

Abstract
Most antibodies display very low brain exposure due to the blood-brain barrier (BBB) preventing their entry into brain parenchyma. Transferrin receptor (TfR) has been used previously to ferry antibodies to the brain by using different formats of bispecific constructs. Tetravalent bispecific tandem immunoglobulin Gs (IgGs) (TBTIs) containing two paratopes for both TfR and protofibrillar forms of amyloid-beta (Aβ) peptide were constructed and shown to display higher brain penetration than the parent anti-Aβ antibody. Additional structure-based mutations on the TfR paratopes further increased brain exposure, with maximal enhancement up to 13-fold in wild-type mice and an additional 4–5-fold in transgenic (Tg) mice harboring amyloid plaques, the main target of our amyloid antibody. Parenchymal target engagement of extracellular amyloid plaques was demonstrated using in vivo and ex vivo fluorescence imaging as well as histological methods. The best candidates were selected for a chronic study in an amyloid precursor protein (APP) Tg mouse model showing efficacy at reducing brain amyloid load at a lower dose than the corresponding monospecific antibody. TBTIs represent a promising format for enhancing IgG brain penetration using a symmetrical construct and keeping bivalency of the payload antibody.
Keywords: brain delivery, bispecific antibodies, transferrin receptor
Graphical Abstract
Tetravalent bispecific tandem immunoglobulin Gs (IgGs) (TBTIs) containing two paratopes for both transferrin receptor (TfR) and amyloid-beta (Aβ) peptide were constructed and shown to display higher brain penetration than the parent Aβ antibody, parenchymal target engagement, and efficacy at reducing brain amyloid load in a chronic study in an amyloid precursor protein (APP) transgenic (Tg) mouse model.
Introduction
Brain tissue is a highly protected tissue. The endothelial cells lining the blood vessels that are at the interface of the blood and brain are, as opposed to peripheral endothelial cells, extremely tightly jointed, non-fenestrated, and equipped with many efflux systems. This blood-brain barrier (BBB) is only permeable to very small lipophilic compounds and is actively preventing most molecules from crossing, in particular, large or polar molecules such as biotherapeutics and antibodies.1,2 Therefore, fewer biologics are in development for the central nervous system (CNS) compared to other therapeutic indications and no biologics have been approved for Alzheimer’s disease (AD) or Parkinson’s disease to date. Huge medical needs remain to be addressed, in particular, for difficult targets for which biologics are the main modality in the therapeutic area, such as neurosciences, CNS lymphoma or glioblastoma, or CNS rare diseases.3 Therefore, strategies to increase brain exposure of biotherapeutics will be key to their success in these indications.
Several strategies to increase the brain exposure of biotherapeutics have been reported. Focused ultrasounds,4 intranasal administration,5 or various formulations, including charge and nanotechnologies,6 have been used for several modalities but, to a lower extent, for antibodies. The “trojan horse” approach, ferrying the biotherapeutics by a ligand or antibody against a receptor which performs transcytosis, is certainly the most used strategy when it comes to antibodies.7,8
Receptor-mediated transcytosis is a specific endogenous process allowing brain transport of selected proteins such as insulin, transferrin, or lipoproteins. At present, the most convincing data on brain enhancement of antibodies have been reported with transferrin receptor (TfR),9, 10, 11 using several antibodies fusions12, 13, 14, 15, 16, 17, 18 or bispecific constructs19, 20, 21, 22 (Figures 1A–1I). The nature of the interaction of these constructs with TfR seems to be key to the extent of brain exposure, with the bispecific formats bivalent for TfR (Figures 1A–1C) using both high13,16, 17, 18,25 and medium17 affinity or avidity14 TfR antibodies and the monovalent for TfR formats (Figures 1D and 1G) using either a low affinity20,22 or low avidity15 TfR antibodies. We have designed novel so-called tetravalent bispecific tandem immunoglobulin Gs (IgGs) (TBTIs; Figures 1H and 1I)24 and report their enhanced brain penetration in vivo. This format, analogous to the dual variable domain (DVD) format,23 is bivalent for both the TfR and the therapeutic target. It should provide a significant advantage in large-scale production since it incorporates only two polypeptidic chains (light and heavy) and its symmetry should prevent chain mispairing. Its bivalency ensures full avidity for targets such as aggregated proteins. The construct affinity for TfR can be modulated by the positional effect of the two paratopes, with the internal position displaying, generally, a poorer affinity26 than the parent TfR antibody. We chose to illustrate the potential of such constructs for brain enhancement and brain parenchymal target engagement using the anti-amyloid-beta (Aβ) antibody 13C327 as a tool (specific for protofibrillar and fibrillar Aβ and therefore without any target in wild-type [WT] animals) and the highly published murine anti-TfR monoclonal antibody (mAb) 8D328 to allow direct comparison with previous literature. In addition to the positional TfR paratope affinity modulation, we engineered a series of structure-based mutations in the complementarity determining regions (CDRs) of the anti-TfR paratopes to further lower their affinity. We report significant improvement in brain exposure for these constructs and for the best one’s efficacy at prevention of cortical and hippocampal Aβ plaque accumulation in an amyloid mouse transgenic (Tg) model, demonstrating target engagement and efficacy at a lower dose than the corresponding monospecific anti-Aβ IgG.
Figure 1.

Bispecific IgG Formats Reported to Enhance Brain Exposure of Proteins
(A) Tetravalent bispecific format fusing two Aβ ScFvs to the C-terminal heavy chains of a high affinity TfR antibody.12 (B) Bivalent high or medium affinity TfR antibody fusing two payload proteins to the C-terminal heavy chains.13,16, 17, 18 (C) Tetravalent bispecific format fusing two Aβ ScFvs to the C-terminal light chains of a therapeutic antibody.14 (D) Brain shuttle format fusing one TfR scFab to one of the IgG heavy chain.15 (E) Divalent bispecific format.22 (F) Therapeutic antibody with TfR affinity engineered in its Fc domain.20 (G) Tribody format linking two anti-Aβ ScFvs to an anti-TfR Fab.21 (H and I) DVDs23 and TBTIs24. Blue, TfR; green, Aβ; pink, BACE1 recognizing paratopes; yellow, payload proteins such as IL-1 receptor antagonist,17 TNF-alpha decoy receptor protein,13 GDNF,18 or lysosomal enzymes such as iduronate sulfatase.16
Results
Tool Anti-Aβ Antibody
To demonstrate the potential of TBTI formats to increase brain penetration, we have used a tool antibody 13C3, the murine IgG1 precursor of SAR228810,27 an anti-protofibrillar Aβ mAb. 13C3 binds preferentially to soluble protofibrillar and fibrillar or aggregated forms of Aβ and not to the monomeric Aβ. In addition, it is selective versus other amyloid structures, such as α-synuclein and islet amyloid polypeptide fibrils. In vivo, 13C3 displayed marked prevention of amyloid plaque development in Tg mice. No change in plasma Aβ after treatment confirmed the specificity versus monomeric Aβ. Preclinical safety studies further demonstrated that 13C3 antigen is present only in Tg and human AD brains and can be classified as a stealth antibody. The lack of antigen in WT mice is useful in the present context since, in normal animals, there is no role of target binding in modulating tissue accumulation. Tissue levels of 13C3 were measured with a sensitive immuno-assay (enhanced chemiluminescence [ECL] detection) based on binding to amyloid. Therefore, only molecules with the active binding site are detected. Quantification of 13C3 in the brain homogenate of WT mice after 35, 70, and 140 nmol/kg intravenous (i.v.) injection showed low, albeit detectable, brain levels of around 0.1% of the plasma levels, consistent with what is generally reported for antibodies29 (Figure 2A). 70 nmol/kg represents 10 mg/kg as used in previous reports, but because the molecular weight of the TBTI constructs are higher than IgG, we used molarity concentration units in the present report to enable a direct comparison.
Figure 2.

Brain Exposure of 13C3 and TBTI1 in WT and APPmutxPS1M146L Transgenic Mice
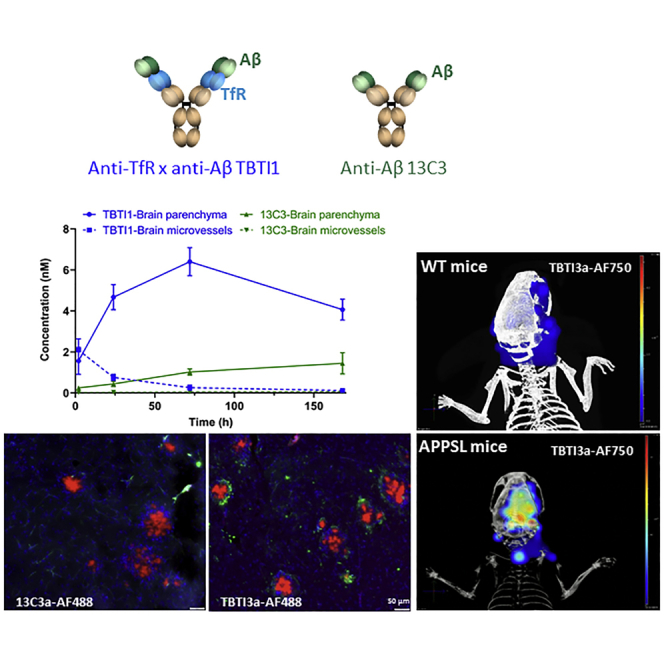
(A) Brain levels of 13C3 in WT mice. Brain levels of 13C3 were determined by specific immunoassay from 5 min to 168 h after i.v. 13C3 administration at 35, 70, and 140 nmol/kg. Values depicted are means ± SD (n = 3 per time point). (B) Time course of TBTI1 and 13C3 in the brain parenchyma and brain microvessels of APPmutxPS1M146L transgenic mice after i.v. administration at 70 nmol/kg. The brain microvessels were separated from brain parenchyma by using a dextran 70-based capillary depletion method. The amount of TBTI1 and 13C3 was measured in capillary and parenchymal fractions from 2 to 168 h after injection using a specific immunoassay with electrochemiluminescent detection. Values depicted are means ± SD (n = 3 per time point).
Engineering TBTIs Bispecific for Aβ and TfR
8D3, a murine IgG1,28,30 was selected as the anti-TfR antibody. This high affinity antibody recognizing an extracellular epitope of the mouse TfR, non-competitive with the endogenous substrate transferrin, has been widely used in several brain enhancement approaches,9 enabling a more direct comparison of the different formats. TBTI proteins were produced by transient transfection in HEK293 cells. TBTIs 1 and 2, carrying the anti-TfR paratope at the internal (Figure 1H) and external (Figure 1I) position, respectively, confirmed the important positional effect on the affinity for the target. TBTI2 (TBTI_8D3x13C3) with TfR at the external position showed a 45-fold higher apparent affinity for TfR than its counterpart, TBTI1 (TBTI_13C3x8D3) with TfR on the internal position (Figure 1H). Of note, the first, TBTI_8D3x13C3 had a 10-fold higher affinity to TfR than the starting 8D3 itself (Table 1), showing that the format has a strong influence on apparent binding affinity. Similarly, the affinities for Aβ followed the same rule, with TBTI bearing the anti Aβ paratope on the external position (TBTI_13C3x8D3, TBTI1) being closer to the value of the initial 13C3.
Table 1.
Relative Affinities of Bispecific Anti-TfR/Anti-Aβ Antibodies (TBTIs) Engineered with 8D3 Paratope at the Internal and External Positions
| EC50 (nM) | 13C3 | 8D3 | TBTI_13C3x8D3 (TBTI1) | TBTI_8D3x13C3 (TBTI2) |
|---|---|---|---|---|
| Aβ | 0.229 | – | 0.647 | 1.833 |
| TfR | – | 3.28 | 15 | 0.33 |
Relative affinities (EC50s) for Aβ determined by ELISA with visible detection and for TfR by immunoassay with electrochemiluminescent detection compared to the monospecific anti-Aβ 13C3 and anti-TfR 8D3 mAbs.
In order to keep maximal affinity for the therapeutic paratope (Aβ in this case) and based on the assumption that lower TfR affinity would be a better start for optimization, we selected TBTI1 (TBTI_13C3x8D3) with TfR paratope in the internal position for further modulation of the TfR affinity.
Design of Mutations Lowering Affinity on a Mouse Anti-TfR Antibody and Engineering the Corresponding TBTIs
Mutations aimed at lowering the affinity of 8D3 for TfR have been previously reported by stochastic alanine scanning outside of CDRs23 or in CDR3.17 We used an approach driven by homology modeling, protein-protein docking, and in silico predictions of changes in both affinity and stability as described in the Materials and Methods. The employed approach must be considered as a coarse evaluation of the interaction potential of the residues in the CDRs. Notably, we do not claim that the docking poses realistically reflected Fab/TfR binding. Potential hotspots were identified at heavy chain (HC) positions 28, 30–32 (CDRH1), 52–54, 56, 59 (CDRH2), and 100–102 (CDRH3) or light chain (LC) positions CDR 3, 27, 28, 31 (CDRL1), 49, 52 (CDRL2), 66, 68, 69 (CDRL4), 92–94, and 96 (CDRL3; Figure 3). Out of these 24 residues, 15 were suggested by at least 2 docking poses. All residues were subjected to alanine scanning, while scanning of all-natural amino acids was performed at LC G68. Predicted loss in affinity by mutation did not necessarily concur with predicted loss in stability. Selection of mutations for experimental evaluation focused on residues with the most frequent or strongest reduction in affinity independent of the predicted change in stability. Furthermore, care was taken to include at least one residue per CDR. The final selection included HC mutations N31A, Y52A, Y53A, T100A, and S101A and LC mutations N31A, Y49A, Y92A, N93A, and G68I (see Table 2).
Figure 3.

Molecular Model of Anti-TfR 8D3 Antibody
Top view on the variable fragment of the Fab model represented as a ribbon diagram with CDRs and heavy and light chains depicted in cyan, blue, and brown, respectively, and including the sidechains of potential paratope residues considered for modulating TfR affinity in ball-and-stick representation. Carbon atoms are depicted in colors corresponding to the ribbon section, oxygen, nitrogen, and hydrogen atoms in red, blue, and light yellow, respectively. Non-polar hydrogen atoms are not represented.
Table 2.
Kinetic Constants of the TBTIs Mutated in Their Anti-TfR CDRs
| Protein Name | KD (nM) | Kon 104 (1/Ms) | Koff 10−4 (1/s) | Entry | Name |
|---|---|---|---|---|---|
| Anti-mTfRC 8D3 | 0.54 | 7.13 | 0.39 | 1 | |
| TBTI_13C3x8D3_mIgG1 | 2.83 | 3.53 | 0.99 | 2 | TBTI1 |
| TBTI_8D3x13C3_mIgG1 | 0.4 | 7.44 | 0.29 | 3 | TBTI2 |
| TBTI_13C3x8D3 LC_N31A_mIgG1 | 2.18 | 2.5 | 0.54 | 4 | |
| TBTI_13C3x8D3 LC_G68I_mIgG1 | 3.84 | 2.72 | 1.05 | 5 | |
| TBTI_13C3x8D3 LC_N93A_mIgG1 | 4.16 | 2.71 | 1.12 | 6 | |
| TBTI_13C3x8D3 LC_Y49A_mIgG1 | 4.46 | 2.21 | 0.99 | 7 | |
| TBTI_13C3x8D3 LC_Y92A_mIgG1 | 6.88 | 2.17 | 1.49 | 8 | TBTI3 |
| TBTI_13C3x8D3 HC_T100A_mIgG1 | 3.83 | 6.08 | 2.33 | 9 | |
| TBTI_13C3x8D3 HC_Y53A_mIgG1 | 4.25 | 2.92 | 1.24 | 10 | |
| TBTI_13C3x8D3 HC_N31A_mIgG1 | 6.61 | 2.4 | 1.59 | 11 | |
| TBTI_13C3x8D3 HC_S101A_mIgG1 | 8.96 | 2.88 | 2.58 | 12 | TBTI4 |
| TBTI_13C3x8D3 HC_Y52A_mIgG1 | 17.9 | 2.92 | 5.21 | 13 | |
| TBTI_13C3x8D3_HC_Y52A/LC_Y92A_mIgG1 | 29.2 | 2.52 | 7.34 | 14 | TBTI5 |
| TBTI_13C3x8D3_HC_S101A/LC_Y92A_mIgG1 | 6.94 | 5.14 | 3.57 | 15 | |
| TBTI_13C3x8D3_HC_N31A/LC_Y92A_mIgG1 | 8.9 | 6.21 | 5.53 | 16 | |
| TBTI_13C3x8D3_HC_Y52A/LC_Y49A_mIgG1 | 0.86 | 79.3 | 6.89 | 17 | |
| TBTI_13C3x8D3_HC_Y52A-S101A/LC_Y49A-Y92A-mIgG1 | 19.64 | 3.26 | 11.1 | 18 | TBTI6 |
The affinity of TBTI antibodies was calculated from kinetic measurements against mouse transferrin receptor protein by BLI. The bold formatting indicates the specific mutations.
In the TBTI 13C3x8D3 format, all single mutations reduced affinity by factors of up to 6-fold versus TBTI1 (4- to 33-fold compared to 8D3, the standard IgG format) (Table 2). Out of all LC mutants, Y92A showed an ∼2.5-fold loss in affinity, while S101A and Y52A HC mutations reduced affinity by about 3- and 6-fold, respectively. HC mutations increased KD mostly by changes in koff, while the affinity of LC mutants was due to about equal changes in koff and kon.
Based on these observations, double mutants were generated by a combination of mutated LC_Y92A with HC_Y52A, HC_S101A, or HC_N31A, leading to changes of KD up to 10-fold (54-fold compared to 8D3). A quadruple mutant HC_Y52A-S101A/LC_Y49A-Y92A combining the two strongest mutations of both HC and LC failed to show a further increase in KD. The resulting TBTIs are displayed in Table 2, along with their binding kinetic constants for TfR using bilayer interferometry (BLI) (Materials and Methods). We selected variants covering a range of TfR affinities (thereafter referred to as TBTI3–6) for further studies.
Pharmacokinetics (PKs) of 13C3 and the Selected TBTIs 1–6 in WT and Tg Mice
PKs of TBTIs in WT Mice
The selected TBTIs were evaluated for their PKs at the dose of 70 nmol/kg after single i.v. injection in WT mice that did not bear the antigen for 13C3 and therefore were not subject to target-mediated elimination and accumulation. Plasma, brain, and total brain levels were analyzed at several time points and the TBTI concentrations were measured.
The results of two time points, 24 h and 72 h, are shown in Table 3 (the full PK profiles can be found in Table S1). All mutant TBTIs demonstrated an increased brain exposure compared to 13C3, with the best one obtained with TBTI3, which showed a 13- and 7-fold enhancement of total brain and membrane unbound (parenchymal) brain exposure, respectively, at 24 h. However, TBTI3 plasma levels were much lower and decreased very significantly at 72 h as compared to 13C3, potentially explained by the moderate TfR affinity and distribution in different TfR-expressing tissues. This short plasma half-life might compromise the progressive blood-to-brain transport of TBTI3 over time. To that avail, TBTI6 also displayed a good enhancement of brain transport at both 24 h and 72 h while having plasma levels similar to the standard IgG1 13C3, in line with its much lower TfR affinity. It represented an overall PK profile different than TBTI3, and both were selected for additional characterization. Of note, there was no apparent correlation between TfR affinity and brain transport in the TBTI format. However, the lower TfR affinity constructs (TBTI5 and TBTI6) displayed the longer plasma half-lives, which could again reflect a much lower distribution in tissues other than brain.
Table 3.
Brain and Plasma Levels of TBTIs 1–6
| Brain (nM) |
Brain (nM) |
Plasma (nM) |
||||
|---|---|---|---|---|---|---|
| Total |
Parenchyma |
|||||
| Name | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h |
| 13C3 | 0.382 | 0.445 | 0.444 | 0.402 | 539 | 495 |
| TBTI1 | 4.42 | 0.646 | 2.095 | 0.5735 | 160 | 3.55 |
| TBTI2 | 4.12 | 1.55 | 1.72 | 0.94 | 95 | 25.1 |
| TBTI3 | 5.08 | 2.23 | 3 | 1.31 | 112 | 6.23 |
| TBTI4 | 2.61 | 2.24 | 1.55 | 1.79 | 513 | 341 |
| TBTI5 | 1.36 | 1.44 | ND | ND | 593 | 447 |
| TBTI6 | 3.61 | 2.05 | 2.91 | 1.89 | 618 | 452 |
Single i.v. injection in C57BL/6J mice (70 nmol/kg, n = 3 mice per group). ND, not determined; brain total, cortex homogenate; brain parenchyma, membrane-unbound fraction (supernatant).
PKs of TBTI1 in Amyloid Precursor Protein (APP)mutxPS1M146L Tg Mice
The brain enhancements shown above are purely TfR-mediated as the anti-Aβ paratope does not recognize any antigen in WT mice. PKs in Tg mice displaying amyloid plaques in the brain was next performed to fully assess the brain enhancement potential of these constructs. 10-month-old APPmutxPS1M146L mice31 (age when brain amyloid plaques are very well established) were injected with the same dose of TBTI1 and 13C3 parent IgG (70 nmol/kg) and brain exposures were assessed. In the in vivo context, once crossing the brain endothelial cell layer, the antibody can bind to its target protofibrillar and fibrillar Aβ in both perivascular and parenchymal spaces. As above, the removal of vessels-bound antibodies is important to quantify only the parenchymal content. However, in APPmutxPS1M146L mice, this could not be done with a simple high-speed centrifugation, as described previously in WT mice, since amyloid plaques would co-pellet with vessels and bring along the antibody construct that would have been bound to plaques. Therefore, a special capillary depletion method based on a dextran-70 density gradient was used to efficiently separate the vessels’ fraction from the brain parenchyma as described previously.32 In addition, an acid treatment was then applied to the preparation to dissociate antigen/antibody complexes and precisely determine the total (bound and unbound) quantity of the antibodies in each compartment. The brain parenchyma amount of both TBTI1 and 13C3 increased with time in APPmutxPS1M146L mice (Figure 2B). The quantities of TBTI1 in the brain parenchyma were 11- and 6-fold higher than those of 13C3 at 24 h and 72 h, respectively. Of note, while in WT mice TBTI1 levels were decreasing at 72 h (compared to 24 h), they were increasing in Tg mice. In contrast the plasma levels of both 13C3 and TBTI1 remained largely unmodified between WT and Tg mice, reflecting the fact that the antigen is mainly in the brain (Table S2). The progressive accumulation in parenchyma in Tg mice as compared to WT mice is in line with the presence of the antigen in Tgs trapping the antibodies. Therefore, the TBTI increase in brain penetration observed in WT mice was further enhanced in Tg mice bearing the antigen. Interestingly, levels of capillary TBTI1, which represented over 50% of the total brain amount at 2 h, decreased with time and accounted for less than 2.5% of the brain’s amount of TBTI1 at 168 h. The capillary level of 13C3 was very low and only represented 5%–10% of the total brain amount of the antibody at all time points (Figure 2B).
Target Engagement in Brain by Imaging
Retention of AF750-Labeled TBTI3a in the Brain of APPSL Mice, Demonstrated by Repeated In Vivo Fluorescence Trans-illumination Technology (FLIT) Bio-imaging Sessions
The differential brain accumulation of TBTI1 in APPmutxPS1M146L31 mice brains versus WT mice observed in the above experiments was further evidenced with the single Tg APPSL mice using in vivo fluorescence imaging. A tomographic fluorescence study was performed concomitantly with the trans-illumination protocol using an X-ray scanning of the whole body (computed tomography [CT]) to reconstruct the topography of the animal. For all in vivo studies (except for single i.v. PK studies), low effector function TBTIs were used and were produced by mutation of the glycosylation site asparagine 289 into alanine33 (referenced as TBTIa). TBTI3a with rapid high brain uptake and short plasma half-life was selected for this study to allow clear differentiation between blood and brain fluorescence. An i.v. injection of TBTI3a-AF750 antibody solution at a dose of 57 nmol/kg was performed in WT and APPSL mice and the fluorescence levels were measured in two regions of interest: one in the extra-cerebral area and another in the intra-cranial part, corresponding to the brain region at different times 24 h before (baseline) and 3, 72, and 168 h after injection. At the last time point, the fluorescence of the antibody was retained in the brain of APPSL mice when it had already disappeared from blood and brain of the WT mice (Figures 4A–4E). Fluorescence was seen in the region of interest (ROI) where the plaques were known to be present. Moreover, blood samples collected in animals before their awakening just after imaging at times 3 h, 72 h, and 168 h demonstrated that the fluorescence (flux in radiance efficiency) followed the same kinetic profiles as the in vivo imaging measures observed in the extra-cerebral part, with no difference between the WT and the Tg mouse strains (Figure S1).
Figure 4.

In Vivo Imaging of AF750-Labeled TBTI3 Retention in the Brain of APPSL Transgenic Mice
3D-reconstruction of the fluorescence signals detected in the head area in WT (A) and APPSL transgenic (B) mice 168 h following i.v. administration of the labeled fluorescent antibody TBTI3a-AF750 using the image of X-ray tomography and fluorescence trans-illumination technology (FLIT). (C) Representative 2D X-ray image of the intracerebral area and extra-cerebral area. Kinetic curves of fluorescent picomoles were detected in the intra-cerebral area (D) or extra-cerebral area (E) of WT and APPSL transgenic mice following i.v. administration of the labeled fluorescent antibody TBTI3a-AF750. The fluorescent molecule was detected by in vivo FLIT imaging procedure (four points of trans-illumination at emission filter: 800 nm; excitation filter: 745 nm) applied at 3, 72, and 168 h following TBTI3a-AF750 i.v. injection. Baseline represents auto-fluorescence levels (zero picomoles of AF750). Each symbol represents the mean (±SEM) of detected picomoles. n = 5 for the WT group and n = 3–5 for the APPSL group. Two-way analysis of variance with repeated factor on time and on ranked transformed values and post hoc Fisher’s test: ∗p < 0.05, ∗∗p < 0.01 for comparison between the WT and the APPSL transgenic mice.
Histology of APPSL Mice Treated with AF488-Labeled TBTIs 3 and 6
Ex vivo fluorescent imaging was also used to determine if AF488-labeled anti-Aβ constructs, when engaging with or without TfR on the BBB, localizes differently in APPSL mice, which were given two 20 mg/kg i.v. doses and sacrificed 72 h after the second administration. Fluorescence emission of administered antibodies over brain tissue sections allowed us to evidence different brain uptake and penetration. No background fluorescence was detectable on tissue sections of the animal injected with PBS (data not shown). Green fluorescence signals emitted by negative control IgG1-AF488 and 13C3a-AF488 antibodies were limited and mainly restricted to cerebral vasculature structures, while, for the two bispecific TBTI 3a and 6a antibodies, a strong signal was detectable over parenchyma as illustrated for the thalamic subarea (Figures 5A–5D), even if some green signal could still be detectable in endothelial cells and/or vasculature (see Figure S2 for TBTI3-AF488 antibody). The parenchymal penetration of TBTI3a-AF488 antibody was particularly high in the thalamus and cortex, two brain areas well characterized for Aβ peptide deposition in this animal model. The pattern is quite similar to classical parenchymal Aβ deposits, as exemplified by Aβ immunolabeling of nearby brain sections (see illustrations of thalamic and cortical subareas in Figures 5E–5J). In additional in vivo experiments, multi-fluorescence imaging, with simultaneous detection of administered antibodies and Angiospark vascular staining34 and/or Congo red35 positive staining on brain sections, demonstrated high localization of 13C3a-AF488 antibody in green, with pink color of vasculature within sites deficient in BBB, such as the circumventricular area (Figure 5K) and, occasionally, in parenchyma at close proximity of vascular amyloid deposits. There was limited evidence that this antibody accessed parenchymal Aβ deposits (Figure 5L). On the opposite side, bispecific TBTI3a antibody frequently colocalized with amyloid plaques, stained with Congo red (Figures 5M and 5N). Overall, these data confirm that bispecific TBTIs efficiently decorated the parenchymal amyloid deposits in vivo, reflecting direct target engagement.
Figure 5.

Brain Uptake and Distribution of AF-488-Labeled Antibodies in Old APPSL Transgenic Mice
Representative images of single green fluorescent of AF-488-labeled administered antibodies (two i.v. injections at 20 mg/kg) or multi-fluorescent emissions over coronal brain sections (counterstained or not with DAPI) of APPSL mice 72 h after second i.v. injection. Green fluorescent signals were restricted to vascular structures with control IgG1-AF488 antibody (A) and anti-Aβ 13C3a-AF488 antibody (B) and predominantly detectable within parenchyma with TBTI3a-AF488 (C) and TBTI6a-AF488 (D). TBTI3a-AF488 administered antibody (E–G) accumulates in parenchymal Aβ deposits as evidenced by comparative analysis of Aβ 4G8 immunolabelling (H–J) over the entire section or both thalamic (single asterisk) and cortical (double asterisks) subareas. Multi-fluorescence emissions confirmed the 13C3a-AF488 green signal was mainly concentrated in vasculature structures of circumventricular areas and parenchymal vessels also stained with Angiospark positive fluorescent signal (pink) (K) and absent over or at proximity of parenchymal Congo red-positive deposits (L). With TBTI3a-AF488 (M and N), marked green signals are surrounding thalamic parenchymal Congo red amyloid deposits, indicative of enhanced brain penetration efficacy as compared to 13C3a administered antibody. The high density of nuclei stained in blue with DAPI around parenchymal Congo red deposits is indicative of characteristic inflammatory response associated with the cerebral Aβ peptide deposition process (L and N). Scale bars, 100 μm (A–D, G, and J) or 50 μm (K–N, F, and I).
Efficacy in a Tg Amyloid Model of AD
Based on the significantly different PK profiles of TBTI3a and TBTI6a, both were next evaluated in chronic treatment in APPSL Tg mice compared to 13C3a antibody to analyze efficacy at prevention of brain Aβ deposition. An anti-IgG1 antibody that recognizes a non-mammalian antigen was also administered as negative control (Ctrl-IgG1 group). Dosing (once weekly intraperitoneally [i.p.] for 4 months) was initiated at 3 months of age when the onset of amyloid deposition in such Tg line is demonstrated.31 In these conditions, 70 nmol/kg of 13C3a decreased Aβ deposition, measured biochemically and by immunohistochemistry (IHC), which was in line with previous results.27 Because of the higher brain penetration of the two TBTIs, a lower dose, 15 nmol/kg (5-fold lower), was selected. After 4 months of treatment, blood, brain, spleens, and livers were collected after transcardial perfusion.
Evaluation of TBTI3a, TBTI6a, and 13C3a Antibodies Treatment on the Level of Aß38, Aß40, and Aß42 in APPSL Mouse Brain
As expected, no Aβ38, Aβ40, and Aβ42 could be detected in the cortex of WT mice (6E10 antibody of the Meso Scale Discovery kit used is human specific). In the cortex of APPSL, 13C3a, TBTI3a, and TBTI6a reduced total brain Aβ peptides (three sub forms analyzed in guanidine extract to fully solubilize aggregated Aβ) significantly, between −53% to −67%, when compared to the control IgG1 group (Figures 6A–6C). No significant difference could be evidenced between the three antibodies, suggesting that TBTI3a and TBTI6a antibodies used at a five times lower dose (15 nmol/kg) are as effective at decreasing Aβ levels in the cortex as the 13C3a antibody at a dose of 70 nmol/kg. A further analysis of each total, insoluble and soluble fractions for Aβ38, 40, and 42, was also performed on the cortical samples from TBTI6a-treated mice in comparison with 13C3a, confirming the effect in all three fractions (data not shown). The results were less marked in the hippocampus. No significant decrease of the minor Aβ38 peptide levels could be seen with any of the three antibodies 13C3a, TBTI3a, and TBTI6a in comparison to the control IgG1 group (Figures 6D–6F). A significant decrease of Aβ40 peptide level was only observed with 13C3a antibody treatment, probably due to high dispersion of the individual data in TBTI3a- and TBTI6a-treated hippocampi (Figures 6E and 6F). This dispersion was consistent across all peptides for all treated mice, i.e., the same mice displayed high or low levels for all three peptides. A trend for the decrease could be seen with TBTI6a, while a non-significant increase of 41% of the Aβ42 level was observed with TBTI3a treatment (Figure 6F).
Figure 6.

Effect of 13C3a and TBTI Antibodies on Brain Aβ Levels in APPSL Mice
Quantification of Aβ38, Aβ40, and Aβ42 in cortical (A–C) and hippocampal (D–F) homogenates of APPSL mice after chronic administration of Ctrl IgG1 (70 nmol/kg), anti-Aβ (13C3a, 70 nmol/kg), or bispecific anti-Aβ/anti-TfR antibodies (TBTI3a and TBTI6a, 15 nmol/kg). Data are means ± SEM. % calculated as means vs Ctrl IgG1. Statistics: one-way ANOVA followed by Newman-Keuls post hoc analysis on Log-transformed data. ∗∗p < 0.01, ∗∗∗p < 0.01 versus Ctrl IgG1, ° p < 0.05 versus 13C3a. No other significant differences between 13C3a and TBTI3a or TBTI6a.
Efficacy and Target Engagement Using Immunohistochemical Markers
Using quantitative image analysis of cortical and hippocampal Aβ peptide immunolabeling, significant preventive activities of all three administered antibodies (ranging from −28% to −63%) against total positive staining surface or total positive deposit number were also demonstrated (Figure 7). In the cortex, TBTI6a had a similar efficacy as 13C3a, while TBTI3a appeared less effective, but there was no statistically significant difference between TBTI3a and TBTI6a (Figures 7A and 7B). The results in the hippocampus confirmed the trends seen with biochemistry, with only TBTI6a and 13C3a showing marked similar significant protective activity, ranging from −51% to −63%, against Aβ deposition (total surface and deposit number) (Figures 7C and 7D). Surprisingly, there was even a trend for TBTI3a to produce a higher hippocampal Aβ deposit number with small size (Figure 7D). Such high density of small deposits was not detectable in mice injected with Ctrl-IgG1 (Figure S3). We have no explanation for this observation and we haven’t seen a similar report in the literature.
Figure 7.

Aβ Load and IgG1 Immunostaining in the Cortex and Hippocampus of APPSL Mice Treated with 13C3a and TBTI Antibodies
Quantitative analysis of Aβ immunostaining (4G8) in the cortex and hippocampus of APPSL mice after chronic administration of anti-Aβ (13C3a, 70 nmol/kg) or bispecific anti-Aβ/anti-TfR antibodies (TBTI3a and TBTI6a, 15 nmol/kg). Total surface and deposit numbers in the cortex (A and B) and hippocampus (C and D) over 8 cortical rostro-caudal anatomical levels. Data are means ± SEM. % calculated as means vs Ctrl IgG1. Statistics: one-way ANOVA followed by Newman-Keuls post hoc analysis on raw data (for cortex, A and B) or on Log-transformed data (for hippocampus, C and D). ∗∗p < 0.01, ∗∗∗p < 0.001 versus Ctrl IgG1. °p < 0.05, °p < 0.001 versus 13C3a. Representative images of cortical and hippocampal subareas of sagittal hemibrain sections immunostained with anti-IgG1 antibody and counterstained with Congo red of APPSL mice administered once a week for 4 months with Ctrl IgG1 (E), 13C3a (G), or bispecific anti-Aβ/anti-TfR (TBTI3a and TBTI6a) antibodies (I and K). Higher magnification views within cortical fields evidenced that remaining parenchymal Congo red-positive deposits could be decorated with IgG1 immunostaining, indicative that 13C3a (H), TBTI3a (J), and TBTI6a (L) reach their target. As expected, no IgG1 labeling was detected over Congo red-positive deposits after chronic treatment with Ctrl IgG1 antibody (F). Scale bar in the cortical insets, 50 μm.
IHC with IgG1 Detection and Congo Red Staining in Brains of APPSL Mice Treated with 13C3a and TBTIs 3a and 6a
Qualitative image analysis of IgG1 IHC/Congo red double staining evidenced that, after 4-month treatment with anti-Aβ or bispecific antibodies, remaining cortical and hippocampal congophilic plaques in the brain of APPSL mice were decorated with IgG1 immunolabeling (Figures 7E–7L). This was indicative of antibodies’ brain penetration and access to their target. A higher intensity of IgG1 labeling (brown staining) on congophilic Aβ deposits (pink color) could be observed with 13C3a compared to TBTI6a and TBTI3a (administered at a 5-fold lower dose). Of note, as expected, no IgG1 labeling over Congo red parenchymal deposits could be observed in control IgG1-treated animals.
Preliminary Safety Analysis of APPSL Mice after a 4-Month Chronic Treatment with 13C3a and TBTIs 3a and 6a
All blood parameters were analyzed using flow cytometry and revealed slightly to moderately lower red blood cell count mass parameters along with moderately higher reticulocyte and neutrophil counts for mice treated with TBTI3a. No difference was observed for TBTI6a (full description in Table S3).
The spleens and livers of treated mice were also subjected to histopathological analysis. In the liver, there was minimal to mild centrilobular hypertrophy (increased size of hepatocytes) observed only in the Ctrl IgG1 (1/9 in APPSL mice and 2/11 in WT mice) and TBTI3a (5/9 animals) groups. The other liver changes were considered to be part of the background. The splenic changes were mostly observed with TBTI6a (consisting of a minimal decreased white pulp cellularity in 5/8 animals), 13C3a (consisting of a minimal decreased white pulp cellularity in 3/10 animals), and TBTI3a (consisting of a minimal or mild extramedullary hemopoiesis in 5/9 animals). All of these observed lesions were of minimal or mild severity (when the usual grading system is minimal-mild-moderate-marked-severe). They were considered of low concern, although TBTI3a seemed to show the most prominent lesions (full description in Table S4).
TFR Expression in Brain Endothelial and Neuronal Cells after Treatment with TBTIs
To evaluate the impact of anti-TfR antibody treatment on brain vascular and parenchymal expression of TfR, we measured TfR levels in the cell surface and whole cell fractions of both bEnd.3 cells and primary mouse cortical neurons following exposure to TBTIs for 24 h. The tested concentrations were chosen based on relevant TBTI plasmatic concentrations at 24 h and on maximal brain concentrations observed in mice following i.v. administration at 70 nmol/kg. In bEnd.3 cells, a significant downregulation of whole cell TfR levels was observed following treatment with the TfR high-affinity TBTI2a at both concentrations, while TBTI3a provoked a downregulation in both whole cell and cell surface fractions only for the highest concentration when compared to its control IgG1 (Figure 8A). On the other hand, the lower affinity TBTI6a did not modify either total or surface cellular TfR levels, regardless of the concentration tested (Figure 8A). In mouse primary neurons, we did not find any significant difference either in the whole cell TfR levels or in the cell surface TfR levels (Figure 8B) following the treatment with TBTIs compared to the control IgG1. These data suggest that, at the studied concentrations, high-affinity TBTIs, but not moderate-affinity TBTI6a, may lead to a decrease in TfR levels in the brain endothelium, while none of the TBTI antibodies modify the TfR levels in primary neurons at 5 nM.
Figure 8.

Effect of TBTI Antibodies on TfR Levels in Brain Endothelial Cells and Neurons
Expression of murine TfR in bEnd.3 cells (A) and in vitro cultured mouse primary cortical neurons (B) following exposure to TBTI2a, TBTI3a, and TBTI6a versus a control IgG1 for 24 h. TfR protein levels from whole cell lysates were normalized to α-tubulin and compared to control IgG1-treated cells, while TfR protein levels from cell surface lysates were normalized to N-cadherin and compared to control IgG1-treated cells. In both whole cell and surface cell lysates, protein levels were assessed by western blot. Bar graphs show means of each group (±SD); p values were obtained by one-way ANOVA with Dunnet’s multiple comparison test versus Ctrl IgG1 groups. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns p > 0.05.
However, PKs of TBTI3 did not evidence a decrease in brain to plasma ratio after repeated administration, suggesting that no downregulation occurred, even if the experiment was not totally conclusive as it was performed at different doses and routes of administration (i.v. versus i.p.) (Table S5).
Discussion
Enhancement of antibody and/or biotherapeutics brain exposure using TfR-mediated transcytosis has been largely described.9 In the present study, we have investigated the potential of a series of TBTIs, recognizing, on one hand, the protofibrillar Aβ peptide and, on the other hand, TfR to enhance brain exposure of the anti-Aβ antibody. This format allows divalent binding to each of the two antigens, which can be useful when higher avidity is required. In addition, this format is fully symmetrical (only two polypeptidic chains), which should improve productivity at scale. The murine TfR-recognizing antibody 8D328 has been widely used and was selected as the starting point to allow direct comparison. Two different bispecific constructs, TBTI1 and TBTI2, were designed bearing the WT TfR paratopes in the internal and external positions of the TBTI, respectively. This simple positional change already conferred largely different TfR binding properties, with TBTI2 having 45-fold lower half maximal binding concentrations (EC50) using ELISA and 7-fold tighter KD using BLI compared to TBTI1. Positional effect has already been observed for this format and has been linked to steric hindrance of the paratopes in the internal position of the construct.19 A few recent studies have shown an inverse correlation between TfR binding affinity and brain exposure when anti-TfR antibody bispecific constructs were administered at therapeutic doses. Yu et al.22 suggested that the dissociation of the antibody from the receptor after receptor-mediated transcytosis will be more likely with a lower-affinity antibody, resulting in higher transfer in the brain parenchyma. It was later shown that this brain accumulation is linked to TfR-dependent cell trafficking.36 In our study, both TBTI1 and TBTI2 showed increased brain exposure (24 h and 72 h) compared to the control monospecific anti-Aβ (13C3) antibody. Surprisingly, despite their differences in TfR binding affinity, both brain and plasma exposures of TBTI1 and TBTI2 were roughly similar at the dose of 70 nmol/kg. Several hypotheses might explain the absence of correlation between TfR binding affinity and brain/plasma exposure between TBTI1 and TBTI2. Both constructs have KDs lower than 5 nM and are in the same range as high affinity anti-TfR antibodies described previously.17,22,37 It is possible that the difference in TfR affinity might not be large enough to modify the subcellular pathways and thus transcytosis of these constructs. However, recent examples of brain exposure enhancements of bispecific anti-TfR antibodies linked to TfR affinities in the same ranges19,22 are challenging this explanation. It’s probable that factors other than TfR affinity, such as conformation or topography of the paratopes in TBTI format, might also have an impact on the transcytosis of bivalent anti-TfR antibodies in brain endothelial cells. Indeed, Hanzatian et al.19 suggested that a difference in the TfR binding conformation could explain the lack of correlation between the TfR affinity and brain exposure of some of their Aβ/TfR-binding DVDs (DVD IgGs). To explain lower affinity and higher brain exposure of their bivalent anti-TfR antibody construct, Hultqvist et al.14 hypothesized that conformational hindrance might prevent interaction with two different TfRs. In our study, the different positions of anti-TfR variable domains between TBTI1 and TBTI2 might impact their binding modes or dimerization pattern with TfR at the cell membrane.15 Further studies are needed to clarify this point, but, overall, current data underline the important impact of antibodies’ structure on their brain and plasma PKs. Based on similar PK properties for the two constructs, we explored further the TBTI1 format because it had minimal impact on the binding properties of the payload paratope (here 13C3).
To further decrease the TfR binding affinities of the constructs, we used homology modeling to design single mutations in the CDR’s LCs or HCs of TBTI1 (entries 4–13). The most effective mutations were combined in both LCs and HCs (entries 14–18), leading to TBTIs with TfR binding affinities spanning from 0.5 nM up to nearly 30 nM using BLI. It is difficult to compare these affinities with the ones previously reported for 8D3 variants since TfR binding measurements are quite sensitive to the technology and the protein source used. In our own studies, TBTI1 versus TBTI2 differ 7-fold using BLI and 45-fold using ELISA. Hanzatian et al.23 resorted to TfR-expressing cells for binding affinity determination of their variants (spanning from 0.12 to 13 nM) after observing that ELISA or surface plasmon resonance (SPR) assays gave artificially low or lack of binding for low affinity TfR antibodies. On the other hand, Webster et al.17 reported affinities using SPR (spanning from 1.2 to 610 nM) because the slow off-rate of the complex between WT 8D3 and murine TfR (mTfR) made it impossible to accurately determine kinetic parameters for this interaction using BLI.
Four TBTIs with a range of TfR affinities were selected for in vivo assessment of their brain and plasma profiles. The mutations in CDRs of anti-TfR variable domains resulted in increased brain exposure of TBTIs compared to the TBTI1. However, no clear correlation between TfR binding affinity and brain exposure was observed. For example, TBTI3, the tighter antibody (KD = 6.88 nM), had the highest total brain concentration at 24 h, but like TBTI1, concentration decreased at 72 h in the brain and more dramatically in plasma. On the contrary, the lower affinity antibodies TBTI4 and TBTI6 had slightly lower brain exposure at 24 h, but their brain concentration was better maintained, displaying even higher brain concentrations at 72 h than TBTI3, possibly due to a longer plasma half-life enabling continuous entry in the brain over time.
TBTI5, which displayed the lowest binding affinity (KD = 30 nM), did not show further enhancement in brain exposure compared to TBTI6, suggesting that the optimal range of affinity might be outran. In agreement with our findings, in some recent studies, the lowest affinity for bivalent anti-TfR antibodies did not always lead to the highest brain penetration.17,19,37 Even though the binding epitopes are not the same, and thus the KD ranges are different between studies, current data indicate that, for bivalent anti-TfR antibodies, the correlation between antibody brain exposure and TfR binding affinity can be bell-shaped and the anti-TfR binding affinity needs to be very fine-tuned to obtain the optimal brain exposure profile. The latter has to be defined by the therapeutic objectives of the intended construct, whether it needs to attain a high brain parenchymal concentration to be effective (Cmax-dependent) or must be maintained at a steady level over time in the brain (area under the curve [AUC] dependent).
In this study, deep brain penetration of our anti-TfR TBTIs was demonstrated with two methodologies. First, high-speed centrifugation was used to determine the antibody concentration in cell-free supernatant as described previously.15,19,22,37 However, this procedure presents some limitations. Capillary-bound antibodies cannot be separated from parenchymal cell-bound antibodies so that the total parenchymal concentration of highly bound antibodies could be underestimated. In addition, the sample treatment procedure could allow free diffusion of the antibodies from capillary and brain cells to the supernatant, making the brain quantification imperfect. These difficulties could be partially overcome by using the capillary depletion technique,32 allowing measurement of the amount of tested molecule in the brain parenchyma as well as in the capillary fraction.15,38 However, the capillary depletion has limitations as well, since, after the separation step, small levels of contaminating brain vessels are still present in the brain parenchyma fraction.39 This might lead to an overestimation of the parenchymal concentration of the studied molecule.
Fortunately, recent studies suggest that the brain penetration of antibodies can be more readily demonstrated in case of the presence of intraparenchymal targets.40, 41, 42 Thus, we tested TBTI1 and parent monospecific 13C3 in 10-month old APPmutxPS1M146L mice in which amyloid plaques were well established. In this situation, the anti-Aβ antibodies, once they have crossed the BBB, will strongly bind to amyloid plaques and Aβ protofibrils in brain parenchyma and will be retained in the brain (target mediated) longer than in WT animals, which are devoid of antigen. Using the capillary depletion method to separate the brain parenchyma from brain vessels as well as an acid washing step to dissociate antibodies from their antigen (in capillary or plaques), over 50% of TBTI1 was found in the brain capillary fraction at an early time point. However, it decreased rapidly while brain parenchyma levels of both TBTI1 and 13C3 increased strongly in later time points. At 168 h, only less than 2.5% of brain TBTI1 was found in the capillary fraction. Even if the capillary depletion procedure might have left a small quantity of vessels in the brain parenchyma fraction, the very low concentration of TBTI1 or 13C3 in brain capillary fraction at this later time point made the contamination from capillary fraction negligible. These data clearly evidenced that the brain amount of TBTI1 and 13C3 in APPmutxPS1M146L mice at extended time points was mostly parenchymal.
The brain parenchyma penetration and brain target engagement of TBTI3a and TBTI6a was demonstrated directly using two orthogonal imaging techniques in another model bearing amyloid plaques, APPSL mice. First, in-life fluorescence analysis of TBTI3 showed brain retention in Tg versus WT mice in the area of the brain known to harvest plaque, such as the frontal cortex. Second, immunohistochemical evaluation evidenced only TBTI3a and TBTI6a antibodies, but not irrelevant 13C3a or IgG1 control antibodies, colocalized with Aβ deposits in different brain structures, including the cortex and hippocampus (13C3a and the control IgG1 antibodies were mostly restricted to the brain vessels).
Even though we have previously shown that 13C3a can be detected on amyloid plaques after chronic exposure,27 the faster and more pronounced brain penetration of the TBTI formats predicts higher efficacy at preventing amyloid plaque accumulation. To confirm pharmacodynamic activity, both TBTI3a and TBTI6a, which have distinct brain and plasma PK profiles, were evaluated in chronic treatment in a mouse model in comparison to the monospecific 13C3a and control IgG1 antibodies. The APPSL mouse model was selected, as previously reported, for 13C3 and the doses were adjusted to 3 mg/kg for the TBTIs corresponding to a dose of 13C3 inactive in biochemical Aβ evaluation.27 Consistent with the increase in brain exposure, both TBTI3a and TBTI6a significantly reduced the cortical amount of Aβ and cortical Aβ deposition to the same extent as the higher dose of 13C3a. To our knowledge, this is the first report of anti-TfR-anti-Aβ antibodies reducing both Aβ plaques and the cerebral pool of Aβ peptides at a lower dose and at the same administration frequency as the control monospecific antibody. Tetravalent bispecific format had been shown to bind to brain amyloid plaques of APPswe mice after a 3-month treatment, but brain exposure enhancement was not quantified and efficacy could only be demonstrated after daily subcutaneous administrations (while monospecific anti-Aβ antibodies are generally administered weekly to show efficacy in such models).25 The “brain shuttle” monovalent for TfR when applied to an anti-Aβ antibody has demonstrated efficacy at a lower dose than the corresponding monospecific Gantenerumab at decreasing amyloid plaque load in a Tg mouse model, but an effect on the cerebral pool of Aβ has not been shown.15 Finally, the analogous DVD format has not demonstrated efficacy in an amyloid mouse model.19 Moreover, in the latter, an increase in brain Aβ was observed after acute treatment.23 In this case, the authors used the 3D6 anti-Aβ antibody, an antibody known to recognize both Aβ protofibrils and Aβ monomers with the same affinity.27 As there is a significant pool of Aβ monomers in the blood, the enhanced transport of the bispecific anti-TfR-anti-Aβ DVDs to the brain could potentially result in an increase in the transport of Aβ monomers from blood to brain.23,43 On the contrary, the monospecific anti-Aβ antibody (13C3a) used in our study has extremely low affinity for the Aβ monomers and much higher preference for Aβ protofibrils and plaques.27
TBTI6a reduced cortical and hippocampal Aβ deposition to the same level as 13C3a. Even though not statistically significant, TBTI3a was less effective than TBTI6a in reducing the cortical Aβ deposition, while no effect could be seen in the hippocampus. The reduced effect of TBTI3a compared to TBTI6a might be due to its higher clearance rate, especially in plasma. With a repeated weekly low-dose regimen, the accumulation factor of TBTI3a in APPSL mice is therefore smaller than for TBTI6a, as shown by their plasma exposures of 0.32 and 49.5 nM, respectively, 1 week after the last administration. However, the terminal brain exposures, 0.28 and 0.37 nM, respectively, did not confirm such accumulation. Another potential reason for the lower efficacy of TBTI3a could be a TfR downregulation linked to its higher affinity for the receptor. It has been shown that high affinity36 and/or avidity15 anti-TfR antibodies led to a dose-dependent reduction of TfR levels in vitro, yielding lower in vivo exposure after repeated administration. This was explained by different cell trafficking of low and high affinity constructs being sorted through the endosome and lysosome respectively. We also investigated the potential of our constructs to induce TfR downregulation. We selected TBTI2 as a TfR high affinity control. While no downregulation was observed in vitro on neurons with any of the constructs, TBTI2 led to a reduction in cell surface and total TfR content of brain endothelial cells at both low and high dose. Interestingly, TBTI3 displayed this effect only at the high concentration and TBTI6 had no effect on TfR levels at any of the tested concentrations. In vivo repeated administration of TBTI3, however, did not suggest downregulation.
Interestingly, contrary to previous claims,44 we obtained the pharmacodynamic effect of our TBTIs with no or strictly minimal effector function, as mutations have been made in the Fc fragment of the antibodies to inhibit their binding to the Fc gamma receptors (FcγRs). Our data confirmed previous findings, with the monospecific anti-Aβ antibody showing that the lack of effector function does not impact the beneficial effect of anti-Aβ antibodies.27 Without effector function, the bispecific anti-Aβ-anti-TfR TBTIs should be safer for use as there should be no or less first infusion reaction44 and lower occurrence of vasogenic edema and micro-hemorrhage associated with amyloid-related imaging abnormalities (ARIAs).45 Our toxicological studies in TBTI-treated mice confirmed the lack of serious adverse effects on both hematological parameters and organ histopathology after chronic treatment.
Overall, our efficacy studies confirmed the brain target engagement and the pharmacodynamic effect of the bispecific anti-TfR-anti-Aβ TBTIs. This could be very promising as, recently, a high dose of the anti-Aβ antibody, aducanumab,46 showed a modest but significant beneficial effect in AD patients in a phase 3 clinical trial and doses for several anti-Aβ antibodies in the clinic are steadily increased, as is the case for gantenerumab (R. Bateman, 2020, AAT-AD/PD 2020 Conference)47,48 or crenezumab, currently administered at 60 mg/Kg.49 The brain enhancement obtained with TBTI antibodies could allow us to reduce the effective dose and thus the cost of goods along with the well-known adverse effects of high-dose anti-Aβ antibodies.
It’s also worth noting that, in the current study, mouse IgG1 framework has been used while most studies have used human IgGs.15,19,22,37,41 Using human IgGs presents a technical advantage for measuring the concentration of studied constructs in animal tissues. However, human IgGs might have a non-negligible impact on the biodistribution of tested constructs in WT mice. First, it has been shown that human IgG1 have 10-fold higher affinity for mouse neonatal Fc receptor (FcRn) than mIgG1 at pH 6.0.50,51 As FcRn plays an important role in extending the half-life of Igs by lowering lysosomal degradation and increasing recycling to the plasma membrane, the plasma PK profile of human IgGs could be overestimated in WT mice.52 Moreover, FcRn is described as the only Fc receptor expressed in brain endothelial cells,53 and some recent studies suggested an involvement of FcRn in the transport of IgG at the BBB.52,54 Even though it’s not clear whether FcRn is involved in the efflux and/or influx of IgG at the BBB, the increased affinity of human IgG (hIgG) for FcRn at the BBB might have an unpredicted impact on the brain exposure of tested molecules in mice. Second, dosing of hIgG in WT mice could induce immunogenic response and generation of anti-drug antibodies.55 The latter could strongly impact the PK profiles of tested antibodies. It also limits the use of such antibodies in chronic efficacy studies unless a prior immunotolerance is induced using anti-CD4 antibodies.15,27,56 Even though this immunotolerance allows the treatment of mice with hIgGs, on its own, it could induce changes in the brain environment as well as pathological conditions in treated animals.57 Overall, current data suggest that precaution should be taken when interpreting the PK and pharmacodynamic data of hIgGs in mice.
In conclusion, our data show that anti-TfR bispecific antibodies of TBTI format can cross the BBB to enter the brain parenchyma. This transport is remarkably enhanced in the presence of a CNS target such as Aβ peptide. In addition, we demonstrate that two factors influence affinity of the TBTIs for TfR: the positional effect of the TfR paratope in the construct and specific mutations on the TfR paratope. Both factors have a direct impact on brain exposure. Brain exposure enhancement resulted in a higher potency of anti-TfR-anti-Aβ TBTI antibodies as a lower dose was sufficient to obtain therapeutic effects in the animal model. The TBTI technology will allow us to develop new biotherapeutics for neurologic diseases with better efficacy and less side effects.
Materials and Methods
Ethics
Experiments were performed at Sanofi in our Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility in full compliance with standards for the care and use of laboratory animals, according to the French and European Community (Directive 2010/63/EU) legislation. All procedures were approved by the local animal ethics committee (Ethical Committee on Animal Experimentation [CEEA] #24) and the French Ministry for Research.
Animals
All mice were produced and provided by Charles River (France). Tg mouse models (APPmutxPS1M146L and APPSL mice) that mimic histological and cognitive markers of AD were obtained by overexpressing APP transgenes bearing mutations linked to familial forms of AD in mice as described.31 After arrival, mice were housed individually in an enriched environment in a pathogen-free facility at a constant temperature of 22 ± 2°C and humidity (50 ± 10%) on a 12-h light/dark cycle with ad libitum access to food and water. For PK studies, 105 C57BL6/J and 24 APPmutxPS1M146L mice were used. 47 male APPSL and 15 male WT mice were used for the efficacy and animal imaging studies.
Design of Antibodies with Reduced Affinity for mTfR
The Maestro58 molecular modeling suite, released in 2015 by Schrödinger, was used for model visualization and design of antibodies with modulated affinity for mTfR, applying recommended standard procedures and parameter settings unless specified otherwise. More specifically, homology models of antibody 8D3, mTfR, and mTf were built by PRIME59 using protein sequences from patent US2010077498, Uniprot: Q62351 and Q921I1, and PDB: 4IJD and 3S9L as templates. The homology models were optimized by PRIME, applying loop prediction for non-template loops and side chain prediction for all residues, followed by minimization. The Fab model was docked to the receptor chains in the model of the heterotetramer (TfR/Tf)2 in BioLuminate.60 Six out of 30 poses were retained by visual inspection for side chain optimization at the Fab/receptor interface (PRIME, options SAMPLE_BACKBONE yes, BACKBONE_LEN 3) to remediate steric clashes. Based on these 6 optimized poses, 23 CDR positions were selected for alanine scanning plus complete residue scanning of LC G68. A consensus evaluation of the predictions on both affinity and stability was deduced for residues implicated in multiple poses, and at least one residue per CDR was selected for experimental testing.
Characterization of TBTIs 1 to 6
TBTI proteins were produced with two different processes: transiently transfected HEK293 cells and stable Chinese hamster ovary (CHO).
For transient transfection of HEK293 cells, TBTIs were encoded by mono-cistronic plasmids. Coding sequences were obtained by gene synthesis and codon usage was optimized for Homo sapiens (GeneArt). HEK293 cells were transfected using 293Fectin as a transfection reagent. Then transfected cells were cultivated in FreeStyle 293 Expression Medium up to 1.5 L. After 7 days of culture, cells were removed by centrifugation and cell supernatants were filtered through a 0.22-μm membrane.
For stable CHO process, TBTIs were encoded by bi-cistronic plasmids. Coding sequences were obtained by gene synthesis and codon usage was optimized for Cricetulus griseus organism (GeneArt). Sanofi’s proprietary CHO cell line was electroporated with the plasmid and cells were grown in CD OptiCHO medium supplemented with FeedB (Gibco) and the selection marker. Once cells recovered a cell viability of 90%, scaling up was performed until a volume of 1.5 L. At a cell viability of 70% (12–14 days), cells were removed by centrifugation and cell supernatants were filtered on a 0.22-μm membrane. Cell supernatants were stored at 2°C–8°C until purification.
Clarified supernatants were applied on Protein A columns equilibrated with Dulbecco’s PBS (DPBS) buffer. TBTI proteins were eluted with an acidic pH buffer (2.8), neutralized, and filtered on a 0.22-μm membrane. Then they were applied on Superdex 200 columns to remove high molecular weight products and concentrated using Centriprep Centrifugal Filter units. The protein batches were stored at 2°C–8°C and a certificate of analysis was generated (liquid chromatography-tandem mass spectrometry [LC-MS], purity, super elongation complex [SEC], endotoxin dosage, and sterility test).
For single-dose i.v. bolus PK studies, glycosylated TBTIs were used. For all other in vivo studies, particularly when a chronic treatment was needed, low effector function TBTIs were produced by mutation of the glycosylation site asparagine 289 into alanine (named TBTIa).
KD Measurement by BLI
The affinity of one or more antibodies was calculated from kinetic measurements against mouse TfR protein using the Octet QKe device from ForteBio. Kinetic parameters (kon, koff, and KD) determination allows the ranking of variant antibodies for associating and/or dissociating to its antigen. In such a kinetic analysis, anti-mouse IgG Fc capture (AMC) biosensors (18-5088, ForteBio) were incubated with variant antibodies diluted in DPBS 1× at 50 nM. Then the biosensors were soaked in a concentration range of mTfR (50741-M07H, Sino Biological) for 300 s and the association rate was measured. Finally, the biosensors were soaked in DPBS 1× for 300 s to measure the dissociation rate. Raw data analysis, including blank subtraction, fitting, and kinetic parameters calculations, were performed with ForteBio Data Analysis 10.0 software.
Relative Affinities of the Bispecific Antibodies for mTfR
The relative affinity of bispecific antibodies for mTfR (EC50) was estimated from immunoassays against the extracellular domain of mouse TfR protein. For these assays, the purified extracellular domain of mTfR (Evotec, Toulouse, France) in PBS was added to the plate (CLS3601, Corning, Sigma) at a concentration of 5 μg/mL and incubated for 1 h at room temperature. Plates were then washed three times with PBS-Tween 0.05% (524653, Calbiochem) and blocked for 1 h at room temperature with 0.1% BSA solution (A7030, Sigma). A titration of each individual bispecific anti-mTfR/brain antigen (anti-mTfR/Aβ) antibody was added to the plate and incubated for 2 h at room temperature. Plates were then washed three times with PBS-Tween 0.05% and a Sulfo-Tag labeled goat anti-mouse IgG (R32AC-1, Meso Scale Discovery) was added and incubated for 1 h at room temperature. Plates were washed three times with PBS-Tween 0.05% and antibody bound to the plate was detected with tripropylamine (TPA) containing read buffer (R92TC-2, Meso Scale Discovery). EC50s were generated using GraphPad Prism version 8.0.2 for Windows (GraphPad Software, San Diego, CA, USA).
Relative Affinities of the Bispecific Antibodies for Aβ
The relative affinity of bispecific antibodies for Aβ (EC50) was estimated from ELISA measurements against Aβ protofibrils. Briefly, the purified Aβ protofibrils (Sanofi, Vitry, France) in PBS were added to the plate (353279, BD Falcon) at a concentration of 1 μg/mL and incubated overnight at 4°C. Plates were then washed three times with TBS-BSA 0.1% (A2153 Sigma)-Tween 0.1% (330195000, Acros Organics) and blocked for 1 h at room temperature with TBS-1% BSA solution. A titration of each TBTI or the monospecific 13C3 was added to the plate and incubated for 1 h at room temperature. Plates were then washed three times with TBS-BSA 0.1%-Tween 0.1% and goat anti-mouse IgG HRP-linked antibody (A0168, Sigma) was added and incubated for 1 h at room temperature. Plates were washed three times with TBS-BSA 0.1%-Tween 0.1% and antibody bound to the plate was detected with tetramethylbenzidine (TMB) peroxidase substrate (T0440, Sigma). EC50s were generated using Biost@t Speed-LTS V2.3PROD (Sanofi).
PKs of TBTIs and Control Antibodies in WT and Tg Mice
TBTIs or control monospecific antibody (13C3) were administered to WT or Tg mice at a dose of 70 nmol/kg. Blood samples were collected in heparinized tubes from posterior vena cava at 5 min and at 2, 5, 24, 72, and, for some molecules, 168 h after single bolus i.v. administration, and plasma was obtained by centrifugation for 10 min at 1,500 g. Immediately after blood sampling, mice were perfused via the left ventricle with 50 mL of ice-cold PBS (524560, Calbiochem, San Diego, CA). The mouse brain cortex was harvested, frozen immediately on dry ice, and stored at −80°C until use.
Quantification of Antibodies in PK Studies
Plasma Antibody Quantification
For plasma concentration determination, standard 96-well sector plates (L15XA-1, Meso Scale Discovery) were coated with 0.5 μg/mL of purified extracellular domain of mTfR (50741-M07H, Sinobiological) or 0.5 μg/mL of purified human Aβ peptide (H-1368, Bachem) in PBS and then incubated for 1 h under agitation at room temperature. After incubation, the plates were washed three times with PBS-Tween 0.05% (524653, Calbiochem) and blocked for 1 h at room temperature with 0.1% BSA solution (A7030, Sigma). After blocking the plates, diluted plasma samples as well as quality controls (QCs) and standards were incubated on plates for 2 h at room temperature. Standards and QCs were prepared using purified antibody spiked in buffer containing the same amount of control plasma as unknown plasma samples. After incubation, plates were washed three times with PBS-Tween 0.05%, and bound antibody was detected with SULFO-TAG conjugated goat anti-mouse antibody (R32AC-1, Meso Scale Discovery) using TPA containing read buffer (R92TC-2, Meso Scale Discovery). Concentrations were determined from standard curve using a four-parameter non-linear regression program (Discovery Workbench version 4.0 software).
Brain Quantification of Antibodies
Samples Treatment for Membrane Unbound Brain Quantification
For the brain quantification of free antibodies, mouse brain samples were homogenized at 4°C in five volumes of 1% NP-40 supplemented with protease inhibitor cocktail (88666, Pierce) using Precellys bead homogenizer coupled to a Cryolys cooling system (Bertin Instruments). Homogenized brain samples were rotated at about 4°C for 1 h before spinning at 20,000 g for 20 min. The supernatant was collected for brain antibody quantification.
Samples Treatment for Total Brain Quantification
For the quantification of total brain antibodies, the mouse brain cortex was first homogenized as described above. Standards and QCs were then prepared using purified antibody spiked in buffer containing the same amount of control brain homogenate as unknown brain homogenate samples. Standards, QCs, and brain homogenate samples were treated for 20 min at room temperature with 9 volumes of 0.2 M glycine (G8790, Sigma) buffer adjusted to pH 3.5 with 1 M HCl solution (H3162, Sigma). After the acid treatment, brain samples, standards, and QCs were centrifuged at 20,000 g for 20 min at 4°C. The supernatants were then harvested and neutralized with 1 M Tris HCl solution (T3253, Sigma) at pH 9.0 for brain antibody quantification.
Sample Treatment for Antibody Quantification in Mouse Brain Microvessels and Mouse Brain Parenchyma
The pre-weighted hemisphere was transferred to pre-chilled glass homogenizer and homogenized (20 up-and-down strokes of the pestle) in 5 volumes of ice-cold Krebs-bicarbonate buffer. The homogenate was then transferred to a pre-weighted 5 mL tube and ice-cold 26% dextran solution (Sigma, MW: 70,000) was added to obtain a final dextran concentration of 16%. The homogenate was mixed well in dextran solution and then centrifuged at 4,800 g for 20 min at 4°C in a swinging-bucket rotor. The capillary-depleted supernatant was removed from the vasculature-containing pellet and harvested in a 5 mL Eppendorf tube for brain parenchymal antibody quantification. The weighted microvessel pellet was incubated overnight in 9 volumes of radioimmunoprecipitation assay (RIPA) lysis buffer (R0278, Sigma) containing EDTA-free protease inhibitor tablets at 4°C. The microvessel suspension was further diluted in 4 volumes of PBS-Tween/BSA 0.1% buffer. Standards and QCs were prepared using purified antibody spiked in buffer containing the same amount of control capillary-depleted brain homogenate or microvessel suspension.
The brain parenchymal supernatant, microvessel suspension, standards, and QCs were then treated for 20 min at room temperature with 9 volumes of 0.2 M glycine (G8790, Sigma) buffer at pH 3.5. After the acid treatment, unknown brain samples, standards and QCs were centrifuged at 20,000 g for 20 min at 4°C. The supernatants were then harvested and neutralized with 1 M Tris HCl solution (T3253, Sigma) at pH 9.0 for antibody quantification.
Immunoassay Method
Standard 96-well sector plates (Meso Scale Discovery) were coated with 0.5 μg/mL of purified human Aβ peptide (H-1368, Bachem) or, for the TBTI2, 0.5 μg/mL of purified extracellular domain of mTfR (50741-M07H, Sinobiological) in PBS and then incubated for 1 h under agitation at room temperature. After incubation, the plates were washed three times with PBS-Tween 0.05% (Calbiochem, 524653) and blocked for 1 h at room temperature with 0.1% BSA solution (A7030, Sigma). After blocking the plates, brain supernatant samples as well as QCs and standards were incubated on plates for 2 h at room temperature. After incubation, plates were washed three times with PBS-Tween 0.05%, and bound antibody was detected with SULFO-TAG conjugated goat anti-mouse antibody (R32AC-1, Meso Scale Discovery) using TPA containing read buffer (R92TC-2, Meso Scale Discovery). Concentrations were determined from the standard curve using a four-parameter non-linear regression program (Discovery Workbench version 4.0 software).
In Vivo and Ex Vivo Imaging Studies
Labeling of Antibodies with AF488 and AF750
Antibodies were fluorescently labeled with Alexa Fluor 488 NHS Ester and Alexa Fluor 750 NHS Ester (Molecular Probes) in PBS. Briefly, a stock solution of the amine-reactive probe AF750 NHS Ester or AF488 NHS Ester dissolved in anhydrous DMSO (5.92 mM) was freshly prepared. The dye was then added to the antibody solution at an appropriate proportion (2.5:1). The mixture was kept in the dark under gentle agitation at room temperature for 1 h. The reaction was stopped by using NH4Cl 1 M solution, and non-conjugated dye was removed by passing the mixture through a short gel filtration column (Zeba Spin Desalting Columns, 40K MWCO) with PBS (PBS 1×, pH 7.4). The absence of free label and the monomeric purity were checked by high performance liquid chromatography on Acquity UPLC columns (200 Å, 1.7 μm, 4.6 mm × 15 mm, Waters).
Fluorescence Optical Tomography Imaging Study (Fluorescence Trans-Illumination Technology [FLIT] 3D)
Six days before the imaging study, 19-week-old WT (n = 5) and APP Tg mice (n = 5) were shaved (head and neck) under anesthesia (oxygen + 2% isoflurane). Mice were then individually placed under a heat lamp to secure their wakeup after anesthesia and were returned to their cage 10 min after full recovery from anesthesia. Fluorescence imaging was performed using the preclinical multimodal imaging device IVIS Spectrum CT (PerkinElmer) dedicated to imaging small animals. This device allows in vivo fluorescence or bioluminescence imaging tomography (real 3D images) under anesthesia in mice.
In trans-illumination mode used for this 3D fluorescence study, the animal was placed on a rectangular platform comprising (9 × 19) holes; all were possible points of trans-illuminations (diameter 5 mm, inter-space 5 mm). Four excitation sites were selected on the grid to trans-illuminate the area of the skull from one end to the other and thus excite the fluorescent agent (TBTI3a-AF750).
The device used, concomitantly with the trans-illumination protocol, an X-ray scanning of the whole body (CT), which allowed the precise anatomical 3D location of fluorescent sources and the calculation of their intensity. In this study, fluorescence was quantified at two ROIs: one in intracranial ROI 9×9×6 mm, (for the brain or intra-cerebral ROI) and another in sub-extracranial ROI 12×10×4 mm (for an extra-cerebral ROI, which included jugular carotids). Quantification of source fluorescence voxels was expressed in pmol, a semiquantitative unit defined for fluorescent sources calibrated on the Living Image software.61,62
Imaging protocol: FLIT sessions
For all FLIT imaging sessions performed in the IVIS Spectrum CT, the mice were placed in ventral decubitus position on the trans-illumination grid and maintained under anesthesia with 2% isoflurane/oxygen. The platform was temperature controlled to maintain the mice at 37°C. The passage of the animal into the IVIS Spectrum CT took a few minutes, including few seconds for exposure to X-rays. After waking up under a heat lamp, the animal was returned to its home cage. Basal FLIT imaging (CTRL) was performed on each mouse 24 h before the injection of TBTI3a-AF750 antibody. The next day at T0 min, the mice received a caudal i.v. injection of a TBTI3a-AF750 antibody solution at a dose of 57 nmol/kg. FLIT imaging identical sessions were performed at 3, 72, and 168 h after administration. Blood samples were retro-orbitally collected after each session and epi-illuminated in a 96-well plate (excitation 745 nm/emission 800 nm) in the IVIS Spectrum CT at the end of the session to measure fluorescence levels in blood expressed in total radiance efficiency.63
Ex Vivo Multi-fluorescence
To determine if the different administered antibodies behave differently when engaging with or without TfR on the BBB, ex vivo fluorescent imaging was used to directly detect injected antibodies coupled with Alexa Fluor dye 488 on the brain tissue section of 10-month-old APPSL mice. Mice were treated with a first dose of 20 mg/kg under a volume of 5 mL/kg of AF488-mAbs using right retro-orbital injection. A second injection of the same dose was administered 7 days after using the left retro-orbital injection. The animals received a right retro-orbital injection of Angiospark 680 (100 μL/25 g mice) 72 h after this second injection. Subsequent to immersion fixation in 4% formaldehyde and cryoprotection, frozen entire brains were cut serially along their latero-medial axis (Cryostat HM560, Microm). A 20-μm-thick floating coronal section prepared as described before for multi-fluorescence analysis was mounted on slides, dehydrated, and coverslipped using Prolong DAPI mounted medium (Molecular Probe, Ref P36930). Some sections were stained using 0.2% Congo red solution in NaCl-saturated 80% ethanol (Accustain amyloid stain Congo Red Kit, HT60, Sigma) before counterstaining with DAPI. Single and multi-fluorescent microscopical analyses were done on an Olympus VS120 scanner for detection of administered antibodies (green fluorescence), nuclei over all tissue (in blue), and intraparenchymal and vascular Congo red deposits (in red). In addition, Angiospark 680 (NEV10149, Perkin Elmer) injected just before in vivo imaging was used to evidence cerebral vasculature with purple fluorescence.
Efficacy Study in APPSL Mice
Animal Treatment
Male APPSL mice (n = 11–14 per group) were weighted and randomized into the different groups using Biost@t Rando V2.1 (Sanofi). They were treated once a week by i.p. administration of antibodies at the following indicated doses dissolved in DPBS (calcium- and magnesium-free) under a volume of 10 mL/kg: 70 nmol/kg for (Ctrl-IgG1) and (13C3a); and 15 nmol/kg for (TBTI3a) and (TBTI6a) and clinical observations were noted. All treatments were started at the age of 3 months. Treatment duration was 4 months. Due to spontaneous mortality in this Tg strain, 8–10 mice survived until the end of the treatment period with no group bias. To avoid any bias, the experiment was double-blinded to the allocation of the mice into treatment groups and assessment for efficacy.
Blood and Tissue Sampling
Sampling was performed 1 week after the last treatment. Mice were deeply anesthetized by i.p. co-administration of xylazine at 10 mg/kg and ketamine at 100 mg/kg under a volume of 10 mL/kg. Whole blood (about 0.5 mL) was collected by posterior vena cava puncture for hematologic evaluation in tubes containing K3EDTA at room temperature. The rest of the blood was centrifuged (4°C × 10,000 g × 10 min) and plasma was harvested for the PK final point. Animals were then intracardially perfused with 40 mL of ice-cold heparinized PBS (−/−) (10 U/mL). Brains were dissected, and one hemi-cortex was kept for the biochemical evaluation of Aβ peptide levels in the cortex while the other hemisphere was prepared for immunohistochemical analysis. For biochemical evaluation of Aβ, cerebral cortices were quickly frozen in cooled isopentane and then stored at −80°C. The hemispheres used for immunohistochemistry were immersion-fixed in 4% formaldehyde (Carlo Erba, Ref 415691) for 3 days at 4°C. Liver and spleen were immersed in 4% formaldehyde for histology.
Biochemical Analysis and Quantification of Aβ38, Aβ40, and Aβ42 in the brain
For biochemical analysis of Aβ38, Aβ40, and Aβ42 in brain, the 4-spot multiplex V-plex Aβ peptide panel 1 (6E10, human specific) kit (ref. K15200G) from Meso Scale Discovery was used. The assay was performed following the manufacturer’s instructions.
Tissues were first lysed in Precellys tubes (CK14) in around 10% (w/v) buffer (1 mL buffer for cortices) containing Tris-HCl 4 mM pH 7.4, sucrose 0.32 M, and supplemented with protease and phosphatase inhibitors (Halt, ThermoScientific). At this step, protein concentration was determined on 1 μL of the lysates using DC Protein Assay (Biorad). To solubilize Aβ, cortical homogenates were then brought to a 6 M final concentration of guanidine-HCl by adding 300 μL guanidine-HCl 8 M (Sigma) per 100 μL of homogenate and sonicated briefly. Solubilized samples were further serially 10-fold diluted in the “Tris-Sucrose-Guanidine” solubilization buffer. The last dilution was done in diluent 35 provided in the Meso Scale Discovery kit. At the end, solubilized cortical homogenates were diluted 1/10,000th for Aβ42 and 1/100th for Aβ38 and Aβ40. Similarly, to brain samples, calibrators used to generate the standard curve were prepared and diluted in the same manner as the tissue lysates. Electrochemiluminescent signals were measured using a MESO QuickPlex SQ120 (Meso Scale Discovery), and collected data were analyzed using Discovery Workbench version 4.0 software. Values are expressed as pg Aβ peptide per μg of total protein.
Immunohistochemistry
Post-fixed brains and sections were prepared as described before for ex vivo immunofluorescence analysis. Randomly selected series of 30-μm-thick hemibrain free-floating sections were used to perform Aβ peptide and IgG1 immunohistochemistry, and, respectively, used to quantify cerebral Aβ peptide deposition and detect the therapeutic antibody that enters the cerebral tissue. After pretreatment with 80% formic acid for 3 min for Aβ immunostaining only, sections were incubated at room temperature for 30 min in 0.1 M PBS-0.15% triton-0.3% hydrogen peroxide and finally in blocking buffer (i.e., in BSA diluted at 3% in 0.1 M PBS) for 30 min. They were incubated overnight at room temperature with primary antibodies: biotinylated mouse monoclonal anti-Aβ peptide antibody (4G8, human Aβ17-24, 800705, Biolegend, dilution 1/2,000 or 9240-10, Signet, dilution 1/2,000) or biotinylated goat anti-mouse IgG1 antibody (1070-08, Southern Biotech, dilution 1/200). Sections were further incubated with peroxidase-coupled avidin complex (Vectastain ABC kit, Vector Laboratories, dilution 1/400) for 30 min. Diaminobenzidine substrate was used for color development. Sections immunostained for IgG1 were counterstained using 0.2% Congo red solution in NaCl-saturated 80% ethanol (Accustain amyloid stain Congo Red Kit, HT60, Sigma) to identify aggregated Aβ peptide deposits.
For quantitative analysis of Aβ immunostaining, images of the entire sections were acquired on an Olympus Dotslide BX scanner and quantitatively analyzed on a computer-based workstation (Mercator system/Explora Nova using Dotslide software). Based on a thresholding procedure allowing for automatic detection of individually stained deposits, total count and total detectable surface area (μm2) over eight anatomical levels representative of the entire latero-medial axis of both hippocampal and cortical areas were measured. For IgG1/Congo red double-staining, qualitative image analysis was performed on an Olympus scanner using bright-field microscopy.
Blood Analysis
Analyses were performed by using an automated hematology analyzer with a veterinary module (ADVIA 2120, Siemens Healthcare Diagnostics) and using the settings for mouse blood (software version 5.9, Siemens). Hematology assessment included the following parameters: hemoglobin (HGB), hematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), red blood cell distribution width (RDW), red blood cell (RBC), white blood cell (WBC), reticulocyte, basophil, neutrophil, eosinophil, lymphocyte, monocyte, platelet, and large unstained (LU) cell counts.
Anatomic Pathology
The liver and spleen were examined. Tissues taken at necropsy were fixed using standard fixation procedures in 4% neutral buffered formalin. They were then paraffin-embedded and sectioned at 4 μm to produce standardized sections of both organs. A routine hematoxylin, eosin, and saffron stain was used to produce the final slides. Slides were evaluated microscopically by a board-certified veterinary pathologist and lesions were graded using the industry standard semiquantitative system (5 grades: minimal, mild, moderate, marked, and severe). The terminology used followed the INHAND (International Harmonization of Nomenclature and Diagnostic Criteria) nomenclature.
TfR Protein Levels in bEnd.3 and Mouse Primary Cortical Neurons Treated with TBTIs
Culture and Treatment of bEnd.3 Cells and Mouse Primary Cortical Neurons
The murine brain endothelial cell line bEnd.3 and mouse primary cortical neurons were used to determine the influence of exposure to TBTIs on TfR cell levels.
bEnd.3 cells plated on rat tail collagen type I and at confluence were exposed to cell media containing TBTI2a, TBTI3a, TBTI6a, or a control monoclonal IgG1 at 25 or 350 nM for 24 h. Mouse primary neuronal cultures were prepared from E16 mouse brains (C57BL/6J, Charles River Laboratories, France) as previously described27 and plated in poly-d-lysine-coated 24-well culture microplates at 5 × 105 cells/mL. On day 6, cells were exposed to cell media containing TBTI2a, TBTI3a, TBTI6a, or a control monoclonal IgG1 at 5 nM for 24 h.
Following antibody incubation, cells were lysed using ice-cold RIPA buffer (CST) containing protease inhibitor cocktail (ThermoFisher), and whole cell lysates were harvested. Alternatively, cell surface proteins were obtained through biotinylation (EZ-link Sulfo-NHS-SS-Biotin, Thermo Fisher).64 Following biotinylation at 4°C for 1 h, cells were rinsed twice with PBS (Ca2+/Mg2+, Gibco), each sample was lysed, and biotinylated proteins were extracted on streptavidin agarose resins (Thermo Fisher) and eluted on denaturating conditions (NuPAGE LDS Sample Buffer 4× + 5% β-mercaptoethanol). Equal amounts of protein from whole cell lysates and cell surface cell lysates were used in western blotting for assessing TfR relative protein levels.
Relative Quantification of TfR Protein Levels by Western Blot
Whole cell lysates and cell surface cell lysates from bEnd.3 and mouse primary neurons were loaded into 4%–12% Tris-glycine SDS-page gels (Invitrogen) and allowed to migrate for 1 h at 180 V. Samples were then transferred onto polyvinylidene fluoride (PVDF) membranes using an iBlot 2 Dry Blotting System (Invitrogen) on the P0 program (20 V for 1 min, 23 V for 4 min, 25 V for 2 min). PVDF membranes were then rinsed with Tris-buffered saline with 0.1% Tween 20 (TBST) and blocked for 1 h in 5% non-fat dry milk in TBST (blocking buffer). Membranes were subsequently probed overnight at 4°C with primary antibodies mouse anti-TfR H68.4 (1:500, Thermo Fisher), rabbit anti-Pan Cadherin (1:1,000, Cell Signaling Technologies), or mouse anti-tubulin (1:1,000, Cell Signaling Technologies) diluted in blocking buffer. Membranes were then rinsed multiple times with TBST before being subjected to probing with secondary antibodies diluted in TBST for 1 h at room temperature (1:10,000 diluted HRP-coupled goat anti-mouse IgG or goat anti-rabbit IgG, GE Healthcare). Following secondary antibody incubation, membranes were rinsed thoroughly with TBST and were subsequently imaged using a LICOR Odyssey Imager and quantified using Multi-Gauge v3.0.
Statistical Analysis
Graphs were generated using GraphPad Prism version 8.0.2 for Windows (GraphPad Software, San Diego, CA, USA) and statistical analyses were conducted using SAS V9.4 (SAS Institute, USA). The significance level was set at 5%. Data were analyzed with one-way analysis of variance (ANOVA) followed by Newman-Keuls post hoc analysis either on raw data (for Aβ immunostaining in cortex) or log-transformed data (for Aβ immunostaining in the hippocampus or biochemical evaluation in the cortex), depending on whether normality assumption and equal variance assumptions held for ANOVA. Whenever possible, boxplots (with jitter points) were used to visualize the data. For in vivo FLIT study, after checking residuals, a two-way ANOVA with factor genotype, repeated factor time, and their interaction was performed on rank-transformed data. When the interaction “Genotype∗Time” or the factors “Genotype” and “Time” were significant, a Fisher’s test was performed to compare the Tg mice versus the WT group on the studied parameter globally and at each time point. The significance level was set to 5%, except for the interaction for which the significant level was set to 10%.
Author Contributions
T.-M.D., C. Capdevila, V.B., M.L.-G., N.S., A.S., J.B., D.B., P.D., N.A., G.D., C. Chaves, E.G., Y.L., F.M., and N.M. conducted the experiments and analyzed the data. P.V., C.L., F.C., T.G., S.E., D.V., O.B., E.R., L.P., and D.L. supervised and analyzed the data. T.-M.D., C. Capdevila, V.B., M.L.-G., N.S., A.S., D.B., C. Chaves, F.M., F.C., and D.L. wrote the manuscript. S.E., D.V., O.B., E.R., L.P., and D.L. edited the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
All authors are current employees of Sanofi. The design and financial support for this research were provided by Sanofi. Sanofi participated in the review and approval of the publication.
Acknowledgments
The authors would like to thank Athanase Bayard and Sophie Ho-Van for their help with imaging studies, Fabienne Gallen, Pascal Vicat, and technicians for their help with pharmacokinetic studies, Agnès Choquart for her work in antibody quantification, Pascal Chaillou for his help with IHC, Francis Duffieux and technicians for their work on protein purification, and Luc Essermant and Jun Luo for statistical analyses. We are also thankful to Jacques Dumas and Pascal Barnéoud for their contributive output throughout this work, critical review of the manuscript, and data cross checking.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.08.014.
Supplemental Information
References
- 1.Banks W.A. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016;15:275–292. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 2.Obermeier B., Daneman R., Ransohoff R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar N.N., Pizzo M.E., Nehra G., Wilken-Resman B., Boroumand S., Thorne R.G. Passive Immunotherapies for Central Nervous System Disorders: Current Delivery Challenges and New Approaches. Bioconjug. Chem. 2018;29:3937–3966. doi: 10.1021/acs.bioconjchem.8b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher D.G., Price R.J. Recent Advances in the Use of Focused Ultrasound for Magnetic Resonance Image-Guided Therapeutic Nanoparticle Delivery to the Central Nervous System. Front. Pharmacol. 2019;10:1348. doi: 10.3389/fphar.2019.01348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z., Xiong G., Tsang W.C., Schätzlein A.G., Uchegbu I.F. Nose-to-Brain Delivery. J. Pharmacol. Exp. Ther. 2019;370:593–601. doi: 10.1124/jpet.119.258152. [DOI] [PubMed] [Google Scholar]
- 6.Saeedi M., Eslamifar M., Khezri K., Dizaj S.M. Applications of nanotechnology in drug delivery to the central nervous system. Biomed. Pharmacother. 2019;111:666–675. doi: 10.1016/j.biopha.2018.12.133. [DOI] [PubMed] [Google Scholar]
- 7.Abdul Razzak R., Florence G.J., Gunn-Moore F.J. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. Int. J. Mol. Sci. 2019;20:20. doi: 10.3390/ijms20123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pulgar V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2019;12:1019. doi: 10.3389/fnins.2018.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnsen K.B., Burkhart A., Thomsen L.B., Andresen T.L., Moos T. Targeting the transferrin receptor for brain drug delivery. Prog. Neurobiol. 2019;181:101665. doi: 10.1016/j.pneurobio.2019.101665. [DOI] [PubMed] [Google Scholar]
- 10.Pardridge W.M. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs. 2017;31:503–519. doi: 10.1007/s40259-017-0248-z. [DOI] [PubMed] [Google Scholar]
- 11.Paterson J., Webster C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today. Technol. 2016;20:49–52. doi: 10.1016/j.ddtec.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 12.Boado R.J., Zhang Y., Zhang Y., Xia C.F., Pardridge W.M. Fusion antibody for Alzheimer’s disease with bidirectional transport across the blood-brain barrier and abeta fibril disaggregation. Bioconjug. Chem. 2007;18:447–455. doi: 10.1021/bc060349x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang R., Knox J., Chang J., Derbedrossian A., Vasilevko V., Cribbs D., Boado R.J., Pardridge W.M., Sumbria R.K. Blood-Brain Barrier Penetrating Biologic TNF-α Inhibitor for Alzheimer’s Disease. Mol. Pharm. 2017;14:2340–2349. doi: 10.1021/acs.molpharmaceut.7b00200. [DOI] [PubMed] [Google Scholar]
- 14.Hultqvist G., Syvänen S., Fang X.T., Lannfelt L., Sehlin D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics. 2017;7:308–318. doi: 10.7150/thno.17155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niewoehner J., Bohrmann B., Collin L., Urich E., Sade H., Maier P., Rueger P., Stracke J.O., Lau W., Tissot A.C. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81:49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 16.Sonoda H., Morimoto H., Yoden E., Koshimura Y., Kinoshita M., Golovina G., Takagi H., Yamamoto R., Minami K., Mizoguchi A. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018;26:1366–1374. doi: 10.1016/j.ymthe.2018.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webster C.I., Hatcher J., Burrell M., Thom G., Thornton P., Gurrell I., Chessell I. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain. 2017;158:660–668. doi: 10.1097/j.pain.0000000000000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Q.H., Boado R.J., Lu J.Z., Hui E.K., Pardridge W.M. Monoclonal antibody-glial-derived neurotrophic factor fusion protein penetrates the blood-brain barrier in the mouse. Drug Metab. Dispos. 2010;38:566–572. doi: 10.1124/dmd.109.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karaoglu Hanzatian D., Schwartz A., Gizatullin F., Erickson J., Deng K., Villanueva R., Stedman C., Harris C., Ghayur T., Goodearl A. Brain uptake of multivalent and multi-specific DVD-Ig proteins after systemic administration. MAbs. 2018;10:765–777. doi: 10.1080/19420862.2018.1465159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kariolis M.S., Wells R.C., Getz J.A., Kwan W., Mahon C.S., Tong R., Kim D.J., Srivastava A., Bedard C., Henne K.R. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci. Transl. Med. 2020;12:12. doi: 10.1126/scitranslmed.aay1359. [DOI] [PubMed] [Google Scholar]
- 21.Syvänen S., Hultqvist G., Gustavsson T., Gumucio A., Laudon H., Söderberg L., Ingelsson M., Lannfelt L., Sehlin D. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimers Res. Ther. 2018;10:49. doi: 10.1186/s13195-018-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Y.J., Zhang Y., Kenrick M., Hoyte K., Luk W., Lu Y., Atwal J., Elliott J.M., Prabhu S., Watts R.J., Dennis M.S. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011;3:84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- 23.Hanzatian D.K., Goodearl A.D., Mueller B.K., Mueller R., Klein C. 2015. Blood-brain barrier (BBB) penetrating dual specific binding proteins for treating brain and neurological diseases. WO 2015/191934 A2, filed November 6, 2015, and published December 17 2015. [Google Scholar]
- 24.Rao E., Mikol V., Li D., Kruip J., Davison M. 2007. Antibodies that bind IL-4 and/or IL-13 and their uses. WO2009052081, filed October 14, 2008, and published April 23, 2009. [Google Scholar]
- 25.Sumbria R.K., Hui E.K., Lu J.Z., Boado R.J., Pardridge W.M. Disaggregation of amyloid plaque in brain of Alzheimer’s disease transgenic mice with daily subcutaneous administration of a tetravalent bispecific antibody that targets the transferrin receptor and the Abeta amyloid peptide. Mol. Pharm. 2013;10:3507–3513. doi: 10.1021/mp400348n. [DOI] [PubMed] [Google Scholar]
- 26.Digiammarino E.L., Harlan J.E., Walter K.A., Ladror U.S., Edalji R.P., Hutchins C.W., Lake M.R., Greischar A.J., Liu J., Ghayur T., Jakob C.G. Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. MAbs. 2011;3:487–494. doi: 10.4161/mabs.3.5.16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pradier L., Blanchard-Brégeon V., Bohme A., Debeir T., Menager J., Benoit P., Barneoud P., Taupin V., Bertrand P., Dugay P. SAR228810: an antibody for protofibrillar amyloid β peptide designed to reduce the risk of amyloid-related imaging abnormalities (ARIA) Alzheimers Res. Ther. 2018;10:117. doi: 10.1186/s13195-018-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H.J., Engelhardt B., Lesley J., Bickel U., Pardridge W.M. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J. Pharmacol. Exp. Ther. 2000;292:1048–1052. [PubMed] [Google Scholar]
- 29.Pepinsky R.B., Shao Z., Ji B., Wang Q., Meng G., Walus L., Lee X., Hu Y., Graff C., Garber E. Exposure levels of anti-LINGO-1 Li81 antibody in the central nervous system and dose-efficacy relationships in rat spinal cord remyelination models after systemic administration. J. Pharmacol. Exp. Ther. 2011;339:519–529. doi: 10.1124/jpet.111.183483. [DOI] [PubMed] [Google Scholar]
- 30.Kissel K., Hamm S., Schulz M., Vecchi A., Garlanda C., Engelhardt B. Immunohistochemical localization of the murine transferrin receptor (TfR) on blood-tissue barriers using a novel anti-TfR monoclonal antibody. Histochem. Cell Biol. 1998;110:63–72. doi: 10.1007/s004180050266. [DOI] [PubMed] [Google Scholar]
- 31.Blanchard V., Moussaoui S., Czech C., Touchet N., Bonici B., Planche M., Canton T., Jedidi I., Gohin M., Wirths O. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp. Neurol. 2003;184:247–263. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- 32.Triguero D., Buciak J., Pardridge W.M. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J. Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- 33.Reddy M.P., Kinney C.A., Chaikin M.A., Payne A., Fishman-Lobell J., Tsui P., Dal Monte P.R., Doyle M.L., Brigham-Burke M.R., Anderson D. Elimination of Fc receptor-dependent effector functions of a modified IgG4 monoclonal antibody to human CD4. J. Immunol. 2000;164:1925–1933. doi: 10.4049/jimmunol.164.4.1925. [DOI] [PubMed] [Google Scholar]
- 34.Buono C., Anzinger J.J., Amar M., Kruth H.S. Fluorescent pegylated nanoparticles demonstrate fluid-phase pinocytosis by macrophages in mouse atherosclerotic lesions. J. Clin. Invest. 2009;119:1373–1381. doi: 10.1172/JCI35548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dapson R.W. Amyloid from a histochemical perspective. A review of the structure, properties and types of amyloid, and a proposed staining mechanism for Congo red staining. Biotech. Histochem. 2018;93:543–556. doi: 10.1080/10520295.2018.1528385. [DOI] [PubMed] [Google Scholar]
- 36.Bien-Ly N., Yu Y.J., Bumbaca D., Elstrott J., Boswell C.A., Zhang Y., Luk W., Lu Y., Dennis M.S., Weimer R.M. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014;211:233–244. doi: 10.1084/jem.20131660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thom G., Burrell M., Haqqani A.S., Yogi A., Lessard E., Brunette E., Delaney C., Baumann E., Callaghan D., Rodrigo N. Enhanced Delivery of Galanin Conjugates to the Brain through Bioengineering of the Anti-Transferrin Receptor Antibody OX26. Mol. Pharm. 2018;15:1420–1431. doi: 10.1021/acs.molpharmaceut.7b00937. [DOI] [PubMed] [Google Scholar]
- 38.Friden P.M., Walus L.R., Musso G.F., Taylor M.A., Malfroy B., Starzyk R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 1991;88:4771–4775. doi: 10.1073/pnas.88.11.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freskgård P.-O., Niewoehner J., Urich E. Time to open the blood–brain barrier gate for biologics? Future Neurol. 2014;9:243–245. [Google Scholar]
- 40.Johnsen K.B., Bak M., Kempen P.J., Melander F., Burkhart A., Thomsen M.S., Nielsen M.S., Moos T., Andresen T.L. Antibody affinity and valency impact brain uptake of transferrin receptor-targeted gold nanoparticles. Theranostics. 2018;8:3416–3436. doi: 10.7150/thno.25228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakano R., Takagi-Maeda S., Ito Y., Kishimoto S., Osato T., Noguchi K., Kurihara-Suda K., Takahashi N. A new technology for increasing therapeutic protein levels in the brain over extended periods. PLoS ONE. 2019;14:e0214404. doi: 10.1371/journal.pone.0214404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sehlin D., Fang X.T., Cato L., Antoni G., Lannfelt L., Syvänen S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat. Commun. 2016;7:10759. doi: 10.1038/ncomms10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webster C.I., Stanimirovic D.B. A gateway to the brain: shuttles for brain delivery of macromolecules. Ther. Deliv. 2015;6:1321–1324. doi: 10.4155/tde.15.78. [DOI] [PubMed] [Google Scholar]
- 44.Weber F., Bohrmann B., Niewoehner J., Fischer J.A.A., Rueger P., Tiefenthaler G., Moelleken J., Bujotzek A., Brady K., Singer T. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018;22:149–162. doi: 10.1016/j.celrep.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 45.Mo J.J., Li J.Y., Yang Z., Liu Z., Feng J.S. Efficacy and safety of anti-amyloid-β immunotherapy for Alzheimer’s disease: a systematic review and network meta-analysis. Ann. Clin. Transl. Neurol. 2017;4:931–942. doi: 10.1002/acn3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sevigny J., Chiao P., Bussière T., Weinreb P.H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 47.Alzforum . 2020. In DIAN-TU, Gantenerumab Brings Down Tau. By a Lot. Open Extension Planned.https://www.alzforum.org/news/conference-coverage/dian-tu-gantenerumab-brings-down-tau-lot-open-extension-planned [Google Scholar]
- 48.Alzoforum . 2017. High-Dose Gantenerumab Lowers Plaque Load.https://www.alzforum.org/news/conference-coverage/high-dose-gantenerumab-lowers-plaque-load [Google Scholar]
- 49.Guthrie H., Honig L.S., Lin H., Sink K.M., Blondeau K., Quartino A., Dolton M., Carrasco-Triguero M., Lian Q., Bittner T Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J Alzheimers Dis. 2020;76:967–979. doi: 10.3233/JAD-200134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwayanagi Y., Igawa T., Maeda A., Haraya K., Wada N.A., Shibahara N., Ohmine K., Nambu T., Nakamura G., Mimoto F. Inhibitory FcγRIIb-Mediated Soluble Antigen Clearance from Plasma by a pH-Dependent Antigen-Binding Antibody and Its Enhancement by Fc Engineering. J. Immunol. 2015;195:3198–3205. doi: 10.4049/jimmunol.1401470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ober R.J., Radu C.G., Ghetie V., Ward E.S. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 2001;13:1551–1559. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- 52.St-Amour I., Paré I., Alata W., Coulombe K., Ringuette-Goulet C., Drouin-Ouellet J., Vandal M., Soulet D., Bazin R., Calon F. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier. J. Cereb. Blood Flow Metab. 2013;33:1983–1992. doi: 10.1038/jcbfm.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlachetzki F., Zhu C., Pardridge W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002;81:203–206. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 54.Abuqayyas L., Balthasar J.P. Application of knockout mouse models to investigate the influence of FcγR on the tissue distribution and elimination of 8C2, a murine IgG1 monoclonal antibody. Int. J. Pharm. 2012;439:8–16. doi: 10.1016/j.ijpharm.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 55.Hwang W.Y., Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Bohrmann B., Baumann K., Benz J., Gerber F., Huber W., Knoflach F., Messer J., Oroszlan K., Rauchenberger R., Richter W.F. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 2012;28:49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- 57.Zarif H., Paquet A., Lebrigand K., Arguel M.J., Heurteaux C., Glaichenhaus N., Chabry J., Guyon A., Petit-Paitel A. CD4+ T Cells Affect the Thyroid Hormone Transport at the Choroid Plexus in Mice Raised in Enriched Environment. Neuroimmunomodulation. 2019;26:59–66. doi: 10.1159/000495987. [DOI] [PubMed] [Google Scholar]
- 58.Releases S. S, LLC; New York, NY: 2019. 2015 and late: Maestro. [Google Scholar]
- 59.Jacobson M.P., Pincus D.L., Rapp C.S., Day T.J., Honig B., Shaw D.E., Friesner R.A. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 60.Beard H., Cholleti A., Pearlman D., Sherman W., Loving K.A. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS ONE. 2013;8:e82849. doi: 10.1371/journal.pone.0082849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leblond F., Davis S.C., Valdés P.A., Pogue B.W. Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications. J. Photochem. Photobiol. B. 2010;98:77–94. doi: 10.1016/j.jphotobiol.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rice B.W., Cable M.D., Nelson M.B. In vivo imaging of light-emitting probes. J. Biomed. Opt. 2001;6:432–440. doi: 10.1117/1.1413210. [DOI] [PubMed] [Google Scholar]
- 63.Suzuki T., Miyazaki C., Ishii-Watabe A., Tada M., Sakai-Kato K., Kawanishi T., Kawasaki N. A fluorescent imaging method for analyzing the biodistribution of therapeutic monoclonal antibodies that can distinguish intact antibodies from their breakdown products. MAbs. 2015;7:759–769. doi: 10.1080/19420862.2015.1038683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crupi M.J., Yoganathan P., Bone L.N., Lian E., Fetz A., Antonescu C.N., Mulligan L.M. Distinct Temporal Regulation of RET Isoform Internalization: Roles of Clathrin and AP2. Traffic. 2015;16:1155–1173. doi: 10.1111/tra.12315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.