits of Aβ and hyperphosphorylated tau [17, 18]. In PD, α-synuclein is recognized as a genetic and pathological link to disease progression. More specifically, this protein is found deposited in Lewy bodies which are abnormal aggregates formed in presynaptic nerve terminals [19, 20]. Unlike malignancies, where inhibition of Hsp90’s function is ideal for treatment, it has been suggested that neurodegenerative diseases would benefit most from the upregulation of chaperone function to increase its cytoprotective role and ultimately decrease protein aggregation [17]. While the development of Hsp90 treatments for AD and PD is warranted, we will focus the remainder of our insights on Hsp90 therapeutic approaches in the context of malignancies.
Here, we provide a comprehensive, up-to-date evaluation of the Hsp90 inhibitors that have entered clinical trials, specifically in malignancies, as well as some observations as to why they have not yet achieved FDA approval. From these observations in trials, researchers have now shifted to alternative approaches to Hsp90 inhibition, including targeting the carboxy terminal domain as well as characterizing and developing isoform-specific Hsp90 inhibitors that can then be selectively targeted in certain disease states.
2. HSP90 FAMILY OF PROTEINS
The mammalian Hsp90 family of proteins is highly conserved and includes four members or isoforms. These proteins are indirectly involved in several cellular processes and found in distinct cellular compartments. In addition to Hsp90’s function as a molecular chaperone, it serves an essential role in overall cellular homeostasis [10, 21, 22]. The majority of Hsp90 isoforms are located in the cytoplasm and in the HSP90A subfamily, which includes inducible Hsp90α and constitutively expressed Hsp90β isoforms. Also, there are the HSP90B and TRAP subfamilies that include the Grp94 isoform in the endoplasmic reticulum and TRAP1 isoform in the mitochondria, respectively [22]. The three Hsp90 subfamilies, their five gene products, and respective cellular locations are listed in Table 1. Despite occupying different cellular compartments, all isoforms share a very similar sequence and structural homology, which are important to understand in order to better evaluate and critique the challenges of current Hsp90 inhibitors.
Table 1.
Hsp90 family members, gene products, and cellular location – There are three Hsp90 subfamilies and each located in different cellular compartments. The majority of isoforms are in the cytoplasm and include Hsp90α and Hsp90β. The endoplasmic reticulum and mitochondria isoforms are Grp94 and TRAP1, respectively.
| Subfamily | Gene(s) | Protein Isoform(s) | Cellular Location |
|---|---|---|---|
| HSP90A | HSP90AA1, HSP90AA2 | Hsp90α1, Hsp90α2 | Cytoplasm |
| HSP90AB1 | Hsp90β | ||
| HSP90B | HSP90B1 | Grp94 | Endoplasmic reticulum |
| TRAP | TRAP1 | TRAP1 | Mitochondria |
Grp94: 94-kDa glucose related protein; TRAP1: TNF receptor associated protein 1
The family of molecular chaperones is biologically active as homodimers with each monomer made up of three structurally and functionally distinct domains: N-terminal, middle, and C-terminal domains (Fig. 1) [21]. The N-terminal domain (NTD) is the site for nucleotide binding and ATPase activity [23]. The middle domain (MD) also plays an important role in the hydrolysis of ATP to ADP – namely, the Arg380 residue, which is part of the catalytic loop for hydrolysis [24]. The NTD and MD are connected or tethered to each other via a charged linker region. Lastly, the C-terminal domain (CTD) is the site for protein dimerization and additional ATP binding [25]. The CTD contains a tetratricopeptide repeat (TPR) motif that increases binding specificity for its substrates, especially co-chaperones with similar TPR-motifs. All three regions have been reported to bind clients and co-chaperones.
Fig. 1.
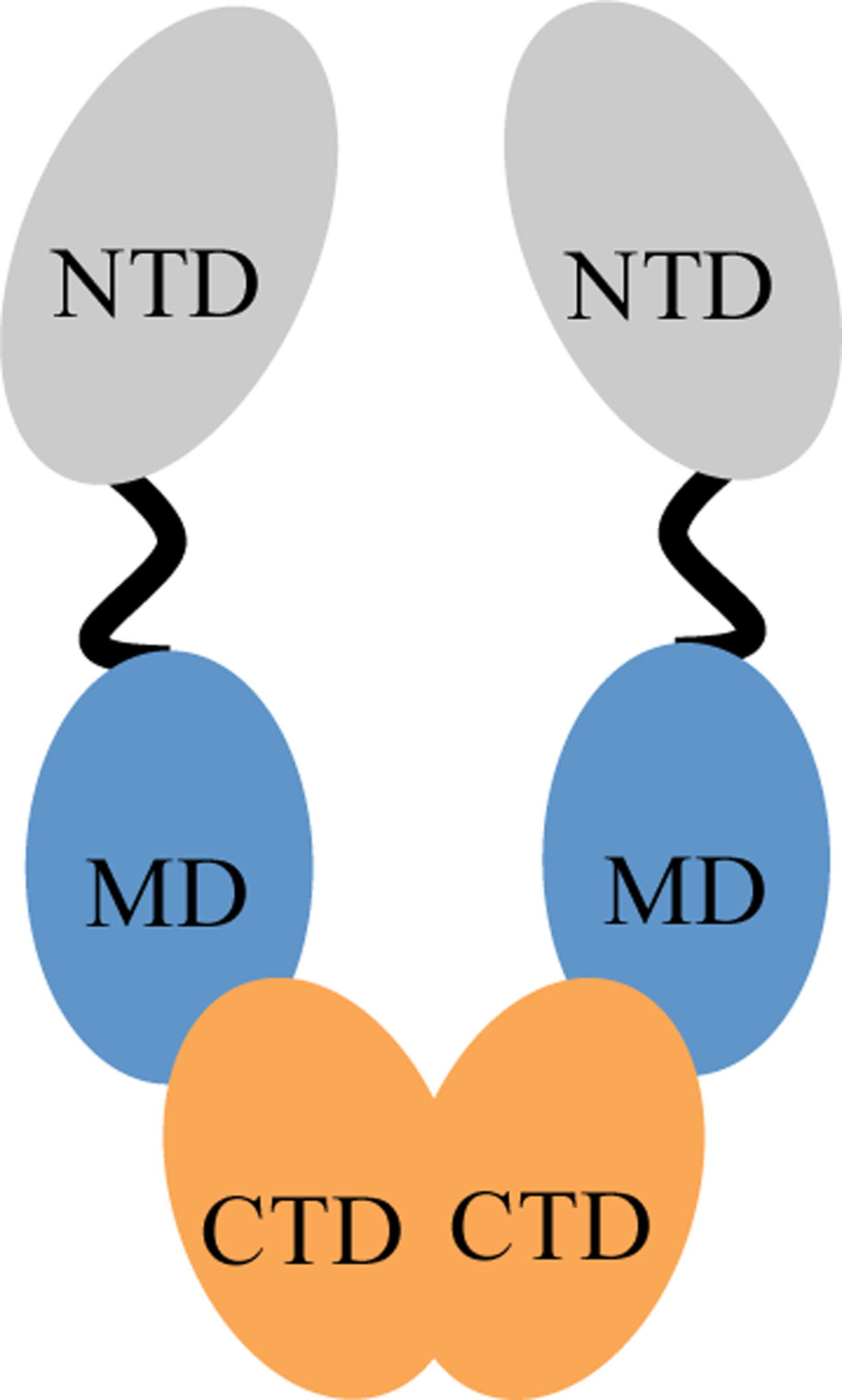
Structure of Hsp90 – A schematic of Hsp90’s homodimer structure. Each monomer has a N-terminal (NTD; green), middle (MD; orange), and C-terminal (CTD; blue) domains. The NTD and MD are connected by a charged linker region (black line). *Original figure created by authors
All inhibitors that have undergone clinical investigation as anticancer drugs, target the NTD and bind competitively to its ATP binding site. Although effective and potent at killing cancer cells preclinically, in clinical trials detrimental dose-limiting toxicities (DLTs) and dose scheduling limitations have prevented NTD Hsp90 inhibitors from gaining FDA approval [26]. One speculation in the field regarding the failure of NTD compounds in clinical trials due to DLTs is that NTD targeting leads to a “pan-inhibitory” effect against all Hsp90 isoforms and induces the HSR, leading to a pro-survival effort by the cell that in turn requires a higher dose of inhibition of Hsp90 to achieve the same cancer cell suppressive effect thereby resulting in progressive dose escalation, and over time, DLTs [21, 27, 28]. Although this remains a major obstacle in the clinic, recent findings at the 9th International Conference on the Hsp90 Chaperone Machine (ICHCM) suggest a new approach to target the chaperone which could decrease DLTs. At the conference, Dr. Brian Blagg presented two newly designed scaffolds of Hsp90β isoform-selective inhibitors that promoted degradation of HSF1 and ultimately led to no HSR induction [29]. This novel approach will be revisited in a subsequent section of the manuscript. As of February 2019, more than 170 clinical trials have taken place to test 18 different Hsp90 inhibitors and their potential uses as therapeutic agents in humans [see ClinicalTrials.gov]. Although the majority of trials are seen through to completion, more than half do not progress past Phase I (Fig. 2). In the following section, we present a comprehensive description of these chaperone inhibitors in clinical trials to date, in order to highlight their strengths and weaknesses. Additionally, we provide insight and recommendations to improve the development of novel Hsp90 inhibitors as anticancer agents.
Fig. 2.
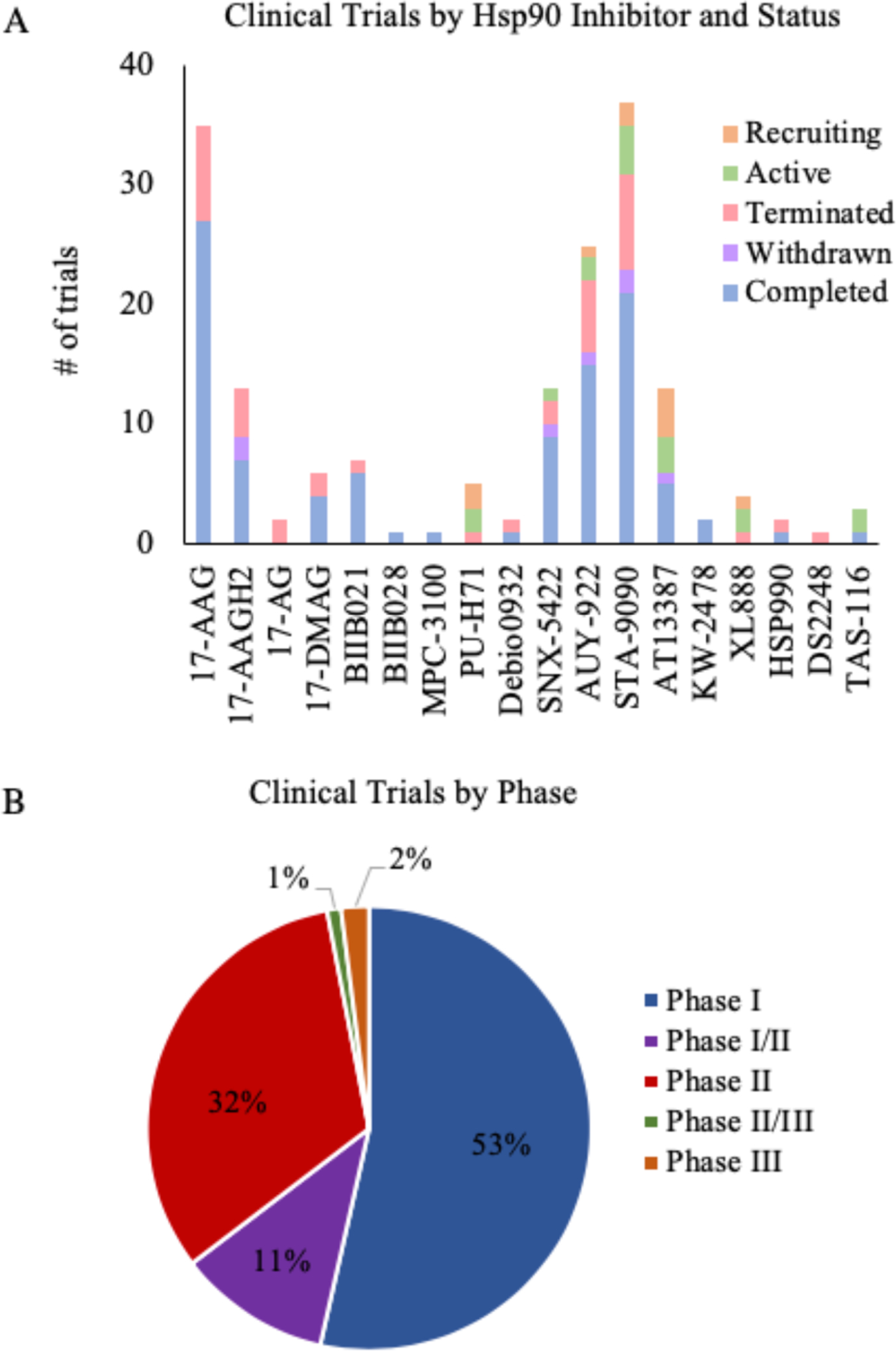
Clinical trial overview of Hsp90 inhibitors – 18 Hsp90 inhibitors have entered the clinical pipeline totaling over 170 trials. A) There are currently 7 inhibitors in active or recruiting trials (green or orange, respectively). B) The majority of inhibitors have not progressed past Phase I. Note: active trials testing TAS-116 are in Japan. *Original figure created by authors
3. CLINICAL LANDSCAPE OF HSP90 INHIBITORS
For over 25 years, the development of Hsp90 inhibitors has predominantly centered around the design of drug compounds that competitively bind the N-terminal ATP-binding sites to disrupt molecular chaperone function [21, 30]. Moreover, all four Hsp90 isoforms share high sequence identity in this region, which decreases the potential to selectively target a specific isoform. This form of pan-inhibition, wherein all isoforms are non-selectively inhibited, has been thought to represent a major limitation in the clinical development and approval of these inhibitors as anticancer agents. For cancer cells, optimal inhibitors should selectively target the cytosolic isoforms Hsp90α and Hsp90β, however this has been very difficult as these isoforms share > 95% sequence identity in the NTD. Perhaps by gaining a better understanding of the chaperone’s enzyme kinetics, it could help to distinguish each isoform and lead to the generation of more durable and effective Hsp90 inhibitors. Lee et al. eloquently present Hsp90 as being a “perfect enzyme” due to the chaperone having an ATP hydrolysis rate of ~1 s−1. Although demonstrated in Escherichia coli, this work emphasizes the indispensable value of gaining a mechanistic understanding of the ATPase activity, especially before optimizing inhibitors that disrupt its function [31]. The 18 Hsp90 inhibitors are characterized by five chemical structure-based categories: (i) natural products and their derivatives, (ii) purine-based, (iii) benzamide, (iv) resorcinol-containing, and (v) miscellaneous. The following five sections give an in-depth evaluation of Hsp90 inhibitors that have entered clinical trials.
3.1. Natural Products and their Derivatives
As mentioned above, in the early 1990s Whitesell and colleagues identified GDA as a ligand of Hsp90, and years later, demonstrated its binding to the N-terminal ATP-binding site. Upon GDA binding, ATPase activity of the molecular chaperone is disrupted and subsequently clientprotein stabilization, which results in client protein degradation and an attack on multiple cellular processes [32]. GDA is a 1,4-benzoquinone ansamycin antibiotic derived from Streptomyces hygroscopicus (Fig. 3A) [33]. Although GDA effectively and potently kills cancer cells, it is not a good clinical candidate due to in vivo toxicity, instability, and poor solubility [34]. Specifically, GDA contains a reactive quinone that produces superoxide radicals causing cell death independent of Hsp90 inhibition [35]. Because of this, various groups have put forth a substantial amount of effort and resources to modify this compound and eliminate its redox potential. In addition to GDA, another natural product, radicicol (RDC), is isolated from the fungus Monosporium bonorden (Fig. 3C) [33]. Similar to GDA, RDC competitively binds Hsp90 at the ATP-binding site. Although GDA and RDC do not have clinical utility, the two natural products support the rationale that inhibiting Hsp90 represents a multi-pronged anticancer therapeutic approach. This resulted in the design and development of several semi-synthetic GDA derivatives. Upon further investigation, it was determined that the C-17 position of GDA was critical for its mechanism of action and toxic reactivity. As a result, 17-N-allylamino17-dimethoxygeldanamycin (17-AAG; tanespi-mycin), 17-AAGH2 (IPI-504; retaspimycin), 17-AG (IPI-493), and 17dimethylamino-17-dimthoxygeldanamycin (17-DMAG; alvespimycin) were designed and synthesized (Fig. 3A and 3B) [32].
Fig. 3.
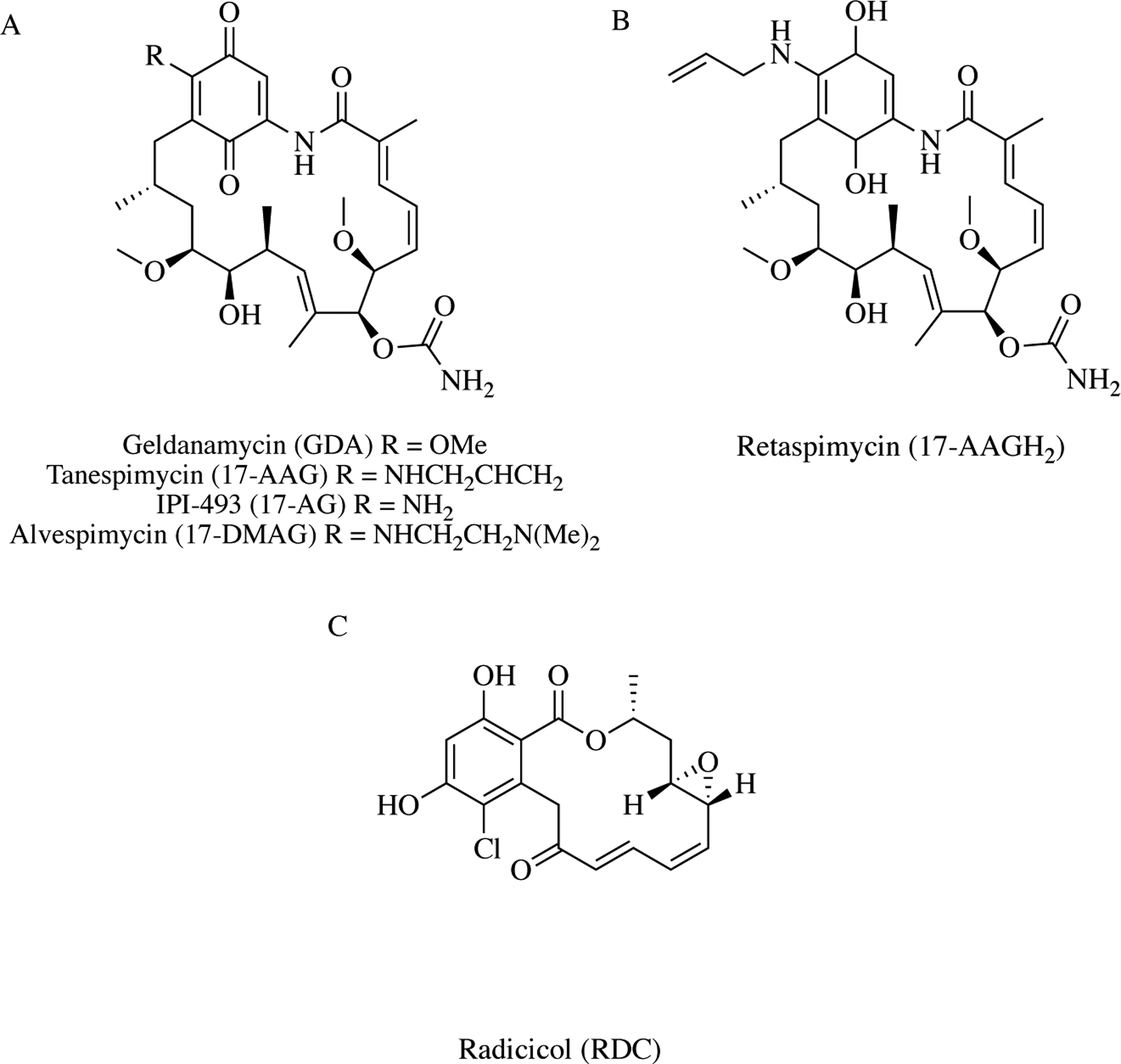
Hsp90 inhibitors derived from natural products – A) Geldanamycin is a benzoquinone ansamycin antibiotic. Three of its derivatives (tanespimycin, IPI-493, and alvespimycin) make substitutions to the C-17 position. B) In addition to a C-17 substitution, retaspimycin is the reduced derivative of GDA. C) Radicicol is an antifungal natural product.
By the end of 2003, the first GDA derivative 17-AAG, which adds an allylamine substituent to the C-17 position, entered eight Phase I clinical trials. Since then, there have been a total of 35 (23% terminated). Of these trials, none progressed past Phase II, which decreased enthusiasm for the development of GDA derivatives. Despite being reasonably effective against cancer tumor growth in early clinical trials, 17-AAG exhibited poor bioavailability. In order to improve this, Infinity Pharmaceuticals designed and synthesized the compound 17-AAGH2 (IPI-504; retaspimycin) [36]. This hydroquinone hydrochloride salt analog improves the metabolic profile of 17-AAG by eliminating the requirement for reduction. Another benefit of this analog was its increased potency of Hsp90 inhibition. The development of 17-AAGH2 resulted in 13 clinical Phase I or II trials with approximately half terminated or withdrawn. Another semi-synthetic benzoquinone ansamycin derivative developed by the same pharmaceutical company is 17-AG (IPI-493) [37]. At the time, 17-AG was a promising clinical candidate since it is the major metabolite of all GDA derivatives and was effective in preclinical xenograft models [38]. There have been only two reported 17-AG clinical trials, which ran simultaneously with retaspimycin trials, both of which were terminated due to superior effect of retaspimycin. Kosan Biosciences designed and developed 17-dimethylamino-17-dimthoxygeldanamycin (17-DMAG; alvespimycin) which improved the physiochemical profile of GDA by remaining protonated at physiological pH. 17-DMAG entered six Phase I clinical trials (17% terminated) and one Phase II that was terminated.
3.2. Purine-based Inhibitors
Similar to all ATPases, Hsp90 binds and hydrolyzes ATP, but the chaperone does so in a unique way. The molecular chaperone belongs to the Gyrase, Hsp90, Histidine kinase, MutL (GHKL) ATPase family, all of which share a special β-α-β fold containing four motifs I-IV. A pair of motifs within the fold interact with a different moiety of the ATP molecule; motifs I and III interact with the phosphate groups while motifs II and IV with the adenine component [39]. Taking advantage of this unique interaction with ATP, Chiosis and colleagues at Memorial Sloan Kettering designed the first reported fully-synthetic small molecule Hsp90 inhibitor, PU-3 (Fig. 4A) [34, 40]. After successfully arresting growth and differentiation in breast cancer cells, researchers began to enhance the purine chemical scaffold in PU-3 and generate derivatives [41]. To-date, there are five purine or purine-like compounds that entered clinicals trials, with none progressing past Phase II. These inhibitors include: BIIB021, BIIB028, MPC-3100, PU-H71, and Debio0932 (Fig. 4B–E). Two defining characteristics of inhibitors in this class are a purine (or purine-like) scaffold with amine and aryl substituents.
Fig. 4.
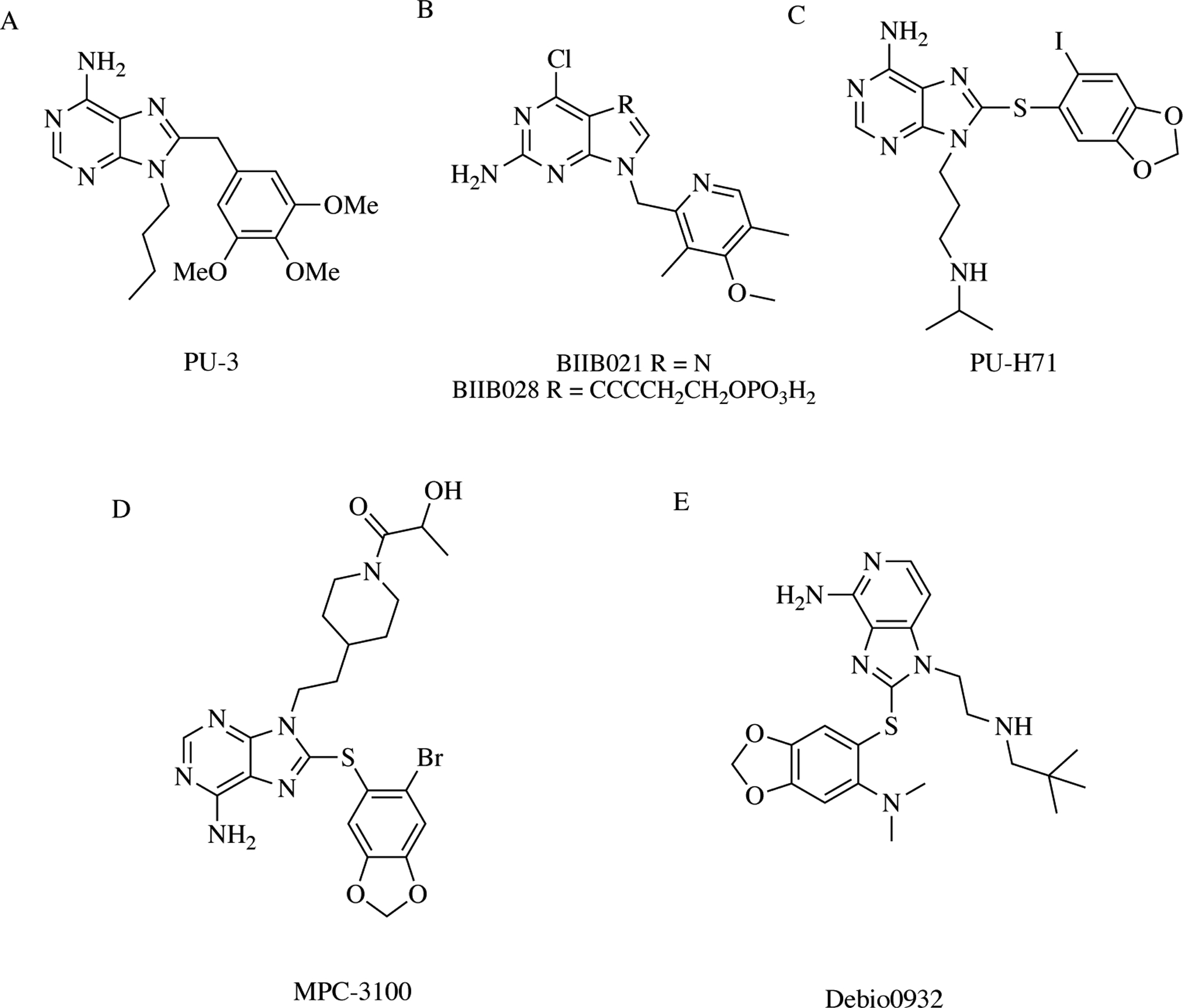
Purine-based Hsp90 inhibitors – A) PU-3 was the first fully synthetic Hsp90 inhibitor and first of the purine-based class. B) BIIB021 and BIIB028 modify the N-7 and N-9 positions of the purine base. C-E) PU-H71, MPC-3100, Debio0932 each add a unique 1,3-benzodioxole moiety to the N-8 position via a thioether bond and a variable substituent to the N-9 position.
Biogen, Inc. developed BIIB021 and BIIB028, wherein the latter is an optimization of the former (Fig. 4B) [34, 42–44]. In 2006, two Phase I clinical trials tested BIIB021 in chronic lymphocytic leukemia and advanced solid tumors. Although BIIB021 eventually progressed to two Phase II clinical trials by 2009, major limitations kept it from moving through the clinical pipeline. BIIB021 is an effective small molecule inhibitor, but lacks potency requiring higher doses to achieve biological effects. Additionally, the chemicals needed to synthesize the molecule on a large scale are toxic and an acceptable intravenous formulation is difficult to obtain. Due to these setbacks, Biogen, Inc. designed and developed BIIB028. In an effort to improve potency, tolerability, and physical properties, the pharmaceutical company used X-ray crystal structure analysis to identify the N-7 position on the purine scaffold as an optimal site for modification [43]. After developing a series of alkynol analogs, BIIB028 was designed as a prodrug and identified as a lead second generation clinical candidate. Although seen through to completion, it entered only one trial in 2008.
MCP-3100 and Debio0932, inhibitors developed by Myrexis and Curtis Pharma respectively, entered three trials collectively, and did not demonstrate any clinical promise to move beyond Phase II [34]. Conversely, a newer compound, PU-H71, discovered by the Chiosis group at Memorial Sloan Kettering, is now listed in five clinical trials as either active, recruiting, or terminated (80% active or recruiting) [45]. The Phase I trial that was terminated resulted from a drug supply shortage and not a dose limiting toxicity.
3.3. Benzamide Inhibitors
Serenx Inc. discovered the pyrazole-containg Hsp90 inhibitor, SNX-5422, using an ATP-affinity column (Fig. 5) [34, 46, 47]. In 2007, the first clinical trial testing SNX-5422 was posted. To-date there have been 13 trials, with one currently on-going. This compound shows clinical promise due to its bioavailability as a prodrug, but a major limitation is its pan-inhibitory activity against all Hsp90 isoforms, with the induction of the HSR and resulting dose escalation challenges.
Fig. 5.
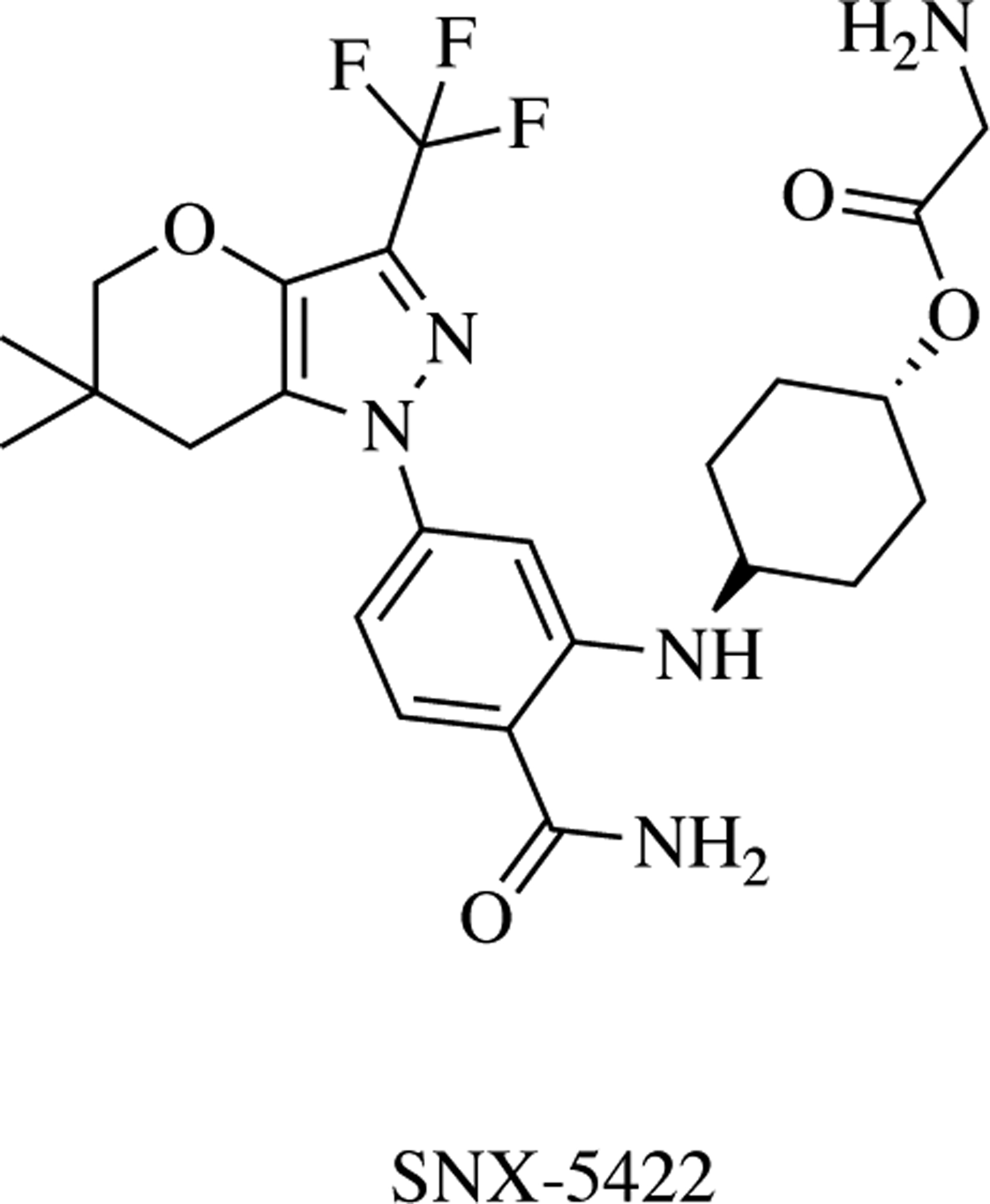
Benzamide Hsp90 inhibitor – SNX-5422 is a pyrazole-containing inhibitor and the only of its class.
3.4. Resorcinol Containing Inhibitors
Using a high-throughput screen, Chueng and colleagues at The Institute for Cancer Research in London were the first to identify resorcinol-containing small molecule Hsp90 inhibitors [48]. In an effort to optimize this class of molecules, scientists at Vernalis and Novartis used structure-based drug design to improve compound solubility by adding substituents, which led to the discovery of AUY922 (luminespib) (Fig. 6A) [34, 49]. Preclinical studies show that this Hsp90 inhibitor is active against tumor growth, angiogenesis, and metastasis in a xenograft mouse model [50, 51]. In fact, AUY922 entered 25 clinical trials, both Phase I and II, with two trials currently on-going and one recruiting as of February 2019. Similarly, scientists at Synta Pharmaceuticals modified the resorcinol scaffold to discover STA-9090 (Fig. 6B) [52]. The Synta group first presented this novel inhibitor at the AACR-NCI-EORT International Conference on Molecular Targets and Cancer Therapeutics. In 2010, they presented preclinical and clinical data at another international cancer conference showing that the second-generation Hsp90 inhibitor demonstrates higher potency in downregulating oncoproteins and pathways than first-generation ansamycinderived inhibitors. Interestingly, this Hsp90 inhibitor progressed into Phase III trials and entered nearly 40 trials in total, making it the most clinically evaluated Hsp90 inhibitor on record. Around the same time, scientists at Astex Therapeutics in the UK discovered another resorcinol-containing Hsp90 inhibitor, AT13387 (Fig. 6C) [53]. This compound was discovered as part of a fragment-based drug design approach by combining NMR and X-ray crystallography. Optimization of the compound series led to a resorcinol scaffold that has both high potency and ideal ligand efficacy [54]. AT13387 is currently in three active and four recruiting clinical trials. Finally, KW-2478 is an Hsp90 inhibitor discovered by scientists at Kyowa Hakko Kirin in Japan (Fig. 6D) [55]. The drug was tested in two clinical trials, one of which was completed in 2014 in combination with bortezomib, a proteasome inhibitor, in multiple myeloma. The primary objectives of the study were to establish safety of the combination therapy and assess overall response rates [56].
Fig. 6.
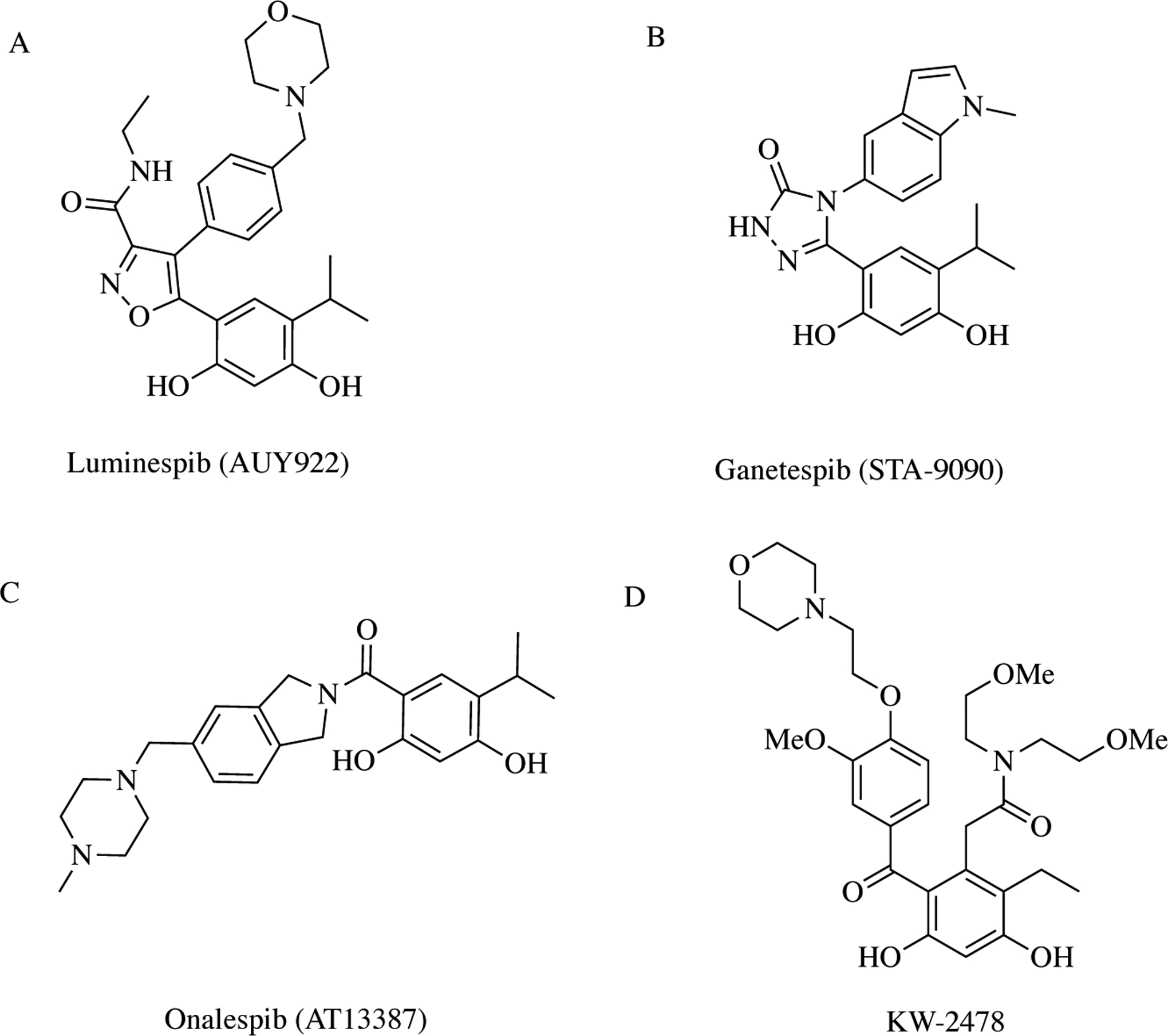
Resorcinol-containing Hsp90 inhibitors – All inhibitors in this class contain a resorcinol moiety. A-C) Luminespib, ganetespib, and onalespib add two substituents to the resorcinol ring at ortho-positions to the hydroxyl groups; one is an isopropanol moiety and the other is unique to each compound. D) KW-2478 adds three substituents to the resorcinol moiety – two ortho and one meta to the hydroxyl groups.
3.5. Miscellaneous Inhibitors
Other research groups have also designed and developed inhibitors of the molecular chaperone, but are unique or do not have publicly available structures and therefore do not fall into one of the general classes of Hsp90 inhibitors above. These compounds include XL888, HSP990, DS2248, and TAS-116 discovered by Exelixis, Inc., Novartis, Daiichi Sanko, Inc., and Taiho Pharmaceutical Co., respectively (Fig. 7) [57–59]. Interestingly, all of these inhibitors manifest the same mechanism of action as the aforementioned inhibitors—binding Hsp90’s NTD—except for TAS-116. This drug is unique, as it is the first reported compound to enter clinical trials that selectively binds to the cytosolic isoforms of Hsp90 (Hsp90α and Hsp90β) [34, 60]. Between 2014 and 2017, 61 patients with advanced solid tumors in Japan and the United Kingdom were enrolled in the first-in-human clinical trial testing of TAS-116. The primary objectives of the study aimed to identify the maximum tolerated dose, safety, and overall response rates in patients with advanced solid tumors taking TAS-116 as a monotherapy intervention [61]. In March 2019, results from the study revealed minor adverse events (e.g. Grade 1 or 2) related to Hsp90 inhibition and a positive antitumor effect against solid tumors including KIT wild-type gastrointestinal stromal tumors (GIST). Additionally, the study identified three oral dosing schedules for future trials testing TAS-116. There are two on-going trials in Japan evaluating this cellular compartment-selective Hsp90 inhibitor. The trials are testing TAS-116 in combination with nivolumab, an immune checkpoint PD-1 inhibitor, in metastatic solid tumors or alone in GIST patients (Phase 1 and Phase II, respectively) [62].
Fig. 7.
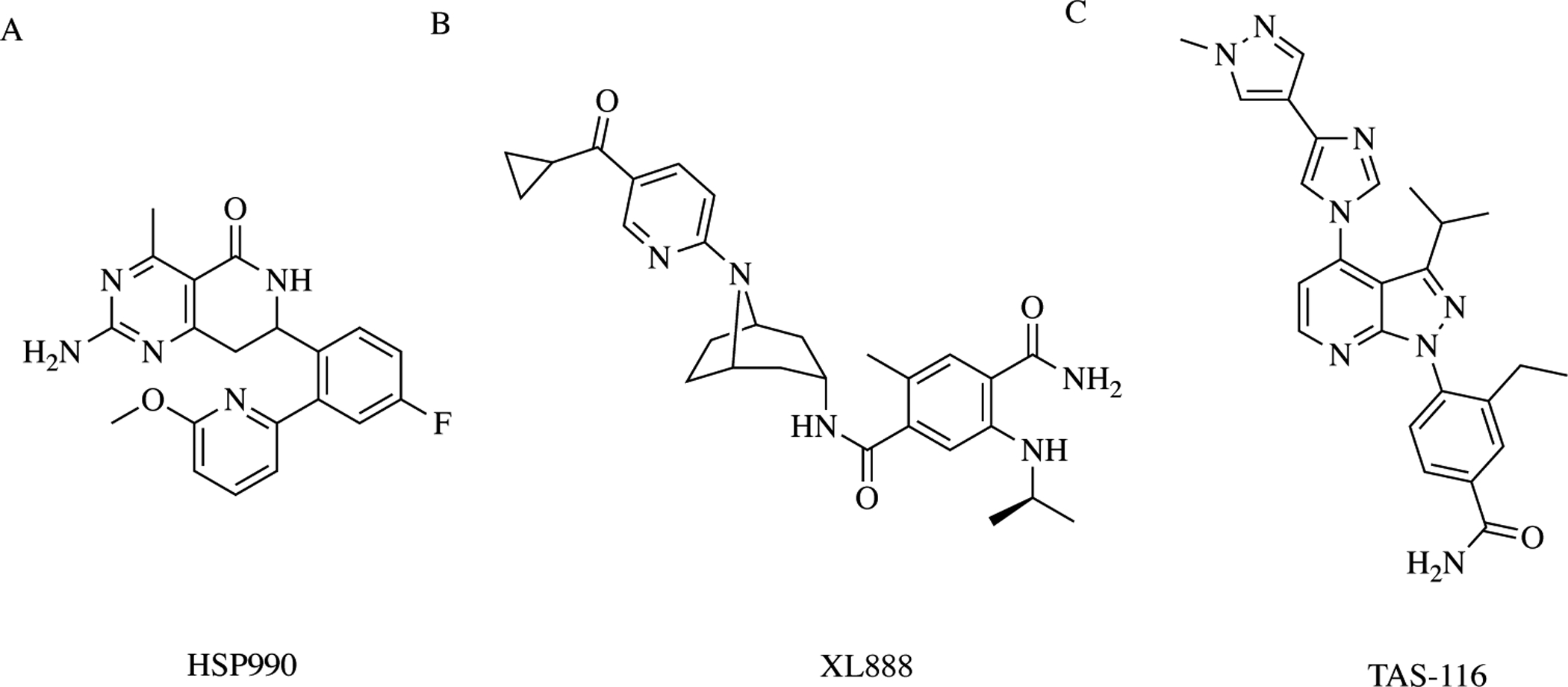
Miscellaneous Hsp90 inhibitors – HSP990 and XL888 do not fall into any of the chemical structure categories listed previously, but still target the N-terminal ATP-binding site. TAS-116 is a Hsp90α/β isoform-selective inhibitor.
Since the early 2000s, all Hsp90 inhibitors, except for TAS-116, that have entered clinical trials target the NTD of Hsp90. Clinical observations and trial data interpretation indicate that this approach carries antitumor efficacy but suffers from toxicities and adverse effects including hepatic, cardio, and ocular toxicities, as well as dose-scheduling limitations after induction of the HSR. Given the clinical efficacy potential Hsp90 inhibition carries, it would be ideal to develop novel Hsp90 inhibitors that can improve upon the pharmacodynamic and toxicity profiles of this therapeutic approach. Various efforts have been put forth to circumvent these setbacks, such as optimizing the approach to inhibit Hsp90’s function. Specifically, certain groups, like Taiho Pharmaceutical Co. and our research group, hypothesize that selectively targeting the cytosolic isoforms (Hsp90α and Hsp90β) will lead to better clinical outcomes and have the potential to be FDA approved. In the following section we elaborate upon the rational for employing a more selective approach to inhibit Hsp90’s function and provide examples to demonstrate recent advancements in the field.
4. INHIBITING THE HSP90 C-TERMINAL DOMAIN
Although GDA and RDC bind to the N-terminal ATP binding motif of Hsp90, recent studies have demonstrated the existence of a C-terminal ATP binding region as well [63–65]. Similarly, Hsp90 contains two different binding sites for proteins, allowing the chaperone to bind both co-chaperones and unfolded clients proteins at the C- and N-terminal regions [10, 23, 64, 66]. Unlike the NTD, inhibitors that competitively bind at the nucleotide binding site to abrogate ATPase activity, small molecules that target and bind the CTD disrupt the association of co-chaperones containing TPR-motifs. This ultimately leads to aberrant chaperone function [17]. A hallmark example of a CTD Hsp90 inhibitor is novobiocin, a DNA gyrase ATP-binding site inhibitor, and its subsequent analogue KU-174 (Fig. 8). Inhibition of the Hsp90 protein folding machinery by novobiocin also leads to destabilization of the multiprotein complex (Fig. 9), which results in ubiquitinylation of the client protein, proteasome mediated hydrolysis and in many cases, induction of apoptosis in numerous cancer cell types [64, 66]. A desirable characteristic of novobiocin is the lack of HSR induction, which is a major clinical drawback to all NTD Hsp90 inhibitors [67]. It is important to note that although novobiocin and most of its derivatives inhibit Hsp90’s function, there has been recent evidence supporting paradoxical ATPase activation. Chatterjee et al. report that the novobiocin derivative, KU-32, binds the CTD which in turn leads to a global change in chaperone structure. This structural shift not only promotes ATP binding, but increases ATPase activity [68]. In this context, KU-32 is viewed as a positive allosteric modulator of Hsp90 function and could be used as a tool to develop AD and PD therapies. In addition to disrupting protein-protein interactions (PPIs) between Hsp90 and co-chaperones with distinct chemical scaffolds, such as novobiocin, Rahimi et al. demonstrated blockade of heterocomplex chaperone PPIs with a unique small molecule, LB76 [69]. This molecule was designed de novo using an amino acid sequence specific to the MEEVD region of the TPR-motif on the CTD.
Fig. 8.
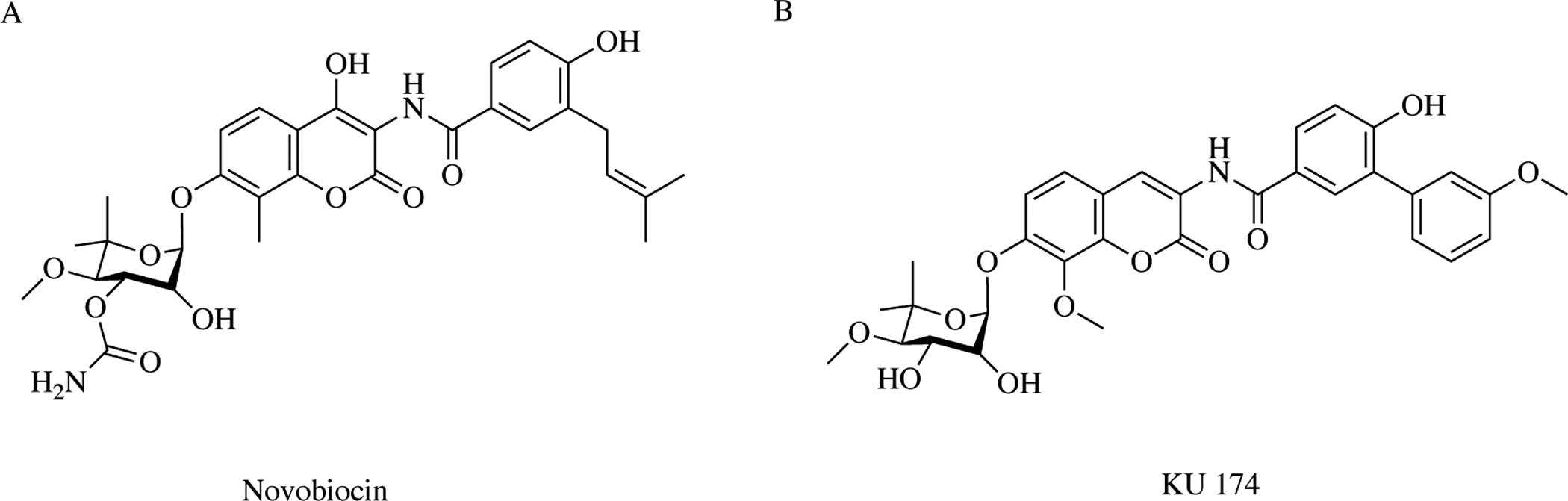
Structures of C-terminal Hsp90 inhibitors – The aminocoumarin antibiotic A) novobiocin derived from Steptomyces niveus and its analogue B) KU174. Compounds in this class share three common moieties: benzoic acid derivative, coumarin residue, and sugar novobiose.
Fig. 9. Hsp90 C-terminal inhibition –
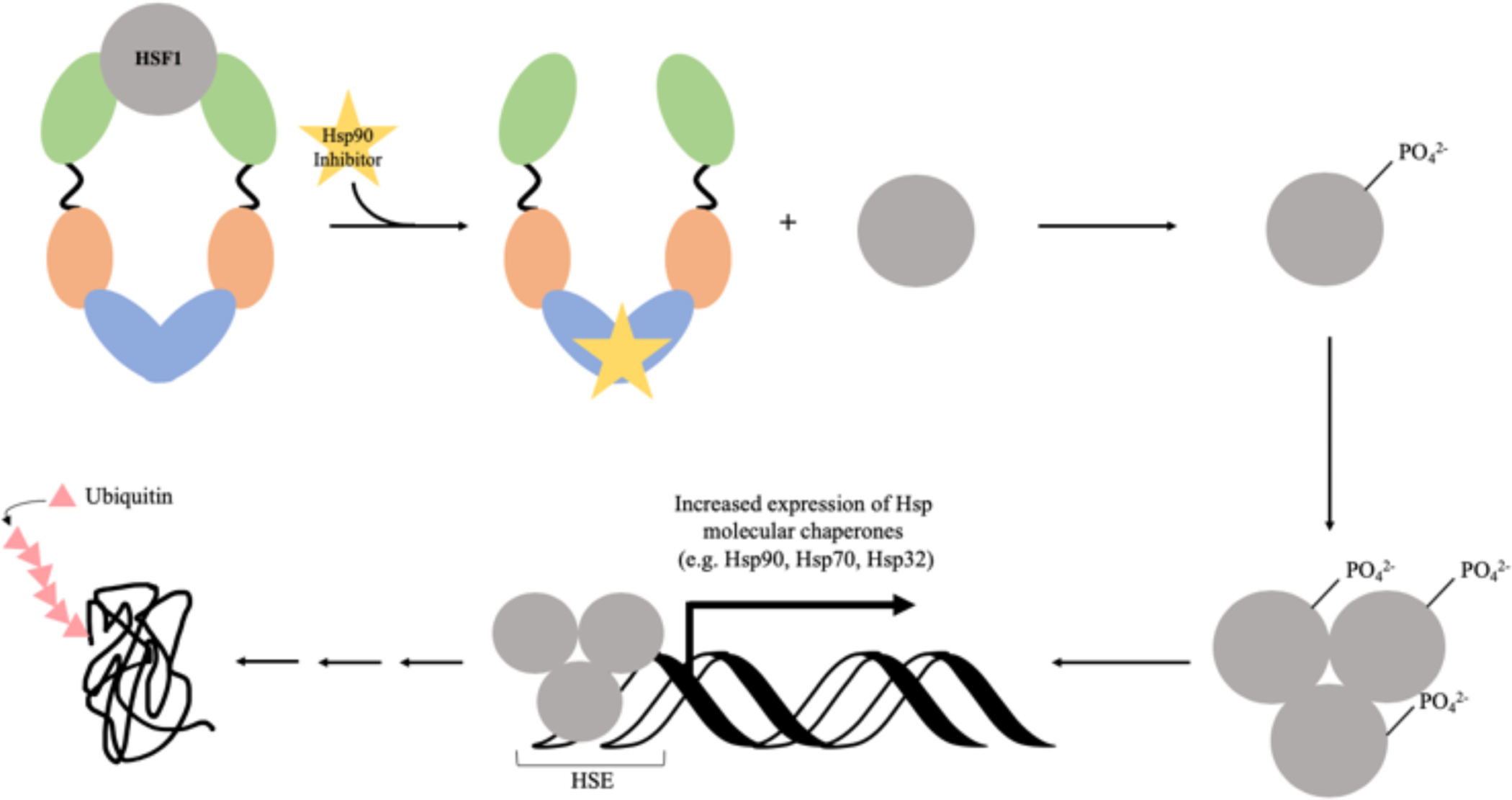
From upper left: Hsp90 chaperone and HSF1 transcription factor interaction is destabilized by C-terminal Hsp90 inhibitor binding. Once HSF1 is phosphorylated it associates with other phosphorylated HSF1 proteins and binds to DNA at its transcription element, HSE. After a tightly regulated process with several steps, this leads to ubiquitinylation of client proteins. *Original figure created by the authors
Prior to studies by Prodromou and coworkers in 2006, no crystal structure of full-length Hsp90 protein had been solved [23]. Two binding sites for ligands are suggested by this structure. One is located in the NTD and the other appears proximal to the dimerization domain. Clearly, a cocrystal structure of Hsp90 bound to novobiocin would be helpful towards further elucidation of the CTD binding site. However, the affinity of novobiocin for the CTD is too low for co-crystallization studies [64, 67, 70].
In 2004, Cox and coworkers demonstrated that the Hsp90-dependent transcription factor, aryl hydrocarbon receptor (AhR), was preferentially sensitive to the effects of NTD inhibitors and p23 concentration, whereas inhibition of the CTD with novobiocin remained unaffected by p23 concentration. Through subsequent studies, they determined that GDA and RDC were unable to overcome the effects of overexpressed p23, because NTD inhibitors competed for the same region. In contrast, they found that inhibition of the CTD with novobiocin was independent of p23 concentration [71].
Our group has since demonstrated these inhibitors have potent in vitro and in vivo efficacy in human xenograft tumors and do not initiate the HSR with Hsp70 upregulation [72–77]. These preclinical proof of concept studies provide supportive evidence that alternatives to NTD Hsp90 inhibition may have the ability to overcome the limitations of prior inhibitors in clinical trials and should be further validated [72–77].
5. ISOFORM-SELECTIVE INHIBITION
As aforementioned, the mammalian Hsp90 family of molecular chaperones is comprised of three subfamilies, with a total of four distinct isoforms – Hsp90α, Hsp90β, Grp94, and TRAP1 Table 1. To better understand the rationale for selective inhibition of Hsp90, the following subsection will provide information on the biological relevance of each isoform.
5.1. Biological Functions of Hsp90 Isoforms
5.1.1. Hsp90α and Hsp90β
As the two most abundant isoforms, Hsp90α and Hsp90β are predominantly found in the cytoplasm of mammalian cells. Despite this, there have also been reports of nuclear localization, albeit minor amounts [78]. A major distinction between the two isoforms is their expression profile. Hsp90α is the induced isoform, whereas Hsp90β is constitutively expressed. The major functions of the cytoplasmic isoforms are to aid in protein folding and prevent protein aggregation; in line with that of the Hsp90 family. Reported client proteins of Hsp90α and Hsp90β are involved in several cellular processes such as signaling pathways, survival, cell cycle, energy metabolism, and epigenetics – to name a few – making cytoplasmic Hsp90 isoforms indirectly involved in the regulation of these processes [79]. Moreover, several of these processes, if not all, are recognized as a hallmark of cancer [80]. Lastly, an extracellular form of Hsp90α (eHsp90), which is secreted from the cell, is implicated in invasion and migration or wound healing [29, 81]. Since eHsp90 has been reported to play a role in wound healing, this extracellular form of the chaperone could be an alternative target for novel anticancer therapeutics.
5.1.2. Grp-94
Unlike its cytoplasmic family members, Grp94, also known as endoplasmin, is found exclusively in the endoplasmic reticulum (ER) [82]. Here, it functions to orchestrate protein quality control on a small subset of proteins either secreted and/or membrane proteins [83]. Proteins that are misfolded in the ER are typically triaged to the Grp94 molecular chaperone machinery for proper refolding. On the contrary, if an ER protein is not properly folded it is translocated to the cytoplasm, where it can be marked for degradation. In addition to serving as a chaperone for a large number of ER-specific proteins, Grp94 also plays an important role as a Ca2+ binding protein[84, 85].
5.1.3. TRAP1
The last Hsp90 isoform, TRAP1 or tumor necrosis factor (TNF) receptor associated protein 1, is located in the mitochondria and was initially reported as the chaperone for TNF receptor 1, hence its name [86, 87]. In recent years, researchers have shown TRAP1’s function in the mitochondria to extend much further than proteostasis, and in fact, play a major role in mitochondrial homeostasis. For example, TRAP1 has been shown to be involved in the regulation of the organelle’s redox state [88]. Additionally, TRAP1’s involvement in diseases, such as cancer, has been speculated to result in the disruption of energy metabolism, which ties into the fact that Hsp90 is the chaperone for two proteins involved in the citric acid cycle [79, 89–91]. Additionally, this alteration or “metabolic rewiring” has been suggested to have an intimate interplay with epigenetics and Condelli et al. provide thorough perspectives on the matter [79]. In fact, Dr. Oliver Kramer, from Johannes Gutenberg University, presented work at the 9th ICHCM that detailed the use of Hsp90 inhibitors in combination with histone deacetylase 6 (HDAC6), an epigenetic “eraser” of acetyl groups from histones, in acute myeloid leukemia [29]. While the exact mechanism(s) in which Hsp90 is involved in altered metabolism and epigenetics has yet to be elucidated, we further emphasize here, the critical and indirect role the chaperone has in various cellular processes.
5.2. Rationale for Selective Inhibition
Now with a fundamental understanding of all four isoforms biological functions, it is apparent there is some overlap between them, but more importantly, it underscores the necessity to move away from “pan-inhibitors” and develop isoform-selective compounds. Moreover, this highlights an opportunity to target a specific isoform whose function is much more crucial in a given a disease state than another isoform. As previously mentioned, Hsp90 is responsible for the conformational maturation of a myriad of client proteins associated with all ten hallmarks of cancer, various neurodegenerative diseases, infections, and other disease states. Many of these client proteins depend upon a single Hsp90 isoform for their maturation, activation, and/or trafficking. Hsp90 has been shown to play an important role in tumorigenesis through the folding/activation of signaling kinases, steroid hormone receptors, tumor suppressors, and more [2]. For example, the mitogen activated protein kinase (MAPK) pathway kinase, B-Raf, is not only mutated in 60% of melanoma patients, but also is a client protein of the Hsp90 heterochaperone complex, suggesting that chaperone function helps to facilitate stabilization of this kinase which ultimately contributes in the mutated and overexpressed state to oncogenesis [92]. As mentioned previously, Hsp90 plays a critical role in neurodegenerative disorders such as AD and PD. In AD, Hsp90 exacerbates the formation of neurofibrillary tangles through the promotion of tau hyperphosphorylation; in PD, pharmacological induction of the chaperone machinery has been shown to prevent the self-assembly of Aβ aggregates [93, 94]. Recall that Hsp90α and Hsp90β are two of the four Hsp90 isoforms that primarily reside in the cytoplasm to fold nascent polypeptides. Grp-94, the Hsp90 isoform localized to the endoplasmic reticulum, has a much smaller client list and includes Toll-like Receptors (TLRs), integrins, insulin-like growth factors, and has been shown to play a role in tumor immunogenicity [95, 96]. TRAP1 is the mitochondrial isoform of Hsp90 that plays a role in the transition to aerobic glycolysis, accumulation of reactive oxygen species, and the maintenance of protein homeostasis [97].
While the mechanism of Hsp90 has been well explored and a variety of inhibitors have been developed, most inhibitors manifest pan-inhibitory activity against all four Hsp90 isoforms. Not only do pan-inhibitors serve little utility for the elucidation of the biological roles played by each Hsp90 isoform, but most have been removed from clinical evaluation due to toxicity concerns including hepatic, cardio, and/or ocular toxicities, as well as induction of the HSR [35, 98, 99]. Recent work has demonstrated that the human Ether a-go-go Related Gene (hERG) product, which is responsible for repolarization of the cardiac action potential, is dependent upon Hsp90α for its maturation and trafficking [99]. Furthermore, pan-inhibition of Hsp90 also induces the pro-survival HSR by activating HSF-1, which then induces the transcription of Hsp27, Hsp70, Hsp90, and other heat shock proteins [100]. The upregulation of Hsp90 requires an escalation in drug-dosing as well as significant scheduling difficulties. As a result of these observations, it was proposed that isoform-selective inhibition of Hsp90 may provide a viable way to mitigate the detriments and complications observed in clinical trials with pan-inhibition of all four Hsp90 isoforms. Furthermore, isoform-selective inhibitors provide a mechanism to elucidate the biological role played by each isoform in various diseases. Given the aforementioned dependence of clients upon individual isoforms, isoform-selective inhibitors could be used to target specific disease states in which one isoform may play a presiding role
6. GRP94-SELECTIVE INHIBITORS
Differences between the N-terminal ATP-binding pockets of Grp94 and Hsp90 were first elucidated using N-ethylcarboxamidoadenosine (NECA) as a competitive inhibitor in an adenosine-based ligand binding assay (Fig. 10) [101]. Despite structural similarities between Grp94 and Hsp90, and the large abundance of Hsp90 as compared to Grp94 within cells, NECA was able to selectively bind suggesting that Grp94 could be selectively targeted [101]. Grp94 was the first Hsp90 isoform for which isoform-selective inhibitors were pursued.
Fig. 10.
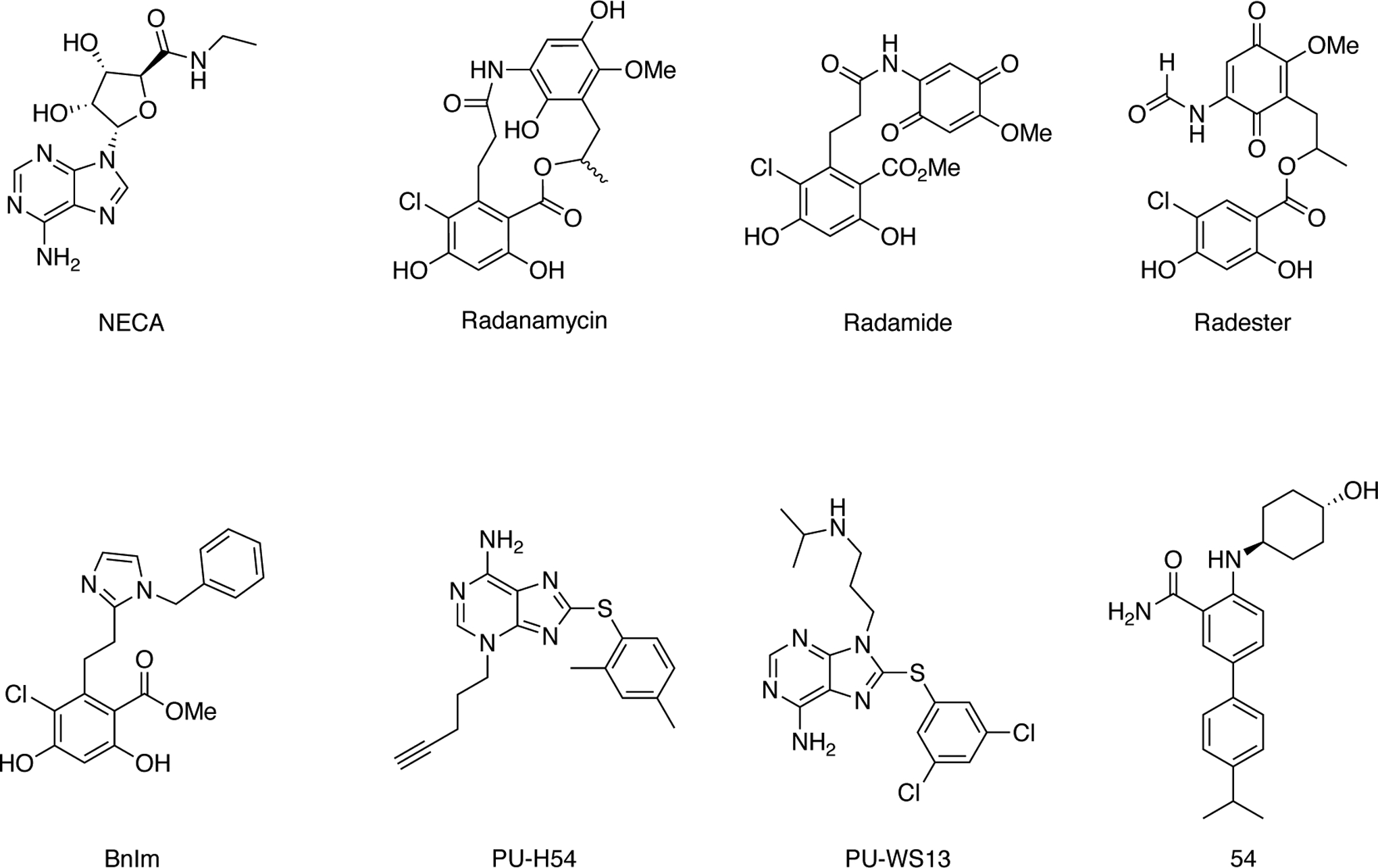
Structures representing various classes of Grp94-selective inhibitors – Radanamycin, radamide, and radester are natural product derived. BnIm maintains the resorcinol moiety but introduces an imidazole ring. The PU compounds represent the purine class, and 54 is a novel inhibitor of the benzamide class.
One approach used to develop pan-Hsp90 inhibitors combined the structural features of GDA and RDC, two well established natural product Hsp90 inhibitors, to form chimeric molecules. These chimeric molecules retained the key binding features of each natural product, but reduced the structural complexity associated with their preparation. The first three chimeric inhibitors produced were radanamycin, radamide, and radester. All three of these chimeric inhibitors manifested anti-proliferative activity against MCF-7 human breast cancer cells and induced the degradation of Hsp90-dependent client proteins, HER2 and Raf (Fig. 10) [102–104]. Co-crystallization of radamide bound to both Grp94 and Hsp90 revealed a unique 5’-extension pocket that was present only in Grp94, which was produced by a five amino acid insertion into the primary sequence that led to formation of this unique pocket. Otherwise, Grp94 and other Hsp90 isoforms share ~85% identity in this region [105]. The cocrystal structures demonstrated that selectivity was conferred through isomerization of the radamide bond, resulting in formation of the cis-amide, which highlighted the need to incorporate a cis-amide bioisostere into the structure of radamide to develop Grp94-selective inhibitors [105].
BnIm was produced by replacement of the cis-amide with a bioisoteric imidazole ring that mimicked the amide heteroatoms, while projecting the two appendages into a cis-orientation (Fig. 10) [106]. A functional assay was developed to evaluate the effect of BnIm on TLR trafficking to the cell surface, which is Grp94-dependent [106, 107]. No cytotoxic effects were observed with BnIm and no degradation of Hsp90α/Hsp90β client proteins were observed at concentrations that affected TLR trafficking, which demonstrated a considerable selectivity of BnIm for Grp94 versus the cytosolic isoforms [106]. Second generation Grp94-selective inhibitors established structure-activity relationships for the BnIm scaffold and replaced the imidazole ring with other heterocycles to improve affinity, while modifying the benzyl appendage to improve selectivity and affinity. It was determined that meta-substitutions were not tolerated, while orthoand para-substitutions improved selectivity [108]. For example, the incorporation of a bromine at the para-position demonstrated efficacy in animal studies for the treatment of glaucoma, as the accumulation of mutant myocilin levels is Grp94-dependent [109, 110]. Modification of the imidazole ring via replacement with a phenyl group resulted in a 2-fold improvement in affinity versus BnIm, and manifested an apparent Kd of 0.63 μM along with a 32-fold selectivity for Grp94 versus Hsp90α [108] Fluorination of this molecule at the para-position yielded a compound that manifested an apparent Kd of 0.54 μM along with 73-fold selectivity for Grp94 [111]. Resorcinol-based Grp94 inhibitors also took advantage of the hydrophobic S2 sub-pocket and ultimately led to compounds that manifested low nanomolar affinity, but unfortunately only ~10-fold selectivity for Grp94 [112].
Because of Grp94’s distinct extension pockets, it accommodated a wide variety of purine-based chemical tools that are also selective for this isoform. Chiosis and co-workers evaluated an in-house library of ~130 purine analogs in a fluorescence polarization assay to identify compounds that bind Grp94 with higher affinity than Hsp90α. While many of the compounds displayed a model of the purine-scaffold ligand PU-H54 bound to both Hsp90 and Grp94, the team demonstrated that the ligand bound to each isoform in a different orientation and caused Phe199 to swing away and expose a major hydrophobic cleft in Grp94 that is not available in Hsp90α [113]. While a similar cleft exists in Hsp90α, access to Hsp90α’s binding pocket is blocked by Phe138. Compounds were found to engage this site and stabilize binding, which conferred Grp94 selectivity [113]. PU-WS13, a Grp94-selective purine derivative bound to this pocket and was shown to be nontoxic in in vivo assays for target modulation (Fig. 10). Furthermore, PU-WS13 disrupted the architecture of the oncoprotein, HER2, at the cell surface. Treatment of HER2-overexpressing SKBr3 human breast cancer cells with PU-WS13 led to a substantial decrease in HER2 levels, highlighting a new role for Grp94 in disease states that are dependent on mutated, modulated, or otherwise modified cell surface receptors. These Grp94-selective inhibitors do not induce the HSR, unlike the pan-inhibitors [113].
Recently, a Grp94 selective inhibitor, Compound 54, with an IC50 of 2 nM and over 1000-fold selectivity for Grp94 versus Hsp90α was developed by Xu and coworkers [114]. In order to exploit the aforementioned Phe199 shift and to confer Grp94 selectivity, the group began with the benzamide moiety and introduced a phenyl ring at the metaposition as their lead compound. SAR investigations led to the introduction of an isopropyl appendage at the fourposition of the benzene ring and a cyclohexanol with an amine linker at the ortho-position of the benzamide scaffold. The compound was shown to induce the “ligand-induced” Phe199 shift, validating the mechanism for Grp94 selective inhibition. The compound was found to be efficacious in mouse models of ulcerative colitis and did not effect Hsp70 expression levels [114].
7. HSP90α AND HSP90β-SELECTIVE INHIBITORS
Cytosolic Hsp90 includes the inducible Hsp90α isoform and constitutively expressed Hsp90β, which are the most conserved among the four isoforms. In an attempt to avoid the profile exhibited by pan-inhibitors, compounds with selectivity toward the cytosolic isoforms of Hsp90 have been pursued with the intent of narrowing the list of clients in an effort to reduce on-target side effects that are observed with pan-Hsp90 inhibitors. Previous studies investigated the efficacy of Hsp90α/β inhibition in Huntington’s Disease and demonstrated that selective siRNA knockdown of cytosolic Hsp90 is sufficient to decrease mutant Huntington protein (mHtt) levels in HEK cells, which illustrates that isoform-selective inhibition leads to the clearance of diseasepromoting aggregates, without affecting the function of the endoplasmic reticulum and mitochondrial chaperones [115]. The cytosolic Hsp90 inhibitor, SNX-0723, demonstrated about 100-fold selectivity for cytosolic Hsp90 isoforms versus Grp94 and about 300-fold selectivity versus TRAP1 (Fig. 11). However, SNX-0723 manifests a similar affinity for both Hsp90α and Hsp90β [115, 116]. Ernst and Conor revealed SNX-0723 as a promising lead compound for further optimization due to its CNS permeability as well as its selectivity for both cytosolic Hsp90 isoforms. Subsequent work by these researchers led to the identification of compound 31, a benzolactam-hydroindolone derivative that contains a cyclopentyl substituent and exhibits similar pharmacokinetics as SNX-0723, but less cellular toxicity as a result of the 1,000-fold selectivity for cytosolic Hsp90s versus Grp94 and TRAP1 [117]. Similarly, TAS-116, which was prepared by researchers at Taiho Pharmaceuticals, Co. Ltd., was shown to be a cytosolic Hsp90 inhibitor that manifested a large selectivity over the non-cytosolic isoforms (Fig. 11). TAS-116 induced the degradation of Hsp90 clients and reduced tumor burden in human xenograft mouse models, indicating that inhibition of cytosolic Hsp90 alone has the potential to exhibit promising anticancer activity. TAS-116 moved into phase I clinical trials to define the maximum tolerated dose, pharmacokinetics, pharmacodynamics and preliminary antitumor activity of this orally available and selective inhibitor. While ocular disturbances are common with Hsp90 inhibitors, those observed in this trial were limited to grade 1 and a partial response was observed in patients with various tumors and mutation statuses [61]. While compounds have been prepared to manifest selectivity for both cytosolic Hsp90 isoforms as compared to Grp94 and TRAP1, the generation of compounds that are selective for either Hsp90α or Hsp90β proved to be a more challenging task. Hsp90α and Hsp90β share approximately 95% identity within the N-terminal ATP binding pocket, and differ by only two amino acids; Hsp90β replaces Hsp90α’s serine and isoleucine residues at positions 52 and 91 with alanine and leucine residues, respectively [118]. It has been demonstrated that there are three conserved water molecules that play different roles in Hsp90α versus Hsp90β due to the replacement of serine with alanine in Hsp90β. Based on this observation, this differential hydrogen bonding network was exploited to develop the first Hsp90β-selective inhibitors. KUNB31 was the first Hsp90β-selective N-terminal inhibitor produced and was shown to manifest low micromolar anti-proliferative activity, induce the degradation of Hsp90β-dependent clients, and did not induce the pro-survival HSR (Fig. 11) [118]. Studies have also suggested that Grp94-selective compounds based on the purine scaffold (PU) also exhibit a 3-to 5-fold selectivity for Hsp90α versus Hsp90β [113]. The proposed mechanism for selectivity of the PU compounds is based on a conserved N-terminus that is intercepted during quaternary transitions between isoforms, suggesting that selectivity must be conferred through a region outside the N-terminus. Further work toward the development of Hsp90α-and Hsp90β-selective inhibitors is ongoing.
Fig. 11.
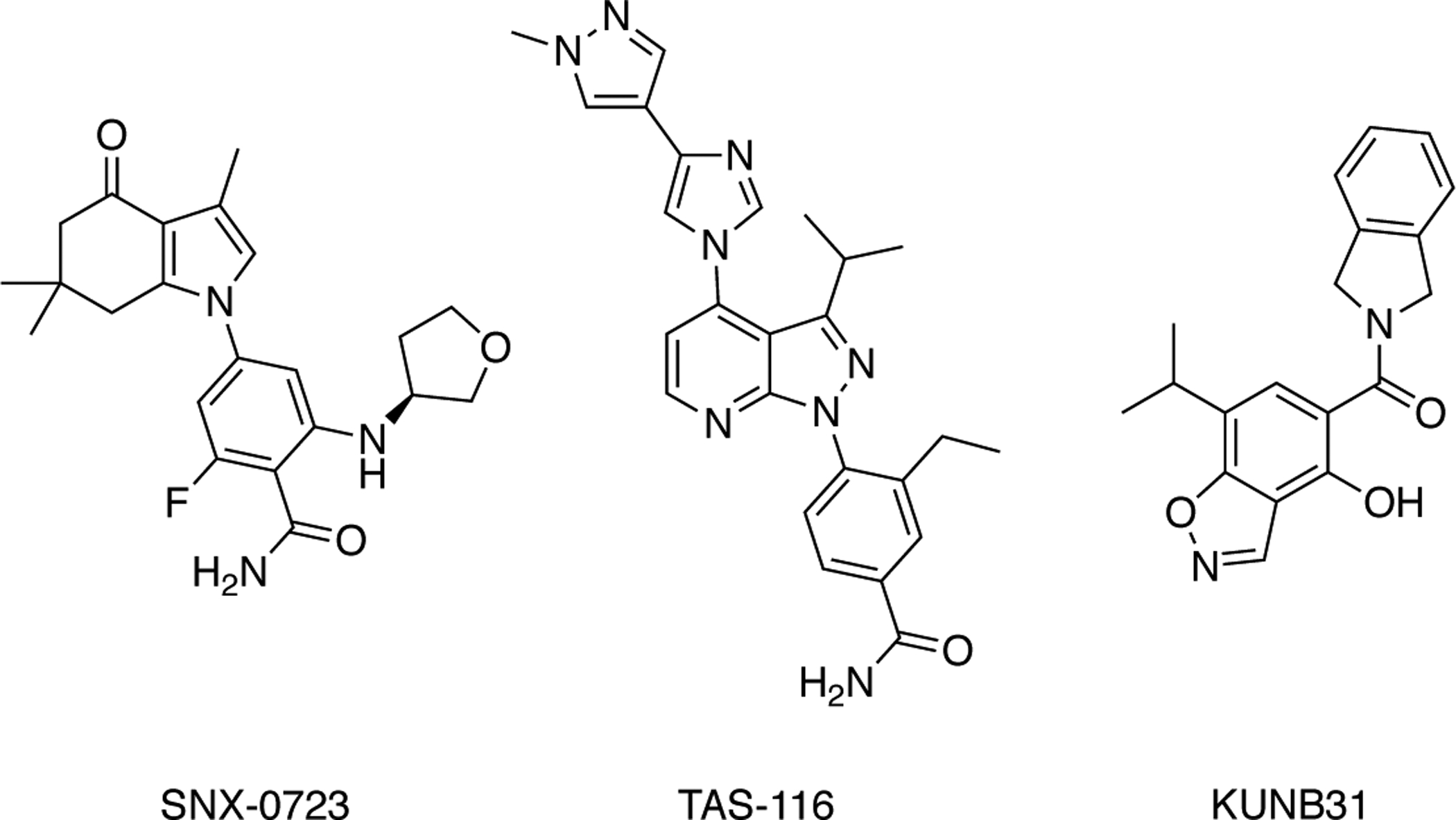
Hsp90α/β-selective inhibitors – TAS-116 and KUNB31, the first N-terminal, Hsp90α/β and Hsp90β isoform-selective inhibitors, respectively.
8. TRAP1-SELECTIVE INHIBITORS
Typical pan-inhibitors of Hsp90 have not shown efficient inhibition of TRAP1 in the mitochondria due to a lack of permeability and drug accumulation within this organelle. The long-standing strategy for TRAP1 inhibition has been to use pan-inhibitors of Hsp90 that are targeted to the mitochondria. Shepherdin, a cell-permeable peptidomimetic, was the first rationally designed molecule to enter the mitochondria and target TRAP1 (Fig. 12) [119]. When a highly positively charged moiety was placed at the N-terminus of shepherdin, it enabled mitochondrial penetration and induced extensive cell death through compromising mitochondrial integrity, which caused swelling, membrane depolarization and subsequent release of cytochrome c [120]. Given the peptidomimetic nature of shepherdin, it manifested a short half-life and was capable of inducing an immunogenic response. Shepherdin also targets cytosolic Hsp90s and induces the degradation of several Hsp90 clients [113, 121]. Limitations associated with shepherdin opened the door for alternative TRAP1 inhibitors that contain a moiety to target pan-inhibitors of Hsp90, such as GDA, to the mitochondria. The first inhibitors of this class were formed by the inclusion of a triphenylphosphonium (TPP) or cyclic guanidinium moiety onto GDA. These compounds disrupted mitochondrial function, induced cell death in cancer cell lines, and affected tumor growth in xenograft mouse models [122]. Other compounds soon followed and utilized inhibitors attached to cationic species, including SMTIN-P01 (Fig. 12). SMTIN-P01 replaced the corresponding ammonium group on PU-H71 with the mitochondrial permeating TPP moiety [123]. SMTIN-P01 induced membrane depolarization and demonstrated cytotoxicity in cancer cells, which implicated TRAP1’s role in carcinogenesis. While this method of inhibitor development can traffic pan-inhibitors to the mitochondria, it is not truly an isoform-selective process. TRAP1 and other isoforms of Hsp90 are highly conserved, but there are still significant differences within the ATP-binding regions that make TRAP1-selective inhibitors possible. However, compounds may not target TRAP1 with the same affinity observed for the cytosolic Hsp90s. A lack of selectivity may also lead to off target effects as compounds may interact with cytosolic chaperones. Kang and co-workers sought to minimize the binding to cytosolic Hsp90 and to maximize affinity for TRAP1 by the incorporation of guanine mimics that contain a piperonyl side chain. Modification of the pyridine ring to provide the pyrazolopyrimidine scaffold with a pyridinyl appendage demonstrated stronger inhibitory activities against TRAP1 than the corresponding piperonylcontaining side chains, but comparable inhibitory activity against cytosolic Hsp90. Further modification of the pyridinyl group led to potent inhibitors of TRAP1 (79 nM) as compared to Hsp90 (698 nM). In fact, DN401, which exhibited ~9-fold selectivity for TRAP1 versus Hsp90, is the most selective TRAP1 inhibitor reported to date (Fig. 12) [124].
Fig. 12.
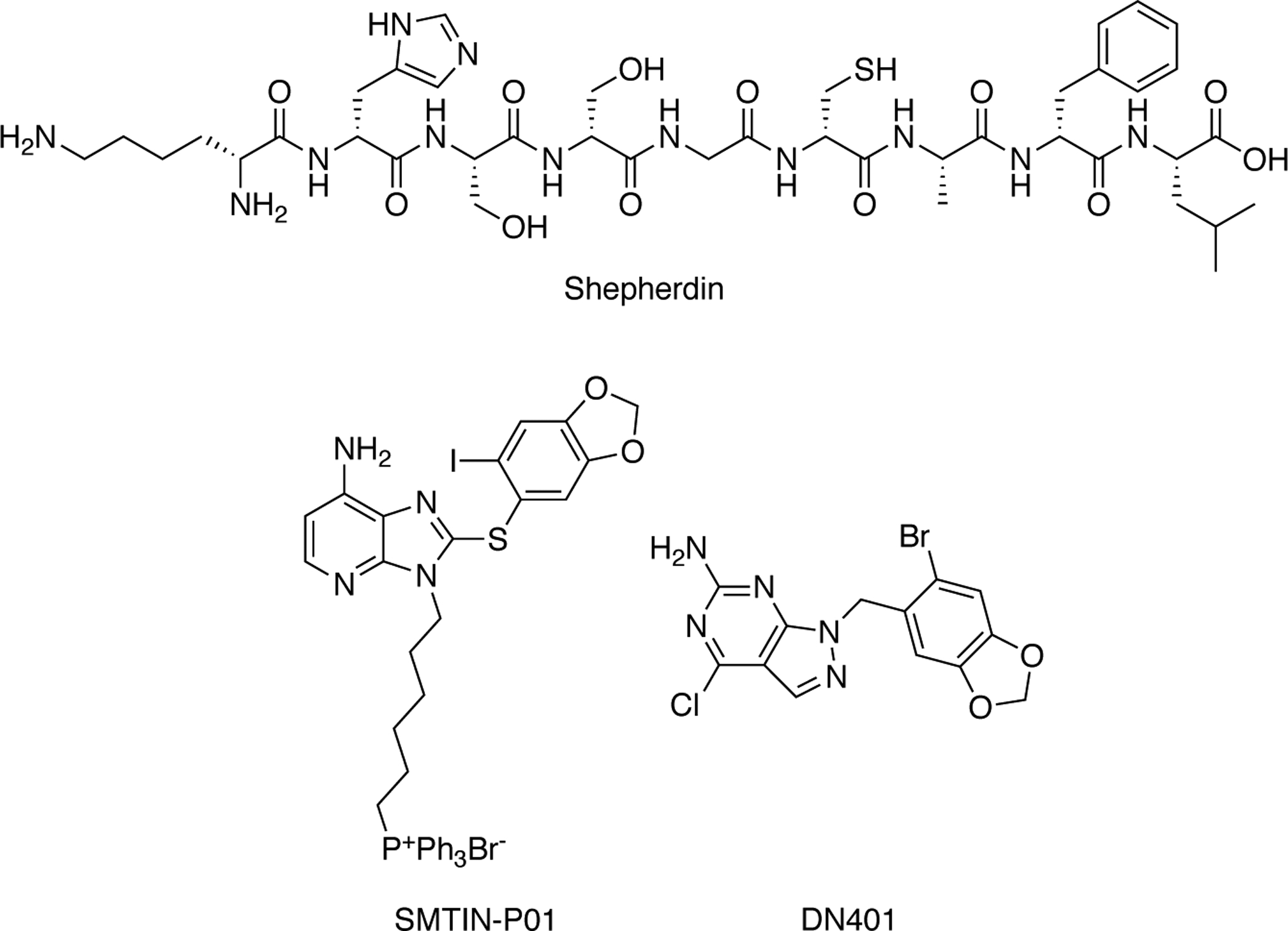
TRAP1-selective inhibitors – Shepherdin is the minimal nine amino acid sequence of survivin named for its binding to the “shepherding” protein Hsp90. SMTIN-PO1 represents compounds conjugated to mitochondrial targeting moieties, and DN401 is the most potent and selective TRAP1 inhibitor reported.
CONCLUSION
Hsp90 and its isoforms are shown to play a critical role in cancer, neurodegenerative disorders, and other disease states, illustrating that its pharmacological targeting can have profound implications for the treatment of these illnesses. Most attempts at Hsp90 inhibition have not seen success in clinical trials. Despite innovative chemistry and targeting these compounds all target the N-terminal binding site and therefore are pan-inhibitors of all four Hsp90 isoforms. These inhibitors—including natural products and their derivatives, purine-based inhibitors, benzamide inhibitors and resorcinol containing inhibitors—have time-and-again failed progression in clinical trials due to the detrimental toxicities associated with Hsp90 pan-inhibition. In fact, over half of the clinical trials with these Hsp90 Inhibitors did not progress past Phase I due to the cardiac, hepatic, and ocular toxicities commonly associated with Hsp90 pan-inhibition, making progress through in-human clinical trials difficult thus far.
Although Hsp90 pan-inhibitors may not be the key to a combinatorial attack on a variety of disease-causing pathways, non-traditional modulation of Hsp90, namely C-terminal inhibition or isoform-selective inhibition, remain a viable method to achieve the desired anticancer effect without harmful side effects seen with pan-inhibition. Inhibition of the cytosolic Hsp90 isoforms maintains the integrity of the endoplasmic reticulum and the mitochondria by leaving Grp94 and TRAP1, respectively, unaffected. Inhibition of Hsp90β in particular leaves the hERG channel unaffected and avoids induction of the pro-survival HSR. Grp94 and TRAP1 inhibition allow for the targeting of particular disease states in which their isoforms are implicated. Further, pre-clinical validation and advancement of these novel C-terminal and isoform-selective inhibitors will generate exciting new anticancer compounds to move into clinical trials that could overcome the challenges of prior Hsp90 inhibitors, providing much-needed novel anti-cancer drug compounds for patients.
ACKNOWLEDGEMENTS
JS and TC contributed equally to writing the manuscript. MSC and BSB revised the text as needed.
FUNDING
The sources of funding that supported in part the work and effort completed on the manuscript include the National Cancer Institutes (R01CA216919 - BSB, MSC; R01CA213566 - BSB, MSC; R01CA120458 - BSB, MSC) and the University of Michigan Genetics Training Program (T32GM007544 - JS).
LIST OF ABBREVIATIONS
- AD
Alzhiemer’s disease
- AhR
aryl hydrocarbon receptor
- CTD
C-terminal domain
- eHsp90
extracellular Hsp90
- ER
endoplasmic recticulum
- GDA
geldanamycin
- GHKL
Gyrase, Hsp90, Histidine kinase, MutL
- GIST
gastrointestinal stromal tumors
- HDAC6
histone deacetylase 6
- hERG
human ether a-go-go related gene
- HSE
heat shock element
- HSF-1
heat shock factor 1
- Hsp
heat shock protein
- Hsp90
90kDa heat shock protein
- HSR
heat shock response
- ICHCM
Int’l Conference Hsp90 Chaperone Machinery
- MAPK
mitogen activated protein kinase
- MD
middle domain
- NECA
N-ethylcarboxamidoadenosine
- NTD
N-terminal domain
- PD
Parkinson’s disease
- PPI
protein-protein interactions
- PU
purine scaffold
- RDC
radicicol
- TLR
Toll-like receptors
- TNF
tumor necrosis factor
- TPP
triphenylphosphonium
- TPR
tetratricopeptide
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
There are no conflicts of interest (JS, MSC, TC, BSB).
REFERENCES
- [1].Miyata Y; Nakamoto H; Neckers L The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des, 2013, 19(3), 347–365. [ 10.2174/138161213804143725] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Whitesell L; Lindquist SL HSP90 and the chaperoning of cancer. Nat. Rev. Cancer, 2005, 5(10), 761–772. [ 10.1038/nrc1716] [DOI] [PubMed] [Google Scholar]
- [3].Ritossa FM A new puffing pattern induced by temperature shock an DNP in Drosophila. Experientia, 1962, 18, 571–573. [ 10.1007/BF02172188] [DOI] [Google Scholar]
- [4].McKenzie SL; Henikoff S; Meselson M Localization of RNA from heat-induced polysomes at puff sites in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA, 1975, 72(3), 1117–1121. [ 10.1073/pnas.72.3.1117] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ritossa FM New puffs induced by temperature shock, DNP and salicilate in salivary chromosomes of D. melanogaster. Drosoph. Inf. Serv, 1963, 37, 122–123. [Google Scholar]
- [6].Ritossa FM Experimental activation of specific loci in ploytene chromosomes of drosophila. Exp. Cell Res, 1964, 35, 601–607. [ 10.1016/0014-4827(64)90147-8] [DOI] [PubMed] [Google Scholar]
- [7].Tissières A; Mitchell HK; Tracy UM Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J. Mol. Biol, 1974, 84(3), 389–398. [ 10.1016/0022-2836(74)90447-1] [DOI] [PubMed] [Google Scholar]
- [8].Bagatell R; Paine-Murrieta GD; Taylor CW; Pulcini EJ; Akinaga S; Benjamin IJ; Whitesell L Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res, 2000, 6(8), 3312–3318. [PubMed] [Google Scholar]
- [9].Yufu Y; Nishimura J; Nawata H High constitutive expression of heat shock protein 90 alpha in human acute leukemia cells. Leuk. Res, 1992, 16(6–7), 597–605. [ 10.1016/0145-2126(92)90008-U] [DOI] [PubMed] [Google Scholar]
- [10].Prodromou C The ‘active life’ of Hsp90 complexes. Biochim. Biophys. Acta, 2012, 1823(3), 614–623. [ 10.1016/j.bbamcr.2011.07.020] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kamal A; Thao L; Sensintaffar J; Zhang L; Boehm MF; Fritz LC; Burrows FJ A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature, 2003, 425(6956), 407–410. [ 10.1038/nature01913] [DOI] [PubMed] [Google Scholar]
- [12].Yano M; Naito Z; Tanaka S; Asano G Expression and roles of heat shock proteins in human breast cancer. Jpn. J. Cancer Res, 1996, 87(9), 908–915. [ 10.1111/j.1349-7006.1996.tb02119.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Whitesell L; Mimnaugh EG; De Costa B; Myers CE; Neckers LM Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA, 1994, 91(18), 8324–8328. [ 10.1073/pnas.91.18.8324] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu Y; Lindquist S Heat-shock protein hsp90 governs the activity of pp60v-src kinase. Proc. Natl. Acad. Sci. USA, 1993, 90(15), 7074–7078. [ 10.1073/pnas.90.15.7074] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xu Y; Singer MA; Lindquist S Maturation of the tyrosine kinase c-src as a kinase and as a substrate depends on the molecular chaperone Hsp90. Proc. Natl. Acad. Sci. USA, 1999, 96(1), 109–114. [ 10.1073/pnas.96.1.109] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim YS; Alarcon SV; Lee S; Lee MJ; Giaccone G; Neckers L; Trepel JB Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem, 2009, 9(15), 1479–1492. [ 10.2174/156802609789895728] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schopf FH; Biebl MM; Buchner J The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol, 2017, 18(6), 345–360. [ 10.1038/nrm.2017.20] [DOI] [PubMed] [Google Scholar]
- [18].Murphy MP; LeVine H III Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis, 2010, 19(1), 311–323. [ 10.3233/JAD-2010-1221] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stefanis L α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med, 2012, 2(2)a009399 [ 10.1101/cshperspect.a009399] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kalia LV; Kalia SK α-Synuclein and Lewy pathology in Parkinson’s disease. Curr. Opin. Neurol, 2015, 28(4), 375–381. [ 10.1097/WCO.0000000000000215] [DOI] [PubMed] [Google Scholar]
- [21].Csermely P; Schnaider T; Soti C; Prohászka Z; Nardai G The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther, 1998, 79(2), 129–168. [ 10.1016/S0163-7258(98)00013-8] [DOI] [PubMed] [Google Scholar]
- [22].Sreedhar AS; Kalmár E; Csermely P; Shen YF Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett, 2004, 562(1–3), 11–15. [ 10.1016/S0014-5793(04)00229-7] [DOI] [PubMed] [Google Scholar]
- [23].Prodromou C; Roe SM; O’Brien R; Ladbury JE; Piper PW; Pearl LH Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell, 1997, 90(1), 65–75. [ 10.1016/S0092-8674(00)80314-1] [DOI] [PubMed] [Google Scholar]
- [24].Meyer P; Prodromou C; Hu B; Vaughan C; Roe SM; Panaretou B; Piper PW; Pearl LH Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell, 2003, 11(3), 647–658. [ 10.1016/S1097-2765(03)00065-0] [DOI] [PubMed] [Google Scholar]
- [25].Minami Y; Kimura Y; Kawasaki H; Suzuki K; Yahara I The carboxy-terminal region of mammalian HSP90 is required for its dimerization and function in vivo. Mol. Cell. Biol, 1994, 14(2), 1459–1464. [ 10.1128/MCB.14.2.1459] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jhaveri K; Taldone T; Modi S; Chiosis G Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta, 2012, 1823(3), 742–755. [ 10.1016/j.bbamcr.2011.10.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yuno A; Lee MJ; Lee S; Tomita Y; Rekhtman D; Moore B; Trepel JB Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol, 2018, 1709, 423–441. [ 10.1007/978-1-4939-7477-1_29] [DOI] [PubMed] [Google Scholar]
- [28].Garg G; Khandelwal A; Blagg BS Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects. Adv. Cancer Res, 2016, 129, 51–88. [ 10.1016/bs.acr.2015.12.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Blair LJ; Genest O; Mollapour M The multiple facets of the Hsp90 machine. Nat. Struct. Mol. Biol, 2019, 26(2), 92–95. [ 10.1038/s41594-018-0177-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soga S; Akinaga S; Shiotsu Y Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr. Pharm. Des, 2013, 19(3), 366–376. [ 10.2174/138161213804143617] [DOI] [PubMed] [Google Scholar]
- [31].Lee BL; Rashid S; Wajda B; Wolmarans A; LaPointe P; Spyracopoulos L The Hsp90 Chaperone: 1H and 19F Dynamic Nuclear Magnetic Resonance Spectroscopy Reveals a Perfect Enzyme. Biochemistry, 2019, 58(14), 1869–1877. [ 10.1021/acs.biochem.9b00144] [DOI] [PubMed] [Google Scholar]
- [32].Gorska M; Popowska U; Sielicka-Dudzin A; Kuban-Jankowska A; Sawczuk W; Knap N; Cicero G; Wozniak F Geldanamycin and its derivatives as Hsp90 inhibitors. Front. Biosci, 2012, 17, 2269–2277. [ 10.2741/4050] [DOI] [PubMed] [Google Scholar]
- [33].Huryn DM; Wipf P Natural Product Chemistry and Cancer Drug Discovery Cancer Drug Design and Discovery, 2nd ed; Neidle S, Ed.; Academic Press: San Diego, 2014, pp. 91–120. [ 10.1016/B978-0-12-396521-9.00003-6] [DOI] [Google Scholar]
- [34].Biamonte MA; Van de Water R; Arndt JW; Scannevin RH; Perret D; Lee WC Heat shock protein 90: inhibitors in clinical trials. J. Med. Chem, 2010, 53(1), 3–17. [ 10.1021/jm9004708] [DOI] [PubMed] [Google Scholar]
- [35].Samuni Y; Ishii H; Hyodo F; Samuni U; Krishna MC; Goldstein S; Mitchell JB Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med, 2010, 48(11), 1559–1563. [ 10.1016/j.freeradbiomed.2010.03.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hanson BE; Vesole DH Retaspimycin hydrochloride (IPI-504): a novel heat shock protein inhibitor as an anticancer agent. Expert Opin. Investig. Drugs, 2009, 18(9), 1375–1383. [ 10.1517/13543780903158934] [DOI] [PubMed] [Google Scholar]
- [37].Lee J IPI-493, a potent, orally bioavailable Hsp90 inhibitor of the ansamycin class, in EORTC-NCI-AACR-International Conference, 2008. Geneva, Switzerland [Google Scholar]
- [38].Floris G; Sciot R; Wozniak A; Van Looy T; Wellens J; Faa G; Normant E; Debiec-Rychter M; Schöffski P The Novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin. Cancer Res, 2011, 17(17), 5604–5614. [ 10.1158/1078-0432.CCR-11-0562] [DOI] [PubMed] [Google Scholar]
- [39].Chène P ATPases as drug targets: learning from their structure. Nat. Rev. Drug Discov, 2002, 1(9), 665–673. [ 10.1038/nrd894] [DOI] [PubMed] [Google Scholar]
- [40].Chiosis G; Timaul MN; Lucas B; Munster PN; Zheng FF; Sepp-Lorenzino L; Rosen N A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol, 2001, 8(3), 289–299. [ 10.1016/S1074-5521(01)00015-1] [DOI] [PubMed] [Google Scholar]
- [41].Wright L; Barril X; Dymock B; Sheridan L; Surgenor A; Beswick M; Drysdale M; Collier A; Massey A; Davies N; Fink A; Fromont C; Aherne W; Boxall K; Sharp S; Workman P; Hubbard RE Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol, 2004, 11(6), 775–785. [ 10.1016/j.chembiol.2004.03.033] [DOI] [PubMed] [Google Scholar]
- [42].Lundgren K; Zhang H; Brekken J; Huser N; Powell RE; Timple N; Busch DJ; Neely L; Sensintaffar JL; Yang YC; McKenzie A; Friedman J; Scannevin R; Kamal A; Hong K; Kasibhatla SR; Boehm MF; Burrows FJ BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol. Cancer Ther, 2009, 8(4), 921–929. [ 10.1158/1535-7163.MCT-08-0758] [DOI] [PubMed] [Google Scholar]
- [43].Lundgren K; Biamonte MA CHAPTER 5 The Discovery of BIIB021 and BIIB028, 2014, 158–179.
- [44].Shi X; Kiesman WF; Walker DG Development of Hsp90 Inhibitors for the Treatment of HER-2 Positive Solid Cancers, in Comprehensive Accounts of Pharmaceutical Research and Development: From Discovery to Late-Stage Process Development; American Chemical Society., 2016, 1, p. 69–100. [ 10.1021/bk-2016-1239.ch003] [DOI] [Google Scholar]
- [45].Caldas-Lopes E; Cerchietti L; Ahn JH; Clement CC; Robles AI; Rodina A; Moulick K; Taldone T; Gozman A; Guo Y; Wu N; de Stanchina E; White J; Gross SS; Ma Y; Varticovski L; Melnick A; Chiosis G Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA, 2009, 106(20), 8368–8373. [ 10.1073/pnas.0903392106] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fadden P; Huang KH; Veal JM; Steed PM; Barabasz AF; Foley B; Hu M; Partridge JM; Rice J; Scott A; Dubois LG; Freed TA; Silinski MA; Barta TE; Hughes PF; Ommen A; Ma W; Smith ED; Spangenberg AW; Eaves J; Hanson GJ; Hinkley L; Jenks M; Lewis M; Otto J; Pronk GJ; Verleysen K; Haystead TA; Hall SE Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem. Biol, 2010, 17(7), 686–694. [ 10.1016/j.chembiol.2010.04.015] [DOI] [PubMed] [Google Scholar]
- [47].Huang KH; Veal JM; Fadden RP; Rice JW; Eaves J; Strachan JP; Barabasz AF; Foley BE; Barta TE; Ma W; Silinski MA; Hu M; Partridge JM; Scott A; DuBois LG; Freed T; Steed PM; Ommen AJ; Smith ED; Hughes PF; Woodward AR; Hanson GJ; McCall WS; Markworth CJ; Hinkley L; Jenks M; Geng L; Lewis M; Otto J; Pronk B; Verleysen K; Hall SE Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents. J. Med. Chem, 2009, 52(14), 4288–4305. [ 10.1021/jm900230j] [DOI] [PubMed] [Google Scholar]
- [48].Cheung KM; Matthews TP; James K; Rowlands MG; Boxall KJ; Sharp SY; Maloney A; Roe SM; Prodromou C; Pearl LH; Aherne GW; McDonald E; Workman P The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett, 2005, 15(14), 3338–3343. [ 10.1016/j.bmcl.2005.05.046] [DOI] [PubMed] [Google Scholar]
- [49].Brough PA; Aherne W; Barril X; Borgognoni J; Boxall K; Cansfield JE; Cheung KM; Collins I; Davies NG; Drysdale MJ; Dymock B; Eccles SA; Finch H; Fink A; Hayes A; Howes R; Hubbard RE; James K; Jordan AM; Lockie A; Martins V; Massey A; Matthews TP; McDonald E; Northfield CJ; Pearl LH; Prodromou C; Ray S; Raynaud FI; Roughley SD; Sharp SY; Surgenor A; Walmsley DL; Webb P; Wood M; Workman P; Wright L 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J. Med. Chem, 2008, 51(2), 196–218. [ 10.1021/jm701018h] [DOI] [PubMed] [Google Scholar]
- [50].Eccles SA; Massey A; Raynaud FI; Sharp SY; Box G; Valenti M; Patterson L; de Haven Brandon A; Gowan S; Boxall F; Aherne W; Rowlands M; Hayes A; Martins V; Urban F; Boxall K; Prodromou C; Pearl L; James K; Matthews TP; Cheung KM; Kalusa A; Jones K; McDonald E; Barril X; Brough PA; Cansfield JE; Dymock B; Drysdale MJ; Finch H; Howes R; Hubbard RE; Surgenor A; Webb P; Wood M; Wright L; Workman P NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res, 2008, 68(8), 2850–2860. [ 10.1158/0008-5472.CAN-07-5256] [DOI] [PubMed] [Google Scholar]
- [51].Jensen MR; Schoepfer J; Radimerski T; Massey A; Guy CT; Brueggen J; Quadt C; Buckler A; Cozens R; Drysdale MJ; Garcia-Echeverria C; Chène P NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res, 2008, 10(2), R33 [ 10.1186/bcr1996] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang Y; Trepel JB; Neckers LM; Giaccone G STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer. Curr. Opin. Investig. Drugs, 2010, 11(12), 1466–1476. [PubMed] [Google Scholar]
- [53].Woodhead AJ; Angove H; Carr MG; Chessari G; Congreve M; Coyle JE; Cosme J; Graham B; Day PJ; Downham R; Fazal L; Feltell R; Figueroa E; Frederickson M; Lewis J; McMenamin R; Murray CW; O’Brien MA; Parra L; Patel S; Phillips T; Rees DC; Rich S; Smith DM; Trewartha G; Vinkovic M; Williams B; Woolford AJ Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem, 2010, 53(16), 5956–5969. [ 10.1021/jm100060b] [DOI] [PubMed] [Google Scholar]
- [54].Murray CW; Carr MG; Callaghan O; Chessari G; Congreve M; Cowan S; Coyle JE; Downham R; Figueroa E; Frederickson M; Graham B; McMenamin R; O’Brien MA; Patel S; Phillips TR; Williams G; Woodhead AJ; Woolford AJ Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J. Med. Chem, 2010, 53(16), 5942–5955. [ 10.1021/jm100059d] [DOI] [PubMed] [Google Scholar]
- [55].Nakashima T; Ishii T; Tagaya H; Seike T; Nakagawa H; Kanda Y; Akinaga S; Soga S; Shiotsu Y New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin. Cancer Res, 2010, 16(10), 2792–2802. [ 10.1158/1078-0432.CCR-09-3112] [DOI] [PubMed] [Google Scholar]
- [56].Cavenagh J; Oakervee H; Baetiong-Caguioa P; Davies F; Gharibo M; Rabin N; Kurman M; Novak B; Shiraishi N; Nakashima D; Akinaga S; Yong K A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br. J. Cancer, 2017, 117(9), 1295–1302. [ 10.1038/bjc.2017.302] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bussenius J; Blazey CM; Aay N; Anand NK; Arcalas A; Baik T; Bowles OJ; Buhr CA; Costanzo S; Curtis JK; DeFina SC; Dubenko L; Heuer TS; Huang P; Jaeger C; Joshi A; Kennedy AR; Kim AI; Lara K; Lee J; Li J; Lougheed JC; Ma S; Malek S; Manalo JC; Martini JF; McGrath G; Nicoll M; Nuss JM; Pack M; Peto CJ; Tsang TH; Wang L; Womble SW; Yakes M; Zhang W; Rice KD Discovery of XL888: a novel tropane-derived small molecule inhibitor of HSP90. Bioorg. Med. Chem. Lett, 2012, 22(17), 5396–5404. [ 10.1016/j.bmcl.2012.07.052] [DOI] [PubMed] [Google Scholar]
- [58].Haarberg HE; Paraiso KH; Wood E; Rebecca VW; Sondak VK; Koomen JM; Smalley KS Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol. Cancer Ther, 2013, 12(6), 901–912. [ 10.1158/1535-7163.MCT-12-1003] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Menezes DL; Taverna P; Jensen MR; Abrams T; Stuart D; Yu GK; Duhl D; Machajewski T; Sellers WR; Pryer NK; Gao Z The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol. Cancer Ther, 2012, 11(3), 730–739. [ 10.1158/1535-7163.MCT-11-0667] [DOI] [PubMed] [Google Scholar]
- [60].Ohkubo S; Kodama Y; Muraoka H; Hitotsumachi H; Yoshimura C; Kitade M; Hashimoto A; Ito K; Gomori A; Takahashi K; Shibata Y; Kanoh A; Yonekura K TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Ther, 2015, 14(1), 14–22. [ 10.1158/1535-7163.MCT-14-0219] [DOI] [PubMed] [Google Scholar]
- [61].Shimomura A; Yamamoto N; Kondo S; Fujiwara Y; Suzuki S; Yanagitani N; Horiike A; Kitazono S; Ohyanagi F; Doi T; Kuboki Y; Kawazoe A; Shitara K; Ohno I; Banerji U; Sundar R; Ohkubo S; Calleja EM; Nishio M First-in-Human Phase I Study of an Oral HSP90 Inhibitor, TAS-116, in Patients with Advanced Solid Tumors. Mol. Cancer Ther, 2019, 18(3), 531–540. [ 10.1158/1535-7163.MCT-18-0831] [DOI] [PubMed] [Google Scholar]
- [62].Kurokawa Y Phase II study of TAS-116, on oral inhibitor of heat shock protein (HSP90), in metastatic or unresectable gastrointestinal stromal tumor refractory to imatinib, sunitinib, and regorafenib, in ESMO 2017 Congress, Annals of OncologyMadrid, Spain: 2017, p. v521–v538. [Google Scholar]
- [63].Söti C; Rácz A; Csermely P A Nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. J. Biol. Chem, 2002, 277(9), 7066–7075. [ 10.1074/jbc.M105568200] [DOI] [PubMed] [Google Scholar]
- [64].Donnelly A; Blagg BS Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem, 2008, 15(26), 2702–2717. [ 10.2174/092986708786242895] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schulte TW; Akinaga S; Soga S; Sullivan W; Stensgard B; Toft D; Neckers LM Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones, 1998, 3(2), 100–108. [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yun BG; Huang W; Leach N; Hartson SD; Matts RL Novobiocin induces a distinct conformation of Hsp90 and alters Hsp90-cochaperone-client interactions. Biochemistry, 2004, 43(25), 8217–8229. [ 10.1021/bi0497998] [DOI] [PubMed] [Google Scholar]
- [67].Marcu MG; Chadli A; Bouhouche I; Catelli M; Neckers LM The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem, 2000, 275(47), 37181–37186. [ 10.1074/jbc.M003701200] [DOI] [PubMed] [Google Scholar]
- [68].Chatterjee BK; Jayaraj A; Kumar V; Blagg B; Davis RE; Jayaram B; Deep S; Chaudhuri TK Stimulation of heat shock protein 90 chaperone function through binding of a novobiocin analog KU-32. J. Biol. Chem, 2019, 294(16), 6450–6467. [ 10.1074/jbc.RA118.002502] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rahimi MN; McAlpine SR Protein-protein inhibitor designed de novo to target the MEEVD region on the C-terminus of Hsp90 and block co-chaperone activity. Chem. Commun. (Camb.), 2019, 55(6), 846–849. [ 10.1039/C8CC07576J] [DOI] [PubMed] [Google Scholar]
- [70].Terracciano S; Russo A; Chini MG; Vaccaro MC; Potenza M; Vassallo A; Riccio R; Bifulco G; Bruno I Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Sci. Rep, 2018, 8(1), 1709 [ 10.1038/s41598-017-14902-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cox MB; Miller CA III Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones, 2004, 9(1), 4–20. [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhao J; Zhao H; Hall JA; Brown D; Brandes E; Bazzill J; Grogan PT; Subramanian C; Vielhauer G; Cohen MS; Blagg BS Triazole Containing Novobiocin and Biphenyl Amides as Hsp90 C-Terminal Inhibitors. MedChemComm, 2014, 5(9), 1317–1323. [ 10.1039/C4MD00102H] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].White PT; Subramanian C; Zhu Q; Zhang H; Zhao H; Gallagher R; Timmermann BN; Blagg BS; Cohen MS Novel HSP90 inhibitors effectively target functions of thyroid cancer stem cell preventing migration and invasion. Surgery, 2016, 159(1), 142–151. [ 10.1016/j.surg.2015.07.050] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Subramanian C; Kovatch KJ; Sim MW; Wang G; Prince ME; Carey TE; Davis R; Blagg BSJ; Cohen MS Novel C-Terminal Heat Shock Protein 90 Inhibitors (KU711 and Ku757) Are Effective in Targeting Head and Neck Squamous Cell Carcinoma Cancer Stem cells. Neoplasia, 2017, 19(12), 1003–1011. [ 10.1016/j.neo.2017.09.003] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Samadi AK; Zhang X; Mukerji R; Donnelly AC; Blagg BS; Cohen MS A novel C-terminal HSP90 inhibitor KU135 induces apoptosis and cell cycle arrest in melanoma cells. Cancer Lett, 2011, 312(2), 158–167. [ 10.1016/j.canlet.2011.07.031] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cohen SM; Mukerji R; Samadi AK; Zhang X; Zhao H; Blagg BS; Cohen MS Novel C-terminal Hsp90 inhibitor for head and neck squamous cell cancer (HNSCC) with in vivo efficacy and improved toxicity profiles compared with standard agents. Ann. Surg. Oncol, 2012, 19(Suppl. 3), S483–S490. [ 10.1245/s10434-011-1971-1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Byrd KM; Subramanian C; Sanchez J; Motiwala HF; Liu W; Cohen MS; Holzbeierlein J; Blagg BS Synthesis and Biological Evaluation of Novobiocin Core Analogues as Hsp90 Inhibitors. Chemistry, 2016, 22(20), 6921–6931. [ 10.1002/chem.201504955] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Langer T; Rosmus S; Fasold H Intracellular localization of the 90 kDA heat shock protein (HSP90alpha) determined by expression of a EGFP-HSP90alpha-fusion protein in unstressed and heat stressed 3T3 cells. Cell Biol. Int, 2003, 27(1), 47–52. [ 10.1016/S1065-6995(02)00256-1] [DOI] [PubMed] [Google Scholar]
- [79].Condelli V; Crispo F; Pietrafesa M; Lettini G; Matassa DS; Esposito F; Landriscina M; Maddalena F HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy. Cells, 2019, 8(6)E532 [ 10.3390/cells8060532] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell, 2011, 144(5), 646–674. [ 10.1016/j.cell.2011.02.013] [DOI] [PubMed] [Google Scholar]
- [81].Li W; Sahu D; Tsen F Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim. Biophys. Acta, 2012, 1823(3), 730–741. [ 10.1016/j.bbamcr.2011.09.009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Marzec M; Eletto D; Argon Y GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta, 2012, 1823(3), 774–787. [ 10.1016/j.bbamcr.2011.10.013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Eletto D; Dersh D; Argon Y GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol, 2010, 21(5), 479–485. [ 10.1016/j.semcdb.2010.03.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Van PN; Peter F; Söling HD Four intracisternal calciumbinding glycoproteins from rat liver microsomes with high affinity for calcium. No indication for calsequestrin-like proteins in inositol 1,4,5-trisphosphate-sensitive calcium sequestering rat liver vesicles. J. Biol. Chem, 1989, 264(29), 17494–17501. [PubMed] [Google Scholar]
- [85].Biswas C; Ostrovsky O; Makarewich CA; Wanderling S; Gidalevitz T; Argon Y The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J, 2007, 405(2), 233–241. [ 10.1042/BJ20061867] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Felts SJ; Owen BA; Nguyen P; Trepel J; Donner DB; Toft DO The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem, 2000, 275(5), 3305–3312. [ 10.1074/jbc.275.5.3305] [DOI] [PubMed] [Google Scholar]
- [87].Song HY; Dunbar JD; Zhang YX; Guo D; Donner DB Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem, 1995, 270(8), 3574–3581. [ 10.1074/jbc.270.8.3574] [DOI] [PubMed] [Google Scholar]
- [88].Hua G; Zhang Q; Fan Z Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J. Biol. Chem, 2007, 282(28), 20553–20560. [ 10.1074/jbc.M703196200] [DOI] [PubMed] [Google Scholar]
- [89].Sciacovelli M; Guzzo G; Morello V; Frezza C; Zheng L; Nannini N; Calabrese F; Laudiero G; Esposito F; Landriscina M; Defilippi P; Bernardi P; Rasola A The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab, 2013, 17(6), 988–999. [ 10.1016/j.cmet.2013.04.019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Masgras I; Sanchez-Martin C; Colombo G; Rasola A The Chaperone TRAP1 As a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol, 2017, 7, 58 [ 10.3389/fonc.2017.00058] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Xiang F; Ma SY; Lv YL; Zhang DX; Song HP; Huang YS Tumor necrosis factor receptor-associated protein 1 regulates hypoxia-induced apoptosis through a mitochondria-dependent pathway mediated by cytochrome c oxidase subunit II. Burns Trauma, 2019, 7, 16 [ 10.1186/s41038-019-0154-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Grbovic OM; Basso AD; Sawai A; Ye Q; Friedlander P; Solit D; Rosen N V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA, 2006, 103(1), 57–62. [ 10.1073/pnas.0609973103] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Evans CG; Wisén S; Gestwicki JE Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J. Biol. Chem, 2006, 281(44), 33182–33191. [ 10.1074/jbc.M606192200] [DOI] [PubMed] [Google Scholar]
- [94].Dickey CA; Kamal A; Lundgren K; Klosak N; Bailey RM; Dunmore J; Ash P; Shoraka S; Zlatkovic J; Eckman CB; Patterson C; Dickson DW; Nahman NS Jr; Hutton M; Burrows F; Petrucelli L The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest, 2007, 117(3), 648–658. [ 10.1172/JCI29715] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Robert J; Ménoret A; Cohen N Cell surface expression of the endoplasmic reticular heat shock protein gp96 is phylogenetically conserved. J. Immunol, 1999, 163(8), 4133–4139. [PubMed] [Google Scholar]
- [96].Ansa-Addo EA; Thaxton J; Hong F; Wu BX; Zhang Y; Fugle CW; Metelli A; Riesenberg B; Williams K; Gewirth DT; Chiosis G; Liu B; Li Z Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Curr. Top. Med. Chem, 2016, 16(25), 2765–2778. [ 10.2174/1568026616666160413141613] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Amoroso MR; Matassa DS; Sisinni L; Lettini G; Landriscina M; Esposito F TRAP1 revisited: novel localizations and functions of a ‘next-generation’ biomarker (review). Int. J. Oncol, 2014, 45(3), 969–977. [ 10.3892/ijo.2014.2530] [DOI] [PubMed] [Google Scholar]
- [98].Renouf DJ; Velazquez-Martin JP; Simpson R; Siu LL; Bedard PL Ocular toxicity of targeted therapies. J. Clin. Oncol, 2012, 30(26), 3277–3286. [ 10.1200/JCO.2011.41.5851] [DOI] [PubMed] [Google Scholar]
- [99].Peterson LB; Eskew JD; Vielhauer GA; Blagg BS The hERG channel is dependent upon the Hsp90α isoform for maturation and trafficking. Mol. Pharm, 2012, 9(6), 1841–1846. [ 10.1021/mp300138n] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Neckers L; Workman P Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res, 2012, 18(1), 64–76. [ 10.1158/1078-0432.CCR-11-1000] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rosser MF; Nicchitta CV Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. I. Evidence for allosteric regulation of ligand binding. J. Biol. Chem, 2000, 275(30), 22798–22805. [ 10.1074/jbc.M001477200] [DOI] [PubMed] [Google Scholar]
- [102].Wang M; Shen G; Blagg BS Radanamycin, a macrocyclic chimera of radicicol and geldanamycin. Bioorg. Med. Chem. Lett, 2006, 16(9), 2459–2462. [ 10.1016/j.bmcl.2006.01.086] [DOI] [PubMed] [Google Scholar]
- [103].Shen G; Blagg BS Radester, a novel inhibitor of the Hsp90 protein folding machinery. Org. Lett, 2005, 7(11), 2157–2160. [ 10.1021/ol050580a] [DOI] [PubMed] [Google Scholar]
- [104].Duerfeldt AS; Brandt GE; Blagg BS Design, synthesis, and biological evaluation of conformationally constrained cis-amide Hsp90 inhibitors. Org. Lett, 2009, 11(11), 2353–2356. [ 10.1021/ol900783m] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Immormino RM; Metzger LE IV; Reardon PN; Dollins DE; Blagg BS; Gewirth DT Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J. Mol. Biol, 2009, 388(5), 1033–1042. [ 10.1016/j.jmb.2009.03.071] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Duerfeldt AS; Peterson LB; Maynard JC; Ng CL; Eletto D; Ostrovsky O; Shinogle HE; Moore DS; Argon Y; Nicchitta CV; Blagg BS Development of a Grp94 inhibitor. J. Am. Chem. Soc, 2012, 134(23), 9796–9804. [ 10.1021/ja303477g] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Yang Y; Liu B; Dai J; Srivastava PK; Zammit DJ; Lefrançois L; Li Z Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity, 2007, 26(2), 215–226. [ 10.1016/j.immuni.2006.12.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Crowley VM; Khandelwal A; Mishra S; Stothert AR; Huard DJ; Zhao J; Muth A; Duerfeldt AS; Kizziah JL; Lieberman RL; Dickey CA; Blagg BS Development of Glucose Regulated Protein 94-Selective Inhibitors Based on the BnIm and Radamide Scaffold. J. Med. Chem, 2016, 59(7), 3471–3488. [ 10.1021/acs.jmedchem.6b00085] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Stothert AR; Suntharalingam A; Tang X; Crowley VM; Mishra SJ; Webster JM; Nordhues BA; Huard DJE; Passaglia CL; Lieberman RL; Blagg BSJ; Blair LJ; Koren J III; Dickey CA Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep, 2017, 7(1), 17951 [ 10.1038/s41598-017-18344-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Stothert AR; Suntharalingam A; Huard DJ; Fontaine SN; Crowley VM; Mishra S; Blagg BS; Lieberman RL; Dickey CA Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum. Mol. Genet, 2014, 23(24), 6470–6480. [ 10.1093/hmg/ddu367] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Crowley VM; Huard DJE; Lieberman RL; Blagg BSJ Second Generation Grp94-Selective Inhibitors Provide Opportunities for the Inhibition of Metastatic Cancer. Chemistry, 2017, 23(62), 15775–15782. [ 10.1002/chem.201703398] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Khandelwal A; Crowley VM; Blagg BSJ Resorcinol-Based Grp94-Selective Inhibitors. ACS Med. Chem. Lett, 2017, 8(10), 1013–1018. [ 10.1021/acsmedchemlett.7b00193] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Patel PD; Yan P; Seidler PM; Patel HJ; Sun W; Yang C; Que NS; Taldone T; Finotti P; Stephani RA; Gewirth DT; Chiosis G Paralog-selective Hsp90 inhibitors define tumorspecific regulation of HER2. Nat. Chem. Biol, 2013, 9(11), 677–684. [ 10.1038/nchembio.1335] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jiang F; Guo AP; Xu JC; You QD; Xu XL Discovery of a Potent Grp94 Selective Inhibitor with Anti-Inflammatory Efficacy in a Mouse Model of Ulcerative Colitis. J. Med. Chem, 2018, 61(21), 9513–9533. [ 10.1021/acs.jmedchem.8b00800] [DOI] [PubMed] [Google Scholar]
- [115].Ernst JT; Liu M; Zuccola H; Neubert T; Beaumont K; Turnbull A; Kallel A; Vought B; Stamos D Correlation between chemotype-dependent binding conformations of HSP90α/β and isoform selectivity-Implications for the structure-based design of HSP90α/β selective inhibitors for treating neurodegenerative diseases. Bioorg. Med. Chem. Lett, 2014, 24(1), 204–208. [ 10.1016/j.bmcl.2013.11.036] [DOI] [PubMed] [Google Scholar]
- [116].Putcha P; Danzer KM; Kranich LR; Scott A; Silinski M; Mabbett S; Hicks CD; Veal JM; Steed PM; Hyman BT; McLean PJ Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alphasynuclein-induced toxicity. J. Pharmacol. Exp. Ther, 2010, 332(3), 849–857. [ 10.1124/jpet.109.158436] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ernst JT; Neubert T; Liu M; Sperry S; Zuccola H; Turnbull A; Fleck B; Kargo W; Woody L; Chiang P; Tran D; Chen W; Snyder P; Alcacio T; Nezami A; Reynolds J; Alvi K; Goulet L; Stamos D Identification of novel HSP90α/β isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington’s disease. J. Med. Chem, 2014, 57(8), 3382–3400. [ 10.1021/jm500042s] [DOI] [PubMed] [Google Scholar]
- [118].Khandelwal A; Kent CN; Balch M; Peng S; Mishra SJ; Deng J; Day VW; Liu W; Subramanian C; Cohen M; Holzbeierlein JM; Matts R; Blagg BSJ Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat. Commun, 2018, 9(1), 425 [ 10.1038/s41467-017-02013-1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Plescia J; Salz W; Xia F; Pennati M; Zaffaroni N; Daidone MG; Meli M; Dohi T; Fortugno P; Nefedova Y; Gabrilovich DI; Colombo G; Altieri DC Rational design of shepherdin, a novel anticancer agent. Cancer Cell, 2005, 7(5), 457–468. [ 10.1016/j.ccr.2005.03.035] [DOI] [PubMed] [Google Scholar]
- [120].Altieri DC; Stein GS; Lian JB; Languino LR TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta, 2012, 1823(3), 767–773. [ 10.1016/j.bbamcr.2011.08.007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Siegelin MD Inhibition of the mitochondrial Hsp90 chaperone network: a novel, efficient treatment strategy for cancer? Cancer Lett, 2013, 333(2), 133–146. [ 10.1016/j.canlet.2013.01.045] [DOI] [PubMed] [Google Scholar]
- [122].Seo YH Organelle-specific Hsp90 inhibitors. Arch. Pharm. Res, 2015, 38(9), 1582–1590. [ 10.1007/s12272-015-0636-1] [DOI] [PubMed] [Google Scholar]
- [123].Lee C; Park HK; Jeong H; Lim J; Lee AJ; Cheon KY; Kim CS; Thomas AP; Bae B; Kim ND; Kim SH; Suh PG; Ryu JH; Kang BH Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc, 2015, 137(13), 4358–4367. [ 10.1021/ja511893n] [DOI] [PubMed] [Google Scholar]
- [124].Park HK; Jeong H; Ko E; Lee G; Lee JE; Lee SK; Lee AJ; Im JY; Hu S; Kim SH; Lee JH; Lee C; Kang S; Kang BH Paralog Specificity Determines Subcellular Distribution, Action Mechanism, and Anticancer Activity of TRAP1 Inhibitors. J. Med. Chem, 2017, 60(17), 7569–7578. [ 10.1021/acs.jmedchem.7b00978] [DOI] [PubMed] [Google Scholar]