

Abstract
Atomoxetine (ATX), a selective and potent inhibitor of the presynaptic norepinephrine transporter, is used mainly to treat attention-deficit hyperactivity disorder. Although multiple adverse effects associated with ATX have been reported including severe liver injuries, the mechanisms of ATX-related toxicity remain largely unknown. Metabolism frequently contributes to adverse effects of a drug through reactive metabolites, and the bioactivation status of ATX is still not investigated yet. Here, we systematically investigated ATX metabolism, bioactivation, species difference in human, mouse, and rat liver microsomes (HLM, MLM, and RLM) and in mice using metabolomic approaches as mice and rats are commonly used animal models for the studies of drug toxicity. We identified thirty one ATX metabolites and adducts in LMs and mice, 16 of which are novel. In LMs , we uncovered two methoxyamine-trapped aldehydes, two cyclization metabolites, detoluene-ATX, and ATX-N-hydroxylation for the first time. Detoluene-ATX and one cyclization metabolite were also observed in mice. Using chemical inhibitors and recombinant CYP enzymes, we demonstrated that CYP2C8 and CYP2B6 mainly contribute to the formation of aldehyde; CYP2D6 is the dominant enzyme for the formation of ATX cyclization and detoluene-ATX; CYP3A4 is major enzyme responsible for the hydroxylamine formation. The findings concerning aldehydes should be very useful to further elucidate the mechanistic aspects of adverse effects associated with ATX from metabolic angles. Additionally, the species differences for each metabolite should be helpful to investigate the contribution of specific metabolites to ATX toxicity and possible drug-drug interactions in suitable models.
Keywords: atomoxetine, cyclization, aldehyde, hydroxylamine, metabolomics
Graphical Abstract
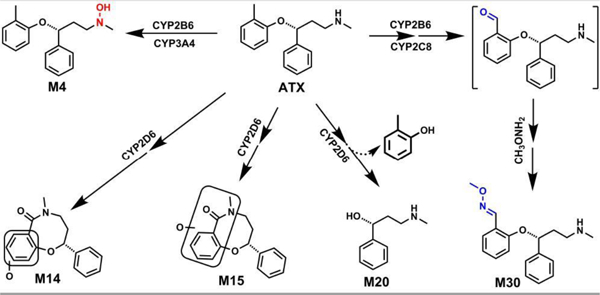
1. Introduction
Atomoxetine (ATX, formally known as tomoxetine) is a selective and potent inhibitor of the presynaptic norepinephrine transporter (Preti, 2002; Simpson and Perry, 2003). ATX is the first non-stimulant medication approved by the US Food and Drug Administration in late 2002 for attention-deficit hyperactivity disorder (ADHD) in children (≥6 years of age), adolescents, and adults (Sauer et al., 2005; Childress, 2016). ATX is generally considered to be safe and effective, and its use is associated with relatively few adverse drug reactions (Simpson and Perry, 2003; Camporeale et al., 2015). Suicidality associated with withdrawal of ATX treatment from children and adolescents has led to a black box warning (Wooltorton, 2005). Although rare, acute liver injuries associated with ATX have been reported, including instances of requiring emergency liver transplantation (Lim et al., 2006; Stojanovski et al., 2007; Erdogan et al., 2011; DiPaola and Molleston, 2018). The labeling of ATX was modified in 2004 to include severe liver injury, based on case reports of hepatotoxicity (Lim et al., 2006; Stojanovski et al., 2007). To date, the mechanisms by which ATX causes toxicity are largely unknown.
In the clinic, the onset of ATX associated liver injury occurs within 3 to 12 weeks, with features resembling acute viral hepatitis with significant increases in serum aminotransferase levels (Stojanovski et al., 2007). Immunoallergic features are not frequently associated with ATX treatment, but in several patients with acute injury, antinuclear antibodies were observed and at least one patient has displayed other features resembling autoimmune hepatitis (Chalasani et al., 2015; Potnis and Wackernah, 2015). These features indicate that ATX–induced liver injury could be caused by metabolic or immunologic idiosyncrasy (Hussaini and Farrington, 2007; Erdogan et al., 2011): rare ATX metabolites might be directly toxic, or they might induce an immunologic reaction (Bleibel et al., 2007; Gunawan and Kaplowitz, 2007; Leise et al., 2014; DiPaola and Molleston, 2018). Reactive metabolites such as epoxides and aldehydes have previously been shown to contribute to the toxicity of other drugs through these mechanisms (Tang and Lu, 2010; Thompson et al., 2016). The biotransformation of ATX has been investigated in human (Cui et al., 2007; Dinh et al., 2016), rats and dogs (Mattiuz et al., 2003), and in human liver microsomes (HLM) in vitro (Ring et al., 2002; Sauer et al., 2003), establishing that the dominant phase I and phase II metabolites are 4-hydroxy-ATX and glucuronic acid conjugated 4-hydroxy-ATX, and that CYP2D6 is the primary enzyme responsible for phase I metabolism. Minor metabolites have been also identified in human and animal models, including N-desmethyl-ATX, 2-hydroxymethyl-ATX, hydroxycarboxy-ATX, 4-hydroxy-N-desmethyl-ATX-O-glucuronide, 2-hydroxymethyl-ATX-O-glucuronide, and dihydroxy-ATX-O-glucuronide (Sauer et al., 2005; Yu et al., 2016). The formation of hydroxycarboxy-ATX suggests that ATX may be oxidized to an aldehyde intermediate (Sauer et al., 2003), which could contribute to toxicity, but the bioactivation and species differences of ATX has not been studied in detail. In this study, we employed metabolomic approaches to profile the metabolism of ATX in human, mouse, rat liver microsomes (HLM, MLM, and RLM) and in mice. MLM and RLM are employed in our current studies because mice and rats are commonly used animal models for the studies of drug toxicity.
Metabolomics has proven to be an effective and powerful tool to investigate drug metabolism (Li et al., 2018; Li et al., 2020), bioactivation (Li et al., 2014; Liu et al., 2015; Liu et al., 2016), and drug toxicity (Li et al., 2013; Lu et al., 2019; Zhao et al., 2019). We identified 31 metabolites and adducts generated from ATX in mice and LMs, of which 16 are novel. In LMs, two methoxyamine-trapped aldehydes (M30 and M31) and one ATX-hydroxylamine (M4) in LMs were identified and characterized for the first time. Two novel cyclization metabolites (M14 and M15) and one detoluene-ATX (M20) were identified and characterized. M15 was also observed in urine, feces and liver samples from ATX-treated mice; M20 was detected in mouse urine and liver. Our studies show that only CYP2D6 contributes to the formation of M14, M15, M20 and M31. Multiple enzymes contribute to the formation of aldehyde M30, but CYP2C8 and CYP2B6 are the major contributors. CYP3A4 and CYP2B6 are the primary enzymes responsible for the ATX-hydroxylamine formation (M4). The aldehydes M30 and M31 were less formed in HLM compared to those in MLM and RLM. These findings may provide valuable information for understanding ATX-related adverse effects from metabolic angles and also for predicting possible drug-drug interactions.
2. Materials and Methods
2.1. Materials
ATX, [(R)-N-Methyl-3-phenyl-3-(o-tolyloxy)propan-1-amine)], quercetin (QT), alpha-naphthoflavone (NF) and nootkatone (NT) were purchased from Cayman Chemical (Ann Arbor, Michigan). Quinidine (Qui), ticlopidine (TCP), ketoconazole (KCZ), methoxyamine hydrochloride, reduced glutathione (GSH), formic acid, titanium(III) chloride (TiCl3, ca. 12 wt. % solution in 12 wt. % hydrochloric acid), and NADPH (β-Nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate) were obtained from Sigma-Aldrich (St. Louis, MO). HLM, MLM, RLM and the recombinant human CYPs (EasyCYP Bactosomes) were purchased from XenoTech (Lenexa, KS). All solvents for liquid chromatography and mass spectrometry were of the highest grade commercially available.
2.2. Animal treatments and sample preparation
All mice (ICR mice, 2–4 months old, male) were maintained under a standard 12-h dark/light cycle with water and chow provided ad libitum. Handling was in accordance with study protocols approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. The mice were treated (200 μl for a 20 g mouse, p.o.) with ATX (12 mg/kg, 1.2 mg/ml in water) and housed separately in metabolic cages. The dose is clinically relevant, which is calculated by dose translation from human studies to mice based on 60 mg/daily for human (Nair and Jacob, 2016) Urine and feces were continually collected for 18 hours. Serum and liver samples were harvested 30 min after the treatment of ATX (p.o., 12 mg/kg). Urinary samples were prepared by mixing 20 μl of urine with 160 μl of 50% ice-cold methanol and serum samples were prepared by adding 60 μl of ice-cold methanol to 20 μl of serum. The resulting mixtures were vortexed and centrifuged at rcf 15,000 for 20 min. Feces and liver were weighed and homogenized in 50% methanol in water (100 mg liver in 500 μl of 50% methanol; 100 mg feces in 1000 μl of 50% methanol). Subsequently, 150 μl of methanol was added to 50 μl of the resulting mixture, followed by centrifugation at rcf 15,000 for 20 min. The supernatant was transferred to a new Eppendorf vial for a second centrifugation (rcf 15,000 for 20 min), and each supernatant was transferred to an auto sampler vial. For analysis, 3.0 μl was injected onto a system combining ultra-high performance liquid chromatography (UHPLC) coupled with Q Exactive Orbitrap mass spectrometer.
2.3. Metabolism of ATX in LMs, recombinant CYPs and aldehyde trapping.
Incubations were conducted in 1X phosphate-buffered saline (1 X PBS, pH 7.4) containing 25 μM ATX and 0.2 mg LMs (HLM, MLM, and RLM) or 2 pmol of each cDNA-expressed CYPs enzyme (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4) in a final volume of 190 μl. After a 5 min pre-incubation at 37 °C, 10 μl of 20 mM NADPH was added (final concentration 1.0 mM) and incubation was continued for 30 min with gentle shaking. Incubations without NADPH were used as controls. Co-incubations of ATX (25 μM) and NF (CYP1A2 inhibitor, 6 μM), or TCP (CYP2B6 inhibitor, 10 μM, pre-incubation for 20 min before adding ATX), or QT (30 μM, CYP2C8 inhibitor), or NT (10 μM, CYP2A6 and CYP2C19 inhibitor), or Qui (2.0 μM, CYP2D6 inhibitor), or KCZ (CYP3A4 inhibitor 2.0 μM) in HLM were performed to determine the role of CYPs in the formation of detoluene-ATX (M20), ATX-hydroxylamine (M4), dimethyl-ATX (M5), cyclization metabolites (M14 and M15), and O-methyl oxime (M30). Reactions were terminated by adding 200 μl of ice-cold methanol and vortexing for 30 seconds, and then centrifuged at rcf 15,000 for 15 min. Each supernatant was transferred to an auto sampler vial, and 3.0 μl was injected on to UHPLC-Q Exactive MS system for analysis. Incubations were performed in duplicate for cDNA-expressed enzymes, and in triplicate for LM experiments. Aldehyde trapping experiments were conducted as above except that the final volume of 190 μl included 2.5 mM methoxyamine. To seek glutathione adducts, the experiments were also performed in the presence of 2.5 mM glutathione. The concentration of ATX (25 μM) was used in the study of LMs according to the data from our enzyme kinetic study of ATX in HLM. ATX at 25 μM in HLM show the maximal Vmax after 30 min incubation (data not shown). In addition, the Cmax of ATX at stead-state are around 0.72 uM (20 mg, twice a day) and 2.1 uM (40 mg, twice a day), respectively, in the healthy adults with normal CYP2D6 activity (Yu et al., 2016). Generally, the drug concentration in liver is at least 20 times higher than that in plasma (Wright et al., 2015). Thus, 25 μM used in LM should be also relevant to ATX concentration in human liver.
2.4. Reduction of N-hydroxylamine of metabolite M4 by TiCl3.
Incubations were conducted in 1X phosphate-buffered saline (1 X PBS, pH 7.4), containing 25 μM ATX, 2 pmol of cDNA-expressed CYP2B6 in a final volume of 190 μl. After 5 min of pre-incubation at 37 °C, the reaction was initiated by adding 10 μl of 20 mM NADPH (final concentration 1.0 mM) and continued for 30 min with gentle shaking. Reactions were terminated by adding 200 μl of ice-cold methanol. After vortex for 30 seconds, the mixture was centrifuged at rcf 15,000 for 15 min. 50 μl of supernatant of each sample was transferred to an Eppendorf vial used as a control and kept on ice. 300 μl of supernatant was transferred to another Eppendorf vial for a reduction experiment. TiCl3 (4.0 μl) was added to an ice-cold 300 μl of supernatant and the resulting mixture was vortexed for 30 seconds and kept on ice for 1.0 hour. An aliquot from each control or reaction mixture was diluted three-fold with ice-cold methanol. After centrifugation at rcf 15,000 for 10 min, the clear supernatant was transferred to an auto sampler vial, and 3.0 μl was injected on to UHPLC-Q Exactive MS system for analysis.
2.5. UHPLC-Q Exactive Orbitrap MS analyses
ATX and its metabolites were resolved, identified and quantified (relatively) using UHPLC coupled with Q Exactive Orbitrap MS (Thermo Fisher Scientific, San Jose, CA) equipped with 100 mm x 2.1 mm column (XDB C-18, Agilent Technologies, Santa Clara, CA). The column temperature was maintained at 40 °C. The flow rate of was 0.3 mL/min with a gradient ranging from 2% to 95% aqueous acetonitrile containing 0.1% formic acid in a 15-min run. Q Exactive MS was operated in positive mode with electrospray ionization. Ultra-pure nitrogen was applied as the sheath (45 arbitrary unit), auxiliary (10 arbitrary unit), sweep (1.0 arbitrary unit) and the collision gas. The capillary gas temperature was set at 275 °C and the capillary voltages was set at 3.7 kV. MS data were acquired from 80 to 1200 Da in profile mode and reference ions at m/z 371.1012, and 445.1200 in the positive mode were used as lock masses during acquisition. The MS/MS of ATX metabolites was performed in targeted mode with an isolation width of 2 m/z with ramp collision energy being set at 15, 20, and 30 eV.
Data analysis
Mass chromatograms and mass spectra were acquired and processed using the Xcalibur software (Thermo Fisher Scientific, San Jose, CA) in profile formats from m/z 50 to 750. The acquired data were processed by Compound Discoverer 3.0 software (Thermo Fisher Scientific, San Jose, CA) to generate a multivariate data matrix. Data matrices were exported into SIMCA14 (Umetrics, Kinnelon, NJ) for multivariate data analysis (Cazanave et al., 2009). Orthogonal projection to latent structures-discriminant analysis (OPLS-DA) was conducted on Pareto-scaled data (Worley and Powers, 2013). For chemometric analysis, matrix data were processed from m/z 100 to 750. Statistical analysis was conducted using Student’s independent t-test. Experimental data are presented as mean ± S.E.M.
3. Results
3.1. Profiling ATX metabolism in mice and MLM using metabolomic approaches
The results of the chemometric analysis on the ions generated from the UHPLC-Q Exactive MS analysis of control and ATX-treated mouse urine are shown in Fig. 1. Principal component analysis revealed two groups corresponding to the control and ATX-treated groups (Fig. 1A). The S-plot (Fig. 1B) generated from OPLS-DA displays the ions that contribute to the group separation; the top-ranking ions identified as ATX metabolites are labeled in the S-plot. The major metabolites of ATX in MLM were labelled in Fig. 1C. ATX and its metabolites were mainly excreted in urine (M2, M3, M5, M8, M10, M16-M18, and M20-M29 (Fig. 2A and Table 1). The metabolites M2, M3, M10, M15, M16, M17 and M21 were also found in feces (Fig. 2B and Table 1). Overall, 20 ATX metabolites from phase I and phase II were identified in mice, including eleven known metabolites (M2, M3, M5, M8, M10, M18, M21-M23, M25, and M26), and nine novel metabolites (M9, M15-M17, M20, M24, M27-M29). In urine, monohydroxylated-ATX (M2 and M3), glucuronide of monohydroxylated-ATX (M21), glucuronide of monohydroxylated-N-demethyl-ATX (M23), glucuronide of dihydroxylated-ATX (M26) and detoluene-ATX (M20) are the dominant ATX metabolites. In feces, M2, ATX-acid (M10), dihydroxylated-ATX (M16), and M21 are the major ATX metabolites (Fig. 2B). The relative abundance of metabolites in mouse urine and feces is presented in Figs. 2A and 2B. M21 (96%) is the most abundant circulating metabolite in the mouse serum; M2 (50%), M21 (43%), and ATX-acid M10 (3%) are major metabolites in the mouse liver (data not shown). The structures of M2, M3 and M21-M23 were determined based on the MS/MS, exact mass and the relative abundances in previous studies as the standards of these metabolites are not available (Lantz et al., 2003; Mattiuz et al., 2003; Yu et al., 2016).
Figure 1. Metabolomic analysis of control and ATX-treated mouse urine.

Wild-type (WT) mice (n = 3) were treated with ATX (12 mg/kg, p.o.). Urine and feces were collected for analysis 18 hours after ATX treatment. (A) Separation of control and ATX-treated mouse urine in OPLS-DA score plots. The t[1] and to[1] values represent the score of each sample in principal component 1 and 2, respectively. (B) Loading S-plot generated by OPLS-DA analysis of urine metabolome of mice treated with ATX. (C) Loading S-plot generated by OPLS-DA analysis of metabolome of ATX in MLM. The X-axis is a measure of the relative abundance of ions and the Y-axis is a measure of the correlation of each ion to the model. These loading plots represent the relationship between variables (ions) in relation to the first and second components present in the OPLS-DA score plot. The top ranking ATX metabolites were labeled in S-plots, respectively. The number of ions (metabolite identification) was accordant with those in Table 1.
Figure 2. Relative abundance of metabolites of ATX in mouse urine and feces.

Urine and feces from mice were continuously collected for 18 hours after the treatment of ATX (12 mg/kg, p.o.). All the samples were analyzed using UHPLC-Q Exactive MS. The relative quantification was conducted based on the peak area. The overall abundance of metabolites was set as 100% in each sample. The data are expressed as mean ± SEM (n = 3). (A) Relative abundance of metabolites in urine. (B) Relative abundance of metabolites in feces.
Table 1.
Summary of metabolites of ATX in HLM, MLM, RLM, and mice.
| RT (min) | Observed m/z [M+H]+ | Calculated m/z [M+H]+ | Mass error (ppm) | Predicted molecular formula | Identification | Metabolite ID* | Source |
|---|---|---|---|---|---|---|---|
| 6.83 | 256.1695 | 256.1696 | −0.39 | C17H22NO | Atomoxetine | ATX | HLM, MLM, RLM, serum, liver, urine, feces |
| 4.43 | 272.1645 | 272.1645 | 0.0 | C17H22NO2 | O+ATX | M1 | HLM, MLM, RLM |
| 5.16 | 272.1647 | 272.1645 | 0.74 | C17H22NO2 | O+ATX | M2 | HLM, MLM, RLM, serum, liver, urine, feces |
| 5.64 | 272.1644 | 272.1645 | −0.37 | C17H22NO2 | O+ATX | M3 | HLM, MLM, RLM, serum liver, urine, feces |
| 7.45 | 272.1646 | 272.1645 | 0.37 | C17H22NO2 | O+ATX (N-hydroxylation) | M4 | HLM, MLM, RLM |
| 6.66 | 242.1540 | 242.1539 | 0.42 | C16H20NO | ATX (−CH3) | M5 | HLM, MLM, RLM, liver, urine |
| 4.50 | 258.1491 | 258.1489 | 0.78 | C16H20NO2 | O+ATX (−CH3) | M6 | HLM, MLM, RLM |
| 4.71 | 258.1490 | 258.1489 | 0.39 | C16H20NO2 | O+ATX (−CH3) | M7 | HLM, MLM, RLM |
| 5.00 | 258.1489 | 258.1489 | 0 | C16H20NO2 | O+ATX (−CH3) | M8 | HLM, MLM, RLM, urine, liver |
| 5.45 | 258.1490 | 258.1489 | 0.39 | C16H20NO2 | O+ATX (−CH3) | M9 | HLM, MLM, RLM, urine, liver |
| 5.93 | 286.1439 | 286.1438 | 0.35 | C17H20NO3 | ATX-Acid | M10 | HLM, MLM, RLM, liver, serum, urine, feces |
| 5.49 | 272.1283 | 272.1281 | 0.73 | C16H18NO3 | ATX-Acid (−CH3) | M11 | HLM, MLM, RLM, liver |
| 4.96 | 302.1390 | 302.1387 | 0.99 | C17H20NO4 | O+ATX-Acid | M12 | MLM |
| 5.18 | 302.1389 | 302.1387 | 0.66 | C17H20NO4 | O+ATX-Acid | M13 | MLM |
| 5.93 | 284.1283 | 284.1281 | 0.70 | C17H18NO3 | Cyclization | M14 | HLM, MLM, RLM |
| 7.80 | 284.1282 | 284.1281 | 0.35 | C17H18NO3 | Cyclization | M15 | HLM, MLM, RLM, liver, urine, feces |
| 4.47 | 288.1593 | 288.1594 | −0.35 | C17H22NO3 | 2O+ATX | M16 | HLM, MLM, RLM, urine, feces, liver |
| 4.73 | 288.1592 | 288.1594 | −0.69 | C17H22NO3 | 2O+ATX | M17 | HLM, MLM, RLM, urine, liver, feces |
| 5.16 | 288.1595 | 288.1594 | 0.35 | C17H22NO3 | 2O+ATX | M18 | HLM, MLM, RLM, urine |
| 5.69 | 288.1594 | 288.1594 | 0.0 | C17H22NO3 | 2O+ATX | M19 | HLM, MLM, RLM |
| 2.48 | 166.1228 | 166.1226 | 1.20 | C10H16NO | De-toluene | M20 | HLM, MLM, RLM, urine, liver |
| 4.15 | 448.1968 | 448.1966 | 0.45 | C23H30NO8 | O+ATX+Glu | M21 | urine, liver, serum |
| 5.43 | 448.1969 | 448.1966 | 0.67 | C23H30NO8 | O+ATX+Glu | M22 | urine, liver, serum |
| 4.01 | 434.1811 | 434.1809 | 0.46 | C22H28NO8 | O+ATX+Glu (−CH3) | M23 | urine, liver, serum |
| 3.73 | 462.1760 | 462.1759 | 0.22 | C23H28NO9 | ATX-Acid+Glu | M24 | urine |
| 3.39 | 464.1918 | 464.1915 | 0.65 | C23H30NO9 | 2O+ATX+Glu | M25 | urine |
| 3.64 | 464.1916 | 464.1915 | 0.2 | C23H30NO9 | 2O+ATX+Glu | M26 | urine, liver, serum |
| 3.97 | 464.1919 | 464.1915 | 0.86 | C23H30NO9 | 2O+ATX+Glu | M27 | urine |
| 4.23 | 464.1915 | 464.1915 | 0.0 | C23H30NO9 | 2O+ATX+Glu | M28 | urine liver, serum |
| 4.09 | 610.2496 | 610.2494 | 0.33 | C29H40NO13 | O+ATX+Glu+Gluc | M29 | urine |
| 6.95 | 299.1756 | 299.1754 | 0.67 | C18H23N2O2 | ATX_hydrazone | M30 | HLM, MLM, RLM |
| 5.53 | 315.1709 | 315.1703 | −1.9 | C18H23N2O3 | O+ATX_hydrazone | M31 | MLM, RLM |
ATX, atomoxetine; Glu, glucuronic acid; Gluc, glucose; −CH3, demethylation; O+, monohydroxylation; 2O+, dihydroxylation; Acid, carboxylic acid; HLM, MLM, RLM, human, mouse, and rat liver microsmes.
, novel metabolites indicated by red.
In the incubations of ATX in HLM, MLM, and RLM, a total of 20 stable phase I metabolites were identified and 10 of them are novel (Table 1). The major metabolites from MLM are labelled in the S-plot generated from the OPLS-DA analysis of control and ATX groups in MLM (Fig. 1C). M2, M3, and M5 are primary metabolites in all LMs. Detoluene-ATX (M20) is observed in all three LMs used in our studies and is relatively more abundant in MLM compare to HLM and RLM (Fig. 3). Cyclization metabolites (M14 and M15) were observed in all three LMs. The relative abundances of these metabolites in LMs are shown in Fig. 3. Their structures were identified based on their exact mass and MS/MS fragments. Additionally, enzymes responsible for ATX metabolic pathways and their relative contribution in HLM were summarized in the supplementary Table S1 based on data from cDNA expressed CYPs and inhibitory experiments using chemical inhibitors.
Figure 3. Relative abundance of metabolites of ATX in HLM, MLM, and RLM.
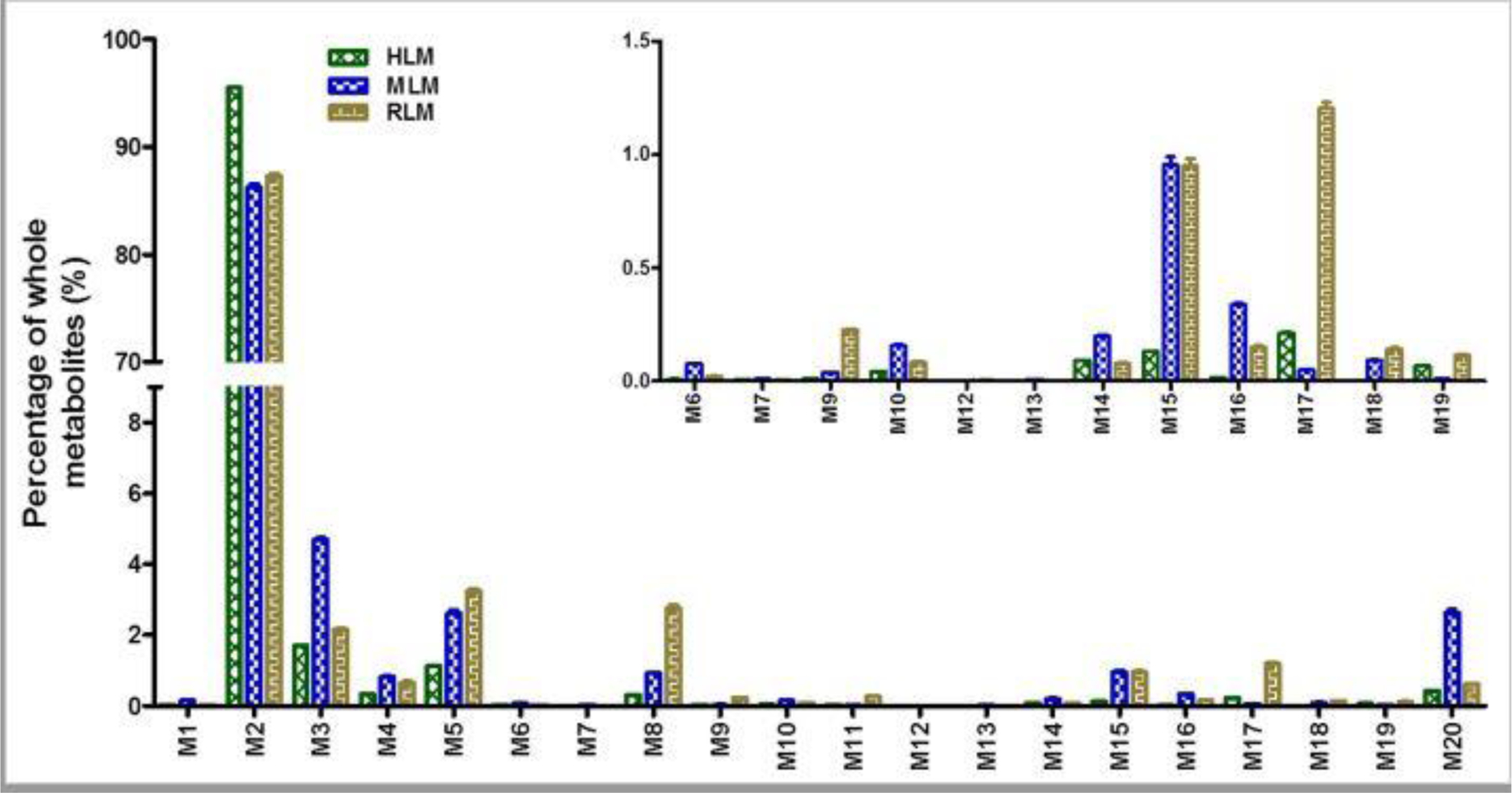
Incubations were conducted in 1X phosphate-buffered saline (1X PBS, pH 7.4), containing 25 µM ATX, 0.2 mg LM in a final volume of 190 µl. After 5 min of pre-incubation at 37 °C, the reaction was initiated by adding 10 µl of 20 mM NADPH (final concentration 1.0 mM) and continued for 30 min with gentle shaking. The relative quantification was conducted based on the peak area. The overall abundance of metabolites was set as 100% in each sample. The data are expressed as mean ± SEM (n = 3).
3.2. Formation of ATX-O-methyl oxime in liver microsomes
Our study also revealed the formation of aldehyde associated with ATX. Their existence was confirmed using methoxyamine as a trapping agent in LMs. The formation of aldehyde M30 was NADPH dependent. The aldehydes M30 and M31 were less formed in HLM compared to those in MLM and RLM, which was inlaid in Fig. 4A. M30 eluted at 6.95 min (Fig. 4A), and was detected as a protonated molecule at m/z 299.1756. The fragmental ions at m/z 152 and 148 were interpreted in the inlaid structural diagram (Fig. 4B). The proposed mechanism and trapping strategy were illustrated in Fig. 4C. Additionally, one O-ATX- O-methyl oxime (M31) was also observed and NADPH-dependent, but with relative lower abundance compared to the M30. M31 eluted at 5.53 min and was detected as a protonated molecule at m/z 315.1709 (theoretical exact mass: m/z 315.1703, Table 1), which was identified as O-ATX-O-methyl oxime based on the exact mass and predicted formula as the MS/MS was not available due to the low abundance. The possible formation mechanism of M31 was described in the Fig. 6.
Figure 4. Identification of ATX-O-methyl oxime (M30).
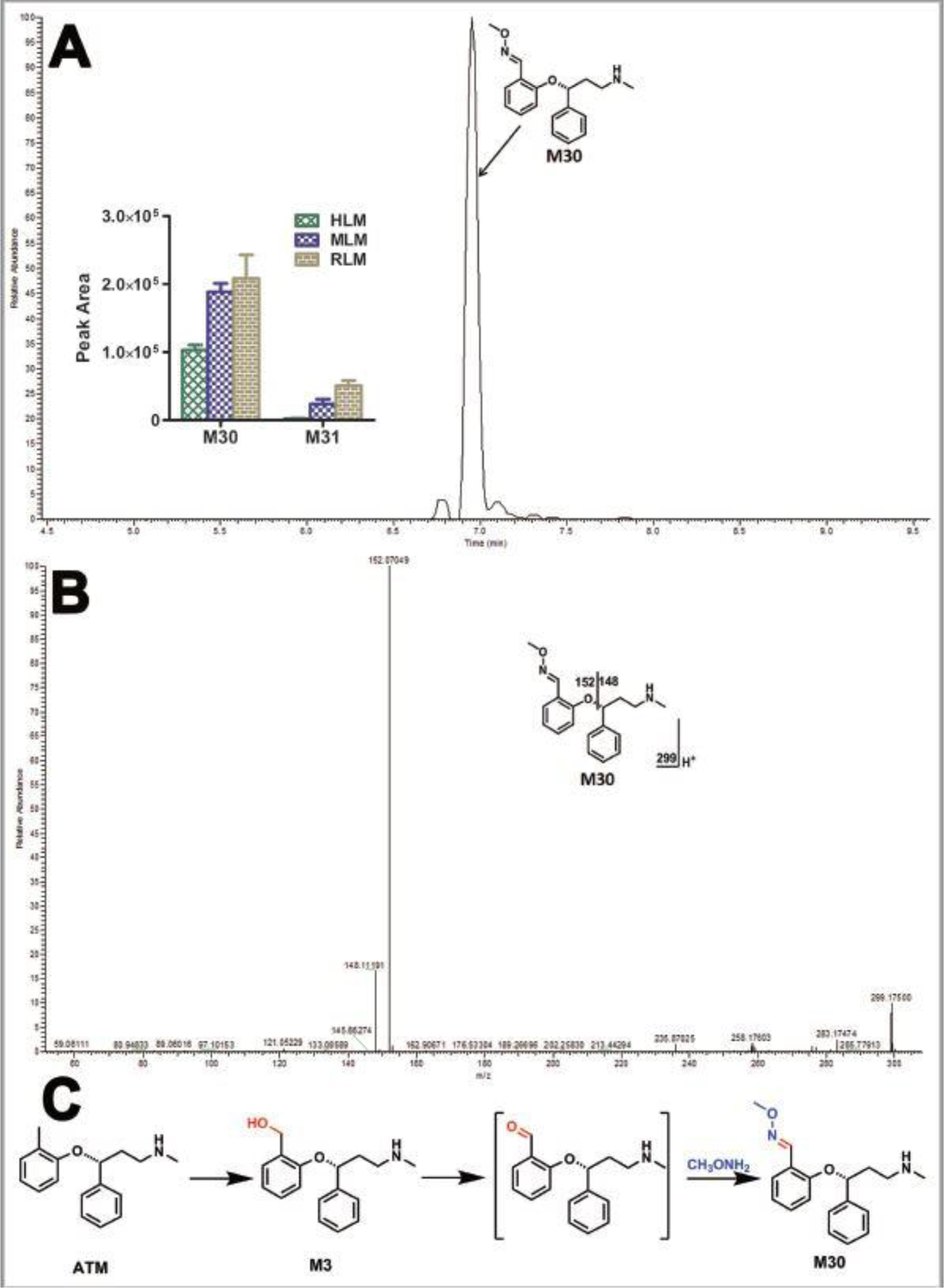
Incubations were conducted in 1X phosphate-buffered saline (1X PBS, pH 7.4), containing 25 µM ATX, 0.2 mg HLM, 2.5 mM methxoyamine, and NADPH (final concentration 1.0 mM) in a final volume of 200 µl. All the samples were analyzed using UHPLC-Q Exactive MS. Structural elucidations were performed based on accurate mass (mass errors less than 5 ppm) and MS/MS fragmentation. MS/MS was performed with collision energy ramping from 10–30 V. The major fragmental ions are interpreted in the insets. (A) Chromatograms of M30 and the relative abundance of M30 and M31 in LMs. (B) MS/MS of M30. (C) Proposed mechanism of the aldehyde formation and trapping strategy. ATX-O-methyl oxime (M30) was produced in three steps: 1) monohydroxylation of ATX to form M3; 2) further oxidation to generate the aldehyde; 3) reacted with methoxyamine to form M30.
Figure 6. Proposed mechanism of the formation of M14 and M15.

M14 and M15 were detected in all the three LMs used in our studies. ATX was first oxidized to O-ATX (M1, M2, or M3), which was further oxidized to form the aldehyde. The aldehyde is cyclized to form the intermediate O-hemiaminal and subsequent oxidation to form the lactam M14. M15 is generated by oxidizing ATX to form M3, followed by the second oxidation to generate aldehyde. The aldehyde was intra-cyclized to form the intermediate hemiaminal, which was further oxidized to form M15.
3.3. Cyclization metabolites of ATX (M14 and M15) and detoluene-ATX (M20) in liver microsomes and mouse urine
Two novel ATX cyclization metabolites (M14 and M15) were identified and characterized in HLM, MLM, and RLM; M15 was identified in mouse urine, feces and liver. Metabolite M14 eluted at 5.93 min (Fig. 5A), and was detected as a protonated molecule at m/z 284.1278 (Fig. 5B). The MS/MS of M14 produced the major fragmental ions at m/z 180, 133, and 117. The fragmental ions are interpreted in the inlaid structural diagram (Fig. 5B). Metabolite M15 eluted at 7.80 min (Fig. 5A), and was detected as a protonated molecule at m/z 284.1272 (Fig. 5C). The MS/MS of M15 produced the major fragmental ions at m/z 180, 152, and 117. The fragmental ions are interpreted in the inlaid structural diagram (Fig. 5C). The possible mechanism of M14 and M15 formation was proposed in Fig. 6. Detoluene-ATX (M20) was observed in liver microsomes, mouse urine and liver. M20 eluted at 2.48 min (Fig. 5D), and was detected as a protonated molecule at m/z 166.1219 (Fig. 5E). The MS/MS of M20 produced the major fragmental ions at m/z 148, 121 and 107. The fragmental ions are interpreted in the inlaid structural diagram (Fig. 5E). A possible route for M20 formation is presented in Fig. 5F.
Figure 5. Identifying cyclized metabolites (M14 and M15) and detoluene-ATX (M20).

Incubations without methxoyamine were conducted as described in Fig. 4. All the samples were analyzed using UHPLC-Q Exactive MS. The metabolite elucidation conditions are described in Fig. 4. (A) Chromatograms of metabolite M14 and M15 in HLM. (B) MS/MS of M14. (C) MS/MS of M15. (D) Chromatograms of metabolite M20 in HLM. (E) MS/MS of M20. (F) Formation of detoluene-ATX.
3.4. Identification of ATX-hydroxylamine (M4)
The NADPH-dependent formation of novel monohydroxylated-ATX (M4, hydroxylamine) was detected in HLM, MLM, and RLM, but not in mice (Table 1 and Fig. 3). M4 eluted at 7.45 min (Fig. 7A), and was detected as a protonated molecule at m/z 272.1633 that is less polar than parent compound ATX (Figs. 7A and 7C). The fragmental ion at m/z 164 from M4 (Fig. 7C) and ion at m/z 148 from ATX (Fig. 6B) indicate that the oxidation did not occur in the toluene motif. The fragmental ion at m/z 60 suggested the formation of hydroxylamine (Fig. 7C). Fragmental ions for ATX and M4 are interpreted in the inlaid structural diagrams in Figs. 7B and 7C, respectively. Previous studies have shown that TiCl3 can efficiently convert hydroxylamine to the corresponding secondary amine (Murahashi and Kodera, 1985). In our TiCl3 treated samples (Fig. 8A), the chromatographic peak for metabolite M4 completely disappeared, while other monohydroxylated metabolites (e.g. M2 and M3) were unchanged (Fig. 8B).
Figure 7. Identification of ATX-hydroxylamine (M4).

Incubations were conducted in 1X phosphate-buffered saline (1X PBS, pH 7.4), containing 25 µM ATX, 2 pmol of CYP2B6 and NADPH (final concentration 1.0 mM) in a final volume of 200 µl. All the samples were analyzed using UHPLC-Q Exactive MS. The metabolite elucidation conditions are described in Fig. 4. (A) Chromatograms of M2, M3 and M4. (B) MS/MS of ATX. (C) MS/MS of M4.
Figure 8. Conformation of hydroxylamine metabolite (M4) using TiCl3.

Incubations were conducted in 1X phosphate-buffered saline (1 X PBS, pH 7.4), containing 25 μM ATX, 2 pmol of cDNA-expressed CYP2B6 and NADPH (final concentration 1.0 mM) in a final volume of 200 μl. After terminating the reactions with 200 μl of ice-cold methanol, 50 μl of each supernatant was used as controls. TiCl3 was added to an ice-cold 300 μl of supernatant and the resulting mixture was kept on ice for 1.0 hour. An aliquot from each control or reaction mixture was diluted 3 times with ice-cold methanol. All samples were analyzed using UHPLC-Q Exactive MS. (A) Chromatograms of M2, M3 and M4. (B) Reaction scheme and relative abundance of M2, M3 and M4. The relative quantification was conducted based on the peak area. The abundance of each metabolite was set as 100% in each sample without TiCl3. The data are expressed as mean (n = 2). ND, not detected. TiCl3, Titanium (III) chloride.
3.5. Role of CYPs in the ATX metabolism and bioactivation
The role of CYPs in the metabolism and bioactivation of ATX was determined by incubating ATX with different human cDNA-expressed P450s (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and CYP3A4) and by chemical inhibitory experiments in HLM. Our data show that CYP2D6 is the major enzyme contributing to the formation of metabolites M14, M15 and M20. The role of CYP2D6 in the formation of M14, M15 and M20 was verified by co-incubation with quinidine, a potent CYP2D6 inhibitor, in HLM. The formation of M14, M15 and M20 was suppressed up to 92%, 93% and 90% (respectively) by quinidine at 2.0 μM (Fig. 9F). Both CYP3A4 and CYP2B6 mediated the formation of ATX-hydroxylamine M4 (Table 2). The roles of CYP3A4 and CYP2B6 of the formation of M4 were verified by co-incubation with KCZ, a potent CYP3A4 inhibitor or TCP, a potent CYP2B6 inhibitor. The formation of M4 was suppressed up to 25 % and 50 % by TCP at 10 μM and KCZ at 2.0 μM (Figs. 9A and 9B), but CYP1A2 (NF, 6.0 μM), CYP2C19 (NT, 10 μM), and CYP2D6 (Qui, 2.0 μM) inhibitors have no effect of the M4 formation in HLM (Table S1). O-methyl oxime (M30) is formed mainly mediated by CYP2C8 and CYP2B6 (Table 2). The roles of CYP2B6 and CYP2C8 in the formation of M30 were further confirmed by co-incubation with TCP or QT, a potent CYP2C8 inhibitor. The formation of M30 was suppressed up to 40% by TCP at 10.0 μM and 61% by QT at 30.0 μM, respectively (Fig. 9A and 9C). NT is also a potent CYP2A6 inhibitor except as CYP2C19 inhibitor (Tassaneeyakul et al., 2000). NT at 10 μM and CYP2D6 inhibitors (Qui, 2.0 μM) suppressed the 14% and 23% formation of M30 in HLM (Figs. 9D & 9E and Table S1). Multiple enzymes are involved in the formation of M5 (Table 2 and Table S1). The formation of M5 was suppressed up to 10%, 13%, 22%, and 50% by CYP1A2, CYP2C19, CYP3A4 and CYP2B6 inhibitors in HLM, respectively. Although cDNA-expressed CYP2D6 generated the metabolite M5, CYP2D6 inhibitor in HLM has no effect on the formation of M5 (data not shown). We also evaluated the roles of CYPs in the formation of other stable metabolites (M1-M3, M8, M9-M11, and M17-M18) using cDNA-expressed CYPs (Table 2). CYP2B6, CYP2D6 and CYP3A4 mediated the formation of M3 (Table S1). CYP2B6 contributes to the formation of M9. CYP2D6 is the dominant enzyme involved in their formation of M1, M2, M8, M10, M11, M17, and M18 (Table S1).
Figure 9. Roles of CYPs in the formation of M4, M14, M15, M20 and M30.

TCP (10.0 µM, CYP2B6 inhibitor), KCZ (2.0 μM, CYP3A4 inhibitor), NF, (6.0 µM, CYP1A2 inhibitor); NT, (10.0 µM, CYP2C19 inhibitor); QT, (30.0 µM, CYP2C8 inhibitor), and Qui (2.0 μM, CYP2D6 inhibitor) were used in the inhibitory tests in HLM. The incubation conditions of ATX in HLM were detailed in experimental procedures. All samples were analyzed by UHPLC-Q Exactive MS. (A) Effects of TCP on the formations of M4 and M30 in HLM. The relative abundance of M4 and M30 from the incubation with HLM in the absence of TCP was set as 100%. (B) Effects of KCZ on the formations of M4 in HLM. The relative abundance of M4 from the incubation in HLM without KCZ was set as 100%. (C-E) Effects of QT, NT, and on the formations of M30 in HLM. The relative abundance of M30 from the incubation in HLM without QT, NT and Qui was set as 100%. (F) Effects of Qui on the formations of M14, M15 and M20 in HLM. The relative abundance of M14, M15 and M20 from the incubation in HLM without Qui was set as 100%. All data are expressed as mean ± SEM (n = 3). P*<0.05, **P<0.01. KCZ, ketoconazole; Qui, quinidine; TCP, ticlopidine; NF, alpha-naphthoflavone; NT, nootkatone; QT, quercetin.
Table 2.
CYPs involved in the formation of metabolites of ATX.
| M1 | M2 | M3 | M4 | M5 | M8 | M9 | M10 | M11 | M14 | M15 | M17 | M18 | M19 | M20 | M30 | M31 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CYP1A2 | 0.0 | 0.52 | 0.0 | 24.3 | 42.6 | 4.7 | 0.0 | 2.6 | 0.0 | 0.0 | 6.2 | 0.0 | 0.0 | 100 | 0.0 | 4.7 | 0.0 |
| CYP2A6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 13.8 | 0.0 |
| CYP2B6 | 0.0 | 0.04 | 100 | 100 | 100 | 0.0 | 100 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100 | 0.0 |
| CYP2C8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 43.1 | 0.0 |
| CYP2C9 | 0.0 | 0.04 | 0.0 | 4.0 | 5.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.5 | 0.0 |
| CYP2C19 | 0.0 | 0.31 | 0.0 | 11.0 | 60.1 | 8.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.6 | 0.0 |
| CYP2D6 | 100 | 100 | 20.7 | 16.7 | 47.4 | 100 | 0.0 | 100 | 100 | 100 | 100 | 100 | 100 | 0.0 | 100 | 10.4 | 100 |
| CYP2E1 | 0.0 | 0.02 | 0.0 | 0.0 | 0.41 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 5.8 | 0.0 |
| CYP3A4 | 0.0 | 0.14 | 6.2 | 82.8 | 26.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 7.1 | 0.0 |
cDNA-expressed CYP450s (control, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4) were used to determine the role of individual CYP450 in ATX metabolism. All samples were analyzed by UHPLC-Q Exactive MS. The largest peak area of each metabolite from CYP enzymes was set as 100%. All data are expressed as mean (n = 2).
4. Discussion
Comprehensive metabolic profiling of a drug is beneficial to the understanding the adverse effect of drugs. Metabolomics-based strategies have been successfully utilized to identify stable metabolites and reactive metabolites (Li et al., 2011b; Li et al., 2012) and has enabled the identification of unpredictable, novel metabolites (Li et al., 2011a; Li et al., 2011c). Using LC-MS-based metabolomic approaches, a total of 31 metabolites and adducts related to ATX were identified, including two aldehyde adducts (M30 and M31), 20 phase I metabolites (M1-M20), and 9 glucuronides (M21-M29). In human studies using 14C-labelled ATX, most of the metabolites are excreted in urine (greater than 80% of the dose) and the dominant metabolites are ATX-O-glucuronides (Sauer et al., 2003; Sauer et al., 2005). Our study in mice found that monohydroxylated-ATX (M2 and M3, 21%), and glucuronides (M21-M29, 77.5%) are the primary metabolites in mouse urine (Fig. 2A). M29 is a novel adduct of ATX with both glucuronic acid and glucose. Detoluene-ATX (M20, 0.78%), monohydroxylated-N-desmethyl-ATX (M8, 0.2%), and dihydroxylated-ATX (M16, 0.18%) are the metabolites excreted in mouse urine. M2, M3, M10, M16 and M21 are also excreted in mouse feces (Fig. 2B). These findings are consistent with previous human studies.
In our studies, we first investigate the metabolism of ATX in HLM, MLM, & RLM and 20 phase I metabolites were identified. Monohydroxylated-ATX (M2) is dominant metabolite in all the species, but species differences are observed for certain minor metabolites (Fig. 3). MLM and RLM produced more cyclization metabolite M15 than that of HLM. MLM have more activity to generate detoluene-ATX (M20) than HLM and RLM. This information should be helpful to investigate the contribution of specific metabolites to ATX toxicity and possible drug-drug interactions in suitable models from a metabolic angle.
The reactive metabolites play a critical role in the pathogenesis of idiosyncratic adverse drug reactions (Gunawan and Kaplowitz, 2007; Thompson et al., 2016). No glutathione adducts were observed in our study (data not shown), suggesting ATX is unlikely to react with free sulfur group in proteins. However, the formation of carboxylic acid in previous studies indicated that the precursor, aldehydes, was produced in the metabolism of ATX (Sauer et al., 2005). The formation aldehydes were confirmed in HLM using methoxyamine as a trapping agent (Fig. 4A). The ATX-aldehyde in all three LMs and trace amount of O-ATX-aldehyde were detected in MLM and RLM using methoxyamine as a trapping agent (Table 1). Previous studies indicated that aldehydes could react with ε-amino groups of lysine residues and exocyclic amino groups of DNA to cause the deoxynucleoside-protein amino acid cross-links (Stein et al., 2006). These interactions may cause toxicity by impairing the function of macromolecules (LoPachin et al., 2009). Aldehydes have been proposed contributing to the adverse effects of several drugs (LoPachin and Gavin, 2014; Laskar and Younus, 2018). For instance, the atropaldehyde produced from felbamate is responsible for felbamate-induced hepatotoxicity (Dieckhaus et al., 2002). Therefore, the aldehyde generated from ATX may be involved in the ATX-induced toxicity.
Cytochrome P450-mediated cyclization reactions have been reported for multiple drugs (Meng et al., 2007; Arfeen et al., 2014), but cyclization metabolites of ATX have not previously been reported. We identified two novel cyclization metabolites M14 and M15 in this study. The proposed mechanisms of M14 and M15 formation were presented in Fig. 6. ATX was first oxidized to M3, followed by the second oxidation to generate the aldehyde. The aldehyde was intra-cyclized to form the intermediate hemiaminal, which was further oxidized to form M14 or M15. Alternatively, the lactam M14 could be also formed by firstly oxidizing ATX to O-ATX (M1, M2, or M3), followed by further oxidation to produce the aldehyde. The aldehyde is cyclized to form the intermediate O-hemiaminal and subsequent oxidation to form M14. The aldehydes are the intermediates of the cyclization metabolites M14 and M15, which may be also related to ATX toxicity. More studied are needed to demonstrate the relevance of these metabolic pathways to ATX toxicity. Additionally, a novel ATX-hydroxylamine (M4) was identified and characterized based on exact mass and the MS/MS fragments. We also conformed the hydroxylamine formation by incubating ATX in CYP2B6, followed by the treatment with TiCl3 (Fig 8), as TiCl3 could efficiently convert hydroxylamine to secondary amines (Murahashi and Kodera, 1985). ATX-hydroxylamine (M4) completely disappeared in the samples treated with TiCl3 (Fig. 8A) and the abundance of other metabolites (M2 and M3) remained unchanged (Fig. 8B).
Using recombinant CYPs, we found that CYP3A4 and CYP2B6 mainly contributed to the formation of hydroxylamine M4. Our inhibitory experiments in HLM indicated that CYP3A4 is the major enzyme contributing to M4 formation, as CYP3A4 is much more abundant than CYP2B6 in HLM (Fig. 9A and 9B). Multiple enzymes are involved in the formation of aldehyde (M30). CYP2C8 and CYP2B6 are major enzymes (Table 2 and Figs. 9A and 9C) as CYP2A6 inhibitor (Tassaneeyakul et al., 2000) and CYP2D6 inhibitor only suppressed 14% and 23 % of M30 formation in HLM (Figs. 9D and 9E). CYP2B6 is involved in producing 2-hydroxymethylatomoxetine (M3) (Table 2), which is the precursor of the aldehyde (M30) (Fig. 4C). In previous study, it appeared that CYP2B6-mediated formation of M3 became a more predominant pathway of metabolism in HLM with lower CYP2D6 activity levels (Dinh et al., 2016). Although CYP2D6 cannot be readily induced, it is highly polymorphic (Ingelman-Sundberg, 2005). Thus, slow CYP2D6 metabolizers with concurrent CYP2B6 induction may be at risk for a high production of M3 and oxidation to the aldehyde (M30), which likely increases ATX-related toxicity. CYP2D6 contributed to the formation of novel cyclization metabolites M14 and M15 (Table 2, and Fig. 9). Ultrarapid CYP2D6 metabolizers could be at risk for the high production of M14 and M15. As discussed above, the intermediates of the M14 and M15 formation are the aldehydes (Fig. 6). Accordingly, ultrarapid CYP2D6 metabolizers may increase the ATX aldehyde formation, which also may increase the potential of ATX toxicity. Consequently, assay of CYP2D6 activity before description may improve the medication safety of ATX. CYP 2B6 and CYP2C8 induction may increase the production of the aldehyde (M30) and potentiate the ATX toxicity. Thus, the co-administrated ATX and the strong inducers of CYP2C8 (e.g., rifampicin and secobarbital) and CYP2B6 (e.g., rifampicin and phenobarbital) should be cautious (Brodie et al., 2013).
Meanwhile, we also evaluated the roles of human CYPs in the formation of other stable metabolites (M1-M3, M5-M11, and M17-M19). CYP2D6 is a dominant enzyme, which is in agreement with the previous report (Sauer et al., 2003). Previous studies reported that CYP2C19 is the primary enzyme responsible for the formation of demethylated-ATX (M5) using recombinant human CYPs, but not detected in the incubation of CYP2D6 (Ring et al., 2002). Our studies suggested that multiple enzymes are involved in the formation of M5 (Table 2) including CYP1A2, CYP2B6, CYP2C19, CYP2D6 and CYP3A4 (Table 2). In our inhibitory experiment with CYP2D6 and HLM, quinidine at 2.0 μM (a CYP2D6 inhibitor) suppressed the 83% formation of M5 in CYP2D6 (data not shown), but it has no effect in the formation of M5 in HLM. These results suggested that CYP2D6 is able to produce M5, but has limited role in ATX metabolism possibly because of the low affinity of ATX with CYP2D6 in HLM compared to the affinities with CYP1A2, CYP2C19, CYP3A4 and CYP2B6. Our inhibitory study in HLM indicated that CYP1A2, CYP2C19, CYP3A4 and CYP2B6 contributed to M5 formation, which is consistent previous report (Ring et al., 2002). In this study, we mainly focus on the identification of novel metabolic pathways and drug metabolizing enzymes involved in ATX metabolism and bio activation. Thus, the effect of CYP2D6 genetic polymorphisms on ATX metabolism was not discussed here, which has been extensively investigated in human (Trzepacz et al., 2008; Todor et al., 2016).
5. Conclusions
In summary, this study provided a global view of ATX metabolism in liver microsomes and mice. The novel pathways related to ATX bioactivation and species difference in LMs were identified. These findings could be utilized for further understanding the mechanism of adverse effects and possible drug-drug interaction associated with ATX. Further studies are suggested to illustrate the possible role of the metabolism in ATX-related adverse effects through increase/decrease the formation of specific metabolites (e.g., aldehyde) in vitro and in vivo.
Supplementary Material
Figure 10. Metabolic map of ATX in mice and LMs.

All structures were determined based on the exact mass (mass error less than 2 ppm), MS/MS fragments and previous metabolism information of ATX. *, M31 was only based on the exact mass and predict formula as the high quality of MS/MS spectrum of M31 is not available.
Acknowledgements
This work was supported by the National Institute of Diabetes and Digestive and Kidney (R01-DK121970) the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R61HD099995) to Dr. Feng Li; Cancer Prevention & Research Institute of Texas (RP160805), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01 HD087157), Welch Foundation Grant (H-Q-0042) and Bill and Melinda Gates Foundation (INV-001902) to Dr. Martin M. Matzuk; the National Institute of General Medical Sciences (R01-GM115622) to Dr. Jin Wang.
Abbreviations
- ATX
atomoxetine
- GSH
reduced glutathione
- KCZ
ketoconazole
- NADPH
β-Nicotinamide adenine dinucleotide 2′-phosphate reduced
- NF
alpha-naphthoflavone
- NT
nootkatone
- QT
quercetin
- Qui
quinidine
- TCP
ticlopidine
- TiCl3
Titanium(III) chloride
- CYP
cytochrome P450
- HLM
human liver microsomes
- MLM
mouse liver microsomes
- RLM
rat liver microsomes
- OPLS-DA
orthogonal projection to latent structures-discriminant analysis
- UHPLC
ultra-high-performance liquid chromatography
- Q Exactive MS
Q Exactive™ Hybrid Quadrupole-Orbitrap™ Mass Spectrometer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None
References:
- https://www.drugbank.ca/categories/DBCAT002615.
- Arfeen M, Patel DS, Abbat S, Taxak N, and Bharatam PV (2014) Importance of cytochromes in cyclization reactions: quantum chemical study on a model reaction of proguanil to cycloguanil. J Comput Chem 35:2047–2055. [DOI] [PubMed] [Google Scholar]
- Bleibel W, Kim S, D’Silva K, and Lemmer ER (2007) Drug-induced liver injury: review article. Dig Dis Sci 52:2463–2471. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, and Schmidt D (2013) Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia 54:11–27. [DOI] [PubMed] [Google Scholar]
- Camporeale A, Porsdal V, De Bruyckere K, Tanaka Y, Upadhyaya H, Deix C, and Deberdt W (2015) Safety and tolerability of atomoxetine in treatment of attention deficit hyperactivity disorder in adult patients: an integrated analysis of 15 clinical trials. J Psychopharmacol 29:3–14. [DOI] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, and Gores GJ (2009) JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem 284:26591–26602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, Reddy KR, Watkins PB, Navarro V, Barnhart H, Gu J, Serrano J, and United States Drug Induced Liver Injury N (2015) Features and Outcomes of 899 Patients With Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 148:1340–1352 e1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress AC (2016) A critical appraisal of atomoxetine in the management of ADHD. Ther Clin Risk Manag 12:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui YM, Teng CH, Pan AX, Yuen E, Yeo KP, Zhou Y, Zhao X, Long AJ, Bangs ME, and Wise SD (2007) Atomoxetine pharmacokinetics in healthy Chinese subjects and effect of the CYP2D6*10 allele. Br J Clin Pharmacol 64:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckhaus CM, Thompson CD, Roller SG, and Macdonald TL (2002) Mechanisms of idiosyncratic drug reactions: the case of felbamate. Chem Biol Interact 142:99–117. [DOI] [PubMed] [Google Scholar]
- Dinh JC, Pearce RE, Van Haandel L, Gaedigk A, and Leeder JS (2016) Characterization of Atomoxetine Biotransformation and Implications for Development of PBPK Models for Dose Individualization in Children. Drug Metab Dispos 44:1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaola F and Molleston JP (2018) Drug-Induced Liver Injury in Children. Current Hepatology Reports 17:283–291. [Google Scholar]
- Erdogan A, Ozcay F, Piskin E, Karaman MG, Bilezikci B, Calik M, Tekin I, and Haberal M (2011) Idiosyncratic liver failure probably associated with atomoxetine: a case report. J Child Adolesc Psychopharmacol 21:295–297. [DOI] [PubMed] [Google Scholar]
- Gunawan BK and Kaplowitz N (2007) Mechanisms of drug-induced liver disease. Clin Liver Dis 11:459–475. [DOI] [PubMed] [Google Scholar]
- Hussaini SH and Farrington EA (2007) Idiosyncratic drug-induced liver injury: an overview. Expert Opin Drug Saf 6:673–684. [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M (2005) Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. The pharmacogenomics journal 5:6–13. [DOI] [PubMed] [Google Scholar]
- Lantz RJ, Gillespie TA, Rash TJ, Kuo F, Skinner M, Kuan HY, and Knadler MP (2003) Metabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjects. Drug Metab Dispos 31:1142–1150. [DOI] [PubMed] [Google Scholar]
- Laskar AA and Younus H (2018) Aldehyde toxicity and metabolism: The role of aldehyde dehydrogenases in detoxification, drug resistance and carcinogenesis. Drug Metab Rev:1–61. [DOI] [PubMed]
- Leise MD, Poterucha JJ, and Talwalkar JA (2014) Drug-induced liver injury. Mayo Clin Proc 89:95–106. [DOI] [PubMed] [Google Scholar]
- Li F, Gonzalez FJ, and Ma X (2012) LC–MS-based metabolomics in profiling of drug metabolism and bioactivation. Acta Pharmaceutica Sinica B 2:118–125. [Google Scholar]
- Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL, Klaassen CD, Gonzalez FJ, and Ma X (2013) Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy. Nat Med 19:418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Lu J, and Ma X (2011a) Metabolomic screening and identification of the bioactivation pathways of ritonavir. Chem Res Toxicol 24:2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Lu J, and Ma X (2011b) Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem Res Toxicol 24:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Lu J, and Ma X (2014) CPY3A4-mediated alpha-hydroxyaldehyde formation in saquinavir metabolism. Drug Metab Dispos 42:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, MacKenzie KR, Jain P, Santini C, Young DW, and Matzuk MM (2020) Metabolism of JQ1, an inhibitor of BET bromodomain proteins, in human and mouse liver microsomesdagger. Biology of reproduction. [DOI] [PMC free article] [PubMed]
- Li F, Miao Y, Zhang L, Neuenswander SA, Douglas JT, and Ma X (2011c) Metabolomic analysis reveals novel isoniazid metabolites and hydrazones in human urine. Drug metabolism and pharmacokinetics 26:569–576. [DOI] [PubMed] [Google Scholar]
- Li F, Zhang N, Gorantla S, Gilbertson SR, and Pati D (2018) The Metabolism of Separase Inhibitor Sepin-1 in Human, Mouse, and Rat Liver Microsomes. Front Pharmacol 9:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JR, Faught PR, Chalasani NP, and Molleston JP (2006) Severe liver injury after initiating therapy with atomoxetine in two children. J Pediatr 148:831–834. [DOI] [PubMed] [Google Scholar]
- Liu X, Lu Y, Guan X, Dong B, Chavan H, Wang J, Zhang Y, Krishnamurthy P, and Li F (2015) Metabolomics reveals the formation of aldehydes and iminium in gefitinib metabolism. Biochem Pharmacol 97:111–121. [DOI] [PubMed] [Google Scholar]
- Liu X, Lu YF, Guan X, Zhao M, Wang J, and Li F (2016) Characterizing novel metabolic pathways of melatonin receptor agonist agomelatine using metabolomic approaches. Biochem Pharmacol 109:70–82. [DOI] [PubMed] [Google Scholar]
- LoPachin RM and Gavin T (2014) Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol 27:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, and Barber DS (2009) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chemical research in toxicology 22:1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhao X-M, Hu Z, Wang L, and Li F (2019) LC–MS-Based Metabolomics in the Study of Drug-Induced Liver Injury. Current Pharmacology Reports 5:56–67. [Google Scholar]
- Mattiuz EL, Ponsler GD, Barbuch RJ, Wood PG, Mullen JH, Shugert RL, Li Q, Wheeler WJ, Kuo F, Conrad PC, and Sauer JM (2003) Disposition and metabolic fate of atomoxetine hydrochloride: pharmacokinetics, metabolism, and excretion in the Fischer 344 rat and beagle dog. Drug Metab Dispos 31:88–97. [DOI] [PubMed] [Google Scholar]
- Meng X, Maggs JL, Pryde DC, Planken S, Jenkins RE, Peakman TM, Beaumont K, Kohl C, Park BK, and Stachulski AV (2007) Cyclization of the acyl glucuronide metabolite of a neutral endopeptidase inhibitor to an electrophilic glutarimide: synthesis, reactivity, and mechanistic analysis. J Med Chem 50:6165–6176. [DOI] [PubMed] [Google Scholar]
- Murahashi S-I and Kodera Y (1985) Titanium (III) induced transformations of N,N-disubstituted hydroxylamines to imines and secondary amines. Tetrahedron Letters 26:4633–4636. [Google Scholar]
- Nair AB and Jacob S (2016) A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potnis D and Wackernah RC (2015) Drug-Induced Liver Injury in Children: Atomoxetine and Nonstimulants for ADHD. Am J Pharm Benefits 7:e15–e20. [Google Scholar]
- Preti A (2002) Tomoxetine (Eli Lilly & Co). Curr Opin Investig Drugs 3:272–277. [PubMed] [Google Scholar]
- Ring BJ, Gillespie JS, Eckstein JA, and Wrighton SA (2002) Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos 30:319–323. [DOI] [PubMed] [Google Scholar]
- Sauer JM, Ponsler GD, Mattiuz EL, Long AJ, Witcher JW, Thomasson HR, and Desante KA (2003) Disposition and metabolic fate of atomoxetine hydrochloride: the role of CYP2D6 in human disposition and metabolism. Drug Metab Dispos 31:98–107. [DOI] [PubMed] [Google Scholar]
- Sauer JM, Ring BJ, and Witcher JW (2005) Clinical pharmacokinetics of atomoxetine. Clin Pharmacokinet 44:571–590. [DOI] [PubMed] [Google Scholar]
- Simpson D and Perry CM (2003) Atomoxetine. Paediatr Drugs 5:407–415; discussion 416–407. [DOI] [PubMed] [Google Scholar]
- Stein S, Lao Y, Yang IY, Hecht SS, and Moriya M (2006) Genotoxicity of acetaldehyde- and crotonaldehyde-induced 1,N2-propanodeoxyguanosine DNA adducts in human cells. Mutation research 608:1–7. [DOI] [PubMed] [Google Scholar]
- Stojanovski SD, Casavant MJ, Mousa HM, Baker P, and Nahata MC (2007) Atomoxetine-induced hepatitis in a child. Clin Toxicol (Phila) 45:51–55. [DOI] [PubMed] [Google Scholar]
- Tang W and Lu AY (2010) Metabolic bioactivation and drug-related adverse effects: current status and future directions from a pharmaceutical research perspective. Drug Metab Rev 42:225–249. [DOI] [PubMed] [Google Scholar]
- Tassaneeyakul W, Guo LQ, Fukuda K, Ohta T, and Yamazoe Y (2000) Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch Biochem Biophys 378:356–363. [DOI] [PubMed] [Google Scholar]
- Thompson RA, Isin EM, Ogese MO, Mettetal JT, and Williams DP (2016) Reactive Metabolites: Current and Emerging Risk and Hazard Assessments. Chem Res Toxicol 29:505–533. [DOI] [PubMed] [Google Scholar]
- Todor I, Popa A, Neag M, Muntean D, Bocsan C, Buzoianu A, Vlase L, Gheldiu AM, and Briciu C (2016) Evaluation of a Potential Metabolism-Mediated Drug-Drug Interaction Between Atomoxetine and Bupropion in Healthy Volunteers. J Pharm Pharm Sci 19:198–207. [DOI] [PubMed] [Google Scholar]
- Trzepacz PT, Williams DW, Feldman PD, Wrishko RE, Witcher JW, and Buitelaar JK (2008) CYP2D6 metabolizer status and atomoxetine dosing in children and adolescents with ADHD. Eur Neuropsychopharmacol 18:79–86. [DOI] [PubMed] [Google Scholar]
- Wooltorton E (2005) Suicidal ideation among children taking atomoxetine (Strattera). CMAJ 173:1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley B and Powers R (2013) Multivariate Analysis in Metabolomics. Curr Metabolomics 1:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DH, Caro L, Cerra M, Panorchan P, Du L, Anderson M, Potthoff A, Nachbar RB, Wagner J, Manns MP, and Talal AH (2015) Liver-to-plasma vaniprevir (MK-7009) concentration ratios in HCV-infected patients. Antivir Ther 20:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Li GF, and Markowitz JS (2016) Atomoxetine: A Review of Its Pharmacokinetics and Pharmacogenomics Relative to Drug Disposition. J Child Adolesc Psychopharmacol 26:314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Zhang T, Xiao XR, Huang JF, Wang Y, Gonzalez FJ, and Li F (2019) Impaired clearance of sunitinib leads to metabolic disorders and hepatotoxicity. Br J Pharmacol. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.