

Abstract
The human epidermal growth factor receptor (EGFR/ERBB1) is a receptor tyrosine kinase (RTK) that forms activated oligomers in response to ligand. Much evidence indicates that EGFR/ERBB1 also forms oligomers in the absence of ligand, but the structure and physiological role of these ligand-independent oligomers remain unclear. To examine these features, we use fluorescence microscopy to measure the oligomer stability and FRET efficiency for homo- and hetero-oligomers of fluorescent protein-labeled forms of EGFR and its paralog, human epidermal growth factor receptor 2 (HER2/ERBB2) in vesicles derived from mammalian cell membranes. We observe that both receptors form ligand-independent oligomers at physiological plasma membrane concentrations. Mutations introduced in the kinase region at the active state asymmetric kinase dimer interface do not affect the stability of ligand-independent EGFR oligomers. These results indicate that ligand-independent EGFR oligomers form using interactions that are distinct from the EGFR active state.
Keywords: epidermal growth factor receptor, receptor tyrosine kinase, signal transduction, membrane protein, oligomerization, fluorescence resonance energy transfer, FRET, EGFR
Human epidermal growth factor receptor (EGFR/ERBB1) and its paralogs human epidermal growth factor receptor 2 (HER2/ERBB2), HER3/ERBB3, and HER4/ERBB4, collectively known as ERBBs, are receptor tyrosine kinases (RTKs) that are essential for normal growth and development (1). Abnormal activation of both EGFR and HER2 is associated with multiple human cancer types, and each is the target of several anticancer agents (2). ERBBs are Type I integral membrane proteins composed of an extracellular domain (ECD) that is made up of four distinct subdomains, an α-helical transmembrane domain (TMD), and an intracellular domain comprising a juxtamembrane region, a tyrosine kinase domain, and a nonglobular ∼230 amino acid C-terminal tail. The canonical mechanism by which ERBBs are thought to act is ligand-dependent oligomerization, which results in stimulation of the intracellular kinase, trans-autophosphorylation, recruitment of effector proteins, and initiation of intracellular signaling cascades (1, 3–5).
The reported levels of endogenous EGFR expression vary by several orders of magnitude depending on cellular context (6–15). At least seven studies have reported that a fraction of cell-surface EGFR is dimeric in the absence of ligand (hereafter called ligand-independent dimers) (16–22) (Table S1), indicating that the receptor concentration in the plasma membrane might specify the ligand-independent oligomeric fraction in accordance with the law of mass action (23, 24). Despite much study, the structure and signaling role of ligand-independent dimers remain unclear.
Mutations or deletions in EGFR and HER2 result in ligand-independent receptor activation, indicating that in addition to being activated in the presence of ligand, ERBBs are actively inhibited in the absence of ligand (25–30). The mechanism for this autoinhibition remains incompletely understood, however. In contrast, much is known about how ligand binding promotes ERBB activation (31, 32). In the absence of ligand, the EGFR, ERBB3, and ERBB4 ECDs adopt a tethered conformation that buries an extended β-hairpin loop (33–36). Binding of ligand to these ECDs stabilizes an extended conformation in which an extended β-hairpin (termed the dimerization arm) is exposed and mediates formation of an active receptor dimer. In this dimer the intracellular kinase domains adopt an asymmetric dimer interaction in which the C-lobe of a donor kinase contacts the N-lobe of a receiver kinase, stabilizing the active conformation of the receiver kinase (37). Comparatively little is known about the structure and physiological role of either the TMD or the intracellular domain in the inactive state, although mechanisms by which the TMD may function in the inactive state have been proposed (35, 38, 39).
Here we report quantitative FRET measurements of homo- and hetero-interactions between near full-length EGFR and HER2 in vesicles derived from plasma membranes. We confirm previous findings that EGFR and HER2 are in dynamic equilibrium between monomeric, homo-oligomeric and hetero-oligomeric forms at physiological receptor concentrations (40). Because we are unable to distinguish dimers from higher-order oligomers (n ≥ 2) by our method we refer to oligomers rather than dimers, although single-molecule photobleaching and fluorescence intensity fluctuation studies indicate that ligand-independent EGFR oligomers are predominantly dimeric (21, 23, 41). We observe that mutations in the asymmetric kinase interface do not preclude formation of ligand-independent EGFR oligomers, indicating that an alternate conformation to the active dimer exists within the ligand-independent oligomer ensemble.
Results
Quantitative imaging FRET microscopy
To enable measurement of homo- and hetero-ERBB interactions in native cell membranes using FRET microscopy, human EGFR and HER2 variants labeled at their C termini with a fluorescent donor (EYFP) or acceptor (mCherry) protein were expressed in Chinese hamster ovary (CHO) cells. ERBBs have ∼200 amino acid disordered regions at their C termini, which, along with 8 amino acid Gly-Ser linkers inserted between each ERBB and its fluorescent protein (fp) fusion partner, are presumed to mitigate effects of fluorescent dipole orientation on observed FRET (Fig. S1) (42). Expression levels of full-length forms of fp-tagged EGFR and HER2 proved too low to characterize interactions with confidence. EGFR and HER2 variants with C-terminal deletions were thus tested to identify minimal deletions that preserve activity and result in improved expression levels. Removal of 107 and 25 residues from the C termini of EGFR and HER2, respectively, generated the variants EGFR-Δ107-fp and HER2-Δ25-fp and resulted in proteins that express well and are functional as judged by anti-phosphotyrosine Western blots (Fig. 1A). EGFR variants with a 237 amino acid C-terminal deletion are able to elicit EGF-dependent cellular functions, indicating that the region deleted in EGFR-Δ107-fp is not essential for activation of downstream effectors (43, 44).
Figure 1.
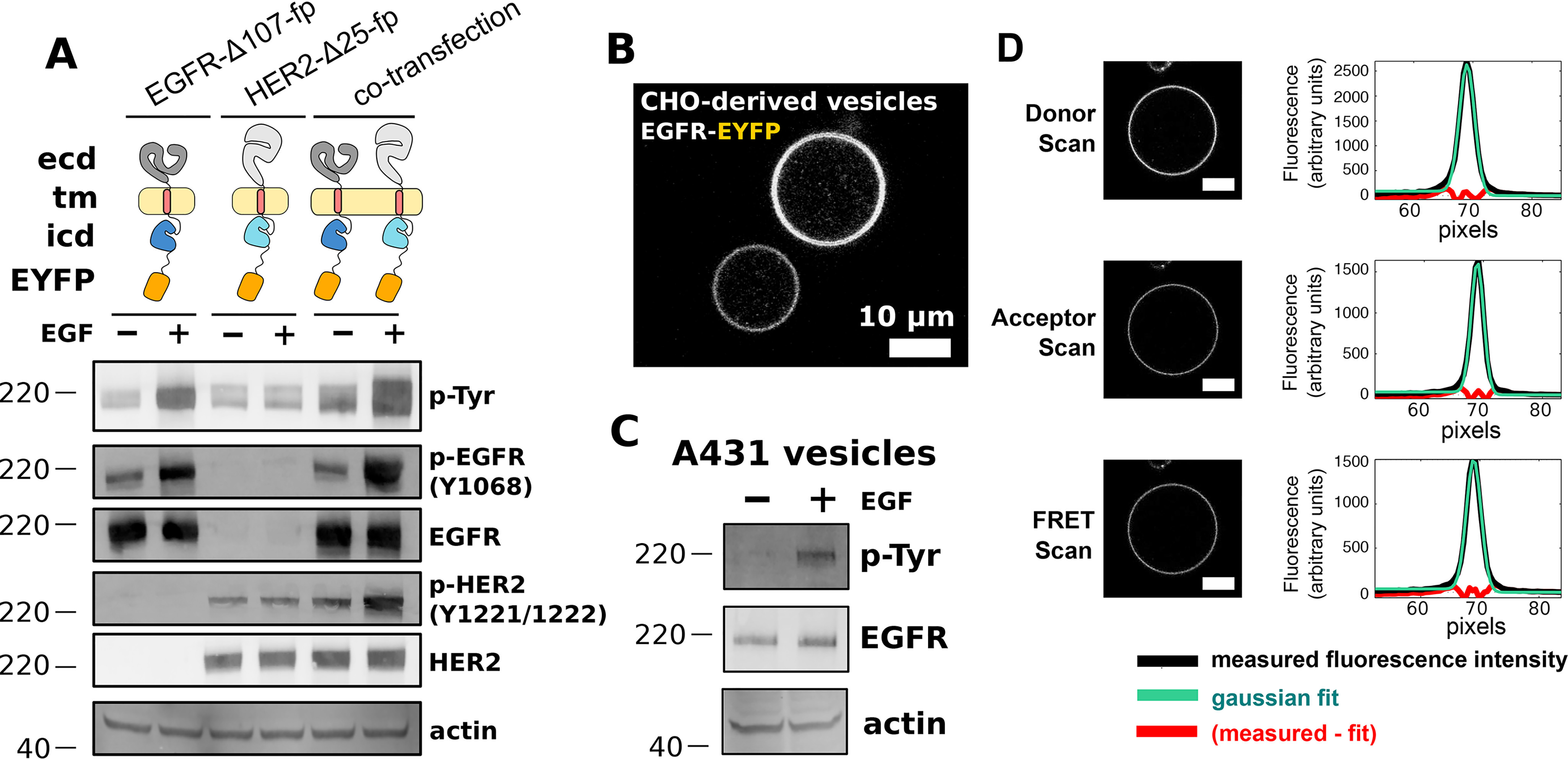
FRET microscopy of near full-length EGFR and HER2 in CHO cell vesicles. A, Western blot analysis of transient expression and EGF-dependent phosphorylation of EGFR-Δ107-FP and HER2-Δ25-FP in CHO cells. Blots are representative of three independent experiments. Primary antibodies are indicated at the right of each blot. ICD = intracellular domain, pTyr = phosphotyrosine. B, confocal image of two vesicles derived from the plasma membranes of CHO cells expressing EGFR-EYFP. Scale bar = 10 μm. C, Western blot analysis of EGFR in vesicles derived from A431 cells. EGFR becomes phosphorylated in response to EGF in the presence of ATP. D, representative scans for a FRET experiment. Scale bars = 10 μm. Plots to the right of each vesicle image show the average fluorescence intensity (y axis) as a function of radius from the center of the vesicle (x axis, in units of pixels). Molecular mass markers (kDa) are indicated to the left of each Western blot.
Homo- and hetero-interactions between EGFR-Δ107-fp and HER2-Δ25-fp were measured using quantitative imaging FRET (QI-FRET) microscopy (45–47). Briefly, plasma membrane vesicles from CHO cells transiently transfected with fp-labeled EGFR and/or HER2 were generated by incubating live cells in a hyperosmotic solution (Fig. 1B) (48, 49). Endogenous EGFR in vesicles derived from A431 cells retains EGF-dependent phosphorylation indicating that vesiculation does not impair ERBB function (Fig. 1C). The concentrations of donor- and acceptor-labeled ERBBs as well as the FRET efficiency between labeled ERBBs in vesicles were obtained from three fluorescence scans (Fig. 1D). Cells in the same transfection experiment display a wide range of receptor expression levels as judged by fluorescence intensity (Fig. 1B), which allows FRET efficiency to be determined as a function of receptor concentration. A wide range (roughly 10-fold) was observed for all variants tested, and the identity of the fluorescent protein (EYFP or mCherry) did not affect the variability in the observed concentration. Following correction of FRET signals for FRET arising from density-dependent proximity (known as proximity or bystander FRET) (50) (Figs. S2–S4) and for the relative fraction of donors and acceptors in each vesicle (45, 46), apparent two-dimensional dissociation constants can be determined.
The oligomeric state of fluorescently labeled proteins affects the FRET signal (51). The FRET changes we observe fit a monomer-dimer model as well as monomer-dimer-tetramer or monomer-trimer models. We thus report fits to a monomer-dimer equilibrium, although the receptors likely form higher-order oligomers in the presence of EGF (23, 41). This fit depends on two parameters: the intrinsic FRET efficiency, E˜, which equals the FRET efficiency within a dimer, and the two-dimensional dissociation constant, Kd, in units of molecules/μm2 (45, 46). In cases where the association is strong at all receptor concentrations, we assume that the intrinsic FRET value (E˜) is equal to the mean observed FRET value. As the presence of higher-order oligomers is likely (41), especially when ligand is present, we use the term oligomer throughout to denote complexes of two or possibly more receptors.
Near full-length EGFR forms ligand-independent oligomers at physiological plasma-membrane concentrations
The FRET efficiency for EGFR-Δ107-fp increased as a function of concentration and fit well to a monomer-dimer equilibrium model, with a two-dimensional Kd of 156 molecules/μm2 (Fig. 2A and B) and a FRET efficiency value of 0.35 ± 0.06 (Table 1), consistent with previous observations (23, 24). Addition of EGF or substitution of the EGFR extracellular domain with a constitutively dimeric immunoglobulin Fc-domain results in constant FRET efficiency values of 0.29 ± 0.03 and 0.29 ± 0.02, respectively, as well as constitutive receptor phosphorylation (Fig. 2E and F, and Fig. S5). The cancer-associated L858R mutation has been shown to promote EGFR oligomerization in the absence of ligand (52), and introduction of L858R into EGFR-Δ107-fp results in higher FRET efficiency values over all concentration ranges measured indicating increased self-association in the absence of ligand (Fig. 2C and D). Observation of higher FRET efficiencies under conditions known to activate EGFR provides confidence that our observations reflect physiological behavior. The FRET efficiency differences become less apparent when the FRET data are binned over all observed receptor concentrations (Fig. 2B, D, and F), underscoring the need for measurements over a range of concentrations.
Figure 2.

Near full-length EGFR forms ligand-independent oligomers at physiological plasma membrane concentrations. FRET efficiency for EGFR-Δ107-fp homo-oligomers. A, C, and E, cartoon representations of fluorescent-protein-linked (A) EGFR WT, (C) L858R, and (E) IgG-Fc/EGFR. B, D, and F, FRET efficiency plots for (B) EGFR-Δ107-fp WT, (D) L858R and (F) IgG-Fc/EGFR. Each panel in B, D, and F contains two graphs. The graph on the left shows the FRET efficiency ((FRETapp − FRETprox)/Xa)) (y axis) as a function of concentration (x axis). The data in B, D and F were projected onto the y axis to yield FRET histograms, plotted at the right. FRETapp is the apparent FRET efficiency, FRETprox is the theoretical FRET efficiency that results from nonspecific interactions, and Xa is the fraction of acceptor molecules in a given vesicle. Binned data points are shown as circles. Error bars represent the S.E. in x and y. The best fit to a monomer-dimer equilibrium model is represented by solid lines. B, data for EGFR-Δ107-fp in the absence (black) and presence (green) of 100 nm EGF. Each dataset was derived from at least three independent biological replicates. The number of vesicles per experiment ranged from 25 to 100.
Table 1.
Statistics for fits to a monomer-dimer equilibrium model
ND, not determined.
| Proteins | (EGF) (nm) | Kd (molecules/μm2) | (95% CI) | E˜ | (95% CI) |
|---|---|---|---|---|---|
| EGFR | 0 | 156 | (49–339) | 0.35 | (0.28–0.41) |
| HER2 | 0 | ND* | (ND) | 0.29 | (0.27–0.31) |
| EGFR + HER2 | 0 | ND* | (ND) | 0.26 | (0.24–0.28) |
| EGFR | 100 | ND* | (ND) | 0.29 | (0.26–0.32) |
| EGFR-L858R | 0 | 27 | (0–190) | 0.37 | (0.26–0.48) |
| IgG-Fc/EGFR chimera | 0 | ND* | (<21) | 0.29 | (0.27–0.31) |
| EGFR-I706Q | 0 | 65 | (8–183) | 0.46 | (0.39–0.53) |
| EGFR-V948R | 0 | 364 | (212–573) | 0.70 | (0.63–0.77) |
* best-fit value for Kd was less than 1 molecule per μm2. Kd is the apparent dissociation constant (molecules/μm2).
HER2 forms homo- and hetero-oligomers in the absence of ligand
FRET efficiency values for self-association of HER2-Δ25-fp and associations between HER2-Δ25-fp and EGFR-Δ107-fp were ∼0.3 and varied little with concentration, consistent with formation of constitutive ligand-independent HER2 homo-oligomers and EGFR/HER2 hetero-oligomers (Fig. 3 and Table 1). These results indicate HER2 self-associates more strongly than EGFR in the absence of ligand.
Figure 3.

HER2 forms ligand-independent homo-oligomers and hetero-oligomers. FRET efficiency for HER2-Δ25-fp homo-oligomers and hetero-oligomers. A and C, cartoon representations of fluorescent protein–linked (A) HER2-Δ25-fp and (C) HER2-Δ25-fp co-expressed with EGFR-Δ107-fp. B and D, FRET efficiency plots for (B) HER2-Δ25-fp and (D) HER2-Δ25-fp co-expressed with EGFR-Δ107-fp. Each panel contains two graphs. The graph on the left shows the FRET efficiency ((FRETapp − FRETprox)/Xa)) (y axis) as a function of concentration (x axis). The data were projected onto the y axis to yield FRET histograms, plotted at the right. FRETapp is the apparent FRET efficiency, FRETprox is the theoretical FRET efficiency that results from stochastic, nonspecific interactions, and Xa is the fraction of acceptor molecules in a given vesicle. Each dataset was derived from at least three independent biological replicates. The number of vesicles per experiment ranged from 25 to 100.
EGF-independent EGFR phosphorylation increases with increasing EGFR expression but has no measurable effect on STAT or ERK phosphorylation
Consistent with previous observations, ligand-independent EGFR oligomer formation increases at higher EGFR concentrations, in accordance with the law of mass action (Fig. 2) (23, 24). To assess the activity of EGFR oligomers formed in the absence of ligand, we generated a panel of CHO cell lines stably expressing full-length EGFR with EYFP at its C terminus (EGFR-EYFP) and sorted cells to select cell lines expressing increasing EGFR-EYFP levels. The apparent EGFR-EYFP surface concentration of selected cell lines was measured by observing fluorescence in vesicles derived from each cell line. Despite extensive effort, we were unable to isolate a cell line stably expressing EGFR-EYFP at levels higher than ∼200 receptors per μm2 on average (Fig. 4A), which is somewhat lower than the 646/μm2 reported for A431 cells, a cell line with the highest known EGFR expression level (53). This concentration is nevertheless similar to the value observed for the dissociation constant for EGFR-Δ107-fp (Table 1) and indicates that a substantial fraction of EGFR-EYFP is likely to exist in oligomers in these cell lines.
Figure 4.
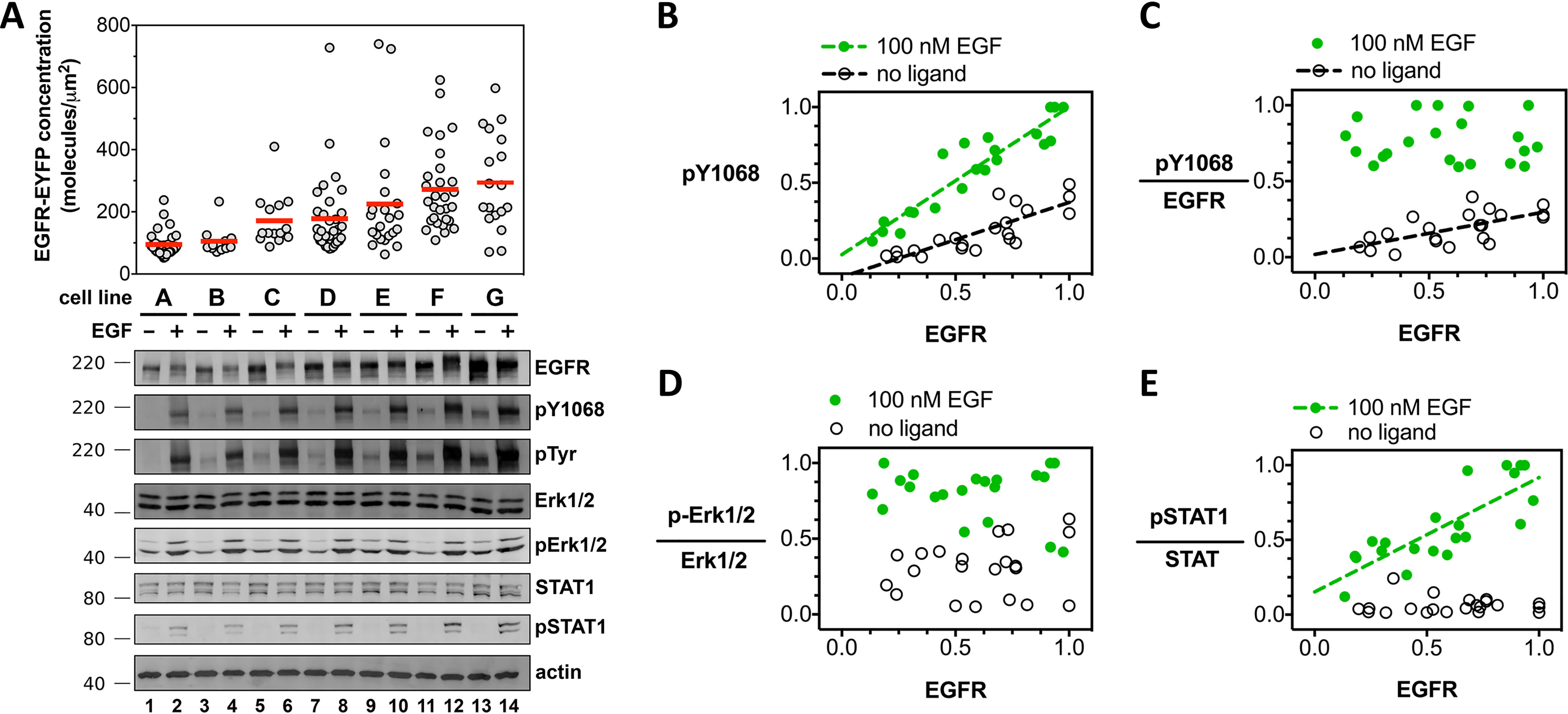
Increased EGFR cell surface concentration correlates with increased EGFR phosphorylation but not increased effector phosphorylation in the absence of ligand. A, confocal microscopy and Western blot analysis of EGF-independent and EGF-dependent EGFR signal transduction. Measurements from seven unique CHO cell lines (cell line A, B, C,… G), each stably expressing full-length EGFR-EYFP. The concentration of EGFR-EYFP, determined by confocal microscopy, is plotted on the y axis. Each gray dot corresponds to a measurement from a single vesicle. Fluorescence data are representative of three independent biological experiments. Primary antibodies are indicated to the right of the Western blots, which are representative of three independent experiments. Molecular mass markers (kDa) are indicated to the left of each Western blotting. Each of the seven stable cell lines (A–G) were treated and analyzed in parallel for a single experiment. Selected Western blot groups were quantified in ImageJ and plotted in panels B–E. All values in panels B–E represent integrated band intensities, and the highest value within the experiment for a particular axis was normalized to equal a value of 1. Green circles depict bands from wells that were treated with 100 nm EGF, white circles represent bands from wells which were left untreated. Data were fit to a straight line. Slopes that deviated significantly from zero (p < 0.002) are shown as dashed lines. B, phosphorylated-EGFR (pY-1068) is plotted on the y axis, total EGFR level on the x axis. C, phosphorylated-EGFR (pY-1068) divided by total EGFR level plotted on the y axis; total EGFR level on the x axis. D and E, phospho-Erk1/2 (D) and phospho-STAT1 (E), divided by the signal for total ERK1/2 and STAT1 proteins, respectively, as a function of total EGFR. Molecular mass markers (kDa) are indicated to the right of each Western blot. Western blots are representative of three independent biological experiments, all of which are plotted in panels B–E.
Seven cell lines selected for increasing EGFR-EYFP expression levels were treated with EGF and analyzed by Western blotting for EGFR and phosphotyrosine (Fig. 4, A–C). In the absence of ligand, EGFR phosphorylation was not detectable at concentration levels below 100 molecules/μm2. Between 100 and 300 molecules/μm2, ligand-independent phosphorylation increased in a linear fashion. Stimulation with EGF resulted in a linear increase in phosphorylation across all measured expression levels (Fig. 4C). Notably, the EGFR-EYFP expression range over which EGF-independent phosphorylation begins to become detectable (100–300 molecules/μm2) is comparable to the best fit value for the two-dimensional dissociation constant for EGFR-Δ107-fp (156 molecules/μm2) (Table 1), suggesting that concentration-dependent oligomerization might underlie ligand-independent EGFR phosphorylation.
To determine whether EGFR phosphorylation correlated with phosphorylation of downstream effectors in CHO cells, the influence of EGFR membrane concentration on two downstream signaling pathways (STAT and MAPK/Erk) was examined by Western blotting (Fig. 4). Increasing EGFR expression had no measurable effect on Erk1/2 phosphorylation or expression in the absence or presence of EGF (Fig. 4, A and D). Total STAT1 expression levels increased modestly with increasing EGFR expression, but, unlike EGFR, phosphorylation of STAT1 did not increase with increasing EGFR in the absence of ligand. The level of EGF-dependent pSTAT1 was enhanced in EGFR-YFP overexpressing cells, however (Fig. 4E). Thus, although higher EGFR concentrations result in higher basal levels of EGFR phosphorylation, this increase in ligand-independent phosphorylation does not correlate with any measurable increase in phosphorylation of downstream effectors.
Asymmetric kinase dimer interface mutations alter but do not destabilize ligand-independent EGFR oligomer ensembles
A key element of EGFR activation is formation of an asymmetric dimer interface between the N-lobe of one kinase domain and the C-lobe of another kinase domain. Single amino acid substitutions at these N-lobe (I706Q) and C-lobe (V948R) interfaces abolish EGF-dependent EGFR activity, but co-expression of these two point mutants restores activity because of their ability to complement each other's defect (37). To determine whether the asymmetric kinase dimer interface is involved in the formation of EGFR oligomers in the absence of ligand, the I706Q and V948R mutations were introduced into the EGFR-Δ107-fp variant and the oligomerization propensity of each variant measured by QI-FRET (Fig. 5). Neither variant had a strong effect on the propensity of EGFR to form ligand-independent oligomers. This is consistent with a previous observation that the EGFR-L704N variant, which disrupts the asymmetric kinase dimer interface and abolishes ligand-dependent EGFR phosphorylation, is 30–40% dimeric in the absence of ligand (54). These results contrast with previous observations that these mutations reduce ligand-dependent oligomers (41). In the absence of EGF, the dissociation constants did not significantly differ between the WT, I706Q, and V948R EGFR-Δ107-fp variants (Fig. 5, A and B and Table 1).
Figure 5.

The EGFR missense variants (I706Q and V948R) form ligand-independent oligomers that are structurally distinct from the WT oligomer. A and B, FRET efficiency for the EGFR-Δ107-fp variants I706Q (A) and V948R (B). The graphs on the left show plots of the FRET efficiency (y axis, FRETapp) as a function of receptor concentration (x axis, units in molecules per μm2) for I706Q (A) and V948R (B). Each protein is indicated above the graph. The black line shows the best fit line for EGFR-Δ107-FP from Fig. 2B. The dotted lines represent the best fit line for the I706Q (red) and V948R (blue) variants. As in Figs. 2 and 3, the FRET data in A and B were projected onto the y axis to yield FRET histograms, plotted at the right. Data were derived from three independent biological replicates, with 25–100 vesicles for each experiment.
Although these mutations did not alter EGFR oligomerization strength, they did alter the FRET efficiency of the oligomer ensemble. The FRET efficiency in the absence of ligand was much higher for both variants relative to WT EGFR-Δ107-fp, with a best-fit parameter for the intrinsic FRET of 0.46 and 0.70 for the I706Q and V948R variants, respectively, versus 0.35 for EGFR-Δ107-fp (Table 1). The FRET efficiency for both variants was also higher in the presence of ligand relative to WT EGFR-Δ107-fp in the presence of ligand (Fig. 5 and Table 1), indicating that disruption of the asymmetric kinase dimer alters the ensemble of intracellular domain interactions in ligand-driven EGFR oligomers.
Discussion
In this study we use QI-FRET to investigate homo- and hetero-interactions between EGFR and HER2 in vesicles derived from cell membranes. This approach allows measurement of FRET efficiency as a function of receptor concentration because of variable ERBB levels in vesicles derived from different cells. We observe concentration-dependent FRET efficiency values for several EGFR variants in the absence of ligand, which indicates dynamic equilibria between different EGFR association states and allows derivation of effective 2-dimensional dissociation constants. The homo-oligomerization strength of EGFR observed here, in both the presence and absence of ligand, is similar to previously published results (23, 24, 55). Mutations at the EGFR asymmetric kinase dimer interface alter the ensemble of ligand-independent EGFR oligomers, implying that ligand-independent EGFR oligomers are distinct from the active state dimer, consistent with previously published results (54). We also show that, although levels of phosphorylated EGFR increase with increasing EGFR concentrations, this phosphorylation does not correlate with any measurable increase in phosphorylation of either Erk1/2 or STAT1, indicating that receptor phosphorylation may be insufficient to trigger pathway activation as recently observed for artificially forced EGFR dimers (56, 57).
Understanding the role of ligand-independent EGFR oligomers in regulating ERBB activity remains the topic of much investigation (18, 19, 41, 54, 58–60). To estimate the oligomeric fraction of unliganded EGFR and HER2 in physiological membranes using the two-dimensional dissociation constants reported here, the concentration of ERBBs in cell membranes must be known. One recent study reported receptor concentrations in cell lines derived from human tumors as ∼150 molecules/μm2 (A549 cells) and ∼650 molecules/μm2 (A431 cells) (53). These cell lines have intermediate (A459, ∼1 × 105) and high (A431, ∼1 × 106) numbers of total receptors per cell. Uncertainty about the total surface area of a cell's plasma membrane precludes direct conversion between receptors per unit area and receptors per cell.
The presence of phosphorylated oligomeric EGFR in the absence of ligand (24, 56, 61) (Fig. 4) raises the question of whether EGFR is sampling the active dimer state, which involves a specific asymmetric dimer interaction between intracellular kinase domains (37). Amino acid substitutions at this interface that impair the ability of EGFR to become phosphorylated in response to ligand and preclude formation of the asymmetric dimer in crystals do not prohibit the formation of ligand-independent EGFR oligomers, however, implying the presence of an inactive EGFR oligomer that is distinct from the active dimer. This inactive oligomer appears to depend on the EGFR intracellular domain as deletion of intracellular domain results in loss or decrease of EGFR association as judged by FRET (62), single-molecule tracking (63), chemical crosslinking (64), and loss of negative cooperativity in a saturation binding experiment (18). The ERBB extracellular domains must also play a role in mediating or favoring this inactive oligomer; deletion of the extracellular domains leads to constitutive receptor phosphorylation (35), and mutations within the interface of the extended ECD dimer destabilize ligand-independent oligomers (23, 63). No conserved interaction between the ERBB extracellular domains has been observed in crystals of tethered forms of ERBB ECDs, however, and it is not clear how they inhibit receptor activity in the absence of ligand.
In contrast, a number of interactions observed in crystal structures of inactive ERBB kinase forms have been suggested as reflective of inactive ERBB dimers (39, 54, 65, 66). A consensus has yet to emerge on the physiological relevance of these dimers, however, and truncation of ERBB C-terminal tail domains in crystallized forms of inactive ERBB kinases may remove essential components of physiological interactions in the inactive state. Regions of the EGFR C-tail immediately following the kinase domain appear to play a role in stabilizing an inactive state as deletions or mutations in this region lead to enhanced or ligand-independent receptor activity (29, 67, 68). An attractive hypothesis is that the kinase distal region interacts with the kinase C-lobe in a fashion that competes with interactions made in the asymmetric kinase dimer interface (68). If so, this contact between the kinase and the tail might stabilize the inactive oligomer, which could underlie the observation that the kinase C-lobe mutation (V948R) alters the inactive oligomer ensemble as reflected in an increase in maximal FRET efficiency.
Conversion from inactive to active forms of ERBBs clearly involves coupled rearrangements and interactions between many structural elements. We provide evidence here that in the absence of ligand a substantial fraction of EGFR exists in an inactive oligomeric state that is distinct from the active state. We observe that HER2 is predominantly oligomeric in the plasma membrane, but our method does not resolve any structural or functional features of this oligomeric state. Future studies are needed to determine the structure and function of ligand-independent EGFR and HER2 oligomers and how they contribute to regulating receptor activity in normal and disease states.
Materials and methods
Plasmid construction
The coding sequences for variants of EGFR, HER2, EYFP, and mCherry were amplified using the PCR and cloned into pCDNA3.1(+). Fluorescent protein coding sequences were first inserted between the KpnI and XbaI sites. The coding sequences for EGFR and HER2 were then inserted between the NheI and KpnI sites. The dimerization-impaired EYFP-A206K variant was used. The missense variants EGFR-I607Q, EGFR-V948R, EGFR-L858R were generated using site-directed mutagenesis.
Cell culture and transfection
CHO cells and A431 cells were maintained in Dulbecco's modified Eagle Medium (DMEM F-12) supplemented with 2 mm glutamine, 5% FBS and were grown at 37°C, 5% CO2. Both cell lines were purchased from ATCC. For imaging experiments, CHO cells were seeded in 35-mm dishes at a density of 2 × 104 cells per well and grown for 24 h prior to transfection. Cells were transfected with plasmids using Lipofectamine 3000 (Life Technologies) according to the manufacturer's protocol.
Generation of monoclonal CHO cell lines expressing EGFR-EYFP
CHO cells stably expressing EGFR-EYFP were generated using the pCDNA3.1(+) manual as a guide. Briefly, cells transfected with EGFR-EYFP in the pCDNA3.1(+) vector were grown for several days in medium without antibiotics. G418 was added to a final concentration of 1 mg/ml and the cells were cultured until individual colonies appeared. All colonies were pooled into a single polyclonal culture, from which monoclonal populations were derived using FACS on a FACSCalibur instrument (BD Biosciences). Resulting cell lines were maintained in DMEM F-12 supplemented with 10% FBS, 1 mg/ml G418, and 25 mm HEPES, pH 7.2. Stable expression of EGFR-EYFP in these cell lines was assayed by confocal microscopy and Western blotting (see below).
Vesiculation, image acquisition, and analysis
All vesiculation procedures were performed as described in Refs. 48, 49, and 69. For ligand addition experiments, EGF was added to the vesicles to a final concentration of 100 nm and incubated for at least 1 h prior to imaging. Vesicle images were acquired using a Nikon C1 laser scanning confocal microscope at 60× magnification (water immersion objective). For each vesicle, three scans were recorded: a donor scan (λexc = 488 nm, λem = 500–530 nm), an acceptor scan (λexc = 543 nm, λem = 650 nm longpass), and a FRET scan (λexc = 488 nm, λem = 565-615 nm). Argon (488 nm) and He-Ne lasers were used as excitation sources. The image size was 512 × 512 pixels. The pixel dwell time was 1.68 μs, and the gains were set to 8. The Förster radius for the EYFP-mCherry pair was calculated to be 53.1 Å. All images were processed using a MATLAB program written in the Hristova laboratory which calculates the FRET efficiency for each vesicle. A detailed description of the analysis is found elsewhere (46, 47). Absolute protein concentration in the membrane was calculated by comparing the fluorescence intensity in vesicles with the intensities measured from a dilution series of fluorescent protein standards (EYFP and mCherry). Bleed-through coefficients were calculated for each experiment (typically ∼0.3 and ∼0.2 for EYFP and mCherry, respectively). Where appropriate, the processed data (FRET versus concentration) were fit to a monomer-dimer equilibrium model using GraphPad Prism:
where the dimer dissociation constant, Kd, is
and the total receptor concentration, [T] is a function of the concentration of monomers, [M], and dimers, [D]
The dimeric fraction for each vesicle, fD, is given by the equation
where apparent Eapp is the apparent measured FRET efficiency, Xa is the fraction of acceptor molecules in a given vesicle, and E˜ is the FRET value within a dimer.
EGF stimulation of ERBB proteins in CHO cells
Recombinant human EGF was expressed in Escherichia coli and purified as described (70). Briefly, human EGF bearing an N-terminal hexa-histidine tag was purified using immobilized metal affinity chromatography. The hexa-histidine tag was removed by proteolysis with tobacco etch virus protease. Soluble, monomeric, human EGF was separated from tobacco etch virus protease by size-exclusion chromatography, concentrated, and flash frozen in liquid nitrogen. CHO cells were grown to 90% confluency, then transfected and grown for 14 h, then serum starved for 7 h at 37°C in Ham's F-12 supplemented with 1 mg/ml BSA. The cells were incubated in starvation media in the presence or absence of 100 nm EGF (5 min, 37°C). The medium was aspirated, the cells were washed twice with cold PBS (1×, plus 1 mm Na3VO4), then lysed for 15–20 min at room temperature in RIPA buffer (150 mm NaCl, 50 mm Tris, pH 8, 1% Nonidet P-40, 0.5% w/v sodium deoxycholate, 0.1% SDS, 1 mm Na3VO4). The lysates were clarified by centrifugation and the total protein concentration of the supernatants was determined by BCA assay (Pierce, Life Technologies). Lysate protein concentrations were normalized, and the samples were analyzed by SDS-PAGE and Western blotting to detect phosphotyrosine (4G10, Millipore), EGFR (D38B1), EGFR pY1068, HER2 (29D8), or HER2-pY1221/1222 (all ERBB antibodies purchased from Cell Signaling Technology).
Data Availability
All data are included in this manuscript.
Supplementary Material
Acknowledgments
Lily Raines, Nuala Del Piccolo and Chris King provided technical support with protein purification and fluorescence microscopy. Xiaoling Zhang of the Ross Flow Cytometry Core (Johns Hopkins University, School of Medicine) assisted with fluorescence activated cell sorting.
This article contains supporting information.
Author contributions—P. O. B. conceptualization; P. O. B. data curation; P. O. B. formal analysis; P. O. B. and K. H. validation; P. O. B. investigation; P. O. B. visualization; P. O. B. and K. H. methodology; P. O. B. writing-original draft; P. O. B. and K. H. project administration; P. O. B., K. H., and D. J. L. writing-review and editing; K. H. software; K. H. supervision; K. H. and D. J. L. funding acquisition; D. J. L. resources.
Funding and additional information—This work was supported by National Institutes of Health Grant 5R01GM099321-17 and Cancer Prevention Research Institute of Texas (CPRIT) Grant RR160023 (to P.O.B. and D.J.L.) and by National Science Foundation (NSF) Grant MCB-1712740 (to K. H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- EGFR
- epidermal growth factor receptor
- RTK
- receptor tyrosine kinase
- HER
- human epidermal grown factor receptor
- ECD
- extracellular domain
- TMD
- transmembrane domain
- CHO
- Chinese hamster ovary
- fp
- fluorescent protein
- QI-FRET
- quantitative imaging FRET.
References
- 1. Lemmon M. A., and Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sacco A. G., and Worden F. P. (2016) Molecularly targeted therapy for the treatment of head and neck cancer: A review of the ErbB family inhibitors. OncoTargets Ther. 9, 1927–1943 10.2147/OTT.S93720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Endres N. F., Engel K., Das R., Kovacs E., and Kuriyan J. (2011) Regulation of the catalytic activity of the EGF receptor. Curr. Opin. Struct. Biol. 21, 777–784 10.1016/j.sbi.2011.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yarden Y., and Schlessinger J. (1987) Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 26, 1443–1451 10.1021/bi00379a035 [DOI] [PubMed] [Google Scholar]
- 5. Yarden Y., and Schlessinger J. (1987) Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 26, 1434–1442 10.1021/bi00379a034 [DOI] [PubMed] [Google Scholar]
- 6. Todaro G. J., De Larco J. E., and Cohen S. (1976) Transformation by murine and feline sarcoma viruses specifically blocks binding of epidermal growth factor to cells. Nature 264, 26–31 10.1038/264026a0 [DOI] [PubMed] [Google Scholar]
- 7. Fabricant R. N., De Larco J. E., and Todaro G. J. (1977) Nerve growth factor receptors on human melanoma cells in culture. Proc. Natl. Acad. Sci. U. S. A. 74, 565–569 10.1073/pnas.74.2.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holley R. W., Armour R., Baldwin J. H., Brown K. D., and Yeh Y. C. (1977) Density-dependent regulation of growth of BSC-1 cells in cell culture: control of growth by serum factors. Proc. Natl. Acad. Sci. U. S. A. 74, 5046–5050 10.1073/pnas.74.11.5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carpenter G., and Cohen S. (1978) Biological and molecular studies of the mitogenic effects of human epidermal growth factor. Symp. Soc. Dev. Biol. 35, 13–31 10.1016/b978-0-12-612981-6.50007-7 [DOI] [PubMed] [Google Scholar]
- 10. Rees A. R., Adamson E. D., and Graham C. F. (1979) Epidermal growth factor receptors increase during the differentiation of embryonal carcinoma cells. Nature 281, 309–311 10.1038/281309a0 [DOI] [PubMed] [Google Scholar]
- 11. Green M. R., Basketter D. A., Couchman J. R., and Rees D. A. (1983) Distribution and number of epidermal growth factor receptors in skin is related to epithelial cell growth. Dev. Biol. 100, 506–512 10.1016/0012-1606(83)90243-9 [DOI] [PubMed] [Google Scholar]
- 12. Filmus J., Pollak M. N., Cailleau R., and Buick R. N. (1985) MDA-468, a human breast cancer cell line with a high number of epidermal growth factor (EGF) receptors, has an amplified EGF receptor gene and is growth inhibited by EGF. Biochem. Biophys. Res. Commun. 128, 898–905 10.1016/0006-291X(85)90131-7 [DOI] [PubMed] [Google Scholar]
- 13. Cowley G. P., Smith J. A., and Gusterson B. A. (1986) Increased EGF receptors on human squamous carcinoma cell lines. Br. J. Cancer 53, 223–229 10.1038/bjc.1986.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davidson N. E., Gelmann E. P., Lippman M. E., and Dickson R. B. (1987) Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol. Endocrinol. 1, 216–223 10.1210/mend-1-3-216 [DOI] [PubMed] [Google Scholar]
- 15. Di Fiore P. P., Pierce J. H., Fleming T. P., Hazan R., Ullrich A., King C. R., Schlessinger J., and Aaronson S. A. (1987) Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell 51, 1063–1070 10.1016/0092-8674(87)90592-7 [DOI] [PubMed] [Google Scholar]
- 16. Kozer N., Henderson C., Jackson J. T., Nice E. C., Burgess A. W., and Clayton A. H. (2011) Evidence for extended YFP-EGFR dimers in the absence of ligand on the surface of living cells. Phys. Biol. 8, 066002 10.1088/1478-3975/8/6/066002 [DOI] [PubMed] [Google Scholar]
- 17. Liu P., Sudhaharan T., Koh R. M., Hwang L. C., Ahmed S., Maruyama I. N., and Wohland T. (2007) Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 93, 684–698 10.1529/biophysj.106.102087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Macdonald J. L., and Pike L. J. (2008) Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U. S. A. 105, 112–117 10.1073/pnas.0707080105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin-Fernandez M., Clarke D. T., Tobin M. J., Jones S. V., and Jones G. R. (2002) Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 82, 2415–2427 10.1016/S0006-3495(02)75585-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moriki T., Maruyama H., and Maruyama I. N. (2001) Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 311, 1011–1026 10.1006/jmbi.2001.4923 [DOI] [PubMed] [Google Scholar]
- 21. Nagy P., Claus J., Jovin T. M., and Arndt-Jovin D. J. (2010) Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. U. S. A. 107, 16524–16529 10.1073/pnas.1002642107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saffarian S., Li Y., Elson E. L., and Pike L. J. (2007) Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 93, 1021–1031 10.1529/biophysj.107.105494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stoneman M. R., Biener G., Ward R. J., Pediani J. D., Badu D., Eis A., Popa I., Milligan G., and Raicu V. (2019) A general method to quantify ligand-driven oligomerization from fluorescence-based images. Nat. Methods 16, 493–496 10.1038/s41592-019-0408-9 [DOI] [PubMed] [Google Scholar]
- 24. Kim D. H., Park S., Kim D. K., Jeong M. G., Noh J., Kwon Y., Zhou K., Lee N. K., and Ryu S. H. (2018) Direct visualization of single-molecule membrane protein interactions in living cells. PLoS Biol. 16, e2006660 10.1371/journal.pbio.2006660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., Louis D. N., Christiani D. C., Settleman J., and Haber D. A. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- 26. Padfield E., Ellis H. P., and Kurian K. M. (2015) Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 5, 5 10.3389/fonc.2015.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pahuja K. B., Nguyen T. T., Jaiswal B. S., Prabhash K., Thaker T. M., Senger K., Chaudhuri S., Kljavin N. M., Antony A., Phalke S., Kumar P., Mravic M., Stawiski E. W., Vargas D., Durinck S., et al. (2018) Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 34, 792–806.e795 10.1016/j.ccell.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penuel E., Akita R. W., and Sliwkowski M. X. (2002) Identification of a region within the ErbB2/HER2 intracellular domain that is necessary for ligand-independent association. J. Biol. Chem. 277, 28468–28473 10.1074/jbc.M202510200 [DOI] [PubMed] [Google Scholar]
- 29. Pines G., Huang P. H., Zwang Y., White F. M., and Yarden Y. (2010) EGFRvIV: A previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 29, 5850–5860 10.1038/onc.2010.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stephens P., Hunter C., Bignell G., Edkins S., Davies H., Teague J., Stevens C., O'Meara S., Smith R., Parker A., Barthorpe A., Blow M., Brackenbury L., Butler A., Clarke O., et al. (2004) Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526 10.1038/431525b [DOI] [PubMed] [Google Scholar]
- 31. Burgess A. W., Cho H. S., Eigenbrot C., Ferguson K. M., Garrett T. P., Leahy D. J., Lemmon M. A., Sliwkowski M. X., Ward C. W., and Yokoyama S. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552 10.1016/S1097-2765(03)00350-2 [DOI] [PubMed] [Google Scholar]
- 32. Diwanji D., Thaker T., and Jura N. (2019) More than the sum of the parts: Toward full-length receptor tyrosine kinase structures. IUBMB Life 71, 706–720 10.1002/iub.2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bouyain S., Longo P. A., Li S., Ferguson K. M., and Leahy D. J. (2005) The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. U. S. A. 102, 15024–15029 10.1073/pnas.0507591102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho H. S., and Leahy D. J. (2002) Structure of the extracellular region of HER3 reveals an interdomain tether. Science 297, 1330–1333 10.1126/science.1074611 [DOI] [PubMed] [Google Scholar]
- 35. Endres N. F., Das R., Smith A. W., Arkhipov A., Kovacs E., Huang Y., Pelton J. G., Shan Y., Shaw D. E., Wemmer D. E., Groves J. T., and Kuriyan J. (2013) Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556 10.1016/j.cell.2012.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H. S., Leahy D. J., and Lemmon M. A. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 10.1016/S1097-2765(03)00047-9 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X., Gureasko J., Shen K., Cole P. A., and Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 10.1016/j.cell.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 38. Arkhipov A., Shan Y., Das R., Endres N. F., Eastwood M. P., Wemmer D. E., Kuriyan J., and Shaw D. E. (2013) Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569 10.1016/j.cell.2012.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landau M., Fleishman S. J., and Ben-Tal N. (2004) A putative mechanism for downregulation of the catalytic activity of the EGF receptor via direct contact between its kinase and C-terminal domains. Structure 12, 2265–2275 10.1016/j.str.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 40. Wehrman T. S., Raab W. J., Casipit C. L., Doyonnas R., Pomerantz J. H., and Blau H. M. (2006) A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. Proc. Natl. Acad. Sci. U. S. A. 103, 19063–19068 10.1073/pnas.0605218103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang Y., Bharill S., Karandur D., Peterson S. M., Marita M., Shi X., Kaliszewski M. J., Smith A. W., Isacoff E. Y., and Kuriyan J. (2016) Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 5, e14107 10.7554/eLife.14107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evers T. H., van Dongen E. M., Faesen A. C., Meijer E. W., and Merkx M. (2006) Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry 45, 13183–13192 10.1021/bi061288t [DOI] [PubMed] [Google Scholar]
- 43. Opresko L. K., and Wiley H. S. (1990) Functional reconstitutional of the human epidermal growth factor receptor system in Xenopus oocytes. J. Cell Biol. 111, 1661–1671 10.1083/jcb.111.4.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reddy C. C., Wells A., and Lauffenburger D. A. (1996) Receptor-mediated effects on ligand availability influence relative mitogenic potencies of epidermal growth factor and transforming growth factor alpha. J. Cell. Physiol. 166, 512–522 [DOI] [PubMed] [Google Scholar]
- 45. Li E., Placone J., Merzlyakov M., and Hristova K. (2008) Quantitative measurements of protein interactions in a crowded cellular environment. Anal. Chem. 80, 5976–5985 10.1021/ac800616u [DOI] [PubMed] [Google Scholar]
- 46. Chen L., Novicky L., Merzlyakov M., Hristov T., and Hristova K. (2010) Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 132, 3628–3635 10.1021/ja910692u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen L., Placone J., Novicky L., and Hristova K. (2010) The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal. 3, ra86 10.1126/scisignal.2001195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sarabipour S., Chan R. B., Zhou B., Di Paolo G., and Hristova K. (2015) Analytical characterization of plasma membrane-derived vesicles produced via osmotic and chemical vesiculation. Biochim. Biophys. Acta 1848, 1591–1598 10.1016/j.bbamem.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kavran J. M., McCabe J. M., Byrne P. O., Connacher M. K., Wang Z., Ramek A., Sarabipour S., Shan Y., Shaw D. E., Hristova K., Cole P. A., and Leahy D. J. (2014) How IGF-1 activates its receptor. Elife 3, e03772 10.7554/eLife.03772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wolber P. K., and Hudson B. S. (1979) An analytic solution to the Forster energy transfer problem in two dimensions. Biophys. J. 28, 197–210 10.1016/S0006-3495(79)85171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. King C., Raicu V., and Hristova K. (2017) Understanding the FRET signatures of interacting membrane proteins. J. Biol. Chem. 292, 5291–5310 10.1074/jbc.M116.764282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valley C. C., Arndt-Jovin D. J., Karedla N., Steinkamp M. P., Chizhik A. I., Hlavacek W. S., Wilson B. S., Lidke K. A., and Lidke D. S. (2015) Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol. Biol. Cell 26, 4087–4099 10.1091/mbc.E15-05-0269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang F., Wang S., Yin L., Yang Y., Guan Y., Wang W., Xu H., and Tao N. (2015) Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal. Chem. 87, 9960–9965 10.1021/acs.analchem.5b02572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zanetti-Domingues L. C., Korovesis D., Needham S. R., Tynan C. J., Sagawa S., Roberts S. K., Kuzmanic A., Ortiz-Zapater E., Jain P., Roovers R. C., Lajevardipour A., van Bergen En Henegouwen P. M. P., Santis G., Clayton A. H. A., Clarke D. T., et al. (2018) The architecture of EGFR's basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 9, 4325 10.1038/s41467-018-06632-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh D. R., King C., Salotto M., and Hristova K. (2020) Revisiting a controversy: The effect of EGF on EGFR dimer stability. Biochim. Biophys. Acta Biomembr. 1862, 183015 10.1016/j.bbamem.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang S. I., van Lengerich B., Eichel K., Cha M., Patterson D. M., Yoon T. Y., von Zastrow M., Jura N., and Gartner Z. J. (2018) Phosphorylated EGFR dimers are not sufficient to activate ras. Cell Rep. 22, 2593–2600 10.1016/j.celrep.2018.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoshida T., Okamoto I., Okabe T., Iwasa T., Satoh T., Nishio K., Fukuoka M., and Nakagawa K. (2008) Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int. J. Cancer 122, 1530–1538 10.1002/ijc.23253 [DOI] [PubMed] [Google Scholar]
- 58. Ward C. W., Lawrence M. C., Streltsov V. A., Adams T. E., and McKern N. M. (2007) The insulin and EGF receptor structures: New insights into ligand-induced receptor activation. Trends Biochem. Sci. 32, 129–137 10.1016/j.tibs.2007.01.001 [DOI] [PubMed] [Google Scholar]
- 59. Macdonald-Obermann J. L., Adak S., Landgraf R., Piwnica-Worms D., and Pike L. J. (2013) Dynamic analysis of the epidermal growth factor (EGF) receptor-ErbB2-ErbB3 protein network by luciferase fragment complementation imaging. J. Biol. Chem. 288, 30773–30784 10.1074/jbc.M113.489534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Purba E. R., Saita E. I., and Maruyama I. N. (2017) Activation of the EGF Receptor by ligand binding and oncogenic mutations: The “rotation model” Cells 6, 13 10.3390/cells6020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. King C., Stoneman M., Raicu V., and Hristova K. (2016) Fully quantified spectral imaging reveals in vivo membrane protein interactions. Integr. Biol. (Camb) 8, 216–229 10.1039/c5ib00202h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Freed D. M., Bessman N. J., Kiyatkin A., Salazar-Cavazos E., Byrne P. O., Moore J. O., Valley C. C., Ferguson K. M., Leahy D. J., Lidke D. S., and Lemmon M. A. (2017) EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell 171, 683–695.e618 10.1016/j.cell.2017.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., and Mellman I. (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787 10.1038/nature08827 [DOI] [PubMed] [Google Scholar]
- 64. Yu X., Sharma K. D., Takahashi T., Iwamoto R., and Mekada E. (2002) Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 13, 2547–2557 10.1091/mbc.01-08-0411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., and Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 10.1016/j.cell.2009.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jura N., Shan Y., Cao X., Shaw D. E., and Kuriyan J. (2009) Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. U. S. A. 106, 21608–21613 10.1073/pnas.0912101106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kovacs E., Das R., Wang Q., Collier T. S., Cantor A., Huang Y., Wong K., Mirza A., Barros T., Grob P., Jura N., Bose R., and Kuriyan J. (2015) Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol. Cell Biol. 35, 3083–3102 10.1128/MCB.00248-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang R. Y., Yang K. S., Pike L. J., and Marshall G. R. (2010) Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chem. Biol. Drug Des. 76, 1–9 10.1111/j.1747-0285.2010.00986.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Del Piccolo N., Placone J., He L., Agudelo S. C., and Hristova K. (2012) Production of plasma membrane vesicles with chloride salts and their utility as a cell membrane mimetic for biophysical characterization of membrane protein interactions. Anal. Chem. 84, 8650–8655 10.1021/ac301776j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qiu C., Tarrant M. K., Boronina T., Longo P. A., Kavran J. M., Cole R. N., Cole P. A., and Leahy D. J. (2009) In vitro enzymatic characterization of near full length EGFR in activated and inhibited states. Biochemistry 48, 6624–6632 10.1021/bi900755n [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in this manuscript.