

Abstract
Regions of the genome with the potential to form secondary DNA structures pose a frequent and significant impediment to DNA replication and must be actively managed in order to preserve genetic and epigenetic integrity. How the replisome detects and responds to secondary structures is poorly understood. Here, we show that a core component of the fork protection complex in the eukaryotic replisome, Timeless, harbours in its C‐terminal region a previously unappreciated DNA‐binding domain that exhibits specific binding to G‐quadruplex (G4) DNA structures. We show that this domain contributes to maintaining processive replication through G4‐forming sequences, and exhibits partial redundancy with an adjacent PARP‐binding domain. Further, this function of Timeless requires interaction with and activity of the helicase DDX11. Loss of both Timeless and DDX11 causes epigenetic instability at G4‐forming sequences and DNA damage. Our findings indicate that Timeless contributes to the ability of the replisome to sense replication‐hindering G4 formation and ensures the prompt resolution of these structures by DDX11 to maintain processive DNA synthesis.
Keywords: DNA replication, fork protection complex, G‐quadruplex, replisome, timeless
Subject Categories: DNA Replication, Repair & Recombination; Structural Biology
Cooperation of a helicase and a component of the fork protection complex allows eukaryotic replisomes to detect and respond to DNA secondary structures.

Introduction
DNA can create significant impediments to its own replication through the formation of secondary structures. When unwound, certain sequences, often repetitive or of low complexity, can adopt a variety of non‐B form structures, including hairpins, cruciforms, triplexes and quadruplexes (Mirkin & Mirkin, 2007). It is increasingly clear that secondary structure formation is a frequent event during replication, even at genomically abundant sequences previously thought not to be a major source of difficulty (Šviković et al, 2019). To prevent such sequences causing havoc with the genetic and epigenetic stability of the genome, cells deploy an intricate network of activities to counteract secondary structure formation and limit its effects. These activities include proteins that bind and destabilise DNA structures and specialised helicases that unwind them (Lerner & Sale, 2019). In addition, the repriming activity of PrimPol can be deployed to confine a structure into a minimal region of single‐stranded DNA (ssDNA), limiting the potential dangers of exposing extensive ssDNA in a stalled replisome (Schiavone et al, 2016; Šviković et al, 2019).
G4s are one of the most intensively studied and potent structural replication impediments. G4s arise in consequence of the ability of guanine to form Hoogsteen base‐paired quartets (Gellert et al, 1962). In favourable sequence contexts, comprising runs of dG separated by variable numbers of non‐G bases, stacks of G quartets form G4 secondary structures. Current estimates suggest that over 700,000 sites in the human genome have the potential to form G4s (Chambers et al, 2015). While some of these G4s may have important roles in genome physiology, all pose a potential threat to DNA replication and sites with G4‐forming potential have been linked to both genetic and epigenetic instability (Šviković & Sale, 2017; Kaushal & Freudenreich, 2019).
Precisely, how DNA structures are detected and resolved by the replication machinery remains unclear. Many of the factors involved in processing G4 secondary structures, for instance FANCJ and REV1 (Kruisselbrink et al, 2008; London et al, 2008; Wu et al, 2008; Youds et al, 2008; Sarkies et al, 2010), do not appear to be constitutive components of the replisome (Dungrawala et al, 2015). It is thus likely that core components of the replisome will act as “first responders” to DNA structures and play an important role coupling their detection with suppressing their deleterious effects on DNA synthesis. Particularly interesting in this context is a subset of replisome components known as the fork protection complex (FPC). The FPC comprises four main proteins—Timeless, Tipin, Claspin and AND‐1—that are conserved from yeast to mammals (Errico & Costanzo, 2012). FPC components associate with the replication fork via direct interactions with the CMG replicative helicase and replicative polymerases α, δ and ε (Nedelcheva et al, 2005; Numata et al, 2010; Cho et al, 2013; Bastia et al, 2016; Kilkenny et al, 2017; Baretić et al, 2020). They also interact with DNA both directly (Tanaka et al, 2010) and indirectly via replication protein A (RPA) (Witosch et al, 2014). These interactions allow the FPC to remain at the fork, which ensures a normal speed of DNA synthesis (Yeeles et al, 2017). Additionally, the FPC has a series of functions that promote normal replisome progression and fork integrity: it is essential to avoid uncoupling of pol ε from the replicative helicase and consequent formation of long stretches of ssDNA (Katou et al, 2003; Lou et al, 2008). It also has a conserved role in S‐phase checkpoint activation in response to DNA damage, including checkpoint kinase activation, cell cycle arrest and maintenance of the integrity of the replication fork (Chou & Elledge, 2006; Gotter et al, 2007; Unsal‐Kaçmaz et al, 2007; Yang et al, 2010). Furthermore, it plays an important, but incompletely understood, role in maintaining sister chromosome cohesion (Chan et al, 2003; Leman et al, 2010).
The FPC is thus well placed to play a role in the detection and metabolism of DNA secondary structures that could impede DNA synthesis. Indeed, deficiency of both TOF1 in yeast and Timeless in human cells leads to replication fork stalling, repeat instability and fragility at secondary structure‐forming sequences (Voineagu et al, 2008, 2009; Leman et al, 2012; Liu et al, 2012b; Gellon et al, 2019), underscoring the potential importance of Timeless in maintaining processive replication through regions of the genome capable of forming secondary structures.
Although Timeless itself does not appear to possess catalytic activity that would process DNA secondary structures, it interacts with the DNA helicase DDX11 (Calì et al, 2016). DDX11 (or CHLR1), is a 5′–3′ Fe–S helicase of the same superfamily 2 as FANCJ, RTEL and XPD (Lerner & Sale, 2019). In humans, mutations in DDX11 cause Warsaw breakage syndrome, an extremely rare autosomal recessive disease characterised by microcephaly, growth retardation, cochlear abnormalities and abnormal skin pigmentation (Alkhunaizi et al, 2018). In vitro, DDX11 has unwinding activity on several non‐duplex DNA structures, such as G4s (Wu et al, 2012a; Bharti et al, 2013), triplex DNA (Guo et al, 2015) and D‐loops (Wu et al, 2012a). Further, the helicase activity of DDX11 is enhanced by Timeless (Calì et al, 2016). However, it remains unclear how Timeless and DDX11 collaborate in vivo in detecting and processing G4s during replication. Here, we provide in vivo evidence that Timeless and DDX11 operate together to ensure processive replication of G4‐forming DNA. We report a previously unappreciated DNA‐binding domain (DBD) in the C‐terminus of Timeless, which exhibits specificity towards G4 structures. We propose that Timeless plays a role in the detection of G4 structures at the replication fork, recruiting DDX11 to unwind them and ensure processive replication is maintained, thereby avoiding G4‐induced genetic and epigenetic instability.
Results
Timeless is required for processive replication of a genomic G4 motif
To address whether Timeless is involved in maintaining processive replication of G4‐forming DNA in vivo, we disrupted the TIMELESS locus in chicken DT40 cells with CRISPR/Cas9‐induced deletions. We isolated several timeless mutants with biallelic disruptions in exon 1, around the guide site (Appendix Fig S1). The timeless mutant cells were sensitive to cisplatin (Fig EV1), as previously observed in human cells depleted of Timeless (Liu et al, 2017). To assess the role of Timeless in the replication of a G4‐forming sequence, we took advantage of the Bu‐1 loss variant assay (Schiavone et al, 2014). The stable expression of the BU‐1 locus in DT40 is dependent on the maintenance of processive replication through a G4 motif located ~ 3.5 kb downstream of the promoter (Fig 11A). Prolonged pausing of leading‐strand replication at this motif leads to loss of epigenetic information around the promoter of the gene and a permanent and heritable change in its expression (Sarkies et al, 2012; Schiavone et al, 2014, 2016; Guilbaud et al, 2017). This stochastic and replication‐dependent generation of Bu‐1 loss variants can be monitored by flow cytometry as BU‐1 encodes a surface glycoprotein. Small pools of Bu‐1high wild‐type and timeless cells were expanded in parallel for ~ 20 divisions (15–21 days), and the proportion of cells in each pool that had lost their Bu‐1high status determined. We detected increased levels of Bu‐1 expression instability in timeless DT40 cells compared to the wild‐type cells, which retained their stable Bu‐1high expression (Fig 11B and C). The instability of Bu‐1 expression in timeless cells was fully reversed by expression of human Timeless (Fig 11C) and is dependent on the +3.5 G4 motif (Fig 11C). Cells deficient in Tipin (Abe et al, 2016), a constitutive interactor of Timeless within the FPC, also exhibit instability of BU‐1 expression (Fig 11D). These results show that Timeless is necessary to maintain processive DNA replication of a genomic G4 motif.
Figure EV1. Sensitivity of wild type (WT), timeless, ddx11 and fancj DT40 mutants to cisplatin (CDDP).
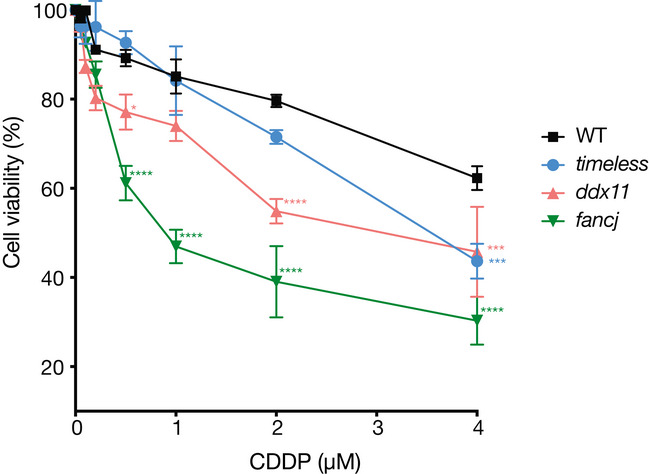
Cell viability, assessed by MTS assay, of DT40 wild type, ddx11, timeless and fancj, after 72 h in presence of cisplatin at the indicated doses. The values represent the means (error bars indicate SD) of two independent experiments performed in triplicate. *P < 0.05, ***P < 0.001 and ****P < 0.0001; one‐way ANOVA compared to the wild type.
Figure 1. Timeless and Tipin are required to maintain processive replication past G4 structures in vivo .
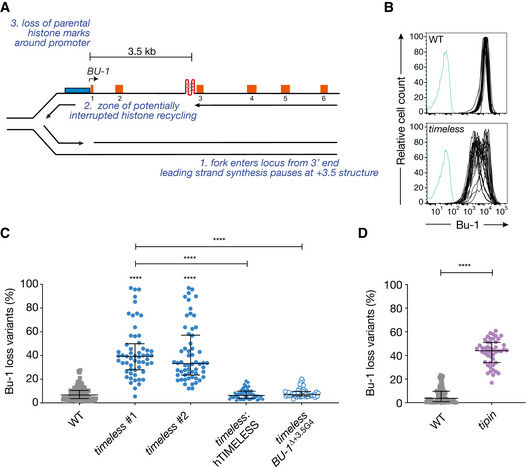
- The BU‐1 locus as a model system to record G4‐dependent replication stalling. The leading strand of a replication fork entering the locus from the 3′ end stochastically stalls at the +3.5 G4, leading to the formation of a region of ssDNA, with interruption of parental histone recycling and of histone modifications necessary to maintain normal expression of the locus (Schiavone et al, 2014).
- Instability of BU‐1 expression in timeless cells. FACS plots of wild‐type and timeless (clone 1) DT40 cells stained with anti‐Bu-1 conjugated with phycoerythrin. Each line represents the Bu‐1 expression profile of an individual clonal population. Unstained controls are shown in blue.
- Fluctuation analysis for Bu‐1 loss in wild‐type DT40 cells and two independent timeless clones generated by CRISPR‐Cas9 targeting (clones 1 and 2; Appendix Fig S1), timeless (clone 1) complemented by expression of human Timeless cDNA and a timeless mutant on a background in which the endogenous +3.5 G4 has been deleted (ΔG4) (Schiavone et al, 2014).
- Fluctuation analysis for Bu‐1 loss in DT40 wild‐type and tipin cells.
Identification and characterisation of a Timeless DNA‐binding domain
As a core component of the replisome, Timeless is intimately associated with DNA synthesis at the replication fork (Yeeles et al, 2017). In vitro data show that the Timeless–Tipin complex can bind to ssDNA through RPA (Witosch et al, 2014), and the Swi1‐Swi3 complex, the fission yeast orthologue of Timeless–Tipin, was also shown to bind DNA (Tanaka et al, 2010). Inspection of the amino acid sequence of human Timeless revealed the presence of a conserved domain in its C‐terminal half (residues 816–954; Fig 22A), with a predicted fold similarity to the myb‐like proteins of the homeodomain‐like superfamily, that bind double‐stranded DNA (dsDNA) with a tandem repeat of 3‐helix bundles (named here N‐term and C‐term).
Figure 2. Identification and characterisation of a DNA‐binding activity in Timeless.
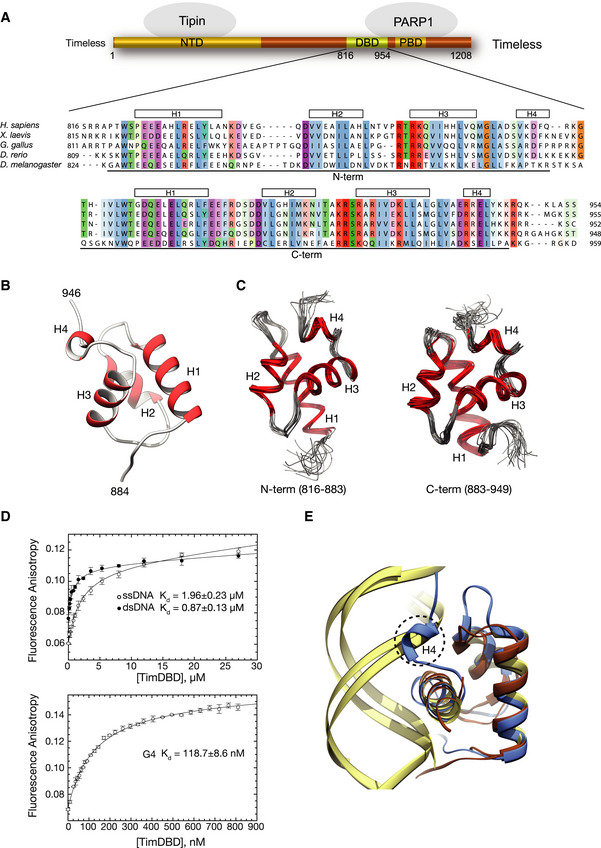
- Schematic drawing of human Timeless and its known domain structure (NTD: N‐terminal domain; DBD: DNA‐binding domain; PBD: PARP‐binding domain). A multiple sequence alignment of vertebrate Timeless sequences is shown underneath, with amino acid conservation coloured according to the Clustal colour scheme. The alignment is annotated with the extent and secondary structure elements of the two helical domains (N‐term and C‐term) composing the DBD.
- Ribbon drawing of the 1.15 Å crystal structure at of the DBD C‐term. Helices are in red and labelled H1–H4.
- Ribbon drawings of the N‐term and C‐term domains of the DBD determined by NMR. The two domains are shown in the same orientation to highlight their high degree of three‐dimensional similarity. The superposition of the 20 lowest energy structures is shown for each domain.
- The DNA‐binding affinity of DBD was measured by fluorescence anisotropy, titrating the DBD protein against Cy3 3′‐labelled ssDNA, dsDNA and G4 DNA (see Appendix Table S2 for sequence details). The top panel shows binding curves for ss‐ and dsDNA, and the bottom panel shows the binding curve for the G4 DNA substrate. The data points represent the mean of at least three independent experiments, and the error bars indicate one standard deviation (SD).
- Ribbon diagram of the superposition of DBD C‐term with the highly similar DNA‐binding domains of telomeric protein TRF1 (PDB ID 1W0T) (Court et al, 2005) and the bacterial cell cycle regulator GcrA (PDB ID 5Z7I) (Wu et al, 2018) in complex with their DNA substrates. A similar DNA‐binding mode by DBD would cause a steric overlap of helix H4 with the phosphate backbone of dsDNA. DBD C‐term is in light blue, TRF1 and GcrA proteins in brown and their DNA substrates in khaki.
We used X‐ray diffraction and NMR spectroscopy to investigate experimentally the structure and dynamics of the newly discovered Timeless domain. The 1.15 Å crystal structure of amino acids 885–947 (C‐term), corresponding to a single myb‐like fold, confirmed the presence of a three‐helix bundle characteristic of the homeodomain superfamily of DNA‐binding proteins, extended by the presence of a fourth C‐terminal alpha helix unique to the Timeless domain (Fig 22B). The NMR structural ensemble of amino acids 816–954 revealed two well‐converged domains (824–880 and 891–944, backbone r.m.s.d. 0.4 and 0.5 Å, respectively) connected by a linker (881–890) that was significantly less well converged, implying a high degree of flexibility between N‐ and C‐term domains (Fig 22C). This observation was confirmed by backbone dynamics measurements (Appendix Fig S2). The N‐ and C‐terminal domains adopted the same three‐dimensional fold; in particular, the N‐terminal repeat shared the presence of an additional fourth helix, H4, as seen in the C‐terminal repeat (Fig 22C).
In keeping with the similarity of its structure to known DNA binding domains (DBDs), we examined the ability of the Timeless domain to interact with DNA. We found that it bound with low micromolar affinity to both ss‐ and dsDNA probes (Fig 22D; top panel). A distinguishing feature of the Timeless DBD is the presence of a fourth alpha helix in both N‐ and C‐terminal 3‐helix bundle repeats. Superposition of the DBD C‐term onto the structurally homologous domains of the telomeric protein TRF1 (Court et al, 2005) and bacterial cell cycle regulator GcrA (Wu et al, 2018) in complex with their dsDNA substrates (Fig 22E) shows that, if the DBD were to adopt a similar mode of dsDNA binding, the fourth helix of both its N‐term and C‐term repeats would likely lead to a steric clash with the phosphate backbone of the DNA.
Although it is conceivable that the DBD might rearrange its conformation upon DNA binding, the fourth helix of both repeats participates in core hydrophobic interactions, mediated by conserved residues L872 and F879 (N‐terminal repeat), and L936, V937 and L943 (C‐terminal repeat), making such rearrangements unlikely. Given the functional context in which Timeless operates as a replisome component and the observations, presented in Fig 11, that it is required to maintain processive replication of the BU‐1 G4, we speculated that the C‐terminal DNA‐binding activity of Timeless might be directed towards recognition of DNA secondary structures that form transiently on the unwound template, such as G4s.
Indeed, when we tested a well‐characterised G4 sequence present in the promoter of the MYC gene (Ambrus et al, 2005), the DBD bound to it with nanomolar affinity, and about one order of magnitude tighter than ds‐ or ssDNA (Fig 22D). This observation prompted us to ask whether the Timeless–Tipin complex, like the isolated DBD, is able to bind to the same G4 motif. To mimic the unwound template DNA, we embedded the G4 within a longer ssDNA sequence (ssG4; Appendix Table S2). We found that the Timeless–Tipin complex bound to ssG4 with low micromolar affinity and in a selective fashion, as it did not show measurable interactions with a hairpin DNA embedded within the same ssDNA (ssHP), or a mutated ssG4 sequence that had lost the ability to fold into a G4 (ss; Figs 33A and EV2, Appendix Table S2). We next tested a series of G4 sequences found in different genomic contexts and with different folding topologies: we found that the Timeless–Tipin complex bound to all of them, albeit with different affinities that varied several fold, whereas it did not show appreciable binding to ss‐ or dsDNA (Fig 33B).
Figure 3. The Timeless–Tipin complex shows a preference for binding G4 DNA .

- ssG4: G4 flanked by single‐stranded DNA; ssHP: hairpin flanked by single‐stranded DNA; ss: single‐stranded DNA (Appendix Table S2 for sequence details).
- Binding affinity of Timeless–Tipin for a range of G4 DNA sequences (see Appendix Table S2 for sequence details and references). Single‐stranded (ss20: 5′‐6FAM-ATAAGAGTGGTTAGAGTGTA) and double‐stranded (ds20: ss20 annealed to complementary sequence) DNA were also tested as controls.
Figure EV2. The Timeless–Tipin complex shows a preference for binding G‐quadruplex DNA structures.
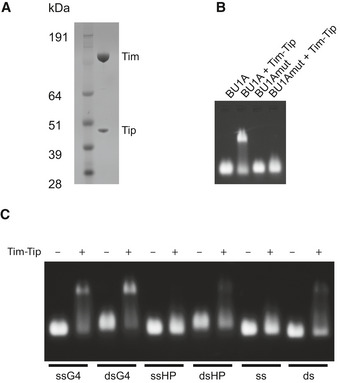
- Coomassie‐stained SDS–PAGE gel of purified Timeless–Tipin complex.
- Electrophoretic mobility shift assay (EMSA) showing the binding of Timeless–Tipin to G‐quadruplex sequence BU1A + 3.5. Mutation of the G‐quadruplex sequence (BU1A + 3.5 mut) disrupts Timeless–Tipin binding (see Appendix Table S2 for sequence details). Timeless–Tipin and DNA are both present at a final concentration of 5 μM.
- EMSA showing the binding of Timeless–Tipin to G‐quadruplex sequences (ssG4, dsG4) but not single‐stranded DNA (ss), double‐stranded DNA (ds) or hairpin‐containing sequences (ssHP, dsHP). Timeless–Tipin and DNA are both present at a final concentration of 5 μM.
These findings show that Timeless contains, in its C‐terminal half, a previously unrecognised DBD, which closely resembles in structure the tandem repeat of three‐helix bundles found in the homeodomain‐like superfamily of transcription factors. While Timeless DBD binds to both ss‐ and dsDNA, it binds with ~ 10‐fold greater affinity to a defined G4 DNA sequence. The preference for G4 DNA is retained by the Timeless–Tipin complex.
The Timeless C‐terminus is crucial for processive G4 replication in vivo
To further explore the in vivo contribution of the Timeless C‐terminus to G4 replication, we generated a DT40 cell line expressing a version of Timeless truncating the gene before the DBD, using CRISPR/Cas9 gene targeting to exon 16. This truncation also removes a domain previously reported to bind PARP1, the PARP1‐binding domain (PBD) (Xie et al, 2015). This cell line exhibited instability of BU‐1 expression comparable to the timeless mutant (Fig 44) suggesting a role for the C‐terminus of the protein in G4 replication. We confirmed this result by expressing human Timeless truncated at amino acid 816, and thus lacking both the DBD and PBD, in the timeless mutant (Appendix Fig S3). To further dissect this observation, we complemented timeless cells with truncated versions of human Timeless, lacking only the DBD (ΔDBD) or the PBD (PARP*). Timeless lacking either the DBD or the PBD largely restored, although not completely, the BU‐1 expression instability of the timeless mutant suggesting that the DBD and PBD act redundantly. Additionally, co‐immunoprecipitation (Co‐IP) experiments showed that neither region is required for binding the DDX11 helicase (Fig EV3), a known binding partner of Timeless and whose ability to unwind G4s is stimulated by Timeless (Calì et al, 2016). These results indicate that the C‐terminus of the Timeless protein has an important role in G4 replication to which both the DBD and the PDB contribute, independently of DDX11 recruitment.
Figure 4. The C‐terminus of Timeless is required for processive G4 replication.
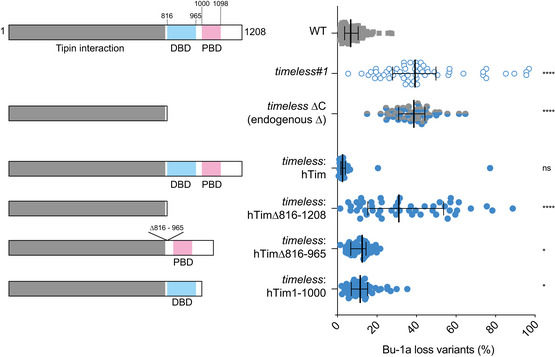
Fluctuation analysis for the generation of Bu‐1 loss variants. Top to bottom: wild type, timeless (clone 1), a timeless mutant (timeless ∆C) generated by CRISPR‐Cas9 targeting exon 16 which truncates the protein removing the CTD containing both the DBD and the PARP‐binding domains. Then, complementation of timeless#1 with human Timeless (hTim), hTim∆816–1,208 (lacking both the DBD and PBD), hTim∆816–965 (lacking the DBD) and hTim[1:1,000], lacking the PBD. At least two independent fluctuation analyses were performed with 24–36 individual clones each cell line per repeat. Bars and whiskers represent median and interquartile range, respectively. *P < 0.05 and ****P < 0.0001; one‐way ANOVA for comparison with the wild‐type cells.
Figure EV3. The C‐terminus of Timeless is not required for its interaction with DDX11.

HEK293T cells were transiently transfected with a plasmid encoding Flag‐hTimeless, or co‐transfected with plasmids encoding hDDX11 and Flag‐Timeless or with Timeless mutated to delete the DNA‐binding domain (ΔDBD: deletion of region S816–S965) or PARP‐binding domain (PARP*: truncation at V1000). Twenty‐four h after transfection, whole‐cell extracts were subjected to immunoprecipitation with anti‐Flag magnetic beads. Western Blot analyses were performed to detect overexpressed DDX11 protein in the pulled down samples using a specific antibody. Upper panel: Input and pulled down samples transfected with different Timeless constructs detected with an anti‐Flag antibody. Bottom panel: Input and pulled down samples transfected with different Timeless constructs detected with an anti‐DDX11 antibody. Tubulin was used as a loading control for the input samples.
DDX11 ensures processive G4 replication in vivo
Since Timeless itself lacks any catalytic activity, we next explored the extent to which the genetic interaction between Timeless and the DDX11 helicase accounted for G4 processing in this system. As noted above, Timeless interacts with DDX11 (Leman et al, 2010; Calì et al, 2016; Cortone et al, 2018), and this interaction has been shown to be important for sister chromatid cohesion (Cortone et al, 2018) and preservation of fork progression in perturbed conditions (Calì et al, 2016). Further, in vitro, DDX11 can unwind several DNA structures including G4s (Wu et al, 2012a; Bharti et al, 2013; Guo et al, 2015) and the activity of DDX11 is stimulated by Timeless (Calì et al, 2016). Recent work has shown that DDX11 interacts with Timeless through a domain encoded in exon 4 (Cortone et al, 2018). Although DDX11 was not identified by iPOND as constitutively present at normal or HU‐stressed replication forks in human cells (Dungrawala et al, 2015), it interacts with several components of the core replisome in addition to Timeless, such as AND‐1, POLD1 (Simon et al, 2020) and PCNA (Farina et al, 2008). We examined whether, in DT40, DDX11 is able to associate with chromatin‐bound PCNA and whether this association is increased following exposure to the G4 ligand pyridostatin (PDS). We therefore performed chromatin immunoprecipitation with PCNA and blotted for FLAG‐DDX11. Treatment with PDS did indeed result in more DDX11 being pulled down with PCNA (Fig 55A and Appendix Fig S4) suggesting DDX11 is recruited to chromatin after G4 stabilisation.
Figure 5. DDX11 is required for processive replication in collaboration with Timeless.
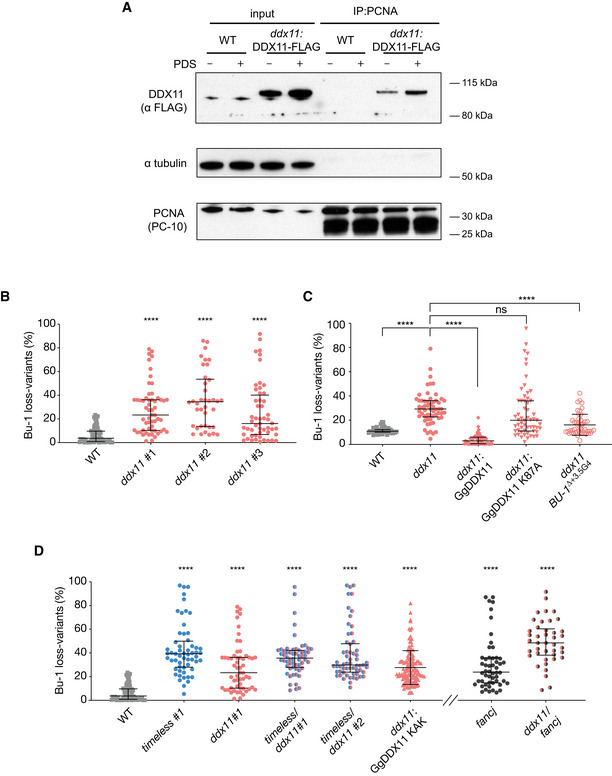
- Enhanced recruitment of DDX11 to chromatin associated PCNA following exposure to 4 μM PDS for 24 h. PCNA was precipitated from cross‐linked chromatin and the immunoprecipitate blotted for FLAG‐DDX11.
- Fluctuation analysis for Bu‐1a loss in wild‐type DT40 cells, two independent ddx11 clones generated by CRISPR‐Cas9 targeting (clones 1 and 2) and one ddx11 clone generated by conventional homologous recombination gene targeting (clone 3). Each symbol represents the percentage of cells in an individual clone expanded for 2–3 weeks that have lost Bu‐1ahigh expression.
- Fluctuation analysis for Bu‐1a loss variant generation in wild‐type cells, ddx11 (clone 1) cells, ddx11 (clone 1) complemented by expression of chicken DDX11 WT cDNA, ddx11 (clone 1) complemented by expression of helicase‐dead form of chicken DDX11 (K87A) cDNA, and a ddx11 clone generated in cells in which the endogenous +3.5 G4 has been deleted (ΔG4).
- Fluctuation analysis for Bu‐1 loss in two independent timeless/ddx11 double‐mutant clones (#1 and #2), ddx11 expressing DDX11KAK (see Appendix Fig S3), and fancj and fancj/ddx11 double mutants. Fluctuation analyses for wild type, timeless #1 (Fig 44) and ddx11 #1 (Fig 55A) are shown for comparison.
We next examined BU‐1 stability both in a ddx11 mutant generated by disrupting exons 7–12 by conventional gene targeting (Abe et al, 2016) and in deletion mutants generated by CRISPR/Cas9 targeting to exon 4 (Appendix Fig S1). The newly generated CRISPR/Cas9‐generated ddx11 mutants are sensitive to cisplatin (Fig EV1), similar to the previously reported mutant generated by gene targeting (Abe et al, 2016). All ddx11 mutants examined exhibited elevated rates of Bu‐1 loss variant generation (Fig 55B). This expression instability was suppressed by complementation with full‐length chicken DDX11, but not with the K87A helicase‐dead form of DDX11 (the chicken equivalent of the human K50A mutation in the Walker A motif), demonstrating that the helicase activity of DDX11 is essential for G4 replication in vivo (Fig 55C). Importantly, the BU‐1 expression instability of ddx11 cells is also dependent on the presence of the +3.5 G4 in BU‐1 (Fig 55C).
DDX11 works with Timeless to prevent G4‐dependent instability of BU‐1 expression
We next asked whether DDX11 and Timeless participate in the same pathway for G4 resolution at the replication fork. We generated ddx11/timeless double mutants by targeting Timeless in the CRISPR/Cas9‐generated ddx11 clone #1 cells. Fluctuation analysis showed that the double‐mutant ddx11/timeless does not exhibit higher rates of Bu‐1 loss variant formation, compared to the single timeless or ddx11 mutants (Fig 55D), suggesting that these proteins act in the same pathway for G4 replication. Consistent with the genetic interaction between Timeless and DDX11 depending on their physical interaction, expression of DDX11 in which the “EYE” motif, required for its interaction with Timeless, is mutated to KAK (Cortone et al, 2018) failed to complement the instability of BU‐1 expression of ddx11 cells (Fig 55D and Appendix Fig S5).
DDX11 is closely related to the FANCJ helicase and, like DDX11, FANCJ is able to unwind G4s and plays a prominent role in their replication (Kruisselbrink et al, 2008; London et al, 2008; Wu et al, 2008; Youds et al, 2008). fancj DT40 exhibit instability of BU‐1 expression (Sarkies et al, 2012) that is dependent on the helicase activity of FANCJ (Fig EV4). However, a double fancj/ddx11 mutant (Abe et al, 2018) exhibits significantly more BU‐1 expression instability than either single mutant (Fig 55D), suggesting that these helicases operate on the BU‐1 G4 motif independently of each other.
Figure EV4. The catalytic activity of FANCJ is required for its role in suppressing G4‐induced instability of BU‐1 expression.
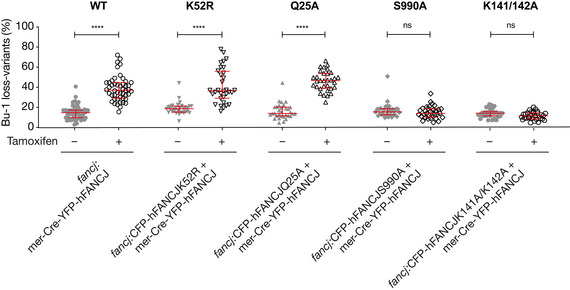
Fluctuation analysis for the generation of Bu‐1 loss variants in an inducible system to study FANCJ function (see Materials and Methods for full details). Briefly, FANCJ‐deficient DT40 cells are rescued with two transgenes, one encoding the wild‐type protein and the other the mutant in question. The wild‐type transgene is flanked by loxP sites and can be deleted by expression of Cre recombinase, induced by treatment of cells with tamoxifen. The K52R and Q25A mutants of FANCJ both disrupt the helicase activity of the enzyme (Cantor et al, 2001; Wu et al, 2012b). S990A disrupts the interaction of FANCJ with BRCA1, which is important for the role of FANCJ in homologous recombination (Xie et al, 2010). K141/142A disrupts the interaction of FANCJ with MutLα, which is needed for efficient interstrand crosslink repair (Peng et al, 2007). The data for each cell line represent the pooled results of at least two independent fluctuation analyses with a minimum of 32 data points per condition. Bars and whiskers represent median and interquartile range, respectively. ****P < 0.0001; one‐way ANOVA for comparison between uninduced and tamoxifen‐induced lines.
Timeless and DDX11 deficiency leads to decreased proliferation and increased activation of DDR in presence of pyridostatin
We next asked whether deficiency in Timeless and DDX11 exacerbates the globally detrimental cellular consequences of G4‐binding ligands. We treated timeless, ddx11 and ddx11/timeless cells with the G4 ligand PDS (Rodriguez et al, 2008). All mutant cell lines, but not the wild type, exhibited an intrinsic reduction in doubling time with cellular proliferation further decreased in the presence of the G4 ligand (Fig 66A). We next looked at the induction of histone H2AX phosphorylation (γ‐H2AX) using permeabilised cell flow cytometry. We have previously observed that PDS induces little or no γ‐H2AX in wild‐type DT40 cells (Guilbaud et al, 2017), and this was also true in our cytometric assay. However, timeless and ddx11 mutants both exhibit increased levels of γ‐H2AX following treatment with 4 μM PDS for 3 days compared with wild‐type cells (Fig 66B and C). We confirmed our results obtained using flow cytometry by direct visualisation of the fixed cells and quantification of γ‐H2AX foci (Appendix Fig S6). γ‐H2AX levels in the double ddx11/timeless mutant after PDS treatment were not significantly higher than the timeless single mutant, again supporting the observation that these two proteins operate in the same pathway for avoiding G4 ligand‐induced DNA damage signalling.
Figure 6. Timeless and DDX11‐deficient cells have impaired growth and increased H2AX phosphorylation in the presence of a G4 ligand.
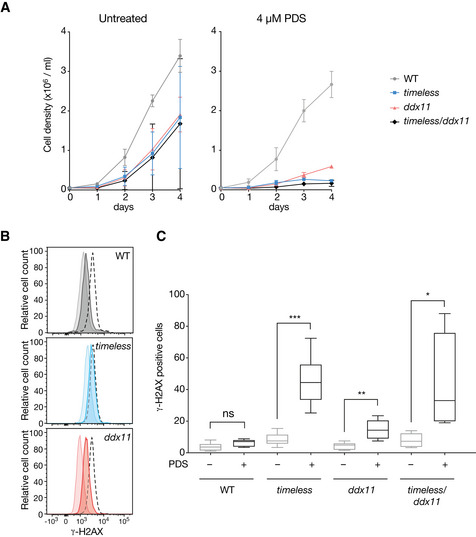
- Growth curves for DT40 wild type, ddx11, timeless and ddx11/timeless cells, with and without 4 μM pyridostatin (PDS). Cells were seeded at 5 × 104 cells/ml on day 0 and the viable cells were counted each 24 h for 4 days. Bars represent SD of two independent experiments performed in duplicate. Doubling times (DMSO): WT 13 h, timeless 18 h, ddx11 16 h, ddx11/timeless 24 h. Doubling times (PDS): WT 13.6 h, timeless 27 h, ddx11 25.7 h, ddx11/timeless 47.5 h.
- DDR signalling detected by phosphorylation of histone H2AX (γ‐H2AX) by flow cytometry in untreated cells or cells exposed to 4 μM PDS for 3 days. Pale histogram, untreated; dark histogram, treated; black dotted line, positive control cells treated with 0.1 μM cisplatin, also for 3 days.
- Quantification of γ‐H2AX in DT40 wild type, ddx11, timeless and ddx11/timeless cells treated with 4 μM PDS for 3 days. The central band represents the median, the box the 25th–75th centile and whiskers the minimum to maximum range of three independent experiments performed in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 unpaired, two‐tailed t‐test for each pairwise comparison ± PDS. See also Appendix Fig S4 for immunofluorescence images of the γ‐H2AX signal.
Deficiency of DDX11 and Timeless leads to dysregulation of a common set of genes harbouring G4s in close proximity to their TSS
We have previously reported that the instability of expression of the BU‐1 locus in mutants defective in G4 replication is observed in other loci across the genome (Sarkies et al, 2012; Papadopoulou et al, 2015). We have shown that correlations between the identity of the genes dysregulated in different mutants, along with the direction and magnitude of those changes, can be used to infer genetic relationships between G4 processing enzymes (Sarkies et al, 2012; Papadopoulou et al, 2015). We applied this approach to the relationship between Timeless and DDX11, assessing gene expression changes with RNA‐seq. Loss of DDX11 or Timeless induced marked changes in gene expression (timeless 1,752; ddx11 821 genes with altered expression at P > 0.95), with an approximately equal number of genes being up‐ or downregulated (Fig EV5A) in both cases. Moreover, there was a significant (P < 2.2 × 10−16) overlap in the identity of the deregulated genes in timeless and ddx11 cells (Fig EV5B) and a significant correlation (Spearman correlation 0.82, P value < 10−16) both in the magnitude and direction of the change in expression at the level of individual genes (Fig 77A). Consistent with our previously proposed model for replication and G4‐dependent epigenetic instability (Sarkies et al, 2010, 2012; Schiavone et al, 2014), genes dysregulated in timeless and ddx11 mutants are more likely to harbour a G4 close to their TSS. Using a regular expression for G4s of (G3N12)3G3, we found that the region 1.5 kb around the TSS has a higher density of G4 motifs in genes dysregulated in timeless and ddx11 (Fig 77B blue & red line), compared to the average for all genes (Fig 77B, black line), an observation that is not simply explained by a higher GC content (Fig EV5C). Further, a greater number of G4s are observed immediately downstream of the TSS in the genes dysregulated in timeless, ddx11 and fancj mutants. These excess G4s are predominantly located on the coding strand (Fig 77C) orientated similarly to the +3.5 G4 in BU‐1 to act as a leading‐strand impediment for a fork entering the locus from the 3′ end (Sarkies et al, 2010; Schiavone et al, 2014; Papadopoulou et al, 2015). Further, the changes in gene expression in timeless and ddx11 also correlated strongly with those observed in fancj cells, which we have previously shown to exhibit G4‐dependent epigenetic instability (Sarkies et al, 2012; Papadopoulou et al, 2015) (Fig EV5D, Spearman correlation ~ 0.9, P value < 10−16). These observations provide further support for a common, G4‐dependent mechanism for the gene dysregulation in these three mutants. Overall, our results suggest that DDX11 and Timeless collaborate to ensure expression stability during DNA replication for a subset of genes with G4s close to their transcription start site.
Figure EV5. Gene expression dysregulation in timeless and ddx11 DT40 cells.
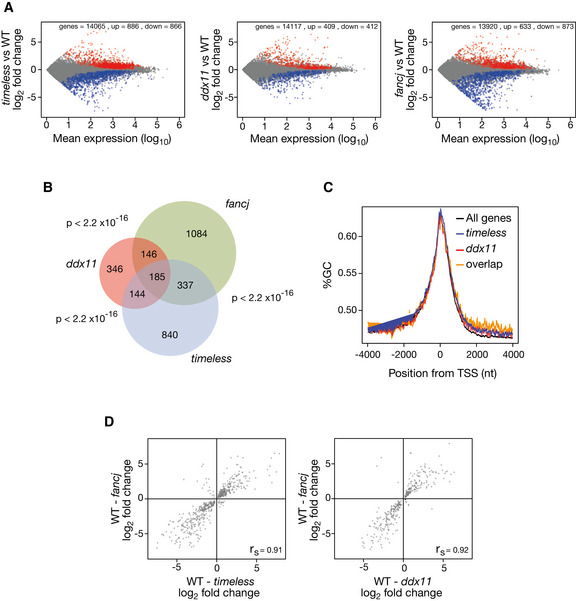
- Dysregulated genes in timeless (left panel), ddx11 (centre panel) and fancj (right panel) mutants relative to wild type. All genes with > 1 transcript per million in both conditions are plotted. Significantly (P ≥ 0.95) upregulated genes shown in red; downregulated in blue.
- Venn diagram showing the overlap in genes deregulated in timeless, ddx11 and fancj relative to wild type. P < 2.2 × 10−16 for each pairwise comparison (Fisher hypergeometric distribution).
- GC content around the TSS in genes dysregulated in timeless (blue), ddx11 (red) and in both mutants (“overlap”, orange) compared with all genes (black).
- Correlation of magnitude and direction of change of genes dysregulated (relative to wild type) in fancj vs. timeless (left panel) and ddx11 (right panel) DT40 cells. r s (Spearman rho) is shown for each correlation.
Figure 7. Loss of DDX11 and Timeless leads to genome wide expression dysregulation of genes with G4s around the transcription start site (TSS).
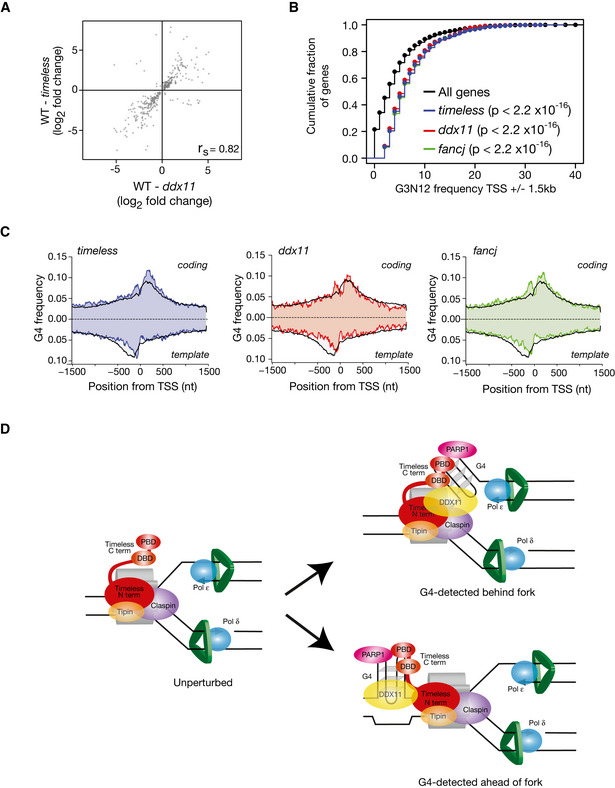
- Correlation of magnitude and direction of change of genes dysregulated (relative to wild type) in timeless vs. ddx11 DT40 cells. r s (Spearman rho) is shown for each correlation.
- Genes dysregulated in timeless, ddx11 and fancj mutants have a higher density of G4s around their TSS. Cumulative fraction of the genes dysregulated in timeless (red), ddx11 (blue) and fancj (green) containing n (x‐axis) G4 motifs within 1.5 kb of the TSS compared with all genes (black). P values calculated with the Kolmogorov–Smirnov test.
- Metagene analysis showing G4 frequency around the TSS of genes dysregulated (up or down) in timeless (left panel), ddx11 (centre panel) and fancj (right panel) compared with all genes (black line). G4 frequency is calculated separately for coding (above the x‐axis) and template strands (below the x‐axis).
- A model for recognition and processing of replisome associated G4s by Timeless/DDX11. Current evidence suggests that Timeless is a constitutive component of the replication fork. We suggest that the C‐terminus of Timeless may help detect G4s in the vicinity of the replisome by a combination of direct recognition through the DNA‐binding domain and indirectly through the PARP‐binding domain. It is not currently possible to distinguish whether this mechanism would operate ahead or behind the fork itself (Lerner & Sale, 2019), although recent structural evidence placing Tof1, the yeast homologue of Timeless, ahead of the fork (Baretić et al, 2020) would appear to make the first possibility more probable. However, for failure to resolve G4s ahead of the fork to result in uncoupling would require the CMG helicase to traverse the structure, as has been suggested for interstrand crosslinks (Sparks et al, 2019).
Discussion
Accumulating genetic evidence suggests that DNA secondary structure formation is a frequent challenge to vertebrate DNA replication (Lerner & Sale, 2019). Numerous factors collaborate to ensure that these structures do not result in persistent stalling of DNA synthesis or that the resulting tracts of single‐stranded DNA formation are limited, for instance by efficient repriming (Schiavone et al, 2016). By limiting uncoupling of DNA unwinding and DNA synthesis, these mechanisms prevent both genetic and epigenetic instability.
A key to ensuring that polymerase pausing at secondary structures does not lead to extensive single‐stranded DNA formation should be efficient sensing of structure formation by the replisome coupled to the recruitment of factors that will remove the structure and, if needed, reprime DNA synthesis. However, it remains to be established how secondary structure formation on the DNA template is detected by the replisome.
Timeless, along with other components of the FPC, appears to be a constitutive component of the core replisome (Gambus et al, 2006). Although not essential for DNA replication, the FPC increases the speed and efficiency of fork progression and DNA synthesis (Yeeles et al, 2017). The intimate association of the FPC with the replisome potentially puts it in an ideal place to coordinate the response of the replisome to secondary structure formation. Indeed, loss of components of the FPC leads to instability of structure‐forming repeat sequences (Voineagu et al, 2008, 2009; Leman et al, 2012; Liu et al, 2012a; Gellon et al, 2019).
Here, we have shown that the C‐terminus of Timeless contains a DBD that is able to bind G4 DNA more avidly than unstructured single‐ and double‐stranded DNA in vitro. Using a sensitive in vivo assay that monitors episodes of replication pausing at a defined secondary structure‐forming sequence, we show that Timeless is required to maintain processive replication of G4 DNA. This requires the C‐terminus of the protein, which contains both the newly identified DBD and an adjacent domain previously shown to bind PARP1 (Xie et al, 2015; Young et al, 2015). The nature of the complex formed between Timeless, Tipin and PARP1 in this context is unclear and requires further study. Timeless/Tipin and Timeless/PARP1 have been reported to form mutually exclusive complexes (Young et al, 2015), yet the interaction between PARP1 and Timeless is required for recruitment of both Timeless and Tipin to sites of DNA damage (Xie et al, 2015). Interestingly, cells expressing Timeless lacking either the DNA‐binding or PARP‐binding domain exhibit significant, although not complete, reversal of BU‐1 instability suggesting that there may be functional redundancy between these two domains. The precise role of PARP1 in G4 sensing during replication also remains to be explored further, but it is noteworthy that PARP1 has been previously reported to bind G4 DNA in vitro (Soldatenkov et al, 2008) suggesting, potentially, that Timeless could recognise G4s both directly through its DBD and indirectly via recruitment of PARP1 to the G4.
Timeless interacts with the G4 helicase DDX11, and this interaction stimulates the helicase activity of DDX11 (Calì et al, 2016). We have shown here that loss of Timeless and DDX11 are epistatic for G4‐induced BU‐1 expression instability and the previously identified interaction between the two proteins (Cortone et al, 2018) is required, consistent with them operating as a unit, possibly with the recruitment of DDX11 to the replisome stimulated by G4 detection by Timeless. Determining the precise conformation of Timeless and DDX11 at a G4‐stalled replisome remains to be determined and will represent a significant experimental challenge. Interestingly, loss of both DDX11 and the related G4‐unwinding Fe–S helicase, FANCJ, results in more rapid generation of Bu‐1 loss variants than either single mutant suggesting that these two helicases independently contribute to replication of the +3.5 BU‐1 G4. Our analysis of genome wide gene expression dysregulation suggests that this division of labour between DDX11/Timeless and FANCJ is likely to be extended to other G4s in the genome.
Much work remains to be done to dissect the interactions between protein factors that process G4s and other fork‐stalling DNA structures, to ensure smooth progress of DNA synthesis. Mechanistically, advances in replisome structural biology should help inform where G4 sensing by Timeless takes place relative to the CMG helicase. It is reasonable to anticipate that it will detect G4s forming transiently in the negatively supercoiled DNA emerging from CMG (Fig 77D). The large size and multidomain structure of Timeless indicates that its DBD could also reach and interact with unwanted secondary structures in the DNA within a wide radius around the fork. This could potentially include those structures present in the DNA before it reaches CMG (Fig 77D). Such structures might then be “traversed” by the helicase, analogous to its proposed behaviour at a DNA‐protein crosslink (Sparks et al, 2019), before stalling the leading‐strand polymerase (Lerner & Sale, 2019). In either case, detection and resolution of the structure will ensure the smooth execution of DNA synthesis, avoiding the generation of potentially deleterious ssDNA.
Materials and Methods
Molecular cloning
Oligonucleotide sequences can be found in Appendix Table S1. The Timeless DBD deletion (ΔDBD), PARP‐binding domain truncation and ∆CTD mutant plasmids were constructed by PCR. pcDNA4‐Flag‐hTimeless was linearised with PCR using outward primers to allow the desired section of the cDNA to be removed. Ten fmol mutant plasmid was recircularised (through the overlapping sequences) via Gibson assembly (1 h, 50°C). Recombinant DNA was treated with 10U of DpnI to minimise carryover of the original plasmid and transformed into DH5α competent bacteria. Correct mutagenesis was confirmed by restriction digest and Sanger sequencing.
FANCJ point mutations were generated by site‐directed mutagenesis using a Quick Change II XL Site‐Directed Mutagenesis Kit (Agilent), according to the manufacturer's instructions, in the pEAK8‐CFP‐hFANCJ plasmid (generated in this study). This plasmid was generated by digesting a CFP‐containing plasmid, a hWT FANCJ cDNA‐plasmid and a pEAK8 backbone plasmid with HindIII and SalI, SalI and NotI and HindIII and NotI, respectively. All fragments were gel‐purified with a QIAEX II Gel Extraction Kit (QIAGEN) and ligated in a three‐way ligation reaction using a Rapid Ligation Kit (Roche) for 30 min at room temperature.
Cell lines and transfections
All DT40 cell lines were cultured at 37°C in a humidified 10% CO2 atmosphere in RPMI 1640 with Glutamax (Thermo Fisher Scientific) supplemented with 7% foetal bovine serum (Thermo Fisher Scientific), 3% chicken serum (Sigma‐Aldrich), 50 μM 2‐mercaptoethanol and 1% penicillin/streptomycin, as previously described (Simpson & Sale, 2003). HEK293T cells were cultured at 37°C in a humidified 5% CO2 atmosphere in DMEM (Thermo Fisher Scientific) supplemented with 10% foetal bovine serum and penicillin/streptomycin.
DT40 ddx11 (CRISPR), timeless and ddx11/timeless mutants were generated in this study by CRISPR‐Cas9‐mediated gene disruption. DT40 WT BU‐1 ΔG4 cells were generated by deleting the +3.5 G4 motif from both alleles of the BU‐1 locus (Schiavone et al, 2014). Other mutant lines have also been described previously: fancj (Sarkies et al, 2012), ddx11, fancj/ddx11 and tipin (Abe et al, 2016, 2018).
Complemented ddx11 cells were obtained by transfecting the KO cell lines with pEGFP‐C1 (Clontech) harbouring chicken DDX11 cDNA or a helicase‐dead variant (K87A) (Abe et al, 2016) with selection of G418‐resistant (2 mg/ml) clones followed by screening for GFP expression by flow cytometry. The EYE motif in DDX11 (Cortone et al, 2018) was mutated to KAK by site‐directed mutagenesis using the Quick Change II XL Site‐Directed Mutagenesis Kit (Agilent) in pcDNA3‐Flag‐hDDX11 plasmid (Cortone et al, 2018), a kind gift from Francesca Pisani. Complementation of timeless was achieved by transfection of plasmids encoding the human Timeless WT cDNA (Addgene plasmid 22887), or truncated versions thereof, selected with zeocin (1 mg/ml) followed by screening for Flag‐positive clones by Western blot (WB). Complemented fancj cells were obtained as follows. First, fancj +/− cells harbouring a tamoxifen‐regulatable Cre recombinase, Mer‐Cre‐Mer (Zhang et al, 1996) were transfected with pXSPN (Ross et al, 2005) with wild‐type human YFP‐FANCJ flanked by loxP sites. The second allele of the FANCJ locus was then disrupted with a previously described targeting construct (Sarkies et al, 2012). For testing the effect of FANCJ mutations, a second pXPSN plasmid was introduced harbouring CFP‐FANCJ[mut] without loxP sites, where “mut” is the desired mutation. Treatment with tamoxifen results in excision of the wild‐type FANCJ transgene leaving the cells either FANCJ‐deficient or solely expressing FANCJ[mut].
Transfections for CRISPR‐Cas9 targeting were performed using a Neon electroporator (Thermo Fisher Scientific), 20 × 106 cells and 20 μg plasmid DNA and three pulses of 1,400 V with intervals of 10 msec. For cDNA expression, either the Neon, with the same conditions, or a BioRad electroporator with 1 pulse of 250 V 950 μF 100 Ω in 4 mm cuvettes was used.
Gene disruption using CRISPR‐Cas9
Guide RNA sequences used for disrupting DDX11 and TIMELESS in DT40 cells are listed in Appendix Table S1. Guide RNAs were designed using the CRISPOR online tool of the Zhang lab (https://zlab.bio/guide-design-resources). Each guide was cloned into the pSpCas9(BB)‐2A‐GFP (PX458) plasmid (Ran et al, 2013) and transfected as described above. Twenty‐four hours after transfection, cells were collected by centrifugation at 400 g for 5 min and sorted at single live (propidium iodide negative), GFP‐positive cell per well in 96‐well plates using a MoFlo (Beckman Coulter) or a Synergy (Sony) cell sorter. Cells were grown for 2 weeks, and clones were collected and genotyped by Sanger sequencing of the targeted region amplified by PCR, gel‐purified with a QIAquick Gel Extraction Kit (QIAGEN) and blunt‐cloned using Zero Blunt TOPO PCR Cloning Kit (Thermo Fisher Scientific). Sequences were aligned against the WT DT40 genome using MacVector software.
Bu‐1 staining and fluctuation analysis for generation of Bu‐1 loss variants
Bu‐1 staining and fluctuation analysis were performed as described previously (Schiavone et al, 2014; Guilbaud et al, 2017). Briefly, cells (confluency between 0.4–2 × 106) were directly stained with anti‐Bu‐1 conjugated with phycoerythrin (Santa Cruz 5K98‐PE 70447 or Invitrogen 21‐1A4‐PE MA5‐28754) at 1:100 dilution for 10 min at room temperature. Cells were analysed by flow cytometry using an LSRII flow cytometer (BD Biosciences) and the FlowJo software. Bu‐1 expression in single live cells was gated using side scatter (SSC) in the y‐axis and PE fluorescence in the x‐axis. To perform fluctuation analysis, Bu‐1‐positive single cells were sorted and grown for ~ 20 generations before staining and flow cytometry analysis as described above.
G‐quadruplex ligand
The small molecule pyridostatin (PDS) (Rodriguez et al, 2008) was purchased from Sigma‐Aldrich.
Cell proliferation and viability assays
5 × 105 cells were seeded in 10 cm dishes containing 10 ml of media with G4 ligand/DMSO. Living cells were counted at 24, 48, 72 and 96 h on a Vi‐Cell cell counter (Beckman Coulter). Doubling time was calculated during the exponential growth phase using the formula Doubling Time = duration *log (2)/(log (Final Concentration) − log (Initial Concentration)).
Cell viability was determined 72 h after PDS or cisplatin treatment using a CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega), according to the manufacturer's instructions. 1 × 105 cells/well were seeded in 96‐well plates in 200 μl media containing PDS or cisplatin. Seventy‐two h later, 20 μl of MTS reagent was added to 100 μl cells and incubated for 2–4 h at 37°C. Absorbance was measured at 492 nm (formazan salt) in a PHERAstar plate reader (BMG Labtech). The percentage of cell viability was calculated as follows: Cell viability (%) = (Test absorbance/Control absorbance)*100.
Flow cytometry and microscopy to monitor γ‐H2AX
Cells were collected, fixed with 1% paraformaldehyde for 15 min in ice, washed with PBS and fixed with 70% ethanol for at least 24 h at −20°C. Cells were blocked and permeabilised in BD wash/permeabilisation buffer (BD Biosciences) and then incubated with anti‐γ‐H2AX antibody (05‐636, Merck, diluted at 1:500 in BD Buffer) for 2 h at room temperature. Samples were washed twice with BD Buffer, incubated with anti‐mouse AF594 antibody (A11062, Thermo Fisher Scientific, diluted at 1:200 in BD Buffer) for 1 h at room temperature in the dark and then washed twice with BD Buffer. Cells were resuspended in PBS containing 0.5% BSA and 1 μg/ml DAPI. Cells were analysed by flow cytometry using an LSRII flow cytometer (Beckman Coulter) and the FlowJo software. A gate for γ‐H2AX‐positive cells was defined based on untreated cells that were considered negative for γ‐H2AX staining (see Fig 66B). Cells for microscopy were prepared in exactly the same way as for cytometry with a drop of the final suspension of fixed and stained cells being directly visualised using a Nikon C1si equipped with a Hamamatsu ORCA‐Flash4.0 LT+ cooled CMOS camera controlled by Nikon NIS Elements Software. γH2AX foci were quantified using Fiji (Schindelin et al, 2012). Briefly, the blue channel (DAPI staining) was used to define masks to outline cell nuclei with the “Threshold” function. Merged nuclei were split with the “Watershed” function. γH2AX foci were identified using the “Find Maxima” function on the green channel (γH2AX staining), and nuclear foci were counted by applying the previously defined masks. A minimum of 50 cells per conditions was considered for statistical analysis. Differences between conditions were tested using Student's t‐tests.
Protein expression and purification
Full‐length human Timeless (1–1,208) was amplified from I.M.A.G.E cDNA clone IRATp970C0576D (Source Bioscience) and cloned into pRSF‐Duet1 (Novagen) with an N‐terminal His14‐SUMO tag. Our previous structural analysis had demonstrated that residues 239–330 comprise a large disordered loop within the folded N‐terminal region of Timeless (Holzer et al, 2017) and their removal improved dramatically the biochemical behaviour of the protein; they were therefore replaced by a short linker consisting of residues (Gly–Ser–Thr)2. All experiments reported here were performed with this optimised Timeless construct. The full‐length human Tipin gene (1–301) was codon‐optimised for Escherichia coli expression (Life Technologies) and cloned into the pGAT3 vector for expression fused to an N‐terminal His6‐GST tag (Peränen et al, 1996). Timeless and Tipin were co‐expressed in E. coli Rosetta2 (DE3). The complex was purified using Ni‐NTA agarose (Qiagen) followed by cleavage of the tags with TEV and SUMO proteases, ion exchange chromatography (HiTrap Q HP, GE Healthcare) and size‐exclusion chromatography (Superdex 200 16/60, GE Healthcare). The DBD of human Timeless (residues 816–954) was cloned into pRSF‐Duet1 (Novagen) with an N‐terminal His14‐SUMO tag and expressed in E. coli Rosetta2 (DE3). Purification entailed Ni‐NTA agarose (Qiagen), His14‐SUMO tag cleavage with SUMO protease, Ni‐NTA recapture of the cleaved tag and size‐exclusion chromatography (Superdex 75 16/60, GE Healthcare). Timeless 883–947 (DBD C‐term) was expressed and purified in the same way as DBD.
X‐ray crystal structure determination
The DBD C‐term was crystallised by vapour diffusion, mixing equal volumes of protein at 800 μM and a solution of 200 mM sodium formate and 18% PEG 3350 at 19°C. For cryoprotection of the crystals, the mother liquor was replaced with a 2:2:1 (volume ratio) mixture of reservoir solution, protein buffer and 100% glycerol. X‐ray diffraction data for a native crystal were collected at the I24 beamline at Diamond Light Source, Oxford, UK. The dataset was processed with XDS (Kabsch, 2010) in space group P65 to a resolution of 1.15 Å.
For structure determination by anomalous scattering, crystals were grown of Se‐Met labelled DBD. Selenomethionine was incorporated in the DBD using metabolic inhibition in the presence of Se‐Met during bacterial expression in minimal media. Diffraction data at the peak wavelength of 0.9793 Å were collected at the Proxima2A beamline of the Soleil Synchrotron, Gif‐sur‐Yvette, France. The data were processed to 2.4 Å with XDS (Kabsch, 2010) in the same space group and cell dimensions as for the native crystal. The structure was solved exploiting the anomalous signal of the peak dataset using Autosol (Phenix) (Zwart et al, 2008). A largely complete Se‐Met model was used to solve the native dataset by molecular replacement using PHASER (McCoy et al, 2007). The model of the resulting solution was subsequently extended by Autobuild (Phenix) (Zwart et al, 2008) and completed by iterative cycles of manual building and model refinement in Coot (Emsley & Cowtan, 2004) and Phenix (Zwart et al, 2008). The structural figures were generated with Chimera (Pettersen et al, 2004). Data collection and refinement statistics are given in Appendix Table S3.
NMR spectroscopy
For expression of 15N‐ or 13C, 15N‐labelled Timeless 816–954, 13C6‐glucose and/or 15NH4Cl was used as the sole carbon/nitrogen source in M9 minimal medium. NMR measurements were made on ~ 0.5 mM solutions in 10% 2H2O, 10 mM phosphate (pH 6.4), 40 mM KCl and 0.5 mM EDTA. Experiments were recorded at 25°C on Bruker AVANCE III 600 or 800 MHz spectrometers equipped with QCI or TXI cryoprobes. Data were processed using the AZARA suite of programs (v. 2.8, © 1993–2019; Wayne Boucher and Department of Biochemistry, University of Cambridge, unpublished). Backbone assignments were derived from established versions (Cavanagh et al, 2006) of HNCA, HN(CO)CA, HN(CO)CACB, HNCACB and HNCO experiments acquired with non‐uniform sampling, and side chains using 3D (H)CC(CO)NH‐TOCSY, TOCSY‐15N‐HSQC, NOESY‐15N‐HSQC, HC(C)H‐TOCSY and NOESY‐13C‐HSQC. Assignment was carried out using CcpNmr Analysis v. 2.4 (Vranken et al, 2005). For the heteronuclear NOE experiments, either 4 s of 1H saturation using a 120° pulse train or a 4‐s delay was employed prior to the first 15N pulse. Inter‐proton distance restraints were derived from NOE peak heights in the 3D NOESY spectra using the relaxation matrix method (which accounts for spin diffusion) and r −6 summation for ambiguous NOEs. Backbone ϕ and ψ torsion‐angle restraints were obtained using TALOS+ (Shen et al, 2009). The restraints provided the input for the iterative assignment protocol ARIA v.1.2 (Linge et al, 2003). For the final iteration, 100 structures were calculated with the violation tolerance set to 0.1 Å. After the last iteration, the 20 lowest energy structures were subjected to a final water refinement to give an ensemble of 20 structures. Torsion‐angle molecular dynamics simulations were performed using CNS (Brünger et al, 1998). The simulated annealing protocol used to calculate each structure included 60,000 molecular dynamics steps, including those for refinement, and two cooling stages (to 1,000 K and 50 K). A summary of the restraints used in the calculation of the structure and characterisation of the ensemble of energy‐minimised structures is presented in Appendix Table S4.
EMSA‐based DNA‐binding experiments
G‐quadruplex sequences (Appendix Table S2) were folded by heating the DNA to 95°C followed by gradual cooling to 10°C, in TE buffer supplemented with 150 mM KCl. Reactions contained 5 μM Timeless–Tipin and 5 μM 6FAM‐labelled DNA in 25 mM HEPES pH 7.1, 150 mM KCl, 5 mM MgCl2 and 1 mM DTT. Reactions were incubated on ice for 30 min, after which native loading dye was added and the samples analysed on a 1% agarose gel in EMSA buffer (0.5× TBE, 10 mM KCl). Gels were run for 1 h at 50 V and 4°C, and bands were visualised under UV light.
Fluorescence anisotropy analysis of DBD domain and Timeless–Tipin complex binding to DNA
DNA sequences were annealed by heating to 95°C followed by gradual cooling to 10°C, using a thermocycler, in TE buffer supplemented with 150 mM KCl. Fluorescence anisotropy measurements were recorded at 25°C in a plate reader (PHERAstar FS; BMG Labtech). Excitation for Cy3‐3′‐labelled DNA was at 540 nm and emission at 590 nm (20 nm bandwidth for both). Excitation for FAM6‐5′‐labelled DNA was at 485 nm and emission at 520 nm (10 nm bandwidth for both). For DBD titrations, each well contained 10 nM 3′Cy3‐labelled DNA and increasing concentrations of DBD protein in assay buffer (25 mM HEPES‐NaOH pH 7.2, 150 mM KCl, 0.5 mM EDTA, 0.005% Tween‐20). For Timeless–Tipin complex titrations, each well contained 10 nM 6FAM‐labelled DNA and increasing concentrations of the complex in assay buffer (25 mM HEPES pH 7.1, 150 mM KCl, 1 mM DTT, 5 mM MgCl2). The voltage gains and focal heights were adjusted using the instrument software using free fluorescein as reference (35 mP), and data collection was set to 200 flashes. Each data point is the mean of at least three independent measurements. The averaged data were analysed using non‐linear fits of ligand‐depletion binding isotherms adapted for fluorescence anisotropy measurements, assuming one to one binding model and using the ProFit software package (Quantum Soft). No corrections were necessary for changes in the quantum yield as a function of protein concentration.
Co‐immunoprecipitation and WB
For whole‐cell extracts, the protein samples were lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 20 mM NaCl, 1 mM MgCl2, 0.1% SDS, protease inhibitors (Complete Protease Inhibitor Cocktail Tablets, Merck), phosphatase inhibitors (Halt Phosphatase Inhibitor Cocktail, Thermo Fisher Scientific) and 250 U/ml benzonase (Merck)) for 15 min at room temperature under agitation and cleared by centrifugation at 16,000 g for 20 min at 4°C. Samples were quantified using the Bradford method, and approximately 30 μg were mixed with 5× SDS–Page Sample Buffer (Jena Bioscience), boiled for 3 min, fractionated in 4–12 or 10% Nu‐PAGE Bis‐Tris gels (Thermo Fisher Scientific) in MOPS (200 mM MOP, 50 mM Sodium Acetate, 10 mM Na2EDTA) and transferred to nitrocellulose membranes using an iBlot2 apparatus (Thermo Fisher Scientific) at 25 V for 7 min. Membranes were blocked for 1 h at room temperature with TBS 0.1% Tween‐20 5% skimmed milk. Membranes were incubated with primary antibodies diluted in TBS‐T 5% milk overnight at 4°C under agitation, washed with three washes of 10 min each with TBS‐T and incubated with HRP‐conjugated secondary antibodies at room temperature for 1 h, followed by three washes with TBS‐T. Membranes were developed with Immobilon Crescendo Western HRP substrate (Merck) for 1 min and exposed to X‐ray films.
For the Co‐IP experiments with Timeless, pcDNA4‐Flag‐Timeless plasmids (WT and truncated versions) were co‐transfected into 2 × 106 HEK293T cells using Lipofectamine 2000 (Thermo Fisher Scientific) and seeded in 10 cm dishes 16 h prior to transfection (80% confluency at transfection) with pcDNA3‐hDDX11 (Abe et al, 2016) (30 μg total DNA). Twenty‐four hours after transfection, cells were detached with trypsin and washed in cold PBS. Pellets were lysed in NETN‐M buffer with glycerol (150 mM NaCl, 50 mM Tris–HCl pH 8, 0.5% Igepal, 1 mM EDTA, 5 mM MgCl2 and 10% glycerol, supplemented with protease and phosphatase inhibitors) and benzonase for 15 min at room temperature under agitation. Samples were sonicated with eight cycles of 30 s with 30‐s intervals at the low setting in a Bioruptor Plus water bath sonicator (Diagenode) and centrifuged for 15 min at 16,000 g at 4°C. Supernatant was collected and quantified, and 2 mg of total cell extract was mixed with 30 μl Flag‐M2 magnetic beads (M8823, Sigma‐Aldrich). Samples were incubated at 4°C for 2 h under agitation. Beads were then washed five times for 5 min each with lysis buffer, and proteins were eluted by boiling with Sample Buffer Laemmli 2× (Sigma‐Aldrich) for 5 min at 95°C. Samples were subjected to analysis by WB as described above, using the indicated antibodies.
For ChIP of DDX11 with PCNA, 24–36 × 106 DT40 cells were cross‐linked with 1% formaldehyde for 5 min at room temperature, quenched with 0.2 M glycine and then lysed with hypotonic lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP40) at room temperature. Crude chromatin extract was resuspended in sonication buffer (50 mM Tris–HCl pH 8.0, 1% SDS, 10 mM EDTA), solubilised by sonication in Bioruptor plus (30 cycles of 30″ On/Off, high setting at 4°C) and then diluted tenfold with IP dilution buffer (16.7 mM Tris–HCl pH 7.4, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X‐100). PCNA‐bound chromatin was immunoprecipitated with 0.5 μg PC‐10 antibody (Santa Cruz Biotechnology, sc‐56) overnight. Immunocomplexes were captured with 40 μl of Protein G magnetic beads (CST, #9006) and washed extensively with low‐salt, high‐salt and LiCl wash buffers and TBS‐T. Crosslinks were reverted and proteins eluted by boiling the beads for 10 min at 95°C with 1.5× Laemmli buffer.
Primary antibodies used: mouse anti‐Flag (M2, F3165, Sigma), mouse anti‐DDX11 (D‐2 271711, Santa Cruz), mouse anti‐α‐tubulin (T6074, Sigma), mouse anti‐human PCNA PC10 (sc‐56, Santa Cruz) and mouse anti‐human PCNA C‐20 (sc‐9857, Santa Cruz). Secondary antibody: goat anti‐mouse HRP (P0447 Dako).
RNA extraction and RNA‐seq library preparation
RNA was extracted from three independent cell populations using the RNeasy Mini Kit (QIAGEN), according to the manufacturer's instructions. RNA sample quality was checked in BioAnalyzer RNA Pico chips (Agilent), and only high‐quality samples (RIN > 7) were used to build the libraries. Seven hundred fifty nanogram RNA was used to build the next‐generation sequencing libraries with the NEBNext Ultra II RNA Library Prep Kit for Illumina (New England BioLabs), NEBNext Poly(A) mRNA Magnetic Isolation Module and NEBNext Multiplex Oligos for Illumina (Index Primers Set 1 and 2), according to the manufacturer's instructions. Library quality was checked in BioAnalyzer DNA High Sensitivity chips (Agilent) and quantified using the KAPA Library Quanti Kit (Illumina) Universal qPCR Mix (KAPA Biosystems) on an ABI Prism ViiA 7 Real‐Time PCR System (Thermo Fisher Scientific). Libraries were sequenced on a HiSeq4000 (Illumina).
RNA‐seq library alignment
RNA sequencing reads were aligned to the GRCg6a chicken genome using Bowtie 2 (Langmead & Salzberg, 2012). Expression estimates (transcripts per million; TPM) were calculated using the rsem‐calculate‐expression command of RSEM (Li & Dewey, 2011) and the version 96 of the chicken genome gene annotation from Ensembl. Only transcripts with an expression above or equal at 1 TPM were considered for further analysis. Gene expression matrixes were built using rsem‐generate‐data‐matrix and three independent biological replicates. Differentially expressed genes were called using EBseq (Leng et al, 2013). Change in gene expression was estimated using the EBseq posterior Fold Change (PostFC) value, and genes were considered differently expressed when the EBseq posterior probability of being differentially expressed (PPDE) was above or equal at 0.95.
Promoter sequence analysis
Gene promoter sequences were recovered from the GRCg6a chicken genome assembly using the getfasta command of BEDTools (Quinlan & Hall, 2010). Sequence analyses were performed using custom scripts in the R environment (http://www.R-project.org/). Assessment of the enrichment of G4s within the promoter of DDX11‐ and TIMELESS‐dependent genes was based on regular expression matching algorithms to monitor the cumulative count of the G3N12 motif within promoters. This was computed by identifying the number of sequences of the form d(G3+N1–12G3+N1–12G3+N1–12G3+), where N is any base, which represents the loose definition of G4 forming sequences (Huppert & Balasubramanian, 2005). Densities of G4‐forming sequences around promoters were computed by assessing the number of promoter sequences containing sequences of the form d(G3+N1+G3+N1+G3+N1+G3+) in windows of 50 nucleotides sliding by 10 nucleotides normalised to the total number of promoter sequences analysed.
Statistical analysis of RNA‐seq data
Data were analysed and statistics performed in the R environment. Overlaps between gene lists were tested using Fisher's exact tests. Differences between distributions were tested using Kolmogorov–Smirnov tests.
Author contributions
JES, LP, LKL and SH conceived the overall project. LKL generated the cell lines and performed the molecular biology experiments (flow cytometry, survival, IP, RNA‐seq). SH expressed and purified the Timeless‐Tipin complex, the DBD and the DBD C‐term domain, determined the X‐ray crystal structure of the C‐term domain, and performed the initial characterisation of the DNA‐binding properties of Timeless‐Tipin and the DBD. SS constructed the truncated versions of Timeless plasmids, performed ChIP experiments and some fluctuation analyses. DS generated the mer‐cre‐mer FANCJ cell line. CBE performed the γ‐H2AX IF microscopy. PM performed the RNA‐seq data analysis. MLK, AB and JDM performed the DNA‐binding experiments. KS and SH determined the NMR structure of the DBD. DB provided key KO cell lines. LKL, JES and LP wrote the initial manuscript with assistance and editing by all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors thank M. Daly and F. Zhang in the LMB flow cytometry facility for cell sorting, Anno Koetje for help with the DNA‐binding experiments, Joe Yeeles for discussions and members of both the Sale and Pellegrini groups for critical reading of the manuscript. Work in the Sale group is supported by a core grant to the LMB by the MRC (U105178808). L.K.L. received a 1‐year Science Without Borders postdoctoral funding from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília, DF, Brazil). Work in the Pellegrini group is supported by a Wellcome Trust investigator award to L.P. (104641/Z/14/Z), a Boehringer‐Ingelheim Fonds PhD fellowship and awards from the Janggen‐Poehn‐Stiftung and the Swiss National Science Foundation to S.H.
The EMBO Journal (2020) 39: e104185
See also: CH Freudenreich (September 2020)
Contributor Information
Luca Pellegrini, Email: lp212@cam.ac.uk.
Julian E Sale, Email: jes@mrc-lmb.cam.ac.uk.
Data availability
The datasets produced in this study are available in the following databases: Coordinates and structure factors have been deposited in the Protein Data Bank (https://www.rcsb.org) under accession codes 6T9Q (http://www.rcsb.org/pdb/explore/explore.do?structureId=6T9Q) for the crystal structure of the C‐terminal repeat of DBD and 6TAZ (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TAZ; BMRB ID 34443) for the NMR structure of the DBD. The RNA sequencing data have been deposited in GEO (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE139256 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139256).
References
- Abe T, Kawasumi R, Arakawa H, Hori T, Shirahige K, Losada A, Fukagawa T, Branzei D (2016) Chromatin determinants of the inner‐centromere rely on replication factors with functions that impart cohesion. Oncotarget 7: 67934–67947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Ooka M, Kawasumi R, Miyata K, Takata M, Hirota K, Branzei D (2018) Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc Natl Acad Sci USA 115: 8412–8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhunaizi E, Shaheen R, Bharti SK, Joseph‐George AM, Chong K, Abdel‐Salam GMH, Alowain M, Blaser SI, Papsin BC, Butt M et al (2018) Warsaw breakage syndrome: further clinical and genetic delineation. Am J Med Genet A 176: 2404–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrus A, Chen D, Dai J, Jones RA, Yang D (2005) Solution structure of the biologically relevant G‐quadruplex element in the human c‐MYC promoter. Implications for G‐quadruplex stabilization. Biochemistry 44: 2048–2058 [DOI] [PubMed] [Google Scholar]
- Baretić D, Jenkyn‐Bedford M, Aria V, Cannone G, Skehel M, Yeeles JTP (2020) Cryo‐EM structure of the fork protection complex bound to CMG at a replication fork. Mol Cell 78: 926–940.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastia D, Srivastava P, Zaman S, Choudhury M, Mohanty BK, Bacal J, Langston LD, Pasero P, O'Donnell ME (2016) Phosphorylation of CMG helicase and Tof1 is required for programmed fork arrest. Proc Natl Acad Sci USA 113: E3639–E3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin‐ya K, Teulade‐Fichou MP, Kisker C, Brosh RM (2013) Specialization among iron‐sulfur cluster helicases to resolve G‐quadruplex DNA structures that threaten genomic stability. J Biol Chem 288: 28217–28229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse‐Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS et al (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54(Pt 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Calì F, Bharti SK, Di Perna R, Brosh RM, Pisani FM (2016) Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res 44: 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA et al (2001) BACH1, a novel helicase‐like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105: 149–160 [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ (2006) Protein NMR spectroscopy, 2nd edn Cambridge, MA: Academic Press; [Google Scholar]
- Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S (2015) High‐throughput sequencing of DNA G‐quadruplex structures in the human genome. Nat Biotechnol 33: 877–881 [DOI] [PubMed] [Google Scholar]
- Chan RC, Chan A, Jeon M, Wu TF, Pasqualone D, Rougvie AE, Meyer BJ (2003) Chromosome cohesion is regulated by a clock gene paralogue TIM‐1. Nature 423: 1002–1009 [DOI] [PubMed] [Google Scholar]
- Cho WH, Kang YH, An YY, Tappin I, Hurwitz J, Lee JK (2013) Human Tim‐Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc Natl Acad Sci USA 110: 2523–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Elledge SJ (2006) Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci USA 103: 18143–18147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortone G, Zheng G, Pensieri P, Chiappetta V, Tatè R, Malacaria E, Pichierri P, Yu H, Pisani FM (2018) Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork‐protection factor Timeless promotes sister chromatid cohesion. PLoS Genet 14: e1007622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court R, Chapman L, Fairall L, Rhodes D (2005) How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high‐resolution crystal structures. EMBO Rep 6: 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Errico A, Costanzo V (2012) Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol 47: 222–235 [DOI] [PubMed] [Google Scholar]
- Farina A, Shin JH, Kim DH, Bermudez VP, Kelman Z, Seo YS, Hurwitz J (2008) Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18‐RFC and Fen1. J Biol Chem 283: 20925–20936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, Jones RC, Sanchez‐Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 8: 358–366 [DOI] [PubMed] [Google Scholar]
- Gellert M, Lipsett MN, Davies DR (1962) Helix formation by guanylic acid. Proc Natl Acad Sci USA 48: 2013–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellon L, Kaushal S, Cebrián J, Lahiri M, Mirkin SM, Freudenreich CH (2019) Mrc1 and Tof1 prevent fragility and instability at long CAG repeats by their fork stabilizing function. Nucleic Acids Res 47: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Suppa C, Emanuel BS (2007) Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork‐associated factors. J Mol Biol 366: 36–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Murat P, Recolin B, Campbell BC, Maiter A, Sale JE, Balasubramanian S (2017) Local epigenetic reprogramming induced by G‐quadruplex ligands. Nat Chem 9: 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Hundseth K, Ding H, Vidhyasagar V, Inoue A, Nguyen CH, Zain R, Lee JS, Wu Y (2015) A distinct triplex DNA unwinding activity of ChlR1 helicase. J Biol Chem 290: 5174–5189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer S, Degliesposti G, Kilkenny ML, Maslen SL, Matak‐Vinkovíc D, Skehel M, Pellegrini L (2017) Crystal structure of the N‐terminal domain of human Timeless and its interaction with Tipin. Nucleic Acids Res 45: 5555–5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33: 2908–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr, 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S‐phase checkpoint proteins Tof1 and Mrc1 form a stable replication‐pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Kaushal S, Freudenreich CH (2019) The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom Cancer 58: 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny ML, Simon AC, Mainwaring J, Wirthensohn D, Holzer S, Pellegrini L (2017) The human CTF4‐orthologue AND‐1 interacts with DNA polymerase α/primase via its unique C‐terminal HMG box. Open Biol, 7: 170217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M (2008) Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ‐defective C. elegans . Curr Biol 18: 900–905 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman AR, Noguchi C, Lee CY, Noguchi E (2010) Human Timeless and Tipin stabilize replication forks and facilitate sister‐chromatid cohesion. J Cell Sci 123: 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman AR, Dheekollu J, Deng Z, Lee SW, Das MM, Lieberman PM, Noguchi E (2012) Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 11: 2337–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C (2013) EBSeq: an empirical Bayes hierarchical model for inference in RNA‐seq experiments. Bioinformatics 29: 1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner LK, Sale JE (2019) Replication of G Quadruplex DNA. Genes (Basel) 10: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge JP, Habeck M, Rieping W, Nilges M (2003) ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics, 19: 315–316 [DOI] [PubMed] [Google Scholar]
- Liu G, Chen X, Gao Y, Lewis T, Barthelemy J, Leffak M (2012a) Altered replication in human cells promotes DMPK (CTG)n middle dot (CAG)n repeat instability. Mol Cell Biol 32: 1618–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Myers S, Chen X, Bissler JJ, Sinden RR, Leffak M (2012b) Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine‐polypyrimidine (Pu‐Py) tract. J Biol Chem 287: 33412–33423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Lin HX, Lin CY, Sun XQ, Ye LP, Qiu F, Wen W, Hua X, Wu XQ, Li J et al (2017) TIMELESS confers cisplatin resistance in nasopharyngeal carcinoma by activating the Wnt/β‐catenin signaling pathway and promoting the epithelial mesenchymal transition. Cancer Lett 402: 117–130 [DOI] [PubMed] [Google Scholar]
- London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K (2008) FANCJ is a structure‐specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem 283: 36132–36139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL (2008) Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell 32: 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM (2007) Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71: 13–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Shevchenko A, Stewart AF, Stoynov SS (2005) Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol 347: 509–521 [DOI] [PubMed] [Google Scholar]
- Numata Y, Ishihara S, Hasegawa N, Nozaki N, Ishimi Y (2010) Interaction of human MCM2‐7 proteins with TIM, TIPIN and Rb. J Biochem 147: 917–927 [DOI] [PubMed] [Google Scholar]
- Papadopoulou C, Guilbaud G, Schiavone D, Sale JE (2015) Nucleotide pool depletion induces G‐quadruplex‐dependent perturbation of gene expression. Cell Rep 13: 2491–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Litman R, Xie J, Sharma S, Brosh RM, Cantor SB (2007) The FANCJ/MutLalpha interaction is required for correction of the cross‐link response in FA‐J cells. EMBO J 26: 3238–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peränen J, Rikkonen M, Hyvönen M, Kääriäinen L (1996) T7 vectors with modified T7lac promoter for expression of proteins in Escherichia coli . Anal Biochem 236: 371–373 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Müller S, Yeoman JA, Trentesaux C, Riou JF, Balasubramanian S (2008) A novel small molecule that alters shelterin integrity and triggers a DNA‐damage response at telomeres. J Am Chem Soc 130: 15758–15759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AL, Simpson LJ, Sale JE (2005) Vertebrate DNA damage tolerance requires the C‐terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res 33: 1280–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, Sale JE (2010) Epigenetic instability due to defective replication of structured DNA. Mol Cell 40: 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, Sale JE (2012) FANCJ coordinates two pathways that maintain epigenetic stability at G‐quadruplex DNA. Nucleic Acids Res 40: 1485–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Guilbaud G, Murat P, Papadopoulou C, Sarkies P, Prioleau M‐N, Balasubramanian S, Sale JE (2014) Determinants of G quadruplex‐induced epigenetic instability in REV1‐deficient cells. EMBO J 33: 2507–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, Doherty AJ (2016) PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol Cell 61: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44: 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AK, Kummer S, Wild S, Lezaja A, Teloni F, Jozwiakowski SK, Altmeyer M, Gari K (2020) The iron‐sulfur helicase DDX11 promotes the generation of single‐stranded DNA for CHK1 activation. Life Sci Alliance 3: e201900547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LJ, Sale JE (2003) Rev1 is essential for DNA damage tolerance and non‐templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J 22: 1654–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatenkov VA, Vetcher AA, Duka T, Ladame S (2008) First evidence of a functional interaction between DNA quadruplexes and poly(ADP‐ribose) polymerase‐1. ACS Chem Biol 3: 214–219 [DOI] [PubMed] [Google Scholar]
- Sparks JL, Chistol G, Gao AO, Räschle M, Larsen NB, Mann M, Duxin JP, Walter JC (2019) The CMG helicase bypasses DNA‐protein cross‐links to facilitate their repair. Cell 176: 167–181.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Sale JE (2017) The effects of replication stress on S phase histone management and epigenetic memory. J Mol Biol 429: 2011–2029 [DOI] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan‐Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE (2019) R‐loop formation during S phase is restricted by PrimPol‐mediated repriming. EMBO J: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yokoyama M, Matsumoto S, Fukatsu R, You Z, Masai H (2010) Fission yeast Swi1‐Swi3 complex facilitates DNA binding of Mrc1. J Biol Chem 285: 39609–39622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal‐Kaçmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro‐Stone M, Sancar A, Kaufmann WK (2007) The human Tim/Tipin complex coordinates an Intra‐S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol 27: 3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, Mirkin SM (2008) Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA 105: 9936–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM (2009) Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol 16: 226–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696 [DOI] [PubMed] [Google Scholar]
- Witosch J, Wolf E, Mizuno N (2014) Architecture and ssDNA interaction of the Timeless‐Tipin‐RPA complex. Nucleic Acids Res 42: 12912–12927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shin‐ya K, Brosh RM (2008) FANCJ helicase defective in Fanconi anemia and breast cancer unwinds G‐quadruplex DNA to defend genomic stability. Mol Cell Biol 28: 4116–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sommers JA, Khan I, de Winter JP, Brosh RM (2012a) Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem 287: 1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sommers JA, Loiland JA, Kitao H, Kuper J, Kisker C, Brosh RM (2012b) The Q motif of Fanconi anemia group J protein (FANCJ) DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J Biol Chem 287: 21699–21716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Haakonsen DL, Sanderlin AG, Liu YJ, Shen L, Zhuang N, Laub MT, Zhang Y (2018) Structural insights into the unique mechanism of transcription activation by Caulobacter crescentus GcrA. Nucleic Acids Res 46: 3245–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Litman R, Wang S, Peng M, Guillemette S, Rooney T, Cantor SB (2010) Targeting the FANCJ–BRCA1 interaction promotes a switch from recombination to polη‐dependent bypass. Oncogene 29: 2499–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Mortusewicz O, Ma HT, Herr P, Poon RY, Poon RR, Helleday T, Qian C (2015) Timeless interacts with PARP‐1 to promote homologous recombination repair. Mol Cell 60: 163–176 [DOI] [PubMed] [Google Scholar]
- Yang X, Wood PA, Hrushesky WJ (2010) Mammalian TIMELESS is required for ATM‐dependent CHK2 activation and G2/M checkpoint control. J Biol Chem 285: 3030–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, Diffley JFX (2017) How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youds JL, Barber LJ, Ward JD, Collis SJ, O'Neil NJ, Boulton SJ, Rose AM (2008) DOG‐1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross‐link repair. Mol Cell Biol 28: 1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LM, Marzio A, Perez‐Duran P, Reid DA, Meredith DN, Roberti D, Star A, Rothenberg E, Ueberheide B, Pagano M (2015) TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the DNA damage response. Cell Rep 13: 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Riesterer C, Ayrall AM, Sablitzky F, Littlewood TD, Reth M (1996) Inducible site‐directed recombination in mouse embryonic stem cells. Nucleic Acids Res 24: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart PH, Afonine PV, Grosse‐Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC et al (2008) Automated structure solution with the PHENIX suite. Methods Mol Biol 426: 419–435 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The datasets produced in this study are available in the following databases: Coordinates and structure factors have been deposited in the Protein Data Bank (https://www.rcsb.org) under accession codes 6T9Q (http://www.rcsb.org/pdb/explore/explore.do?structureId=6T9Q) for the crystal structure of the C‐terminal repeat of DBD and 6TAZ (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TAZ; BMRB ID 34443) for the NMR structure of the DBD. The RNA sequencing data have been deposited in GEO (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE139256 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139256).