

Abstract
In recent years, the field of natural products has seen an explosion in the breadth, resolution and accuracy of profiling platforms for compound discovery, including many new chemical and biological annotation methods. With these new tools come opportunities to examine extract libraries using systematized profiling approaches that were not previously available to the field, and which offer new approaches for the detailed characterization of the chemical and biological attributes of complex natural products mixtures. This review will present a summary of some of these untargeted profiling methods, and provide perspective on the future opportunities offered by integrating these profiling methods for novel natural products discovery.
Graphical Abstract
INTRODUCTION
Natural products have historically played a major role in the discovery and development of a diverse array of therapeutics including antibiotics, anticancer agents, antifungal drugs and analgesics. The modern era of natural products discovery has been driven in large part by continued innovation in both bioassay screening systems and analytical methods for the discovery of secondary metabolites with unique structures and biological properties. These efforts have led to an impressive diversity of new drugs, and the discovery of countless bioactive small molecules with value as chemical probes and sources of inspiration for medicinal chemistry campaigns. However, despite significant developments in these areas, natural products discovery is still challenged by a number of issues that have hampered the field for over 50 years.
In 1981, Drs. Matthew Suffness and John Douros from the U.S. National Cancer Institute published an opinion piece in Trends in Pharmacological Sciences,1 in which they presented some of the problems and solutions associated with the then “new” field of anticancer drug discovery from natural sources, and discussed their outlook for the future. Reading their paper, it is remarkable how many of the challenges they identified remain substantial barriers to efficient discovery of bioactive natural products today. In this review of strategies for high-content biological and chemical characterization in natural product discovery, we will begin by revisiting some of the issues raised by Drs. Suffness and Douros in 1981, and briefly discuss our interpretations of these issues for the field as we see them in 2015.
“…most active materials are undetectable, and those that are tend to be discovered repeatedly.”
The issue of re-isolation was a problem then, and remains a significant challenge today. Despite dramatic advances in analytical hardware (high-field cryoprobe NMR and benchtop accurate mass LC-MS systems) it is a rare student that has not isolated a known compound at some time during their Ph.D. studies. Owing to of the large number of compounds now isolated from natural sources, rediscovery is becoming the norm rather than the exception in many instances. A number of metabolomics approaches have been developed to circumvent this issue, as will be discussed in more detail below, but new methods are still required to integrate these approaches with biological data in order to identify compounds with the highest value as novel bioactive lead compounds.
“…cytotoxicity tests are sensitive to any cell killing substance and give many false leads.”
Traditional colorimetric live/dead assays say nothing about target, with the result that active extracts from these assays must be selected based on raw potency, rather than mechanistic behavior. Given that even some new compounds will likely hit targets for which there are already drugs on the market, it is important that modern natural products discovery programs take advantage of multi-parametric profiling tools for screening where possible, and use these methods for the targeted discovery of compounds with novel biological functions. A number of unbiased biological profiling platforms are discussed below, including examples of their use for the discovery or characterization of natural products with unique biological properties.
“The design and development of in vitro screens which are specific for detection of key mechanisms of drug action is a challenging task.”
This issue has largely been resolved, thanks to the development of a vast array of target- and pathway-based high-throughput screening platforms. However, because many of these assay platforms are relatively complex or time-consuming to run, it is still true that mechanistic assays are hard to implement broadly. There is therefore still a need for the creation of new unbiased screening tools that characterize bioactive extracts in terms of broad mode of action (MOA) classifications, as a complement to the two extremes of live/dead cytotoxicity, and target-based screening methods.
“The isolation and purification of active compounds present in minute quantities in a crude extract is a time consuming and difficult task…”
Just as was true in 1981, natural products discovery remains difficult! Despite the advances in hardware mentioned above and the development of numerous derivatization, labeling, and analytical methods for compound identification, detailed and unequivocal determination of the constitution and configuration of complex natural products is a time-consuming task that typically requires a significant investment of resources and material. The development of integrated tools that consider both biological MOA predictions and chemical constitution of natural products extracts is beginning to provide solutions to this issue by ensuring that compounds selected for full structural characterization are of the highest priority in terms of both structural and/or biological novelty. The third section of this review will discuss this integrated approach, including both the advantages and current limitations of these strategies.
If the 20th century was the age of structure-driven natural products discovery, then the 21st century promises to be the age of function-driven natural products research. There remains a high degree of value in “old” natural products for which the biological attributes remain poorly characterized, but deriving accurate functional information for natural products libraries on a global scale remains a major challenge. This review will cover methods for untargeted chemical and biological characterization, and will present a perspective on future directions for the integration of these analytical platforms for the de novo prediction of natural product structures and MOAs from complex screening libraries.
CHEMICAL CHARACTERIZATION STRATEGIES
Preamble.
Chemical characterization of natural products has progressed dramatically from early studies, which relied heavily on degradation, derivatization and the synthesis of structural subunits to solve chemical structures2 to the modern scenario where even the largest and most complex structures can be determined using microscale analytical techniques.3,4 Although many of these methods have seen incredible development since the creation of the earliest instruments5–7 this review will focus on the broad characterization of natural product libraries, rather than the development of techniques to aid in the structure determination for individual compounds. For recent reviews of the development of MS technologies and the use of NMR-based metabolomics in natural products, see Carter,8 Jarmusch and Cooks,7 and Robinette et al.9
Thin-layer chromatography (TLC) emerged as the first method for parallelized characterization of natural product extracts, and is still widely used as a rapid, low-resolution method for profiling chemical constitution of natural product extracts; however, high-performance liquid chromatography (HPLC) and hyphenated techniques have all but completely replaced TLC for most natural products discovery applications, because of their increased resolution and greater information content.10–12 The use of HPLC retention time in combination with ultraviolet and visible absorbance spectra allows the profiling and comparison of extracts within any screening library and has been used widely by industry and academia. In an early example Miller et al. used stream splitting and automated fraction collection in a compound-by-compound bioactivity and dereplication process for the discovery of clavulanic acid,13,14 paving the way for adoption of this approach by many other research groups. The primary disadvantages of these techniques are that HPLC protocols are time-consuming, analysis and dereplication are performed on a compound-by-compound basis, and saving fractions is not practical for large libraries.10
The rapid improvement in resolution and throughput introduced by ultra-performance liquid chromatography (UPLC), bench-top HRMS, and advances in NMR experimentation and technologies like 1.7 mm cryogenic NMR probes have recently changed the chemical characterization landscape of natural products libraries from a compound-by-compound dereplication process to a situation where analysis can reveal an unbiased global view of all metabolites in a given library, as will be described below.
Mass Spectrometric Profiling Methods.
Owing to its sensitivity and relatively high throughput, MS-based techniques have come to the forefront of rapid chemical characterization. Studies have demonstrated the coverage and accuracy of such techniques for representative fungal compound libraries.15 The use of multivariate statistical methods such as principal component analysis has also been used to discover unique compounds from MS-based untargeted analysis of libraries of Myxococcus xanthus strains and Ascidian-associated Actinomycetales.16,17 Similarly, traditional metabolomics platforms including versions of XCMS have been used to discover novel compounds from organisms as well studied as Streptomyces coelicolor.18 In this last study structural characterization was assisted by the use of tandem MS, which allowed structural information to be incorporated into MS-based dereplication and discovery. More recently, MS2 fragmentation pattern matching has been used to develop Molecular Networking as a dereplication strategy for identifying known compounds and ascribing structural classes to unknown metabolites.19,20 The use of MS fragmentation patterns for compound identification is a standard tool in traditional metabolomics analysis (e.g., electron impact fragmentation in most GC-MS systems). However, the use of relative mass differences in fragmentation spectra to connect compounds from a given structural family, coupled with network analysis to visualize the relatedness of analytes in a given sample set, provide new opportunities for the rapid characterization and visualization of the metabolic capacity of sets of samples regardless of source origin or the availability of pure compound standards for every analyte. Finally, Müller and co-workers have developed a new approach to the acquisition of MS2 data for complex natural product samples, which generates a “scheduled precursor list” of features present in extracts of microbial cultures but not the corresponding medium blanks, and uses this list to direct subsequent MS2 data acquisition.21 Advances such as this improve the coverage of relevant molecules over traditional MS2 selection methods that rely on signal intensity for fragmentation selection, and are indicative of the new approaches to data acquisition being developed and are moving the field towards the comprehensive untargeted metabolomics profiling of complex natural products mixtures.
Nuclear Magnetic Resonance Profiling Methods.
While less common than MS-based techniques, developments in NMR experimentation and instrumentation have led to a significant rise in the use of NMR-based metabolomics for the profiling of crude extracts in recent years. The discovery of iotrochotrazine by 1H NMR comparisons of extracts enriched for compounds obeying Lipinski’s “rule of five” exemplifies the utility of this strategy.22 Similar to MS approaches, standardized acquisition and databases can be used to identify chemical constituents from crude mixtures.23 The primary advantages of NMR-based chemical profiling over MS-based strategies are that (1) the analysis is quantitative, unlike MS-based approaches where poor ionization or ion suppression by other metabolites can preclude the observation of all constituents in an extract, and that (2) structural information is more readily derived from the data, particularly if 1H spectra are augmented with TOCSY or phase-sensitive HSQC experiments. The structural information inherent in two-dimensional (2D) experiments has been used extensively for the characterization of chemical components of insect and spider venom, fireflies, and ladybugs.24–27 Integration of NMR spectroscopy with biological data has been used to identify pheromones in Caenorphabditis elegans through differential analysis by 2D NMR spectroscopy (DANS).28 Similar to MS-based metabolomics strategies, this study was able to identify specific signals corresponding to the ascarosides that have synergistic effects with other pheromones and were therefore unidentifiable by activity-guided fractionation. This elegant approach lays the foundation for integrating biological and chemical profiling for the discovery of molecules correlated with a specific phenotype in a given biological assay.
PROFILING STRATEGIES FOR BIOLOGICAL CHARACTERIZATION
Initial Remarks.
Natural product screening has made significant progress since the early development of disk diffusion assays for microbial pathogens and colorimetric live/dead screens for mammalian cell lines. Recent developments in screening hardware and informatics now offer a wealth of readily accessible tools for the detailed biological characterization of compound libraries against almost any target system. These advances are providing opportunities for the early mode of action (MOA) prediction for bioactive compounds, which in turn is driving a “function-first” selection process for lead discovery and development (Figure 2).29
Figure 2.
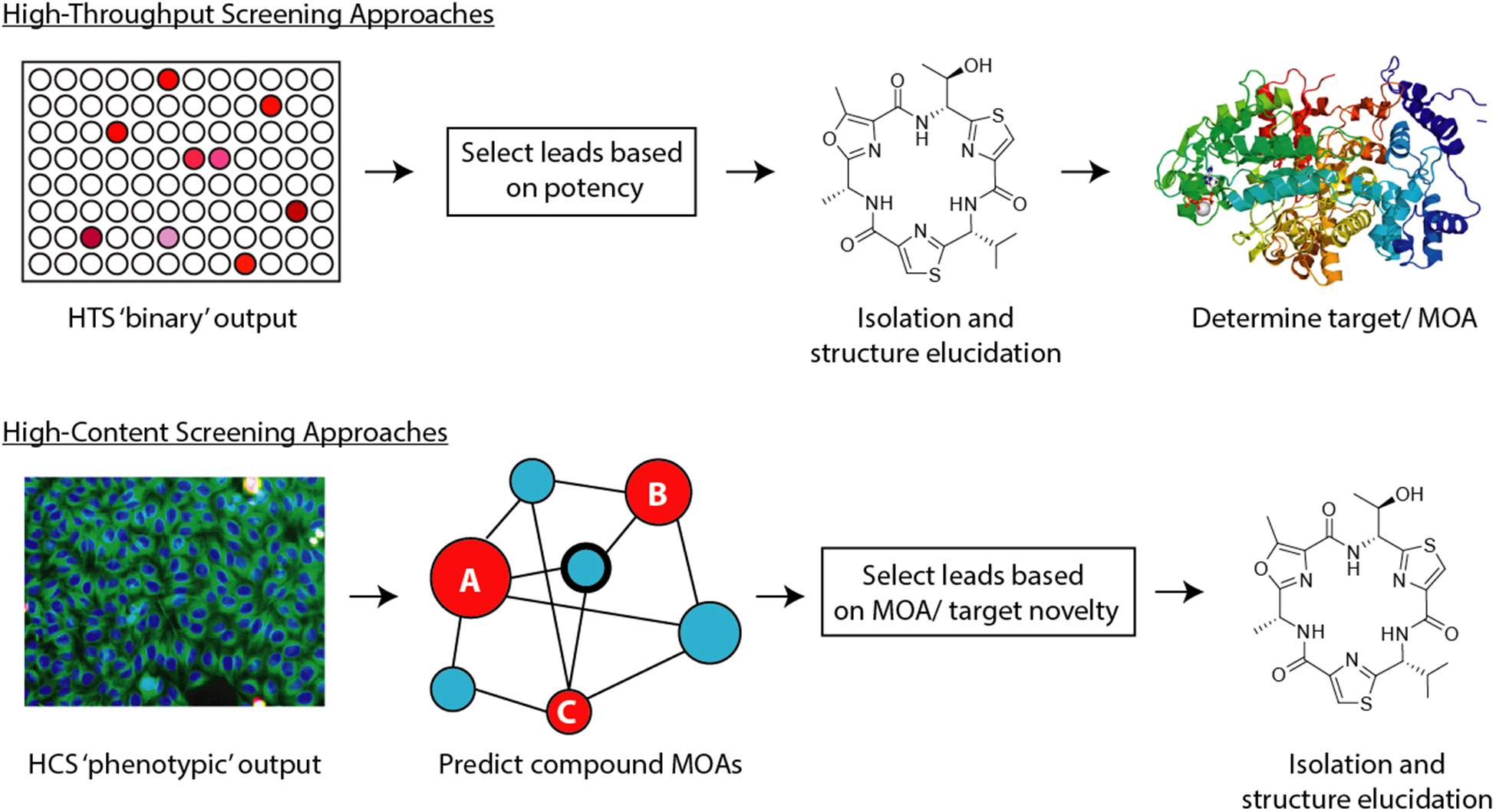
High-throughput versus multiparametric screening strategies, illustrating the differences in lead selection approaches and discovery workflows between live/dead and multiparametric screening methods.
Although there are many examples of innovative screening systems for specific molecular targets and processes, we will restrict our focus in this review to unbiased assay systems that offer tools for the broad classification of bioactive compounds independent of specific MOAs, because of the inherent value that these tools offer the natural products chemist in terms of early global characterization of complex natural product libraries. Within this general area, the majority of development has been focused on four main target systems: mammalian cancer cell lines, yeast, bacteria and early vertebrate models. Each of these will be discussed in turn, highlighting recent advances and the advantages and limitations of each system for natural products research.
Mammalian Cell Screening.
Multiparametric screening in mammalian cells was first pioneered as a systematic strategy for the evaluation of compound mode of action by the development of the NCI 60-cell-line screen from 1985–1990.30 This platform is the original “high-content” screening platform for natural products research, and has been used successfully to determine the MOAs of numerous natural products. For example, extracts containing salicylihalamides, potent vacuolar ATP-ase inhibitors, were first identified based on their particular NCI 60-cell-line profile.31 This platform is still in regular use and is very information rich, but is logistically impractical for widespread library screening, given the quantities of material required to screen against the entire 60-cell-line panel, and the inherently low throughput of such a system.
In recent years, cytological profiling, broadly defined as multiparametric evaluation of cellular response to compound treatment, has gained increasing attention as a complement to target-based and colorimetric live/dead screening assays. Cytological profiling is most commonly performed on mammalian cell lines, and can incorporate a variety of analytical techniques, including microarrays, MS-based metabolomics, gene signatures, and high-content automated microscopy.32–36 Several of these approaches have been employed for the investigation of natural product libraries, as outlined below.
Image-Based Screening.
Image-based screening was first widely adopted in industry because early systems were expensive, and required substantial informatics support to analyze the resulting image files. More recently, the hardware cost has dropped and the analytical software has improved, making this a routine tool in academic screening centers. Image-based screening has been used to develop a number of unbiased whole cell phenotypic screening platforms.32,37 In our own laboratory we have developed a modified version of the platform initially reported by Altschuler and co-workers32 in order to create a tool suitable for the examination of complex natural product libraries.38 This tool characterizes cell morphology using a set of structural and cell cycle fluorescent stains to extract hundreds of size and shape metrics for cells under drug pressure at sub-lethal concentrations. Subsequent informatics analysis compares these size and shape metrics to those for untreated control cells, and uses the differences in these values to create a numerical fingerprint that provides a graphical representation of the phenotypic differences between treated and control cells.
We have demonstrated that this tool can be used to classify the MOA of active constituents from complex natural product mixtures. Subsequent image-guided peak library fractionation can be used to pinpoint active compounds, and directly verify the cytological profiling signatures of these individual constituents, making the platform a powerful one for the discovery of natural products with unique phenotypic profiles (Figure 3).
Figure 3.
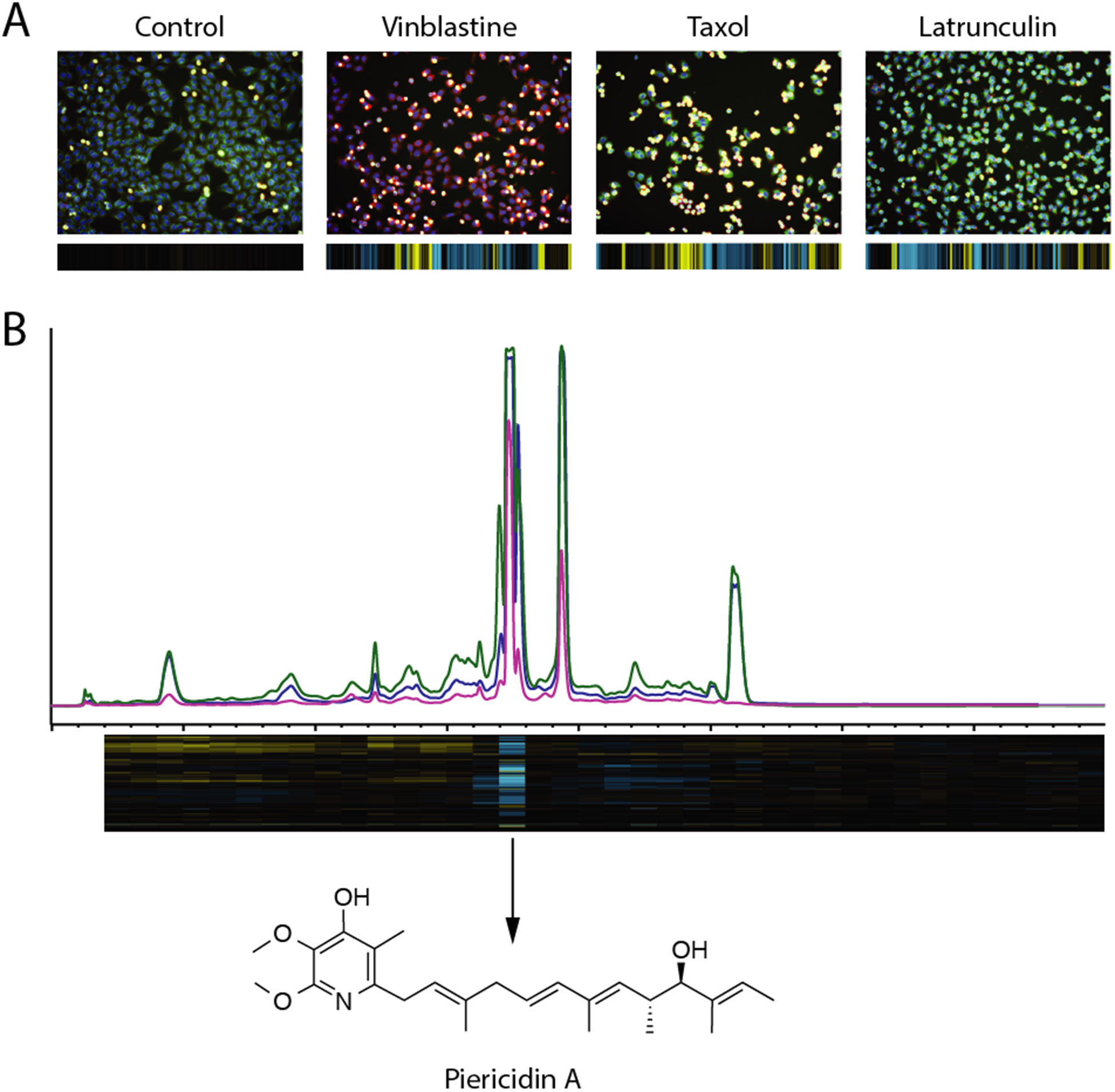
(A) Images of control cells, test cells treated with purified natural products, and their corresponding cytological profiles (B) Example of the use of cytological profiling-driven peak library screening and bioactive compound discovery for piericidin A, an inhibitor of the mitochondrial electron transport chain.
A complementary approach that uses a combination of fluorescence and brightfield imaging for the characterization of cellular phenotypes was recently reported by Osada and co-workers.39 This platform, termed MorphoBase, uses imaging data for two cell lines (HeLa and srcts-NRK) to characterize the phenotypic effect of compounds on cell development, and compares these phenotypic profiles to those of over 200 reference compounds of known mode of action to make direct predictions about the pathways or processes being disrupted by test compounds/extracts. MorphoBase has been used in conjunction with a proteomic profiling platform termed ChemProteoBase40 for the de novo prediction of the mode of action of a new fungal metabolite, pyrrolizilactone.41 In this work, both MorphoBase and ChemProteoBase identified strong clustering between pyrrolizilactone and test compounds known to inhibit proteasome function. Subsequent in vitro evaluation of 20S proteasome function confirmed this prediction, with the strongest inhibition of trypsin-like activity, providing an elegant demonstration of the use of unbiased profiling platforms for the direct prediction of bioactive natural products of unknown MOA.
Overall, image-based screens offer a large amount of biological annotation for natural product screening libraries in a format and timeframe that is appropriate for medium-throughput primary screens that number in the thousands of wells. We expect that the continuing improvements in screening hardware and software tools (e.g., the ability to perform high-throughput live cell imaging) will further lower the barrier to entry for these screening platforms, and that image-based profiling is likely to become a mainstay of future natural product discovery programs.
Gene Expression Profiling Platforms.
In addition to image-based approaches, a number of powerful gene profiling methods have been developed that are of relevance to the natural products community. The “Connectivity Map”, developed by researchers at the Broad Institute, was the first MOA profiling tool to compare the gene expression profiles of test compounds to a set of known bioactive molecules.42 This platform is finding widespread use in the biomedical community beyond the prediction of compound MOAs, and has already been cited over 1000 times since its publication in 2006. In the natural products area, this system has been used to profile compounds from a range of sources, including a recent study that used Connectivity Map profiles to compare the bioactivity of intact Gila monster venom to the drug Byetta®, which is a synthetic derivative of a lead compound derived from this venom mixture currently in clinical use to treat diabetes.43
Another gene profiling method recently applied to the characterization of natural product modes of action is the Functional Signal Ontology (FUSION) system developed by researchers at the University of Texas Southwestern Medical Center.44 This powerful platform uses the gene expression signatures of six key genes in HCT116 cells, as well as two genes with low variance as internal controls, to map the effect of treatment with either miRNAs, siRNAs, or natural product extracts. The team was able to demonstrate that these selected genes displayed non-colinearity of response under different treatment conditions, but that treatments of siRNAs or miRNAs from related pathways gave related FUSION signatures, and that FUSION signature matching can be used to accurately characterize the pathways targeted by specific bioactive natural products. More recently, this platform has been used to identify DDR2 as the molecular target of a new family of alkaloid natural products, discoipyrroles A – D, demonstrating the power of this untargeted approach for molecular target determination for natural products.45
Yeast Profiling.
The baker’s yeast, Saccharomyces cerevisiae, is a popular model system for studying mammalian cell biology thanks to the conservation of many of the genes implicated in human disease.46 Saccharomyces cerevisiae has therefore become a powerful model organism for studying the mode of action of bioactive small molecules.47 This has been aided by the creation of an ordered 5100-member gene deletion mutant library for all non-essential genes,48–50 that permits the systematic evaluation of the effect of test compounds on gene deletion mutants for the prediction of compound MOAs. Coupled with the systematic evaluation of synthetic interactions between 5.4 million gene-gene pairs that has created a comprehensive gene interaction network map for S. cerevisiae,50–52 this platform now represents a mature and powerful strategy for exploring chemical genetic properties of small molecules, including natural products.
Synthetic lethality screening uses the hypersensitivity of single gene deletion mutants to treatment with test compounds to indirectly report on compound molecular targets. If a single gene is non-essential, but treatment of that deletion strain with a bioactive small molecule causes lethality, then the small molecule must disrupt a compensatory pathway that is complementary to the function of the deleted gene product. By using the susceptibility of gene deletion mutants to test compounds in conjunction with the global genetic interaction network map, it is therefore in theory possible to determine the specific target of any individual compound, provided that this target has a homologue in S. cerevisiae, and that the compound is active against this yeast protein.
There have been several recent examples of the use of this technology for the determination of natural product MOAs, including the discovery that the macrocyclic lipopeptide papuamide B targets phosphatidylserine in yeast,53 and the determination that the marine sponge metabolite girolline targets Elongation Factor 2, and therefore exerts its anti-inflammatory activity through inhibition of protein synthesis at the elongation step.54
Antibiotic Screening.
Antimicrobial assays were some of the earliest assays used in natural products discovery, including the original serendipitous discovery of penicillin, and are still in widespread use around the world for the early characterization of natural product extract libraries. Although simple assays such as disk diffusion, cross streak, and well-plate liquid culture growth assays against individual pathogens are rapid and cheap, the number of published natural products with antibiotic activities now means that rates of rediscovery using these methods are extremely high. To overcome this limitation, a number of unbiased antibiotic screening platforms have been developed that provide multi-parametric characterization of the effects of natural product extracts on bacterial cell development. These tools provide direct information about compound class and/or MOA for active constituents, and can be used to rapidly triage large natural product libraries so that development effort is focused on those few extracts with highest potential for the discovery of new classes of antibiotics.
BioMAP Screening.
The BioMAP screening platform, developed in our laboratory in 2012, uses a panel of Gram-positive and Gram-negative bacterial pathogens to create activity profiles across the panel, in analogous fashion to the NCI 60-cell-line screen described above.55 By comparing these BioMAP profiles to profiles for a suite of commercially available antibiotics, it is possible to identify extracts that contain members of known classes of antibiotics, and to prioritize extracts with unique BioMAP signatures for further development. We have used this platform to discover new classes of antibiotics,55 and to profile large numbers of pure compounds and extracts from collaborative partners from academia and industry. This technology is readily transferable to any research laboratory with access to basic microbiology facilities and a standard plate reader, and has successfully been implemented by other research groups, including institutions in developing nations such as Indonesia.
Bacterial Cytological Profiling.
Although BioMAP profiling is very efficient at identifying extracts with unique antibiotic profiles, it does not provide information about the molecular targets or MOAs of these active constituents. To address this issue, a number of research groups have turned to image-based screening to explore antibiotic MOA profiles. MOA determination using cell imaging is challenging for bacterial targets, because bacterial cells are typically 100 times smaller than mammalian targets such as HeLa cells, making it technically difficult to acquire images of high enough resolution for cytological profiling in a high-throughput manner. In addition, most automated microscopy systems do not have pre-programmed modules to directly score images of bacterial cells, complicating the analytical component of this approach. Notwithstanding these challenges, two bacterial cytological profiling strategies have recently been reported.
The first, developed in our laboratory, uses high-throughput imaging of a chromosomally GFP-tagged strain of V. cholerae at 40 x magnification and a bespoke image analysis software platform to quantify cell size and shape features.56 These size and shape features are used to provide a numerical description of the phenotypes of individual cells under varying concentrations of either test extracts or training set antibiotics of known MOA. The progression of phenotypes is then compared to those for the training set antibiotics and these phenotypic “trajectories” used to predict compound MOAs. In the initial study 58 antibiotics were profiled to generate the training set phenotypic trajectories. Comparing these trajectories to those of a set of natural product extracts identified four bioactive compounds with predicted MOAs. Of these, three (novobiocin, cosmomycin D, cycloprodigiosin) had predicted MOAs that concurred with previous literature, while the fourth (pentachloropseudilin) had its MOA predicted for the first time.
In a second study, cells were examined at higher magnification, using FM4–64 to stain cell membranes, DAPI to stain the nucleus, and SYTOX green to stain cells with permeabilized membranes.57 The platform was used to examine the effects on cell morphology of 41 antibiotics from 26 separate structural classes, and was able to demonstrate a strong clustering of compounds by phenotype that closely paralleled the known MOAs for these compounds. In addition, the authors examined a novel antibiotic natural product, spirohexenolide A, and proved that it rapidly collapses the proton motor force using a combination of bacterial cytological profiling and complementary secondary assays. This approach provides more detailed information about cell shape and the fate of specific cellular components, but at a lower throughput than the previous study. The development of motorized SCLM stages and automated 100 x water immersion objectives offer new opportunities for further method development in this area, though this has yet to be applied to natural product MOA determination.
Zebrafish Imaging.
In vivo imaging represents another substantial advance for natural product screening. Just as the early conotoxin screening in whole animals revealed a wealth of neurological activities for individual components of these complex mixtures,58 so in vivo screening in zebrafish (Danio rerio) is providing a new strategy for the broad evaluation of natural products libraries. Advantages of this strategy include: a whole animal response; the ability to simultaneously measure both efficacy and off-target toxicity; the identification of developmental defects; the measurement of neurological and behavioral factors; and the ability to perform live animal time-resolved assays that look at temporal effects of compounds on animal health and survival.59
Although zebrafish have now been used for a wide array of targeted assays,60–62 and as a tool for downstream target identification or validation,63 there are still few examples of untargeted phenotypic screening in zebrafish, particularly for natural products.
One innovative system that has recently been developed incorporates both in vivo zebrafish screening and micro-scale fractionation for the simultaneous bioassay and physical characterization of plant extracts.64,65 This system has been used to identify both angiogenesis inhibitors from African plant extracts,66 and anticonvulsant compounds from Philippine medicinal plant Solanum torvum.67
Zebrafish screening has also been developed in industry, with Novartis reporting the results from profiling their in-house collection of 12,000 purified natural products.68 This impressive study, likely the largest of its kind, identified 114 phenotypic hits from this primary screen, including 50 compounds that caused developmental arrest without necrosis. This set of compounds contained molecules known to disrupt the mitochondrial electron transport chain, leading the authors to hypothesize a similar mechanism for other compounds displaying this phenotype. Subsequent transcriptional profiling of these compounds revealed that many of them did indeed target specific complexes of the mitochondrial electron transport chain, but also revealed instances where these two profiling systems did not agree, highlighting the importance of careful secondary screening for MOA predictions derived from high-throughput multiparametric profiling primary screens.
Certainly, the development of new screening systems in live animal models offers the potential for the rapid and detailed profiling of complex libraries, with the capacity to examine broader physiological characteristics of extracts and lead compounds than is possible using simple cell-based or enzyme assays. It will be interesting to see how these tools continue to evolve in the coming years as assay platforms develop in terms of liquid handling and image/ phenotype analysis.
Overall, multi-parametric screening tools are offering new opportunities to the natural products community for the rapid and efficient classification of complex natural product libraries. These tools provide new methods for the early prioritization of extracts and compounds with unique biological properties, and are a valuable complement to traditional live/dead screening systems for the discovery of next-generation therapeutic lead compounds. With the widespread availability of screening centers in academic institutions, development and implementation of these screening tools is well within the reach of most natural products research groups. Given the obvious benefit that such screening methods offer for natural products discovery, we expect that these approaches will enjoy increasing prominence within the natural products community in the coming years.
INTEGRATING CHEMICAL AND BIOLOGICAL DATASETS
Bioinformatics tools are becoming essential in natural products research, as advances in experimental throughput and the complexity of data obtained from genomic, chemical, and biological profiling make manual interpretation difficult or impossible. As previously mentioned, many laboratories have now developed sophisticated platforms to discover and classify biosynthetic gene clusters, to connect biosynthetic gene clusters to their gene products, and to classify complex small molecule libraries based on their chemical signatures.20,28,69–71 Recently, the integration of proteomics, metabolomics, and genomics has allowed genes, enzymes, and their small molecule products to be connected informatically for the discovery of bioactive compounds,72 providing examples of how integrated multiparametric profiling can be used to solve complex analytical problems, such as the connection of genes to molecules. While these techniques are powerful and have significantly advanced our understanding of natural products genomics and biosynthesis, there are a number of difficulties that preclude the facile integration of multiparametric chemical and biological screening information for natural products discovery.
Challenges with Multiparametric Data Integration.
The requirement for the integration of chemical constitution and biological screening techniques favors MS based chemical profiling strategies because of their throughput, resolution, and sensitivity; however, most developed metabolomics techniques require binary control and experimental groups looking for the correlation of genes with a defined outcome. Therefore, these analyses require the library to be manually curated. Instead, integrated profiling strategies require the use of untargeted metabolomics approaches that report on the presence of all constituents, whether or not the structures of all of these components are known. These tools can be developed with relative ease to create lists of individual components (defined by retention time and HRMS properties) and their distribution throughout the natural products library, but, connecting these components to specific structures is a much more challenging task which currently hampers the use of this approach for broad scale library characterization.
Concentrations, Timescales and the Analysis of Mixtures.
Since natural products libraries are extremely complicated mixtures, often with large variations in the concentration of different analytes, dynamic range is an issue for both screening and metabolomics platforms. This large variation in concentrations requires both the chemical profiling strategy and the biological screen to be sensitive, but to have the ability to characterize compounds at a range of concentrations. Typically, this is done by selecting a concentration for profiling that gives useful data for the majority of extracts, and performing a second profiling experiment at higher dilution factor for extracts that give either a strongly cidal readout in the profiling assay, or a saturated signal in the chemical analysis (typically a problem for accurate mass analyses such as ESI-qTOF). Furthermore, it is important that the analyses are configured such that the lower limits of detection are similar for the two systems. This is important because without this bioactive compounds can be ignored, either because they were observed in the metabolomics system but not classified as active, or because the extract was classified as active, but the compound concentration was below the detection limit in the chemical analysis.
Technical Requirements for the Integration of High-Content Datasets.
Some of the major challenges in integrating high-content datasets involve how the data are processed and integrated. Generalizable strategies for either chemical or biological annotation such as those described above are useful; however, directly integrating data from these analytical platforms is often difficult or impossible using existing tools. For example, while multivariate statistical methods such as principal component analysis are effective for discovering unique compounds from MS-based metabolomics libraries, it is difficult to confidently assign biological information to the resulting components when these statistical methods are extended to include high-content screening.
It is our opinion that an integration strategy should aim to correlate every detectable chemical feature with undefined phenotypes or screening profiles. In this way, the data should draw hypotheses about the biological activity of each detectable compound in the library for a global view of the chemical and chemical-genetic potential in the library. This resource would be invaluable for dereplicating known compounds, identifying modes of action, finding new biological activities using orthogonal screens, and discovering new compounds. For example, when newly developed biological screens are relatively low throughput, we can avoid re-screening samples containing frequent nuisance molecules like hydroxamic acid-containing metal chelators, pan-specific kinase inhibitors like staurosporine, or grossly cytotoxic anthracyclines by cherry picking the natural products library to avoid extracts previously annotated by multiparametric screening systems as containing these compound classes. The prediction of the broad MOAs of bioactive molecules can also be useful to avoid inclusion of extracts containing compounds with potential negative host interactions such as those associated with DNA damage, highlighting just a couple of situations where the target-independent characterization of biological and chemical properties of natural products libraries can be used to improve the discovery workflow for next-generation natural products-based therapeutics.
As an example of the ways in which such a strategy can be applied, we have recently developed a new integrated profiling platform, termed Compound Activity Mapping, which profiles natural products libraries using a combination of image-based cytological profiling and untargeted UPLC-TOF metabolomics to directly identify and characterize all bioactive constituents of any natural products screening library against HeLa cells (Figure 4). This tool is capable of generating networks that cluster extracts and their bioactive constituents based on biological and chemical similarities, such that each cluster contains a list of related compounds predicted to cause a specific phenotypic effect on HeLa cell development, and the extracts that contain these bioactive constituents. Using this tool we are discovering a wealth of new bioactive constituents from our microbially-derived natural products library, as well as providing phenotypic annotations for a large number of known compounds, some of which have not previously been characterized in terms of mammalian cell MOA.
Figure 4.
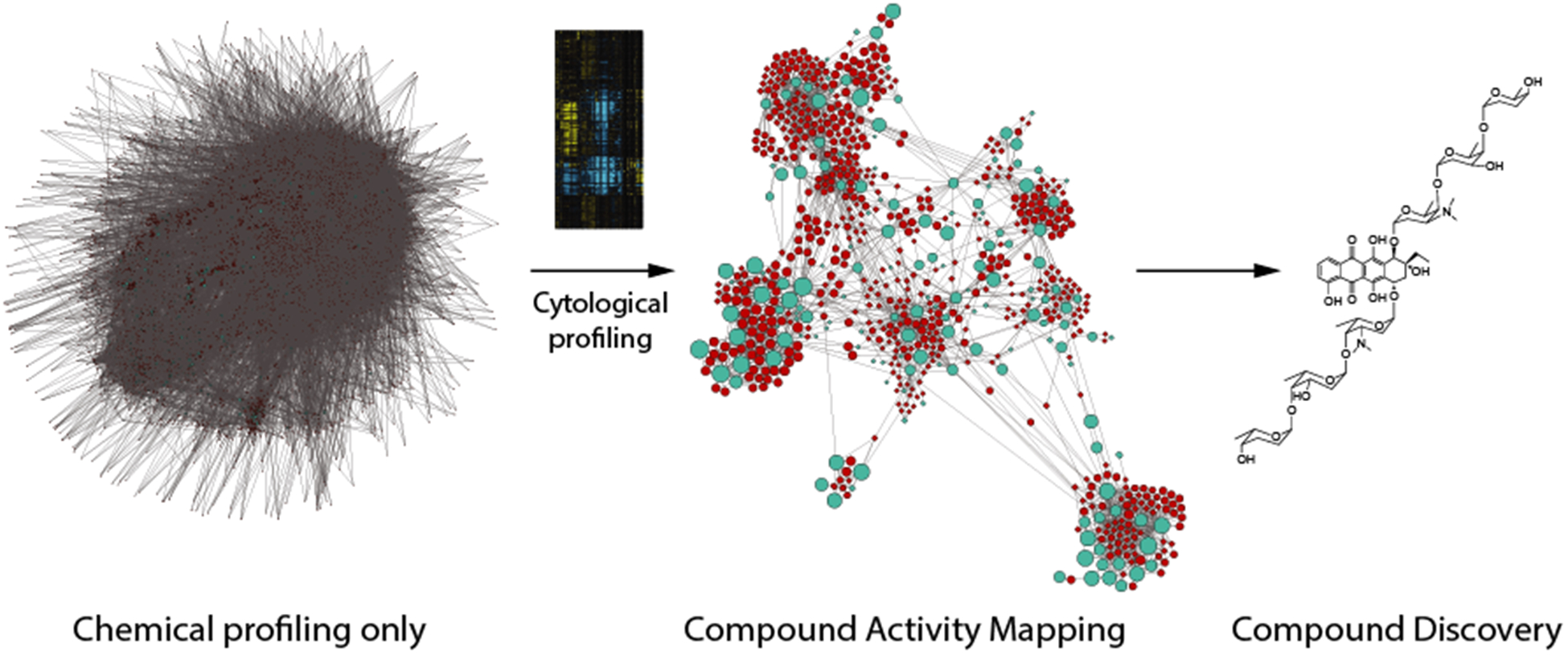
Output from Compound Activity Mapping platform, indicating the network generated using metabolomics data alone (left), the resulting Compound Activity Map generated by the integration of chemical and biological profiling datasets (center) and an example of a bioactive compound annotated using this platform (right).
FUTURE PERSPECTIVE
A rapid expansion in the resolution and throughput of academic screening data is currently taking place as high-throughput screening centers become more prevalent in universities and research institutes. Coupled with increasingly affordable and reliable MS tools and advances in the use of NMR methods for direct analysis of complex mixtures, we are poised to “open the box” on natural product discovery and transition from the traditional “grind and find” model, to a scenario in which we possess a priori knowledge about the constitution and MOA of all bioactive constituents of any screening library in advance of the isolation and detailed biological evaluation of individual compounds. Expansion of this approach to include whole genome sequence data for producing organisms is an obvious next step for improving the accuracy and coverage of molecular identification, and is close to becoming a reality as robust and affordable sequencing and genome assembly methods come of age. By extending this strategy from single profiling approaches to the integration of multiple profiling methods, each of which provides complementary but orthogonal information about the constitution and function of secondary metabolites from natural products libraries, we can now consider the possibility of developing universal characterization methods that describe the precise constitutions and biological activities of all members of any complex natural product library. The implications of developing such tools are widespread, with many fields set to benefit. Areas of future application of these technologies include chemotaxonomy, chemical ecology and interspecies interactions, botanicals research, natural product drug discovery, and human microbiome research, to name a few. The era of “Big Data” is here for natural products; it is already changing the field, and we are only beginning to see the impact that multiparametric biological and chemical evaluation of will have on natural products discovery. It is an exciting time to be involved in natural products research, and we are fascinated to see what new discoveries this next generation of sophisticated tools will bring.
Figure 1.
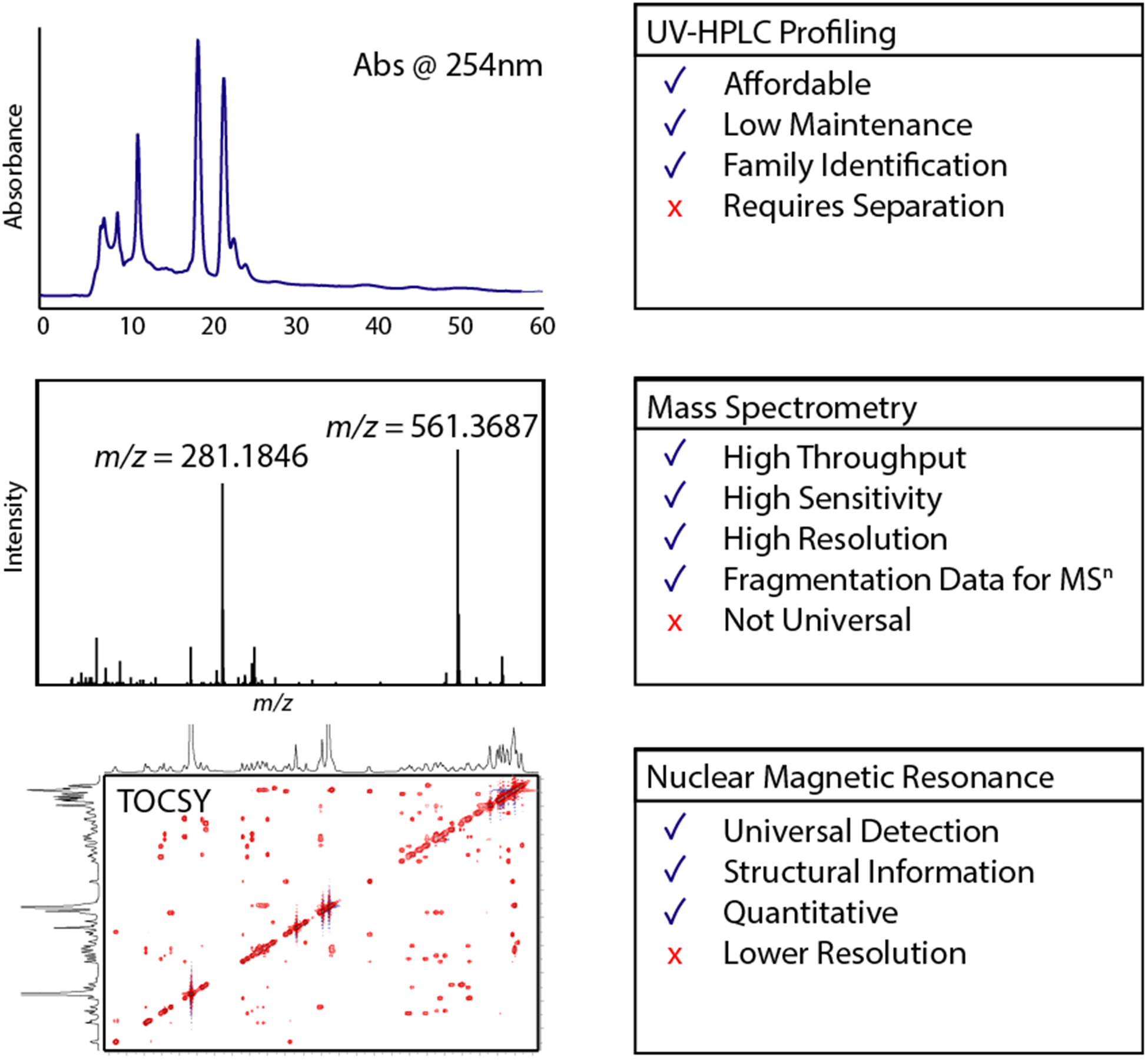
Summary of advantages and limitations of common chemical profiling strategies for natural products libraries.
Funding Sources
This work was supported by NIH grant TW006634.
Footnotes
Dedicated to Dr. William Fenical of Scripps Institution of Oceanography, University of California-San Diego, for his pioneering work on bioactive natural products
Adapted from a Matthew Suffness Award address, 55th Annual Meeting of the American Society of Pharmacognosy, Oxford, Mississippi, August 2–6, 2014
REFERENCES
- (1).Suffness M; Douros JD Trends Pharmacol. Sci 1981, 2, 307–310. [Google Scholar]
- (2).Woodward RB; Brehm WJ; Nelson AL J. Am. Chem. Soc 1947, 69, 2250–2250. [DOI] [PubMed] [Google Scholar]
- (3).Hamada T; Matsunaga S; Fujiwara M; Fujita K; Hirota H; Schmucki R; Güntert P; Fusetani NJ Am. Chem. Soc 2010, 132, 12941–12945. [DOI] [PubMed] [Google Scholar]
- (4).Molinski TF Nat. Prod. Rep 2010, 27, 321–329. [DOI] [PubMed] [Google Scholar]
- (5).Purcell EM Science 1953, 118, 431–436. [DOI] [PubMed] [Google Scholar]
- (6).Bloch F Science 1953, 118, 425–430. [DOI] [PubMed] [Google Scholar]
- (7).Jarmusch AK; Cooks RG Nat. Prod. Rep 2014, 31, 730–738. [DOI] [PubMed] [Google Scholar]
- (8).Carter GT Nat. Prod. Rep 2014, 31, 711–717. [DOI] [PubMed] [Google Scholar]
- (9).Robinette SL; Brüschweiler R; Schroeder FC; Edison AS Acc. Chem. Res 2011, 45, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hook DJ; Pack EJ; Yacobucci JJ; Guss J J. of Biomol. Screening 1997, 2, 145–152. [Google Scholar]
- (11).Wong MY; Steck PA; Gray GR J. Biol. Chem 1979, 254, 5734–5740. [PubMed] [Google Scholar]
- (12).Miller RD; Huckstep LL; McDermott JP; Queener SW; Kukolja S; Spry DO; Elzey TK; Lawrence SM; Neuss N J. Antibiot 1981, 34, 984–993. [DOI] [PubMed] [Google Scholar]
- (13).Miller RD; Neuss N J. Antibiot 1978, 31, 1132–1136. [DOI] [PubMed] [Google Scholar]
- (14).Hook DJ; More CF; Yacobucci JJ; Dubay G; O’Connor S J. Chromatogr. A 1987, 385, 99–108. [DOI] [PubMed] [Google Scholar]
- (15).Nielsen KF; Månsson M; Rank C; Frisvad JC; Larsen TO J. Nat. Prod 2011, 74, 2338–2348. [DOI] [PubMed] [Google Scholar]
- (16).Cortina NS; Krug D; Plaza A; Revermann O; Müller R Angew. Chem. Int. Ed 2011, 51, 811–816. [DOI] [PubMed] [Google Scholar]
- (17).Hou Y; Braun DR; Michel CR; Klassen JL; Adnani N; Wyche TP; Bugni TS Anal. Chem 2012, 84, 4277–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sidebottom AM; Johnson AR; Karty JA; Trader DJ; Carlson EE ACS Chem. Biol 2013, 8, 2009–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Watrous J; Roach P; Alexandrov T; Heath BS; Yang JY; Kersten RD; van der Voort M; Pogliano K; Gross H; Raaijmakers JM; Moore BS; Laskin J; Bandeira N; Dorrestein PC Proc. Natl. Acad. Sci. U.S.A 2012, 109, E1743–E1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang JY; Sanchez LM; Rath CM; Liu X; Boudreau PD; Bruns N; Glukhov E; Wodtke A; de Felicio R; Fenner A; Wong WR; Linington RG; Zhang L; Debonsi HM; Gerwick WH; Dorrestein PC J. Nat. Prod 2013, 76, 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hoffmann T; Krug D; Hüttel S; Müller R Anal. Chem 2014, 86, 10780–10788. [DOI] [PubMed] [Google Scholar]
- (22).Grkovic T; Pouwer RH; Vial ML; Gambini L; Noël A; Hooper JNA; Wood SA; Mellick GD; Quinn RJ Angew. Chem. Int. Ed 2014, 53, 6070–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bingol K; Bruschweiler Li L; Li D-W; Brüschweiler R Anal. Chem 2014, 86, 5494–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang F; Dossey AT; Zachariah C; Edison AS; Brüschweiler R Anal. Chem 2007, 79, 7748–7752. [DOI] [PubMed] [Google Scholar]
- (25).Taggi AE; Meinwald J; Schroeder FC J. Am. Chem. Soc 2004, 126, 10364–10369. [DOI] [PubMed] [Google Scholar]
- (26).Gronquist M; Meinwald J; Eisner T; Schroeder FC J. Am. Chem. Soc 2005, 127, 10810–10811. [DOI] [PubMed] [Google Scholar]
- (27).Deyrup ST; Eckman LE; McCarthy PH; Smedley SR; Meinwald J; Schroeder FC Proc. Natl. Acad. Sci. U.S.A 2011, 108, 9753–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pungaliya C; Srinivasan J; Fox BW; Malik RU; Ludewig AH; Sternberg PW; Schroeder FC Proc. Natl. Acad. Sci. U.S.A 2009, 106, 7708–7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schulze CJ; Linington RG In Natural Products: Discourse, Diversity, and Design; Osbourn A; Goss RJ; Carter GT, Eds.; Wiley: Oxford, 2014; pp. 373–396. [Google Scholar]
- (30).Alley MC; Scudiero DA; Monks A; Hursey ML; Czerwinski MJ; Fine DL; Abbott BJ; Mayo JG; Shoemaker RH; Boyd MR Cancer Res 1988, 48, 589–601. [PubMed] [Google Scholar]
- (31).Erickson KL; Beutler JA; Cardellina JH; Boyd MR J. Org. Chem 1997, 62, 8188–8192. [DOI] [PubMed] [Google Scholar]
- (32).Perlman ZE; Slack MD; Feng Y; Mitchison TJ; Wu LF; Altschuler SJ Science 2004, 306, 1194–1198. [DOI] [PubMed] [Google Scholar]
- (33).Young DW; Bender A; Hoyt J; McWhinnie E; Chirn G-W; Tao CY; Tallarico JA; Labow M; Jenkins JL; Mitchison TJ; Feng Y Nat. Chem. Biol 2008, 4, 59–68. [DOI] [PubMed] [Google Scholar]
- (34).Feng Y; Mitchison TJ; Bender A; Young DW; Tallarico JA Nat. Rev. Drug. Discov 2009, 8, 567–578. [DOI] [PubMed] [Google Scholar]
- (35).Mitchison TJ ChemBioChem 2005, 6, 33–39. [DOI] [PubMed] [Google Scholar]
- (36).Lorang J; King RW Genome Biol 2005, 6, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tanaka M; Bateman R; Rauh D; Vaisberg E; Ramachandani S; Zhang C; Hansen KC; Burlingame AL; Trautman JK; Shokat KM; Adams CL PLoS Biol 2005, 3, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schulze CJ; Bray WM; Woerhmann MH; Stuart J; Lokey RS; Linington RG Chem. Biol 2013, 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Futamura Y; Kawatani M; Kazami S; Tanaka K; Muroi M; Shimizu T; Tomita K; Watanabe N; Osada H Chem. Biol 2012, 19, 1620–1630. [DOI] [PubMed] [Google Scholar]
- (40).Muroi M; Kazami S; Noda K; Kondo H; Takayama H; Kawatani M; Usui T; Osada H Chem. Biol 2010, 17, 460–470. [DOI] [PubMed] [Google Scholar]
- (41).Futamura Y; Kawatani M; Muroi M; Aono H; Nogawa T; Osada H ChemBioChem 2013, 14, 2456–2463. [DOI] [PubMed] [Google Scholar]
- (42).Lamb J; Crawford ED; Peck D; Modell JW; Blat IC; Wrobel MJ; Lerner J; Brunet J-P; Subramanian A; Ross KN; Reich M; Hieronymus H; Wei G; Armstrong SA; Haggarty SJ; Clemons PA; Wei R; Carr SA; Lander ES; Golub TR Science 2006, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- (43).Aramadhaka LR; Prorock A; Dragulev B; Bao Y; Fox JW Toxicon 2013, 69, 160–167. [DOI] [PubMed] [Google Scholar]
- (44).Potts MB; Kim HS; Fisher KW; Hu Y; Carrasco YP; Bulut GB; Ou Y-H; Herrera-Herrera ML; Cubillos F; Mendiratta S; Xiao G; Hofree M; Ideker T; Xie Y; Huang LJ-S; Lewis RE; MacMillan JB; White MA Sci. Signal 2013, 6, ra90–ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hu Y; Potts MB; Colosimo D; Herrera-Herrera ML; Legako AG; Yousufuddin M; White MA; MacMillan JB J. Am. Chem. Soc 2013, 135, 13387–13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Karathia H; Vilaprinyo E; Sorribas A; Alves R PLoS ONE 2011, 6, e16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Luesch H; Wu TYH; Ren P; Gray NS; Schultz PG; Supek F Chem. Biol 2005, 12, 55–63. [DOI] [PubMed] [Google Scholar]
- (48).Winzeler EA; Shoemaker DD; Astromoff A; Liang H; Anderson K; Andre B; Bangham R; Benito R; Boeke JD; Bussey H; Chu AM; Connelly C; Davis K; Dietrich F; Dow SW; El Bakkoury M; Foury F; Friend SH; Gentalen E; Giaever G; Hegemann JH; Jones T; Laub M; Liao H; Liebundguth N; Lockhart DJ; Lucau-Danila A; Lussier M; M’Rabet N; Menard P; Mittmann M; Pai C; Rebischung C; Revuelta JL; Riles L; Roberts CJ; Ross-MacDonald P; Scherens B; Snyder M; Sookhai-Mahadeo S; Storms RK; Véronneau S; Voet M; Volckaert G; Ward TR; Wysocki R; Yen GS; Yu K; Zimmermann K; Philippsen P; Johnston M; Davis RW Science 1999, 285, 901–906. [DOI] [PubMed] [Google Scholar]
- (49).Giaever G; Chu AM; Ni L; Connelly C; Riles L; Véronneau S; Dow S; Lucau-Danila A; Anderson K; André B; Arkin AP; Astromoff A; Bakkoury, El M; Bangham R; Benito R; Brachat S; Campanaro S; Curtiss M; Davis K; Deutschbauer A; Entian K-D; Flaherty P; Foury F; Garfinkel DJ; Gerstein M; Gotte D; Güldener U; Hegemann JH; Hempel S; Herman Z; Jaramillo DF; Kelly DE; Kelly SL; Kötter P; LaBonte D; Lamb DC; Lan N; Liang H; Liao H; Liu L; Luo C; Lussier M; Mao R; Menard P; Ooi SL; Revuelta JL; Roberts CJ; Rose M; Ross-Macdonald P; Scherens B; Schimmack G; Shafer B; Shoemaker DD; Sookhai-Mahadeo S; Storms RK; Strathern JN; Valle G; Voet M; Volckaert G; Wang C-Y; Ward TR; Wilhelmy J; Winzeler EA; Yang Y; Yen G; Youngman E; Yu K; Bussey H; Boeke JD; Snyder M; Philippsen P; Davis RW; Johnston M Nature 2002, 418, 387–391. [DOI] [PubMed] [Google Scholar]
- (50).Ooi SL; Pan X; Peyser BD; Ye P; Meluh PB; Yuan DS; Irizarry RA; Bader JS; Spencer FA; Boeke JD Trends Genet 2006, 22, 56–63. [DOI] [PubMed] [Google Scholar]
- (51).Tong AH; Evangelista M; Parsons AB; Xu H; Bader GD; Pagé N; Robinson M; Raghibizadeh S; Hogue CW; Bussey H; Andrews B; Tyers M; Boone C Science 2001, 294, 2364–2368. [DOI] [PubMed] [Google Scholar]
- (52).Costanzo M; Baryshnikova A; Bellay J; Kim Y; Spear ED; Sevier CS; Ding H; Koh JLY; Toufighi K; Mostafavi S; Prinz J; St Onge RP; VanderSluis B; Makhnevych T; Vizeacoumar FJ; Alizadeh S; Bahr S; Brost RL; Chen Y; Cokol M; Deshpande R; Li Z; Lin Z-Y; Liang W; Marback M; Paw J; San Luis B-J; Shuteriqi E; Tong AHY; van Dyk N; Wallace IM; Whitney JA; Weirauch MT; Zhong G; Zhu H; Houry WA; Brudno M; Ragibizadeh S; Papp B; Pál C; Roth FP; Giaever G; Nislow C; Troyanskaya OG; Bussey H; Bader GD; Gingras A-C; Morris QD; Kim PM; Kaiser CA; Myers CL; Andrews BJ; Boone C Science 2010, 327, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Parsons AB; Lopez A; Givoni IE; Williams DE; Gray CA; Porter J; Chua G; Sopko R; Brost RL; Ho CH; Wang J; Ketela T; Brenner C; Brill JA; Fernandez GE; Lorenz TC; Payne GS; Ishihara S; Ohya Y; Andrews B; Hughes TR; Frey BJ; Graham TR; Andersen RJ; Boone C Cell 2006, 126, 611–625. [DOI] [PubMed] [Google Scholar]
- (54).Fung S-Y; Sofiyev V; Schneiderman J; Hirschfeld AF; Victor RE; Woods K; Piotrowski JS; Deshpande R; Li SC; de Voogd NJ; Myers CL; Boone C; Andersen RJ; Turvey SE ACS Chem. Biol 2014, 9, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wong WR; Oliver AG; Linington RG Chem. Biol 2012, 19, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Peach KC; Bray WM; Winslow D; Linington PF; Linington RG Mol. BioSyst 2013, 9, 1837–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Nonejuie P; Burkart M; Pogliano K; Pogliano J Proc. Natl. Acad. Sci. U.S.A 2013, 110, 16169–16174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Olivera BM; Rivier J; Clark C; Ramilo CA; Corpuz GP; Abogadie FC; Mena EE; SR W; Hillyard DR; Cruz LJ Science 1990, 249, 257–263. [DOI] [PubMed] [Google Scholar]
- (59).Zon LI; Peterson RT Nat. Rev. Drug. Discov 2005, 4, 35–44. [DOI] [PubMed] [Google Scholar]
- (60).Basu S; Sachidanandan C Chem. Rev 2013, 113, 7952–7980. [DOI] [PubMed] [Google Scholar]
- (61).Rihel J; Prober DA; Arvanites A; Lam K; Zimmerman S; Jang S; Haggarty SJ; Kokel D; Rubin LL; Peterson RT; Schier AF Science 2010, 327, 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Raldúa D; Piña B Expert Opin. Drug Metab. Toxicol 2014, 10, 685–697. [DOI] [PubMed] [Google Scholar]
- (63).Crawford A; Esguerra C; de Witte P Planta Med 2008, 74, 624–632. [DOI] [PubMed] [Google Scholar]
- (64).Challal S; Bohni N; Buenafe OE; Esguerra CV; de Witte PAM; Wolfender J-L; Crawford AD Chimia 2014, 1–4. [DOI] [PubMed] [Google Scholar]
- (65).Bohni N; Cordero-Maldonado ML; Maes J; Siverio-Mota D; Marcourt L; Munck S; Kamuhabwa AR; Moshi MJ; Esguerra CV; de Witte PAM; Crawford AD; Wolfender J-L PLoS ONE 2013, 8, e64006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Crawford AD; Liekens S; Kamuhabwa AR; Maes J; Munck S; Busson R; Rozenski J; Esguerra CV; de Witte PAM PLoS ONE 2011, 6, e14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Challal S; Buenafe OEM; Queiroz EF; Maljevic S; Marcourt L; Bock M; Kloeti W; Dayrit FM; Harvey AL; Lerche H; Esguerra CV; de Witte PAM; Wolfender J-L; Crawford AD ACS Chem. Neurosci 2014, 5, 993–1004. [DOI] [PubMed] [Google Scholar]
- (68).Lai K; Selinger DW; Solomon JM; Wu H; Schmitt E; Serluca FC; Curtis D; Benson JD ACS Chem. Biol 2013, 8, 257–267. [DOI] [PubMed] [Google Scholar]
- (69).Medema MH; Blin K; Cimermancic P; de Jager V; Zakrzewski P; Fischbach MA; Weber T; Takano E; Breitling R Nucleic Acids Res 2011, 39, W339–W346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Cimermancic P; Medema MH; Claesen J; Kurita K; Brown LCW; Mavrommatis K; Pati A; Godfrey PA; Koehrsen M; Clardy J; Birren BW; Takano E; Sali A; Linington RG; Fischbach MA Cell 2014, 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Bumpus SB; Evans BS; Thomas PM; Ntai I; Kelleher NL Nat. Biotechnol 2009, 27, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Gubbens J; Zhu H; Girard G; Song L; Florea BI; Aston P; Ichinose K; Filippov DV; Choi YH; Overkleeft HS; Challis GL; van Wezel GP Chem. Biol 2014, 1–12. [DOI] [PubMed] [Google Scholar]