

Abstract
Solution-state NMR is an important tool for studying protein structure and function. The ability to probe methyl groups has substantially expanded the scope of proteins accessible by NMR spectroscopy, including facilitating study of proteins and complexes greater than 100 kDa in size. While the toolset for studying protein structure and dynamics by NMR continues to grow, a major rate-limiting step in these studies is the initial resonance assignments, especially for larger (> 50 kDa) proteins. In this practical review, we present strategies to efficiently isotopically label proteins, delineate NMR pulse sequences that can be used to determine methyl resonance assignments in the presence and absence of backbone assignments, and outline computational methods for NMR data analysis. We use our experiences from assigning methyl resonances for the aromatic biosynthetic enzymes tryptophan synthase and chorismate mutase to provide advice for all stages of experimental set-up and data analysis.
Keywords: NMR, Large proteins, Methyl labeling, Isotopic labeling, Peak assignment, NOESY
1. Introduction
NMR has provided critical insights into protein structure, dynamics and function. Solution-state NMR has a protein size limitation owing to the relationship between the slow tumbling of large proteins in solution and its effects on relaxation properties and signal acquisition. Several advancements over many years have lifted protein size limitations, including the development of methods to analyze 1H–13C methyl resonances [1–4]. These studies have allowed solution-state NMR studies to be carried out on proteins and complexes several hundred kilodaltons to up to 1 megadalton in size [5–9].
Methyl labeling offers several advantages over 1H–15N backbone amide labeling schemes for large proteins. Methyl protons have threefold degeneracy due to rapid rotation on the methyl axis, meaning that all three protons contribute to the same NMR signal [3]. Methyl groups also have enhanced sensitivity for any HMQC (heteronuclear multiple quantum coherence) type experiments. HMQC experiments have a field-independent transverse relaxation optimized (TROSY) effect for methyl groups, and so provide signals with narrow linewidths even for proteins > 100 kDa [3]. Additionally, the 1H–13C methyl NMR spectrum will often have fewer overlapping peaks compared to 15N backbone labeling because on average only 30% of the amino acids in proteins contain methyl groups [10].
Methyl-containing amino acids are well-distributed throughout the protein structure, and so they are excellent probes for structural characterization of large proteins. Consistent with the theme of the present issue, there are also several NMR methods to gain insight into biomolecular structural dynamics across many timescales. Experiments for studying events on the microsecond-to-second timescale such as DEST (Dark state Exchange Saturation Transfer) [11], CEST (Chemical Exchange Saturation Transfer) [12–14], CPMG (Carr-Purcell-Meiboom-Gill) relaxation-dispersion [15–17], and paramagnetic relaxation enhancement (PRE) [18] have been developed for methyl groups. The sensitivity of methyl groups is also useful for studying dynamics by line shape analysis involving micromolar affinity ligand binding since expedient 2D correlation spectra can be obtained even on low (< 50 μM) concentration protein samples in as short a time as 10 min [19].
There are several reviews that explore the merits of protein methyl NMR and the scientific insights resulting from these studies (e.g. [7,9,20]). Here, we focus on perhaps the rate-limiting step of these studies – the initial assignment of the methyl resonances. We use our own experiences with enzymes from the aromatic biosynthetic pathway, namely tryptophan synthase and chorismate mutase, to guide the reader through this process. For tryptophan synthase, we studied the ~30 kDa alpha subunit (αTS), where we could base the 1H–13C methyl assignments on the previously established 1H–15N amide backbone resonance assignments [21,22]. For the ~60 kDa chorismate mutase, there were no previous NMR assignments, and so we used a NOESY (Nuclear Overhauser Effect SpectroscopY) based method [23] along with a known X-ray crystal structure [24] to determine assignments. We have also previously assigned methyl resonances for the 52 kDa poliovirus RNA-dependent RNA polymerase [25] through mutagenesis (i.e. look for the disappearance of a resonance after changing a methyl bearing residue to a different residue that will not be similarly isotopically labeled), but this method can be tedious, potentially very expensive and assignments are not always conclusive, although there have been efforts to streamline this approach [26]. We note that there are other exciting, perhaps complementary methods, that take advantage of paramagnetic probes to assign methyl groups [27,28], but we do not go into detail with those strategies here. In this Review, we will discuss different isotopic labeling schemes and their merits, and practical considerations for NMR data acquisition, processing and analysis for methyl assignments. We first discuss assigning methyl resonances based on previously acquired 1H–15N amide backbone assignments, and then have a deeper discussion on methyl assignment strategies in the absence of backbone assignment data based on NOESY methods.
2. Assigning side chain methyl resonances based on previous backbone assignments
For smaller proteins (< 20 kDa), methyl resonances can often be assigned based on backbone resonance assignments (following classical 1H/13C/15N triple resonance experiments [29–32]) using TOCSY (total correlation spectroscopy) type transfer along the aliphatic side chain [33,34]. However, the slow overall tumbling times for larger proteins lead to diminished magnetization during the TOCSY mixing period. The presence of 1H groups near methyl side chain groups of interest can also contribute to 1H–1H dipole-dipole relaxation. As such, it is common to replace non-exchangeable 1H groups with 2H by heterologous production of proteins in 2H2O-based minimal media, sometimes supplemented with 2H-labeled carbon sources. Within this background, backbone amide groups can be 15N labeled, while desired aliphatic groups can be appropriately 1H, 13C-labeled.
2.1. Production and purification of methyl labeled αTS
We have been especially interested in how the subunits of tryptophan synthase communicate and coordinate their catalytic activities [35]. Tryptophan synthase is composed of two subunits, αTS and βTS, and catalyzes the final two steps of tryptophan biosynthesis. The indole product of αTS is channeled directly to the active site of βTS through a 25 Å hydrophobic tunnel. Our previous backbone 1H–15N NMR studies had suggested that there is an amino acid interaction network connecting the αTS active site and the αTS/βTS binding interface that serves as an information conduit between αTS and βTS [22,36,37]. However, X-ray crystal structures have suggested that there are little backbone structural changes in αTS that might help to explain the amino acid interaction networks. So to complement our previous 1H–15N NMR studies [22,36,37], we were interested in gaining insight on side chain structural dynamics. We note that all of these NMR studies were performed on αTS alone (~30 kDa) in the absence of βTS. The methyls of isoleucine in αTS were selectively 1H, 13C-labeled at the δ1 position while the rest of the protein was uniformly 2H, 13C, 15N-labeled. We note that the buffers used throughout the αTS purification process and for the NMR experiments are 1H2O-based, which effectively ensures that all exchangeable 2H amide protons are exchanged back to 1H. The protein was heterologously expressed in Escherichia coli BL21(DE3) cells in minimal M9 media containing 2 g/L of U-[2H, 13C]-glucose and 1 g/L of U-[15N] labeled ammonium chloride as sole sources of carbon and nitrogen respectively. An hour before protein expression was induced through the addition of 1 mM IPTG (isopropyl β-D-1-thiogalactopyranoside), 120 mg/L of 2-keto-3,3-d2–1,2,3,4-13C-butyrate (Cambridge Isotope Laboratories, CIL Catalog # CDLM-4611) was added to the bacterial growth as described [38]. If the labeling of valine and leucine {L(13CH3,12CD3), V(13CH3,12CD3)} is desired, one would add 200 mg/L of 2-keto-3-methyl-d3–3-d1–1,2,3,4-13C-butyrate (CIL Catalog # CDLM-8100). Other than this modification in the labeling scheme for the isoleucine δ1 methyl groups, the rest of the protocol for the bacterial growth and purification was performed as previously described [22].
2.2. Connecting methyl resonances with 15N backbone amide assignments
The methyl protonated δ1-isoleucine samples were used to collect two sets of 3D experiments. The first experiment involved connecting the correlations between the methyl carbon chemical shifts (ΩCm[i]) and amide chemical shifts (ΩN[i], ΩHN[i]) using the pulse sequence described as 3D-I,L-(HM)CM(CGCBCA)NH [38] (Fig. 1). Sometimes correlation of methyl carbons with neighboring amide residues were also observed owing to very similar values for 1JCα[i]N[i] and 2JCα[i]N[i+1] values. A complementary experiment on the same sample was conducted where correlations between chemical shifts of methyl protons (ΩHm[i]) and amides (ΩN[i], ΩHN[i]) were observed using the pulse sequence described as 3D-I,L-HM(CMCGCBCA)NH [38] (Fig. 1). In addition to these 3D experiments, high-resolution constant time 2D 1H, 13C HMQC and 2D 1H, 15N-HSQC (heteronuclear single quantum coherence) spectra were collected with 512 points in the indirect dimensions. The 2D spectra act as templates to which the 3D-I,L-(HM)CM(CGCBCA)NH and 3D-I,L-HM(CMCGCBCA)NH can be linked for isoleucine methyl assignments (Fig. 2).
Fig. 1.
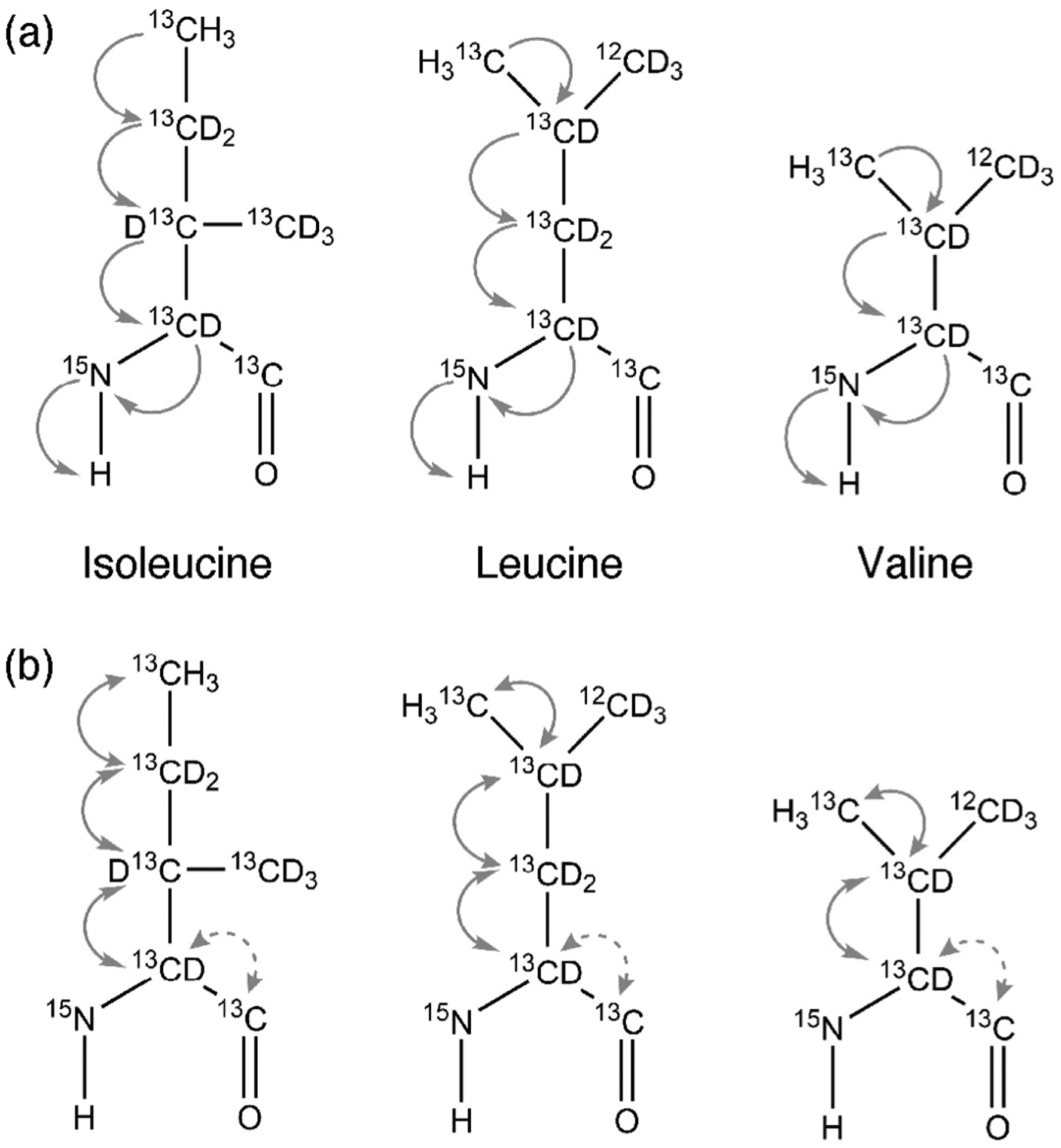
Magnetization transfer pathway for (a) 3D-HM(CMCGCBCA)NH and 3D-(HM)CM(CGCBCA)NH and (b) 3D-HMCM(CG)CBCA and 3D-HMCM (CGCBCA)CO for isoleucine, leucine and valine residues.
Fig. 2.

Assignments of methyl resonances with known backbone assignments. (a) 1H–15N HSQC signal for I240 which is correlated to (b) 13C methyl using HM (CMCGCBCA)NH and (c) 1H methyl using the (HM)CM(CGCBCA)NH pulse sequence. These resonances are also correlated to the methyl resonances on (d) 1H–13C methyl HMQC making methyl assignments straightforward.
2.3. Connecting methyl resonances with C′, Cα and Cβ backbone assignments
Most of the methyl assignments can be accomplished through connections to the backbone amide resonances, but transfer of magnetization from side chains to amides can be weak for larger proteins. In most cases, the carbonyl (C′) and aliphatic (Cα and Cβ) carbon assignments are present from standard amide detect TROSY 3D experiments [39–41], which can be used for assignment of ambiguous methyl residue assignment. There are often overlaps in the chemical shifts for leucine and valine methyl groups, making assignments challenging. However, leucine and valine residues possess unique aliphatic (Cα and Cβ) carbon chemical shifts owing to the different chemical environments and different branched carbons, and so these aliphatic resonances can be used as anchor points for starting assignments. The {I (δ1), L(13CH3,12CD3), V(13CH3,12CD3)} U-[1H, 13C, 15N] labeled sample can be used to collect HMCM[CG]CBCA and HMCM(CGCBCA)CO as described [38]. The pulse sequences used for leucine and valine assignments are different because of the different carbon branched chain and length, which can also help delineate the ambiguous assignments between leucine and valine methyl groups.
2.4. Analysis of data for methyl assignments with backbone assignments
All αTS spectra were processed in Bruker’s TOPSPIN 3.5 software were zero-filled with twice the number of points, and linear prediction was performed in the indirect dimensions [42,43]. A squared sine function was used for apodization before Fourier transformation. The processed NMR spectra were directly imported into SPARKY software where methyl and amide chemical shifts were synced across all 2D and 3D experiments described above for isoleucine assignments (see Fig. 2). The 1H–15N amide chemical shift assignments for αTS were loaded from previous studies [22]. Armed with this knowledge, every unassigned resonance from the methyl 1H–13C HMQC was connected to a previously assigned 1H–15N amide chemical shift via the 3D experiments described above [38]. For complete assignment of the δ1-isoleucine resonances of αTS, it was sufficient to collect just the 3D-I,L-(HM)CM (CGCBCA)NH and 3D-I,L-HM(CMCGCBCA) spectra. In order to resolve some assignment ambiguities, one could also collect 3D HMCM [CG]CBCA, HMCM(CGCBCA)CO and correlate the methyl resonances with preassigned aliphatic and carbonyl carbon.
3. Assigning side chain methyl resonances in the absence of backbone assignments
Methyl resonances can also be assigned in the absence of any backbone resonance assignments. This strategy may be especially important for larger proteins (> 50 kDa), where it can be very challenging to assign backbone resonances using 1H, 13C, 15N triple resonance methods. One strategy is to assign methyl resonances using NOESY methods, taking advantage of through-space nuclear spin polarization transfer via cross-relaxation between NMR active nuclei. The NOE pattern predicted by a 3D protein structure can then be compared against experimental data to begin a “methyl” walk (see Fig. 3). In this section, we discuss practical aspects of this approach.
Fig. 3.
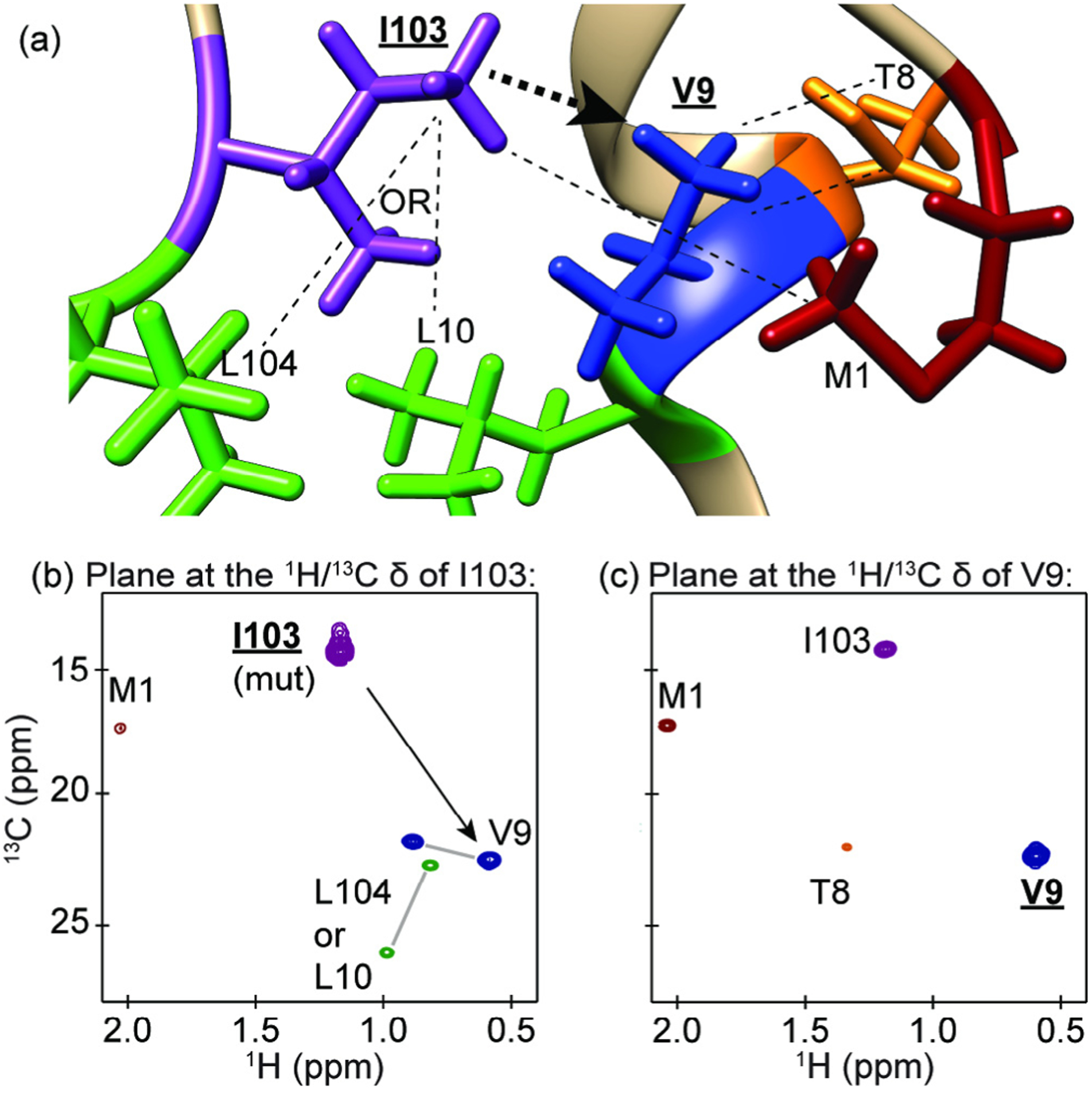
An example of the methyl walk strategy for assigning methyl groups using NOESY data. (a) A depiction of methyl containing residues near V9 from the PDB 1CSM X-ray crystal structure of ScCM. (b) The plane at the methyl proton and carbon resonance frequencies of I103 from a 4D HCCH NOESY spectrum, which was confirmed as an assignment through mutagenesis. The NOE cross peaks shown in this plane for V9 and M1 were readily assigned because these are the closest methionine and valine residues in the 3D protein structure, while the leucine cross peaks are ambiguous since L10 and L104 are equally close to I103 and both are close to V9. Leucine and valine geminal methyl resonance pairs were determined from a short mixing time CCH 3D NOESY experiment (data not shown) and are indicated with a gray line between the peaks. (c) The plane at the proton and carbon resonance frequencies of V9. This plane allowed for the additional assignment of T8 since this is the closest threonine to V9. The residues and resonances for leucine, isoleucine, valine, threonine and methionine are coloured green, purple, blue, orange and dark red, respectively. The black arrows in (a) and (b) indicate the direction of the methyl walk.
3.1. Considerations for choosing a methyl labeling scheme for methyl NOESY experiments
There are currently methods described in the literature for producing proteins labeled with any of the six naturally occurring methyl-containing amino acids [8,44]. The labeling scheme of choice should depend upon the assignment method and desired information. NOE assignment strategies rely on “walking” from one methyl residue to another through space (Fig. 3). The closer the residues are in space the greater the chance that an intense NOE cross-peak will show up in a NOESY spectrum.
Higher diversity in the methyl residue types can help reduce assignment ambiguity, as this can create a greater number of unique NOE cross-peaks for more residues to expedite the assignment process. Common labeling schemes include those that combine methyl labeling on the following groups of amino acid residues: ILV [2], ILVA [45], IT [46], and ILVMA [47], although this list is not exhaustive (see Table 1 for considerations on combining methyl schemes). Incorporation of rarer methyl groups, such as those from threonine and methionine, can aid in assignments because they provide good starting points for the “methyl walk” strategy [48], and are easier to assign because of their scarcity in the amino acid sequence. When choosing a labeling scheme, it is also important to decide which methyl group(s) will be most interesting to study based on previous functional and structural decides, which methyl containing residues are the most numerous in order to generate an information rich NOE network, and how to avoid isotopic scrambling into other methyl groups if undesired (see Table 1).
Table 1.
Isotopically labeled methyl residue precursors for NOESY experiments.
| Site of 13CH3 | Precursor Name | Precursor Structure | 1L single density growth | Additional Supplements | Notes/Potential issues | Suppliers | Ref. |
|---|---|---|---|---|---|---|---|
| I-δ1 | α-Keto-butyrate | 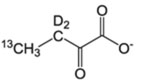 |
60 mg/L | – | Incompatible with A labeling. | CIL, Isotec, NMR-Bio | [1] |
| 2-(S)-Hydroxy-2-ethyl-3-keto-butyrate | 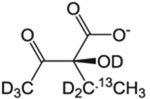 |
60 mg/L | – | Compatible with A labeling | NMR-Bio | [45] | |
| M-ε | l-Methionine |  |
250 mg/L | – | – | CIL, Isotec, NMR-Bio | [72] |
| L-δ/V-γ racemic pro-R pro-S | α-Keto-isovalerate | 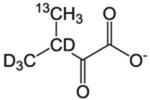 |
120 mg/L | – | – | CIL, Isotec, NMR-Bio | [4] |
| L-δ1, δ2/V-γ1, γ2 | α-Keto-isovalerate | 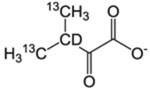 |
120 mg/L | – | This labeling scheme works best to establish intra-residue resonance pairings for L and V, less so for long range NOEs | Isotec | [2] |
| V-γ | α-Keto-isovalerate | Either of the above two precursors | 120 mg/L | 10% deuterated Bioexpress or Isogro | Addition of Bioexpress/Isogro has been observed to decrease I-δ1 and T-γ2 incorporation. Use to distinguish L/V resonances If also labeling I-δ1, add α-acetolactate 1 h before induction and α-ketobutyrate 20 min before induction | CIL, Isotec, or NMR-BioCIL, Isotec for supplement NMR-Bio | [52] |
| L-δ/V-γ pro-S | 2-(S)-Aceto-lactate |  |
300 mg/L | – | [50] | ||
| A-β | l-Alanine | 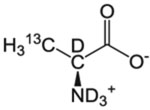 |
600 mg/L | 200 mg/L deuterated α-ketoisovalerate, 60 mg/L deuterated isoleucine (unless labeling L/V or I) | Scrambling to I-γ2 makes this label incompatible with α-ketobutyrate I labeling for NOE studies. 2-(S)-hydroxy-2-ethyl-3-ketobutyrate must be used instead for this scenario | CIL, Isotec, NMR-Bio | [45,49] |
| T-γ2 | l-Threonine | 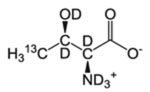 |
50 mg/L | 60 mg/L deuterated α-ketobutyrate (unless also labeling I-δ1) and 100mg/L deuterated Gly | – | CIL, Isotec, NMR-Bio | [46] |
3.1.1. Situations where isotope scrambling may be an issue and other incompatibilities
While it is possible to choose to incorporate several different isotopically labeled methyl residues at once, caution should be exercised, as not all combinations are compatible (Table 1). For example, attempting to incorporate isoleucine Cδ and alanine Cβ methyl labels simultaneously with the labeled alanine and the isoleucine precursor α-ketobutyrate may result in the incorporation of 1H, 13C at the γ2 position and the δ position because alanine can be converted to pyruvate by several enzymes and then to the isoleucine precursor oxaloacetate via the citric acid cycle [45,49]. This situation creates problems for methyl NOESY experiments because the isoleucine γ2 and δ methyls are only 3 Å apart, leading to a significant reduction in inter-residue methyl NOE cross peaks for isoleucine [45]. Kerfah et al. have described a solution to alanine scrambling that makes use of 2-(S)-hydroxy-2-ethyl-3-keto-butyrate as an alternative to the α-ketobutyrate precursor [45] (available from NMRBio in France). Alanine also has the potential for scrambling into the terminal methyl groups of leucine and valine residues, though scrambling is not a problem if leucine and valine residues are also being labeled. There are other solutions to this scrambling effect otherwise [49].
Threonine can also cause issues as it can be scrambled to glycine directly via threonine aldolase or in steps by threonine dehydrogenase followed by 2-amino-3-ketobutyrate ligase, which has the effect of diluting the isotope label [46]. The threonine methyl label will also be significantly scrambled to the δ1 methyl of isoleucine. Scrambling can be prevented in both cases with addition of relatively inexpensive deuterated glycine to growth media and either fully deuterated α-ketobutyrate or, if isoleucine labeling is desired, 3,3 deuterated, δ1 13CH3 α-ketobutyrate [45].
3.1.2. Special case of leucine and valine residues
Leucine and valine residues present extra challenges for NOE based assignments due to the geminal nature of their methyl groups. It is recommended that these groups are either labeled at the pro-R or pro-S methyl group, or racemic at both positions as these labeling schemes maximize the potential NOE interaction distance. The incorporation of pro-R or pro-S 13CH3 and otherwise deuterated LV residues can be achieved by supplementing 2H2O M9 growth media with isotope-labeled 2-acetolactate [45,50]. The more readily available LV precursor α-ketoisovalerate can be purchased from CIL as a racemic mixture of pro-R and pro-S 13CH3, and otherwise deuterated. These labeling patterns prevent cross relaxation between the geminal methyl groups and can increase the intensity of the LV methyl resonances two to four fold [4,50]. An additional sample where both terminal methyl groups in leucine and valine are 13CH3 labeled can be used to collect a short mixing time NOE experiment (~40 ms) [51] to measure the intra-methyl NOE and assign the methyl pairs to the same residue. This method substantially decreases the complexity of assigning both methyl groups of leucine and valine residues.
3.1.3. Determination of residue type
While isoleucine and methionine methyl groups tend to appear in distinct areas of the NMR spectrum, the methyl groups of alanine, threonine, valine and leucine all display varying degrees of overlap. As such, resonance assignments are aided by the collection of 1H–13C 2D spectra on samples with only individual residues types isotopically labeled (e.g. see Fig. 4). Perhaps the simplest way to distinguish leucine and valine residues when starting from the α-ketoisovalerate precursor is to add 10% Bioexpress or Isogro deuterated rich media to suppress leucine incorporation [52]. Bioexpress and Isogro are proprietary isotope-labeled rich growth media blends from CIL and Sigma Aldrich’s Isotec brand, respectively.
Fig. 4.
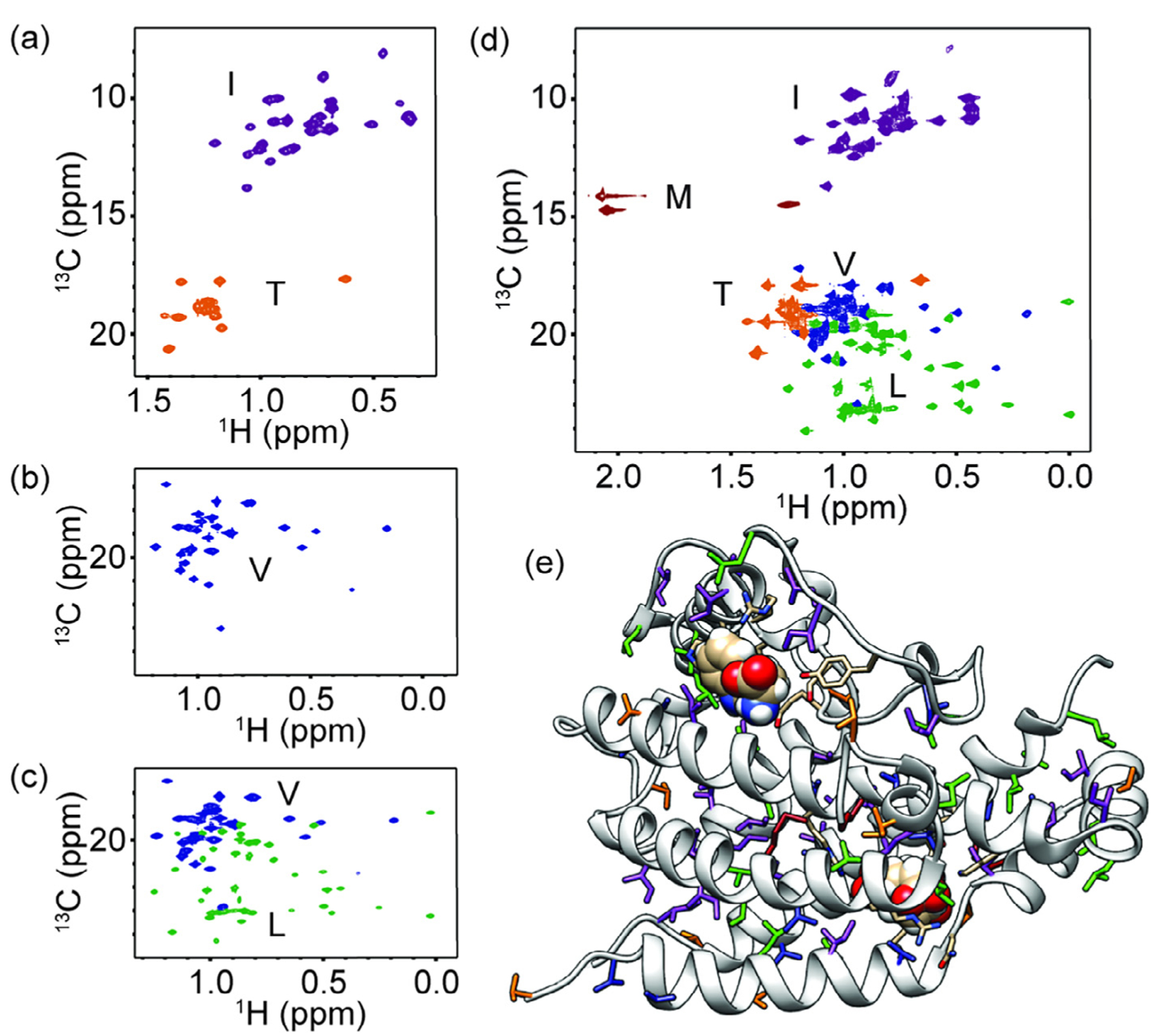
Methyl NMR spectra for yeast chorismate mutase. (a–d) 1H–13C 2D NMR spectra for (a) IT labeling, (b) V labeling, (c) LV labeling, and (d) ILVMT labeling. (e) The 3D protein structure for the monomer of yeast chorismate mutase (PDB 3CSM [24]) with ILVMT residues shown as sticks. The resonances and residues are coloured as follows: purple for isoleucine, green for leucine, blue for valine, dark red for methionine and orange for threonine. All spectra were collected on 0.6–1.8 mM ScCM protein in 50 mM sodium phosphate buffer pH 6.8 with 2 mM β-mercaptoethanol, 0.02% sodium azide in 2H2O and saturating amounts of tryptophan (i.e. 5 mM) added at 30 °C on a Bruker Avance-III 850 MHz spectrometer.
3.2. Isotope labeling scheme for the NOESY-based methyl assignments of chorismate mutase
As an example, we describe the labeling scheme used to gain methyl resonance assignments for Saccharomyces cerevisiae chorismate mutase (ScCM). This protein is a 60 kDa homodimer and has been a model system for understanding allostery [53]. ScCM catalyzes the conversion of chorismate to prephenate via a Claisen rearrangement. The prephenate product is a precursor to phenylalanine and tyrosine, while the substrate chorismate can also be used by a competing pathway to generate tryptophan. ScCM is allosterically inhibited by phenylalanine and tyrosine, and allosterically activated by tryptophan using the same allosteric binding pocket.
The labeling scheme for ScCM was decided with the goal in mind of gaining full assignments for isoleucine and threonine methyl resonances. Isoleucine residues comprise ~10% of the recombinant ScCM construct, and are spatially well-distributed (Fig. 4). Threonine residues only comprise 4.5% of the protein but are positioned to interact with both the substrate chorismate and with the allosteric effectors tryptophan and tyrosine. Leucine and valine methyls were also isotopically labeled, as a way to “fill in the gaps” for potential NOE networks. Finally, because there are only three positions with methionine residues, these were also incorporated to provide excellent starting points for methyl walks. In total, this ILVMT labeling scheme accounts for 29.5% of the 264 residues in ScCM (Fig. 4). Alanine labeling was avoided to prevent scrambling into the γ2 position of isoleucine since 3,3 deuterated, terminal methyl 13CH3 α-ketobutyrate was locally available.
To collect all of the necessary information for assignments, five samples were prepared. Since leucine, threonine and valine methyl resonances overlap in the ScCM 1H–13C 2D spectrum, IT-, LV-, and V-only labeled samples were generated to allow those residues to be distinguished from one another (Fig. 4). For each sample a 1H, 13C HMQC spectrum was collected with saturating amounts of free tryptophan in the NMR buffer, since this ligand allows for the greatest number of resonances to be resolved in the isoleucine and threonine 1H, 13C 2D spectrum. Next, the ILVMT sample was generated to distinguish methionine methyl resonances from threonine and isoleucine methyl resonances (Fig. 4), and this sample was later used in the 4D NOESY experiment to determine an NOE network for assignments. The LV precursor used for both residue type identification and in ILVMT sample for the 4D NOESY experiment was α-ketoisovalerate as a racemic mixture of pro-R and pro-S 13CH3, and was otherwise deuterated. Finally, an LV-labeled sample for a short mixing time intra-residue 3D NOESY experiment was generated using α-ketoisovalerate (from Isotec) labeled as 13CH3 at both methyl positions and deuterated at the 3 position to prevent cross relaxation.
3.2.1. Production of highly deuterated methyl labeled chorismate mutase samples
The ScCM gene was optimized for overexpression in E. coli, synthesized, and cloned into a pEX-C-HIS expression vector with a TEV cleavage site prior to the His-tag by Blue Heron Biotech, LLC. The most widely used method for growing highly deuterated methyl labeled proteins is that of Tugarinov et al. [54], but arguably one of the most time and cost-effective ways of producing highly deuterated protein samples is through the use of ‘quad-density’ cultures based on the method of Marley et al. [55]. Therefore a method of producing highly deuterated, methyl labeled ScCM was developed as a modification of the method of Marley et al. for methyl probe incorporation as detailed in the Supplementary Material.
3.3. Data collection for assignment of methyl resonances by NOESY methyl walk
3.3.1. NMR experiments for residue type identification and linewidth measurement
To identify residue type, HMQC or HSQC type experiments can be used (Fig. 4). The HMQC has the advantage of producing a higher S/N ratio spectrum compared to the HSQC due to the methyl TROSY effect in highly deuterated proteins [3], but both will likely work fine for residue type identification. What is more important is to ensure that enough data points are collected to accurately measure the 1H and 13C resonance frequencies of each peak, fully resolve as many peaks as possible, and provide a rough measure of the linewidth of the methyl groups for estimation of the transverse relaxation time, T2 (or T2* due to magnetic field inhomogeneity). This rough T2 value becomes important for setting up later 3D and 4D experiments. Choosing a long enough acquisition time in the direct proton dimension is quite easy because overestimating this value will not significantly increase acquisition time or lower S/N. As an example, the direct dimension acquisition time typically used to measure linewidths in the proton dimension for ILVMT labeled ScCM on an 850 MHz spectrometer is 400 ms (2048 points) with a spectral width of 3 ppm (2550 Hz), which results in a maximum resolution of 2.5 Hz that is much narrower than the expected proton linewidth for a protein. For the indirect carbon dimension on the same spectrometer, an acquisition time of ~100 ms (1048 points) with a spectral width of 25 ppm (5345 Hz) might be used as the initial value for linewidth measurement. This value corresponds to ~10 Hz of resolution. It is best to choose a resolution less than or equal to half the natural linewidth of a peak in order to properly measure the natural linewidth of that peak. For reference, the relationship between resolution, number of points, spectral width, and acquisition time is described as follows:
3.3.2. TROSY-based NOESY experiments for methyl resonance assignments
There are currently many NOESY-based experiments available for measuring NOEs between methyl groups. The experiments that will be discussed in this section take advantage of the methyl TROSY effect to have a sufficiently high S/N ratio to observe NOE cross peaks in larger proteins. Even though the methyl region of a spectrum is less crowded than the amide region, there can still be significant peak overlap. The potential for overlap is especially problematic for samples with some combination of ATLV labeling. In order to reduce ambiguity in assignments, it is desirable to measure four chemical shifts: the proton and carbon chemical shifts of the methyl groups the experiment starts on (here referred to as H1–C1), and the proton and carbon chemical shifts of the methyl groups that magnetization was transferred to during the mixing period (here referred to as H2–C2). There are two ways to accomplish this process: run two 3D experiments (Fig. 5) or run a single 4D experiment (Fig. 6).
Fig. 5.
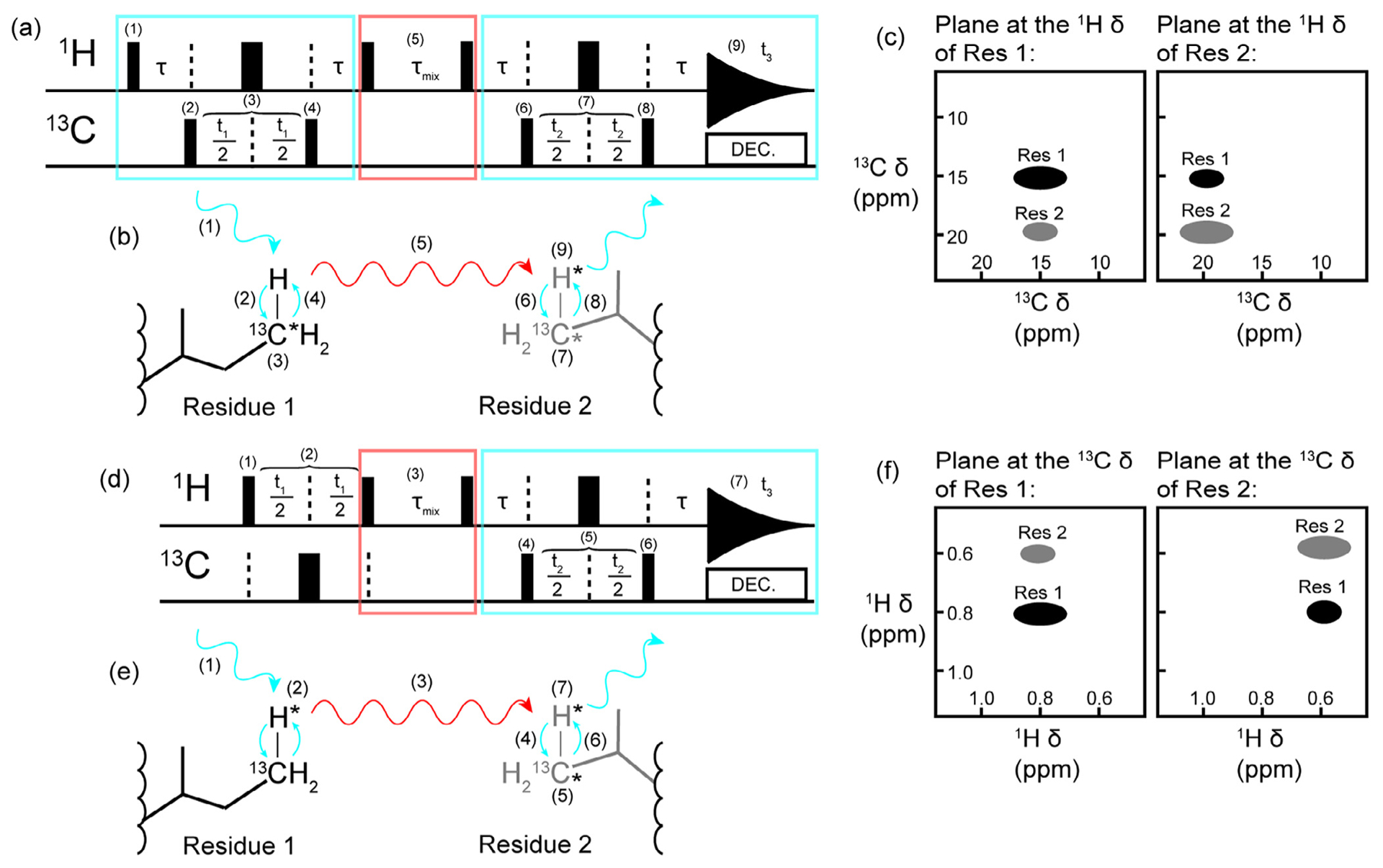
Methyl resonance assignments using two complementary 3D NOESY spectra. Schematics of a (a) a 3D CCH HMQC-NOESY-HMQC, (b) the CCH NOESY magnetization transfer pathway, (c) the associated CCH spectrum, (d) a 3D HCH NOESY-HMQC, (e) the HCH NOESY magnetization transfer pathway, and (f) the associated HCH spectrum. The HMQC elements are indicated by the blue rectangles and arrows while the NOESY elements are indicated by the red rectangles and arrows. In (a) and (d), the narrow black bars represent 90° pulses and the thick black bars represent 180° pulses. All pulses can be considered to have a phase of x. The pathway for the first HMQC block in (a) is as follows: (1) the 90° excitation pulse on the proton creates transverse magnetization which becomes anti-phase with respect to carbon during τ (τ = 1/(2JCH), (2) a 90° pulse on carbon creates multiple quantum transverse magnetization, (3) the carbon chemical shift evolves during the delay time t1 while a 180° refocusing pulse decouples the proton, (4) a 90° pulse transfers magnetization back to the proton and magnetization is returned to in-phase proton during τ. Next a 90° pulse on the proton creates in-phase longitudinal magnetization so that (5) magnetization can transfer via the NOE during τmix. This delay is followed by a 90° proton pulse which leads into another HMQC block: the τ delay creates anti-phase proton magnetization before (6) a 90° carbon pulse creates transverse multiple quantum magnetization so that (7) the carbon chemical shift can be recorded during t2 with proton decoupling achieved with a 180° refocusing pulse. Finally, (8) a 90° carbon pulse creates anti-phase proton magnetization which is refocused during τ and (9) the proton chemical shift is recorded during t3. The NOESY-HMQC depicted in (d) starts with a block to record the proton chemical shift in contrast to the HMQC-NOESY-HMQC experiments. In (d), (1) an excitation pulse creates transverse proton magnetization so that (2) the proton chemical shift can evolve during t1. Carbon decoupling is achieved with a 180° refocusing pulse. Next, the transverse proton magnetization becomes longitudinal with a 90° proton pulse before the NOE mixing period τmix (3). Following τmix, a 90° pulse creates transverse proton magnetization that becomes anti-phase with respect to carbon during τ. A 90° carbon pulse (4) then creates multiple quantum transverse magnetization so that the carbon chemical shift can be recorded during t2 (5). Proton decoupling is achieved during t2 with a 180° refocusing pulse. Anti-phase proton magnetization is created with (6) a 90° carbon pulse. The delay period τ allows the anti-phase magnetization to become in-phase and (7) the chemical shift of the second proton dimension is recorded during t2. The combination of the spectra depicted in (c) and (f) are required to keep ambiguity low when using 3D experiments for assignments instead of a single 4D experiment. We note that the pulse sequences presented are simplified and for illustrative purposes only. Readers are referred to the original manuscript for additional details (e.g. gradient pulses, phase cycling, and so on) [57].
Fig. 6.
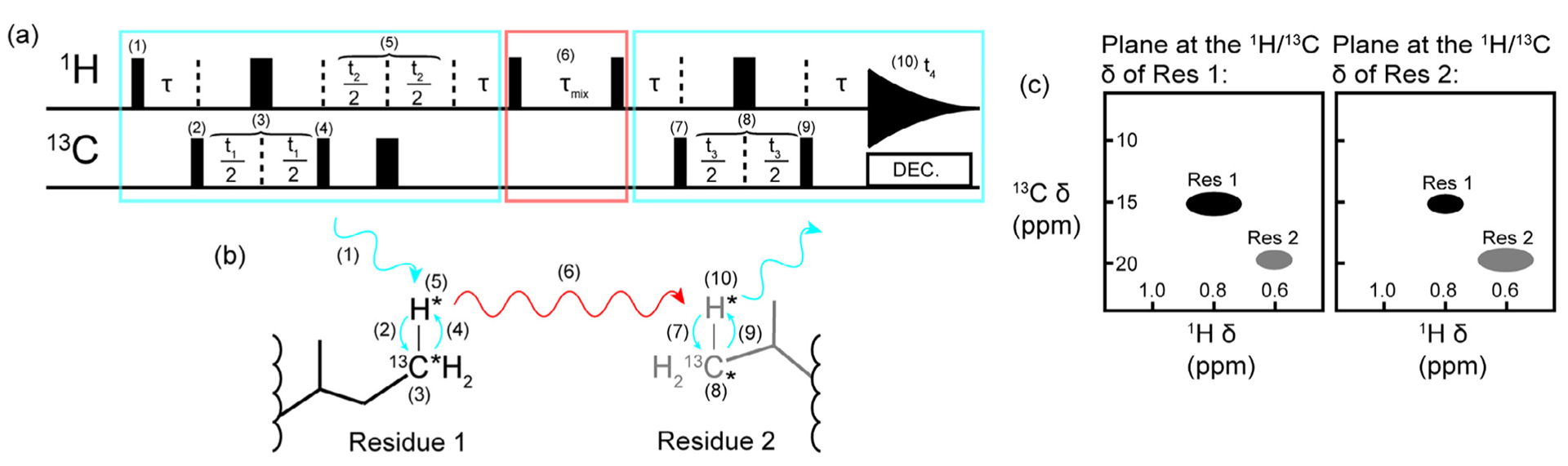
Methyl resonance assignments using 4D NOESY. (a) A schematic of a basic HMQC-NOESY-HMQC pulse sequence and (b) an approximation of the magnetization transfer pathway. Blue rectangles and arrows indicate the HMQC portions of the experiment where chemical shifts are recorded, and red arrows and rectangles indicate where magnetism is transferred through space via an NOE mechanism between the methyl groups of Residue 1 and Residue 2. The narrow black bars represent 90° pulses and the thick black bars represent 180° pulses in (a). All pulses can be considered to have a phase of x. Nuclei that have their chemical shifts recorded during the experiment are indicated by an asterisk (*) in (b). The pathway for the first HMQC block is as follows: (1) the 90° excitation pulse on the proton creates transverse magnetization which becomes anti-phase with respect to carbon during τ (τ = 1/(2JCH), (2) a 90° pulse on carbon creates multiple quantum transverse magnetization, (3) the carbon chemical shift evolves during the delay time t1 while a 180° refocusing pulse decouples the proton, (4) a 90° pulse transfers magnetization back to the proton so that the proton chemical shift can evolve during (5) t2 while a 180° pulse decouples the carbon, and magnetization is returned to in-phase proton during τ. Next a 90° pulse on the proton creates in-phase longitudinal magnetization so that (6) magnetization can transfer via the NOE during τmix. This delay is followed by a 90° proton pulse which leads into another HMQC block: the τ delay creates anti-phase proton magnetization before (7) a 90° carbon pulse creates transverse multiple quantum magnetization so that (8) the carbon chemical shift can be recorded during t3 with proton decoupling achieved with a 180° refocusing pulse. Finally, (9) a 90° carbon pulse creates anti-phase proton magnetization that is refocused during τ and (10) the final proton chemical shift is recorded during t4. (c) The expected appearance of the plane at the proton and carbon resonance frequencies of Residue 1 and Residue 2. Each residue is resolved through four dimensions, resulting in low ambiguity for assignments gained from a 4D HCCH NOESY spectrum. We note that the pulse sequence presented is simplified and for illustrative purposes only. Readers are referred to the original manuscript for additional details (e.g. gradient pulses, phase cycling, and so on) [23].
The pair of 3D NOESY experiments that should be collected is a proton-resolved NOESY-HMQC for H1, H2, and C2, and a carbon-resolved HMQC-NOESY-HMQC for C1, H2, and C2 (Fig. 5). The advantage of this method is that it is possible to collect a high-resolution dataset in less than three days for each experiment on a > 1 mM concentrated sample, especially if NUS (non-uniform sampling) or a SOFAST 3D NOESY is used. In the event of lower sample concentrations down to 500 μM, the SOFAST 3D experiments might still be sensitive enough to obtain useful data with a longer acquisition time. NUS saves time by essentially allowing for fewer points to be collected while keeping the same amount of resolution, while SOFAST pulse sequences have a very short inter-scan delay and so there is less dead time between collecting data points (see below). One advantage of using a pair of 3D experiments is that there will be less loss of signal due to field drifting, which increases with experiment time. The disadvantage is that there can still be some ambiguity even with two 3D NOESY experiments if there is a large amount of spectral overlap. The main advantage to the 4D HMQC-NOESY-HMQC experiment is that only one experiment is required to gain most of the information required for assignments, but it can require 5–7 days of experiment time for even a concentrated (> 1 mM) sample. The 4D NOESY also has the advantage of lower ambiguity because each cross peak has four associated chemical shifts rather than three.
3.3.3. The resolution problem
In theory, having a greater amount of methyl probes (e.g. as in the ILV/ILVA labeling schemes) results in many more NOEs and less ambiguity in assignments. However, there is also a greater potential for overlapping peaks. A solution to this issue is to increase the number of points in the indirect dimensions. The maximum indirect dimension evolution time should be approximately the average T2 for that dimension [56]. Sampling beyond T2 results in less and less S/N in each transient collected and is not generally recommended. For example, the average linewidth at half height in the indirect carbon dimension is ~28 Hz in an HMQC spectrum for ScCM. Linewidth gives a very rough estimate of 1/πT2. From this calculation, we determined that the maximum indirect evolution period for transverse multiple quantum CH magnetization should be ~11 ms. A 3D HCH NOESY which reached this maximum evolution period would take about a day to collect, a 3D CCH NOESY would take ~2 days, while a 4D HCCH NOESY would take ~100 days assuming a minimum four step phase cycling regime. Two ways to gain higher resolution in a reasonable amount of time are the SOFAST NOESY experiments and NUS schemes.
3.3.4. SOFAST makes collecting high-resolution datasets less expensive
Rossi et al. [57] have recently published a suite of 3D SOFAST NOESY experiments that take advantage of the methyl TROSY effect. The SOFAST experiments use an optimized flip angle (~120° for methyl groups) for the excitation pulse instead of the typical hard 90° pulse, and replace all proton pulses with bandwidth-selective pulses to excite only the region of interest. The overall result is a decrease in the effective longitudinal relaxation time, T1, and much shorter relaxation delays, typically ~0.2 s compared to up to 2 s for conventional experiments. The experiments work particularly well for large proteins due to their higher spin relaxation rates, and may not require NUS to fully sample the indirect dimensions, since transients are collected so quickly.
3.3.5. Nonuniform sampling is a necessity for 4D NOESY datasets
The number of points in an indirect dimension in NMR spectroscopy really refers to the number of delay increments used to build the free induction decay (FID) in the indirect dimension (Fig. 7). These time domain delay increments are uniformly sampled in most cases in 2D NMR spectroscopy because of much shorter collection times. When NUS is used for higher dimensional experiments, the idea is that many of the increments are unnecessary and these time domain data points can instead be accurately reconstructed at a later point to save time and have longer maximum indirect signal evolution times (Fig. 7). There are many excellent articles detailing how NUS is achieved [58–62].
Fig. 7.
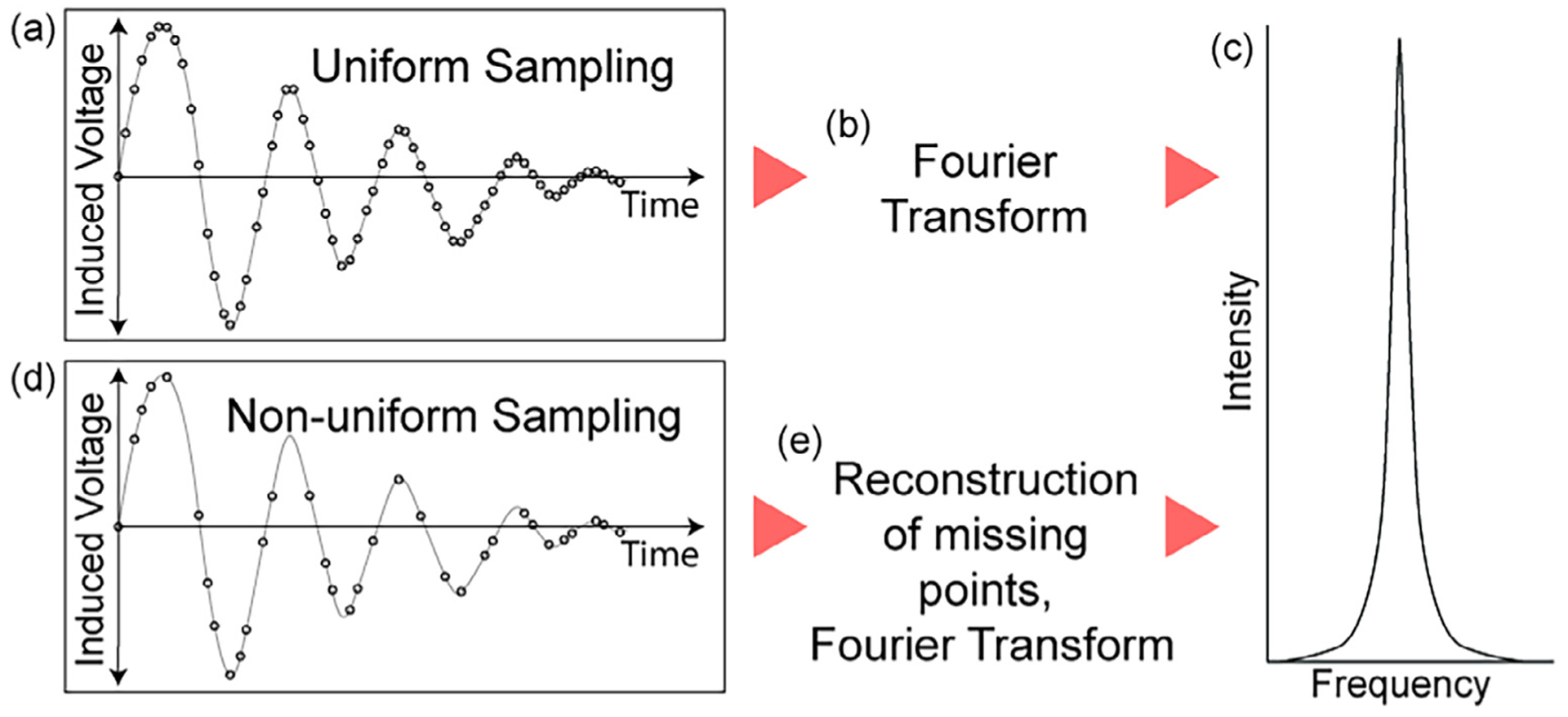
Conceptual understanding for nonuniform sampling in NMR. (a) A uniformly sampled FID (free induction decay) in the indirect dimension is straightforward to (b) Fourier transform to obtain (c) a frequency domain signal. In contrast, (d) a non-uniformly sampled FID in the indirect dimension requires fewer time increments without a loss of resolution but a potential loss of some S/N and the introduction of intense artifacts. These artifacts are mostly removed with a good reconstruction program (e), after which the final result is (c) a frequency domain signal with a similar appearance to the uniformly sampled signal.
Tugarinov et al. [23] introduced the use of high-resolution 4D HCCH NOESY datasets in 2005 by using NUS and multidimensional decomposition (MDD) for reconstruction. Prior to that publication, the wide use of a 4D HCCH NOESY for assignments was impractical due to the prohibitively long acquisition time. Since then there have been a number of different NUS sampling schemes, guidelines for setting up experiments, and reconstruction methods with their own strengths and weaknesses [58–62]. What has been easiest to implement for us and which has provided high quality datasets is the use of Bruker’s random sampling scheme, which can be exponentially weighted for S/N gains in non-constant time dimensions, and SMILE [56] (Spare Multidimensional Iterative Lineshape-Enhanced reconstruction) for reconstruction of the indirect dimensions. The main advantage of either exponentially weighted or non-weighted random sampling is fewer artifacts than other methods such as Poisson-Gap sampling, although Poisson-Gap sampling tends to result in modestly higher S/N [59]. The random sampling scheme is also very convenient because it is included in Bruker Topspin 3.0 and above. The main advantages of using SMILE are that it is relatively fast and easy to use, there is an in-depth manual [63] with many examples, and the resulting dataset is often very similar in appearance to a fully sampled dataset. In the publication associated with SMILE, Ying et al. thoroughly reviewed SMILE’s performance with random, exponentially weighted and Poisson-Gap weighted sampling schemes and found that the random and exponentially weighted schemes had the best performance in terms of reconstruction of peak position, intensity, number of peaks and artifact suppression [56]. These advantages are especially crucial for NOESY experiments that have a very high dynamic range between the self-peaks and the NOE cross-peaks.
When setting up a 4D NUS experiment, it is best to use the minimum number of scans required by the phase cycling and instead increase the number of points sampled to the maximum allowed for the reserved spectrometer time. The signal will increase with the square root of the number of transients so long as the maximum indirect evolution time is less than T2, so by spreading those transients over more sampled points, instead of number of scans, the number of artifacts in the reconstructed spectrum will be lower and the S/N higher. The number of unique time increment transients is much more important than the total percentage of indirect hypercomplex points sampled. At the same time, good reconstruction requires that the number of indirect points be greater than the number of expected peaks and cross-peaks. We suggest that most 3D experiments be scheduled for ~2–3 days and 4D experiments be scheduled for ~5–7 days, erring on a longer collection period for sample concentrations < 1 mM. In general, longer experiments (within the suggested timeframes) and higher sample concentrations lead to better data, barring any aggregation or other sample degradation issues. We have some other suggestions to consider when running these multi-day experiments in the Supplementary Material.
3.3.6. Optimization of the NOE mixing time
The NOE mixing time is the period when magnetization is transferred between nuclei through space. There is a sample-dependent optimal amount of time for NOE buildup after which the signal will start to decay. For a protein molecule, the methyl-methyl NOE mixing time typically ranges from 100 ms to 500 ms, though shorter mixing times might be preferable for observing intra-residue NOEs. Larger proteins with longer tumbling times and higher densities of incorporated methyl groups tend to have a shorter optimal NOE mixing times, while smaller proteins with higher levels of overall deuteration tend to have longer optimal NOE mixing times. Additionally, methyl probes located further apart will require a longer mixing time in order to observe NOE cross-peaks. The mixing time should be optimized for each protein sample by creating a series of short 3D or 4D methyl NOESY experiments that vary the mixing time but have only one point in each carbon dimension. The mixing time that results in the greatest number of cross peaks should likely be used for the full 3D or 4D experiment.
3.3.7. 4D HCCH NOESY on the ILVMT labeled chorismate mutase
The pulse sequence used to collect the ILVMT labeled ScCM HCCH NOESY spectrum was obtained from the Bax lab, but permission has been granted to distribute this pulse sequence upon request. Data was collected on a Bruker Avance III 850 MHz equipped with a cryoprobe and operated with Topspin 3.2 software. In the F4 (direct proton), F3 (indirect proton), F2 (indirect carbon), and F1 (indirect carbon) dimensions, 1536 × 88 × 96 × 96 points were indicated corresponding to maximum evolution times of 70 ms, 20 ms, 11 ms, and 11 ms respectively. Of the potential 811,008 (88 × 96 × 96) indirect points, 7400 (7.3%) randomly selected points were collected over a period of 130 h collection time. The spectral widths were 13 ppm (F4), 2.6 ppm (F3), 20 ppm (F2), and 20 ppm (F1). A relaxation delay of 1.67 s and an NOE mixing period of 175 ms were used. The sample conditions were 1.8 mM ScCM (monomer concentration) in 50 mM sodium phosphate buffer pH 6.8 with 2 mM β-mercaptoethanol, 0.02% sodium azide, and 5 mM tryptophan in 2H2O in a 4 mm NMR tube, and the spectrum was collected at 35 °C.
3.4. NMR data processing for NOESY-based methyl assignments
The two most widely used methods for processing NMR data collected on currently available Bruker instruments are TopSpin and NMRPipe. In our experience NMRPipe produces superior results and handles reconstruction of NUS data more effectively with the built-in SMILE plugin. We have detailed our NMR data processing scripts in the Supplementary Material. For more examples and information on the use of NMRPipe and SMILE, we refer to the detailed manuals available online or the initial publications for each [56,64]. For easy access to all of the software described here, we recommend creating an NMRbox account on NMRbox.org for access to a virtual machine [65]. NMRbox is a cloud-based computing platform currently in its fourth iteration which gives each user access to 32 CPUs, 188 GBs of RAM, and 2 Nvidia GPUs [66].
3.5. NMR data analysis for NOESY-based methyl assignments
At this point, we have covered the data collection stages of assigning methyl resonances using NOESY. The data available includes methyl-methyl NOEs, intramethyl LV NOEs, residue type information, and a 3D protein structure. To finally obtain confident assignments one must go through the iterative process of “walking” through the NOE data for tentative assignments, compare these assignments with those made with the latest semi-automated methyl assignment tools, and potentially perform mutagenesis and analyze protein variants to validate any uncertain assignments.
3.5.1. Methyl walk through NOESY data
One method to assign methyl resonances based on NOESY data, especially for those datasets with low complexity and ambiguity, is the manual methyl walk strategy [48]. In this case, the NOESY dataset is compared with a 2D reference spectrum, and reference spectra for individual amino acid types, so that a “walk” from one methyl resonance to the next through an NOE network can be conducted (Fig. 3). Another 3D NOE spectrum containing information on LV geminal methyl signal pairings should also be viewed if geminal methyl pair information is desired. By comparing the NOE network to a 3D protein structure, and keeping in mind that the typical NOE cutoff should be ~10 Å, full methyl assignments can be potentially obtained.
The initial assignment process for ScCM involved a manual methyl walk strategy. Since ScCM has only three methionine residues per monomer, and these are each expected to have very unique NOE patterns, we decided to start the walk from these methionines. The walk consisted of comparing the stronger NOEs in the 4D NOESY viewed in CCPNMR to the 1CSM X-ray crystal structure of ScCM viewed in the protein molecular modeling program Chimera [67]. The expected NOE cross-peaks can be ascertained from the 3D protein structure and compared to the experimental data (Fig. 3).
If many methyl residues are labeled, the resulting NOE network can become very complex. In this case, we recommend the use of one of the newer automated methyl assignment programs, MAGMA from the Baldwin lab [68] or MAGIC from the Kalodimos lab [51]. What follows is a brief explanation of the theory and use of MAGMA, as well as example inputs and outputs from data worked up from a 4D ScCM HCCH NOESY dataset.
3.5.2. Theoretical basis for MAGMA
MAGMA, or Methyl Assignment by Graph MAtching, is a python-based program that uses graph theory to determine methyl resonance assignments. The driving philosophy behind MAGMA is that even a single incorrect resonance assignment is enough to derail assignments, and so its goal is to return only correct assignments and indicate any ambiguous assignments with multiple possibilities [68]. In its current form, the required inputs are the 3D protein structure and the NOE peak network information gained from associating NOE peaks with self, or diagonal, peak in 3/4D methyl NOESY datasets. The MAGMA algorithm treats the NOE cross-peaks from NMR data as one graph, G1, and attempts to fit this network to a graph of potential methyl NOEs from the 3D protein structure, G2 (Fig. 8) [68]. However, for a large system with many methyl peaks, calculation time speeds up considerably with residue type information, information on whether LV peaks are proR or proS, the geminal methyl LV pairings, and residues that were assigned through mutagenesis.
Fig. 8.
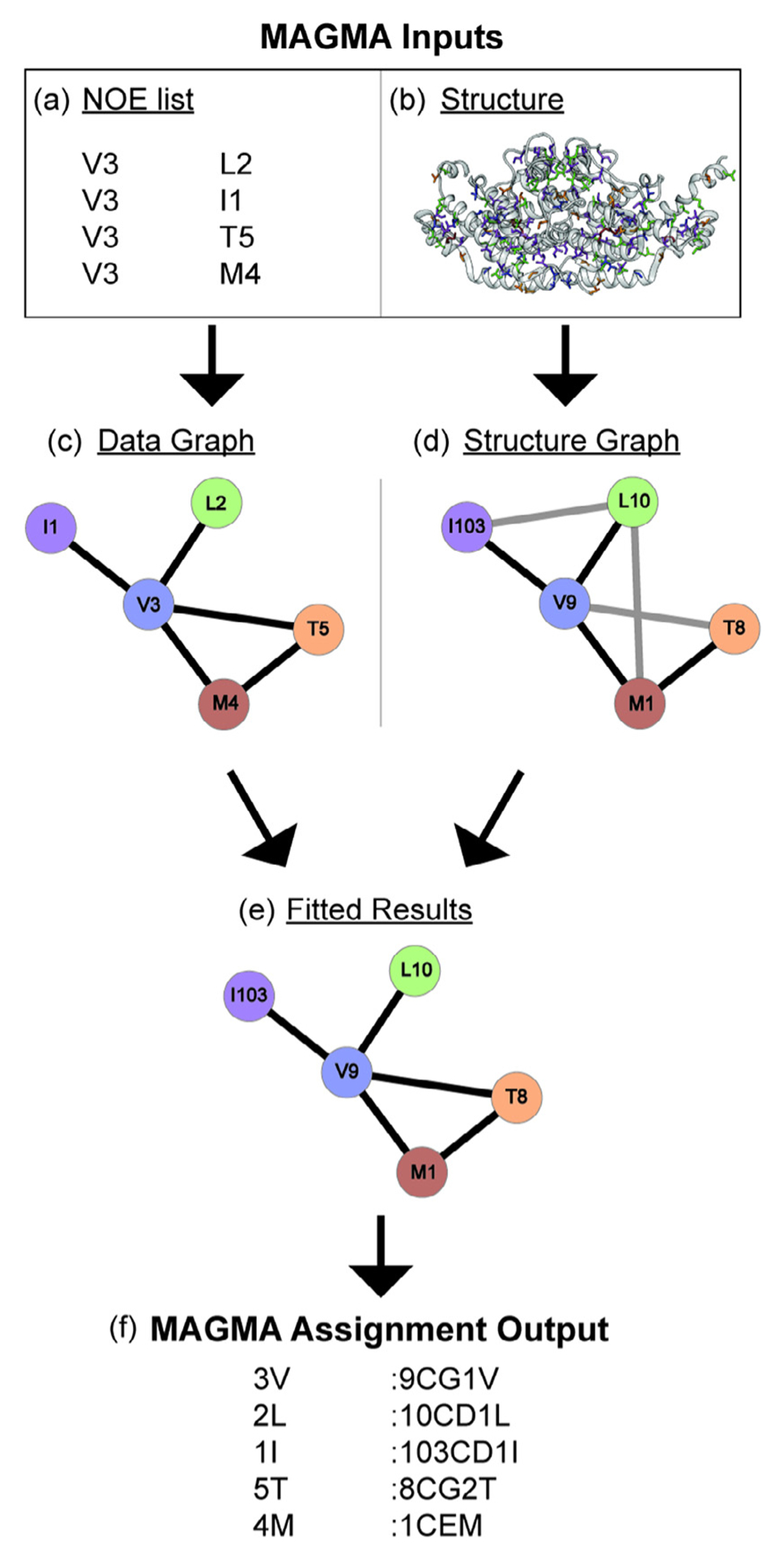
A basic explanation for the use of MAGMA to gain resonance assignments. MAGMA takes a (a) list of peaks with residue types and peak names that have the diagonal peaks in one column and the associated NOE cross-peak residue type and peak names from another column, and (b) a 3D structure as its inputs. A data graph (c) is created from the NOE list showing all observed NOE interactions and (d) a structure graph is created from the structure showing all possible NOE interactions. NOE interactions not present in both graphs are depicted with gray lines and interactions present in both graphs are depicted with black lines. (e) The data graph and structure graph are then fit to associate the peak names from the NOE list with assignments, and (f) MAGMA creates a text file with these assignments as its output with the peak name in the first column and the residue identity in the second column.
MAGMA was able to consistently outperform the Monte Carlo-based methyl assignment programs MAP-XSII [69] and FLAMEnGO2.0 [70] in terms of accuracy, and performed similarly in terms of total number of assignments [68]. The systems used for these benchmarks were methyl labeled ubiquitin, MsrB, the ATCase R2 dimer, MBP, MSG, and the α7α7 double ring of the proteasome. Pritišanac et al. attributed the lower accuracy of FLAMEnGO2.0 and MAP-XSII to their use of chemical shifts in assignment calculations, which is difficult to implement properly for methyl groups due to the high mobility of side chains and the narrow chemical shift range for methyl groups [68]. Nonetheless, the associated research groups are continuing to update their algorithms, and so, best practices may change over time.
3.5.3. Practical aspects of MAGMA
The workflow for creating MAGMA inputs is as follows. First, all of the peaks in the 2D correlation spectrum should be labeled with the letter for the residue type and a unique number. Next, peaks are ‘picked’ in the 3D or 4D NOESY spectrum (e.g. in Sparky or CCPNMR), where it might be necessary to manually curate the lists by removing potential artifact peaks and/or any peaks that are not reciprocated. This conservative peak picking strategy helps to ensure that MAGMA only considers real NOE constraints when fitting the NOE list to the 3D protein structure. Finally, the NOE peaks are associated such that the unique number for that peak in the 2D spectrum will correlate with the unique number for the diagonal peak. The NOE list will then need to be formatted in such a way that it can be read by MAGMA. The reformatting of the peak list can be done either using a python or MATLAB script, or even manually in Excel.
It is strongly recommended that if a sample was used where both the proR and the proS methyl groups of leucines and/or valines were labeled, a short mixing time (~40 ms) NOESY experiment [51] is run to show which LV methyls belong to the same residue. The LV geminal methyl groups should then be merged in the MAGMA input. Doing this merger will significantly cut down on calculation time because MAGMA will treat the pair of methyl groups as a single pseudo-atom [68]. The merging of LV pairs was particularly important for ScCM as the MAGMA calculations did not initially run to completion due to the complexity of the racemic LV labeling in the ILVMT labeling scheme.
The 3D protein structure used as MAGMA input should ideally have resulted from sample conditions similar to those under which the NOESY data was collected. For ScCM, the best data fits and assignments in MAGMA were obtained with the tryptophan-bound PDB 1CSM X-ray crystal structure [71] since the 4D NOESY was obtained with tryptophan in the sample buffer. Additionally, all potential assignments for the methyl residues should show up in the structure. The 1CSM structure contains a threonine to isoleucine point mutation at position 226 that had to be changed back to threonine in the biomolecular structure visualization program Chimera [67]. MAGMA will output two useful lists once the calculation is complete: confident.res and combine-dResults.res. The former contains only the assignments that MAGMA is most certain are correct (Fig. 8). The latter list also contains information on assignments of which MAGMA is not certain that will show two or more possible assignments for these peaks. This information is helpful for determining which peaks to potentially assign via mutagenesis. These mutagenesis assignments can then be fixed in MAGMA using craic mode, potentially reducing the ambiguity for other assignments as well. For more detailed information on the use of MAGMA, we direct the reader to the associated manuscript by Pritišanac et al. [68] and the detailed instruction manual that comes in a compressed document with MAGMA.
As an example, the initial MAGMA output for ScCM indicated ambiguity in the assignment of I74 and I98. Two peaks in the NOE graph could be fit equally well to either I74 or I98. In order to reduce this ambiguity the I74L variant was produced and used to confirm the identity of both peaks (Fig. 9). These mutagenesis-based assignments were then fixed in later runs of MAGMA by including a list with the arbitrary peak number and its actual assignment in a format identical to the output format (Fig. 8) and turning on craic mode. Using this methodology of the manual methyl walk, MAGMA, and mutagenesis to confirm differences between the MAGMA and methyl walk results, all methionine, threonine, and isoleucine peaks for ScCM were confidently assigned (Fig. 10). Additional experiments and analyses are necessary to confirm the valine and leucine assignments due to the racemic labeling scheme used.
Fig. 9.
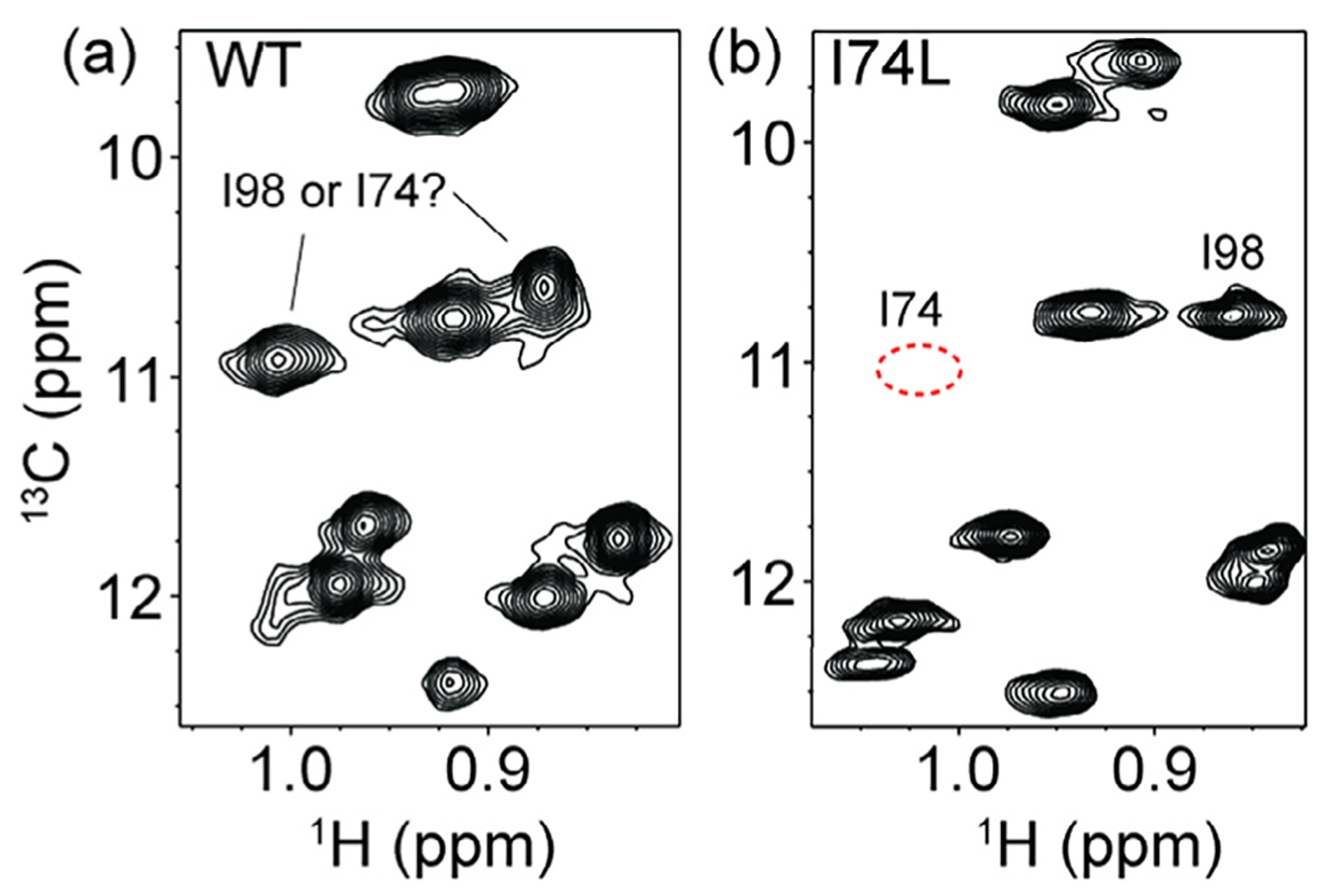
Resolving ambiguous assignments through site-directed mutagenesis. A comparison between 1H–13C 2D NMR spectra of (a) wild-type ScCM and (b) the I74L variant of ScCM. The peaks that could be assigned to either I74 or I98 based on MAGMA results are indicated in (a). (b) The I74L variant effectively removes the I74 resonance, such that the resonances of I74 (i.e. the expected resonance position is indicated by the red dashed circle) and I98 could be assigned. However, other isoleucine resonances had slight changes in position due to the I74L substitution, indicating a potential pitfall for relying on mutagenesis alone for assignments, especially in crowded areas of the spectrum.
Fig. 10.

Tryptophan-bound ScCM methyl resonance assignments for isoleucine δ1, threonine, and methionine methyl groups. Resonances with an asterisk (*) were assigned or confirmed through mutagenesis, while all others were assigned by the manual methyl walk and MAGMA results from the 4D NOESY dataset.
4. Summary
Solution-state NMR experiments on methyl groups have brought great insight into the structure and function of proteins. Here, we have used our experiences with assigning the methyl resonances of aromatic amino acid biosynthetic enzymes to help guide similar studies on other proteins of interest. Further advances in pulse sequence development, data analysis and alternative methods may make methyl assignments possible for even larger protein complexes or proteins under less optimal conditions (e.g. lower concentrations, less stable, membrane proteins, and so on).
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jinfa Ying for providing the methyl NOESY pulse sequence and for help setting up the methyl NOESY experiment and SMILE scripts, Dr. Frank Delaglio for technical support with NMRpipe and Dr. Andrew Baldwin for help with MAGMA. The authors would also like to thank Dr. Erik Cook, Rebecca D’Amico, Grace Usher and Dennis Winston for feedback on this manuscript. This work was supported by the National Science Foundation [MCB-1615032]; and the National Institutes of Health [R01AI104878].
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ymeth.2018.06.010.
References
- [1].Gardner KH, Kay LE, Production and incorporation of 15N, 13C, 2H (1H-δ1 methyl) isoleucine into proteins for multidimensional NMR studies, J. Am. Chem. Soc 119 (1997) 7599–7600, 10.1021/ja9706514. [DOI] [Google Scholar]
- [2].Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE, A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methylprotonated15N-,13C-,2H-labeled proteins, J. Biomol. NMR 13 (1999) 369–374, 10.1023/A:1008393201236. [DOI] [PubMed] [Google Scholar]
- [3].Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE, Cross-correlated relaxation enhanced 1H–13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes, J. Am. Chem. Soc 125 (2003) 10420–10428, 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- [4].Tugarinov V, Kay LE, An isotope labeling strategy for methyl TROSY spectroscopy, J. Biomol. NMR 28 (2004) 165–172, 10.1023/B:JNMR.0000013824.93994.1f. [DOI] [PubMed] [Google Scholar]
- [5].Sprangers R, Kay LE, Quantitative dynamics and binding studies of the 20S proteasome by NMR, Nature 445 (2007) 618–622, 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- [6].Religa TL, Sprangers R, Kay LE, Dynamic regulation of archaeal proteasome gate opening as studied by trosy NMR, Science (80-) 328 (2010) 98–102, 10.1126/science.184991. [DOI] [PubMed] [Google Scholar]
- [7].Rosenzweig R, Kay LE, Bringing dynamic molecular machines into focus by methyl-TROSY NMR, Annu. Rev. Biochem 83 (2014) 291–315, 10.1146/annurev-biochem-060713-035829. [DOI] [PubMed] [Google Scholar]
- [8].Kerfah R, Plevin MJ, Sounier R, Gans P, Boisbouvier J, Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins, Curr. Opin. Struct. Biol 32 (2015) 113–122, 10.1016/j.sbi.2015.03.009. [DOI] [PubMed] [Google Scholar]
- [9].Wiesner S, Sprangers R, Methyl groups as NMR probes for biomolecular interactions, Curr. Opin. Struct. Biol 35 (2015) 60–67, 10.1016/j.sbi.2015.08.010. [DOI] [PubMed] [Google Scholar]
- [10].Gerstein M, A structural census of genomes: comparing bacterial, eukaryotic, and archaeal genomes in terms of protein structure, J. Mol. Biol 274 (1997) 562–576, 10.1006/jmbi.1997.1412. [DOI] [PubMed] [Google Scholar]
- [11].Fawzi NL, Libich DS, Ying J, Tugarinov V, Clore GM, Characterizing methyl-bearing side chain contacts and dynamics mediating amyloid β protofibril interactions using 13C(methyl)-DEST and lifetime line broadening, Angew. Chem. Int. Ed. Engl 53 (2014) 10345–10349, 10.1002/anie.201405180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bouvignies G, Kay LE, A 2D 13C-CEST experiment for studying slowly exchanging protein systems using methyl probes: An application to protein folding, J. Biomol. NMR 53 (2012) 303–310, 10.1007/s10858-012-9640-7. [DOI] [PubMed] [Google Scholar]
- [13].Rennella E, Huang R, Velyvis A, Kay LE, 13CHD2-CEST NMR spectroscopy provides an avenue for studies of conformational exchange in high molecular weight proteins, J. Biomol. NMR 63 (2015) 187–199, 10.1007/s10858-015-9974-z. [DOI] [PubMed] [Google Scholar]
- [14].Yuwen T, Huang R, Kay LE, Probing slow timescale dynamics in proteins using methyl 1H CEST, J. Biomol. NMR 68 (2017) 215–224, 10.1007/s10858-017-0121-x. [DOI] [PubMed] [Google Scholar]
- [15].Lundström P, Vallurupalli P, Religa TL, Dahlquist FW, Kay LE, A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity, J. Biomol. NMR 38 (2007) 79–88, 10.1007/s10858-007-9149-7. [DOI] [PubMed] [Google Scholar]
- [16].Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE, Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme, J. Am. Chem. Soc 126 (2004) 3964–3973, 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- [17].Yuwen T, Vallurupalli P, Kay LE, Enhancing the sensitivity of CPMG relaxation dispersion to conformational exchange processes by multiple-quantum spectroscopy, Angew. Chem. Int. Ed 55 (2016) 11490–11494, 10.1002/anie.201605843. [DOI] [PubMed] [Google Scholar]
- [18].Ceccon A, Marius Clore G, Tugarinov V, Towards interpretation of intermolecular paramagnetic relaxation enhancement outside the fast exchange limit, J. Biomol. NMR 66 (2016) 1–7, 10.1007/s10858-016-0053-x. [DOI] [PubMed] [Google Scholar]
- [19].Schanda P, Kupĉe E, Brutscher B, SOFAST-HMQC experiments for recording two-dimensional deteronuclear correlation spectra of proteins within a few seconds, J. Biomol. NMR 33 (2005) 199–211, 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- [20].Jiang Y, Kalodimos CG, NMR studies of large proteins, J. Mol. Biol 429 (2017) 2667–2676, 10.1016/j.jmb.2017.07.007. [DOI] [PubMed] [Google Scholar]
- [21].Vadrevu R, Falzone CJ, Matthews CR, Partial NMR assignments and secondary structure mapping of the isolated alpha subunit of Escherichia coli tryptophan synthase, a 29-kD TIM barrel protein, Protein Sci 12 (2003) 185–191, 10.1110/ps.0221103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Axe JM, Boehr DD, Long-range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling, J. Mol. Biol 425 (2013), 10.1016/j.jmb.2013.01.030. [DOI] [PubMed] [Google Scholar]
- [23].Tugarinov V, Kay LE, Ibraghimov I, Orekhov VY, High-resolution four-dimensional 1H–13C NOE spectroscopy using methyl-TROSY, sparse data acquisition, and multidimensional decomposition, J. Am. Chem. Soc 127 (2005) 2767–2775, 10.1021/ja044032o. [DOI] [PubMed] [Google Scholar]
- [24].Sträter N, Schnappauf G, Braus G, Lipscomb WN, Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures, Structure 5 (1997) 1437–1452, 10.1016/S0969-2126(97)00294-3. [DOI] [PubMed] [Google Scholar]
- [25].Yang X, Welch JL, Arnold JJ, Boehr DD, Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance, Biochemistry 49 (2010) 9361–9371, 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Amero C, Asunción Durá M, Noirclerc-Savoye M, Perollier A, Gallet B, Plevin MJ, Vernet T, Franzetti B, Boisbouvier J, A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies, J. Biomol. NMR 50 (2011) 229–236, 10.1007/s10858-011-9513-5. [DOI] [PubMed] [Google Scholar]
- [27].Venditti V, Fawzi NL, Clore GM, Automated sequence- and stereo-specific assignment of methyl-labeled proteins by paramagnetic relaxation and methyl-methyl nuclear overhauser enhancement spectroscopy, J. Biomol. NMR 51 (2011) 319–328, 10.1007/s10858-011-9559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lescanne M, Skinner SP, Blok A, Timmer M, Cerofolini L, Fragai M, Luchinat C, Ubbink M, Methyl group assignment using pseudocontact shifts with PARAssign, J. Biomol. NMR 69 (2017) 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grzesiekt S, Bax A, Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR, J. Am. Chem. Soc 114 (1992) 6291–6293, 10.1021/ja00042a003. [DOI] [Google Scholar]
- [30].Wittekind M, Mueller L, HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins, J. Magn. Reson. Ser. B 101 (1993) 201–205, 10.1006/jmrb.1993.1033. [DOI] [Google Scholar]
- [31].Muhandiram DR, Kay LE, Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity, J. Magn. Reson. Ser. B 103 (1994) 203–216, 10.1006/jmrb.1994.1032. [DOI] [Google Scholar]
- [32].Yamazaki T, Kay LE, Lee W, Revington M, Arrowsmith CH, Mattiello DL, Dahlquist FW, An HNCA pulse scheme for the backbone assignment of 15N,13C,2H-labeled proteins: application to a 37-kDa Trp repressor-DNA complex, J. Am. Chem. Soc 116 (1994) 6464–6465, 10.1021/ja00093a069. [DOI] [Google Scholar]
- [33].Kay LE, Ikura M, Bax A, Proton-proton correlation via carbon-carbon couplings: a three-dimensional NMR approach for the assignment of aliphatic resonances in proteins labeled with carbon-13, J. Am. Chem. Soc 112 (1990) 888–889, 10.1021/ja00158a070. [DOI] [Google Scholar]
- [34].Montelione GT, Lyons BA, Emerson SD, Tashiro M, Montelione GT, Tashiro M, An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins, J. Am. Chem. Soc 114 (1992) 10974–10975, 10.1021/ja00053a051. [DOI] [Google Scholar]
- [35].Dunn MF, Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex, Arch. Biochem. Biophys 519 (2012) 154–166, 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Axe JM, O’Rourke KF, Kerstetter NE, Yezdimer EM, Chan YM, Chasin A, Boehr DD, Severing of a hydrogen bond disrupts amino acid networks in the catalytically active state of the alpha subunit of tryptophan synthase, Protein Sci 24 (2015), 10.1002/pro.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Axe JM, Yezdimer EM, Orourke KF, Kerstetter NE, You W, Chang CEA, Boehr DD, Amino acid networks in a (β/α)8 barrel enzyme change during catalytic turnover, J. Am. Chem. Soc 136 (2014) 6818–6821, 10.1021/ja501602t. [DOI] [PubMed] [Google Scholar]
- [38].Tugarinov V, Kay LE, Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods, J. Am. Chem. Soc 125 (2003) 13868–13878, 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- [39].Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K, TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins, J. Am. Chem. Soc 121 (1999) 844–848, 10.1021/ja9834226. [DOI] [Google Scholar]
- [40].Hu K, Eletsky A, Pervushin K, Backbone resonance assignment in large protonated proteins using a combination of new 3D TROSY-HN(CA)HA, 4D TROSY-HACANH and 13C-detected HACACO experiments, J. Biomol. NMR 26 (2003) 69–77, 10.1023/A:1023008719248. [DOI] [PubMed] [Google Scholar]
- [41].Ritter C, Lührs T, Kwiatkowski W, Riek R, 3D TROSY-HNCA coded CB and TROSY-HNCA coded CO experiments: triple resonance NMR experiments with two sequential connectivity pathways and high sensitivity, J. Biomol. NMR 28 (2004) 289–294, 10.1023/B:JNMR.0000013698.89582.dc. [DOI] [PubMed] [Google Scholar]
- [42].Zhu G, Bax A, Improved linear prediction for truncated signals of known phase, J. Magn. Reson 90 (1990) 405–410, 10.1016/0022-2364(90)90150-8. [DOI] [Google Scholar]
- [43].Zhu G, Bax A, Two-dimensional linear prediction for signals truncated in both dimensions, J. Magn. Reson 98 (1992) 192–199, 10.1016/0022-2364(92)90124-P. [DOI] [Google Scholar]
- [44].Lundstrom P, Ahlner A, Blissing AT, Isotope labeling methods for large systems, Adv. Exp. Med. Biol 992 (2012) 3–15, 10.1007/978-94-007-4954-2_1. [DOI] [PubMed] [Google Scholar]
- [45].Kerfah R, Plevin MJ, Pessey O, Hamelin O, Gans P, Boisbouvier J, Scrambling free combinatorial labeling of alanine-β, isoleucine-δ1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs, J. Biomol. NMR 61 (2015) 73–82, 10.1007/s10858-014-9887-2. [DOI] [PubMed] [Google Scholar]
- [46].Velyvis A, Ruschak AM, Kay LE, An economical method for production of 2H,13CH3-threonine for solution NMR studies of large protein complexes: application to the 670 kDa proteasome, PLoS One 7 (2012) 1–8, 10.1371/journal.pone.0043725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Saio T, Guan X, Rossi P, Economou A, Kalodimos CG, Structural basis for protein antiaggregation activity of the trigger factor chaperone, Science (80-.) 344 (2014) 597–610, 10.1126/science.1254064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Proudfoot A, Frank AO, Ruggiu F, Mamo M, Lingel A, Facilitating unambiguous NMR assignments and enabling higher probe density through selective labeling of all methyl containing amino acids, J. Biomol. NMR 65 (2016) 15–27, 10.1007/s10858-016-0032-2. [DOI] [PubMed] [Google Scholar]
- [49].Ayala I, Sounier R, Usé N, Gans P, Boisbouvier J, An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein, J. Biomol. NMR 43 (2009) 111–119, 10.1007/s10858-008-9294-7. [DOI] [PubMed] [Google Scholar]
- [50].Gans P, Hamelin O, Sounier R, Ayala I, Durá MA, Amero CD, Noirclerc-Savoye M, Franzetti B, Plevin MJ, Boisbouvier J, Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins, Angew. Chem. Int. Ed 49 (2010) 1958–1962, 10.1002/anie.200905660. [DOI] [PubMed] [Google Scholar]
- [51].Monneau YR, Rossi P, Bhaumik A, Huang C, Jiang Y, Saleh T, Xie T, Xing Q, Kalodimos CG, Automatic methyl assignment in large proteins by the MAGIC algorithm, J. Biomol. NMR 69 (2017) 215–227, 10.1007/s10858-017-0149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tzeng S-R, Pai M-T, Kalodimos CG, NMR studies of large protein systems, Methods Mol. Biol (2012) 133–140, 10.1007/978-1-61779-480-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin SL, Xu D, Li A, Nussinov R, Electrostatics, allostery, and activity of the yeast chorismate mutase, Proteins Struct. Funct. Genet 31 (1998) 445–452, . [DOI] [PubMed] [Google Scholar]
- [54].Tugarinov V, Kanelis V, Kay LE, Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy, Nat. Protoc 1 (2006) 749–754, 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- [55].Marley J, Lu M, Bracken C, A method for efficient isotopic labeling of recombinant proteins, J. Biomol. NMR 20 (2001) 71–75, 10.1023/A:1011254402785. [DOI] [PubMed] [Google Scholar]
- [56].Ying J, Delaglio F, Torchia DA, Bax A, Sparse multidimensional iterative line-shape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data, J. Biomol. NMR 68 (2017) 101–118, 10.1007/s10858-016-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rossi P, Xia Y, Khanra N, Veglia G, Kalodimos CG, 15N and 13C-SOFASTHMQC editing enhances 3D-NOESY sensitivity in highly deuterated, selectively [1H,13C]-labeled proteins, J. Biomol. NMR 66 (2016) 259–271, 10.1007/s10858-016-0074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schuyler AD, Maciejewski MW, Stern AS, Hoch JC, Nonuniform sampling of hypercomplex multidimensional NMR experiments: dimensionality, quadrature phase and randomization, J. Magn. Reson 254 (2015) 121–130, 10.1002/aur.1474.Replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hyberts SG, Arthanari H, Wagner G, Applications of non-uniform sampling and processing, Top. Curr. Chem 316 (2012) 125–148, 10.1007/128_2011_187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Linnet TE, Teilum K, Non-uniform sampling of NMR relaxation data, J. Biomol. NMR 64 (2016) 165–173, 10.1007/s10858-016-0020-6. [DOI] [PubMed] [Google Scholar]
- [61].Hyberts SG, Arthanari H, Robson SA, Wagner G, Perspectives in magnetic resonance: NMR in the post-FFT era, J. Magn. Reson 241 (2014) 60–73, 10.1016/j.jmr.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hyberts SG, Robson SA, Wagner G, Exploring signal-to-noise ratio and sensitivity in non-uniformly sampled multi-dimensional NMR spectra, J. Biomol. NMR 55 (2013) 167–178, 10.1007/s10858-012-9698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ying J, Bax A, SMILE User’s Manual, (2016). https://spin.niddk.nih.gov/bax/software/SMILE/smile_manual.pdf.
- [64].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6 (1995) 277–293, 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- [65].Maciejewski MW, Schuyler AD, Gryk MR, Moraru II, Romero PR, Ulrich EL, Eghbalnia HR, Livny M, Delaglio F, Hoch JC, NMRbox: a resource for biomolecular NMR computation, Biophys. J 112 (2017) 1529–1534, 10.1016/j.bpj.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].NMRbox, (n.d.). https://www.nmrbox.org/.
- [67].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera – a visualization system for exploratory research and analysis, J. Comput. Chem 25 (2004) 1605–1612, 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [68].Pritišanac I, Degiacomi MT, Alderson TR, Carneiro MG, Ab E, Siegal G, Baldwin AJ, Automatic assignment of methyl-NMR spectra of supramolecular machines using graph theory, J. Am. Chem. Soc 139 (2017) 9523–9533, 10.1021/jacs.6b11358. [DOI] [PubMed] [Google Scholar]
- [69].Xu Y, Matthews S, MAP-XSII: an improved program for the automatic assignment of methyl resonances in large proteins, J. Biomol. NMR 55 (2013) 179–187, 10.1007/s10858-012-9700-z. [DOI] [PubMed] [Google Scholar]
- [70].Chao FA, Kim J, Xia Y, Milligan M, Rowe N, Veglia G, FLAMEnGO 2.0: an enhanced fuzzy logic algorithm for structure-based assignment of methyl group resonances, J. Magn. Reson 245 (2014) 17–23, 10.1016/j.jmr.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xue Y, Lipscomb WNWN, Graf R, Schnappauf G, Braus G, Graft R, Schnappauft G, Braus G, The crystal structure of allosteric chorismate mutase at 2.2-A resolution, Proc. Natl. Acad. Sci 91 (1994) 10814–10818, 10.2210/PDB1CSM/PDB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gelis I, Bonvin AMJJ, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG, Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR, Cell. 131 (2007) 756–769, 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.