

Abstract
The long lag times and subsequent rapid growth of Alzheimer’s Aβ42fibrils can be explained by a secondary nucleation step, in which existing fibril surfaces are able to nucleate formation of new fibrils via an autocatalytic process. The molecular mechanism of secondary nucleation, however, is still unknown. Here we investigate the first step, namely adsorption of the Aβ42 peptide monomers onto the fibril surface. Using long all-atom molecular simulations and an enhanced sampling scheme, we are able to generate a diverse ensemble of binding events. The resulting thermodynamics of adsorption are consistent with experiment, as well as with the requirements for effective autocatalysis determined from coarse-grained simulations. We identify the key interactions stabilizing the adsorbed state, which are predominantly polar in nature, and relate them to the effects of known disease-causing mutations.
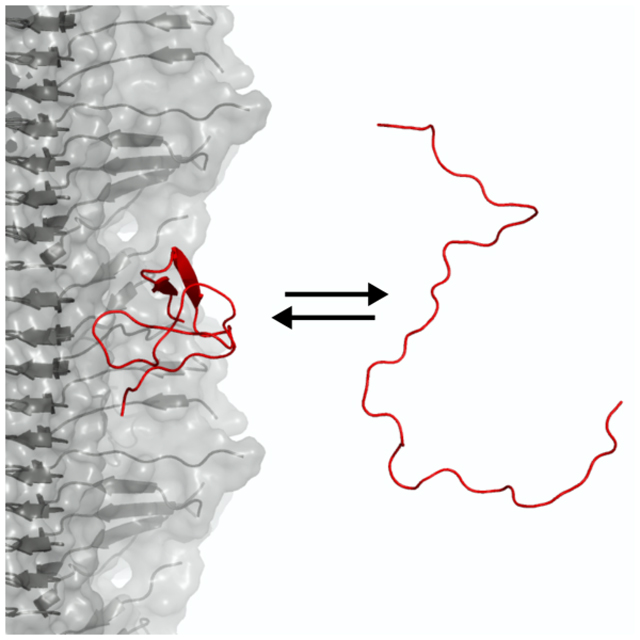
Graphical Abstract
The most common form of dementia is Alzheimer’s Disease (AD), which is related at the cellular and clinical levels to neuropathy and subsequent psychiatric decline involving loss of memory, cognition and motor control. At the present time, this ultimately fatal disease has no cure and is poised to present an intense challenge to a globally aging population. In the United States, for example, it has been predicted that the prevalence of the disease could reach 16 million and could cost $1.1 trillion by 2050.1 At the molecular level, AD has been correlated with the aggregation of the Aβ peptide into amyloid fibrils, which ultimately constitute the insoluble extracellular plaques found in patient autopsies.2,3 Note that Aβ oligomers have also been implicated in the disease, and there is evidence that they are more toxic than the fibrils.4–7 In vitro, amyloid fibril formation is characterized by a long lag phase, followed by rapid growth, which can be quantitatively described by a kinetic model involving both primary nucleation and elongation of fibrils, as well as secondary nucleation.8–10 In secondary nucleation, the surfaces of existing amyloid fibrils catalyze the nucleation of new aggregates; in fact this is how the majority of new oligomers are produced.11 This process gains added significance with the recognition that it may be small soluble oligomeric species produced en route to assembly that are toxic in humans, rather than the final fibrillar aggregates themselves.12–14 In support of this theory, there is evidence that selectively blocking secondary nucleation results in a steep reduction of toxicity in cell and in worm models.15–17
Despite the clear medical importance of secondary nucleation, the molecular details of the process remain unclear as it has only been possible to draw macroscopic inferences about this reaction from experiment, using bulk chemical kinetics. Simulation models using phenomenological coarse-grained models have been able to provide insights into the general nature of the reaction: a fine balance between peptide-peptide and peptide-fibril affinity is necessary if secondary nucleation is to be dominant, and there should be a conformational rearrangement within the peptide for detachment from the fibril to occur.18 All-atom computational approaches could potentially provide additional predictive insights into the reaction mechanism as they over a level of resolution inaccessible to experiments. As a first step towards understanding the molecular mechanism of secondary nucleation, we have studied the adsorption of Aβ42peptides onto fibrils using all-atom molecular simulations in explicit solvent.
In our simulations, we model the fibril based on a structure of the Aβ42fibril recently determined by solid-state NMR.19 We explicitly include 10 layers of peptides, but the fibril is infinitely replicated in the z direction through an appropriate choice of periodic boundaries. An additional free Aβ42 monomer is included to interact with the fibril, as well as 150 mM NaCl and explicit water. To ensure that our simulations were physically accurate, we took steps to address the two main limitations of all-atom simulations: the force field20,21 and sampling problems. We chose to use the Amber03ws force field, which has been shown to describe well the behaviors of intrinsically disordered peptides,22–25 in particular single-molecule measurements of the Aβ42 monomer.26 Long equilibrium simulations alone are unable to sample a wide variety of protein-protein binding interactions, as the time scale for dissociation is long compared to that accessible in simulation, such that complexes become kinetically trapped in the first stable encounter pose they find. We therefore used a novel Hamiltonian replica exchange enhanced sampling scheme in which 16 parallel simulations were run, each differing slightly in the protein-solvent interaction energy, controlled by a parameter λ. When λ is low, peptide-fibril binding is favored, while the highest λ values favor dissociation. This scheme is similar to one that has been used to study cryptic binding pockets in folded proteins,27 though here it is applied to an intrinsically disordered peptide. As with temperature replica-exchange schemes, configurations between neighboring windows are periodically exchanged based on a Metropolis criterion, such that the peptide is encouraged to come on and off the fibril several times, allowing us to sample a range of binding modes at equilibrium. We have verified that the fibril is stable in this scheme, by computing the fraction of monomer-monomer contacts that would be present in the experimental structure, as a function of time. As can be seen in Fig. S2, this is stable over the course of the simulation. This sampling scheme is summarized in Fig 1(b), showing schematics of the solvation state of the monomer as a function of λ, which controls the hydrophilicity or proteophilicity of the system. Fig. 1(c) shows a representative trajectory of a continuous simulation (one that has been followed through λ swaps), and demonstrates that this scheme allows us to sample several independent binding and unbinding events (as measured by the minimum distance between fibril and peptide atoms rmin), and that the the peptide-fibril contacts fluctuate while bound. To test the extent of our sampling, we ran two independent 16-copy simulations of our system started from different initial conditions: one in which the peptide was already in contact with the fibril, and the other in which it was free in solution.
Figure 1:
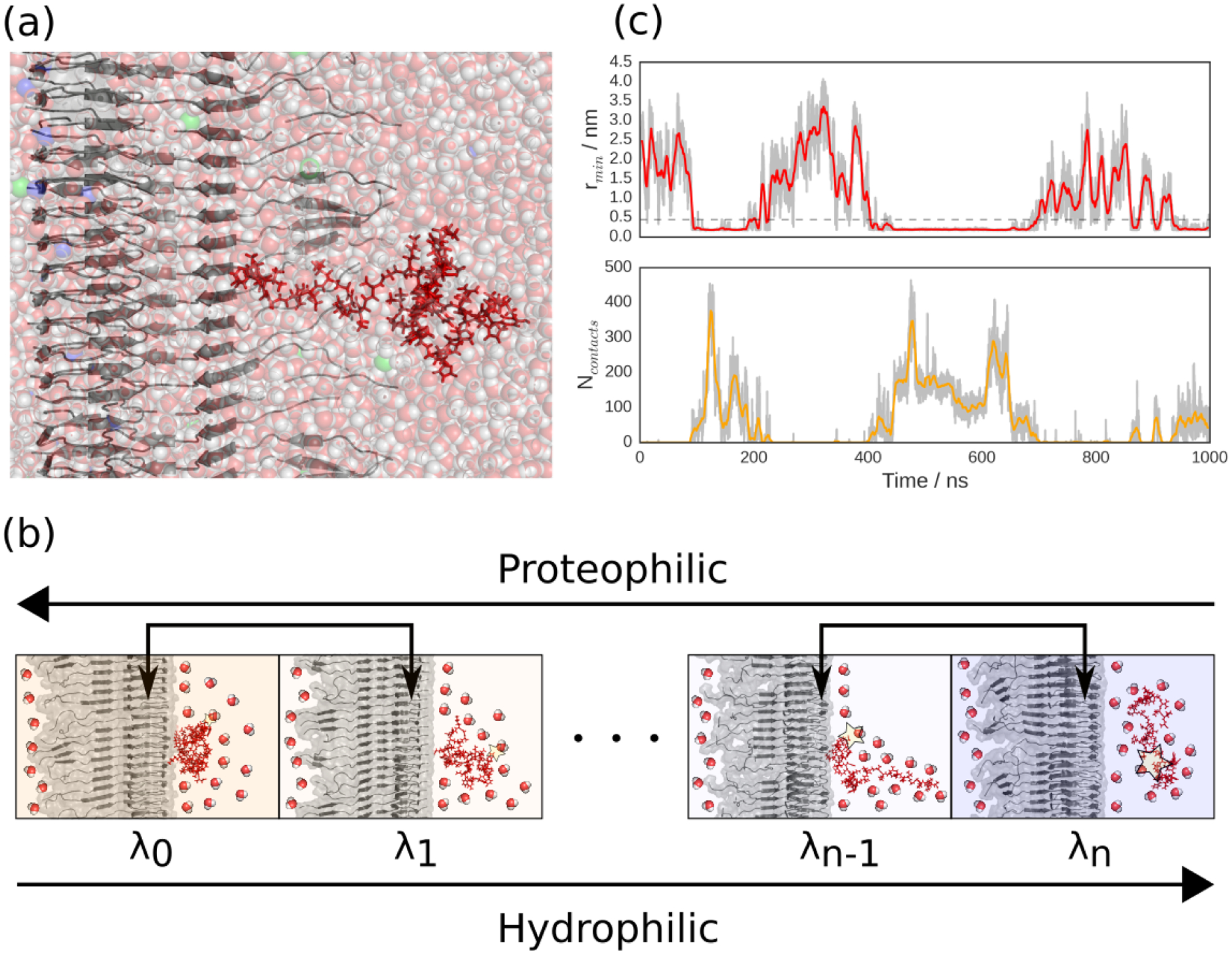
Simulation setup. (a) Starting configuration for one of the independent simulations, with the infinite Aβ42 fibril shown in gray, the Aβ42 monomer in red, water molecules as translucent red and white spheres, sodium ions as translucent violet spheres and chloride ions as translucent green spheres. (b) Schematic view of the Hamiltonian replica exchange enhanced sampling scheme, in which several parallel simulations run, each with a protein water interaction energy scaled by a factor λi, resulting in low λ windows favoring peptide adsorption and high λ windows favoring peptide solvation. Protein-water interactions are depicted schematically by a star of increasing size. (c) Representative fibril-peptide minimum distance (top) and number of contacts (bottom) trajectories, where the simulations have been followed through exchanges and hence made geometrically continuous. Gray curves are raw data, solid colors are moving averages over 7.5 ns windows and horizontal dashed line defines the bound state.
A fundamental parameter characterizing fibril-peptide association is the binding free energy. To obtain this from the simulations, we compute the free energy surface as a function of the order parameter rmin. We use rmin rather than, for example, the distance between centers of mass, because it is able to discriminate clearly whether or not the fibril and peptide species are in contact. This free energy (Fig. 2) shows that adsorption is spontaneous. Furthermore, the results from the two independent simulations are consistent with each other. The residual difference between the curves is due to the challenge of obtaining a representative sampling of the binding equilibrium. The free energy surfaces allow us to estimate a standard free energy of adsorption of ΔblindG⦵ = −19±2 kJ.mol−1, averaged across both simulations. This value is quantitatively in line with experimental surface plasmon resonance measurements which give an estimate of ΔblindG⦵ = −27 kJ.mol−1.28 Our access to all-atom energies also allows us to estimate the standard enthalpy of adsorption as ΔblindH⦵ = −90 ± 40 kJ.mol−1, implying an entropic contribution to adsorption of −TΔblindS⦵ = 70 ± 40 kJ.mol−1. These results are in reasonable agreement with the corresponding experimental estimates, although the errors on the enthalpic and entropic components are large (Table 1). In our work we have used a force field with specifically tuned protein-water interactions, since our work and that of others had suggested that protein-protein interactions were too favourable in standard force fields.22,29,30 An extrapolation of the adsorption free energy from the different λ windows sampled to λ = 1.0 (corresponding to a more standard force field, Amber ff03w31,32) suggests that binding would be significantly tighter (Fig. S4).
Figure 2:
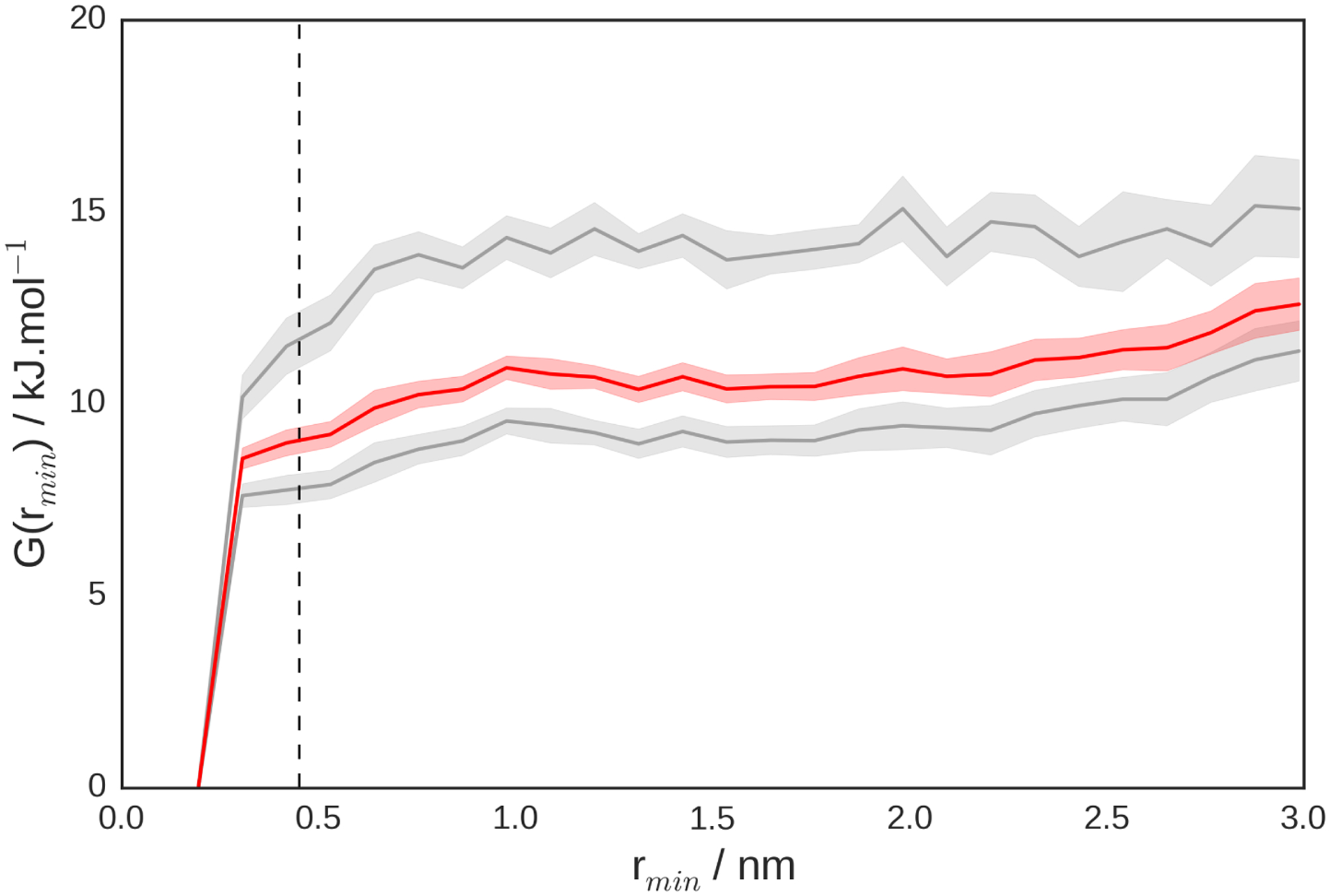
Peptide adsorption free energy surfaces, with gray curves showing results for each individual simulation and the red curve showing the Boltzmann average. Shadings denote standard errors and vertical dashed line defines the bound state. These curves were calculated at the simulation monomer concentration, then corrected to a standard state concentration of c⦵ = 1 M to yield the values shown in Table 1.
Table 1:
Thermodynamics of Aβ42 peptide-fibril adsorption. All values are in units of kJ.mol−1 and refer to a standard state of 1 M monomer and T = 300 K.
| ΔbindG⦵ | ΔbindH⦵ | TΔbindS⦵ | |
|---|---|---|---|
| Simulation | −19 ± 2 | −90 ± 40 | 70 ± 40 |
| Experiment28 | −27 | −67 | 40 |
Our value for the binding energy is also consistent with coarse-grained molecular simulations, from which it was concluded that a binding energy on the order of 20 kJ.mol−1 results in maximally efficient autocatalysis.18 This optimal interaction strength is reminiscent of the Sabatier principle of inorganic heterogeneous catalysis, which states that the the affinity between a catalyst and its substrate should be favorable, but not overly so, for efficient catalytic turnover.33
Encouraged by our ability to reproduce experimental binding thermodynamics, we went on to investigate in more detail the molecular interactions stabilizing binding. The numbers of intermolecular contacts formed by each residue of the fibril and the peptide (Fig. 3) reveal that there are definite interaction hotspots on the fibril, including residues Gln 15 and Lys 16. In contrast, the peptide is more promiscuous in binding, albeit with elevated contact probabilities at the termini and the residues Glu 22 and Asp 23. To obtain representative major binding motifs, we performed a cluster analysis of the intermolecular contact maps. This revealed that the largest cluster is characterized by interactions between the positive Lys 16 of the fibril surface and the negative Glu 22, Asp 23 and Ala 42 carboxyl group of the peptide. The next two important clusters also showed largely polar interactions between charged sidechains and between hydrogen bond donors and acceptors. The favorable charged and polar interactions between peptide and fibril suggest that salt should modulate the binding affinity, and reweighting our simulations using a Debye-Hückel model for screening suggests that binding is indeed tighter at lower ionic strength (Fig. S5) – in spite of the long-range repulsion between peptide and fibril due to their net charge. In our work, we focussed on a single filament model of the Aβ42 fibril, partly as we were already at the limit of what is computationally feasible. Several subsequent structural studies have found that Aβ42 fibrils consist of two filaments related by an approximately two-fold symmetry axis.34–36 Nonetheless, most of the fibril residues interacting with the peptide are not in regions involved in binding the symmetry-related fiber, a prominent exception being Gln-15, which does form contacts with the neighbouring fiber. In addition, it should be noted that although the structures determined experimentally are generally in good agreement, there may be some dependence on the chosen structure, which should be investigated in future work.
Figure 3:
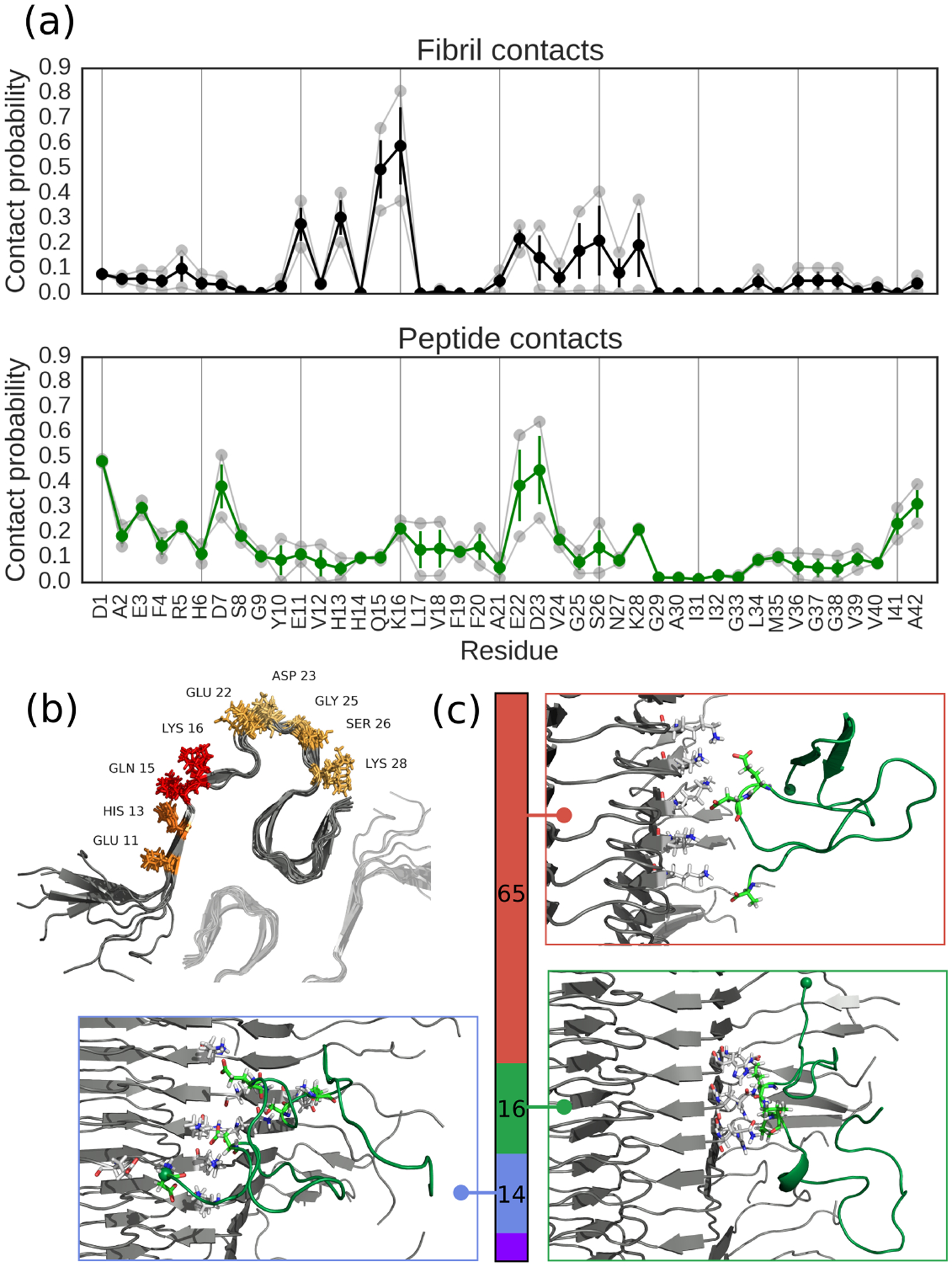
Peptide-fibril contact analysis. (a) Probabilities of individual amino acids being involved in adsorption interactions in (top) the fibril surface and (bottom) the peptide. Translucent curves are results from the independent simulations, full colored curves are averages and error bars are standard errors. (b) Structural visualization of adsorption hotspots on the fibril, with the color of the amino acid getting more red as the residue is more likely to be involved. Translucent fibril shows approximate location of second protofibril in a C2 symmetric arrangement, as suggested is appropriate for Aβ42 fibrils by other structural studies.34–36 (c) Clustering analysis of binding modes showing top three interaction motifs, with colored bar denoting statistical weight of each cluster and inset structures showing structures closest to cluster centroids. N-termini shown as spheres, and all figures made using PyMOL.
Interestingly, the residues Glu 22 and Asp 23 that we identify in our analysis of intermolecular contacts are those that are mutated to neutral ones in the familial Aβ Iowa, Dutch and Arctic mutations. These mutations result in clinical phenotypes similar to Alzheimer’s Disease, often accompanied by cerebral hemorrhage, and in vitro have been found to accelerate Aβ aggregation.37–40 These observations may be explained if desorption were rate-limiting in secondary nucleation, such that destabilizing the monomer-fibril interaction would result in faster turnover, though the mutations could very well affect other determinants of aggregation such as monomer β-propensity or inter-peptide interactions. Rate-limiting detachment has, however, been observed in vitro for Aβ42 at high salt concentrations or low pH conditions.41,42 Furthermore Lys 16, the major fibril residue identified in our analysis, has also been shown to be a key residue in the aggregation process, with K16A mutants showing delayed fibril formation and reduced cytotoxicity.43 Our finding that peptide adsorption is mediated by polar interactions is in contrast with results from other molecular simulation studies, which have found that hydrophobic interactions dominate. Most of these studies have focused on Aβ40;44,45 a recent paper, however, also found that an Aβ1−42 monomer interacts with a truncated Aβ17−42 fibril polymorph with structure modelled on that of an Aβ40 fibril through hydrophobic contacts.46 The main difference from our work in all of these studies is that they are based on the structure of an Aβ40 fibril, either because they were concerned with that peptide, or because they were done before an experimental structure of Aβ42 was published. This is the most likely reason for any differences regarding which residues are important. An additional factor may be that the force field we use, which is optimized for protein-protein interactions, is significantly less hydrophobic overall. The adsorption mechanism involving polar interactions which we observe does, however, appear most consistent with the experimental enthalpy-driven binding thermodynamics in which the positive value of −TΔbindS suggests that the hydrophobic effect is not the major driving force of adsorption.28
We also examined the effect that adsorption has on the monomer configuration by building the intramolecular contact maps shown in Fig. 4(a). As demonstrated by the similarity in the free and bound states, adsorption is accompanied by only modest structural changes in the monomer. This suggests that interactions with other adsorbed peptides may be necessary for formation of ordered structure in secondary nuclei, although it is also possible that structural conversion could occur in the monomer on a longer time scale than is accessible in our simulations. The small amount of local structure formation induced by adsorption appears to be due to formation of β-hairpins (Fig. 4(b)). We confirmed that adsorption was in fact related to the appearance of β structure from the average increase population (≈2–3 % per residue) of residues assigned to β-sheet secondary structure.
Figure 4:
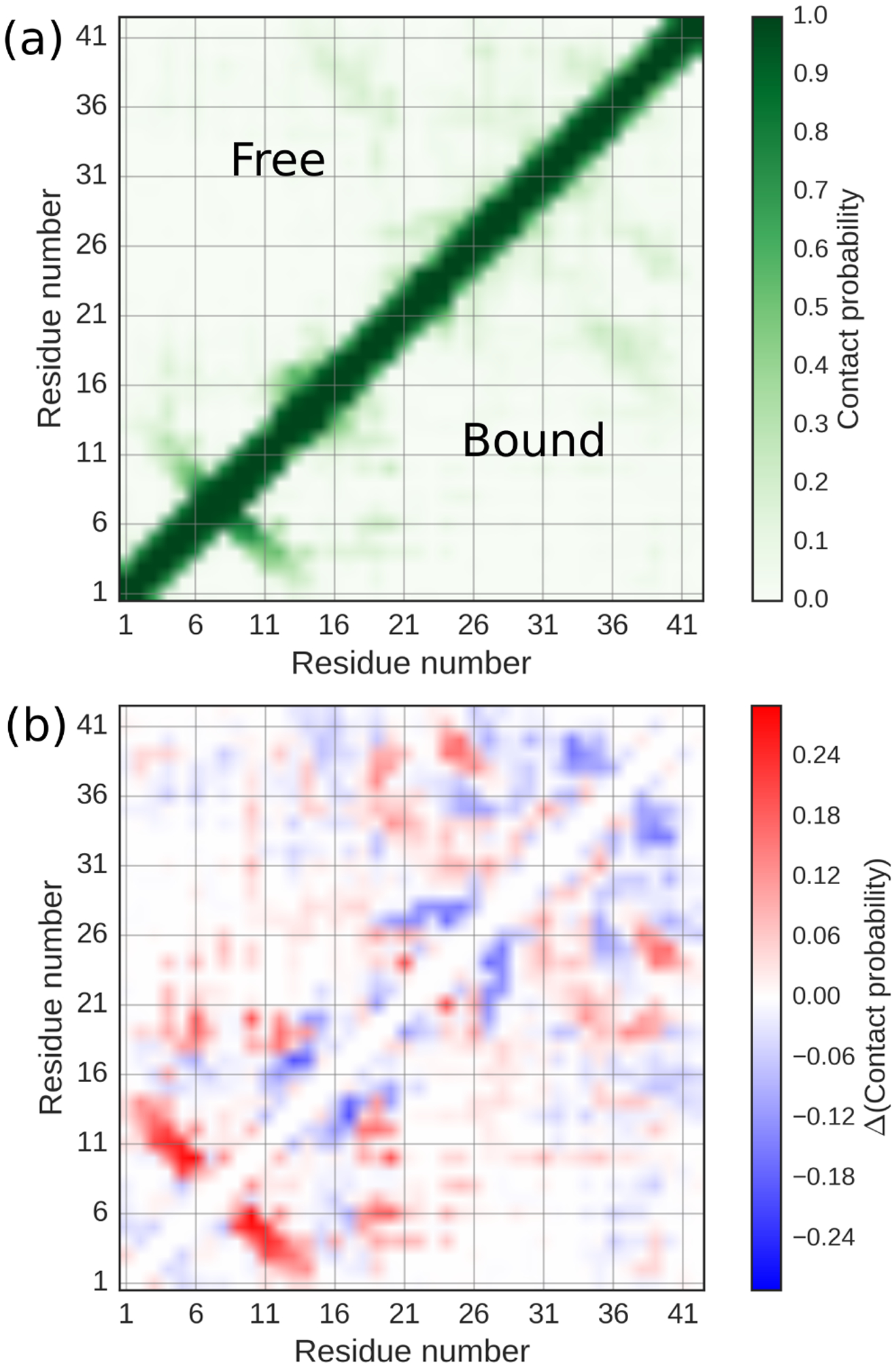
Effect of adsorption on monomer configuration. (a) Intramolecular contact maps for free (upper triangle) and bound (lower triangle) monomers. (b) Difference contact map between bound and free states.
While initial adsorption of monomers to a fibril is clearly only part of the secondary nucleation mechanism, we can nonetheless ask to which mutations it would be most sensitive. How can the adsorption mechanism proposed be tested experimentally? One of the most powerful tools for dissecting protein interactions is site-directed mutagenesis. We therefore designed appropriate mutants by assessing the effect of residue substitutions after reweighting the configurations sampled in our trajectories using a simple contact potential, as described in the Supplementary Information. We chose only those constructs which are expected to reduce catalytic efficiency, either by decreasing or increasing the adsorption strength in accord with the Sabatier principle, while also being compatible with incorporation into the experimental fibril structure. Our analysis suggested several candidates, in particular mutations to residues 15, 16, 22, 23, 28, which should alter peptide-fibril interactions; it will be interesting to test the effect of mutations to these residues experimentally in the future.
In summary, we have used molecular simulations to probe at atomistic resolution the first step of Aβ42 secondary nucleation, namely the adsorption of a monomer onto a fibril surface. The computed thermodynamic properties of binding quantitatively match experimental measures, and are consistent with estimates from coarse-grained simulations of the most efficient binding energy for autocatalysis. In addition, the structural detail of our work has allowed us to identify major binding modes of adsorption, which mostly consist of polar interactions, involving particularly the fibril residue Lys 16 and the peptide residues Glu 22 and Asp 23. These are all residues which have been identified as critical to the aggregation process either by in vitro studies or through patient mutations, providing further evidence that our simulations capture the correct binding mechanism. In principle, our work can also be used to design inhibitors targeting the interactions between the fibril and peptide.
Supplementary Material
Acknowledgement
We thank A. Šarić, A. Buell, T. P. J. Knowles, M. Vendruscolo and S. Linse for helpful discussions. This study utilized the high performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (NIH), Bethesda, MD. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH). M.M.J.B. was supported by the NIH-Oxford/Cambridge Scholars Program, the Cambridge Commonwealth, European and International Trust and the Centre for Misfolding Diseases.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting Information Available
Supporting text including detailed methods, five supporting figures and five supporting tables are available online.
References
- (1).Thies W; Bleiler L 2012 Alzheimers disease facts and figures. Alzheimer’s & Dementia 2012, 8, 131–168. [DOI] [PubMed] [Google Scholar]
- (2).Alzheimer A Über eigenartige Krankheitsfälle des späteren Alters: (On certain peculiar diseases of old age). History of Psychiatry 1991, 2, 74–101. [DOI] [PubMed] [Google Scholar]
- (3).Eisenberg D; Jucker M The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hsia AY; Masliah E; McConlogue L; Yu G-Q; Tatsuno G; Hu K; Kholodenko D; Malenka RC; Nicoll RA; Mucke L Plaque-independent disruption of neural circuits in Alzheimers disease mouse models. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mucke L; Masliah E; Yu G-Q; Mallory M; Rockenstein EM; Tatsuno G; Hu K; Kholodenko D; Johnson-Wood K; McConlogue L High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci 2000, 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dodart J-C; Bales KR; Gannon KS; Greene SJ; DeMattos RB; Mathis C; DeLong CA; Wu S; Wu X; Holtzman DM et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat. Neurosci 2002, 5, 452. [DOI] [PubMed] [Google Scholar]
- (7).Williams TL; Johnson BR; Urbanc B; Jenkins ATA; Connell SD; Serpell LC Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside-containing lipid membranes. Biochem. J 2011, 439, 67–77. [DOI] [PubMed] [Google Scholar]
- (8).Hellstrand E; Boland B; Walsh DM; Linse S Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci 2009, 1, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cohen SI; Linse S; Luheshi LM; Hellstrand E; White DA; Rajah L; Otzen DE; Vendruscolo M; Dobson CM; Knowles TP Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Meisl G; Yang X; Hellstrand E; Frohm B; Kirkegaard JB; Cohen SI; Dobson CM; Linse S; Knowles TP Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Arosio P; Knowles TP; Linse S On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys 2015, 17, 7606–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Haass C; Selkoe DJ Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell. Bio 2007, 8, 101. [DOI] [PubMed] [Google Scholar]
- (13).McLean CA; Cherny RA; Fraser FW; Fuller SJ; Smith MJ; Bush AI; Masters CL Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol 1999, 46, 860–866. [DOI] [PubMed] [Google Scholar]
- (14).Gong Y; Chang L; Viola KL; Lacor PN; Lambert MP; Finch CE; Kra t GA; Klein WL Alzheimer’s disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cohen SI; Arosio P; Presto J; Kurudenkandy FR; Biverstål H; Dolfe L; Dunning C; Yang X; Frohm B; Vendruscolo M et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol 2015, 22, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Aprile FA; Sormanni P; Perni M; Arosio P; Linse S; Knowles TP; Dobson CM; Vendruscolo M Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv 2017, 3, e1700488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Habchi J; Chia S; Limbocker R; Mannini B; Ahn M; Perni M; Hansson O; Arosio P; Kumita JR; Challa PK et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimers disease. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E200–E208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Šarić A; Buell AK; Meisl G; Michaels TC; Dobson CM; Linse S; Knowles TP; Frenkel D Physical determinants of the self-replication of protein fibrils. Nat. Phys 2016, 12, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Aβ (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol 2015, 22, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Carballo-Pacheco M; Strodel B Comparison of force fields for Alzheimer’s Aβ42: a case study for intrinsically disordered proteins. Protein Sci 2017, 26, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rosenman DJ; Wang C; García AE Characterization of Aβ monomers through the convergence of ensemble properties among simulations with multiple force fields. J. Phys. Chem. B 2015, 120, 259–277. [DOI] [PubMed] [Google Scholar]
- (22).Best RB; Zheng W; Mittal J Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput 2014, 10, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Henriques J; Cragnell C; Skepö M Molecular dynamics simulations of intrinsically disordered proteins: force field evaluation and comparison with experiment. J. Chem. Theor. Comput 2015, 11, 3420–3431. [DOI] [PubMed] [Google Scholar]
- (24).Conicella AE; Zerze GH; Mittal J; Fawzi NL ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Graen T; Klement R; Grupi A; Haas E; Grubmuller H Transient secondary and tertiary structure formation kinetics in the intrinsically disordered state of α-synuclein from atomistic simulations. Chem. Phys. Chem 2018, 19, 1–6. [DOI] [PubMed] [Google Scholar]
- (26).Meng F; Bellaiche MM; Kim J-Y; Zerze GH; Best RB; Chung HS Highly Disordered Amyloid-β Monomer Probed by Single-Molecule FRET and MD Simulation. Biophys. J 2018, 4, 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Oleinikovas V; Saladino G; Cossins BP; Gervasio FL Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J. Am. Chem. Soc 2016, 138, 14257–14263. [DOI] [PubMed] [Google Scholar]
- (28).Cohen SI; Cukalevski R; Michaels TC; Šarić A; Törnquist M; Vendruscolo M; Dobson CM; Buell AK; Knowles TP; Linse S Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-β peptide. Nat. Chem 2018, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Petrov D; Zagrovic B Are current atomistic force fields accurate enough to study proteins in crowded environments? PLoS Comput. Biol 2014, 10, e1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Piana S; Donchev AG; Robustelli P; Shaw DE Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 2015, 119, 5113–5123. [DOI] [PubMed] [Google Scholar]
- (31).Duan Y; Wu C; Chowdhury S; Lee MC; Xiong G; Zhang W; Yang R; Cieplak P; Luo R; Lee T et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum chemical calculations. J. Comp. Chem 2003, 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- (32).Best RB; Mittal J Protein simulations with an optimized water model: cooperative helix formation and temperature-induced unfolded state collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [DOI] [PubMed] [Google Scholar]
- (33).Boudart M; Djéga-Mariadassou G Kinetics of heterogeneous catalytic reactions; Princeton University Press: Princeton, U.S.A., 2014. [Google Scholar]
- (34).Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A; Güntert P; Meier BH; Riek R Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev I; Rosay M; Donovan KJ; Michael B; Wall J; Linse S et al. Atomic resolution structure of monomorphic Aβ 42 amyloid fibrils. J. Am. Chem. Soc 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gremer L; Schölzel D; Schenk C; Reinartz E; Labahn J; Ravelli RB; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H et al. Fibril structure of amyloid-β (1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Grabowski TJ; Cho HS; Vonsattel JPG; Rebeck GW; Greenberg SM Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol 2001, 49, 697–705. [DOI] [PubMed] [Google Scholar]
- (38).Van Nostrand WE; Melchor JP; Cho HS; Greenberg SM; Rebeck GW Pathogenic effects of D23N Iowa mutant amyloid β-protein. J. Biol. Chem 2001, 276, 32860–32866. [DOI] [PubMed] [Google Scholar]
- (39).Levy E; Carman MD; Fernandez-Madrid IJ; Power MD; Lieberburg I; van Duinen SG; Bots G; Luyendijk W; Frangione B Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- (40).Nilsberth C; Westlind-Danielsson A; Eckman CB; Condron MM; Axelman K; Forsell C; Stenh C; Luthman J; Teplow DB; Younkin SG et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci 2001, 4, 887. [DOI] [PubMed] [Google Scholar]
- (41).Meisl G; Yang X; Frohm B; Knowles TP; Linse S Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Aβ-peptide. Sci. Rep 2016, 6, 18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Meisl G; Yang X; Dobson CM; Linse S; Knowles TP Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Aβ42 peptide and its variants. Chem. Sci 2017, 8, 4352–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sinha S; Lopes DH; Bitan G A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem. Neurosci 2012, 3, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jiang X; Cao Y; Han W In Silico Study of Recognition between Aβ40 and Aβ40 Fibril Surfaces: An N-Terminal Helical Recognition Motif and Its Implications for Inhibitor Design. ACS Chem. Neurosci 2017, [DOI] [PubMed] [Google Scholar]
- (45).Schwierz N; Frost CV; Geissler PL; Zacharias M From Aβ Filament to Fibril: Molecular Mechanism of Surface-Activated Secondary Nucleation from All-Atom MD Simulations. J. Phys. Chem. B 2017, 121, 671–682. [DOI] [PubMed] [Google Scholar]
- (46).Barz B; Strodel B Understanding Amyloid-β Oligomerization at the Molecular Level: The Role of the Fibril Surface. Chem.-Eur. J 2016, 22, 8768–8772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.