

Significance
Heterostyly is an adaptation to promote outbreeding in plants. In heterostylous primroses, plants form flowers either with long styles and low anthers or with short styles and high anthers. This difference is due to a chromosomal segment containing five predicted genes, yet their roles and the evolution of this segment remain unclear. Here we identify the gene responsible for raising the anthers in short-styled flowers. This gene arose by duplication from a classical floral-organ identity gene and gained a novel function. Surprisingly, the responsible chromosomal segment appears to have evolved by stepwise gene duplications rather than duplication of an entire chromosomal block. These findings thus provide detailed insight into the evolution of complex polymorphisms involving different individual traits.
Keywords: heterostyly, Primula, supergene, gene duplication, neofunctionalization
Abstract
Heterostyly represents a fascinating adaptation to promote outbreeding in plants that evolved multiple times independently. While l-morph individuals form flowers with long styles, short anthers, and small pollen grains, S-morph individuals have flowers with short styles, long anthers, and large pollen grains. The difference between the morphs is controlled by an S-locus “supergene” consisting of several distinct genes that determine different traits of the syndrome and are held together, because recombination between them is suppressed. In Primula, the S locus is a roughly 300-kb hemizygous region containing five predicted genes. However, with one exception, their roles remain unclear, as does the evolutionary buildup of the S locus. Here we demonstrate that the MADS-box GLOBOSA2 (GLO2) gene at the S locus determines anther position. In Primula forbesii S-morph plants, GLO2 promotes growth by cell expansion in the fused tube of petals and stamen filaments beneath the anther insertion point; by contrast, neither pollen size nor male incompatibility is affected by GLO2 activity. The paralogue GLO1, from which GLO2 arose by duplication, has maintained the ancestral B-class function in specifying petal and stamen identity, indicating that GLO2 underwent neofunctionalization, likely at the level of the encoded protein. Genetic mapping and phylogenetic analysis indicate that the duplications giving rise to the style-length-determining gene CYP734A50 and to GLO2 occurred sequentially, with the CYP734A50 duplication likely the first. Together these results provide the most detailed insight into the assembly of a plant supergene yet and have important implications for the evolution of heterostyly.
Flowering plants have evolved many different adaptations to promote efficient cross-pollination (1, 2). Some of the most fascinating of these are found in the group of stylar polymorphisms that often combine reciprocal herkogamy with self-incompatibility (1). Individuals of species with stylar polymorphism fall into two or three classes, or morphs, that differ in their arrangement of male and female reproductive organs. In particular, male and female organs are physically separated within a flower, and this herkogamy is reciprocal between the morphs in most species, with anthers in one morph located in the analogous position to the style in the other, and vice versa. As a result, pollen from the different morphs is deposited in a spatially segregated manner on the bodies of pollinators (3), resulting in preferential cross-pollination between the morphs and preventing pollen wastage and the formation of less fit inbred progeny. Often, reciprocal herkogamy is complemented by self-incompatibility to prevent self-fertilization; with only two or three compatibility types corresponding to the different morphs this manifests as an intramorph incompatibility (4, 5).
An intensively studied stylar polymorphism is heterostyly (6–8). In the simplest case, distyly, individuals either form flowers with short styles and high anthers (S-morph) or flowers with long styles and low anthers (l-morph). Additional polymorphisms can be associated with this reciprocal herkogamy; for example, in many species S-morph flowers produce larger, but fewer, pollen grains compared to l-morph flowers (9). In addition, self-incompatibility is present in many distylous species (9). In all examined cases, distyly is under simple Mendelian genetic control, with a dominant and a recessive haplotype at the single S locus determining the morphs (10). With a few exceptions, the dominant S haplotype causes S-morph and the recessive s haplotype l-morph flowers. Rather than a single gene, the S locus represents a supergene, that is, a chromosomal region with at least two tightly linked genes that determine the different aspects of a coadapted set of phenotypes (10–12). Similar supergenes underlie many fascinating coadapted phenotypes in plants and animals (13–16), and their evolutionary buildup is often only incompletely understood.
The primrose genus Primula is the most studied model for distyly (17). Primula flowers show reciprocal herkogamy, pollen polymorphism, and self-incompatibility, with the dominant S and recessive s haplotypes producing S- and l-morph flowers, respectively (3, 18, 19). Based on classical genetic studies, the S locus supergene has been proposed to contain at least three separable individual loci: G, controlling style length; A, determining anther position; and P, controlling pollen size and number (10). Style length and female-compatibility type have never been genetically separated (20, 21); by contrast, male compatibility is likely controlled by a locus distinct from A and P (21, 22). At the molecular level, the dominant S-locus haplotype represents a roughly 280-kb-long genomic segment that is hemizygous; specifically, it is present only on the chromosome with the S haplotype but missing from the chromosome with the recessive s haplotype (23–25). Five genes have been predicted to reside in this 280-kb segment based on its sequence in Primula vulgaris and the closely related Primula veris (25). This architecture of the S locus with a large hemizygous region appears to be conserved in five species belonging to different clades of the genus Primula (25), yet whether all five predicted genes are present at the S locus throughout the different Primula species is unknown. The five genes predicted in P. vulgaris and P. veris are CYP734A50, encoding a cytochrome P450; GLOBOSA2 (also known as GLOT), coding for a B-class homoeotic MADS-domain transcription factor; a KELCH F-BOX PROTEIN (KFBT) gene; a gene encoding a PUMILIO RNA-binding protein (PUMT); and a gene encoding a CONSERVED C-TERMINAL DOMAIN (CCMT) protein. [Naming of the genes follows priority regarding their first publications (23, 26, 27).] Of these, CYP734A50 has been shown to correspond to the G locus (26). The encoded cytochrome P450 monooxygenase inactivates brassinosteroids, a class of phytohormones promoting cell elongation, and thus suppresses style elongation in S-morph flowers.
Despite this important progress, a number of key questions remain unanswered. One concerns the identity of the other two genetically defined loci, A and P, which determine anther position and pollen size, respectively. The GLO2 gene represents a plausible candidate for the A locus; in fact, a transposon insertion in GLO2 of P. vulgaris has been reported to result in a short-homostyle phenotype (23). However, no description of the mutant phenotype has been provided, nor has this finding been confirmed by an independent allele. Thus, the identity of the GLO2 gene with the A locus remains to be firmly established. As for P, no information is currently available about its molecular nature.
The second major unresolved question concerns the evolutionary buildup of the S locus, both regarding its chromosomal assembly and the sequence of trait changes. Clear paralogues of CYP734A50, GLO2, and CCMT are found outside of the S locus, indicating that genes at the S locus arose by duplication (23, 24, 26). Based on available genome-sequence information, the paralogues of CYP734A50 and GLO2 (termed CYP734A51 and GLO1) are not directly adjacent to each other (24), yet their precise locations are unknown—whether close by on the same chromosome or further apart, possibly on different chromosomes. Thus, two scenarios are conceivable for how duplication(s) could have resulted in the hemizygous S locus (28). It could have arisen by a segmental duplication encompassing the genes now present at the S locus, and other genes, followed by loss of those other genes. Under a segmental-duplication scenario, the paralogues outside of the S locus should be physically and genetically close to each other, and segmental duplication would have automatically provided linkage between the S-locus genes. Alternatively, the S locus could have been assembled by stepwise duplications, with unlinked genes duplicated next to each other to build up the S locus. If all paralogues are unlinked, this would argue strongly against a segmental duplication and lend support to a stepwise scenario. In both cases, the duplicated copies would acquire novel functions in controlling heterostyly. Concerning the sequence of trait changes, both existing models for the evolution of heterostyly argue that the first morphological trait to evolve was the polymorphism in style length, followed by the change in anther height (29, 30); however, the models differ in whether the morphological or the physiological component of heterostyly evolved first.
A third unsolved question concerns the way in which the duplicated paralogues at the S locus have acquired their functions in determining heterostyly. It seems plausible that the S-locus copies have undergone neofunctionalization to gain novel activities in modifying floral-organ growth. This could have occurred either by a change in gene-expression pattern or by altering the activity of the encoded proteins, or a combination thereof.
In the present work, we address these three questions, focusing on GLO2. We demonstrate that GLO2 determines the position of the anthers by modulating cell expansion yet it does not appear to influence either pollen size or male incompatibility behavior. Genetic mapping indicates that the CYP734A51 and GLO1 loci are unlinked, arguing against the segmental-duplication model for the origin of the S locus. A stepwise buildup of the S locus is supported by molecular dating of the duplications, suggesting that the CYP734A50/51 duplication occurred earlier than the GLO1/2 duplication. Finally, silencing of GLO1 demonstrates that GLO2 underwent neofunctionalization after being duplicated.
Results
Silencing of the S-Locus-Linked GLO2 Gene Results in Short-Homostylous Flowers.
The S-locus assembly of P. vulgaris contains a duplicated paralogue of GLO, originally termed GLO2 in the P. veris genome-assembly study and GLOT in the P. vulgaris S-locus sequence (23, 27). To confirm experimentally that GLO2 is linked to the S locus also in other species of Primula, we genetically mapped the gene in both a large seminatural population of P. veris in Park Sanssouci, Potsdam, and an experimental cross of the distantly related Primula forbesii. In both populations we detected complete linkage between the presence of GLO2 and the S-morph (68/68 S-morph plants in P. veris; 16/16 in P. forbesii), while the gene was absent from all l-morph plants (72/72 l-morph plants in P. veris; 16/16 in P. forbesii; SI Appendix, Table S1). This confirms that GLO2 resides on the dominant S haplotype.
To determine the function of GLO2, we down-regulated its expression by virus-induced gene silencing (VIGS) in P. forbesii, using a 333-bp fragment from its 3′ end where the highest nucleotide divergence to GLO1 is found (SI Appendix, Fig. S1). Strongly affected VIGS-GLO2–treated S-morph plants formed short-homostylous flowers with the anthers low in the petal tube, at around the same height as the stigmas (Fig. 1A). Such short-homostylous flowers were seen in 144 out of 200 VIGS-GLO2–treated S-morph plants (Table 1), with an average reduction of anther height of 30% in the affected flowers (Fig. 1D), bringing the anthers to the same position as in l-morph flowers. By contrast, no changes in anther position or any other floral traits were seen in 16 control S-morph plants treated with an empty VIGS construct; also, VIGS-GLO2 treatment of l-morph plants did not affect flower morphology (n = 48; Fig. 1A and Table 1), indicating that the change in anther position seen in S-morph plants is specifically due to down-regulation of GLO2, rather than silencing of GLO1. This was confirmed by qRT-PCR using TaqMan probes to distinguish expression of the highly similar GLO1 and GLO2 genes (Fig. 1 B and C). Consistent with a primary effect of the VIGS construct on expression of GLO2, qRT-PCR indicated a sixfold down-regulation of GLO2 yet no significant effect on GLO1 expression in S-morph plants; in l-morph plants, GLO1 expression was similarly unaltered by VIGS-GLO2 treatment, consistent with the lack of any phenotypic change in their flowers. In addition to the position of the anthers, overall corolla-tube length was slightly reduced in the VIGS-GLO2–treated flowers, but the width of the corolla mouth, which is smaller in l- than in S-morph flowers (31), was not affected by silencing GLO2 (Fig. 1 E and F).
Fig. 1.

VIGS of GLO2 results in short-homostylous flowers. (A) Short-homostylous phenotypes in VIGS-GLO2–treated S (short-styled)-morph plants. Images were taken in dissected flowers (Top) and from the top of flowers (Bottom). Arrow indicates the position of anthers. (Scale bars, 1 mm.) (B) Expression of GLO2 in untreated S- and L (long-styled)-morph plants and in VIGS-GLO2–treated S- and l-morph plants. Values represent the mean ± SD from three biological replicates. Asterisk indicates significant difference from S-morph flowers by Student’s t test with ***P < 0.001. (C) Expression of GLO1 in untreated S- and l-morph plants and in VIGS-GLO2–treated S- and l-morph plants. Values represent the mean ± SD from three biological replicates. Student’s t test showed no significant difference (ns) between samples. (D–F) Anther height (D), length of corolla tube (E), and diameter of corolla mouth (F) as measured in untreated S- and l-morph and in VIGS-GLO2–treated S-morph plants. The lines in the boxes indicate the median, the boxes show the interquartile range and the whiskers indicate the largest and smallest values within 1.5x interquartile ranges above the 75th or below the 25th percentile, respectively, from n = 10 flowers. Asterisks indicate significant difference from S-morph flowers by Student’s t test with *P < 0.05 and ***P < 0.001. (G and H) Length of corolla cells above (G) and below (H) the anther in untreated S- and l-morph plants and in VIGS-GLO2–treated S- and l-morph plants. Ten corolla tubes from each phenotype were measured and values are the mean ± SD. Asterisks indicate significant difference from S-morph flowers by Student’s t test with ***P < 0.001.
Table 1.
Phenotypes of VIGS-treated plants
| Treatment and morph | No. of plants | Phenotype | ||
| VIGS-GLO2–treated | Short homostyle | Wild type | ||
| S | 200 | 144 | 56 | |
| L | 48 | 0 | 48 | |
| VIGS-GLO1–treated | Petal–sepal conversion | |||
| S | 48 | 15 | 33 | |
| L | 48 | 20 | 28 | |
For the VIGS-GLO2 treatment, three independent experiments were performed and plants showing short-homostylous flowers were counted. For VIGS-GLO1 treatment, two independent experiments were performed and plants showing petal–sepal converted flowers were counted.
The stamen filaments and petals are fused to form a corolla tube in Primula. Developmental characterization in P. vulgaris had previously suggested that the difference in anther position between the morphs is largely due to increased cell proliferation in the corolla tube beneath the anther insertion point in S-morph compared to l-morph flowers (31), while in P. veris both differences in cell number and in cell size appear to contribute (32). To determine the cellular basis of the altered anther position in GLO2-silenced S-morph flowers, we quantified the length of the corolla and of corolla cells below and above the anther insertion point (Fig. 1 G and H). This indicated that the corolla tubes of VIGS-GLO2–treated S-morph plants were about 14% shorter than those of untreated plants, yet the cells below the anther insertion point were on average 43% shorter, while those above the anther insertion point were almost twice as long as in untreated flowers. Thus, the reduction in cell length below the anther insertion point of 43% explains the reduction in the height of the anthers of 30%. VIGS-GLO2 silencing thus closely mimics the difference between S- and l-morph P. forbesii flowers, with l-morph flowers also having 30% shorter corolla cells below the anther insertion point (Fig. 1H). Together, this indicates that in P. forbesii GLO2 acts to promote cell elongation below the anther insertion point.
Silencing GLO2 Does Not Affect Pollen Traits.
The classical genetic dissection of heterostyly had indicated that control of anther height by the A locus is separable from the control of pollen size and number by P and from male incompatibility (10, 21, 22). We therefore asked whether GLO2 activity also affects the pollen, either regarding its size and number or its incompatibility behavior. If GLO2 indeed represents the A locus as defined by the above genetic studies, it should not alter these other traits. Measuring the size of pollen grains from VIGS-GLO2–treated and control S- and l-morph plants indicated pollen size was not affected by GLO2 silencing; similarly, also the number of pollen grains was indistinguishable between GLO2-silenced and control S-morph plants (Fig. 2 A and B).
Fig. 2.

Silencing of GLO2 does not affect pollen traits. (A and B ) Pollen number and pollen size in untreated S- and l-morph plants and in VIGS-GLO2–treated S- and l-morph plants. Anthers from 10 flowers for each phenotype were used and boxes are as defined in legend to Figure 1. Asterisks indicate significant difference from S-morph flowers by Student’s t test with *P < 0.05, **P < 0.01 and ***P < 0.001. (C) Seed set from crosses between untreated and VIGS-GLO2–treated S-morph plants with either S- or l-morph plants (male × female). Pollen grains from indicated genotypes were used and boxes are as defined above, with n = 12 different crosses per combination. Asterisk indicates the significant difference from the crosses between S- and l-morph plants and between VIGS-GLO2–treated S- and l-morph plants by Student’s t test with ***P < 0.001.
To test whether silencing of GLO2 also affected the pollen incompatibility behavior, we performed controlled crosses using pollen from VIGS-GLO2-treated and control S-morph plants (Fig. 2C). When pollen from both groups of plants was applied to stigmas of l-morph flowers, abundant seed set resulted. By contrast, there was only very limited seed set when pollen from these plants was applied to S-morph stigmas. Importantly, no difference in seed set was observed between pollinations using control or VIGS-GLO2 S-morph pollen. This indicates that the pollen from GLO2-silenced plants has retained its S-morph-like male incompatibility behavior.
One concern for the interpretation of the above pollen experiments could be whether VIGS in the sporophyte can affect the male gametophyte. Unfortunately, no positive control genes with a well-established specific function in Primula pollen are available to test this issue experimentally. However, this concern is allayed by two arguments. First, while initial reports suggested only a low silencing activity with tobacco-rattle virus (TRV)–mediated VIGS in pollen, recent experiments using TRV-VIGS have found strong effects on pollen development, either by affecting genes that appear to act directly in the pollen (33) or genes indirectly required for pollen development in the tapetal cells of the anthers (34, 35). Second, the genetics of heterostyly in Primula suggests that the P gene acts in diploid sporophytic cells rather than in the haploid pollen, because S-morph flowers generate uniform pollen, both regarding its size and male incompatibility type, rather than the 1:1 segregation expected if the P gene acted in the haploid pollen itself. Therefore, taken together, these results indicate that the GLO2 gene represents the A locus that determines anther position but is distinct from the P locus and the gene determining male incompatibility type.
The Ancestral GLO1 Paralogue Has Retained the Function as a Canonical B-Class Floral-Organ Identity Gene.
To determine the function of the ancestral GLO1 paralogue, we suppressed its expression using VIGS. In both S- and l-morph genetic backgrounds, silencing of GLO1 resulted in homoeotic conversions typical of B-class mutants in model species (Fig. 3 and Table 1). In moderately affected flowers, petals were converted to organs resembling sepals; this was most evident from their absence of purple pigmentation and the presence of farina, a white secretion containing flavones and wax that in the flower is only produced by sepals, even though the shape of the modified organs still resembled that of petals (Fig. 3 A–D). In more severely affected flowers, no stamens were visible, and these may have been converted to carpels and fused with the central ovary (SI Appendix, Fig. S2). Of note, even though petals of VIGS-GLO2–treated plants occasionally formed jagged, irregular edges, no petal-to-sepal conversions of comparable strength were ever observed upon silencing of GLO2. Thus, together, this indicates that the ancestral paralogue GLO1 has retained its function as a canonical B-class floral-organ identity gene; by contrast, GLO2 has lost this activity and has acquired its role in determining the height of the anthers by modulating cell length below the anther insertion point by neofunctionalization after its duplication. The role of GLO1 as a classical B-function gene is also supported by the hose-in-hose mutant of P. vulgaris that shows a sepal-to-petal transformation resulting from ectopic expression of GLO1 in the sepals (36).
Fig. 3.

GLO1 fulfills the ancestral function as a B-class floral-organ identity gene. (A and B) Phenotype of untreated S- (A) and l-morph (B) flowers. (C and D) Phenotype of VIGS-GLO1–treated S- (C) and l-morph (D) flowers. Images were taken from above (Top) or from the side (Bottom) on dissected flowers. (Scale bars, 1 mm.)
Neofunctionalization can occur by changes in gene-expression patterns or in the activity of the encoded proteins. Based on our previous transcriptomic analyses on dissected styles and corolla tubes of mature P. veris flowers (26), both GLO1 and GLO2 are abundantly expressed in the corolla, but not in the style (SI Appendix, Fig. S3 A and C). Despite numerous attempts, we were unable to specifically detect GLO1 or GLO2 expression patterns in flower buds by RNA in situ hybridization. Therefore, we compared GLO1 and GLO2 expression patterns in dissected floral organs using RT-PCR. The expression of GLO1 and GLO2 was observed in corolla tubes consistent with the RNA-sequencing (RNA-seq) data (SI Appendix, Fig. S3 B and D). GLO2 also showed weak expression in other parts of the flower except for sepals. Thus, in the absence of more spatially resolved analyses, we suggest that neofunctionalization of GLO2 is unlikely to have occurred at the level of gene expression but rather is based on changes to the encoded protein.
To identify candidate changes at the protein level, we compared aligned protein sequences of GLO1 and GLO2 from multiple Primula species and of GLO1 orthologs from a large number of angiosperms (SI Appendix, Fig. S4). This indicated that there is a single substitution in the MADS domain responsible for DNA binding that distinguishes all GLO2 from all GLO1 proteins. This substitution replaces an otherwise invariant lysine (K) with asparagine (N) (red arrow in SI Appendix, Fig. S4); the lysine in question is the first amino acid of a predicted nuclear-localization signal (37), and PSORT (38) predicts GLO2 to be cytoplasmic (certainty 0.65) in contrast to the predicted nuclear GLO1 (certainty 0.76). In addition, all GLO2 proteins differ from all GLO1 proteins by a 13-amino-acid deletion of a poorly conserved domain between the K-domain and the PI-motif in the C-terminal region. This functionally important PI-motif also shows a systematic difference between GLO2 and GLO1 proteins, with a highly conserved phenylalanine (F) replaced by a cysteine (C) (black triangle in SI Appendix, Fig. S4). Whereas no straightforward methods for assessing the function of the PI-motif are available, the functionality of nuclear-localization signals can be tested by transient expression of fusion proteins in plant cells. We therefore coexpressed GLO1-mCherry and GLO2-mGFP fusion proteins in tobacco leaves (SI Appendix, Fig. S5). As predicted, GLO1-mCherry fluorescence was detected in the nucleus, but GLO2-mGFP fluorescence was also restricted to the nucleus. Thus, the K-to-N exchange does not appear to disrupt nuclear localization of GLO2 proteins.
The Non-S-Locus Paralogues GLO1 and CYP734A51 Are Unlinked.
Like GLO2, the CYP734A50 gene representing the G locus also arose by duplication from a non-S-locus paralogue. As described in the Introduction, two models are conceivable for the chromosomal assembly of the S-locus supergene, either via a segmental duplication followed by gene loss or by stepwise duplications (28). These models differ in their predictions for the location of the non-S-locus paralogues. While stepwise duplication would be possible from paralogues located anywhere in the genome, the segmental-duplication model predicts them to be close to each other and thus genetically linked, at least unless the paralogues were also involved in a chromosomal rearrangement after the duplication.
In the absence of a highly contiguous reference genome assembly for any Primula species, we used genetic mapping to test the above prediction. No suitable sequence polymorphisms were found for the CYP734A51 locus in our experimental P. forbesii population, and we therefore used a P. veris population generated by crossing two distantly related S- and l-morph plants. PCR-based molecular markers were established for both GLO1 and CYP734A51, and 127 F1 plants were genotyped for both loci and phenotyped for the S locus (Fig. 4A). Consistent with previous mapping of the S locus in P. vulgaris (39), the GLO1 gene was closely linked to the S locus, with a recombination frequency of 2.36%. By contrast, the CYP734A51 locus was unlinked to either (recombination frequency of 55.11%), indicating that it is located very far from GLO1 and the S locus on the same chromosome or on a different chromosome altogether (Fig. 4A). We analyzed the location of putative GLO1 and CYP734A51 orthologs in high-quality genomes from the orders most closely related to the Ericales, containing Primula, to determine whether the absence of linkage between the two genes is likely to be ancestral (SI Appendix, Table S2). This indicated that the putative orthologs of GLO1 and CYP734A51 are located on different chromosomes in tomato (Solanales), Mimulus guttatus (Lamiales), and the lettuce Lactuca sativa (Asterales), while in the carrot genome (Daucus carota; Apiales) the GLO1 ortholog and one of two putative CYP734A51 orthologs are located almost 5 Mb apart on the same chromosome. This analysis therefore argues that these genes were unlinked in the ancestral state. Thus, the segmental duplication model would have to assume that a structural rearrangement first brought GLO1 and CYP734A51 close together in an ancestor of Primula, and another one broke up their linkage again after the segmental duplication. Against this, the stepwise duplication model provides a more parsimonious explanation.
Fig. 4.

CYP734A50 and GLO2 arose by stepwise duplications. (A) Analysis of linkage between CYP734A51 and GLO1 in a mapping population of P. veris. Colors represent different genotypes as indicated. (B) Relationship of Ks values for comparisons of CYP734A-like sequences and GLO-like sequences for different species pairs (red and orange) and for S-locus to non-S-locus paralogues within the four species P. veris, P. forbesii, P. oreodoxa, and P. maximowiczii. The Ks values on which this plot is based can be found in Table 2. (C) Schematic phylogeny of the species used for the analyses in D and E based on refs. 17, 40, and 67. The species abbreviations used in D and E are given in brackets. (D and E) Phylogenies of CYP734A-like (D) and GLO-like sequences (E). For the Primula species, paralogues located at the S locus are indicated in cyan, while paralogues located outside of the S locus are shown in orange. Numbers on branches indicate bootstrap support from 1,000 runs. Bootstrap values below 50% are not shown.
Phylogenetic Analysis Indicates That CYP734A Duplicated before GLO.
Besides the genomic architecture, another corollary of the segmental-duplication model is that all duplicated copies are of the same age, whereas their ages could differ considerably in the stepwise duplication model. In addition, dating the ages of the duplications also has implications for models of trait evolution regarding heterostyly. We therefore sought to date the duplications that gave rise to CYP734A50 and GLO2 relative to each other. For this we identified and assembled CYP734A- and GLO-like sequences from two additional Primula species (Primula oreodoxa and Primula maximowiczii) and for the most closely related species to the genus Primula for which whole-genome sequences are available. These latter two species, Embelia ribes and Embelia sessiliflora, are from the subfamily Myrsinoideae, which is sister to the Primuloideae subfamily in the Primulaceae (40); in addition, we identified the most closely related homologous sequences from Maesa indica and Maesa japonica from the more distantly related subfamily Maesoideae in the Primulaceae and used these as outgroup sequences (Fig. 4C and Dataset S1; note that while we identified two CYP734A51-like genes from whole-genome sequencing data, we could only amplify one of them by PCR from M. japonica).
One method for dating duplications is to compare the divergence at synonymous sites (Ks) between paralogues (41). This indicated that Ks was indeed higher for the pair CYP734A50/CYP734A51 than for the GLO1/GLO2 pair in all four Primula species (Table 2), suggesting that the CYP734A duplication occurred before the GLO duplication. However, one concern with this interpretation is that the rate of mutation and thus Ks may differ between the loci in question, for example related to differences in their gene-expression pattern (42). To assess this possibility, we determined Ks values for each pair of orthologs between all possible species pairs among the four Primula species (Table 2) and between the two Embelia and Maesa species (SI Appendix, Table S3). This analysis found that CYP734A ortholog pairs show higher Ks values than GLO ortholog pairs for each species comparison performed. When plotting the Ks values of CYP734A ortholog pairs versus Ks values of GLO ortholog pairs for all comparisons performed in the genus Primula, a consistent linear relationship was observed (Fig. 4B), suggesting that a higher rate of synonymous substitutions in CYP734A than in GLO genes is a consistent feature in our dataset and applies throughout the Primulaceae as a family. The values for the S-locus versus non-S-locus paralogues closely followed this trend (Fig. 4B) yet were all higher than the between-species comparisons of orthologs. Thus, this analysis supports the notion that the duplications giving rise to the S locus occurred before the divergence of the different Primula species; however, we also conclude that these Ks-value comparisons are not suitable for relative dating of the CYP734A versus GLO duplications.
Table 2.
Pairwise synonymous-site divergence of CYP734A and GLO genes
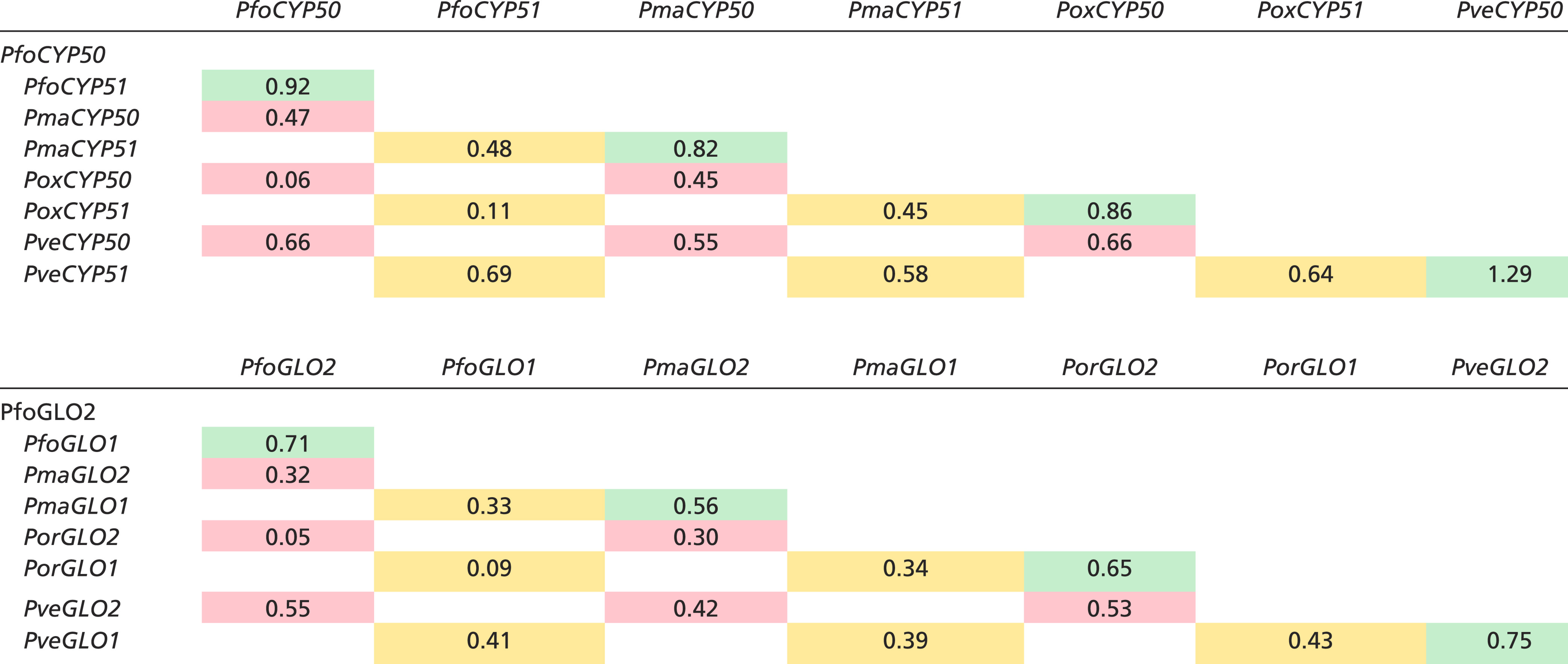 |
Ks values of pairwise synonymous-site divergence for the indicated comparisons are shown. Green shading indicates comparisons of S-locus and non-S-locus paralogues within the four species. Red and yellow shading shows comparisons of S-locus genes (red) and non-S-locus genes (yellow) between the species.
As an alternative, we performed phylogenetic analysis of CYP734A- and GLO-like sequences (Fig. 4 C–E). In the four Primula species, this identified one clade containing the paralogues located at the S locus (cyan in Fig. 4 D and E) and one clade with the non-S-locus paralogues (orange in Fig. 4 D and E) for both genes. This confirms that both genes duplicated before the divergence of the four Primula species. We next considered when these duplications occurred relative to the evolutionary divergence of all of the species analyzed. For CYP734A-like sequences, the phylogeny indicated with robust statistical support that the duplication giving rise to CYP734A50 genes had occurred before the Primuloideae and Myrsinoideae diverged (Fig. 4D). By contrast, for GLO-like sequences, the branches leading to Primula GLO1, Primula GLO2, and the most closely related Myrsinoideae GLO genes could not be resolved, while the internal branches were similarly well-supported as for the CYP734A phylogeny. This suggests that the duplication giving rise to the GLO paralogues only occurred around the time of the divergence of Primuloideae and Myrsinoideae. Thus, these phylogenies support the notion that the duplication giving rise to CYP734A50 occurred before the one resulting in GLO2.
Discussion
The evolution and genetic control of heterostyly have fascinated biologists for decades (6, 8, 28). Here, we have demonstrated that the GLO2 gene represents the genetically defined A locus, that GLO2 underwent neofunctionalization from a B-class homoeotic-gene activity that is still fulfilled by the ancestral GLO1 locus, and that the Primula S locus arose by stepwise duplication, with CYP734A50 likely duplicating before GLO2.
How does GLO2 lead to elevated anthers? In GLO2-silenced S-morph flowers in P. forbesii the reduced anther height was due to shorter corolla cells beneath the anther insertion point. This finding differs from the observation in P. vulgaris that the difference in anther insertion height between S- and l-morph results mostly from altered cell proliferation in the corolla tube below the anthers (31), while in P. veris both increased cell proliferation and cell elongation appear to underlie the higher anther insertion in S-morph plants (32). It is thus possible that the relative effect of GLO2 on cell proliferation versus elongation has shifted in the mentioned Primula species since their evolutionary divergence around 20 Ma (43). In contrast to this effect of GLO2 on the distribution of growth in the corolla, the ancestral GLO1 paralogue has retained the function as a B-class floral-organ identity gene. Given the similar expression pattern of both genes, neofunctionalization of GLO2 has likely occurred at the level of protein activity, either by changing its DNA-binding specificity or its interactions with other proteins. All available Primula GLO2 protein sequences differ in the MADS DNA-binding domain and in the C-terminal PI-motif from all GLO1 sequences in Primula and beyond. Whichever of these differences causes the altered function of GLO2, it is likely that the transcriptional output, that is, the set of regulated genes, differs between GLO1 and GLO2. Neofunctionalization of MADS-domain proteins by changes in the C-terminal region outside of the DNA-binding domain has been described before, both at the level of evolution of the entire gene family (44) or in individual, taxon-specific cases (45). Transcriptome studies, ideally in a stable GLO2 mutant, analysis of directly bound target genes, and systematic investigation of interaction partners will be required to understand this change at the molecular level.
The results of genetic mapping and comparisons with the most closely related high-quality genome assemblies to Primula indicate that the ancestral CYP734A and GLO paralogues were unlinked and potentially located on different chromosomes. Therefore, a model of stepwise duplication of CYP734A and GLO genes provides the most parsimonious explanation for the chromosomal assembly of the S locus. Such a stepwise duplication is also consistent with the results of our phylogenetic analysis. These suggest that the CYP734A locus duplicated before the divergence between the Myrsinoideae and Primuloideae subfamilies and thus earlier than the GLO locus, for which the duplication only seems to have occurred around this time.
Two models have been proposed to explain the evolutionary build-up of heterostyly. While the Charlesworth and Charlesworth (29) model suggests that self-incompatibility evolved before reciprocal herkogamy, the model by Lloyd and Webb (30) proposes that reciprocal herkogamy arose first, possibly, but not always followed by the evolution of self-incompatibility. Unfortunately, we cannot discriminate between these two possibilities, as the molecular control of self-incompatibility is largely unknown. However, the above results have implications for the likely sequence of morphological trait evolution. In this respect, both models agree that style-length dimorphism should have evolved before the anther-height polymorphism. It is now clear that the genes controlling these two traits arose by duplications, followed by neofunctionalization. Even though we do not know and may never find out when the critical mutations leading to neofunctionalization occurred in the duplicated copies, our results implying that CYP734A50 arose earlier than GLO2 are fully consistent with the suggestion from both models that the stylar polymorphism evolved first and anther height was only adjusted later to provide for better reproductive-organ reciprocity between the two morphs (29, 30).
Supergenes underlie many complex polymorphisms in plants and animals, including homomorphic self-incompatibility systems in plants, mimicry in Heliconius numata butterflies, behavioral morphs in white-throated sparrows, migratory behavior in rainbow trout, and the social system in fire ants (11, 13, 14, 46–48). Central to the concept of a supergene is the notion that it contains two or more individual genes controlling separate coadapted aspects of a complex phenotype (11, 46). Indeed, the supergenes in the systems mentioned above, especially in the animal cases, are often large and contain dozens to hundreds of different genes. However, although plausible candidates have been suggested, identifying which of them are responsible for which aspects of the phenotype, and thus functionally confirming the central notion that separate genes control distinct aspects of the phenotype, has proven difficult (49, 50). An exception are the homomorphic self-incompatibility loci in plants, where the function of the male and female specificity genes are understood in molecular detail (51). Against this background, our identification of two separate genes that control style length and anther height in distylous primroses provides a rare example of a functionally validated supergene architecture underlying a complex phenotype.
In conclusion, we have shown that the GLO2 gene representing the A locus arose by duplication and neofunctionalization, with the duplication likely occurring after an earlier duplication had given rise to the CYP734A50 gene at the proto-S locus. To our knowledge, this provides the most detailed insight yet into the evolutionary steps leading to a supergene in plants. These findings should also facilitate comparative studies into independently evolved heterostyly supergenes in other plant lineages to determine whether such stepwise assembly is a common feature of these systems.
Materials and Methods
Plant Material and Growth Conditions.
The P. veris plants that were used for the mapping experiment of GLO2 are from a seminatural population in the Park Sanssouci, Potsdam (26). The F1 mapping population of P. veris that was used for the linkage assay between GLO1 and CYP734A51 was generated by crossing two distantly related S- and l-morph plants of P. veris raised from seeds and cultivated at the Botanical Garden of the University of Zurich. Seeds for the S- and l-morph plants were collected in a natural population in the Thunersee-Region, BE, Switzerland and obtained from the Botanical Garden Jardin Alpin, Meyrin, Switzerland (accession no. 20031402), respectively.
P. forbesii plants were raised from seeds obtained from the Botanical Garden at the University of Zurich (accession no. 20050032). Plants were grown in a growth room at 18 °C, initially under short-day conditions (8 h light, 16 h dark) to stimulate flowering, then under long-day conditions (16 h light, 8 h dark).
Nicotiana benthamiana plants for transient expression of GLO1 and GLO2 proteins were raised from seeds obtained from the Bäurle laboratory at the University of Potsdam. Plants were grown in the growth chamber at 21 °C under long-day conditions (16 h light, 8 h dark).
Dried leaf material of M. japonica was obtained from the Botanical Garden at the University of Bayreuth, Germany.
Genetic Mapping of GLO2.
Genetic mapping of GLO2 in P. veris and P. forbesii was done using PCR-based genotyping as described (26) Genomic DNA was extracted from leaf material using the DNeasy Plant Mini Kit (Qiagen). PCR reactions were performed using primers that bind specifically to the intron region of GLO2 indicated in the SI Appendix, Table S4. PCR products were run on agarose gels and visualized by ethidium bromide staining. The presence or absence of GLO2 in different morphs tested was detected.
Linkage Assay of GLO1 and CYP734A51 in P. veris.
The linkage between GLO1 and CYP734A51 was estimated using PCR-based genotyping. Single-nucleotide polymorphisms (SNPs) on GLO1 and CYP734A51 between S- and l-morphs were detected by sequencing the two parents of the F1 individuals using primers indicated in the SI Appendix, Table S4. dCAPs primers for these SNPs were designed using dCAPS Finder 2.0 (52) and are indicated in SI Appendix, Table S4. Genomic DNA from F1 individuals and parents was extracted from leaf material using a modified cetyltrimethylammonium bromide (CTAB) method (53). After PCR reactions, PCR products were digested and separated on 2% agarose gels. Genotypes and phenotypes of F1 individuals and parents were recorded and the linkage between GLO1, CYP734A51, and the S locus were estimated by OneMap v. 2.1.3 (54) using a maximum recombination fraction of 0.5, the Kosambi mapping function, and arker types D2.15 and D2.18 for the SNPs and the S locus, respectively.
VIGS Assay.
Plasmids for the VIGS assay were prepared as described (55). Due to the highest nucleotide divergence, sequences at the 3′ end of GLO1 and GLO2 were amplified from complementary DNA using primers indicated in SI Appendix, Table S4. Products were digested with EcoRI and BamHI and inserted into pTRV2 vector to produce constructs pTRV2-GLO1 and pTRV2-GLO2, respectively. These constructs were then introduced into Agrobacterium strain GV3101. The VIGS experiments were done as described (26). Briefly, Agrobacterium cultures containing empty pTRV2, pTRV2-GLO1, or pTRV2-GLO2 were mixed with an Agrobacterium culture containing pTRV1 at a 1:1 ratio. These mixtures were infiltrated into leaves of the five-leaf stages of P. forbesii plants. Infiltrated plants were kept in the dark overnight at 18 °C and then followed normal growing conditions until plants flowered.
Plant Phenotyping.
For the cell length measurements, corolla tubes of fully opened flowers of S- and l-morph and VIGS-GLO2–treated S-morph plants were divided into two parts at the anther attachment site. Imprints from the upper and lower parts were made by dried-gel method as described in (56). Images of gel cast were taken under a differential phase contrast microscope. The length of cells of upper and lower parts was measured manually by ImageJ.
For the measurements of the corolla mouth diameter, fully opened flowers of S- and l-morph and VIGS-GLO2–treated S-morph plants was photographed from the top using a SteREO Discovery V.12 microscope (Zeiss). The corolla mouth diameter was measured manually by ImageJ. After taking the images for corolla mouth measurements, flowers of different phenotypes were dissected vertically into two parts and half of the flower was photographed on a black background. The length of the corolla tube and position of the anther insertion point were measured manually by ImageJ.
Pollen counting and pollen size measurements followed the procedures described in ref. 57. At first, flower buds just before opening were collected from S- and l-morph and VIGS-GLO2–treated S-morph plants. From these, anthers were dissected and dried in an oven at 55 °C overnight. This step stimulates the anthers to open. Thirty microliters of 5% Tween 80 was added into the tubes and sonication was applied for 20 min in a water bath. For each flower bud, 10 out of 30 µL was used to photograph on a hemocytometer (Neubauer) and images were taken under the microscope. The number of pollen grains was counted and the pollen size was measured by ImageJ using the “Analyze Particles” function.
In order to check the self-incompatible behavior of VIGS-GLO2–treated S-morph plants, reciprocal crosses with untreated S- and l-morph plants were performed. Pollen grains from VIGS-GLO2–treated S-morph plants were used to hand-pollinate stigmas of flowers of S- and l-morph plants. At the same time, control reciprocal crosses between S- and l-morph plants were done as well. The number of seeds developing from each cross was then counted.
Gene Expression Analysis.
To determine expression of GLO1 and GLO2 in S- and l-morph and VIGS-GLO2–treated S-morph flowers, petals of recently opened flowers of different phenotype plants were manually dissected and immediately frozen in liquid nitrogen. Total RNA was extracted using RNeasy Plant Mini Kit (Qiagen). Reverse transcription was performed using oligo(dT) priming and SuperScript III (Invitrogen). qRT-PCR reactions using TaqMan probes which bind specifically to GLO1 and GLO2 were performed on a LightCycler LC480 (Roche). The Primula TUBULIN gene was used as a reference. Primers and probes used for qRT-PCR reactions are indicated in SI Appendix, Table S4.
The expression patterns of GLO1 and GLO2 in dissected flower organs from P. veris were determined as described (26). Organs including sepals, petals, stamens, style, and ovary from young flower buds were dissected and total RNA was extracted using RNeasy Plant Mini Kit (Qiagen). Reverse transcription was performed using oligo(dT) priming and SuperScript III (Invitrogen). RT-PCR products were separated on agarose gels and visualized by ethidium bromide staining. The Primula TUBULIN gene was used as a reference. Primers for RT-PCR reactions are indicated in SI Appendix, Table S4.
Transient Expression of GLO1 and GLO2 Proteins.
For testing the subcellular localization of GLO1 and GLO2 proteins, coding sequences of GLO1 and GLO2 genes were cloned into binary 2-in-1 vectors (58) using Gateway technology (Invitrogen). Primers are indicated in SI Appendix, Table S4. For detecting protein localization, GLO1 was fused to mCherry and GLO2 was fused to mGFP, respectively. The final construct carrying GLO1::mCherry and GLO2::mGFP under the control of a 35S promoter each was introduced into Agrobacterium GV3101 and the infiltration into tobacco N. benthamiana plants was performed as described (58). The subcellular localization of GLO1 and GLO2 proteins was detected under a confocal microscope LSM 700 (Zeiss).
Phylogenetic Analysis and Synonymous Divergence Estimates.
Genome sequences of Solanum lycopersicum, M. guttatus, D. carota, and L. sativa were accessed via Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html). Orthologs of CYP734A51 and GLO1 were identified via mutual best BLAST matches.
Public DNA-seq and RNA-seq date were downloaded from ENA (https://www.ebi.ac.uk/ena). Species names with identifiers leading to the data are P. oreodoxa (PRJNA544868), P. maximowiczii (PRJNA437902), E. ribes (PRJNA343414, PRJNA397122), E. sessiliflora (PRJNA438407), and M. indica (PRJNA438407). Data were mapped against P. forbesii and P. veris CYP734A50, CYP734A51, GLO1, and GLO2 coding sequence exons using bwa mem (59) and further processed using samtools (60). Mappings were visualized in IGV (61) to manually extract coding sequence exons for other species. In cases where there were only partial mappings, those were used as seed points for edge extension assembly. Raw fastq data were searched for perfect matches to 20 to 30 bordering nucleotides to extend sequences. Fully assembled exons were linked using RNA-seq reads, if data were available and if the transcript was expressed there. Otherwise, whole introns were assembled using the edge extension approach. Repetitive parts were resolved, if possible, using spanning reads or read pairs, if available. Pairwise Ks values were calculated using KaKs_Calculator 2.0 (62) after multiple sequence alignment done with TranslatorX (63) in combination with MUSCLE (64).
For M. japonica, DNA was extracted from dried leaf material using CTAB, and CYP734A-like and GLO-like genomic loci were amplified by PCR and Sanger-sequenced. Primers are indicated in SI Appendix, Table S4. Coding sequence exons were defined by similarity to M. indica.
Sequences used to infer the phylogenetic trees in Fig. 4 are given in Dataset S1. Only coding sequences were used for the phylogenetic analysis. Phylogenetic trees were generated using RAxML, version 8.2.12 with the GTRCAT model and 500 bootstrap runs (65). Visualizations were done using SeaView version 4.0 (66).
Supplementary Material
Acknowledgments
We thank Christiane Schmidt and Doreen Mäker for plant care; and Marianne Lauerer and Michael Burkart for providing material of M. japonica. We are grateful to members of the M.L. laboratory, and to Isabel Bäurle and Ralph Tiedemann for discussion and comments. This work was funded by grants Le1412/19-1 from the Deutsche Forschungsgemeinschaft to M.L., 31003A_175556 from the Schweizerischer Nationalfonds to E.C., and from the Claraz Foundation to B.K. and E.C.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2006296117/-/DCSupplemental.
Data Availability.
M. japonica sequences generated in this work have been deposited in GenBank with accession numbers MT799180 and MT799181. All study data are included in the paper, SI Appendix, and Dataset S1.
References
- 1.Barrett S. C., The evolution of plant sexual diversity. Nat. Rev. Genet. 3, 274–284 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Barrett S. C., Understanding plant reproductive diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 99–109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keller B., Thomson J. D., Conti E., Heterostyly promotes disassortative pollination and reduces sexual interference in Darwin’s primroses: Evidence from experimental studies. Funct. Ecol. 28, 1413–1425 (2014). [Google Scholar]
- 4.Lloyd D. G., Webb C. J., . “The selection of heterostyly” in Evolution and Function of Heterostyly, Barrett S. C. H., Ed. (Springer, 1992), pp. 179–208. [Google Scholar]
- 5.Wedderburn F., Richards A. J., Variation in within-morph incompatibility inhibition sites in Heteromorphic Primula L. New Phytol. 116, 149–162 (1990). [Google Scholar]
- 6.Barrett S. C. H., Evolution and Function of Heterostyly, (Springer, 1992). [Google Scholar]
- 7.Barrett S. C. H., Shore J. S., . “New insights on heterostyly: Comparative biology, ecology and genetics” in Self-Incompatibility in Flowering Plants: Evolution, Diversity and Mechanisms, Franklin-Tong V. E., Ed. (Springer, 2008), pp. 3–32. [Google Scholar]
- 8.Barrett S. C. H., “A most complex marriage arrangement”: Recent advances on heterostyly and unresolved questions. New Phytol. 224, 1051–1067 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Dulberger R., . “Floral polymorphisms and their functional significance in the heterostylous syndrome” in Evolution and Function of Heterostyly, Barrett S. C. H., Ed. (Springer, 1992), pp. 41–84. [Google Scholar]
- 10.Lewis D., Jones D. A., . “The genetics of heterostyly” in Evolution and Function of Heterostyly, Barrett S. C. H., Ed. (Springer, 1992), pp. 129–150. [Google Scholar]
- 11.Schwander T., Libbrecht R., Keller L., Supergenes and complex phenotypes. Curr. Biol. 24, R288–R294 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Charlesworth D., The status of supergenes in the 21st century: Recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evol. Appl. 9, 74–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuttle E. M.et al., Divergence and functional degradation of a sex chromosome-like supergene. Curr. Biol. 26, 344–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joron M.et al., Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477, 203–206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Küpper C.et al., A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 48, 79–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamichhaney S.et al., Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat. Genet. 48, 84–88 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Richards J., Primula, (Timber Press, 2003). [Google Scholar]
- 18.Bateson W., Gregory R. P., On the inheritance of heterostylism in Primula. Proc. R. Soc. London, Ser. B 76, 581–586 (1905). [Google Scholar]
- 19.Ernst A., Zur Vererbung der morphologischen Heterostyliemerkmale. Ber. Dtsch. Bot. Ges. 46, 573–588 (1928). [Google Scholar]
- 20.Ernst A., Heterostylie-Forschung: Versuche zur genetischen Analyse eines Organisations-und „Anpassungs“merkmales. Z. Indukt. Abstamm. Vererbungsl. 71, 156–230 (1936). [Google Scholar]
- 21.Dowrick V. P. J., Heterostyly and homostyly in Primula obconica. Heredity 10, 219–236 (1956). [Google Scholar]
- 22.Kurian V., Richards A. J., A new recombinant in the heteromorphy “S” supergene in Primula. Heredity 78, 383–390 (1997). [Google Scholar]
- 23.Li J.et al., Genetic architecture and evolution of the S locus supergene in Primula vulgaris. Nat. Plants 2, 16188 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Burrows B. A., McCubbin A. G., Sequencing the genomic regions flanking S-linked PvGLO sequences confirms the presence of two GLO loci, one of which lies adjacent to the style-length determinant gene CYP734A50. Plant Reprod. 30, 53–67 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Cocker J. M.et al., Primula vulgaris (primrose) genome assembly, annotation and gene expression, with comparative genomics on the heterostyly supergene. Sci. Rep. 8, 17942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huu C. N.et al., Presence versus absence of CYP734A50 underlies the style-length dimorphism in primroses. eLife 5, e17956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nowak M. D.et al., The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol. 16, 12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kappel C., Huu C. N., Lenhard M., A short story gets longer: Recent insights into the molecular basis of heterostyly. J. Exp. Bot. 68, 5719–5730 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Charlesworth D., Charlesworth B., A model for the evolution of distyly. Am. Nat. 114, 467–498 (1979). [Google Scholar]
- 30.Lloyd D. G., Webb C. J., . “The evolution of heterostyly” in Evolution and Function of Heterostyly, Barrett S. C. H., Ed. (Springer, 1992), pp. 151–178. [Google Scholar]
- 31.Webster M. A., Gilmartin P. M., Analysis of late stage flower development in Primula vulgaris reveals novel differences in cell morphology and temporal aspects of floral heteromorphy. New Phytol. 171, 591–603 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Stirling J., Studies of flowering in heterostyled and allied species. Part 1. The Primulaceae. Publ Hartley Bot Lab 8, 2–42 (1932). [Google Scholar]
- 33.Gong P., Li J., He C., Exon junction complex (EJC) core genes play multiple developmental roles in Physalis floridana. Plant Mol. Biol. 98, 545–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T.et al., Basic helix-loop-helix transcription factor BcbHLHpol functions as a positive regulator of pollen development in non-heading Chinese cabbage. Funct. Integr. Genomics 14, 731–739 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Chen C.et al., CaMF2, an anther-specific lipid transfer protein (LTP) gene, affects pollen development in Capsicum annuum L. Plant Sci. 181, 439–448 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Li J.et al., Hose in Hose, an S locus-linked mutant of Primula vulgaris, is caused by an unstable mutation at the Globosa locus. Proc. Natl. Acad. Sci. U.S.A. 107, 5664–5668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theißen G., Gramzow L., “Structure and evolution of plant MADS domain transcription factors” in Plant Transcription Factors, Gonzalez D. H., Ed. (Academic Press, 2016), chap. 8, pp. 127–138. [Google Scholar]
- 38.Nakai K., Kanehisa M., Expert system for predicting protein localization sites in gram-negative bacteria. Proteins 11, 95–110 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Li J.et al., Integration of genetic and physical maps of the Primula vulgaris S locus and localization by chromosome in situ hybridization. New Phytol. 208, 137–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens P. F., Angiosperm Phylogeny Website (Version 14, July 2017 [and more or less continuously updated since]). http://www.mobot.org/MOBOT/research/APweb/. Accessed 19 August 2020.
- 41.Fawcett J. A., Maere S., Van de Peer Y., Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. U.S.A. 106, 5737–5742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park S. G., Choi S. S., Expression breadth and expression abundance behave differently in correlations with evolutionary rates. BMC Evol. Biol. 10, 241 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Vos J. M., Hughes C. E., Schneeweiss G. M., Moore B. R., Conti E., Heterostyly accelerates diversification via reduced extinction in primroses. Proc. Biol. Sci. 281, 20140075 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vandenbussche M., Theissen G., Van de Peer Y., Gerats T., Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 31, 4401–4409 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erdmann R., Gramzow L., Melzer R., Theissen G., Becker A., GORDITA (AGL63) is a young paralog of the Arabidopsis thaliana B(sister) MADS box gene ABS (TT16) that has undergone neofunctionalization. Plant J. 63, 914–924 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Black D., Shuker D. M., Supergenes. Curr. Biol. 29, R615–R617 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Pearse D. E.et al., Sex-dependent dominance maintains migration supergene in rainbow trout. Nat. Ecol. Evol. 3, 1731–1742 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Wang J.et al., A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493, 664–668 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Saenko S. V.et al., Unravelling the genes forming the wing pattern supergene in the polymorphic butterfly Heliconius numata. Evodevo 10, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horton B. M., Michael C. M., Prichard M. R., Maney D. L., Vasoactive intestinal peptide as a mediator of the effects of a supergene on social behaviour. Proc. Biol. Sci. 287, 20200196 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujii S., Kubo K., Takayama S., Non-self- and self-recognition models in plant self-incompatibility. Nat. Plants 2, 16130 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Neff M. M., Turk E., Kalishman M., Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 18, 613–615 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Doyle J. J., Isolation of plant DNA from fresh tissue. Focus 12, 13–15 (1990). [Google Scholar]
- 54.Margarido G. R. A., Souza A. P., Garcia A. A., OneMap: Software for genetic mapping in outcrossing species. Hereditas 144, 78–79 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Liu Y., Schiff M., Dinesh-Kumar S. P., Virus-induced gene silencing in tomato. Plant J. 31, 777–786 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Fujikura U.et al., Variation in splicing efficiency underlies morphological evolution in Capsella. Dev. Cell 44, 192–203.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Sicard A.et al., Genetics, evolution, and adaptive significance of the selfing syndrome in the genus Capsella. Plant Cell 23, 3156–3171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hecker A.et al., Binary 2 in1 vectors improve in planta (co)localization and dynamic protein interaction studies. Plant Physiol. 168, 776–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 (16 March 2013).
- 60.Li H.et al.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thorvaldsdóttir H., Robinson J. T., Mesirov J. P., Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang D., Zhang Y., Zhang Z., Zhu J., Yu J., KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Dev. Reprod. Biol. 8, 77–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abascal F., Zardoya R., Telford M. J., Translator X., TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–W13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gouy M., Guindon S., Gascuel O., SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Mast A. R., Kelso S., Conti E., Are any primroses (Primula) primitively monomorphic? New Phytol. 171, 605–616 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
M. japonica sequences generated in this work have been deposited in GenBank with accession numbers MT799180 and MT799181. All study data are included in the paper, SI Appendix, and Dataset S1.