

SUMMARY
The fidelity of intracellular signaling hinges on the organization of dynamic activity architectures. Spatial compartmentation was first proposed over 30 years ago to explain how diverse G-protein-coupled receptors achieve specificity despite converging on a ubiquitous messenger, 3’,5’-cyclic adenosine monophosphate (cAMP). However, the mechanisms responsible for spatially constraining this diffusible messenger remain elusive. Here, we reveal the type I regulatory subunit of cAMP-dependent protein kinase (PKA), RIα, undergoes liquid-liquid phase separation (LLPS) as a function of cAMP signaling to form biomolecular condensates enriched in cAMP and PKA activity, critical for effective cAMP compartmentation. We further show that a PKA fusion oncoprotein associated with an atypical liver cancer potently blocks RIα LLPS and induces aberrant cAMP signaling. Loss of RIα LLPS in normal cells increases cell proliferation and induces cell transformation. Our work reveals LLPS as a principal organizer of signaling compartments and highlights the pathological consequences of dysregulating this activity architecture.
Graphical Abstract
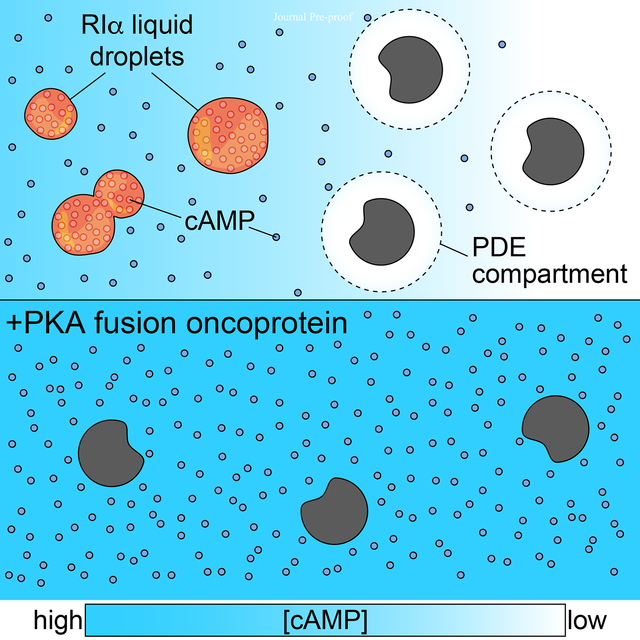
cAMP-responsive condensate formation by PKA’s RIα subunit controls local signaling and disruption of phase separation in this context contributes to tumorigenesis.
INTRODUCTION
3’,5’-cyclic adenosine monophosphate (cAMP) is a universal regulator of cellular function and behavior across evolution. In eukaryotes, cAMP production is canonically triggered in response to hormone signaling via the G protein-coupled receptor (GPCR)-mediated activation of transmembrane adenylyl cyclases (ACs), which catalyze the synthesis of cAMP from ATP. cAMP signals are transduced by a number of well-studied effector proteins, most prominently the cAMP-dependent protein kinase (PKA), a tetrameric holoenzyme consisting of a regulatory subunit dimer bound to a pair of catalytic subunits. Binding of cAMP to the PKA regulatory subunit unleashes the activity of the PKA catalytic subunit (PKAcat), which then phosphorylates a myriad of targets throughout the cell. Together, cAMP and PKA exert tight control over numerous physiological processes, from cell growth and survival (Gottesman and Fleischmann, 1986; Jhala et al., 2003; Li et al., 2000) to cardiac (Boularan and Gales, 2015) and neuronal (Kandel, 2012) functions.
The functional diversity of cAMP signaling is driven by hundreds of GPCR inputs (Kroeze et al., 2003) capable of elevating cAMP levels to produce distinct cellular responses (Patra and Brady, 2018). This remarkable specificity may be achieved through compartmentation of cAMP, a concept first proposed over 35 years ago (Brunton et al., 1981; Buxton and Brunton, 1983; Steinberg and Brunton, 2001). Indeed, cAMP gradients (Bacskai et al., 1993; Gorshkov et al., 2017; Lim et al., 2008; Nikolaev et al., 2006) and microdomains (Maiellaro et al., 2016; Terrin et al., 2012; Zaccolo and Pozzan, 2002) have been observed experimentally in various contexts. While compartmentalized AC activity is involved in forming these cAMP microdomains (Cooper, 2003; Willoughby and Cooper, 2007), cAMP-hydrolyzing phosphodiesterases (PDEs) are widely considered the primary diffusional barrier responsible for fencing local cAMP pools (Baillie, 2009; Houslay, 2010; Zaccolo, 2006).
However, this longstanding model of PDE-controlled cAMP compartmentation is at odds with reports describing almost unrestricted (e.g., 270–780 μm2/s) cAMP diffusion in cells (Bacskai et al., 1993; Chen et al., 1999; Nikolaev et al., 2004). Indeed, various computational studies have failed to reproduce the formation of cAMP gradients through the sole action of PDEs (Boras et al., 2014; Lohse et al., 2017; Rich et al., 2000, 2001; Saucerman et al., 2014), whose catalytic activity appears to be insufficient to constrain such a rapidly diffusing messenger (Boras et al., 2014; Lohse et al., 2017), suggesting that experimentally observed cAMP microdomains instead require substantially (~100- to 10,000-fold) slower cAMP diffusion. Notably, more recent investigations have in fact reported significantly lower cytosolic cAMP diffusion rates that are more conducive of cAMP compartmentalization than the original estimates (Agarwal et al., 2016; Richards et al., 2016). However, the specific mechanisms responsible for spatially constraining this critical second messenger remain to be elucidated.
Here, we identify the formation of biomolecular condensates of the type I regulatory subunit of PKA, RIα, as a key driver of cAMP compartmentation. RIα undergoes liquid-liquid phase separation (LLPS) at endogenous levels as a function of dynamic cAMP signaling to form RIα bodies harboring high levels of cAMP and PKA activity, and this dynamic cAMP sequestration is required to spatially constrain cAMP in cells. Importantly, RIα LLPS is explicitly disrupted by a PKAcat fusion oncoprotein present in the atypical liver cancer fibrolamellar carcinoma (FLC) (Honeyman et al., 2014), leading to aberrant cAMP signaling and cell transformation. Our work reveals LLPS as an essential coordinator of signaling compartments and provides critical mechanistic clues into the etiology of FLC, highlighting the pathological consequences of dysregulated signaling activity architecture.
RESULTS
RIα undergoes liquid-liquid phase separation at endogenous levels
Of the four non-redundant PKA regulatory subunits, only RIα is ubiquitously expressed and it is essential for proper regulation of PKA activity (Cadd and McKnight, 1989). To visualize the dynamics of RIα expressed at endogenous levels, we introduced the 11th β-strand of GFP (FP11) (Leonetti et al., 2016) at the C-terminus of RIα via CRISPR/Cas9 in HEK293A cells, yielding the 293-RIα cell line. This small segment permits efficient knock-in and enables targeted reconstitution of intact GFP when the remaining strands (GFP1–10) are co-expressed. By doing so in 293-RIα cells, we observed fluorescent puncta (Figure 1A) similar to those seen with overexpressed RIα (Day et al., 2011; Mavrakis et al., 2006). These puncta are highly dynamic, with coalescence of dispersed puncta occurring on the minute scale (Figure 1B and Video 1). Fluorescence recovery after photobleaching (FRAP) experiments indicated that labeled RIα can dynamically exhange between the puncta and diffuisble pools, as indicated by similar fluorescence recovery kinetics (t1/2 of 7 s ± 0.44 s, mean ± SEM indicated throughout the paper, n = 12 puncta vs. t1/2 of 7.8 s ± 0.41 s, n = 5 cytosolic regions) (Figure 1C and Video 2), although labeled RIα showed decreased mobility within puncta (t1/2: 35 s ± 1.3 s, n = 9 regions inside RIα puncta; P < 0.0001; Figure S1A). In addition, treatment with 2.5% 1,6-hexanediol, which disrupts weak intermolecular forces present in liquid-like assemblies (Peskett et al., 2018), reduced the number of endogenous RIα puncta per cell by 68% ± 8.6% (Figure 1D).
Figure 1. Endogenous PKA regulatory subunit RIα undergoes phase separation.
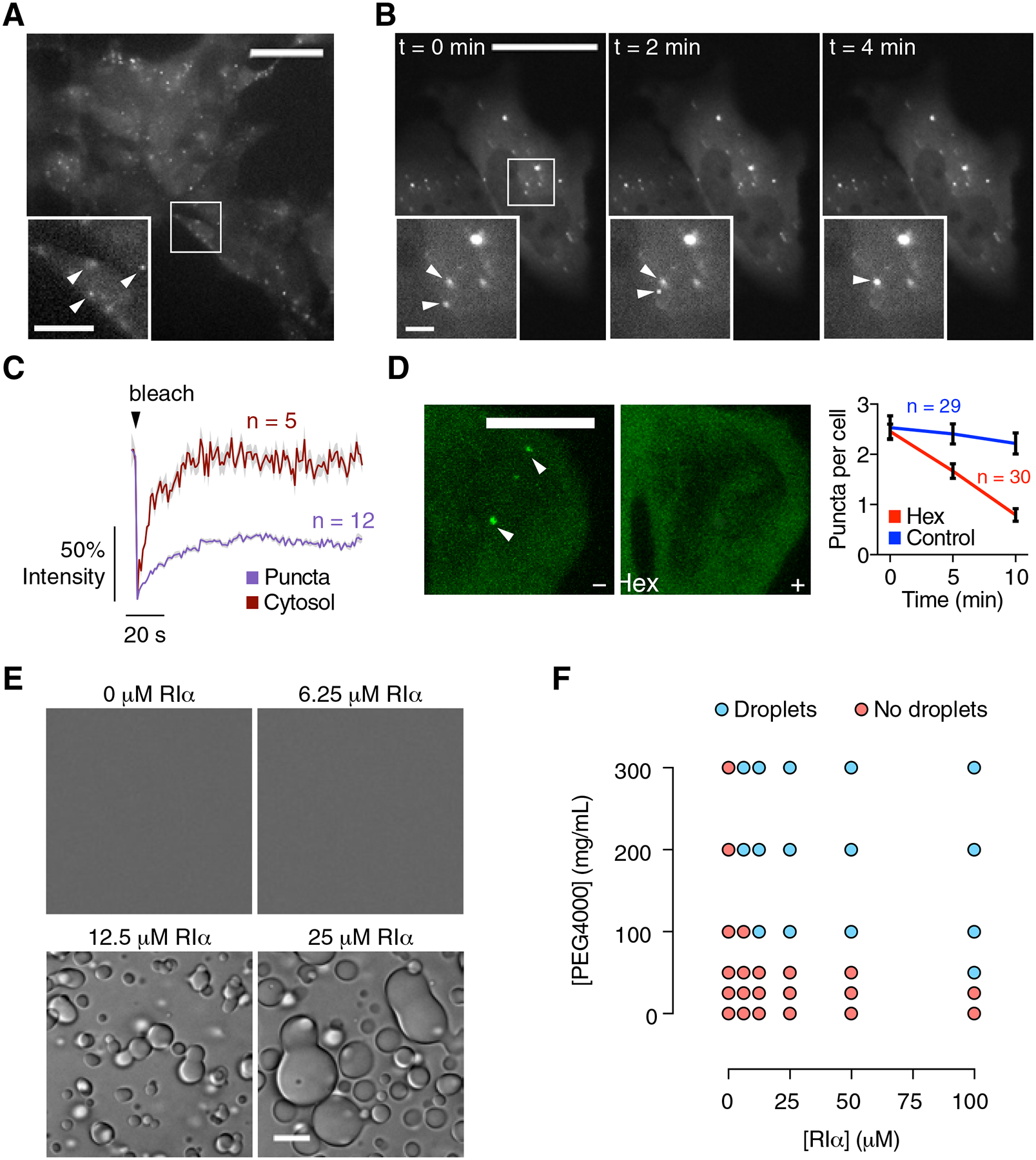
(A) Observing the localization of endogenously expressed RIα. The 11th β-strand of GFP (FP11) was knocked-in at the C-terminus of RIα in HEK293A cells. Transfecting these 293-RIα cells with the remaining GFP β-strands (GFP1–10) and imaging them in the GFP channel revealed the formation of fluorescent RIα puncta. (B) Representative GFP fluorescence images of 293-RIα cells transfected with GFP1–10 show merging of endogenous RIα puncta. (C) Monitoring the dynamics of labeled RIα. FRAP of RIα puncta (blue curve) compared with diffuse RIα (red curve) in GFP1–10-transfected 293-RIα cells. Curves show average time course of normalized fluorescence intensity. Solid lines indicate the mean; shaded areas, SEM. (D) RIα puncta disruption by 1,6-hexanediol. Representative GFP fluorescence images of GFP1–10-transfected 293-RIα cells before (t = 0 min; left) and after (t = 10 min; middle) 2.5% 1,6-hexanediol addition. Quantification of the number of RIα puncta per cell at the indicated times with (Hex; red curve) or without (Control; blue curve) 1,6-hexanediol addition. Error bars indicate ± SEM. (E) Representative DIC images showing liquid droplet formation by purified RIα at the indicated concentrations in vitro. (F) Representative in vitro phase diagram of RIα liquid droplet formation at varying concentrations of PEG 4000. Each condition was assessed at least twice. Scale bars: (A) 30 μm (inset, 10 μm); (B) 30 μm; (inset, 1 μm); (E) 10 μm.
Strikingly, purified RIα by itself formed liquid droplets in vitro (Figure 1E). Increasing molecular crowding with increasing concentrations of PEG decreased the concentrations of RIα needed for liquid droplet formation (Figure 1F), while increasing salt (KCl) concentrations increased the concentrations of RIα needed for liquid droplet formation (Figure S1B). RIα droplets varied in size both in vitro and in cells (Figure S1C), and while endogenous RIα puncta account for only 0.09% ± 0.02% of the entire cellular volume, we estimated the RIα concentration inside these puncta to be about 5.5 μM (Figure S1D and S1E). We also observed similar fluorescent puncta in various other cell types expressing RIα-GFP, such as cardiomyocytes, astrocytes, and neurons (Figure S1F–H). Together, these data indicate that RIα is capable of forming biomolecular condensates and does so at endogenous expression levels.
RIα phase separation is inhibited by PKA catalytic subunit and enhanced by cAMP
RIα forms an obligate dimer via its N-terminal dimerization and docking (D/D) domain, which bridges its binding to A-Kinase Anchoring Proteins (AKAPs). Connecting the D/D domain and cAMP binding domains is a linker region that is relatively disordered (see Methods) and contains an inhibitory sequence that acts as a pseudosubstrate for PKAcat (Figure 2A) (Kim et al., 2005). To probe the role of these domains in RIα LLPS, we generated a panel of EGFP-tagged RIα truncation mutants and monitored their ability to form puncta when overexpressed in wild-type HEK293T cells. No puncta were observed with mutants lacking the D/D domain or the linker region (Figures 2B and S2A–E), in contrast to the wild-type control, whereas fluorescent puncta were observed in cells expressing a truncation mutant containing only these two regions (Figure 2B). These data suggest that a segment containing the D/D domain and linker region is both necessary and, to some extent, sufficient for RIα LLPS.
Figure 2. Regulation of RIα phase separation by PKA catalytic subunit and cAMP.
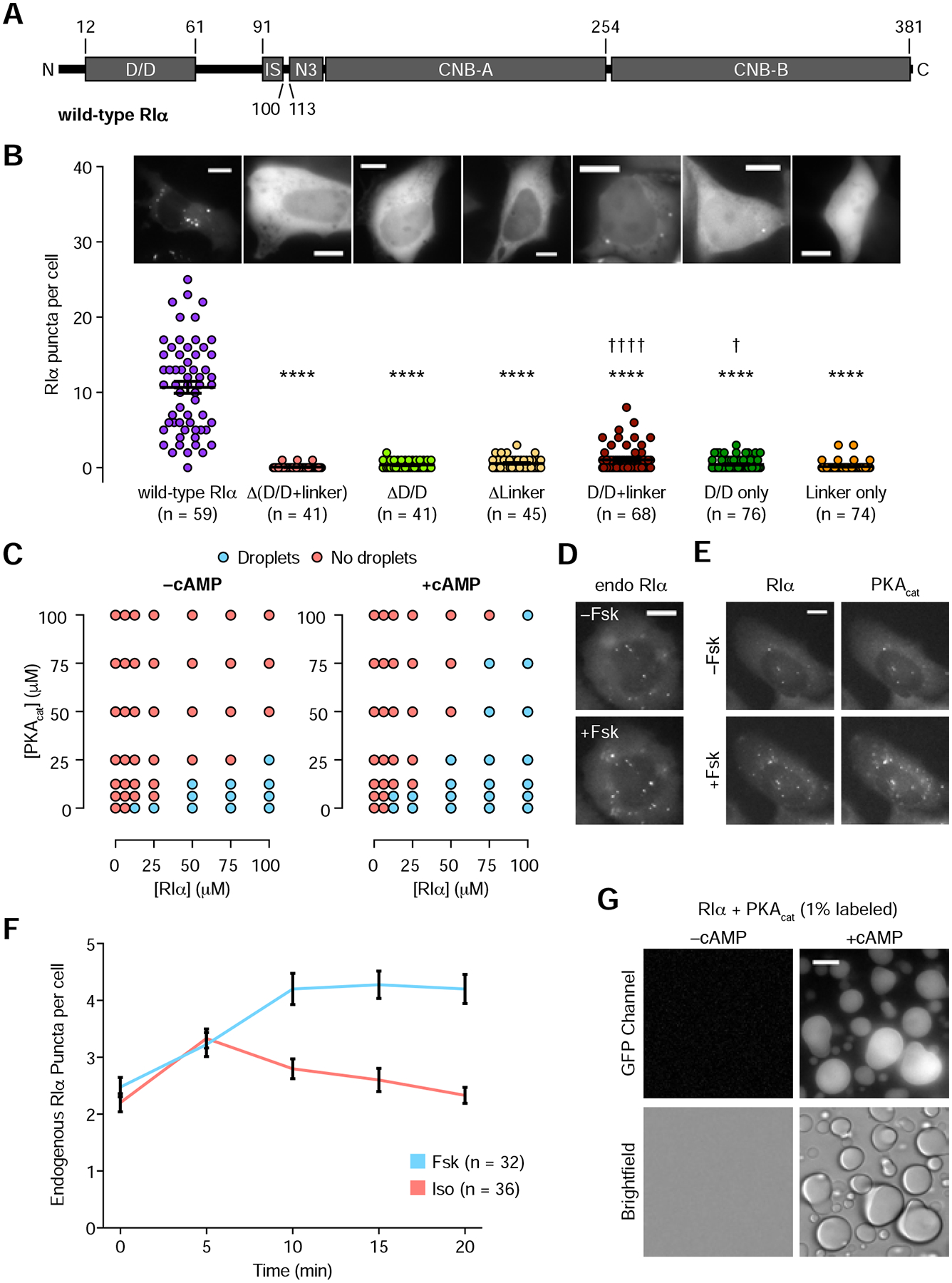
(A) Domain structure of full-length, wild-type RIα. (B) Comparison of RIα puncta number in wild-type HEK293T cells expressing EGFP-tagged wild-type or mutant RIα. The D/D domain (residues 12–61), the linker region (62–113), or both (12–113) were either deleted or overexpressed. Horizontal lines indicate mean ± SEM. Representative fluorescence images of HEK293T cells transfected with the corresponding EGFP-tagged RIα constructs are shown above each bar. (C-G) cAMP enhances RIα phase separation in the presence of PKAcat. (C) Representative in vitro phase diagram of RIα liquid droplet formation as a function of RIα and PKAcat concentration in the presence (right) or absence (left) of 10 μM cAMP. Each condition was assessed at least twice. (D) Representative fluorescence images of GFP1–10-transfected 293-RIα cells before (t = 0; top) and after (t = 10 min; bottom) addition of 50 μM Fsk. (E) Representative fluorescence images of wild-type HEK293T cells transfected with EGFP-RIα (left) and mTagBFP2-PKAcat (right) shown before (t = 0; top) and after (t = 10 min; bottom) addition of 50 μM Fsk. (F) Average time courses of the number of RIα puncta per cell in 293-RIα cells transfected with GFP1–10 and treated with 50 μM Fsk (blue curve) or 10 μM isoproterenol (Iso) (red curve). Error bars indicate ± SEM. (G) Representative GFP (top) and DIC (bottom) images of 50 μM RIα mixed with 25 μM PKAcat (1% GFP-tagged), showing PKAcat in RIα liquid droplets without (left) and with (right) 10 μM cAMP. All scale bars, 10 μm.
The inhibitory sequence and a portion of the linker region become disordered when cAMP-bound RIα is dissociated from PKAcat (Kim et al., 2005). Because this region is involved in RIα LLPS, we hypothesized that PKAcat and cAMP may directly influence this process. Indeed, increasing concentrations of purified PKAcat increased the minimal RIα concentration required for liquid droplet formation in vitro (Figure 2C). Moreover, overexpression of PKAcat in GFP1–10-expressing 293-RIα cells decreased the number of endogenous RIα puncta per cell by 67% (P = 0.001; −PKAcat: n = 18 cells, +PKAcat: n = 16 cells) in the basal state (Figure S2F).
On the other hand, cAMP directly enhances RIα LLPS in the presence of PKAcat, as addition of cAMP attenuated the inhibitory effect of PKAcat on RIα liquid droplet formation in vitro and allowed liquid droplet formation at lower RIα concentrations (Figure 2C). In 293-RIα cells expressing GFP1–10, stimulation with the AC activator forskolin (Fsk) to elevate cAMP induced an acute increase in endogenous RIα puncta (70% ± 12%, n = 32 cells) (Figures 2D and 2F). Furthermore, increasing cAMP levels through the β-adrenergic receptor agonist isoproterenol transiently increased the number of endogenous RIα puncta per cell (Figure 2F), consistent with the cAMP dynamics induced by this GPCR agonist (DiPilato and Zhang, 2009; Rich et al., 2001). These data suggest that cAMP-bound RIα is more prone to undergo LLPS, consistent with an increase in disorder within the inhibitory sequence and linker region when cAMP-bound RIα dissociates from PKAcat. To observe the localization of PKAcat in this process, we stimulated HEK293T cells overexpressing both RIα and PKAcat with Fsk and observed a 164% ± 32% increase in the number of RIα puncta per cell, with PKAcat co-localizing with RIα puncta (Figure 2E, S2G, and S2H and Video 3, n = 22 cells). Consistent with this observation, PKAcat (1% GFP-tagged) formed liquid droplets in vitro when mixed with RIα and cAMP (Figure 2G), but did not form liquid droplets on its own (Figure S5B). These results suggest that cAMP dynamics dictate the formation and dissolution of RIα phase-separated bodies and that PKAcat co-phase separates with RIα.
RIα condensates recruit and retain high cAMP levels and PKA activity
The observed enrichment of PKAcat in RIα phase-separated bodies prompted us to directly probe PKA activity in these bodies. To perform these measurements at the endogenous level, we designed Fluorescent Sensors Targeted to Endogenous Proteins (FluoSTEPs), a class of FRET-based biosensors that contain sensing domains sandwiched between mRuby2 (acceptor) and GFP1–10 (partial donor). The FRET donor fully reconstitutes in the presence of GFP11 fused to an endogenous protein of interest, resulting in the assembly of functional biosensors only at endogenous protein loci. We designed a FluoSTEP A kinase Activity Reporter (FluoSTEP-AKAR) based on a previously established PKA activity sensing domain consisting of a surrogate PKA substrate sequence (LRRATLVD) and forkhead associated domain 1 (FHA1) as the phosphoamino acid-binding domain (Figure 3A) (Zhang et al., 2005). When PKA is active, phosphorylation of the PKA substrate and its subsequent binding to FHA1 are expected to induce a conformational change and an increase in the red/green emission ratio (Figure S3A).
Figure 3. Endogenous RIα condensates form cAMP/PKA compartments and enable PDE-mediated cAMP compartmentation.
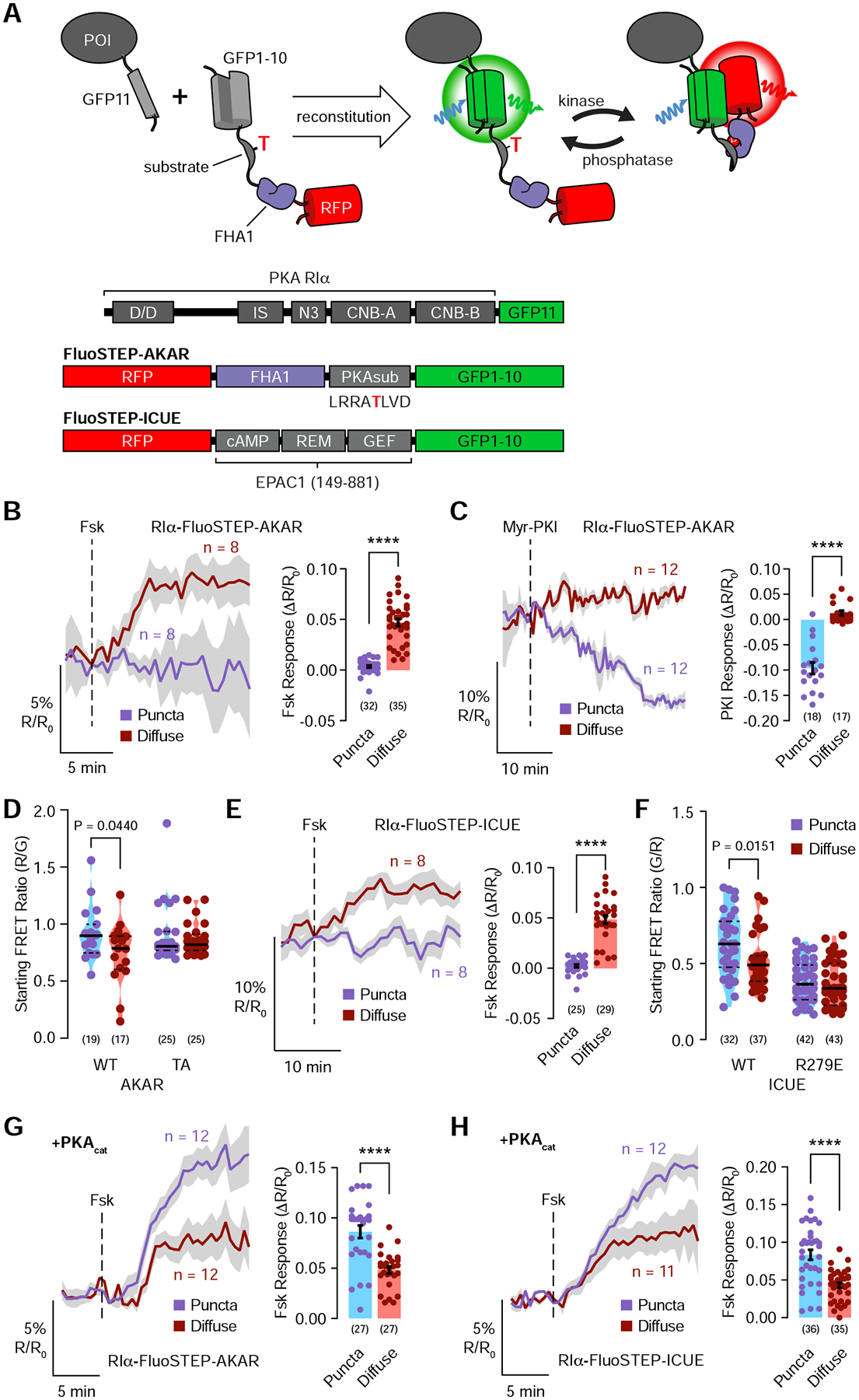
(A) Left: Fluorescent Sensors Targeted to Endogenous Proteins (FluoSTEPs) utilize split-GFP complementation to recruit a biosensor (e.g., FluoSTEP-AKAR) to a protein of interest (POI) expressed at endogenous levels. Right: Domain structures of RIα-GFP11, FluoSTEP-AKAR, and FluoSTEP-ICUE. (B-F) Basal PKA activity and cAMP levels within RIα phase-separated bodies are high enough to saturate FluoSTEP responses prior to stimulation. (B and C) Left: Red/green (R/G) emission ratio changes in 293-RIα cells transfected with FluoSTEP-AKAR and stimulated with either 50 μM Fsk (B) or 20 μM myristoylated-PKI (Myr-PKI) (C). RIα puncta (blue curve) and non-puncta regions (red curve) were analyzed separately. Right: Response to Fsk (B) (n = 32 puncta and 35 diffuse regions from 32 cells) or Myr-PKI (C) treatment. (D) Raw starting emission ratios for FluoSTEP-AKAR and FluoSTEP-AKAR T/A. RIα puncta and non-puncta regions were analyzed separately (WT AKAR: n = 19 puncta and 17 diffuse regions from 17 cells; AKAR T/A: n = 25 puncta and 25 diffuse regions from 25 cells). (E) Left: Green/red (G/R) emission ratio changes in 293-RIα cells transfected with FluoSTEP-ICUE and stimulated with 50 μM Fsk. RIα puncta (blue curve) and non-puncta regions (red curve) were analyzed separately. Right: Response to Fsk stimulation. (F) Raw starting emission ratios for FluoSTEP-ICUE and FluoSTEP-ICUE R279E (WT ICUE: n = 32 puncta and 37 diffuse regions from 32 cells; ICUE R279E: n = 42 puncta and 43 diffuse regions from 42 cells). (G and H) Left: R/G (G) or G/R (H) emission ratio changes in 293-RIα cells transfected with FluoSTEP-AKAR (G) or FluoSTEP-ICUE (H) plus mTagBFP2-PKAcat and stimulated with 50 μM Fsk. Newly formed RIα puncta regions (blue curve) and non-puncta regions (red curve) were analyzed separately (FluoSTEP-AKAR: n = 12 new puncta and 12 diffuse regions from 12 cells; FluoSTEP-ICUE: n = 11 new puncta and 12 diffuse regions from 11 cells). Right: Responses to Fsk stimulation (FluoSTEP-AKAR: n = 27 new puncta and 27 diffuse regions from 27 cells; FluoSTEP-ICUE: n = 35 new puncta and 36 diffuse regions from 35 cells). Solid lines in B, C, E, G, and H indicate representative average time courses of either R/G (B, C, and G) or G/R (D and H) emission ratio changes; shaded areas, SEM. Bar graphs in B, C, E, G, and H show maximum emission ratio changes upon drug addition, with bars indicating mean ± SEM. Violin plots in D and F show the median and quartiles as solid and dashed lines, respectively.
In 293-RIα cells expressing FluoSTEP-AKAR, Fsk induced a 4.7% ± 0.35% increase in the red/green emission ratio (raw emission ratios (R): Rt=0 to Rt=end: 0.82 to 0.86; n = 35 cells) in diffuse RIα regions (Figure 3B) but no detectable changes within RIα puncta (Rt=0 to Rt=end: 0.88 to 0.86; n = 32 cells), thus indicating clear differences in PKA activity despite the limited dynamic range of this first-generation technology. Conversely, the cell-permeable PKA inhibitor myristoylated-PKI induced no detectable ratio changes in diffuse RIα regions (Rt=0 to Rt=end: 0.79 to 0.81; n = 17 cells) under these same conditions but induced a 9.6% ± 1.1% (Rt=0 to Rt=end: 0.89 to 0.81; n = 18 cells) decrease in the red/green emission ratio in RIα puncta (Figure 3C), suggesting that basal PKA activity within RIα phase-separated bodies is high enough to saturate FluoSTEP-AKAR prior to stimulation. Indeed, prior to any stimulation, the initial red/green emission ratio was higher in RIα puncta regions compared with diffuse RIα regions (Figure 3D). As a control, 293-RIα cells transfected with FluoSTEP-AKAR T/A, which contains a non-phosphorylatable PKA substrate (Zhang et al., 2005), showed no significant red/green ratio difference between RIα puncta and diffuse regions (Figure 3D). The observed high basal emission ratio is consistent with the enrichment of PKA activity in RIα phase-separated bodies, although exclusion of phosphatases from RIα condensates could also contribute to the observed effect.
To similarly probe cAMP dynamics within RIα puncta, we utilized a FluoSTEP Indicator of cAMP Using Epac (FluoSTEP-ICUE) designed based on the same split biosensor approach (Figure 3A). Following reconstitution of the donor fluorophore, cAMP binding to a truncated fragment of Epac1, a previously established cAMP sensing domain (DiPilato and Zhang, 2009) which includes the cAMP binding domain (cAMP), Ras exchange motif (REM), and guanine exchange factor domain (GEF), induces a conformational change and an increase in the green/red emission ratio (Figure S3B and S3C). In 293-RIα cells transfected with FluoSTEP-ICUE, Fsk induced no detectable changes in RIα puncta regions (Rt=0 to Rt=end: 0.61 to 0.61; n = 25 cells) but induced a 4.9% ± 0.4% increase in the green/red emission ratio in diffuse RIα regions (Rt=0 to Rt=end: 0.57 to 0.59; n = 29 cells) (Figure 3E), suggesting that basal cAMP levels in RIα phase-separated bodies are also able to saturate FluoSTEP ICUE prior to stimulation. As an indication of basal cAMP levels, the initial green/red emission ratio for FluoSTEP-ICUE was higher in RIα puncta regions compared with diffuse RIα regions, while introducing an R279E mutation to inhibit cAMP binding (DiPilato et al., 2004) abolished this difference (Figure 3F). Importantly, the dynamic range of our cAMP biosensor was not affected by localization to RIα puncta, as demonstrated by identical green/red emission ratio increases both inside and outside puncta in cells expressing RIα-tethered green/red-ICUE and treated with the AC inhibitor MDL-12330A followed by a cAMP analogue (Figure S3D). Notably, the emission ratio of the puncta-localized sensor gradually decreased to that in diffuse regions following MDL addition (Figure S3D), while a fluorescently labeled cAMP analogue (8-Φ−450-cAMP) localized almost exclusively to RIα liquid droplets in vitro (Figure S3E–G), further supporting high basal cAMP levels within these puncta.
Next, we examined how PKA activity and cAMP dynamics changed during the formation of RIα puncta. In PKAcat-expressing 293-RIα cells that showed Fsk-induced puncta formation (Figure S2F), Fsk induced a larger increase in the FluoSTEP-AKAR red/green emission ratio (8.6% ± 0.63%; Rt=0 to Rt=end: 0.8 to 0.87; n = 27 cells) in newly formed RIα puncta compared with the constantly diffuse RIα regions (4.8% ± 0.35%; Rt=0 to Rt=end: 0.8 to 0.82; n = 27 cells) (Figure 3G). Similarly, Fsk-induced FluoSTEP-ICUE responses were larger in newly formed RIα puncta (8.4% ± 0.66%; Rt=0 to Rt=end: 0.55 to 0.59; n = 35 cells) compared with the constantly diffuse RIα regions (4.3% ± 0.34%; Rt=0 to Rt=end: 0.55 to 0.57; n = 36 cells) (Figure 3H). These results suggest that RIα phase-separated bodies recruit and retain active PKAcat and cAMP.
Dynamic cAMP buffering by RIα condensates drives cAMP compartmentation
While cAMP degradation by PDEs has been shown to help create cAMP compartments inside cells, mathematical modeling suggests that PDE activity alone appears to be insufficient to restrict cAMP, given the current understanding of cAMP diffusion characteristics (Boras et al., 2014; Lohse et al., 2017; Yang et al., 2016). Key mechanisms that enable cAMP compartmentation therefore await discovery. Given the high level of cAMP observed in RIα condensates, we hypothesized that these bodies help compartmentalize cAMP by serving as a dynamic buffering system. Using an improved cAMP sensor (ICUE4; Figure S4A and S4B) fused to the catalytic portion of PDE4D2 (PDE4D2cat) (PDE4D2cat-ICUE4; Figure 4A, 4B, and S4C) to monitor local cAMP within PDE compartments (Bock et al., 2020) as a direct assay for cAMP compartmentation, we found that Fsk induced a small increase in the normalized cyan/yellow emission ratio of only 6.2% ± 0.23% (n = 55 cells), whereas blocking PDE activity using the PDE4-selective inhibitor rolipram and a general PDE inhibitor IBMX rescued the response, suggesting that PDE4D2cat can form a cAMP sink under control conditions when the cAMP compartmentation system in intact (Figure 4B). However, when RIα LLPS was disrupted with 2.5% 1,6-hexanediol pretreatment (Figure 4B, S4D–F), Fsk induced much greater cAMP accumulation around PDE4D2, indicated by a larger increase in the normalized cyan/yellow emission ratio (30% ± 0.76%, P < 0.0001; n = 50 cells) of the PDE4D2cat-ICUE4 probe, suggesting that disrupting RIα condensates leads to decreased cAMP buffering and loss of effective cAMP compartmentation. In cells with no RIα LLPS as RIα is homozygously knocked out, Fsk induced similar changes with and without 1,6-hexanediol pretreatment (Figure 4C), suggesting the effect on cAMP compartmentation is mediated by RIα. The effect of disrupting RIα condensates was even stronger when RIα was overexpressed. In control cells, Fsk induced no detectable changes in the normalized PDE4D2cat-ICUE4 emission ratio (−0.40% ± 0.055%, n = 72 cells), suggesting that RIα overexpression further enhances cAMP compartmentation. In sharp contrast, when RIα LLPS was disrupted by 1,6-hexanediol pretreatment, Fsk stimulation induced a large 65% ± 2.0% (n = 30 cells) increase in the normalized emission ratio (Figure 4D, P < 0.0001). Moreover, RIα LLPS decreased basal cAMP levels around the PDE4D2 compartment, as the initial cyan/yellow emission ratios for PDE4D2cat-ICUE4 were lower when RIα was present versus when RIα was knocked out (Figure S4G).
Figure 4. Dynamic cAMP buffering by RIα condensates drives cAMP compartmentation.
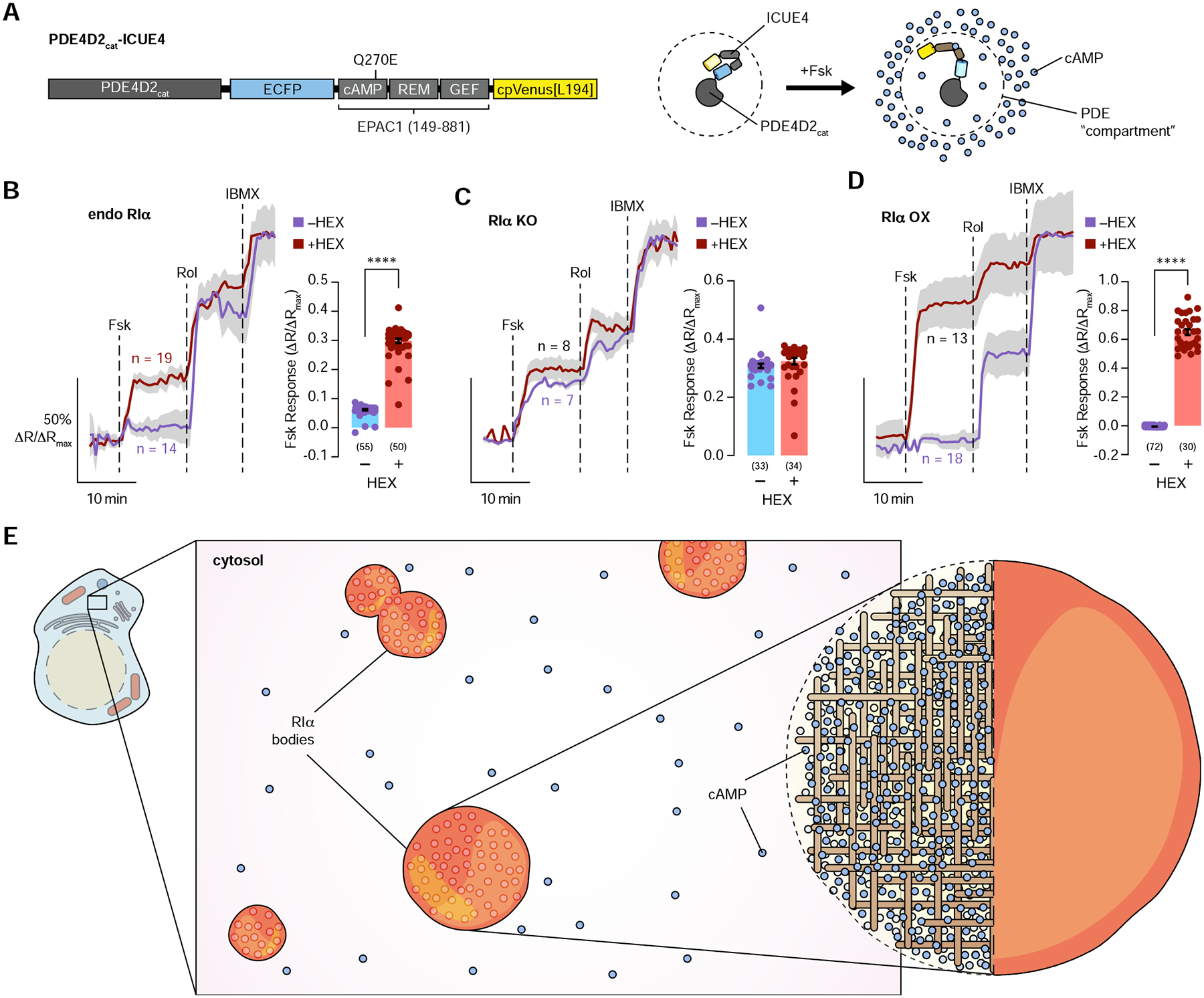
(A) Domain structure of the PDE4D2cat-ICUE4 sensor, which is used to measure cAMP levels within the PDE4D2 compartment. (B-D) Investigating the formation of PDE-mediated cAMP sinks with and without RIα phase separation. Left: Representative average time courses of cyan/yellow (C/Y) emission ratio changes (normalized to maximum) in wild-type HEK293T cells transfected with PDE4D2cat-ICUE4 (endo RIα) (B), RIα null HEK293T cells transfected with PDE4D2cat-ICUE4 (RIα KO) (C), or HEK293T cells co-transfected with PDE4D2cat-ICUE4 and mRuby2-RIα (RIα OX) (D). Cells with (red curve) or without (blue curve) 2.5% 1,6-hexanediol pretreatment were stimulated with 50 μM Fsk, 1 μM rolipram (Rol), and 100 μM IBMX. Right: Maximum normalized emission ratio upon Fsk stimulation. Solid lines in B-D indicate the mean; shaded areas, SEM. Bars in B-D indicate mean ± SEM. (E) Schematic illustration of cAMP buffering via RIα phase separation. RIα droplets dynamically sequester cAMP, effectively buffering cAMP in the cytosol.
To identify the key determinants of RIα-mediated cAMP compartmentation, we expressed mRuby2-tagged RIαD/D+linker, which can phase separate but does not bind cAMP, and mRuby2-tagged RIαΔD/D+linker, which can bind to cAMP but cannot phase separate, in RIα null cells and measured cAMP levels around PDE4D2cat. In both cases, Fsk treatment induced significant increases in cAMP levels around PDE4D2cat irrespective of 1,6-hexanediol pretreatment, while basal cAMP levels around the PDE4D2 compartment were also elevated, as the initial cyan/yellow emission ratios for PDE4D2cat-ICUE4 were higher for both mutants compared with wild-type RIα (Figures S4G–L). These data suggest that both the formation of RIα condensates and the ability of RIα to sequester cAMP are required for effective cAMP compartmentalization. Moreover, experiments using dye-labeled cAMP suggest that cAMP is essentially “trapped” inside the RIα condensates (Supplementary Figure 3E–F and Figure 4E). Overall, these results highlight a key mechanism of cAMP compartmentation wherein RIα condensates enable PDEs to function as local cAMP sinks to drive cAMP signaling specificity.
An oncogenic PKA fusion abolishes RIα phase separation
Disruption of RIα phase-separation leads to defective cAMP compartmentation, and the aberrant cAMP/PKA signaling caused by altered cAMP compartmentation is linked to various diseases (Caretta and Mucignat-Caretta, 2011; Perera and Nikolaev, 2013; Steinberg and Brunton, 2001). FLC is an atypical liver cancer that primarily affects young adults with no pre-existing liver conditions (Sergi, 2015), making the etiology of this cancer enigmatic. While the DnaJB1-PKAcat fusion oncogene is detected in nearly all FLC patients, the mechanism by which this fusion protein drives FLC is completely unknown (Honeyman et al., 2014; Kastenhuber et al., 2017). Intriguingly, we observed an almost complete absence of RIα puncta formation in HEK293T cells overexpressing DnaJB1-PKAcat and RIα compared with cells overexpressing wild-type PKAcat and RIα (Figures 5A, 5B, and S5A). A kinase-dead, ATP-binding deficient mutant of DnaJB1-PKAcat, DnaJB1-PKAcatK72H, which does not induce FLC in animal models (Kastenhuber et al., 2017), restored RIα LLPS when co-expressed (Figure 5A and 5B), presumably due to a reduced affinity for RIα (Lu et al., 2019). Fsk also failed to induce any significant increases in RIα LLPS when DnaJB1-PKAcat or DnaJB1-PKAcatK72H were co-expressed with RIα, in contrast to wild-type PKAcat (Figures 5C and S5C and Video 4), with little RIα LLPS observed in the case of DnaJB1-PKAcat and many RIα puncta observed in the case of DnaJB1-PKAcatK72H.
Figure 5. The FLC oncoprotein DnaJB1-PKAcat disrupts RIα phase separation and cAMP compartmentation, resulting in increased cell proliferation and transformation.
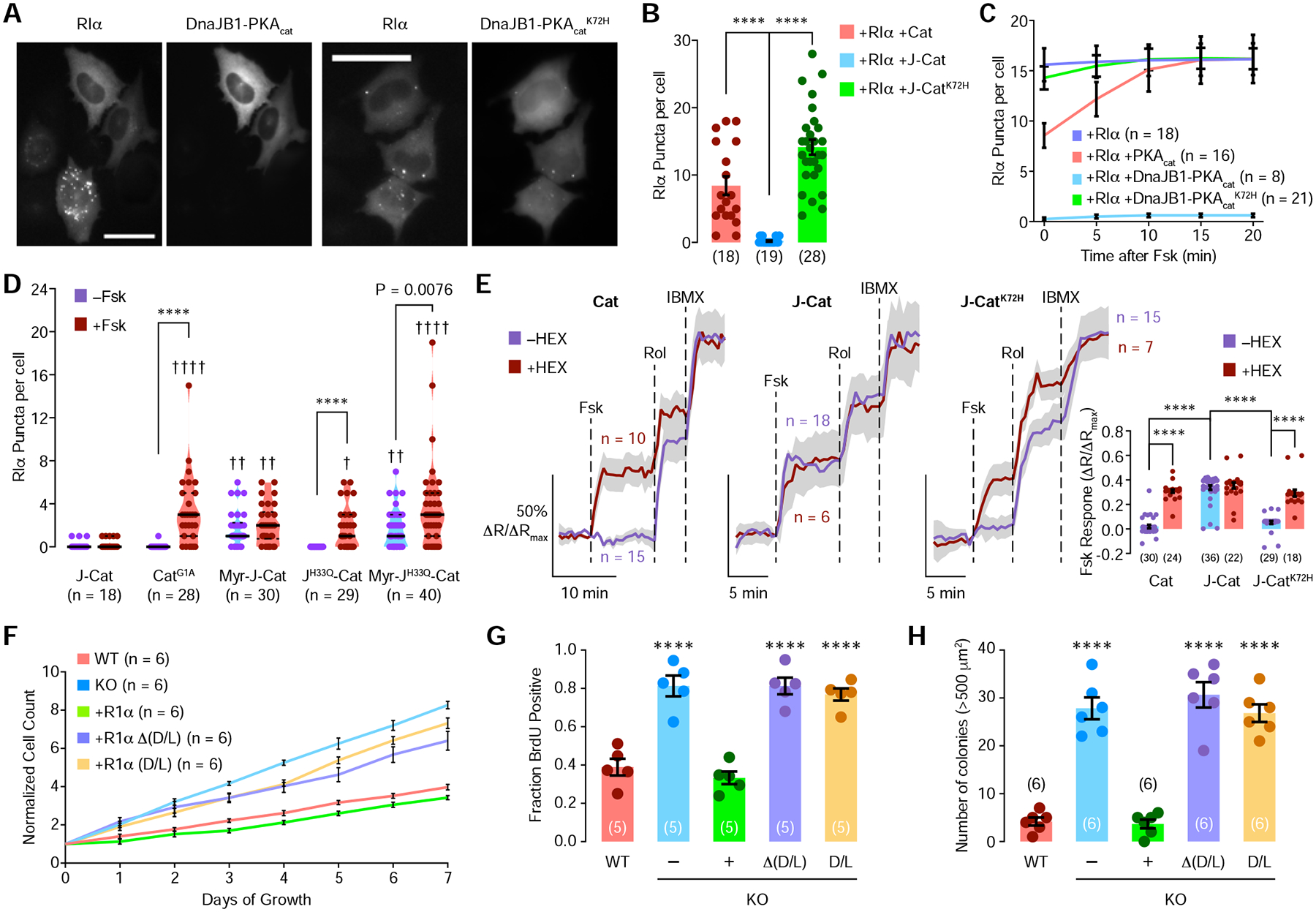
(A) Representative fluorescence images of HEK293T cells transfected with EGFP-tagged RIα and either mTagBFP2-tagged DnaJB1-PKAcat (left) or DnaJB1-PKAcatK72H (right). Scale bars, 40 μm. (B) Average number of RIα puncta per cell in HEK293T cells co-transfected with EGFP-RIα and mTagBFP2-tagged PKAcat (Cat), DnaJB1-PKAcat (J-Cat), or DnaJB1-PKAcatK72H (J-CatK72H). (C) Average time course of the number of RIα puncta per cell following 5 μM Fsk addition to HEK293T cells transfected with EGFP-RIα alone (dark blue curve) or EGFP-RIα plus mTagBFP2-tagged PKAcat (red curve), DnaJB1-PKAcat (light blue curve), or DnaJB1-PKAcatK72H (green curve). (D) Comparison of RIα puncta number between cells expressing RIα plus DnaJB1-PKAcat (J-Cat), wild-type PKAcat with no myristoylation (CatG1A), DnaJB1-PKAcat with myristoylation consensus sequence at N-terminus (Myr-J-Cat), DnaJB1-PKAcat which cannot bind to Hsp70 (JH33Q-Cat), or DnaJB1-PKAcat with both myristoylation and no Hsp70 binding (Myr-JH33Q-Cat). Cells were then stimulated with 50 μM Fsk. (E) Representative average time courses of cyan/yellow (C/Y) emission ratio changes (normalized to maximum) in HEK293T cells transfected with PDE4D2cat-ICUE4 and mTagBFP2-RIα plus mCherry-tagged PKAcat (Cat), DnaJB1-PKAcat (J-Cat), or DnaJB1-PKAcatK72H (J-CatK72H). Cells with (red curves) or without (blue curves) 2.5% 1,6-hexanediol pretreatment were stimulated with 50 μM Fsk, 1 μM rolipram (Rol), and 100 μM IBMX. Solid lines indicate the mean; shaded areas, SEM. Inset: Maximum normalized emission ratio change upon Fsk stimulation for each condition. (F-H) Dysfunctional RIα phase promotes tumorigenic phenotypes in AML12 hepatocytes. RIα phase separation was achieved by knocking out RIα or expressing either RIαD/D+Linker, which permits RIα phase separation but lacks cAMP binding, or RIαΔ(D/D+Linker), which retains cAMP binding but lacks phase separation, in RIα null cells. (F) Average time courses of the cell count for AML12 cells under different conditions. Error bars indicate SEM. (G) Average percentage of BrdU+ AML12 cells. (H) Average number of colonies larger than 500 μm2 grown in soft agar. Bars in B, E, G, and H indicate mean ± SEM. Violin plot in D show the median and quartiles as solid and dashed lines, respectively.
N-terminal fusion of the J-domain abolishes PKAcat myristoylation (Bastidas et al., 2012) and recruits the Hsp70 chaperone to the fusion protein (Turnham et al., 2019). Therefore, we next tested the effect of PKAcat myristoylation and Hsp70 recruitment by the J-domain on RIα LLPS. Mutating the myristoylation site in wild-type PKAcat (PKAcatG1A) significantly decreased the numbers of RIα puncta under both basal and Fsk-stimulated conditions (Figure 5D). Intriguingly, adding an N-terminal sequence to restore myristoylation to DnaJB1-PKAcat partially reversed the abolishing effect of DnaJB1-PKAcat on basal RIα LLPS (Figure 5D). Furthermore, disrupting the ability of the J-domain to recruit the Hsp70 chaperone by introducing an H33Q mutation (Turnham et al., 2019) in DnaJB1-PKAcat restored the cAMP-responsive formation of RIα biomolecular condensates (Figure 5D). Combining these two alterations partially restored both basal and Fsk-stimulated puncta formation (Figure 5D). Collectively, these results show that DnaJB1-PKAcat strongly suppresses both basal and cAMP-responsive RIα LLPS, which are partly mediated by the loss of myristoylation and binding to Hsp70, respectively.
Loss of RIα phase separation disrupts cAMP compartmentation and leads to increased cell proliferation and transformation
Given that RIα LLPS is essential to enable cAMP compartmentation, we expect that the DnaJB1-PKAcat-induced loss of RIα LLPS should lead to defective cAMP compartmentation. We therefore tested the effect of DnaJB1-PKAcat using our PDE4D2cat-ICUE4 cAMP compartmentation assay. In cells expressing wild-type PKAcat and RIα, Fsk induced no detectable changes in the normalized cyan/yellow emission ratio of PDE4D2cat-ICUE4 (2.0% ± 1.5%, n = 30 cells), but induced a large 31% ± 1.3% (n = 24 cells) increase in the normalized cyan/yellow emission ratio under these same conditions when RIα LLPS was disrupted by 1,6-hexanediol pretreatment (Figure 5E, P < 0.0001). On the other hand, cells overexpressing DnaJB1-PKAcat showed similar Fsk-induced PDE4D2cat-ICUE4 responses regardless of 1,6-hexanediol pretreatment (Figure 5E). However, in the presence of DnaJB1-PKAcatK72H, which restores RIα LLPS, Fsk again induced only a small increase in the normalized PDE4D2cat-ICUE4 emission ratio (5.4% ± 1.2%, n = 29 cells) versus a much larger 29% ± 2.8% (n = 18 cells) increase when RIα LLPS was disrupted by 1,6-hexanediol pretreatment (Figure 5E, P < 0.0001). Altogether, these data show that the disruption of RIα LLPS by DnaJB1-PKAcat dramatically impairs cAMP compartmentation, providing a mechanistic clue for aberrant signaling caused by this oncoprotein fusion.
A critical question is whether loss of RIα LLPS and subsequent disruption of cAMP compartmentation could have any functional impact on cellular processes, particularly with respect to tumorigenesis. To test the functional consequences of loss of RIα LLPS, we therefore generated an RIα null cell line using non-tumorigenic hepatocytic AML12 cells in which we then stably expressed wild-type or various mutant forms of RIα and measured their proliferation rates and transformation capabilities. Strikingly, loss of RIα increased cell proliferation and DNA synthesis by 2-fold compared with wild-type cells. In RIα null cells, re-expressing wild-type RIα rescued the wild-type phenotype, whereas stably expressing RIα mutants that were defective in either LLPS or cAMP binding failed to rescue (Figure 5F and 5G, P < 0.0001). Moreover, loss of RIα led to the formation of detectable colonies on soft agar (Figure 5H and S5D, P < 0.0001), suggesting that loss of RIα leads to anchorage-independent growth, a hallmark for cancer cells. Similar to what was observed for cell proliferation, the restoration of wild-type RIα in RIα null cells inhibited colony formation, whereas RIα null cells expressing RIα mutants that were defective in either LLPS or cAMP binding continued to exhibit anchorage-independent growth (Figure 5H and S5D, P < 0.0001). These results suggest that loss of RIα LLPS and disrupted cAMP compartmentation lead to tumorigenic phenotypes in hepatocytes. Similar results were also observed with mouse embryonic fibroblasts (Figure S5E–G), suggesting that the tumor-suppressive nature of RIα LLPS has broad implications.
DISCUSSION
cAMP compartmentation is crucial for our understanding of how this pathway achieves signaling specificity (Baillie, 2009; Steinberg and Brunton, 2001); however, the mechanisms responsible for compartmentalizing this ubiquitous second messenger are unclear (Lohse et al., 2017; Yang et al., 2016). Local degradation of cAMP has been suggested as a key mechanism in spatially constraining cAMP and forming cAMP compartments (Conti et al., 2014; Stangherlin and Zaccolo, 2012), yet the discrepancy between the modest catalytic capabilities of these enzymes (Conti and Beavo, 2007; Goraya et al., 2008) and the reportedly rapid diffusion of cAMP in cells (Bacskai et al., 1993; Chen et al., 1999; Nikolaev et al., 2004) calls into question the dominant role assigned to PDEs (Lohse et al., 2017). In contrast, our companion paper Bock et al. estimated a PDE4A1 turnover number of ~160 molecules/s in intact cells, ~30 times faster than in vitro estimates (Lohse et al., 2017). They further revealed that cAMP appears largely immobile in the basal state, exhibiting buffered diffusion which results in low free cAMP concentrations and ultimately allows PDEs to create functionally relevant nanodomains of low cAMP concentration. Here, we showed that RIα biomolecular condensates act as a dynamic “sponge” in recruiting and retaining cAMP and active PKAcat, processes that are required for cAMP compartmentation, as disrupting these condensates leads to the loss of PDE-mediated cAMP sinks. This work thus fills a key gap in our understanding of this fundamental process by uncovering a key mechanism to enable cAMP compartmentation. Thermodynamically, both the RIα phase-separated bodies and the surrounding cytosol can be considered as mixtures of cAMP, RIα, and all other components present. The formation of droplets via LLPS and the non-stoichiometric increase of cAMP in these droplets point to differences in chemical potential (Denbigh, 1951; De Groot and Mazur, 2013; Hyman et al., 2014; Kuiken, 1994) underlying the cAMP concentration difference between RIα bodies and the cytosol. This discovery of a critical cAMP compartmentation system mediated by LLPS may ultimately redefine our understanding of how cAMP compartmentation shapes the cAMP/PKA signaling landscape to achieve functional diversity. Future studies will further characterize RIα biomolecular condensates to determine whether other components such as AKAPs and phosphatases are present.
Increasing evidence suggests that LLPS acts as a principal organizer of numerous cellular processes such as actin polymerization (Li et al., 2012), transcription (Boija et al., 2018; Gibson et al., 2019; Sabari et al., 2018), and stress responses (Brangwynne et al., 2009; Jain et al., 2016; Molliex et al., 2015). Meanwhile, emerging studies have shown that many signaling molecules also undergo LLPS (Cai et al., 2019; Su et al., 2016). Although many macromolecules have been shown to undergo LLPS in cells, how LLPS impacts their biochemical activities and functions is often unclear. By engineering fluorescent biosensors that can reconstitute at endogenous loci (FluoSTEPs), we can measure enzyme activity and small molecule dynamics directly within biomolecular condensates without perturbing the expression levels of their individual constituents. Here, we targeted FluoSTEPs to endogenous RIα and measured high cAMP levels and PKA activity in RIα bodies. cAMP levels and PKA activity were particularly enriched and retained in newly formed RIα bodies, suggesting that these condensates dynamically buffer cAMP. These live-cell activity measurements were essential for generating our hypothesis, which led us to discover the cellular function of RIα bodies as a key cAMP compartmentation system, critical for signaling specificity in the cAMP/PKA pathway. Because FluoSTEPs share the same modular design as all FRET-based sensors (Greenwald et al., 2018; Zhou et al., 2012), their application is expandable to monitor other signaling activities and should thus aid in further elucidating the organizing principles of cellular activity architectures, including the role of phase-separated enzymatic assemblies in other systems.
FLC is atypical among liver cancers as it is not correlated with age, cirrhosis, or common markers of liver disease (Sergi, 2015). Although the DnaJB1-PKAcat fusion oncoprotein is reported to be present in the majority of FLC patients (Dinh et al., 2017; Honeyman et al., 2014), the pathological mechanisms of this oncogenic fusion are completely unknown. From a structural and biochemical standpoint, DnaJB1-PKAcat is largely indistinguishable from wild-type PKAcat with respect to interaction interface and binding affinity for PKA regulatory subunits (Cao et al., 2019; Cheung et al., 2015), cAMP activation (Cao et al., 2019), and catalytic activity (Cao et al., 2019; Honeyman et al., 2014; Riggle et al., 2016). Furthermore, although DnaJB1-PKAcat is expressed at approximately 10-fold higher levels than wild-type PKAcat due to promoter alterations (Riggle et al., 2016), overexpressing wildtype PKAcat does not induce tumor formation in mice (Kastenhuber et al., 2017), suggesting that expression differences alone are not likely the determining factor. Our study provides a mechanistic link between DnaJB1-PKAcat and tumorigenesis. DnaJB1-PKAcat abolishes RIα LLPS, disrupting cAMP compartmentation and deregulating cAMP/PKA signaling. Furthermore, loss of RIα LLPS in non-tumorigenic hepatocytes and fibroblasts leads to tumorigenic phenotypes such as increased cell proliferation and anchorage-independent growth. Interestingly, a subset of FLC patients lack the DnaJB1-PKAcat oncogene but exhibit loss of RIα protein expression (Graham et al., 2018), corroborating our model that loss of RIα LLPS is a key driver of FLC. Mechanistically, loss of myristylation and gain of Hsp70 binding (Turnham et al., 2019) by DnaJB1-PKAcat are partially responsible for blocking RIα LLPS, though other mechanisms may also be involved. Intriguingly, both DnaJB1-PKAcat and the related fusion oncoprotein ATP1B1-PKAcat have been detected in intraductal oncocytic papillary neoplasms (Singhi et al., 2019); thus, our findings that loss of cAMP compartmetnation drives tumorigenic signaling may be applicable to other cancers. While multiple studies have shown that the emergence or enhancement of LLPS is linked to stress conditions or neurological disease states (Jain et al., 2016; Markmiller et al., 2018; Molliex et al., 2015; Peskett et al., 2018; Wegmann et al., 2018), our work provides a distinct example of LLPS being necessary for normal cellular function, with the loss of LLPS leading to disease phenotypes, where only a limited number of examples exist (Bouchard et al., 2018).
In summary, we have discovered a membraneless organelle that shapes the PKA signaling landscape. Our results represent a conceptual leap forward in understanding how the cAMP/PKA pathway is dynamically organized. Given the universal nature of cAMP/PKA signaling and the ubiquitous expression of RIα (Cadd and McKnight, 1989), our findings have far-reaching physiological implications for various biological systems such as cardiomyocytes (Buxton and Brunton, 1983; Nikolaev et al., 2006; Scholten et al., 2007; Yin et al., 2008) and neurons (Gorshkov et al., 2017; Park et al., 2014; Shelly et al., 2010), in which the cAMP/PKA pathway plays diverse roles and RIα puncta formation can be observed (Figure S1). Furthermore, we have discovered a link between spatially dysregulated cAMP/PKA signaling and cancer. Overall, our findings showcase the intricacies of signaling activity architectures and the importance of biomolecular condensates in their construction.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jin Zhang (jzhang32@ucsd.edu).
Materials Availability
Plasmids generated in this study will be made available through Addgene and can be shared upon request.
Data and Code Availability
The sequences for FluoSTEP-ICUE and FluoSTEP-AKAR have been uploaded onto GenBank (FluoSTEP-ICUE: (MT800777.1); FluoSTEP-AKAR: (MT800778.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
HEK293A, HEK293T, and MEF cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco) containing 1 g L−1 glucose and supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma) and 1% (v/v) penicillin–streptomycin (Pen-Strep, Sigma-Aldrich). AML12 cells were cultured in DMEM:F12 medium (Thermo Fisher) containing 10% FBS, 10 μg mL−1 insulin, 5.5 μg mL−1 transferrin, 5 ng mL−1 selenium (ITS liquid media supplement, Sigma-Aldrich), and 40 ng mL−1 dexamethasone (Sigma-Aldrich). All cells were maintained in a 37°C incubator with a humidified 5% CO2 atmosphere. Before transfection, HEK293A and HEK293T were plated onto sterile poly-D-lysine coated 35-mm glass-bottomed dishes and grown to 50–70% confluence. HEK293A and HEK293T were then transfected using Polyjet (Signagen) and grown for an additional 16–24 h before imaging.
Neonatal rat ventricular myocytes were isolated from cardiac ventricles of 1- to 2-day-old male and female Sprague-Dawley rat pups (Charles River) as described previously (Miyamoto et al., 2010). Neonatal myocytes were plated at a density of 3.0 ×104 cm−2 on laminin-coated 35-mm glass-bottomed dishes and maintained in DMEM with 15% FBS overnight. Cells were transfected 24 h later using Polyjet for 48 h, with the media changed 24 h after transfection. Primary rat hippocampal neurons and glial cells were harvested from male and female E19 Sprague-Dawley rat pups (Charles River) in ice-cold Hank’s Balanced Salt Solution (HBSS, Gibco) and were dissociated using the Papain Dissociation System with MgSO4 and DNase I according to the manufacturer’s instructions. Dissociated neurons and glial cells were diluted to 200,000 cells mL−1 and resuspended in Neurobasal medium (Thermo Fisher) with 2% SM1 supplement (STEMCELL Technologies). Cells were plated onto 35-mm glass-bottom dishes coated with 100 μg mL−1 poly-D-lysine (Sigma) and cultured in a 37°C incubator with a humidified 5% CO2 atmosphere. At 1 or 3 days in vitro, cells were transfected with Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions in Neurobasal media and imaged after 48 h. All animals were treated in accordance with the UC San Diego Animal Care and Use Committee guidelines.
Generation of stable cell lines
To generate 293-RIα cells, HEK293A cells were plated in 6-well plates. After 24 h, 1 μg of px459 plasmid (Ran et al., 2013) (gift of Feng Zhang, Addgene plasmid #62988) (encoding Cas9, gRNA, and puromycin resistence gene) and 20 pmol of ssDNA ultramer HDR template (Integrated DNA Technologies; Table S1) were transfected using Polyjet following the manufacturer’s protocol. Cells were passaged 1 day after transfection into a 60-mm dish, and 1 μg mL−1 puromycin was added 24 h later. When no viable cells remained in the untransfected dish (around 2–3 days), the media was replenished without puromycin. Cells were passaged 24 h later and resuspended in sorting buffer (1 x DPBS with 0.5% BSA, 25 mM HEPES, 1 mM EDTA, pH 7, 2.5 μg mL−1 of DNase I (Thermo Fisher)) with 0.1μg mL−1 DAPI (Thermo Fisher). Cells were sorted for DAPI-negative staining and plated as single cells in a 96-well plate using a BD FACS Aria II Cell Sorter. After 3 weeks of incubation, wells containing single-cell colonies were passaged, and DNA was extracted for genotyping using the DNeasy Blood & Tissue Kit (Qiagen). Genomic PCR was performed using the Q5 High-Fidelity Kit (New England Biolabs) and with primers listed in Table S1. To evaluate the copy number of correct gene edits, PCR products were gel extracted using PureLink Quick Gel Extraction kit (Invitrogen), cloned into TOPO PCR vectors (Invitrogen), and subjected to Sanger sequencing (Genscript).
RIα null HEK293T cells were generated similarly to methods previously described (Ran et al., 2013), but with slight modifications. When cells reached 70% confluency, cells were co-transfected with two px458 plasmids (Ran et al., 2013) (gift of Feng Zhang, Addgene plasmid #48138) (Cas9, gRNA, and GFP) that target RIα. After 24 h of transfection, the cells were aspirated, washed with DPBS, and filtered through a 35-μm cell strainer. Cells with GFP signals were sorted into single cells in a 96-well plate using a BD FACSJazz™ cell sorter. After single cells had grown into colonies, the cells were transferred to 60-mm cell culture plates. The corresponding genomic DNA segment was PCR-amplified using primers listed in Table S1 to verify correct gene editing. Each colony was validated via western blotting and DNA sequencing.
RIα null MEF cells were generated previously (Day et al., 2011). RIα null AML12 (ATCC) cells were generated similarly to RIα knock-out HEK293T cells, except the gRNAs differ, and the corresponding genomic DNA segment to verify correct gene editing was PCR amplified using primers listed in Table S1. To generate stable RIα null MEF and AML12 cells with exogenously expressed RIα mutants, lentiviruses were made by transfection of pLenti backbone versions of RIα mutants with the packaging vectors pMD2.G (gift of Didier Trono, Addgene plasmid #12259) and psPAX2 (gift of Didier Trono, Addgene plasmid #12260) (Dull et al., 1998) into HEK293T cells. At 24 h after transfection, HEK293T cells were replenished with fresh media.
After an additional 2 days, supernatant was collected and sterile-filtered through a 0.45 μm filter. RIα null MEF and AML12 cells were infected with lentiviruses and underwent FACS.
METHOD DETAILS
Plasmid construction
RIα CRISPR constructs
All plasmids are in the pcDNA3.1 backbone unless specified and all primers used to generate plasmids are listed in Table S1. All constructs were verified by Sanger sequencing (Genscript). The vector expressing both gRNA and Cas9 in the px459 v2.0 backbone (px459) (Ran et al., 2013) was generated using Golden Gate cloning as previously described (Adikusuma et al., 2017). To construct gRNA expression vectors for generation of the 293-RIα cell line, the 20-bp target sequence was sub-cloned into px459. To generate RIα null HEK293T cells, two designed guide sequences that specifically target the human RIα gene were each cloned into the sgRNA scaffold in px458 (Ran et al., 2013). For RIα null AML12 cells and MEFs, two designed guide sequences that specifically target the mouse RIα gene were each cloned into the sgRNA scaffold in px458.
Fluorescent protein-tagged RIα constructs
EGFP-RIα and mCherry-PKAcat were generated previously (Day et al., 2011). mRuby2-RIα was generated via PCR amplification of mRuby2 from pcDNA3-AKAR2-CR (Lam et al., 2012) (gift of Michael Lin, Addgene plasmid #40255) and RIα from the RIα-EGFP plasmid backbone, followed by Gibson assembly using the NEBuilder Hi-Fi DNA Assembly Cloning Kit (New England Biolabs). mTagBFP2-RIα was generated by Gibson assembly of PCR products amplified from pBAD-mTagBFP2 (Subach et al., 2011) (gift of Vladislav Verkusha, Addgene plasmid #34632). RIα mutants C-terminally tagged with either EGFP or mRuby2 were generated via Gibson assembly of PCR products amplified from RIα-EGFP or RIα-mRuby2. mTagBFP2-PKAcat was constructed by Gibson assembly of PCR products amplified from pBAD-mTagBFP2 (Subach et al., 2011).
FluoSTEP sensor constructs
pcDNA3.1-GFP1–10 (Leonetti et al., 2016) was a gift from Bo Huang (Addgene plasmid #70219). To construct FluoSTEP-AKAR, mRuby2 was PCR-amplified from AKAR2-CR (Lam et al., 2012) and the FHA1 and PKA substrate were PCR-amplified from AKAR4 (Depry et al., 2011) (Addgene plasmid #61619). The resulting PCR fragments were Gibson assembled into HindIII-and EcoRI-digested pcDNA3.1 GFP1–10. FluoSTEP-ICUE was constructed similarly, except that Epac1149−881 was PCR-amplified from ICUE3 (DiPilato and Zhang, 2009) (Addgene plasmid #61622). FluoSTEP-AKAR (T/A) was constructed by Gibson assembly of PCR products amplified from FluoSTEP-AKAR. FluoSTEP-ICUE R279E was constructed by Gibson assembly of PCR products amplified from FluoSTEP-ICUE.
ICUE4 and related constructs
To construct ICUE4, the Q270E point mutation was introduced using site-directed mutagenesis of ICUE3 (DiPilato and Zhang, 2009). To construct PDE4D2cat-ICUE4, PDE4D286−418 was PCR-amplified from PDE4D2 cDNA (gift from Hengming Ke, UNC, Chapel Hill, NC), and the ICUE4 backbone was PCR-amplified. The resulting PCR fragments were Gibson assembled. RIα-ICUE4 was constructed by Gibson assembly of PCR products amplified from RIα-EGFP and ICUE4. RIαD/D+Linker-ICUE4 was constructed similarly to RIα-ICUE4 using PCR products amplified from RIαD/D+Linker-EGFP.
Red-green ICUE sensor constructs
To construct red-green ICUE (RG-ICUE), mRuby2 was PCR-amplified from AKAR2-CR (Lam et al., 2012), sfGFP was PCR-amplified from pcDNA3.1 GFP1–10, and Epac1149−881 was PCR-amplified from ICUE3 (DiPilato and Zhang, 2009). The resulting PCR fragments were Gibson assembled. To tag RIα with the RG-ICUE sensor, RIα was PCR-amplified from RIα-EGFP construct and RG-ICUE was PCR-amplified. The resulting PCR fragments were Gibson assembled.
PKAcat mutant constructs
mCherry-DnaJB1-PKAcat was generated using DNA gBlock segments (Table S1) designed and synthesized with connecting sequences at the 5’ ends, connecting sequences at the 3’ ends, and - GSGS- linkers in between DnaJB1-PKAcat and mCherry, which were Gibson assembled into HindIII- and XbaI-digested pcDNA3.1. mCherry-tagged DnaJB1-PKAcatK72H was constructed by Gibson assembly of PCR products amplified from mCherry-tagged DnaJB1-PKAcat. mTagBFP2-tagged DnaJB1-PKAcat and DnaJB1-PKAcatK72H plasmids were constructed by Gibson assembly of PCR products amplified from mCherry-tagged DnaJB1-PKAcat or DnaJB1-PKAcatK72H, respectively, and from pBAD-mTagBFP2 (Subach et al., 2011). mCherry-tagged PKAcatG1A was generated via Q5 site-directed mutagenesis (New England Biolabs). mCherry-tagged myristoylated-DnaJB1-PKAcat, which contains only the first 8 amino acids of wild-type PKAcat (first 8 amino acids of Cα1, an isoform of PKAcat, aligns with the consensus myristoylation sequence (Tillo et al., 2017; Udenwobele et al., 2017)), was constructed by Gibson assembly of PCR products amplified from PKAcat. mCherry-tagged DnaJB1H33Q-PKAcat (Turnham et al., 2019) was constructed by Gibson assembly of PCR products amplified from mCherry-DnaJB1-PKAcat. mCherry-myristoylated DnaJB1H33Q-PKAcat was constructed by Gibson assembly of PCR products amplified from mCherry-tagged myristoylated DnaJB1-PKAcat using the same primers described above for the H33Q J-domain mutation.
Lentiviral constructs
pLenti backbone versions of RIα mutants tagged with EGFP were constructed by Gibson assembly of PCR products amplified from pLenti-puro (Guan et al., 2011) (gift of Ie-Ming Shih, Addgene plasmid #39481) and the respective pcDNA3.1 versions of RIα mutants.
Construct to purify superfolder green fluorescent protein
sfGFP in pRSET B was constructed by Gibson assembly of PCR products amplified from pEvolvR-enCas9-PolI3M-TBD (Halperin et al., 2018) (gift from John Dueber & David Schaffer, Addgene plasmid #113077) to amplify sfGFP and from prSET B (Invitrogen) to amplify the pRSET B backbone.
Disorder and charge predictions of RIα
Disorder of full-length human RIα was predicted using PONDR (http://www.pondr.com/), which predicted that residues 63–105 and 264–320 are intrinsically disordered regions. The single-amino-acid and average (sliding window of 10 AA) charge distribution along the primary sequence were analyzed using EMBOSS (http://www.bioinformatics.nl/cgi-bin/emboss/charge), which predicted various regions to have high charge imbalance, such as the highly positively charged region of residues 81–96.
Cell counting to measure cell proliferation
MEF and AML12 stable cell lines were seeded in 6-wells plates at 10,000 cells/well. Cell numbers were quantified using a Countess II cell counter (Life Technologies) each day for 7 days.
BrdU staining to measure cell proliferation
MEF and AML12 stable cell lines were seeded on 35-mm glass-bottomed dishes at 10,000 cells/dish. At 48 h after plating, cells were treated with 10 μM BrdU (Invitrogen) for 4 h. Cells were washed twice with PBS and fixed with 3.7% formaldehyde in PBS. MEF and AML12 cells were imaged following application of standard immunofluorescence protocols: Triton X-100 permeabilization, 1 N and 2 N HCl addition, anti-BrdU primary antibody addition (1:100, Invitrogen), anti-mouse Alexa Fluor 647 (1:1000, Invitrogen), and 100 ng mL−1 DAPI nuclear staining.
Soft agar colony formation assay to assess cell transformation
Soft agar colony formation assays were performed as described previously (Borowicz et al., 2014). Briefly, 6-well plates were prepared containing 0.5% Noble Agar (Thermo Fisher) and 2X concentration of the respective cell media. After the agar solidified, 0.3% Noble Agar containing 5000 cells was applied on top of the 0.5% Noble Agar layer. MEF s and AML12 cells in soft agar were cultured in a 5% CO2 incubator for several weeks with 200 μL of the respective culture media added on top of the gel twice per week. Visible colonies appeared after 3 (MEFs) to 4 weeks (AML12 cells) and were photographed using a Canon EOS 5D Mark III DSLR camera (Canon USA).
Protein purification
Recombinant RIα and RIIβ were purified as described previously (Bastidas et al., 2012; Bruystens et al., 2014) with slight modifications. Constructs were transformed into Escherichia coli BL21 (DE3) cells and inoculated in LB media with 100 μg mL−1 ampicillin. Cultures were induced at OD600 = 0.6–0.8. After 16 h of expression under 0.5 mM IPTG at 16°C, the cell pellets were collected and then re-suspended and lysed in lysis buffer (20 mM MES, pH 6.5, 100 mM NaCl, 2 mM EDTA, 2 mM EGTA, and 5 mM DTT plus protease inhibitors and 10 μM 3-isobutyl-1-methylxanthine (IBMX)). The supernatant was collected after high-speed centrifugation (13,000 rpm, 1 h) and incubated overnight with cAMP-resin at 4°C. After centrifugation (3,000 rpm, 10 min) and removal of the supernatant, the resin was then washed sequentially with lysis buffer, wash buffer (20 mM MES, pH 6.5, 600 mM NaCl, 2 mM EDTA, 2 mM EGTA, and 5 mM DTT), and lysis buffer again. The proteins were eluted using elution buffer (20 mM MES, pH 5.5, 100 mM NaCl, 30 mM cGMP, 2 mM EDTA, 2 mM EGTA, and 5 mM DTT). The eluted proteins were then concentrated and further purified on an S-200 gel filtration column in 50 mM MES, pH 5.8, 200 mM NaCl, and 5 mM DTT.
PKAcat and DnaJB1-PKAcat were each generated previously (Lu et al., 2019). The constructs were transformed into Rosetta pLysS (DE3) cells and inoculated in LB media with 50 μg mL−1 kanamycin. Cultures were induced at OD600 = 0.6–0.8. After 16 h of expression under 0.5 mM IPTG at 18°C, the pellets were collected and the re-suspended and lysed in lysis buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, and 5 mM β-mercaptoethanol (BME)). The supernatant was collected after high-speed centrifugation (13,000 rpm, 1 h) and then passed through Ni-resin. The resin was then washed with 3 column volumes (CVs) of wash buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, 10 mM imidazole, and 5 mM BME), and the proteins were eluted by adding 3 CVs of elution buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, 500 mM imidazole, and 5 mM BME). The eluent was collected and supplemented with His6-tagged Ulp1 (Guerrero et al., 2015) (gift of Hideo Iwai, Addgene plasmid #64697), Ubiquitin-like-specific protease 1 (molar ratio SUMO-PKAcat or SUMO-DnaJB1-PKAcat:Ulp1 = 200:1). The solution was dialyzed (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, and 5 mM BME) overnight at 4°C. The cleaved tag, His6-tagged Ulp1, and uncleaved protein were removed by passing the solution back through the Ni-resin. After collection of the flow-through, the proteins were further purified by S-75 gel filtration in 20 mM MES, pH 6.5, 300 mM NaCl, and 5 mM BME.
To purify EGFP-PKAcat, we used pET-His6-EGFP-PKAcat, which was constructed previously (Lu et al., 2019) and fuses EGFP to His6 with a -GSS- linker and EGFP to PKAcat with a -GSAGSAAGSGEF- linker. The plasmid was transformed into Rosetta pLysS (DE3) cells and inoculated in LB media with 50 μg mL−1 kanamycin. Cultures were induced at OD600 = 0.6–0.8. After 16 h of expression under 0.5 mM IPTG at 18°C, the pellets were collected, re-suspended, and lysed in lysis buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, and 5 mM BME). The supernatant was collected after high-speed centrifugation (13,000 rpm, 1 h) and then passed through Ni-resin. The resin was then washed with 3 CVs of wash buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, 10 mM imidazole, and 5 mM BME), and proteins were eluted by adding 3 CVs of each elution buffer (20 mM Tris-Cl, pH 8.0, 300 mM NaCl, 50–500 mM imidazole, and 5 mM BME). After collection of the eluent, the protein was further purified via S-75 gel filtration in 20 mM MES, pH 6.5, 300 mM NaCl, 5 mM BME.
After purification, all proteins aforementioned were dialyzed into liquid droplet preparation buffer (150 mM KCl, 1 mM MgCl2, 20 mM HEPES, pH 7.0, 1 mM EGTA, 1 mM DTT, 0.5 mM ATP, final pH 7.0) and concentrated using Amicon Ultra-15 centrifugal filters (Millipore). Protein concentrations were measured using the Pierce BCA protein assay kit (Thermo Fisher).
To purify sfGFP, the pRSET B sfGFP construct was transformed into Escherichia coli BL21 (DE3) cells and inoculated in LB media with 100 μg mL−1 ampicillin. Cultures were induced at OD600 = 0.6–1.0. After 6 h of expression under 0.5 mM IPTG at 37°C, the cell pellets were collected, re-suspended in lysis buffer (50 mM Tris, pH 7.4, 300 mM NaCl) containing 1 mM PMSF and Complete EDTA-free Protease Inhibitor Cocktail (Roche), and lysed by sonication. Following centrifugation at 25,000 × g for 30 min at 4°C, the clarified lysate was loaded onto an Ni-NTA column, washed with wash buffer (50 mM Tris, pH 7.4, 300 mM NaCl, 10 mM imidazole), and then eluted using an imidazole gradient (20–200 mM in lysis buffer). The eluted proteins were then concentrated, and the sfGFP protein concentration was measured via BCA assay and absorbance.
In vitro liquid droplet assays
All liquid droplet formation assays were performed in 150 mM KCl (unless specified), 5 mM MgCl2, 10 μM cAMP (as indicated), 20 mM HEPES, pH 7.0, 1 mM EGTA, 1 mM DTT, 0.5 mM ATP, 100 mg/ml Polyethylene Glycol 4000 (unless specified), and a final pH of 7.0. Purified proteins were incubated at different stoichiometries and at various concentrations at room temperature for 1 h and imaged under DIC and/or fluorescence microscopy.
Fluorescent protein intensity calibration to measure RIα concentrations
Puncta RIα concentrations were estimated based on calibration of fluorescent protein intensity on the same imaging system used to generate the data shown in Figures 1A, 1B, 2–5, and S1–5. Briefly, known concentrations of purified sfGFP were loaded in glass capilary tubes and imaged under the same illumination conditions used for live-cell imaging experiments. The resulting intensity images were used to construct a standard curve and calculate a calibration constant (i.e., number of sfGFP molecules per camera count) for the system. Using this value, we then estimated the RIα concentration in each fluorescent puncta in each cell based on the measured area and the mean intensity value, assuming spherical puncta.
Fluorescence recovery after photobleaching
Cells were imaged using a Nikon A1R HD confocal with a four-line (405 nm, 488 nm, 561 nm, and 640 nm) LUN-V laser engine and DU4 detector using bandpass and long-pass filters for each channel (450/50, 525/50, 595/50 and 700/75) mounted on a Nikon Ti2 using an Apo 100× 1.49 NA objective and operated using NIS Elements software. Image stacks were acquired in Galvano mode with unidirectional scanning with a 488 nm laser at 1.5% power with a frame size of 512×512 at scan zoom, 1 frame per second (fps), and 97.1 μm pinhole size. Small regions of interest (ROIs) for stimulation were drawn over the punctate structures and in the cytosol. The total FRAP series contained 3 images before bleaching (obtained at 2 s intervals), 2 cycles of ROI bleaching with the 488 nm laser at 100% laser power (5 frames at 1 fps), and 2 min of continuous acquisition to monitor fluorescence recovery.
Time-lapse epifluorescence imaging
Cells were washed twice with HBSS and subsequently imaged in HBSS in the dark at 37°C. Forskolin (Fsk; Calbiochem), 3-isobutyl-1-methylxanthine (IBMX; Sigma), rolipram (Rol; Alexis), myristoylated PKI 14–22 amide (Myr-PKI; Tocris), isoproterenol (Iso; Sigma), and 1,6-hexanediol (Hex; Sigma-Aldrich) were added as indicated. Epifluorescence imaging was performed either on a Zeiss Axiovert 200M microscope (Carl Zeiss) equipped with a xenon lamp, a 40x/1.3 NA objective and a cooled CCD or on a Zeiss AxioObserver Z1 microscope (Carl Zeiss) equipped with a 40x/1.3 NA objective and a Photometrics Evolve 512 EMCCD (Photometrics), both controlled by METAFLUOR 7.7 software (Molecular Devices). For the Zeiss Axiovert 200M, the following excitation/emission filter combinations (center/bandwidth in nm) were used: BFP - EX380/10, EM475/25; CFP - EX420/20, EM475/25; GFP - EX480/30, EM535/45; YFP - EX495/10, EM535/25; RFP - EX568/55, EM653/95; CFP/YFPFRET -EX420/20, EM535/25; GFP/RFPFRET - EX480/30, EM653/95. For the Zeiss AxioObserver Z1, the following excitation/emission filter combinations were used: GFP - EM480/30, EX535/45. All filter sets were alternated using a Lambda 10–2 filter-changer (Sutter Instruments). Exposure times were 50 (for acceptor direct channel) and 500 ms (for all other channels), with the EM gain set to 20 for the AxioObserver Z1 microscope, and images were acquired every 30 s. All epifluorescence experiments were subsequently analyzed using METAFLUOR 7.7 software. DIC images were acquired on the Zeiss Axiovert 200M microscope. Brightfield images were acquired on an eVos FL cell imaging system (Thermo Fisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
FRET biosensor analysis
Raw fluorescence images were corrected by subtracting the background fluorescence intensity of a cell-free region from the emission intensities of biosensor-expressing cells. Green/red, red/green, or cyan/yellow emission ratios were then calculated at each time point (R). For some curves, the resulting time courses were normalized by dividing the emission ratio at each time point by the basal ratio value at time zero (R/R0), which was defined as the emission ratio at the time point immediately preceding drug addition (R0). Normalized-to-time-zero ratio changes (R/R0) from drug stimulation (ΔR/R0 (drug)) were reported for some of the bar graphs and were calculated as (Rdrug – R0/R0), where Rdrug is the emission ratio at the last time point after the corresponding drug addition. For PDE4D2cat-ICUE4 curves, the resulting time courses were normalized to the maximum ratio change (ΔR/ΔRmax) by calculating (R – R0)/(Rmax – R0), where Rmax is the maximum emission ratio value recorded after all stimulations. Maximum normalized-to-max ratio changes (ΔR/ΔRmax) from Fsk stimulation (Max ΔR/ΔRmax (Fsk)) were reported for the PDE4D2cat-ICUE4 bar graphs and were calculated as (Rmax from Fsk – R0)/(Rmax–R0), where Rmax from Fsk is the maximum emission ratio value recorded after Fsk addition. Graphs were plotted using GraphPad Prism 8 (GraphPad).
Quantification of cellular puncta
For analysis of puncta number, cell images were individually thresholded and underwent particle analysis with circularity and size cut-offs in Image J.
Calculations for the apparent diffusivity of cAMP
The circular FRAP regions were saved and the radius (r) was calculated from the area. Time-to-half maximum values (t1/2) were acquired from Image J data processing tools. Since the FRAP regions were circular, the apparent diffusivity (Dapp) is calculated from the following equation (Kang et al., 2012):
Statistics and reproducibility
Statistical analyses were performed in GraphPad Prism 8 (GraphPad). All data were assessed for normality. For normally distributed data, pairwise comparisons were performed using unpaired two-tailed Student’s t-tests, with Welch’s correction for unequal variances used as indicated. Comparisons between three or more groups were performed using ordinary one-way analysis of variance (ANOVA). For data that were not normally distributed, pairwise comparisons were performed using the Mann-Whitney U test, and comparisons between multiple groups were performed using the Kruskal-Wallis test. Statistical significance was set at P < 0.05. Average time courses shown in Figures 1C; 3B, C, E, G, and H (curves); 4B–D (curves); 5E (curves); S1A; S3A, B, D, and F (curves); and S4B, D, E, and H–K (curves) are representative of at least 3 independently repeated experiments. Average time courses and bar graphs shown in Figures 1D; 2B and F; 3B–H (bar graphs); 4B–D (bar graphs); 5B–D, E (bar graphs), and F–H; S1C–E; S2B–H; S3C, D (bar graph), F, and G; S4B (bar graph), F–J (bar graphs), and L; and S5E–G combined data sets from at least 3 independent experiments, unless otherwise stated.
Throughout the paper, ****P < 0.0001 and ††††P < 0.0001. Kruskal-Wallis test followed by Dunn’s multiple comparisons test was performed for Figures 2B (* vs. wild-type RIα and † vs. Δ(D/D+linker)), 5D (†, vs. the corresponding DnaJB1-PKAcat + RIα column), S2B (vs. wild-type RIα), and S5F (vs. WT). Unpaired two-tailed Student t-tests were performed for Figures 3D, 3F, 5E, S3D, S4B, S4F, and S4L, and Welch’s correction was applied for Figures 3B, 3C, 3E, 3G, 3H, 4B–D, 5B, and S4H–J. Unpaired two-tailed Mann-Whitney U-tests were performed for Figures 5D (*, −Fsk vs. +Fsk), S2D, and S2F. Ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test was performed for Figures 5G, 5H, and S5G (all vs. WT) and followed by Tukey’s multiple comparisons test for Figure S4G. A one-sample t-test versus a hypothetical value of 1 was performed for Figure S5E. In Figure 2B, †P = 0.0258. In Figure 5D, †P = 0.0198, JH33Q-Cat +Fsk vs. J-Cat +Fsk; ††P = 0.0012, Myr-J-Cat −Fsk vs. J-Cat −Fsk; ††P = 0.0021, Myr-J-Cat +Fsk vs. J-Cat +Fsk; ††P = 0.0039, Myr-JH33Q-Cat −Fsk vs. J-Cat − Fsk; ††††P<0.0001 vs. J-Cat +Fsk.
Supplementary Material
Video 1. Mobility and merging of RIα phase-separated bodies, Related to Figure 1. Confocal imaging of endogenous RIα phase-separated bodies in 293-RIα cells expressing GFP1–10. Arrowhead indicates two puncta that merge. Scale bar, 10 μm.
Video 2. Fluorescence recovery after photobleaching of RIα phase-separated bodies, Related to Figure 1. Confocal imaging of endogenous RIα phase-separated bodies in 293-RIα cells expressing GFP1–10 showing photobleaching and recovery of fluorescence. Arrowheads indicate the photobleached puncta. Scale bar, 10 μm.
Video 3. Forskolin induces the formation of RIα phase-separated bodies and co-phase separation of PKAcat, Related to Figure 2. Epifluorescence imaging of HEK293T cells expressing EGFP-RIα (left) and mCherry-PKAcat (right). During imaging, cells were stimulated with 50 μM Fsk to observe RIα puncta formation. Scale bar, 10 μm.
Video 4. DnaJB1-PKAcat inhibits both forskolin-induced RIα phase separation and co-phase separation of DnaJB1-PKAcat, Related to Figure 5. Epifluorescence imaging of HEK293T cells expressing EGFP-RIα (left) and mCherry-DnaJB1-PKAcat (right). During imaging, cells were stimulated with 50 μM Fsk to observe RIα puncta formation. Scale bar, 10 μm.
Figure S1. Additional characterization of RIα phase-separated bodies, Related to Figure 1. (A) FRAP of a region within an RIα puncta (blue curve) compared with an entire RIα puncta (red curve) in HEK293T cells transfected with RIα-EGFP. Curves show average time course of normalized fluorescence intensity. Solid lines indicate the mean; shaded areas, SEM. (B) Representative in vitro phase diagram of RIα liquid droplet formation at varying concentrations of KCl. Each condition was assessed at least twice. (C-E) Average size of (C), percent volume occupied by (D), and RIα concentrations inside RIα droplets (E) in 293-RIα cells transfected with GFP1–10 (endogenous) or in vitro RIα droplets (in vitro) (50 μM RIα + 12.5 μM PKAcat). Analyses were performed before and after 50 μM Fsk stimulation (endogenous) or 10 μM cAMP addition (in vitro) (endogenous: −cAMP: n = 21 puncta from 8 cells, +cAMP: n = 34 puncta from 8 cells; in vitro: −cAMP: 93 droplets, +cAMP: 34 droplets). Violin plots show the median and quartiles as solid and dashed lines, respectively. (F-H) RIα phase separation occurs in various tissues. Representative fluorescence images of EGFP-RIα-expressing (F) neonatal rat ventricular myocytes, (G) astrocytes, and (H) dissociated primary embryonic rat hippocampal neurons grown for 3 days in vitro (outline indicates cell shape; inset, zoomed image) showing RIα puncta formation. Scale bars, 10 μm.
Figure S2. RIα phase separation requires the D/D domain and linker region and is regulated by PKAcat and cAMP, Related to Figure 2. (A) Domain structure of RIα (D/D: docking/dimerization domain, IS: inhibitory sequence, CNB-A: cAMP binding domain A, CNB-B: cAMP binding domain B) and the various truncation mutants used in this study. (B) Comparison of the number of basal RIα puncta per cell in cells expressing PKAcat plus either wild-type RIα or various RIα mutants. Deletion of either the D/D domain or linker region greatly reduced number of RIα puncta per cell. (C-E) Comparison of the number of basal and Fsk-stimulated RIα puncta in HEK293T cells expressing mCherry-tagged PKAcat plus EGFP-tagged RIαΔD/D+Linker (C), RIαΔD/D (D), or RIαΔLinker (E) and stimulated with 50 μM Forskolin (Fsk). (F) Comparison of the number of RIα puncta per cell in 293-RIα cells with or without PKAcat overexpression and stimulated with 50 μM Fsk. Overexpressing PKAcat decreases the basal RIα puncta number. (G and H) Comparison of the number of RIα (G) and PKAcat puncta (H) per cell in HEK293T cells expressing EGFP-RIα and mCherry-PKAcat and treated with 50 μM Fsk, 10 μM isoproterenol (Iso), or 20 μM myrisotylated-PKI (Myr-PKI) 20 min after 50 μM Fsk addition. Fsk and Iso dynamically increase the numbers of both RIα and PKAcat puncta while Myr-PKI treatment following Fsk stimulation has no effect. Horizontal lines in B-F indicate mean ± SEM. Bars in G and H indicate mean ± SEM.
Figure S3. Additional characterization of FluoSTEP-AKAR and FluoSTEP-ICUE, Related to Figure 3. (A and B) Testing the specificity of FluoSTEP-AKAR and FluoSTEP-ICUE. Representative whole-cell average time courses showing (A) the red/green (R/G) emission ratio of either FluoSTEP-AKAR (red curve) or FluoSTEP-AKAR T/A (blue curve) or (B) the green/red (G/R) emission ratio of either FluoSTEP-ICUE (red curve) or FluoSTEP-ICUE R279E (blue curve) in 293-RIα cells stimulated with 50 μM Fsk. (C) Characterizing the dose-response behavior of G/R-ICUE. HEK293T cells were pretreated with 100 μM MDL-12330A followed by the indicated concentrations of 8-Br-2’-O-Me-cAMP-AM. (D) Investigating the effect of puncta localization on the G/R-ICUE response. Left: Representative average time courses of G/R emission ratio changes in HEK293T cells transfected with RIα-G/R-ICUE and stimulated with 100 μM MDL-12330A (MDL) followed by 200 μM 8-Br-2’-O-Me-cAMP-AM (n = 10 puncta and 10 diffuse regions from 10 cells). RIα puncta regions (blue curve) and non-puncta regions (red curve) were analyzed separately. Right: Maximum raw emission ratio changes upon 8-Br-2’-O-Me-cAMP-AM treatment (n = 24 puncta and 24 diffuse regions from 24 cells). (E) When added to 50 μM purified RIα, 10 μM dye-labeled cAMP analogue (8-Φ−450-cAMP) preferentially localized within RIα droplets, with 99% ± 0.33% of cAMP in the droplets. Scale bar, 10 μm. (F) cAMP FRAP experiments using dye-labeled cAMP reveal much slower fluorescence recovery within RIα droplets (Puncta) versus when cAMP is alone in solution (Diffuse; inset). (G) Apparent diffusion coefficients of puncta-localized or diffuse cAMP calculated from FRAP experiments. Solid lines in A, B, D, and F indicate the mean; shaded areas, SEM. Error bars in C, D and G depict mean ± SEM.
Figure S4. RIα phase separation plays a crucial role in maintaining PDE-mediated cAMP compartmentation, Related to Figure 4. (A and B) Characterizing the improved cAMP sensor ICUE4. (A) Domain structures of ICUE3 and ICUE4. (B) Representative average time courses of the cyan/yellow (C/Y) emission ratio in HEK293T cells expressing either ICUE3 (blue curve) or ICUE4 (red curve) and stimulated with 25 μM Fsk. Inset: Comparison of the maximum C/Y emission ratio responses of ICUE3 and ICUE4 after 25 μM Fsk stimulation. (C) PDE4D2cat tethering does not affect the ICUE4 response. HEK293T cells expressing ICUE4 (blue curve) or PDE4D2cat-ICUE4 (red curve) were stimulated with different doses of forskolin (Fsk) in the absence (ICUE4) or presence (PDE4D2-ICUE4) of the PDE4 inhibitor rolipram. Points indicate mean ± SEM. (D and E) Representative average time courses of the C/Y emission ratio in HEK293T cells expressing ICUE4 (D) or ICUE4 plus mRuby2-RIα (E) with (red curve) and without (blue curve) 1,6-hexanediol treatment (added at t = 0 in the red curve) followed by 50 μM Fsk and then 100 μM IBMX. Hexanediol treatment induces a gradual increase in the ICUE4 C/Y emission ratio compared with control (blue curve) but has little effect on the maximum stimulated ICUE4 responses (see insets showing responses normalized to Fsk addition). (F) Comparison of the raw C/Y emission ratio of cytosolic ICUE4 immediately prior to Fsk stimulation with and without hexanediol treatment in HEK293T cells with endogenously expressed RIα (endo RIα) or RIα overexpression (RIα OX). (G) Left: Comparison of the raw initial C/Y emission ratios of PDE4D2cat-ICUE4 in HEK293T cells with endogenously expressed RIα (endo RIα), RIα overexpression (RIα OX), or RIα knockout (RIα KO). Right: Comparison of the raw initial C/Y emission ratios of PDE4D2cat-ICUE4 in RIα knockout HEK293T cells expressing wild-type RIα, RIαD/D+Linker, or RIαΔD/D+Linker. (H-J) Left: Representative average time courses of the C/Y emission ratio (normalized to max) of PDE4D2cat-ICUE4-transfected RIα KO HEK293T cells co-expressing mRuby2-RIα (+RIα) (H), mRuby2-RIαD/D+Linker (+RIαD/D+Linker) (I), or mRuby2-RIαΔD/D+Linker (+RIαΔD/D+Linker) (J) with (red curve) and without (blue curve) hexanediol pretreatment and stimulation with 50 μM Fsk, 1 μM rolipram (Rol), and 100 μM IBMX. Right: Bar graphs showing the average normalized-to-max PDE4D2cat-ICUE4 (C/Y) emission ratio after Fsk stimulation for each condition. (K and L) Additional examination of cAMP levels inside and outside RIα droplets. RIα KO HEK293T cells expressing ICUE4 tethered to either full-length RIα (RIα-ICUE4) (left) or RIαD/D+Linker (RIαD/D+Linker-ICUE4) (right), which phase separates but does not bind to cAMP, were stimulated with 50 μM Fsk and then 100 μM IBMX. RIα puncta (red curve) and diffuse regions (blue curve) were analyzed separately. Representative average time courses of C/Y emission ratio (K) and maximum Fsk-stimulated ratio changes (L) are shown. Consistent with FluoSTEP imaging, these results suggest that cAMP levels are substantially higher in RIα droplets, which depends on the cAMP-binding capabilities of RIα. Solid lines in B, D, E, and H-K indicate the mean; shaded areas, SEM. Error bars in C, G, H-J, and L depict mean ± SEM. Violin plots in B and F show the median and quartiles as solid and dashed lines, respectively
Figure S5. DnaJB1-PKAcat abolishes RIα phase separation, and loss of RIα phase separation leads to tumorigenic phenotypes, Related to Figure 5. (A) Representative in vitro phase diagram of RIα liquid droplet formation as a function of RIα and DnaJB1-PKAcat concentration in the absence (top) or presence (bottom) of 10 μM cAMP, showing that DnaJB1-PKAcat disrupts RIα phase separation. Each condition was assessed at least twice. (B) Purified RIα, PKAcat, PKA type II regulatory subunit (RIIβ), or DnaJB1-PKAcat at the indicated concentrations were incubated in liquid droplet buffer to assess in vitro liquid droplet formation. Only RIα showed droplet formation. Scale bars, 10 μm. (C) DnaJB1-PKAcat disrupts RIα puncta formation in cells. Representative fluorescence images of HEK293T cells transfected with EGFP-RIα and either mTagBFP2-tagged DnaJB1-PKAcat (upper images) or DnaJB1-PKAcatK72H (JB1-CatK72H, lower images) before (t = 0 min, top) or after (t = 20 min; bottom) 50 μM Fsk addition. Scale bars, 10 μm. (D) Representative photographs of AML12 cell (top) or mouse embryonic fibroblast (MEF) colonies (bottom) embedded in soft-agar in 6-well plates for each condition. Mutations that disrupt RIα phase separation also promote anchorage independent cell growth. Scale bar, 10 mm. (E-G) Dysfunctional RIα phase separation promotes tumorigenic phenotypes in MEFs. RIα phase separation was achieved by knocking out RIα or expressing either RIαD/D+Linker, which permits RIα phase separation but lacks cAMP binding, or RIαΔ(D/D+Linker), which retains cAMP binding but lacks phase separation, in RIα null cells. (E) Average fold-change in cell count for MEFs under different conditions after 1 week of growth. Data were analyzed using ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test. (F) Average percentage of BrdU+ MEFs. Data were analyzed using the Kruskal-Wallis test followed by Dunn’s multiple comparisons test (vs. WT). (G) Average number of colonies larger than 500 μm2 grown in soft agar. Bars in E-G indicate mean ± SEM.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| DH5a competent bacteria | NEB | Cat# C2987I |
| RIα-EGFP lentivirus | This paper | N/A |
| RIαD/D+Lmker-EGFP lentivirus | This paper | N/A |
| RIαΔ(D/D+Linker)-EGFP lentivirus | This paper | N/A |
| BL21(DE3) competent bacteria | Agilent | Cat# 200131 |
| Rosetta (DE3) pLysS competent bacteria | Millipore Sigma | Cat# 70956 |
| Biological Samples | ||
| Sprague-Dawley rat pups | Charles River | Strain Code 400 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BbsI | NEB | Cat# R3539 |
| Q5 High Fidelity Polymerase | NEB | Cat# M0491S |
| HiFi DNA Assembly Kit | NEB | Cat# E5520S |
| HindIII | NEB | Cat# R0104S |
| EcoRI | NEB | Cat# R0101S |
| XbaI | NEB | Cat# R0145S |
| Q5 Site Directed Mutagenesis Kit | NEB | Cat# E0554 |
| DMEM:F12 | ThermoFisher | Cat# 12634010 |
| ITS Liquid Media Supplement | Sigma Aldrich | Cat# I3146 |
| Dexamethasone | Sigma Aldrich | Cat# D1159 |
| Polyjet | Signagen | Cat# SL100688 |
| DNase I | ThermoFisher | Cat# EN0525 |
| Neurobasal media | ThermoFisher | Cat# 21103049 |
| SM1 Supplement | STEMCELL Technologies |
Cat# 05711 |
| Poly-D-Lysine | Sigma Aldrich | Cat# P6407 |
| Lipofectamine LTX | Invitrogen | Cat# 15338500 |
| Puromycin | Sigma Aldrich | Cat# P8833 |
| BSA | Roche | Cat# 10738328103 |
| HEPES | Sigma Aldrich | Cat# H3375 |
| EDTA | Sigma Aldrich | Cat# E6758 |
| DAPI | ThermoFisher | Cat# D21490 |
| Noble Agar | ThermoFisher | Cat# AAJ1090722 |
| Powdered DMEM | ThermoFisher | Cat# 12800017 |
| IPTG | Sigma Aldrich | Cat# I6758 |
| MES | Sigma Aldrich | Cat# M3671 |
| EGTA | Sigma Aldrich | Cat# E3889 |
| DTT | Sigma Aldrich | Cat# D0632 |
| Protease inhibitors | Roche | Cat# 11873580001 |
| cGMP | Sigma Aldrich | Cat# G7504 |
| β-mercaptoethanol | Sigma Aldrich | Cat# M6250 |
| Imidazole | Sigma Aldrich | Cat# 1336500 |
| ATP | Sigma Aldrich | Cat# A26209 |
| cAMP | Sigma Aldrich | Cat# A9501 |
| PEG4000 | Sigma Aldrich | Cat# 1546569 |
| Forskolin | CalBioChem | Cat# 344281 |
| IBMX | Sigma Aldrich | Cat# I7018 |
| Rolipram | Alexis | Cat# 61413-54-5 |
| 1,6-hexanediol | Sigma Aldrich | Cat# 240117 |
| Myr-PKI | Tocris | Cat# 2546 |
| Isoproterenol | Sigma Aldrich | Cat# 1351005 |
| Critical Commercial Assays | ||
| BrdU kit | Invitrogen | Cat# B23151 |
| MDL-12330A | Sigma Aldrich | Cat# M182 |
| 8-Br-2’-O-Me-cAMP-AM | Biolog | Cat# B028–01 |
| 8-[Φ−450]-cAMP | Biolog | Cat# P024–001 |
| Deposited Data | ||
| FluoSTEP-ICUE sequence | GenBank | MT800777.1 |
| FluoSTEP-AKAR sequence | GenBank | MT800778.1 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATTC Cat# CRL-11268 |
| HEK293A | ThermoFisher | Cat# R70507 |
| MEF | GS McKnight Lab, University of Washington, Seattle, WA, USA | N/A |
| AML12 | ATCC | ATCC Cat# CRL-2254 |
| 293-RIα | This paper | N/A |
| 293T-RIα KO | This paper | N/A |
| MEF-RIα KO | (Day et al., 2011) | N/A |
| AML12-RIα KO | This paper | N/A |
| AML 12-RIα KO-RIα-EGFP | This paper | N/A |
| AML12-RIα KOD/D+Linker-RIα-EGFP | This paper | N/A |
| AML12-RIα KOΔ(D/D+Linker)-RIα-EGFP | This paper | N/A |
| MEF-RIα KO-RIα-EGFP | This paper | N/A |
| MEF-RIα KO-RIαD/D+Linker-EGFP | This paper | N/A |
| MEF-RIα KO-RIαΔ(D/D+Linker)-EGFP | This paper | N/A |
| Oligonucleotides | ||
| Primers for plasmid construction, see Table S1 | This paper | N/A |
| RIα-FP11 HDR ssDNA, see Table S1 | IDT, for this paper | N/A |
| DnaJB1 exon 1 gBlock, see Table S1 | IDT, for this paper | N/A |
| Recombinant DNA | ||
| px458 | (Ran et al., 2013) | Addgene plasmid #48138 |
| px459 | (Ran et al., 2013) | Addgene plasmid #62988 |
| px458 mouse RIα gRNA | This paper | N/A |
| px459 human RIα gRNA | This paper | N/A |
| pcDNA3 AKAR2-CR | (Lam et al., 2012) | Addgene plasmid #40255 |
| pBAD-mT agBFP2 | (Subach et al., 2011) | Addgene plasmid #34632 |
| pcDNA3.1 EGFP-RIα | (Day et al., 2011) | N/A |
| pcDNA3.1 mCherry-PKAcat | (Day et al., 2011) | N/A |
| pcDNA3.1 mRuby2-RIα | This paper | N/A |
| pcDNA3.1 mTagBFP2-RIα | This paper | N/A |
| pcDNA3.1 RIαD/D+Linker-EGFP | This paper | N/A |
| pcDNA3.1 RIαD/D-EGFP | This paper | N/A |
| pcDNA3.1 RIαLinker-EGFP | This paper | N/A |
| pcDNA3.1 RIαΔ(D/D+Linker)-EGFP | This paper | N/A |
| pcDNA3.1 RIαΔD/D-EGFP | This paper | N/A |
| pcDNA3.1 RIαΔLinker-EGFP | This paper | N/A |
| pcDNA3.1 mTagBFP2-PKAcat | This paper | N/A |
| pcDNA3.1 GFP1–10 | (Leonetti et al., 2016) | Addgene plasmid #70219 |
| pcDNA3 AKAR4 | (Depry et al., 2011) | Addgene plasmid #61619 |
| pcDNA3 ICUE3 | (DiPilato and Zhang, 2009) | Addgene plasmid #61622 |
| pcDNA3.1 FluoSTEP-AKAR | This paper | N/A |
| pcDNA3.1 FluoSTEP-ICUE | This paper | N/A |
| pcDNA3.1 Fluo STEP-AKAR(T/A) | This paper | N/A |
| pcDNA3.1 Fluo STEP-ICUE(R279E) | This paper | N/A |
| pcDNA3.1 ICUE4 | This paper | N/A |
| PDE4D2 cDNA | gift from Hengming Ke, UNC, Chapel Hill, NC | N/A |
| pcDNA3.1 PDE4D2cat-ICUE4 | This paper | N/A |
| pcDNA3.1 RIα-ICUE4 | This paper | N/A |
| pcDNA3.1 RIαD/D+Linker-ICUE4 | This paper | N/A |
| pcDNA3.1 RG-ICUE | This paper | N/A |
| pcDNA3.1 RIα-RG-ICUE | This paper | N/A |
| pcDNA3.1 mCherry-DnaJB 1-PKAcat | This paper | N/A |
| pcDNA3.1 mCherry-DnaJB1-PKAcatK72H | This paper | N/A |
| pcDNA3.1 mTagBFP2-DnaJB1-PKAcat | This paper | N/A |
| pcDNA3.1 mT agBFP2-DnaJB 1-PKAcatK72H | This paper | N/A |
| pcDNA3.1 mCherry-PKAcatG1A | This paper | N/A |
| pcDNA3.1 mCherry-Myr-DnaJB1-PKAcat | This paper | N/A |
| pcDNA3.1 mCherry-DnaJB 1-PKAcat | This paper | N/A |
| pcDNA3.1 mCherry-DnaJB1H33Q-PKAcat | This paper | N/A |
| pcDNA3.1 mCherry-Myr-DnaJB 1 H33Q-PKAcat | This paper | N/A |
| pLenti-puro | (Guan et al., 2011) | Addgene plasmid #39481 |
| pLenti RIα-EGFP | This paper | N/A |
| pLenti RIαD/D+Linker-EGFP | This paper | N/A |
| pLenti RIαΔ(D/D+Linker)-EGFP | This paper | N/A |
| pRSET B sfGFP | This paper | N/A |
| pRSET B | Invitrogen | Cat# V35120 |
| pEvolvR-enCas9-PolI3M-TBD | (Halperin et al., 2018) | Addgene plasmid #113077 |
| pMD2.G | (Dull et al., 1998) | Addgene plasmid #12259 |
| psPAX2 | (Dull et al., 1998) | Addgene plasmid #12260 |
| pET-His6-SUMO-TEV-LIC-PKAcat | (Lu et al., 2019) | N/A |
| pET-His6-SUMO-TEV-LIC-DnaJB 1-PKAcat | (Lu et al., 2019) | N/A |
| pET-His6-EGFP-PKAcat | (Lu et al., 2019) | N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| PRISM | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator |
| ImageJ | NIH | https://imagej.nih.gov |
PKA RIα undergoes liquid-liquid phase separation in response to cAMP dynamics
RIα condensates dynamically sequester cAMP and retain high PKA activity
RIα phase separation is necessary for effective cAMP compartmentation
A PKA oncoprotein disrupts RIα bodies, leading to aberrant signaling and growth
ACKNOWLEDGMENTS
We thank A. Hong, K.L. Guan, and S. Banerjee for their guidance with CRISPR; E. Griffis and D. Bindels for their confocal microscopy expertise; D.L. Schmitt for help with neuron and glial experiments; C. Brand for help with cardiomyocyte experiments; Y.L. Ma and Q. Ni for insightful discussions; and A. Nguyen and M. Tong for help with cloning. This work was supported by a National Science Foundation predoctoral fellowship DGE-1650112 (to J.Z.Z), Taiwan MOE Scholarship (to T.W.L), AFOSR FA9550-18-1-0051 (to P.R.), NIH R35 GM130389 (to S.S.T), and NIH R35 CA197622 and R01 DK073368 (to J.Z).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Authors declare no competing interests.
REFERENCES
- Adikusuma F, Pfitzner C, and Thomas PQ (2017). Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS One 12, e0187236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Clancy CE, and Harvey RD (2016). Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep 6, 19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, and Tsien RY (1993). Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 260, 222–226. [DOI] [PubMed] [Google Scholar]
- Baillie GS (2009). Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J 276, 1790–1799. [DOI] [PubMed] [Google Scholar]
- Bastidas AC, Deal MS, Steichen JM, Keshwani MM, Guo Y, and Taylor SS (2012). Role of N-terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J. Mol. Biol 422, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A, Annibale P, Konrad C, Hannawacker A, Anton S, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M, and Lohse MJ (2020). Optical mapping of cAMP signaling at the nanometer scale. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boras BW, Kornev A, Taylor SS, and McCulloch AD (2014). Using Markov state models to develop a mechanistic understanding of protein kinase A regulatory subunit RIalpha activation in response to cAMP binding. J. Biol. Chem 289, 30040–30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, and Winn RA (2014). The soft agar colony formation assay. J. Vis. Exp 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, et al. (2018). Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 72, 19–36.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boularan C, and Gales C (2015). Cardiac cAMP: production, hydrolysis, modulation and detection. Front. Pharmacol 6, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, and Hyman AA (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- Brunton LL, Hayes JS, and Mayer SE (1981). Functional compartmentation of cyclic AMP and protein kinase in heart. Adv. Cyclic Nucleotide Res 14, 391–397. [PubMed] [Google Scholar]
- Bruystens JGH, Wu J, Fortezzo A, Kornev AP, Blumenthal DK, and Taylor SS (2014). PKA RIalpha homodimer structure reveals an intermolecular interface with implications for cooperative cAMP binding and Carney complex disease. Structure 22, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton IL, and Brunton LL (1983). Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem 258, 10233–10239. [PubMed] [Google Scholar]
- Cadd G, and McKnight GS (1989). Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron 3, 71–79. [DOI] [PubMed] [Google Scholar]
- Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat-Shliom N, Sukenik S, Liu Z, and Lippincott-Schwartz J (2019). Phase separation of YAP reorganizes genome topology for long-term, YAP target gene expression. Nat. Cell Biol 21, 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Lu TW, Martinez Fiesco JA, Tomasini M, Fan L, Simon SM, Taylor SS, and Zhang P (2019). Structures of the PKA RIα Holoenzyme with the FLHCC Driver J-PKAcα or Wild-Type PKAcα. Structure 27, 816–828.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretta A, and Mucignat-Caretta C (2011). Protein kinase a in cancer. Cancers (Basel) 3, 913–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Nakamura T, and Koutalos Y (1999). Cyclic AMP diffusion coefficient in frog olfactory cilia. Biophys. J 76, 2861–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J, Ginter C, Cassidy M, Franklin MC, Rudolph MJ, Robine N, Darnell RB, and Hendrickson WA (2015). Structural insights into mis-regulation of protein kinase a in human tumors. Proc. Natl. Acad. Sci. U. S. A 112, 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M, and Beavo J (2007). Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem 76, 481–511. [DOI] [PubMed] [Google Scholar]
- Conti M, Mika D, and Richter W (2014). Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol 143, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DMF (2003). Regulation and organization of adenylyl cyclases and cAMP. Biochem. J 375, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day ME, Gaietta GM, Sastri M, Koller A, Mackey MR, Scott JD, Perkins GA, Ellisman MH, and Taylor SS (2011). Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol 193, 347–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denbigh KG (1951). The thermodynamics of the steady state (Wiley; ). [Google Scholar]
- Depry C, Allen MD, and Zhang J (2011). Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst 7, 52–58. [DOI] [PubMed] [Google Scholar]
- Dinh TA, Vitucci ECM, Wauthier E, Graham RP, Pitman WA, Oikawa T, Chen M, Silva GO, Greene KG, Torbenson MS, et al. (2017). Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep 7, 44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPilato LM, and Zhang J (2009). The role of membrane microdomains in shaping beta2-adrenergic receptor-mediated cAMP dynamics. Mol. Biosyst 5, 832–837. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, and Zhang J (2004). Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. U. S. A 101, 16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goraya TA, Masada N, Ciruela A, Willoughby D, Clynes MA, and Cooper DMF (2008). Kinetic properties of Ca2+/calmodulin-dependent phosphodiesterase isoforms dictate intracellular cAMP dynamics in response to elevation of cytosolic Ca2+. Cell. Signal 20, 359–374. [DOI] [PubMed] [Google Scholar]
- Gorshkov K, Mehta S, Ramamurthy S, Ronnett GV, Zhou F-Q, and Zhang J (2017). AKAP-mediated feedback control of cAMP gradients in developing hippocampal neurons. Nat. Chem. Biol 13, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, and Fleischmann RD (1986). The role of cAMP in regulating tumour cell growth. Cancer Surv 5, 291–308. [PubMed] [Google Scholar]
- Graham RP, Lackner C, Terracciano L, Gonzalez-Cantu Y, Maleszewski JJ, Greipp PT, Simon SM, and Torbenson MS (2018). Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 68, 1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald EC, Mehta S, and Zhang J (2018). Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev 118, 11707–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot SR, and Mazur P (2013). Non-equilibrium thermodynamics (Courier Corporation; ). [Google Scholar]
- Guan B, Wang T-L, and Shih I-M (2011). ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res 71, 6718–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero F, Ciragan A, and Iwai H (2015). Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif 116, 42–49. [DOI] [PubMed] [Google Scholar]
- Halperin SO, Tou CJ, Wong EB, Modavi C, Schaffer DV, and Dueber JE (2018). CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252. [DOI] [PubMed] [Google Scholar]
- Honeyman JN, Simon EP, Robine N, Chiaroni-Clarke R, Darcy DG, Lim IIP, Gleason CE, Murphy JM, Rosenberg BR, Teegan L, et al. (2014). Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD (2010). Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci 35, 91–100. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Julicher F (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, and Parker R (2016). ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, and Montminy M (2003). cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 17, 1575–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Day CA, Kenworthy AK, and DiBenedetto E (2012). Simplified equation to extract diffusion coefficients from confocal FRAP data. Traffic 13, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, Lalazar G, Houlihan SL, Tschaharganeh DF, Baslan T, Chen CC, Requena D, Tian S, Bosbach B, Wilkinson JE, et al. (2017). DNAJB1–PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A 114, 13076–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Xuong N-H, and Taylor SS (2005). Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science 307, 690–696. [DOI] [PubMed] [Google Scholar]
- Kroeze WK, Sheffler DJ, and Roth BL (2003). G-protein-coupled receptors at a glance. J. Cell Sci 116, 4867 LP–4869. [DOI] [PubMed] [Google Scholar]
- Kuiken GD (1994). Thermodynamics of irreversible processes: applications to diffusion and rheology (Wiley; ). [Google Scholar]
- Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, McKeown MR, Wiedenmann J, Davidson MW, Schnitzer MJ, et al. (2012). Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, and Huang B (2016). A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. U. S. A 113, E3501–E3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, and Heidenreich KA (2000). Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol. Cell. Biol 20, 9356–9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CJ, Kain KH, Tkachenko E, Goldfinger LE, Gutierrez E, Allen MD, Groisman A, Zhang J, and Ginsberg MH (2008). Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol. Biol. Cell 19, 4930–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse C, Bock A, Maiellaro I, Hannawacker A, Schad LR, Lohse MJ, and Bauer WR (2017). Experimental and mathematical analysis of cAMP nanodomains. PLoS One 12, e0174856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T-W, Wu J, Aoto PC, Weng J-H, Ahuja LG, Sun N, Cheng CY, Zhang P, and Taylor SS (2019). Two PKA RIα holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc. Natl. Acad. Sci 116, 16347 LP–16356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiellaro I, Lohse MJ, Kittel RJ, and Calebiro D (2016). cAMP Signals in Drosophila Motor Neurons Are Confined to Single Synaptic Boutons. Cell Rep 17, 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M, Lippincott-Schwartz J, Stratakis CA, and Bossis I (2006). Depletion of type IA regulatory subunit (RIα) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum. Mol. Genet 15, 2962–2971. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, Castillo R, Glembotski CC, Sussman MA, Newton AC, et al. (2010). PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res 107, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, and Taylor JP (2015). Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, and Lohse MJ (2004). Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem 279, 37215–37218. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bünemann M, Schmitteckert E, Lohse MJ, and Engelhardt S (2006). Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ. Res 99, 1084–1091. [DOI] [PubMed] [Google Scholar]
- Park AJ, Havekes R, Choi JH, Luczak V, Nie T, Huang T, and Abel T (2014). A presynaptic role for PKA in synaptic tagging and memory. Neurobiol. Learn. Mem 114, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra C, and Brady MF (2018). Biochemistry, cAMP (StatPearls Publishing; ). [PubMed] [Google Scholar]
- Perera RK, and Nikolaev VO (2013). Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. (Oxf) 207, 650–662. [DOI] [PubMed] [Google Scholar]
- Peskett TR, Rau F, O’Driscoll J, Patani R, Lowe AR, and Saibil HR (2018). A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell 70, 588–601.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, and Karpen JW (2000). Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol 116, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, and Karpen JW (2001). A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. U. S. A 98, 13049–13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards M, Lomas O, Jalink K, Ford KL, Vaughan-Jones RD, Lefkimmiatis K, and Swietach P (2016). Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc. Res 110, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggle KM, Riehle KJ, Kenerson HL, Turnham R, Homma MK, Kazami M, Samelson B, Bauer R, McKnight GS, Scott JD, et al. (2016). Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res 80, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Greenwald EC, and Polanowska-Grabowska R (2014). Mechanisms of cyclic AMP compartmentation revealed by computational models. J. Gen. Physiol 143, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten A, Van Veen TAB, Vos MA, and Heck AJR (2007). Diversity of cAMP-dependent protein kinase isoforms and their anchoring proteins in mouse ventricular tissue. J. Proteome Res 6, 1705–1717. [DOI] [PubMed] [Google Scholar]
- Sergi CM (2015). Hepatocellular Carcinoma, Fibrolamellar Variant: Diagnostic Pathologic Criteria and Molecular Pathology Update. A Primer. Diagnostics (Basel, Switzerland) 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, and Poo M (2010). Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327, 547–552. [DOI] [PubMed] [Google Scholar]
- Singhi AD, Wood LD, Parks E, Torbenson MS, Felsenstein M, Hruban RH, Nikiforova MN, Wald AI, Kaya C, Nikiforov YE, et al. (2019). Recurrent Rearrangements in PRKACA and PRKACB in Intraductal Oncocytic Papillary Neoplasms of the Pancreas and Bile Duct. Gastroenterology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangherlin A, and Zaccolo M (2012). Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol. - Hear. Circ. Physiol 302. [DOI] [PubMed] [Google Scholar]
- Steinberg SF, and Brunton LL (2001). Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol 41, 751–773. [DOI] [PubMed] [Google Scholar]
- Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, and Vale RD (2016). Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, and Verkhusha VV (2011). An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 6, e28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC, Mongillo M, Sawa A, Jordanides NE, Mountford JC, et al. (2012). PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J. Cell Biol 198, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo SE, Xiong W-H, Takahashi M, Miao S, Andrade AL, Fortin DA, Yang G, Qin M, Smoody BF, Stork PJS, et al. (2017). Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep 19, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnham RE, Smith FD, Kenerson HL, Omar MH, Golkowski M, Garcia I, Bauer R, Lau HT, Sullivan KM, Langeberg LK, et al. (2019). An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. Elife 8, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udenwobele DI, Su R-C, Good SV, Ball TB, Varma Shrivastav S, and Shrivastav A (2017). Myristoylation: An Important Protein Modification in the Immune Response. Front. Immunol 8, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, Laskowski PR, MacKenzie D, Kamath T, Commins C, et al. (2018). Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J 37, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, and Cooper DMF (2007). Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev 87, 965–1010. [DOI] [PubMed] [Google Scholar]
- Yang PC, Boras BW, Jeng MT, Docken SS, Lewis TJ, McCulloch AD, Harvey RD, and Clancy CE (2016). A Computational Modeling and Simulation Approach to Investigate Mechanisms of Subcellular cAMP Compartmentation. PLoS Comput. Biol 12, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Jones GN, Towns WH 2nd, Zhang X, Abel ED, Binkley PF, Jarjoura D, and Kirschner LS (2008). Heart-specific ablation of Prkar1a causes failure of heart development and myxomagenesis. Circulation 117, 1414–1422. [DOI] [PubMed] [Google Scholar]
- Zaccolo M (2006). Phosphodiesterases and compartmentalized cAMP signalling in the heart. Eur. J. Cell Biol 85, 693–697. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, and Pozzan T (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, and Tsien RY (2005). Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature 437, 569–573. [DOI] [PubMed] [Google Scholar]
- Zhou X, Herbst-Robinson KJ, and Zhang J (2012). Visualizing dynamic activities of signaling enzymes using genetically encodable FRET-based biosensors from designs to applications. Methods Enzymol 504, 317–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Mobility and merging of RIα phase-separated bodies, Related to Figure 1. Confocal imaging of endogenous RIα phase-separated bodies in 293-RIα cells expressing GFP1–10. Arrowhead indicates two puncta that merge. Scale bar, 10 μm.
Video 2. Fluorescence recovery after photobleaching of RIα phase-separated bodies, Related to Figure 1. Confocal imaging of endogenous RIα phase-separated bodies in 293-RIα cells expressing GFP1–10 showing photobleaching and recovery of fluorescence. Arrowheads indicate the photobleached puncta. Scale bar, 10 μm.
Video 3. Forskolin induces the formation of RIα phase-separated bodies and co-phase separation of PKAcat, Related to Figure 2. Epifluorescence imaging of HEK293T cells expressing EGFP-RIα (left) and mCherry-PKAcat (right). During imaging, cells were stimulated with 50 μM Fsk to observe RIα puncta formation. Scale bar, 10 μm.
Video 4. DnaJB1-PKAcat inhibits both forskolin-induced RIα phase separation and co-phase separation of DnaJB1-PKAcat, Related to Figure 5. Epifluorescence imaging of HEK293T cells expressing EGFP-RIα (left) and mCherry-DnaJB1-PKAcat (right). During imaging, cells were stimulated with 50 μM Fsk to observe RIα puncta formation. Scale bar, 10 μm.
Figure S1. Additional characterization of RIα phase-separated bodies, Related to Figure 1. (A) FRAP of a region within an RIα puncta (blue curve) compared with an entire RIα puncta (red curve) in HEK293T cells transfected with RIα-EGFP. Curves show average time course of normalized fluorescence intensity. Solid lines indicate the mean; shaded areas, SEM. (B) Representative in vitro phase diagram of RIα liquid droplet formation at varying concentrations of KCl. Each condition was assessed at least twice. (C-E) Average size of (C), percent volume occupied by (D), and RIα concentrations inside RIα droplets (E) in 293-RIα cells transfected with GFP1–10 (endogenous) or in vitro RIα droplets (in vitro) (50 μM RIα + 12.5 μM PKAcat). Analyses were performed before and after 50 μM Fsk stimulation (endogenous) or 10 μM cAMP addition (in vitro) (endogenous: −cAMP: n = 21 puncta from 8 cells, +cAMP: n = 34 puncta from 8 cells; in vitro: −cAMP: 93 droplets, +cAMP: 34 droplets). Violin plots show the median and quartiles as solid and dashed lines, respectively. (F-H) RIα phase separation occurs in various tissues. Representative fluorescence images of EGFP-RIα-expressing (F) neonatal rat ventricular myocytes, (G) astrocytes, and (H) dissociated primary embryonic rat hippocampal neurons grown for 3 days in vitro (outline indicates cell shape; inset, zoomed image) showing RIα puncta formation. Scale bars, 10 μm.
Figure S2. RIα phase separation requires the D/D domain and linker region and is regulated by PKAcat and cAMP, Related to Figure 2. (A) Domain structure of RIα (D/D: docking/dimerization domain, IS: inhibitory sequence, CNB-A: cAMP binding domain A, CNB-B: cAMP binding domain B) and the various truncation mutants used in this study. (B) Comparison of the number of basal RIα puncta per cell in cells expressing PKAcat plus either wild-type RIα or various RIα mutants. Deletion of either the D/D domain or linker region greatly reduced number of RIα puncta per cell. (C-E) Comparison of the number of basal and Fsk-stimulated RIα puncta in HEK293T cells expressing mCherry-tagged PKAcat plus EGFP-tagged RIαΔD/D+Linker (C), RIαΔD/D (D), or RIαΔLinker (E) and stimulated with 50 μM Forskolin (Fsk). (F) Comparison of the number of RIα puncta per cell in 293-RIα cells with or without PKAcat overexpression and stimulated with 50 μM Fsk. Overexpressing PKAcat decreases the basal RIα puncta number. (G and H) Comparison of the number of RIα (G) and PKAcat puncta (H) per cell in HEK293T cells expressing EGFP-RIα and mCherry-PKAcat and treated with 50 μM Fsk, 10 μM isoproterenol (Iso), or 20 μM myrisotylated-PKI (Myr-PKI) 20 min after 50 μM Fsk addition. Fsk and Iso dynamically increase the numbers of both RIα and PKAcat puncta while Myr-PKI treatment following Fsk stimulation has no effect. Horizontal lines in B-F indicate mean ± SEM. Bars in G and H indicate mean ± SEM.
Figure S3. Additional characterization of FluoSTEP-AKAR and FluoSTEP-ICUE, Related to Figure 3. (A and B) Testing the specificity of FluoSTEP-AKAR and FluoSTEP-ICUE. Representative whole-cell average time courses showing (A) the red/green (R/G) emission ratio of either FluoSTEP-AKAR (red curve) or FluoSTEP-AKAR T/A (blue curve) or (B) the green/red (G/R) emission ratio of either FluoSTEP-ICUE (red curve) or FluoSTEP-ICUE R279E (blue curve) in 293-RIα cells stimulated with 50 μM Fsk. (C) Characterizing the dose-response behavior of G/R-ICUE. HEK293T cells were pretreated with 100 μM MDL-12330A followed by the indicated concentrations of 8-Br-2’-O-Me-cAMP-AM. (D) Investigating the effect of puncta localization on the G/R-ICUE response. Left: Representative average time courses of G/R emission ratio changes in HEK293T cells transfected with RIα-G/R-ICUE and stimulated with 100 μM MDL-12330A (MDL) followed by 200 μM 8-Br-2’-O-Me-cAMP-AM (n = 10 puncta and 10 diffuse regions from 10 cells). RIα puncta regions (blue curve) and non-puncta regions (red curve) were analyzed separately. Right: Maximum raw emission ratio changes upon 8-Br-2’-O-Me-cAMP-AM treatment (n = 24 puncta and 24 diffuse regions from 24 cells). (E) When added to 50 μM purified RIα, 10 μM dye-labeled cAMP analogue (8-Φ−450-cAMP) preferentially localized within RIα droplets, with 99% ± 0.33% of cAMP in the droplets. Scale bar, 10 μm. (F) cAMP FRAP experiments using dye-labeled cAMP reveal much slower fluorescence recovery within RIα droplets (Puncta) versus when cAMP is alone in solution (Diffuse; inset). (G) Apparent diffusion coefficients of puncta-localized or diffuse cAMP calculated from FRAP experiments. Solid lines in A, B, D, and F indicate the mean; shaded areas, SEM. Error bars in C, D and G depict mean ± SEM.
Figure S4. RIα phase separation plays a crucial role in maintaining PDE-mediated cAMP compartmentation, Related to Figure 4. (A and B) Characterizing the improved cAMP sensor ICUE4. (A) Domain structures of ICUE3 and ICUE4. (B) Representative average time courses of the cyan/yellow (C/Y) emission ratio in HEK293T cells expressing either ICUE3 (blue curve) or ICUE4 (red curve) and stimulated with 25 μM Fsk. Inset: Comparison of the maximum C/Y emission ratio responses of ICUE3 and ICUE4 after 25 μM Fsk stimulation. (C) PDE4D2cat tethering does not affect the ICUE4 response. HEK293T cells expressing ICUE4 (blue curve) or PDE4D2cat-ICUE4 (red curve) were stimulated with different doses of forskolin (Fsk) in the absence (ICUE4) or presence (PDE4D2-ICUE4) of the PDE4 inhibitor rolipram. Points indicate mean ± SEM. (D and E) Representative average time courses of the C/Y emission ratio in HEK293T cells expressing ICUE4 (D) or ICUE4 plus mRuby2-RIα (E) with (red curve) and without (blue curve) 1,6-hexanediol treatment (added at t = 0 in the red curve) followed by 50 μM Fsk and then 100 μM IBMX. Hexanediol treatment induces a gradual increase in the ICUE4 C/Y emission ratio compared with control (blue curve) but has little effect on the maximum stimulated ICUE4 responses (see insets showing responses normalized to Fsk addition). (F) Comparison of the raw C/Y emission ratio of cytosolic ICUE4 immediately prior to Fsk stimulation with and without hexanediol treatment in HEK293T cells with endogenously expressed RIα (endo RIα) or RIα overexpression (RIα OX). (G) Left: Comparison of the raw initial C/Y emission ratios of PDE4D2cat-ICUE4 in HEK293T cells with endogenously expressed RIα (endo RIα), RIα overexpression (RIα OX), or RIα knockout (RIα KO). Right: Comparison of the raw initial C/Y emission ratios of PDE4D2cat-ICUE4 in RIα knockout HEK293T cells expressing wild-type RIα, RIαD/D+Linker, or RIαΔD/D+Linker. (H-J) Left: Representative average time courses of the C/Y emission ratio (normalized to max) of PDE4D2cat-ICUE4-transfected RIα KO HEK293T cells co-expressing mRuby2-RIα (+RIα) (H), mRuby2-RIαD/D+Linker (+RIαD/D+Linker) (I), or mRuby2-RIαΔD/D+Linker (+RIαΔD/D+Linker) (J) with (red curve) and without (blue curve) hexanediol pretreatment and stimulation with 50 μM Fsk, 1 μM rolipram (Rol), and 100 μM IBMX. Right: Bar graphs showing the average normalized-to-max PDE4D2cat-ICUE4 (C/Y) emission ratio after Fsk stimulation for each condition. (K and L) Additional examination of cAMP levels inside and outside RIα droplets. RIα KO HEK293T cells expressing ICUE4 tethered to either full-length RIα (RIα-ICUE4) (left) or RIαD/D+Linker (RIαD/D+Linker-ICUE4) (right), which phase separates but does not bind to cAMP, were stimulated with 50 μM Fsk and then 100 μM IBMX. RIα puncta (red curve) and diffuse regions (blue curve) were analyzed separately. Representative average time courses of C/Y emission ratio (K) and maximum Fsk-stimulated ratio changes (L) are shown. Consistent with FluoSTEP imaging, these results suggest that cAMP levels are substantially higher in RIα droplets, which depends on the cAMP-binding capabilities of RIα. Solid lines in B, D, E, and H-K indicate the mean; shaded areas, SEM. Error bars in C, G, H-J, and L depict mean ± SEM. Violin plots in B and F show the median and quartiles as solid and dashed lines, respectively
Figure S5. DnaJB1-PKAcat abolishes RIα phase separation, and loss of RIα phase separation leads to tumorigenic phenotypes, Related to Figure 5. (A) Representative in vitro phase diagram of RIα liquid droplet formation as a function of RIα and DnaJB1-PKAcat concentration in the absence (top) or presence (bottom) of 10 μM cAMP, showing that DnaJB1-PKAcat disrupts RIα phase separation. Each condition was assessed at least twice. (B) Purified RIα, PKAcat, PKA type II regulatory subunit (RIIβ), or DnaJB1-PKAcat at the indicated concentrations were incubated in liquid droplet buffer to assess in vitro liquid droplet formation. Only RIα showed droplet formation. Scale bars, 10 μm. (C) DnaJB1-PKAcat disrupts RIα puncta formation in cells. Representative fluorescence images of HEK293T cells transfected with EGFP-RIα and either mTagBFP2-tagged DnaJB1-PKAcat (upper images) or DnaJB1-PKAcatK72H (JB1-CatK72H, lower images) before (t = 0 min, top) or after (t = 20 min; bottom) 50 μM Fsk addition. Scale bars, 10 μm. (D) Representative photographs of AML12 cell (top) or mouse embryonic fibroblast (MEF) colonies (bottom) embedded in soft-agar in 6-well plates for each condition. Mutations that disrupt RIα phase separation also promote anchorage independent cell growth. Scale bar, 10 mm. (E-G) Dysfunctional RIα phase separation promotes tumorigenic phenotypes in MEFs. RIα phase separation was achieved by knocking out RIα or expressing either RIαD/D+Linker, which permits RIα phase separation but lacks cAMP binding, or RIαΔ(D/D+Linker), which retains cAMP binding but lacks phase separation, in RIα null cells. (E) Average fold-change in cell count for MEFs under different conditions after 1 week of growth. Data were analyzed using ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test. (F) Average percentage of BrdU+ MEFs. Data were analyzed using the Kruskal-Wallis test followed by Dunn’s multiple comparisons test (vs. WT). (G) Average number of colonies larger than 500 μm2 grown in soft agar. Bars in E-G indicate mean ± SEM.
Data Availability Statement
The sequences for FluoSTEP-ICUE and FluoSTEP-AKAR have been uploaded onto GenBank (FluoSTEP-ICUE: (MT800777.1); FluoSTEP-AKAR: (MT800778.1).