

Abstract
Riback et al. (Reports, 13 October 2017, p. 238) use SAXS experiments to infer a degree of compaction for unfolded proteins in water versus chemical denaturant that is highly consistent with the results from FRET experiments. There is thus no “contradiction” between the two methods, nor evidence to support their claim that commonly used FRET fluorophores cause protein compaction.
Riback et al. (1) have recently presented a “molecular form factor” (MFF) method addressing the well-known challenges (2) of analyzing small-angle X-ray scattering (SAXS) data for unfolded or intrinsically disordered proteins (IDPs) (3, 4). Combined with the precision of SAXS measurements coupled to size exclusion chromatography, they find: (i) unfolded proteins in water have a polymer scaling exponent v ≈ 1/2, near the theta-solvent condition where protein-protein and protein-solvent interactions are balanced; in denaturant, this increases to v ≈ 3/5, the limit where the protein-solvent interactions dominate. (ii) This change of scaling exponent is accompanied by an increase in radius of gyration (Rg) of 10–20 %, depending on the sequence. We are pleased that these findings are in overall agreement with SAXS and Förster resonance energy transfer (FRET) studies from our laboratories (3, 5, 6) and others (4).
The chain expansion observed by Riback et al. helps to resolve a longstanding controversy between SAXS and FRET experiments (7): with increasing denaturant concentration, FRET experiments had generally shown chain expansion, while until recently (3, 4) most SAXS studies observed no statistically significant change of Rg (8, ibid.). Their results are consistent with our recent collaborative study in which we compared SAXS and FRET estimates of Rg for each of two proteins (necessary since chain dimensions can be sequence-dependent). We found that the results are mutually consistent if both data types are analyzed with state-of-the-art methods (3, 6) (Fig. 1). A second study of a large group of IDPs reached a similar conclusion (4).
Fig. 1.
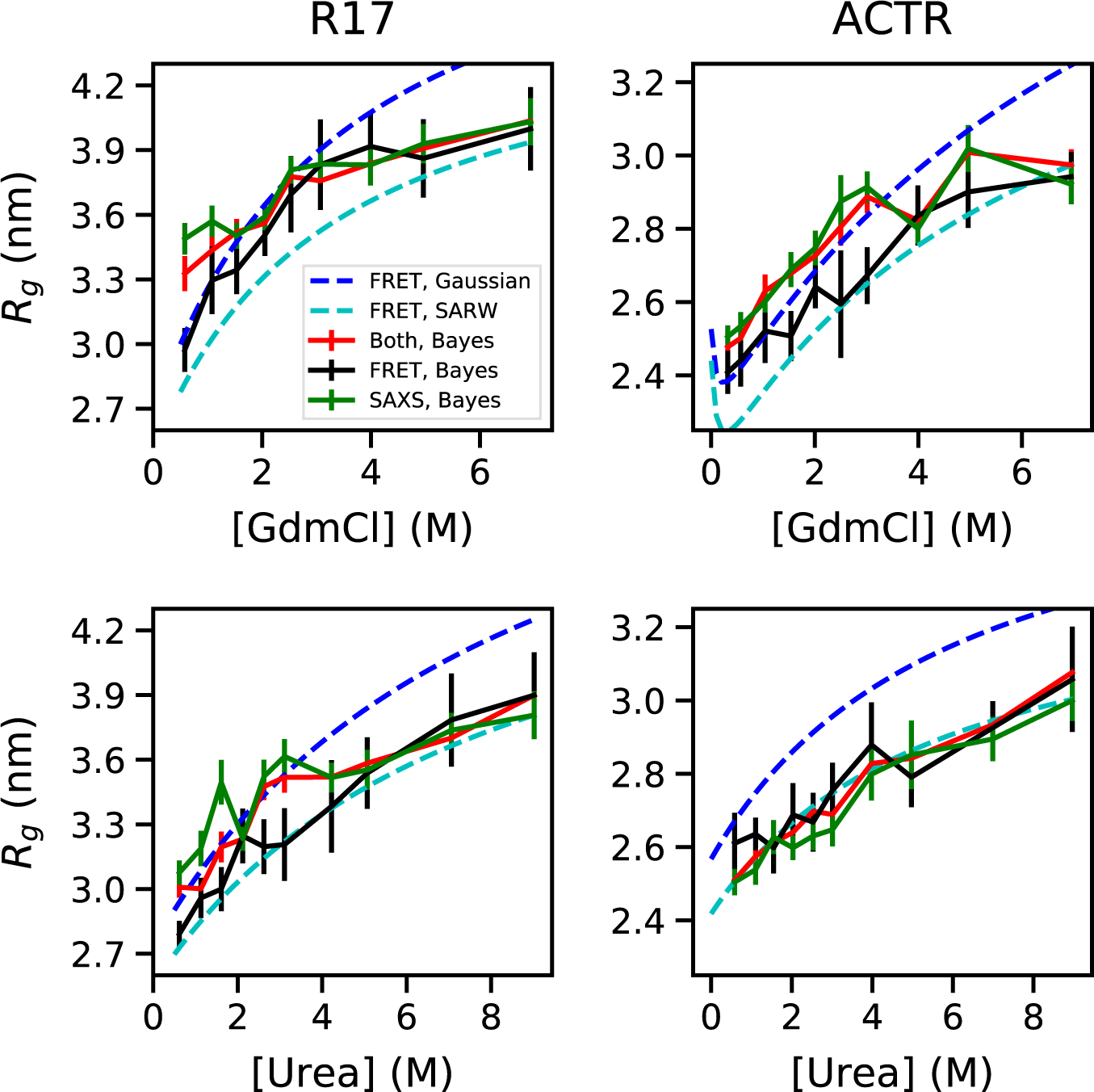
Rg from Bayesian ensemble refinement against experimental data for unfolded proteins in denaturant using FRET, SAXS or both experiments. Results shown for two proteins (ACTR, R17) in urea and guanidinium chloride (GdmCl) (3). Rg from FRET using Gaussian chain or SARW models (3) shown for reference.
The main reasons for the discrepancy were deficiencies in the analysis of both SAXS and FRET data. Earlier SAXS experiments underestimated expansion because the unfolded state of the foldable sequences studied could only be accessed above a certain denaturant concentration (3), as also pointed out by Riback et al. (1), and challenges in obtaining precise and accurate Rg values from SAXS data of IDPs using the Guinier approximation (1, 3, 4). The former difficulty has been overcome by studying destabilized or intrinsically disordered proteins (1, 3, 4), the latter by improved analysis such as Bayesian ensemble refinement (3, 4, 6) or the closely related MFF method (1). On the FRET side, interpreting experiments using polymer models, such as a Gaussian chain or self-avoiding random walk (SARW), can overestimate the change in Rg (3, 9, 10) (Fig. 1), largely because the relative change of Rg upon chain expansion is intrinsically less than of the end-to-end distance most commonly measured by FRET (3, 4, 9, 10). With ensemble refinement applied to either SAXS or FRET, or both combined, the data from both experiments yield consistent distributions of conformations, considering statistical error (3, 4, 6) , as shown in Fig. 1 (3).
We therefore dispute the authors’ claim that their results are “in apparent contradiction to a variety of FRET experiments”, considering that FRET experiments have not been reported on the sequences they study. Their results are consistent with the magnitude of the change in both Rg and v with denaturant inferred from recent FRET studies (3, 4), including the larger change in Rg below 2 M GdmCl (Fig. 2A). Their v values of 0.48–0.54 in water or low denaturant concentration are within the range (reflecting sequence-dependent variation of v) obtained based on ensemble refinement of data from previous FRET studies (5)(3, 4, 6) (Fig. 2B).
Fig. 2.
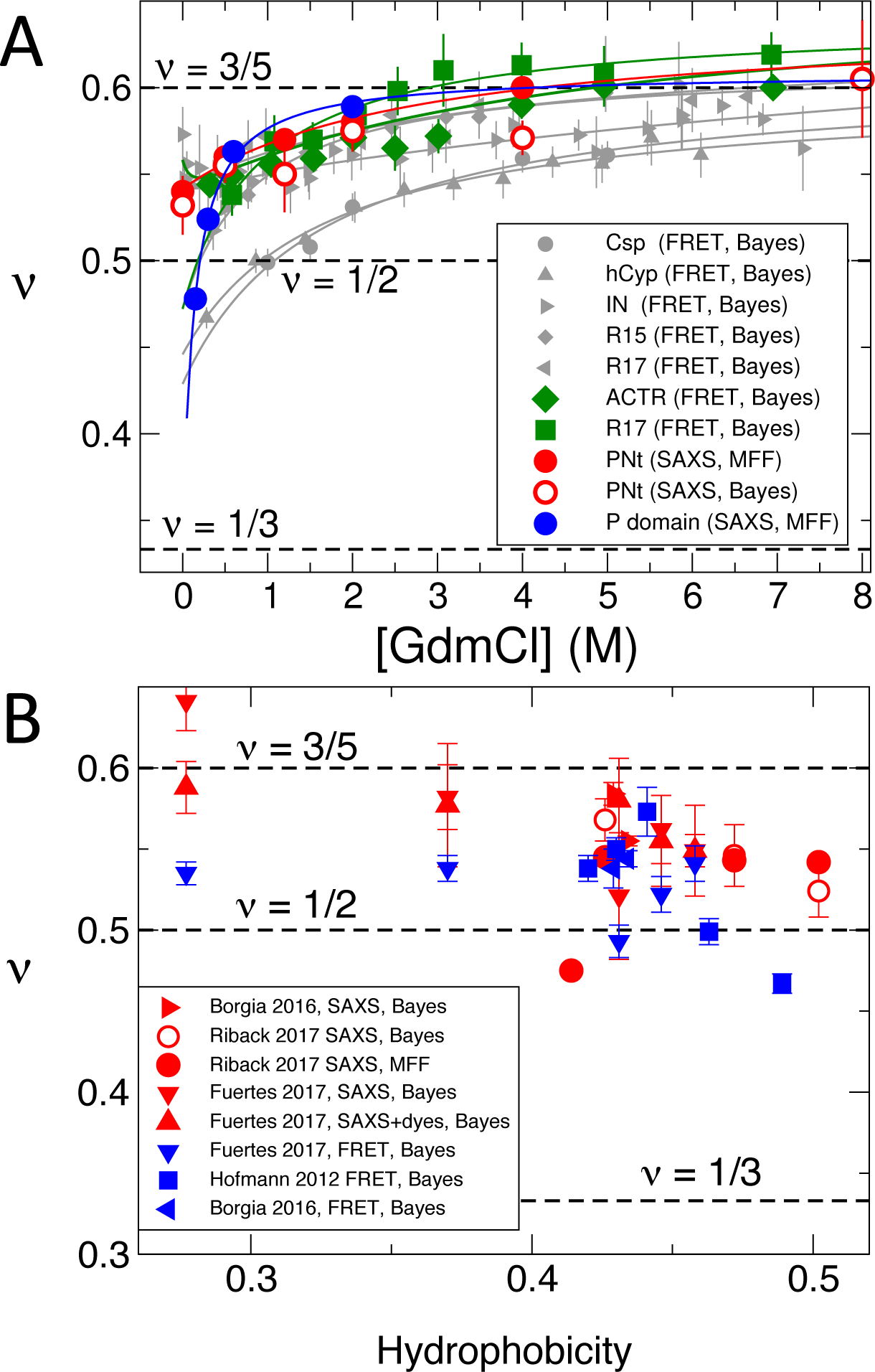
Polymer scaling exponents, ν, for unfolded or disordered proteins. (A) Denaturant-dependent ν from SAXS data of Riback et al. for PNt (red) and P domain (blue) (1) compared with those from FRET data for a variety of unfolded and disordered proteins (grey) (5) and for the IDP ACTR and a destabilized Spectrin R17 domain (green) (3). Exponents were obtained from Bayesian ensemble refinement (3) of primary FRET or SAXS data (‘Bayes’), or MFF analysis where indicated. Curves are fits to a binding model (5), or a polyelectrolyte model for IN and ACTR (3) . (B) Scaling exponents versus Kyte-Doolittle hydrophobicity (11) (rescaled between 0 and 1) for the same proteins in water or low denaturant concentration, as well as additional data for a set of IDPs in water from Fuertes et al. (4) Results for MFF (1) and Bayesian ensemble refinement (3) are highly consistent. Error bars indicate statistical error.
Despite this consistency, the authors suggest that “addition of fluorophores with hydrophobic character may lead to chain compaction and may contribute to FRET signal changes” (1). While some extremely hydrophobic FRET fluorophores can indeed cause additional compaction under native conditions, ensemble refinement identified the inconsistency of the resulting FRET data with SAXS (3). However, results for the more hydrophilic fluorophores most commonly used were in good agreement with SAXS (3) (Fig. 1). Furthermore, a recent tour-de-force SAXS study of proteins with and without fluorophores showed only small perturbations and no systematic changes of Rg and v upon labelling (4) (Fig. 2B). The evidence presented by Riback et al. to support their claim comes not from a protein but earlier SANS/FRET measurements on polyethylene glycol (PEG) (12). PEG lacks complications from a folded state, such as those which previously (13) caused these authors to overlook ubiquitin expansion (1). The PEG study, however, used old protocols to analyze the data. Applying such earlier methods to a protein lacking a folded state, the authors had determined that “fully reduced Ribonuclease A does not expand at high denaturant concentration” (14) – but now find an expansion for the same protein (Fig. 3C in (1)).
Riback et al. thus do not provide a convincing basis for their assertion that the conclusions of FRET and SAXS experiments are contradictory. Rather, their results add to the increasingly consistent picture of unfolded and intrinsically disordered proteins that has been emerging in recent years.
ACKNOWLEDGMENTS
We thank Devarajan Thirumalai, Hue Sun Chan, Rohit Pappu, William Eaton and Ad Bax for helpful comments and Patricia Clark, Tobin Sosnick, Dmitri Svergun, Edward Lemke and Rohit Pappu for sharing their SAXS data. Our analysis utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). R.B. was supported by the Intramural Research Program of the NIDDK, National Institutes of Health. Work at the University of Zurich was supported by funding from the Swiss National Science foundation. W.Z. thanks Arizona State University for start-up support.
References
- 1.Riback JA et al. , Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 358, 238–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calmettes P et al. , Structure of proteins unfolded by guanidinium chloride. J. Phys IV 3, 253–256 (1993). [Google Scholar]
- 3.Borgia A et al. , Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J. Am. Chem. Soc 138, 11714–11726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuertes G et al. , Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proc. Natl. Acad. Sci. U. S. A 114, E6342–E6351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann H et al. , Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U. S. A 109, 16155–16160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng W et al. , Probing the action of chemical denaturant on an intrinsically disordered protein by simulation and experiment. J. Am. Chem. Soc 138, 11702–11713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA, Characterizing the unfolded states of proteins using single molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. U.S.A 104, 1528–1533 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo TY et al. , Small-angle X-ray scattering and single-molecule FRET spectroscopy produce highly divergent views of the low-denaturant unfolded state. J. Mol. Biol 418, 226–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hummer G, Köfinger J, J. Chem. Phys 143, 243150 (2015) 10.1063/1.4937786 [DOI] [PubMed] [Google Scholar]
- 10.O’Brien E, Morrison G, Brooks BR, Thirumalai D, How accurate are polymer models in the analysis of Förster resonance energy transfer experiments on proteins? J. Chem. Phys 130, 124903 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song J, Gomes G-N, Gradinaru CC, Chan H-S, An adequate account of excluded volume is necessary to infer compactness and asphericity of disordered proteins by Förster resonance energy transfer. J. Phys. Chem. B 119, 15191–15202 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Watkins HM et al. , Random coil negative control reproduces the discrepancy between scattering and FRET measurements of denatured protein dimensions. Proc. Natl. Acad. Sci. U. S. A 112, 6631–6636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacob J, Krantz B, Dothager RS, Thiyagarajan P, Sosnick TR, Early collapse is not an obligate step in protein folding. J. Mol. Biol 338, 369–382 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Jacob J, Dothager RS, Thiyagarajan P, Sosnick TR, Fully reduced ribonuclease A does not expand at high denaturant concentration or temperature. J. Mol. Biol 367, 609–615 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Kyte J, Doolittle RF, A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 157, 105–132 (1982). [DOI] [PubMed] [Google Scholar]