

Abstract
ADP-ribosylation (ADPr) is a post-translational modification that plays pivotal roles in a wide range of cellular processes. Mass spectrometry (MS)-based analysis of ADPr under physiological conditions, without relying on genetic or chemical perturbation, has been hindered by technical limitations. Here, we describe the applicability of activated ion electron transfer dissociation (AI-ETD) for MS-based proteomics analysis of physiological ADPr using our unbiased Af1521 enrichment strategy. To benchmark AI-ETD, we profile 9,000 ADPr peptides mapping to >5,000 unique ADPr sites from a limited number of cells exposed to oxidative stress and identify 120% and 28% more ADPr peptides compared to contemporary strategies using ETD and electron-transfer higher-energy collisional dissociation (EThcD), respectively. Under physiological conditions, AI-ETD identifies 450 ADPr sites on low-abundant proteins, including in vivo cysteine modifications on poly(ADP-ribosyl)polymerase (PARP) 8 and tyrosine modifications on PARP14, hinting at specialist enzymatic functions for these enzymes. Collectively, our data provide insights into the physiological regulation of ADPr.
Keywords: ADP-ribosylation, AI-ETD, proteomics, mass spectrometry, physiological ADPr, PARP8, PARP14, Golgi apparatus, endoplasmic reticulum, glycosylation
Graphical Abstract
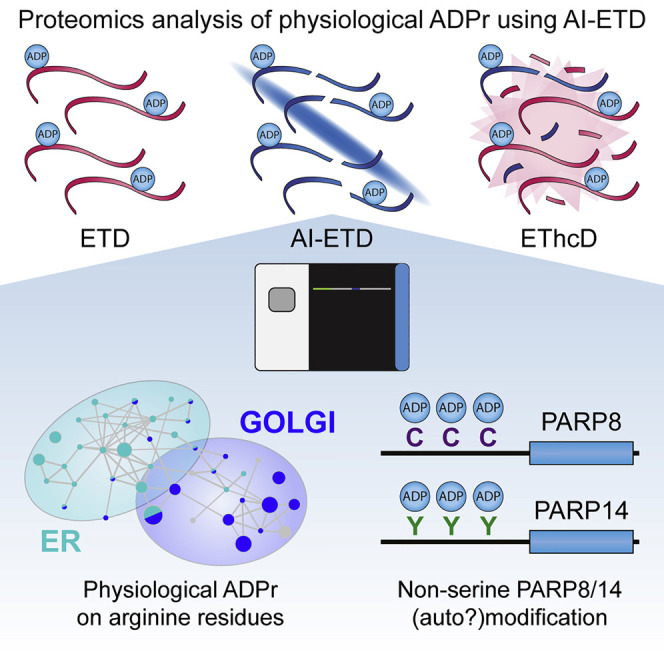
Buch-Larsen et al. investigate the ADP-ribosylome in a physiological context by combining AI-ETD with unbiased proteomics enrichment of ADP-ribosylation. The authors demonstrate that physiological ADPr mainly targets serine residues, specifically targets arginine residues in ER and Golgi proteins, and find that PARP8 and PARP14 are exclusively ADP-ribosylated on cysteine and tyrosine residues, respectively.
Introduction
ADP-ribosylation (ADPr) is an emerging post-translational modification (PTM) regulating a variety of biological processes, including cell signaling and DNA damage repair, by modifying proteins with either one ADPr moiety (mono-ADP-ribosylation [MARylation]) or several ADPr moieties (poly-ADP-ribosylation [PARylation]). ADP-ribosyltransferases (ARTs) catalyze the modification by transferring ADPr units from NAD+ onto target proteins. The group of responsible enzymes can be divided into two groups depending on the conserved structural features: ARTCs (cholera toxin-like) and ARTDs (diphtheria toxin-like, better known as poly[ADP-ribosyl]polymerases [PARPs]), of which the latter is the largest and most characterized group consisting of 17 members in human (Lüscher et al., 2018). Within the ARTDs, 4 and 12 enzymes are reported to possess PARylation and MARylation activity, respectively, while PARP13 is reported to be catalytic inactive (Lüscher et al., 2018).
The most thoroughly studied PARP enzyme is PARP1, which is predominantly investigated in relation to DNA damage repair (De Vos et al., 2012). Under genotoxic stress, PARP1 modifies serine residues when complexed to the co-factor histone PARylation factor 1 (HPF1) (Bonfiglio et al., 2017b), with this induction abrogated by PARP inhibitors (Larsen et al., 2018). ADPr is a reversible PTM, allowing it to adapt to cellular stimuli in a rapid and temporal manner, with poly(ADP-ribose) glycohydrolase (PARG) being one of the key enzymes degrading PAR chains into MAR (D’Amours et al., 1999).
Mass spectrometry (MS)-based proteomics methods have become the primary approach for studying ADPr in a global and unbiased manner. ADPr modifies proteins at very low levels, making enrichment methods necessary. MS-based analysis is further complicated by the dynamic nature of ADPr, with a rapid enzymatic turnover of the modification (Alvarez-Gonzalez and Althaus, 1989), numerous modifiable amino acid residues (Altmeyer et al., 2009; Bonfiglio et al., 2017b; McDonald and Moss, 1994; Moss and Vaughan, 1977; Ogata et al., 1980; Sekine et al., 1989; Van Ness et al., 1980), and the highly labile nature of the modification (Bonfiglio et al., 2017a).
Several methods have been established for enrichment of ADPr-modified peptides for MS analysis (Daniels et al., 2014; Gagné et al., 2018; Hendriks et al., 2019; Larsen et al., 2018; Zhang et al., 2013). For large-scale studies of ADPr, the two most widely used strategies are based either on affinity enrichment using the Af1521 macrodomain or on a combination of boronate-affinity enrichment coupled with chemical conversion of ester-bound ADPr modifications into hydroxyamides using hydroxylamine. Whereas the Af1521-based enrichment strategy identifies the entire ADPr moiety on any amino acid residue (Larsen et al., 2018), the chemical-based strategy identifies chemical marks only on aspartate and glutamate residues as a trace of ADPr (Li et al., 2019; Zhang et al., 2013). Moreover, although Af1521 is capable of identifying ADPr under endogenous conditions, knockdown of PARG is required to increase sensitivity for the chemical-based method, rendering investigation of ADPr under physiological conditions impossible (Li et al., 2019; Zhang et al., 2013).
We previously demonstrated that tandem MS acquisition relying on beam-type collisional activation (e.g., higher-energy collisional dissociation [HCD]) cannot be used for confidently localizing ADPr sites because of the labile nature of the bond between the modification and the amino acid residue (Larsen et al., 2018). However, the non-ergodic fragmentation propensity of the radical-driven electron transfer dissociation (ETD) methodology preserves labile PTMs and can thus allow confident determination of the exact amino acid residue being modified with ADPr. Electron transfer reactions, however, often result in non-dissociative electron transfer (ETnoD), especially for precursors with low mass-to-charge ratio like ADPr-modified peptides (Larsen et al., 2018; Ledvina et al., 2010). To overcome the high degree of ETnoD, supplemental activation can be applied with electron-transfer higher-energy collisional dissociation (EThcD), which has been proved to be more effective than ETD for identification of ADPr sites (Bilan et al., 2017; Hendriks et al., 2019). However, despite that EThcD utilizes supplementary activation in order to counter ETnoD, this effect did not improve dissociation of charge-reduced product ions resulting from fragmentation of ADPr peptides (Hendriks et al., 2019).
By contrast, activated ion ETD (AI-ETD) uses infrared photoactivation during the ETD reaction to disrupt the gas-phase non-covalent interactions that prevent formation of c and ⋅ z-type product ions, thereby greatly reducing ETnoD. This benefits glycoproteomics (Riley et al., 2019), top-down proteomics (McCool et al., 2019; Riley et al., 2018a, 2018b), and phosphoproteomics (Riley et al., 2017a). We therefore set out to investigate the utility of AI-ETD for identification and confident localization of ADPr sites. Here, AI-ETD proved advantageous for analyzing ADPr-enriched samples, with AI-ETD mapping 79% and 23% more ADPr sites compared with ETD and EThcD, respectively. This allowed us to obtain a comparable depth of sequencing from markedly less input material and enabled identification of 450 physiological ADPr sites, of which 60% were not previously identified under physiological conditions. Taken together, we demonstrate that AI-ETD is a superior dissociation technique for analysis of ADPr.
Results
Evaluation of AI-ETD
To investigate the ability of AI-ETD to increase dissociation of ADPr peptides, we utilized our established Af1521 method for efficient and high-purity enrichment of peptides modified by ADPr (Figure 1 A). Because the binding affinity of the macrodomain is toward the terminal moiety of ADP-ribose (Karras et al., 2005), the Af1521 strategy is capable of detecting modification of any amino acid residue type by ADPr (Larsen et al., 2018). For the purpose of initial optimization and direct comparison of AI-ETD with other dissociation methodologies, we purified ADPr peptides from H2O2-treated HeLa cells, because H2O2 is known to induce ADPr via activation of PARP1 and PARP2. This sample was used as a technical standard for quadruplicate comparison of different dissociation types, with each experiment corresponding to a comparatively low amount of ADPr peptides purified from only two million cells. We compared standard ETD and EThcD with AI-ETD at five different laser power (LP) settings spanning the range of routinely used power settings (Ledvina et al., 2010; Riley et al., 2017b).
Figure 1.

Comparison of AI-ETD versus ETD and EThcD for Mapping ADP-Ribosylation
(A) Overview of the strategy employed to enrich ADPr peptides. All samples were analyzed in technical quadruplicate; n = 4.
(B) Overview of the number of ADPr peptide-spectrum matches (PSMs) identified and localized (>90% probability) for each dissociation method and supplemental activation energy (SAE; laser power for AI-ETD, normalized collision energy for EThcD). Significance was determined using two-tailed Student’s t testing. Error bars represent SD; n = 4 technical replicates.
(C) As in (B), but displaying the number of ADPr sites identified.
(D) As in (B), but displaying the spectral quality (in Andromeda score) of all identified ADPr-modified peptides. Distribution of data points is visualized: line limits, 1.5× interquartile range; box limits, 3rd and 1st quartiles; white dot, mean. Number of data points (n) is visualized below the distributions.
(E) ADP-ribose localization probability plotted against the ranked fraction of all PSMs. Note that although all probabilities are displayed, only those over 0.9 were used for assignment of ADPr peptides and sites.
(F) Visualization of the average relative degree of non-dissociative electron transfer (ETnoD). Derived from all peptide-identified MS/MS spectra, and separately visualized for unmodified and ADPr peptides. Error bars represent 5× SEM.
(G) Visualization of the average degree of precursor fragmentation, calculated by dividing observed fragment ion peak intensity by the sum of non-ETD, ETnoD, and all fragment ion peak intensities. Error bars represent 5× SEM.
(H) Spectral quality plotted against the average degree of precursor fragmentation for localized ADPr peptides. Coloring represents the relative density of dots in the plot, with higher values corresponding to higher density.
We observed a higher number of identified and localized unique modified ADPr peptides for all five AI-ETD LP settings, compared with ETD (Figure 1B; Table S1A). The optimum was 20% LP, with a gradual falloff in identification rate for both lower and higher LP. AI-ETD identified 120% and 28% more ADPr peptides than ETD and EThcD, respectively. When considering the number of unique ADPr sites on proteins, we found the same trend (Figure 1C; Table S2A), with AI-ETD mapping 79% and 23% more sites compared with ETD and EThcD, respectively.
In terms of spectral quality, AI-ETD demonstrated the highest scores at relatively low LP, with an optimum at 15% (Figure 1D). Using 25% LP and above, or EThcD, resulted in a decline in spectral quality, suggesting excessive peptide fragmentation under these circumstances. When considering localization of the ADP-ribose to specific amino acid residues, we found that on average AI-ETD at 15% and 20% LP and EThcD significantly outperformed ETD (Figure 1E). However, when filtering ADPr for a stringent (>90%) localization, AI-ETD at 20% LP was superior.
Previously, we demonstrated complementarity of trypsin and Lys-C digestions for identification of unique ADPr sites (Hendriks et al., 2019). Thus, we purified ADPr peptides as described above, but using Lys-C instead of trypsin digestion. We subjected these peptides to technical quadruplicate analysis with a similar experimental setup, using the three optimal AI-ETD LP settings. Overall, the aforementioned findings could be recapitulated, with the best performance of AI-ETD at 20% LP, identifying 134% and 30% more ADPr peptides than ETD and EThcD, respectively (Figure S1A; Table S1A). At the site-specific level, AI-ETD mapped 118% and 28% more ADPr sites than ETD and EThcD, respectively (Figure S1B; Table S2A), and spectral quality was likewise improved by AI-ETD (Figure S1C).
Based on these observations, we wondered whether the enhanced performance of AI-ETD resulted from increased dissociation of reduced charge-state precursors, i.e., decreased levels of ETnoD and an overall more complete fragmentation of the ADPr peptides. Intriguingly, AI-ETD considerably reduced the amount of ETnoD even at low LP and dramatically reduced ETnoD to practically zero at the highest LP (Figure 1F; Figure S1D), whereas EThcD failed to resolve ETnoD product ions to the same extent. Concomitantly, unmodified peptides within the same samples were only modestly affected by AI-ETD, because these were generally short peptides that did not display high levels of ETnoD to start with. This highlights a striking susceptibility of ADPr peptides to AI-ETD, with a notably increased fragmentation of ADPr peptides at higher AI-ETD LP compared with unmodified peptides (Figure 1G; Figure S1E). In contrast, EThcD did not greatly or explicitly increase fragmentation of ADPr peptides. Overall, AI-ETD reduced ETnoD and led to more complete fragmentation of ADPr peptides specifically.
Whereas AI-ETD at the highest LP greatly fragmented ADPr peptides, it did not result in the highest number of identifications. To investigate this, we compared the degree of peptide fragmentation with spectral score. For unmodified peptides, higher degrees of fragmentation globally correlated to higher spectral quality (Figure S1F). For localized ADPr peptides, the highest quality spectra resided between 30% and 45% fragmentation, with a strong decline in spectral quality when fragmentation exceeded 50% (Figure 1H). Indeed, when considering identified but non-localized ADPr peptides, a considerably higher number of these were >50% fragmented (Figure S1G), suggesting that very high degrees of ADPr peptide fragmentation are detrimental to faithful identification and localization. Collectively, we show that AI-ETD is superior to ETD and EThcD for confident profiling of ADPr sites.
Benchmarking AI-ETD for In-Depth Profiling of ADPr
To gauge the performance of AI-ETD in the context of deep analysis of the ADP-ribosylome, we employed our previously described high-pH fractionation on ADPr peptides purified from H2O2-treated HeLa cells (Hendriks et al., 2019; Larsen et al., 2018), using AI-ETD at the optimal 20% LP for all further experiments. Additionally, to address the large amount of starting material usually required for proteomic profiling of ADPr (Abplanalp et al., 2017; Hendriks et al., 2019; Larsen et al., 2018; Li et al., 2019; Zhang et al., 2013), we used a considerably smaller amount of sample. In total, 8,939 unique ADPr peptides were identified (Table S1B), mapping to 5,206 confidently localized ADPr sites (Figure 2 A; Table S2B). Compared with previous studies, the identified number of ADPr sites fell just short of the current single-study record of 7,000 (Hendriks et al., 2019). Nevertheless, the relative amount of input material analyzed here was only ∼20%, yet still facilitated identification of ∼75% of the total number of sites (Figure 2A). Additionally, our AI-ETD profiling of the ADP-ribosylome from limited input revealed 1,800 ADPr target proteins (Table S3), corresponding to 84% of identifications from the largest study to date (Hendriks et al., 2019). Over half of the ADPr target proteins were modified on two or more distinct ADPr sites, demonstrating deep coverage of this low-stoichiometry PTM (Figure S2).
Figure 2.

Benchmarking of AI-ETD
(A) Overview of the number of ADPr sites identified in this experiment, as compared with previous Af1521 screens (Hendriks et al., 2019; Larsen et al., 2018). “Input” corresponds to the relative amount of the summed total ion current (observed at the MS1 level) across the analytical gradients of all experiments and correlates with effective sample load on the column.
(B) Scaled Venn diagram visualizing distribution of identified ADPr sites in this experiment compared with other ADPr studies (Hendriks et al., 2019; Larsen et al., 2018).
(C) Pie chart overview of the amino acid residue distribution of all ADPr sites identified in this experiment (left pie chart) and ADPr sites exclusively identified in this study (right pie chart) as compared with other ADPr studies (Hendriks et al., 2019; Larsen et al., 2018).
(D) Term enrichment analysis using Gene Ontology annotations and UniProt keywords, comparing proteins identified to be ADPr-modified in this study with the total proteome. Relative score is based on multiplication of logarithms derived from the enrichment ratio and the q value. Terms were significant with q < 0.02, as determined through Fisher exact testing with Benjamini-Hochberg correction. The full-term enrichment analysis is available in Table S5A.
Overall, 1,798 (35%) and 2,045 (39%) sites were previously described by either or both of our other studies, respectively (Figure 2B). Moreover, we identified 1,363 (26%) previously undescribed ADPr sites (Table S4). We observed the large majority of modification to occur on serine residues (83%), followed by modification of arginine (5.9%), histidine (4.5%), and tyrosine (2.8%) residues (Figure 2C, left). Considering the subset of ADPr sites exclusively identified in this study, the majority of these still resided on serine residues (58%), but with a relatively larger fraction of ADPr targeting arginine (17%), histidine (8.7%), tyrosine (4.8%), lysine (4.7%), cysteine (3.2%), threonine (2.1%), aspartate (1.1%), and glutamate (0.7%) residues (Figure 2C, right).
The H2O2-induced ADP-ribosylome profiled using AI-ETD largely resided on proteins with functional annotations that are well-known in the context of ADPr biology (Figure 2D; Table S5A). Taken together, AI-ETD is congruous with deep analysis of the ADP-ribosylome from a limited pool of starting material.
Improved Analysis of Physiological ADPr Using AI-ETD
ADPr is frequently investigated in a non-physiological manner, e.g., by genetic perturbation of ADP eraser enzymes (Daniels et al., 2014; Zhang et al., 2013) or by exposure of cells to high levels of stress, which both boost cellular levels of ADPr to facilitate detection. Levels of ADPr are >30-fold lower under physiological conditions (Larsen et al., 2018), which makes physiological ADPr technically challenging to study, despite the biomedical importance of elucidating how ADPr is regulated in, e.g., clinical samples or patient biopsies. Therefore, we set out to evaluate the ability of AI-ETD to profile the physiological ADP-ribosylome.
We purified ADPr-modified peptides from unperturbed HeLa cells growing under standard conditions, and minimized MS machine time by performing single-shot analyses. In total, we identified 703 unique ADPr peptides, corresponding to 450 ADPr sites residing on 295 proteins (Figure 3 A). We identified up to 79% more ADPr sites compared with our previous study (Larsen et al., 2018) and up to 387 physiological ADPr sites in 50 min (i.e., 8 sites/min) of MS time.
Figure 3.

Analysis of Physiological ADPr Using AI-ETD
(A) Overview of the number of physiological ADPr sites identified in this study, compared with a previous study that also included physiological ADPr (Larsen et al., 2018). Total: unique ADPr sites identified across all four cell culture replicates; Best rep.: unique ADPr sites identified in the best replicate; SS: single-shot analysis; all: high-pH fractionated analysis.
(B) Left: pie chart overview of the numerical amino acid residue distribution of all physiological ADPr sites identified in this study. Right: scaled Venn diagram visualizing distribution of identified physiological ADPr sites in this study compared with our previous study (Larsen et al., 2018).
(C) Average localization probability across all ADPr PSMs that were at least partially localized (>51%) to each specific amino acid residue type. Only amino acids with >10 partially localized PSMs were included. Error bars represent SEM; number of data points (n) is indicated.
(D) Depth of sequencing analysis, plotting number of identified physiological ADPr target proteins versus their known copy numbers. Proteins ADP-ribosylated in response to H2O2 were derived from this study, as well as various other studies (Bilan et al., 2017; Jungmichel et al., 2013; Larsen et al., 2018; Martello et al., 2016; Zhang et al., 2013). Protein copy numbers were derived from a deep HeLa proteome study (Bekker-Jensen et al., 2017), which covers the vast majority (>99%) of all ADPr target proteins identified. Dotted lines with color indicator below represent the median protein copy number for ADPr target proteins within each respective subset.
(E) “Fidget spinner” analysis, composed of scaled Venn diagram (center) visualizing distribution of H2O2-induced and physiological ADPr sites; pie chart overviews (around the center) of the amino acid residue distribution of each subset of ADPr sites, as indicated by background coloring; term enrichment analyses (outermost graphs) using Gene Ontology annotations and UniProt keywords, comparing proteins identified to be ADP-ribosylated in each subset of ADPr sites as compared with the other subsets, indicated by background coloring. Relative score is based on multiplication of logarithms derived from the enrichment ratio and the q value. Terms were significant with q < 0.02, as determined by Fisher exact testing with Benjamini-Hochberg correction. The full-term enrichment analysis is available in Table S5B. Background and Venn coloring: red, H2O2 only; purple, both H2O2 and physiological; blue, physiological only.
Of the 450 physiological ADPr sites we identified, 286 (64%) were not previously mapped under unperturbed conditions (Figure 3B, right), and 70 were not at all found in proteomics screens (Table S4). Intriguingly, serine residues (55%) remained the most commonly ADP-ribosylated under physiological conditions (Figure 3B, left). Nonetheless, we also observed frequent modification of arginine (26%), histidine (12%), and cysteine (3.3%) residues. In terms of total modification abundance, we observed 86.6%, 9.5%, and 2.5% of all physiological ADPr to reside on serine, arginine, and histidine residues, respectively (Figure S3A). Physiological ADPr could be confidently localized on cysteine, histidine, arginine, serine, threonine, and tyrosine residues (Figure 3C), although we did not observe consistent ADPr of lysine, glutamate, and aspartate residues. Under physiological conditions, >50% of serine ADPr resided in lysine-serine (KS) motifs (Figure S3B), an established phenomenon for serine ADPr (Larsen et al., 2018). Adherence to KS motifs was considerably lower in response to oxidative stress (Figure S3B), suggesting that under these conditions serine residues may be targeted more promiscuously. Physiological ADPr predominantly targeted nuclear proteins (71%; Figure S3C), although not to the same extent as H2O2-induced ADPr (85%; Figure S3D).
Next, we investigated the expression levels of ADPr target proteins (Figure 3D). With inclusion of H2O2-induced ADPr, protein copy numbers of ADPr target proteins spanned six orders of magnitude. Physiological ADPr target proteins identified here covered a somewhat lower range of expression levels. Nonetheless, compared with physiological ADPr target proteins identified in our previous study (Larsen et al., 2018), we achieved a 57% greater depth of sequencing (Figure 3D).
Combining previous proteomics-based knowledge on ADPr (Bilan et al., 2017; Bonfiglio et al., 2017b; Hendriks et al., 2019; Larsen et al., 2018) with the additional insight gained in this study, we set out to investigate systemic differences between physiological and H2O2-perturbed ADPr biology. Specifically, we compared 9,029 H2O2 ADPr sites with 643 physiological ADPr sites (Figure 3E) and found the vast majority of ADPr sites (93%) were exclusive to H2O2 treatment, 6% of ADPr sites were detected in both cases, and 1% of ADPr sites were exclusive to physiological conditions. In terms of amino acid distribution, H2O2-exclusive ADPr predominantly modified serine residues (84%), in agreement with previous reports (Hendriks et al., 2019; Larsen et al., 2018; Palazzo et al., 2018). ADPr detected under all conditions still targeted a majority of serine residues (62%), but moreover modified significant fractions of arginine (13%), histidine (13%), cysteine (5%), and lysine (4%) residues. Intriguingly, physiologically exclusive ADPr primarily modified arginine residues (66%), with further modification of histidine (15%) and cysteine (10%) residues, and only a small fraction of modification on serine residues (5.7%). Overall, our data reveal a propensity for ADPr to be targeted to different amino acid residues dependent on cellular conditions.
Next, we assessed the relative differences between physiological and non-physiological ADPr (Figure 3E; Table S5B). H2O2-specific ADPr was more frequently observed to target proteins involved in rRNA-binding, DNA replication, and cell cycle. Physiologically specific ADPr preferentially targeted proteins with sulfotransferase activity, glycoproteins, and membrane proteins. Finally, proteins ADP-ribosylated under all conditions represented terms canonically associated with ADPr, including predominant modification of the ADP enzymatic machinery, histone proteins, and DNA repair proteins. Collectively, our comparison between physiological and H2O2-induced ADPr demonstrates a considerable shift in the targeting of this PTM, not only at the substrate level, but also in terms of which amino acid residue types are modified.
Investigation of Residue-Specific Physiological ADPr
We constructed a database containing all 385 proteins that are ADP-ribosylated under physiological conditions (Table S6), based upon physiological ADPr target proteins identified here and existing proteomics evidence (Larsen et al., 2018). Within this database, we investigated which proteins were predominantly modified on distinct amino acid residue types (Table 1 ). In line with previous reports (Hendriks et al., 2019; Larsen et al., 2018; Palazzo et al., 2018), proteins primarily ADP-ribosylated on serine residues were nucleus-localized or chromatin-centric proteins with functions in chromosome organization, DNA replication, and cell-cycle regulation, despite their ADPr being observed under physiological conditions. In contrast, proteins mainly modified on arginine residues had enzymatic functions, including isomerase and hydrolase activity, and were largely localized to the endoplasmic reticulum (ER) and the Golgi apparatus. Proteins physiologically modified on histidine, cysteine, or tyrosine residues did not show a particular trend in their annotated biological process or cellular compartment.
Table 1.
An Overview of Residue-Specific Physiological ADPr Target Proteins
| Gene Name | AA | Intensity | No. of Sites | Primary ADPr | Fraction | Secondary ADPr | Fraction | Tertiary ADPr | Fraction | GOBP | GOCC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PARP1 | S | 1.1E+10 | 11 | S-499 | 67.1% | S-507 | 27.7% | S-519 | 4.3% | ADP-ribosylation | nucleus |
| Histone H2B∗ | S | 8.8E+9 | 8 | S-7 | 99.1% | R-80 | 0.6% | S-15 | 0.3% | nucleosome | chromosome |
| Histone H3∗ | S | 7.7E+9 | 2 | S-11 | 87.6% | S-29 | 12.4% | nucleosome | chromosome | ||
| NPM1 | S | 1.2E+9 | 8 | S-207 | 48.1% | S-139 | 26.3% | S-195 | 11.3% | cell cycle | nucleus |
| HIST1H4A | S | 1.1E+9 | 2 | S-2 | 99.5% | S-48 | 0.5% | nucleosome | chromosome | ||
| HNRNPU | S | 5.4E+8 | 2 | S-187 | 98.6% | S-695 | 1.4% | mRNA processing | nucleus | ||
| NOLC1 | S | 3.5E+8 | 6 | S-303 | 78.2% | S-580 | 19.8% | S-508 | 1.1% | cell cycle | nucleolus |
| HIST1H1E | S | 2.5E+8 | 3 | S-150 | 68.6% | S-55 | 26.5% | S-27 | 4.9% | nucleosome | chromosome |
| RFC1 | S | 2.1E+8 | 6 | S-1126 | 48.1% | S-302 | 34.1% | S-384 | 6.6% | DNA replication | nucleus |
| WDHD1 | S | 1.2E+8 | 3 | S-942 | 83.6% | S-900 | 10.0% | S-946 | 6.4% | DNA replication | nucleus |
| P4HB | R | 1.2E+9 | 1 | R-97 | 100.0% | isomerase | ER | ||||
| PDIA3 | R | 5.9E+8 | 1 | R-62 | 100.0% | isomerase | ER | ||||
| ASPH | R | 5.2E+8 | 1 | R-437 | 100.0% | hydrolase | ER | ||||
| GALNT2 | R | 2.1E+8 | 6 | R-93 | 59.4% | R-128 | 17.9% | R-113 | 10.7% | glycosyltransferase | Golgi |
| TXNDC5 | R | 1.5E+8 | 1 | R-394 | 100.0% | isomerase | ER | ||||
| PTPRG | R | 1.4E+8 | 1 | R-311 | 100.0% | hydrolase | membrane | ||||
| GOLIM4 | R | 1.2E+8 | 2 | R-110 | 66.3% | R-445 | 33.7% | transport | Golgi | ||
| PDIA6 | R | 1.0E+8 | 3 | R-142 | 55.2% | R-235 | 41.0% | R-153 | 3.8% | isomerase | ER |
| PIGS | R | 9.5E+7 | 1 | R-176 | 100.0% | transamidase | ER | ||||
| B2M | R | 8.7E+7 | 1 | R-32 | 100.0% | immunity | secreted | ||||
| HMGB1 | H | 2.4E+8 | 2 | H-117 | 99.8% | S-181 | 0.2% | DNA repair | nucleus | ||
| CPSF6 | H | 1.2E+8 | 1 | H-478 | 100.0% | mRNA processing | nucleus | ||||
| PCMT1 | H | 1.0E+8 | 1 | H-15 | 100.0% | methyltransferase | cytoplasm | ||||
| PPIA | H | 6.9E+7 | 3 | H-126 | 83.1% | C-52 | 15.4% | R-148 | 1.4% | isomerase | cytoplasm |
| PHIP | H | 5.4E+7 | 1 | H-779 | 100.0% | insulin signaling | nucleus | ||||
| FAM118B | H | 3.6E+7 | 1 | H-105 | 100.0% | Cajal body | nucleus | ||||
| XRCC5 | H | 3.0E+7 | 1 | H-152 | 100.0% | DNA repair | chromosome | ||||
| CAMSAP2 | H | 2.7E+7 | 1 | H-24 | 100.0% | microtubule | Golgi | ||||
| CELF1 | H | 2.6E+7 | 1 | H-89 | 100.0% | mRNA processing | nucleus | ||||
| MARF1 | H | 2.0E+7 | 1 | H-1205 | 100.0% | differentiation | peroxisome | ||||
| GMDS | C | 1.2E+8 | 1 | C-8 | 100.0% | dehydratase | cytoplasm | ||||
| NSUN2 | C | 1.1E+8 | 1 | C-271 | 100.0% | methyltransferase | mitochondrion | ||||
| PARP8 | C | 2.3E+7 | 3 | C-376 | 46.1% | C-395 | 39.4% | C-332 | 14.4% | ADP-ribosylation | ER |
| ASCC3 | C | 1.3E+7 | 1 | C-208 | 100.0% | hydrolase | nucleus | ||||
| NUMA1 | C | 5.5E+6 | 2 | C-2009 | 71.7% | H-1689 | 28.3% | cell cycle | nucleus | ||
| AP1G1 | C | 3.3E+6 | 1 | C-539 | 100.0% | transport | Golgi | ||||
| TXN | C | 9.9E+5 | 1 | C-73 | 100.0% | transport | mitochondrion | ||||
| NONO | C | 7.6E+5 | 1 | C-145 | 100.0% | mRNA processing | nucleus | ||||
| GSTM3 | C | 7.6E+5 | 1 | C-190 | 100.0% | transferase | cytoplasm | ||||
| PEX3 | Y | 1.4E+7 | 1 | Y-56 | 100.0% | peroxisome | peroxisome | ||||
| PARP14 | Y | 1.2E+7 | 3 | Y-895 | 51.1% | Y-1088 | 27.0% | Y-59 | 21.9% | ADP-ribosylation | nucleus |
| RPS3A | Y | 2.0E+6 | 1 | Y-155 | 100.0% | differentiation | nucleus |
A listing of the top 10 physiological ADPr target proteins mainly modified (>2/3rd intensity) on specific amino acid residue types. Only 9 and 3 proteins were mainly modified on cysteine and tyrosine, respectively. Residue types with less than 5 total ADPr sites were excluded. The top 3 most abundant ADPr sites are indicated. Multiple variants of the same histone type were merged, indicated with an asterisk (*). AA, main modified residue type; fraction, relative contribution of this ADPr site to the total ADPr modification of the protein; GOBP, Gene Ontology Biological Process; GOCC, Gene Ontology Cellular Compartment.
Within the 51 proteins exclusively found to be ADP-ribosylated under physiological conditions (Figure 4 A; Table S6), only one protein was observed to be modified on a serine residue. Strikingly, 36 of these proteins were solely modified on arginine residues, suggesting a potential downregulation of the arginine ADP-ribosylome in response to oxidative stress. To gain more insight into their biological interconnectivity and functionality, we performed STRING network analysis on all proteins with preponderant arginine ADPr (Figure 4B). The majority (76%) of these physiological ADPr target proteins were interconnected and frequently localized to either the ER (47%, p = 2.1 × 10−12) or the Golgi (36%, p = 5.2 × 10−10). Intriguingly, 80% of the arginine ADPr-modified proteins were also annotated as glycoproteins, representing a highly significant enrichment (p = 1.8 × 10−18), in agreement with site-specific analysis (Figure 3E). To further investigate a potential overlap in protein targets of arginine ADPr and glycosylation, we performed a comparison of our physiological ADPr data with a recent compendium of N-linked glycosylation proteomics data (Table S6) (Sun et al., 2019). In total, 166 physiological ADPr target proteins were found to also be targeted by N-glycosylation. We observed that for proteins predominantly ADP-ribosylated on arginine residues, the propensity for these proteins to also be N-glycosylation targets was significantly greater as compared with all other physiological ADPr target proteins (Figure 4C), hinting at the existence of crosstalk between arginine ADPr and N-glycosylation.
Figure 4.

Analysis of Arginine-Specific ADP-Ribosylation
(A) Pie chart of the amino acid residue distribution of ADPr target proteins exclusively modified under physiological conditions.
(B) STRING network visualizing functional interactions between proteins exclusively ADPr modified on arginine residues. Default STRING clustering confidence was used (p > 0.4), and disconnected proteins were omitted from the network unless they were identified by 2+ ADPr sites. Proteins were significantly interconnected, with a protein-protein interaction enrichment p value of 1.0 × 10−16.
(C) Distribution of the number of N-linked glycosylation sites per protein, as derived from a composite study (Sun et al., 2019). Line limit, 95th percentile; box limits, 3rd and 1st quartiles; black bar, median; blue bar, average. Number of data points (n) is visualized below the distributions. Significance was determined using two-tailed Student’s t testing.
ER, endoplasmic reticulum; Glyco, glycoprotein; Golgi, Golgi apparatus.
Overall, our database with physiological ADPr target proteins comprises a vast proteomics resource, underlining a significant presence of serine ADPr even in unperturbed cell systems, while concomitantly highlighting potential spatial preferences for physiological ADPr focused on other residue types.
Detection of PARP8 and PARP14 Modification in Cells
PARP1 modifies itself in response to DNA binding (Bolderson et al., 2019), and our physiological profiling corroborated the main modification of PARP1 to target three serine residues (S499, S507, and S519) located in the auto-modification domain of the enzyme (Figure 5 A; Table 1).
Figure 5.

ADP-Ribosylation of Different PARP Family Members
(A) Overview of the most abundantly ADPr-modified residues within PARP family members, as a fraction of total protein ADPr modification, either under physiological conditions (Phys.) or in response to H2O2. All physiologically detected ADPr sites, or otherwise up to four of the most abundant total ADPr sites, are indicated on the graphical representation of the PARP family members. Blue lines, physiological ADPr sites; red lines, H2O2-induced ADPr sites.
(B) Overview of the total amount of ADPr detected on each PARP family member, in relation to their known expression level (IBAQ). Both axes are logarithmic, and graph scaling was normalized to PARP1 and PARP2 values.
(C) STRING network visualizing functional interactions between proteins identified to be ADPr modified on cysteine residues in this study or in our previous study (Larsen et al., 2018). Default STRING clustering confidence was used (p > 0.4), and disconnected proteins were omitted from the network. Proteins were significantly interconnected, with a protein-protein interaction enrichment p value of 1.1 × 10−11. Distribution of color is relative to the number of sites within each category; sites detected both physiologically and in response to H2O2 were colored as physiological.
Cys, modification on cysteine residues; other, modification on non-cysteine residues.
Based on deep proteomic profiling of HeLa cells, 10 PARP family members are expressed at different levels in unperturbed HeLa cells (Bekker-Jensen et al., 2017). Using AI-ETD, we could detect ADPr modification of PARP1, PARP2, PARP8, PARP12, and PARP14 (Figure 5B; Tables S3 and S4). Strikingly, even though protein copy numbers of PARP8 and PARP14 are 250–500 times lower than PARP1, we could still detect their ADPr under physiological conditions (Table 1).
On PARP2, we observed modification of only serine residues, similar to PARP1 (Figure 5A), corroborating the known inter-connected functions of the two enzymes (Ménissier de Murcia et al., 2003; Schreiber et al., 2002). ADPr on PARP12 was found only in response to H2O2 treatment and at relatively low abundance. Intriguingly, PARP8 was ADP-ribosylated on three cysteine residues (C332, C376, and C395) under physiological conditions (Table 1; Table S2C; Data S1), whereas a fourth cysteine residue (C367) was modified in response to H2O2. This exclusive modification on cysteine residues could hint at PARP8 auto-modification, implying that PARP8 is capable of ADP-ribosylating cysteine residues. Analogously, we observed physiological modification of three tyrosine residues (Y59, Y895, and Y1088) in PARP14 (Table 1; Table S2C; Data S1), which could likewise imply auto-modification and a potential function of PARP14 as the catalyst for tyrosine ADPr.
We were intrigued by the ADPr modification of cysteine residues, with PARP8 modified on four cysteine residues, and knowledge regarding cysteine ADPr being relatively sparse. Altogether, we could map 123 cysteine ADPr sites to 112 proteins (Tables S3 and S4), and generated an interconnected STRING network based on all proteins with cysteine ADPr (Figure 5C). 85 out of 112 proteins (76%) were connected to the network, with significant (p = 1.1 × 10−11) enrichment for protein-protein interactions. Beyond PARP8, cysteine ADPr modification occurred on a considerable number of interconnected catalytic ADPr enzymes, including PARP1, PARP5A (TNKS), PARP7 (TIPARP), PARP12, PARP13 (ZC3HAV1), and PARP14. When considering only the 60 proteins predominantly ADP-ribosylated on cysteines, there was a significant (p = 0.012) presence of proteins involved in RNA metabolism, including DDX6, HNRNPH1, NSUN2, PAIP1, PARN, PCBP1, PPP2R1A, PSMB2, TNKS1BP1, and TRNT1. Taken together, our sensitive MS-based proteomics screening of ADPr provides insight into modification of non-serine residue types and may hint at specificity of certain PARP enzymes toward distinct amino acids.
Discussion
Here, we investigated the potential of AI-ETD for sensitive identification and confident localization of ADPr sites. To evaluate the ability of AI-ETD for profiling the ADP-ribosylome, we performed a comparison of ETD, EThcD, and AI-ETD at five different LP settings.
ADP-ribosylated peptides are highly labile when subjected to collisional activation, with the ADP-ribose itself presenting the lowest energy pathway for vibrational energy redistribution (Bonfiglio et al., 2017a; Larsen et al., 2018). In contrast, ADPr peptides are very stable when subjected to ETD, and here we observed ADPr peptides to be much more prone to ETnoD, compared with unmodified peptides within the same samples. EThcD failed to completely overcome ETnoD and resulted in decreased spectral quality at higher energies. Intriguingly, we observed that ADPr peptides are highly susceptible to AI-ETD, much more so than small unmodified peptides within the same sample. This susceptibility to AI-ETD resulted in a near-complete abolishing of ETnoD at 25% LP and higher, although these greater energies also resulted in over-fragmentation of the peptides and a sharp drop in spectral quality. At a modest level of energy, AI-ETD resulted in higher quality spectra, improved sensitivity, and more confident localization of ADPr to acceptor amino acids, overall doubling the number of identifications compared with pure ETD.
Recent advances in MS-based proteomics have largely focused on high-throughput analyses, essentially by minimizing time or input material required to achieve the same depth of sequencing (Bache et al., 2018; Batth et al., 2019). Compared with relatively abundant PTMs, such as phosphorylation and glycosylation, for which MS methodology is well established (Hogrebe et al., 2018; Kelstrup et al., 2018; Riley et al., 2019), the study of ADPr with MS-based proteomics remains daunting. Using AI-ETD, we identified >500 ADPr sites from single-shot analyses of purified ADPr material originating from only ∼5 million cells. Moreover, from fractionated samples originating from ∼100 million cells, we identified >5,000 ADPr sites, reaching a great depth of sequencing while using considerably less starting material than other studies (Hendriks et al., 2019; Larsen et al., 2018; Leutert et al., 2018; Li et al., 2019; Martello et al., 2016; Zhang et al., 2013).
Compared with ADPr induced by oxidative stress, the identification of ADPr in unperturbed cells is exceptionally challenging, with the modification occurring at very low abundance and dynamically regulated (Daniels et al., 2015). Our previous identification of physiological ADPr sites required an extensive investment of mass spectrometric machine time and a larger amount of starting material, with a considerable portion of identified ADPr sites relying on pre-fractionation of samples prior to mass spectrometric analysis (Larsen et al., 2018). With the increased analytical sensitivity of AI-ETD, we mapped 450 physiological ADPr sites from single-shot analyses, while using limited starting material and a total of 200 minutes of analytical gradient time for four replicates.
Our compilation of physiological ADPr target proteins (Table 1; Table S6) should serve as a valuable knowledge base for further investigation of the role of ADPr under endogenous and unperturbed conditions. Interestingly, we found that proteins exclusively ADP-ribosylated under physiological conditions were preferentially modified on arginine residues, and these ADPr target proteins were primarily localized to the ER and the Golgi (Figure 4B), suggesting that modification of these proteins is not mediated by PARP1 or PARP2. The ARTCs are known to catalyze arginine ADPr, and even though ARTC4 and ARTC5 were detected in a deep HeLa proteomics study (Bekker-Jensen et al., 2017), ARTC1 and ARTC3 were below the limit of detection. However, ARTC1 has been demonstrated to be localized to the ER (Fabrizio et al., 2015), and in the same study ARTC1 was shown to be expressed in HeLa cells. Therefore, we speculate that sub-stoichiometric levels of ARTC1 could be responsible for the arginine ADPr we observed to be localized to the ER and the Golgi in this study. In support of this, a recent proteomics study that profiled ADPr in heart and skeletal muscle from wild-type and ARTC1 knockout mice (Leutert et al., 2018) also demonstrated that ARTC1 is responsible for the majority of arginine ADPr. Indeed, out of the 78 proteins with physiological arginine ADPr, 31 were also detected by Leutert et al. (2018) to be modified on arginines, with 28 of these no longer detected in ARTC1-deficient mice (Table S6).
Auto-modification of PARPs is a known phenomenon (Vyas et al., 2014), with especially auto-modification of PARP1 being thoroughly studied (Chapman et al., 2013; Muthurajan et al., 2014). Here, we provide evidence that ADPr occurs on PARP1, PARP2, PARP8, PARP12, and PARP14. For PARP1 and PARP2, serine was the main acceptor residue. This is consistent with PARP1 and PARP2 catalyzing ADPr of serine residues upon oxidative stress, in combination with HPF1 (Bonfiglio et al., 2017b; Larsen et al., 2018; Palazzo et al., 2018). Although we observed a relatively high number of serine residues being ADP-ribosylated under physiological conditions, many of those were also detectable in response to oxidative stress. In contrast, ADPr sites exclusively detected under physiological conditions were only rarely modifying serine residues. Taken together, this suggests that serine ADPr observed under physiological conditions could be a result of baseline DNA damage, potentially as a consequence of culturing cells under atmospheric oxygen conditions.
Intriguingly, we observed PARP8 to be ADP-ribosylated only on cysteine residues. This exclusive modification of cysteine residues supports previous in vitro observations of the auto-modification of PARP8 (Vyas et al., 2014), and we confirmed all four hitherto reported cysteine residues, of which three were detectable under physiological conditions. In the case of PARP14, physiological ADPr targeted only tyrosine residues, similarly setting it apart from PARP1 and PARP2, which were primarily modified on serine residues. Auto-modification of PARP14 has been described previously (Qin et al., 2019), and the interactome of PARP14 suggests a role in regulating RNA stability (Carter-O’Connell et al., 2018). Furthermore, PARP14 plays a critical role in innate immunity by preventing ADPr-mediated inhibition of coronavirus replication (Grunewald et al., 2019). Thus, insight into physiological and site-specific PARP14 ADPr may aid in further understanding the functional role that PARP14 plays in the interferon response and RNA metabolism. Overall, the auto-modification of PARP8 and PARP14 could suggest that these enzymes may have the ability to specifically ADP-ribosylate cysteine and tyrosine residues, respectively. However, further investigation is required to elucidate whether, and how, such specificity would be mediated.
Taken together, the combination of the AI-ETD technology with our Af1521 strategy for unbiased purification of ADPr peptides has culminated in a highly sensitive MS-based proteomics approach that can confidently identify large numbers of ADPr events from limited starting material. Moreover, this increased sensitivity makes it feasible to profile ADPr in an entirely physiological context, taking a final step toward biomedical and clinical applicability.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| BL21(DE3) | New England BioLabs | Cat#C2527I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hydrogen peroxide (H2O2) | Sigma Aldrich | Cat#H1009 |
| Lysyl Endopeptidase (Lys-C) | Wako Chemicals | Cat#129-02541 |
| Trypsin, Proteomics Grade | Sigma Aldrich | Cat#T6567 |
| Recombinant PARG enzyme | Prof. Dr. Michael O. Hottiger | N/A |
| Recombinant GST-tagged Af1521 | Martello et al., 2016 | N/A |
| Deposited Data | ||
| Raw and analyzed ADP-ribosylation MS data | This paper | ProteomeXchange: PXD017417 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | CCL-2 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Lund Nielsen (michael.lund.nielsen@cpr.ku.dk).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD017417.
Experimental Model and Subject Details
Cell lines
HeLa cells (CCL-2, female) were acquired via the American Type Culture Collection, and cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Invitrogen) supplemented with 10% fetal bovine serum and a penicillin/streptomycin mixture (100 U/mL; GIBCO). Cells were routinely tested for mycoplasma. Cells were not routinely authenticated.
Bacteria
BL21(DE3) Competent E.coli were acquired from New England BioLabs. Details regarding culture conditions were reported previously (Larsen et al., 2018).
Method Details
Cell treatment
For initial optimization and benchmarking experiments, ADP-ribosylation was induced in HeLa by treatment of the cells with 1 mM H2O2 (Sigma Aldrich) for 10 min in PBS at 37°C. Five cell culture replicates were prepared, each corresponding to approximately 100 million HeLa cells, of which one was used for optimization, and four for benchmarking. For physiological experiments, cells were left untreated, and four cell culture replicates were prepared, each corresponding to approximately 100 million HeLa cells.
Cell lysis and protein digestion
The full procedure for enrichment of ADPr from cells was done as described previously (Hendriks et al., 2019; Larsen et al., 2018). Briefly, cells were washed twice with ice-cold PBS, and gently scraped at 4°C in a minimal volume of PBS. Cells were pelleted by centrifugation at 500g, and lysed in 10 pellet volumes of Lysis Buffer (6 M guanidine-HCl, 50 mM TRIS, pH 8.5). Complete lysis was achieved by alternating vigorous shaking with vigorous vortexing, for 30 s, prior to snap freezing of the lysates using liquid nitrogen. Frozen lysates were stored at −80°C until further processing. Lysates were thawed and sonicated at 30 W, for 1 s per 1 mL of lysate, spread across 3 separate pulses. Tris(2-carboxyethyl)phosphine and chloroacetamide were added to a final concentration of 5 mM, and the lysate was incubated for 1 hour at 30°C. Proteins were digested using Lysyl Endopeptidase (Lys-C, 1:100 w/w; Wako Chemicals) for 3 hours, and diluted with three volumes of 50 mM ammonium bicarbonate. Half of the samples was further digested overnight using modified sequencing grade Trypsin (1:200 w/w; Sigma Aldrich), and the other half was re-digested overnight using Lys-C. Digested samples were acidified by addition of trifluoroacetic acid (TFA) to a final concentration of 0.5% (v/v), cleared by centrifugation, and purified using reversed-phase C18 cartridges (SepPak Classic, 360 mg sorbent, Waters) according to the manufacturer’s instructions. Elution of peptides was performed with 30% or 40% ACN in 0.1% TFA, for peptides digested with either trypsin or Lys-C, respectively. Eluted peptides were frozen overnight at −80°C, and lyophilized.
Purification of ADP-ribosylated peptides
GST-tagged Af1521 macrodomain was produced in-house using BL21(DE3) bacteria, and coupled to glutathione Sepharose 4B beads (Sigma-Aldrich), essentially as described previously (Hendriks et al., 2019; Larsen et al., 2018). Lyophilized peptides were dissolved in IP buffer (50 mM TRIS pH 8.0, 1 mM MgCl2, 250 μM DTT, and 50 mM NaCl), after which long ADP-ribose polymers were reduced to monomers using recombinant PARG at a concentration of 1:10,000 (w/w), overnight and at room temperature. Subsequently, 100 μL of Sepharose beads with GST-tagged Af1521 were added to the samples, and mixed at 4°C for 3 h. Beads were sequentially washed four times with ice-cold IP Buffer, two times with ice-cold PBS, and two times with ice-cold MQ water. On the first wash, beads were transferred to 1.5 mL LoBind tubes (Eppendorf), and LoBind tubes were exclusively used from this point on to minimize loss of peptide. Additional tube changes were performed every second washing step to minimize carryover of background. ADP-ribosylated peptides were removed from the beads using two elution steps with 200 μL ice-cold 0.15% TFA, and the pooled elutions were cleared through 0.45 μm spin filters (Ultrafree-MC, Millipore) and subsequently through pre-washed 100 kDa cut-off filters (Vivacon 500, Sartorius). The filtered ADP-ribosylated peptides were purified using C18 StageTips at high pH (Hendriks et al., 2019), and eluted either as single shot samples (optimization and physiological experiments), or eluted as seven fractions (benchmarking experiments). Briefly, samples were basified by adding ammonium solution to a final concentration of 20 mM, and loaded onto StageTips carrying four layers of C18 disc material (punch-outs from 47mm C18 3M extraction discs, Empore). Elution was performed with 25% of acetonitrile (ACN) in 20 mM ammonium hydroxide for single shot samples, or sequentially performed with 2% (F1), 4% (F2), 7% (F3), 10% (F4), 15% (F5), and 25% (F6) of ACN in 20 mM ammonium hydroxide for fractionated samples. The seventh fraction (F0) was prepared by performing StageTip purification at low pH on the flowthrough fraction from sample loading at high pH. All samples were completely dried using a SpeedVac at 60°C, and dissolved in a small volume of 0.1% formic acid. Final samples were frozen at −20°C until measurement.
Mass spectrometric analysis
All MS samples were analyzed using a Fusion Lumos Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) modified for AI-ETD, coupled to a Dionex UltiMate 3000 nano-UHPLC system (Thermo Fisher, Sunnyvale, CA, USA). For AI-ETD implementation, a Firestar T-100 Synrad 60 Watt (W) CO2 continuous wave laser (Mukilteo, WA, USA) was mounted at the back of the instrument. The IR photon beam was guided into the traps using focusing lenses and a multi-mode hollow-core fiber (Opto-Knowledge Systems, Torrance, CA, USA) prior to a zinc selenide window that was placed in the vacuum manifold. Chromatographic separation of peptides was performed on a 30-cm long analytical column, with an internal diameter of 1.7 μm, packed in-house as described previously (Shishkova et al., 2018) using 130 Å pore size Bridged Ethylene Hybrid C18 particles (Waters, Milford, USA). The analytical column was heated to 50°C using an in-house made column oven. Peptides were eluted from the column using a gradient of Buffer A (0.1% formic acid) and Buffer B (80% ACN in 0.1% formic acid). The primary gradient ranged from 3% buffer B to 24% buffer B over 50 minutes, preceded by sample loading time (5 minutes for evaluation experiments, 20 minutes for benchmarking and physiological experiments), and followed by a 20 minute washing block. Electrospray ionization (ESI) was achieved using a Nanospray Flex Ion Source (Thermo). Spray voltage was set to 2.25 kV, capillary temperature to 275°C, and RF level to 30%. Full scans were performed at a resolution of 60,000, with a scan range of 300 to 1,750 m/z, a maximum injection time of 60 ms, and an automatic gain control (AGC) target of 600,000 charges. Precursors were isolated with a width of 1.3 m/z, with an AGC target of 200,000 charges, and precursor fragmentation was accomplished using either electron transfer dissociation (ETD), electron transfer disassociation with supplemental higher-collisional disassociation (EThcD) at an NCE of 20, or activated-ion electron transfer dissociation (AI-ETD); all using calibrated charge-dependent ETD parameters (Rose et al., 2015). The laser powers were the percentage of power (W) from the Firestar T-100 Synrad 60-W CO2, continuous wave laser. For AI-ETD evaluation, laser power settings of 10% (6 W), 15% (9 W), 20% (12 W), 25% (15 W), and 30% (18 W) were used, and for benchmarking and physiological experiments a laser power setting of 20% was used. Precursors with charge state 2-5 were isolated for MS/MS analysis, and prioritized from charge 3 (highest), to 4, to 5, to 2 (lowest), using the decision tree algorithm. For benchmarking and physiological experiments, only precursors with charge state 3-5 were isolated. Precursors were excluded from repeated sequencing by setting a dynamic exclusion time of 75 s for evaluation experiments, and 90 s for benchmarking and physiological experiments, with an exclusion mass tolerance of 10 ppm. MS/MS spectra were measured in the Orbitrap, with 5 data-dependent MS/MS scans per full MS scan, a maximum precursor injection time of 120 ms, and a scan resolution of 60,000. First mass for MS/MS scans was set to 100 for ETD and EThcD measurements, and variable for AI-ETD measurements, with the first mass defining the laser power applied. MS/MS first masses for AI-ETD were 141, 131, 142, 132, and 143, corresponding to application of laser powers 10%, 15%, 20%, 25%, and 30%, respectively.
Data analysis
Analysis of the mass spectrometry raw data was performed using MaxQuant software (version 1.5.3.30). MaxQuant default settings were used, with exceptions outlined below. Two separate computational searches were performed, one for evaluation and physiological data, and the other for benchmarking data. For generation of the theoretical spectral library, a HUMAN.fasta database was extracted from UniProt on the 24th of May, 2019. N-terminal acetylation, methionine oxidation, cysteine carbamidomethylation, and ADP-ribosylation on all amino acid residues known to potentially be modified (C, D, E, H, K, R, S, T, and Y), were included as variable modifications. For analysis of trypsin samples, up to 6 missed cleavages were allowed. For analysis of Lys-C samples, up to 3 missed cleavages were allowed. A maximum allowance of 4 variable modifications per peptide was used. Second peptide search was enabled (default), and matching between runs was enabled with a match time window of 42 s and an alignment time window of 20 minutes. Mass tolerance for precursors was set to 20 ppm in the first MS/MS search and 4.5 ppm in the main MS/MS search after mass recalibration. For fragment ion masses, a tolerance of 20 ppm was used. Modified peptides were filtered to have an Andromeda score of > 40 (default), and a delta score of > 20. Data was automatically filtered by posterior error probability to achieve a false discovery rate of < 1% (default), at the peptide-spectrum match, the protein assignment, and the site-specific levels.
Data filtering
Beyond automatic filtering and FDR control as applied by MaxQuant, the data was manually filtered in order to ensure proper identification and localization of ADP-ribose. PSMs modified by more than one ADP-ribose were omitted. PSMs corresponding to unique peptides were only used for ADP-ribosylation site assignment if localization probability was > 0.90, with localization of > 0.75 accepted only for purposes of intensity assignment of further evidences. Erroneous MaxQuant intensity assignments were manually corrected in the sites table, and based on localized PSMs only (> 0.90 best-case, > 0.75 for further evidences). For the ADP-ribosylation target proteins table, the proteinGroups.txt file generated by MaxQuant was filtered to only contain those proteins with at least one ADP-ribosylation site detected and localized post-filtering as outlined above, with cumulative ADP-ribosylation site intensities based only on localized evidences.
Determination of fragmentation efficiency
An in-house Python script was used to determine the intensities of non-reduced precursors (no ETD), and the corresponding non-dissociated charge-reduced precursor ions (ETnoD) for each MS/MS spectrum. For that, the centroided, de-isotoped, and charge-deconvoluted peak lists from the MaxQuant Andromeda Peak List (APL) files were used. Second peptide search APLs were not considered. “No ETD” precursor peaks were expected at:
“ETnoD” peaks were expected at:
This compensates for MaxQuant charge deconvolution, which goes by the assumption that the charge state is purely dependent on added protons. The expected peaks were matched to peaks from the MaxQuant APL files, with a mass tolerance of 0.05 Da, and their intensities were stored. Intensity information was mapped to the msms.txt output file from MaxQuant using the scan number. Total fragmentation of peptides was calculated by division of fragment product ion intensities with the sum of all peak intensities.
Comparison to other studies
Data from other studies was retrieved from several publications and online databases. For ADP-ribosylation sites; Larsen et al. (2018), Hendriks et al. (2019), Bonfiglio et al. (2017b), Bilan et al. (2017), Leslie Pedrioli et al. (2018). For ADP-ribosylation proteins; Zhang et al. (2013), Jungmichel et al. (2013), Martello et al. (2016), Bilan et al. (2017), Larsen et al. (2018), Hendriks et al. (2019). For total proteome; Bekker-Jensen et al. (2017). For comparison of proteins between studies, all protein identifiers were mapped to the human proteome as downloaded from Uniprot on the 24th of May, 2019. In case data sources did not include Uniprot IDs, ID mapping on Uniprot was used to convert other IDs to Uniprot IDs, and otherwise gene names were used. Non-existent, non-human, and redundant entries, were discarded from the analysis. For comparison of sites between studies, all parent proteins were mapped to Uniprot IDs as described above, and afterward the reported positions of modification were used to extract 51 amino acid sequence windows (modified residue ± 25 amino acids), with the sequence windows ultimately used to directly compare identified sites. Sites mapping to non-existent, non-human, or redundant proteins, were discarded. Sites not correctly aligning to the reported amino acid residues were discarded.
Quantification and Statistical Analysis
Details regarding the statistical analysis can be found in the respective figure legends. Statistical handling of the data was primarily performed using the freely available Perseus software (Tyanova et al., 2016), and includes term enrichment analysis through FDR-controlled Fisher Exact testing, and density estimation for highly populated scatterplots. Protein Gene Ontology annotations and UniProt keywords used for term enrichment analysis were concomitantly downloaded from UniProt with the HUMAN.fasta file used for searching the RAW data. Boxplots and violin plots were generated using the BoxPlotR web tool (Spitzer et al., 2014). The online STRING database (version 11) was used for generation of protein interaction networks (Szklarczyk et al., 2017), and Cytoscape (version 3.7.1) was used for manual annotation and visualization of the STRING networks (Shannon et al., 2003).
Acknowledgments
The work carried out in this study was in part supported by the Novo Nordisk Foundation Center for Protein Research, the Novo Nordisk Foundation (grant agreement numbers NNF14CC0001 and NNF13OC0006477), The Danish Council of Independent Research (grant agreement numbers 4002-00051, 4183-00322A, 8020-00220B, and 0135-00096B), and The Danish Cancer Society (grant agreement R146-A9159-16-S2). B.F. is the recipient of a fellowship from the Novo Nordisk Foundation as part of the Copenhagen Bioscience PhD Programme (grant agreement number NNF19SA0035442). The AI-ETD technology was funded by the National Institutes of Health-supported National Center for Quantitative Biology of Complex Systems (P41GM108538). The proteomics technology applied was part of a project that has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement EPIC-XS-823839. We would like to acknowledge the lab of Michael O. Hottiger for the expression and purification of recombinant human PARG (University of Zurich), and we thank members of the NNF-CPR Mass Spectrometry Platform for instrument support and technical assistance.
Author Contributions
S.C.B.-L. and I.A.H. prepared all samples. M.S.W., J.M.L., E.S., and J.J.C. established AI-ETD technology and infrastructure. S.C.B.-L., I.A.H., and J.M.L. measured all samples on the mass spectrometer. I.A.H. and S.C.B.-L. processed all MS raw data. I.A.H., S.C.B.-L., and M.R. performed bioinformatics and statistical analyses. M.R. and B.F. wrote the Python script for determination of fragmentation efficiency. M.L.N. and J.J.C. supervised the project. I.A.H., S.C.B.-L., and M.L.N. wrote the manuscript with input from all authors.
Declaration of Interests
The authors declare no competing interests.
Published: September 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108176.
Supplemental Information
(A) A list of all 1,925 unique human ADPr-modified peptides identified in the evaluation and physiological experiments, complete with qualitative information. (B) A list of all 8,939 unique human ADPr-modified peptides identified in the benchmarking experiments, complete with qualitative information.
A list of all 5,344 human ADPr sites identified in this study, complete with qualitative information and fractional modification analysis. (A) 993 ADPr sites identified in the evaluation experiments. (B) 5,206 ADPr sites identified in the benchmarking experiments. (C) 450 ADPr sites identified in the physiological experiments.
A thorough comparison of the 1,801 ADPr target proteins identified in this study to 6 other ADPr proteomics studies, which represent a mixture of different enrichment strategies performed at the protein-level or site-level. The entire human proteome was used as a background.
A thorough comparison of the 5,344 human ADPr sites identified in this study to 4 other site-specific ADPr proteomics studies, which all include serine residues as potential ADPr targets, and do not exclude any specific residue types because of methodological limitations.
(A) Term enrichment analysis comparing ADPr target proteins in the benchmarking experiments with the total proteome, including statistical information. (B) Term enrichment analyses for subsets of ADPr target proteins specifically detected only in H2O2 samples, both in H2O2 and physiological samples, or only in physiological samples, using the other subsets as a statistical background.
A list of all 385 human ADPr target proteins modified under physiological and unperturbed conditions, complete with qualitative information, and an overview of which amino acids are modified.
Six fully annotated MS/MS spectra corresponding to physiologically ADPr-modified peptides, directly identified with the full ADPr mass remnant modifying the peptide. Coloring: z-ions (and y-ions) from target peptide (red), c-ions from target peptide (blue), unassigned (gray).
References
- Abplanalp J., Leutert M., Frugier E., Nowak K., Feurer R., Kato J., Kistemaker H.V.A., Filippov D.V., Moss J., Caflisch A., Hottiger M.O. Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 2017;8:2055. doi: 10.1038/s41467-017-02253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M., Messner S., Hassa P.O., Fey M., Hottiger M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Gonzalez R., Althaus F.R. Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat. Res. 1989;218:67–74. doi: 10.1016/0921-8777(89)90012-8. [DOI] [PubMed] [Google Scholar]
- Bache N., Geyer P.E., Bekker-Jensen D.B., Hoerning O., Falkenby L., Treit P.V., Doll S., Paron I., Müller J.B., Meier F. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell. Proteomics. 2018;17:2284–2296. doi: 10.1074/mcp.TIR118.000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batth T.S., Tollenaere M.A.X., Rüther P., Gonzalez-Franquesa A., Prabhakar B.S., Bekker-Jensen S., Deshmukh A.S., Olsen J.V. Protein Aggregation Capture on Microparticles Enables Multipurpose Proteomics Sample Preparation. Mol. Cell. Proteomics. 2019;18:1027–1035. doi: 10.1074/mcp.TIR118.001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen D.B., Kelstrup C.D., Batth T.S., Larsen S.C., Haldrup C., Bramsen J.B., Sørensen K.D., Høyer S., Ørntoft T.F., Andersen C.L. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017;4:587–599.e4. doi: 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilan V., Leutert M., Nanni P., Panse C., Hottiger M.O. Combining Higher-Energy Collision Dissociation and Electron-Transfer/Higher-Energy Collision Dissociation Fragmentation in a Product-Dependent Manner Confidently Assigns Proteomewide ADP-Ribose Acceptor Sites. Anal. Chem. 2017;89:1523–1530. doi: 10.1021/acs.analchem.6b03365. [DOI] [PubMed] [Google Scholar]
- Bolderson E., Burgess J.T., Li J., Gandhi N.S., Boucher D., Croft L.V., Beard S., Plowman J.J., Suraweera A., Adams M.N. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat. Commun. 2019;10:5501. doi: 10.1038/s41467-019-13167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio J.J., Colby T., Matic I. Mass spectrometry for serine ADP-ribosylation? Think o-glycosylation! Nucleic Acids Res. 2017;45:6259–6264. doi: 10.1093/nar/gkx446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio J.J., Fontana P., Zhang Q., Colby T., Gibbs-Seymour I., Atanassov I., Bartlett E., Zaja R., Ahel I., Matic I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell. 2017;65:932–940.e6. doi: 10.1016/j.molcel.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I., Vermehren-Schmaedick A., Jin H., Morgan R.K., David L.L., Cohen M.S. Combining Chemical Genetics with Proximity-Dependent Labeling Reveals Cellular Targets of Poly(ADP-ribose) Polymerase 14 (PARP14) ACS Chem. Biol. 2018;13:2841–2848. doi: 10.1021/acschembio.8b00567. [DOI] [PubMed] [Google Scholar]
- Chapman J.D., Gagné J.P., Poirier G.G., Goodlett D.R. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2013;12:1868–1880. doi: 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- D’Amours D., Desnoyers S., D’Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- Daniels C.M., Ong S.E., Leung A.K. Phosphoproteomic approach to characterize protein mono- and poly(ADP-ribosyl)ation sites from cells. J. Proteome Res. 2014;13:3510–3522. doi: 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels C.M., Ong S.E., Leung A.K. The Promise of Proteomics for the Study of ADP-Ribosylation. Mol. Cell. 2015;58:911–924. doi: 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos M., Schreiber V., Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Fabrizio G., Di Paola S., Stilla A., Giannotta M., Ruggiero C., Menzel S., Koch-Nolte F., Sallese M., Di Girolamo M. ARTC1-mediated ADP-ribosylation of GRP78/BiP: a new player in endoplasmic-reticulum stress responses. Cell. Mol. Life Sci. 2015;72:1209–1225. doi: 10.1007/s00018-014-1745-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagné J.P., Langelier M.F., Pascal J.M., Poirier G.G. Hydrofluoric Acid-Based Derivatization Strategy To Profile PARP-1 ADP-Ribosylation by LC-MS/MS. J. Proteome Res. 2018;17:2542–2551. doi: 10.1021/acs.jproteome.8b00146. [DOI] [PubMed] [Google Scholar]
- Grunewald M.E., Chen Y., Kuny C., Maejima T., Lease R., Ferraris D., Aikawa M., Sullivan C.S., Perlman S., Fehr A.R. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019;15:e1007756. doi: 10.1371/journal.ppat.1007756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks I.A., Larsen S.C., Nielsen M.L. An Advanced Strategy for Comprehensive Profiling of ADP-ribosylation Sites Using Mass Spectrometry-based Proteomics. Mol. Cell. Proteomics. 2019;18:1010–1026. doi: 10.1074/mcp.TIR119.001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogrebe A., von Stechow L., Bekker-Jensen D.B., Weinert B.T., Kelstrup C.D., Olsen J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018;9:1045. doi: 10.1038/s41467-018-03309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M.O., Nielsen M.L. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell. 2013;52:272–285. doi: 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- Karras G.I., Kustatscher G., Buhecha H.R., Allen M.D., Pugieux C., Sait F., Bycroft M., Ladurner A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelstrup C.D., Bekker-Jensen D.B., Arrey T.N., Hogrebe A., Harder A., Olsen J.V. Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J. Proteome Res. 2018;17:727–738. doi: 10.1021/acs.jproteome.7b00602. [DOI] [PubMed] [Google Scholar]
- Larsen S.C., Hendriks I.A., Lyon D., Jensen L.J., Nielsen M.L. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018;24:2493–2505.e4. doi: 10.1016/j.celrep.2018.07.083. [DOI] [PubMed] [Google Scholar]
- Ledvina A.R., Beauchene N.A., McAlister G.C., Syka J.E., Schwartz J.C., Griep-Raming J., Westphall M.S., Coon J.J. Activated-ion electron transfer dissociation improves the ability of electron transfer dissociation to identify peptides in a complex mixture. Anal. Chem. 2010;82:10068–10074. doi: 10.1021/ac1020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie Pedrioli D.M., Leutert M., Bilan V., Nowak K., Gunasekera K., Ferrari E., Imhof R., Malmström L., Hottiger M.O. Comprehensive ADP-ribosylome analysis identifies tyrosine as an ADP-ribose acceptor site. EMBO Rep. 2018;19:e45310. doi: 10.15252/embr.201745310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutert M., Menzel S., Braren R., Rissiek B., Hopp A.-K., Nowak K., Bisceglie L., Gehrig P., Li H., Zolkiewska A. Proteomic Characterization of the Heart and Skeletal Muscle Reveals Widespread Arginine ADP-Ribosylation by the ARTC1 Ectoenzyme. Cell Rep. 2018;24:1916–1929.e5. doi: 10.1016/j.celrep.2018.07.048. [DOI] [PubMed] [Google Scholar]
- Li P., Zhen Y., Yu Y. Site-specific analysis of the Asp- and Glu-ADP-ribosylated proteome by quantitative mass spectrometry. Methods Enzymol. 2019;626:301–321. doi: 10.1016/bs.mie.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher B., Bütepage M., Eckei L., Krieg S., Verheugd P., Shilton B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018;118:1092–1136. doi: 10.1021/acs.chemrev.7b00122. [DOI] [PubMed] [Google Scholar]
- Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S.C., Young C., Hottiger M.O., Nielsen M.L. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016;7:12917. doi: 10.1038/ncomms12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool E.N., Lodge J.M., Basharat A.R., Liu X., Coon J.J., Sun L. Capillary Zone Electrophoresis-Tandem Mass Spectrometry with Activated Ion Electron Transfer Dissociation for Large-scale Top-down Proteomics. J. Am. Soc. Mass Spectrom. 2019;30:2470–2479. doi: 10.1007/s13361-019-02206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald L.J., Moss J. Enzymatic and nonenzymatic ADP-ribosylation of cysteine. Mol. Cell. Biochem. 1994;138:221–226. doi: 10.1007/BF00928465. [DOI] [PubMed] [Google Scholar]
- Ménissier de Murcia J., Ricoul M., Tartier L., Niedergang C., Huber A., Dantzer F., Schreiber V., Amé J.C., Dierich A., LeMeur M. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J., Vaughan M. Mechanism of action of choleragen. Evidence for ADP-ribosyltransferase activity with arginine as an acceptor. J. Biol. Chem. 1977;252:2455–2457. [PubMed] [Google Scholar]
- Muthurajan U.M., Hepler M.R., Hieb A.R., Clark N.J., Kramer M., Yao T., Luger K. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc. Natl. Acad. Sci. USA. 2014;111:12752–12757. doi: 10.1073/pnas.1405005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata N., Ueda K., Hayaishi O. ADP-ribosylation of histone H2B. Identification of glutamic acid residue 2 as the modification site. J. Biol. Chem. 1980;255:7610–7615. [PubMed] [Google Scholar]
- Palazzo L., Leidecker O., Prokhorova E., Dauben H., Matic I., Ahel I. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife. 2018;7:e34334. doi: 10.7554/eLife.34334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W., Wu H.J., Cao L.Q., Li H.J., He C.X., Zhao D., Xing L., Li P.Q., Jin X., Cao H.L. Research Progress on PARP14 as a Drug Target. Front. Pharmacol. 2019;10:172. doi: 10.3389/fphar.2019.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N.M., Hebert A.S., Dürnberger G., Stanek F., Mechtler K., Westphall M.S., Coon J.J. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem. 2017;89:6367–6376. doi: 10.1021/acs.analchem.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N.M., Westphall M.S., Hebert A.S., Coon J.J. Implementation of Activated Ion Electron Transfer Dissociation on a Quadrupole-Orbitrap-Linear Ion Trap Hybrid Mass Spectrometer. Anal. Chem. 2017;89:6358–6366. doi: 10.1021/acs.analchem.7b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N.M., Sikora J.W., Seckler H.S., Greer J.B., Fellers R.T., LeDuc R.D., Westphall M.S., Thomas P.M., Kelleher N.L., Coon J.J. The Value of Activated Ion Electron Transfer Dissociation for High-Throughput Top-Down Characterization of Intact Proteins. Anal. Chem. 2018;90:8553–8560. doi: 10.1021/acs.analchem.8b01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N.M., Westphall M.S., Coon J.J. Sequencing Larger Intact Proteins (30-70 kDa) with Activated Ion Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2018;29:140–149. doi: 10.1007/s13361-017-1808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N.M., Hebert A.S., Westphall M.S., Coon J.J. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 2019;10:1311. doi: 10.1038/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C.M., Rush M.J., Riley N.M., Merrill A.E., Kwiecien N.W., Holden D.D., Mullen C., Westphall M.S., Coon J.J. A calibration routine for efficient ETD in large-scale proteomics. J. Am. Soc. Mass Spectrom. 2015;26:1848–1857. doi: 10.1007/s13361-015-1183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V., Amé J.C., Dollé P., Schultz I., Rinaldi B., Fraulob V., Ménissier-de Murcia J., de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- Sekine A., Fujiwara M., Narumiya S. Asparagine residue in the rho gene product is the modification site for botulinum ADP-ribosyltransferase. J. Biol. Chem. 1989;264:8602–8605. [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkova E., Hebert A.S., Westphall M.S., Coon J.J. Ultra-High Pressure (>30,000 psi) Packing of Capillary Columns Enhancing Depth of Shotgun Proteomic Analyses. Anal. Chem. 2018;90:11503–11508. doi: 10.1021/acs.analchem.8b02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M., Wildenhain J., Rappsilber J., Tyers M. BoxPlotR: a web tool for generation of box plots. Nat. Methods. 2014;11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Hu Y., Ao M., Shah P., Chen J., Yang W., Jia X., Tian Y., Thomas S., Zhang H. N-GlycositeAtlas: a database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteomics. 2019;16:35. doi: 10.1186/s12014-019-9254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Van Ness B.G., Howard J.B., Bodley J.W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem. 1980;255:10710–10716. [PubMed] [Google Scholar]
- Vyas S., Matic I., Uchima L., Rood J., Zaja R., Hay R.T., Ahel I., Chang P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang J., Ding M., Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods. 2013;10:981–984. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) A list of all 1,925 unique human ADPr-modified peptides identified in the evaluation and physiological experiments, complete with qualitative information. (B) A list of all 8,939 unique human ADPr-modified peptides identified in the benchmarking experiments, complete with qualitative information.
A list of all 5,344 human ADPr sites identified in this study, complete with qualitative information and fractional modification analysis. (A) 993 ADPr sites identified in the evaluation experiments. (B) 5,206 ADPr sites identified in the benchmarking experiments. (C) 450 ADPr sites identified in the physiological experiments.
A thorough comparison of the 1,801 ADPr target proteins identified in this study to 6 other ADPr proteomics studies, which represent a mixture of different enrichment strategies performed at the protein-level or site-level. The entire human proteome was used as a background.
A thorough comparison of the 5,344 human ADPr sites identified in this study to 4 other site-specific ADPr proteomics studies, which all include serine residues as potential ADPr targets, and do not exclude any specific residue types because of methodological limitations.
(A) Term enrichment analysis comparing ADPr target proteins in the benchmarking experiments with the total proteome, including statistical information. (B) Term enrichment analyses for subsets of ADPr target proteins specifically detected only in H2O2 samples, both in H2O2 and physiological samples, or only in physiological samples, using the other subsets as a statistical background.
A list of all 385 human ADPr target proteins modified under physiological and unperturbed conditions, complete with qualitative information, and an overview of which amino acids are modified.
Six fully annotated MS/MS spectra corresponding to physiologically ADPr-modified peptides, directly identified with the full ADPr mass remnant modifying the peptide. Coloring: z-ions (and y-ions) from target peptide (red), c-ions from target peptide (blue), unassigned (gray).
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD017417.