

Supplemental Digital Content is available in the text.
Keywords: basement membrane, cardiovascular diseases, endarterectomy, extracellular matrix, proteomics
Abstract
Rationale:
Plaque rupture is the proximate cause of most myocardial infarctions and many strokes. However, the molecular mechanisms that precipitate plaque rupture are unknown.
Objective:
By applying proteomic and bioinformatic approaches in mouse models of protease-induced plaque rupture and in ruptured human plaques, we aimed to illuminate biochemical pathways through which proteolysis causes plaque rupture and identify substrates that are cleaved in ruptured plaques.
Methods and Results:
We performed shotgun proteomics analyses of aortas of transgenic mice with macrophage-specific overexpression of urokinase (SR-uPA+/0 mice) and of SR-uPA+/0 bone marrow transplant recipients, and we used bioinformatic tools to evaluate protein abundance and functional category enrichment in these aortas. In parallel, we performed shotgun proteomics and bioinformatics studies on extracts of ruptured and stable areas of freshly harvested human carotid plaques. We also applied a separate protein-analysis method (protein topography and migration analysis platform) to attempt to identify substrates and proteolytic fragments in mouse and human plaque extracts. Approximately 10% of extracted aortic proteins were reproducibly altered in SR-uPA+/0 aortas. Proteases, inflammatory signaling molecules, as well as proteins involved with cell adhesion, the cytoskeleton, and apoptosis, were increased. ECM (Extracellular matrix) proteins, including basement-membrane proteins, were decreased. Approximately 40% of proteins were altered in ruptured versus stable areas of human carotid plaques, including many of the same functional categories that were altered in SR-uPA+/0 aortas. Collagens were minimally altered in SR-uPA+/0 aortas and ruptured human plaques; however, several basement-membrane proteins were reduced in both SR-uPA+/0 aortas and ruptured human plaques. Protein topography and migration analysis platform did not detect robust increases in proteolytic fragments of ECM proteins in either setting.
Conclusions:
Parallel studies of SR-uPA+/0 mouse aortas and human plaques identify mechanisms that connect proteolysis with plaque rupture, including inflammation, basement-membrane protein loss, and apoptosis. Basement-membrane protein loss is a prominent feature of ruptured human plaques, suggesting a major role for basement-membrane proteins in maintaining plaque stability.
In This Issue, see p 949
Human atherosclerosis is usually an indolent disease and can be asymptomatic for decades.1 However, coronary and cerebral atherosclerosis can become unstable, precipitating angina, myocardial infarction, transient ischemic attacks, strokes, and death.2,3 The most common cause of instability of both coronary and cerebral atherosclerosis is plaque rupture, the physical disruption of an atherosclerotic plaque that exposes thrombogenic plaque contents, leading to luminal thrombosis, thromboembolism, and vessel occlusion.2,4 Despite the importance of plaque rupture as the proximate cause of major adverse cardiovascular events, the molecular mechanisms that cause plaque rupture remain poorly understood. A better understanding of these mechanisms could facilitate recognition of rupture-prone plaques and hasten the development of therapies that prevent plaque rupture, thereby reducing myocardial infarctions, strokes, and cardiovascular deaths.
Proposed causes of plaque rupture include hemodynamic forces,5,6 plaque cell apoptosis or senescence,7–9 abnormal plaque microvasculature,10 endoplasmic reticulum stress,11 plaque microcalcifications,12 inflammatory cytokines that alter plaque ECM (extracellular matrix) metabolism,13 and increased vascular protease activity.9,13–15 Increased protease activity has received significant attention as a cause of plaque rupture, with candidate proteases including cathepsins, serine proteases, and MMPs (matrix metalloproteinases).16 A role for proteases in plaque rupture is supported by detection of both increased protease activity and increased collagen cleavage in advanced human plaques.17,18 Preclinical data that support a role for proteases in plaque rupture include studies in which MMP deficiency or inhibition in mice increased plaque collagen content (interpreted as an indication of plaque stability),19–21 and a study in which overexpression of constitutively active MMP9 in vascular wall cells of atherosclerotic mice produced histological features that are shared with ruptured human plaques.22 However, these studies have not produced a consensus that plaque rupture is caused primarily by proteolysis,23 and the pathways that connect plaque proteolysis with plaque rupture remain hypothetical.24 Identification of these pathways could provide new drug targets and could also reveal markers of unstable atherosclerosis.25,26
To further investigate a role for proteases in plaque rupture, we have developed a transgenic mouse model in which the human scavenger receptor promoter drives macrophage-specific overexpression of the mouse urokinase-type plasminogen activator (Plau [urokinase-type plasminogen activator]) gene.27 These SR-uPA+/0 mice have accelerated atherosclerosis; however, they die suddenly between 10 and 30 weeks of age, before developing a plaque rupture phenotype. We overcame this obstacle by transplanting bone marrow (BM) from SR-uPA+/0 Apoe−/− (apolipoprotein E) donors into 35-week-old nontransgenic Apoe−/− recipients (that already have rupture-prone innominate artery lesions).28 Innominate artery lesions in SR-uPA+/0 bone marrow transplant (BMT) recipients have an increased prevalence of histological features of plaque rupture, including frequent intraplaque hemorrhage (61%) and fibrous cap disruption (78%). Moreover, aortas of SR-uPA+/0 BMT recipients have significantly elevated MMP activity.29
Because the SR-uPA+/0 mouse model convincingly connects increased artery wall protease activity with histological features of plaque rupture, we hypothesized that atherosclerotic arteries of SR-uPA+/0 mice and of recipients of SR-uPA+/0 BMT would be informative experimental settings for (1) unbiased proteomic studies aimed at elucidating the pathways through which elevated vascular protease activity causes plaque rupture and (2) identifying specific protein or peptide markers of plaque rupture caused by elevated vascular protease activity. Accordingly, we performed proteomic analyses of aortas of SR-uPA+/0 mice and of SR-uPA+/0 BMT recipients, and we applied bioinformatics tools to identify protein networks that are altered by increased aortic protease activity and could plausibly connect increased protease activity with plaque rupture. To help accomplish these goals, we also analyzed extracts of atherosclerotic aortas of SR-uPA+/0 BMT recipients with protein topography and migration analysis platform (PROTOMAP), a proteomic analysis methodology designed to identify proteolytic events and peptide fragments in complex mixtures of proteins.30 To explore the clinical relevance of findings from this mouse model, further test its validity as a model of human plaque rupture, and gain insights into mechanisms of human plaque rupture, we also performed shotgun proteomics, bioinformatics analyses, and PROTOMAP studies on extracts of ruptured and stable areas of freshly harvested human carotid atherosclerotic plaques (using both discovery and validation cohorts) and we compared our results to those obtained from the SR-uPA+/0 mouse. Our results show that basement-membrane protein loss is a reproducible finding in ruptured human plaques and is also a prominent feature of the SR-uPA+/0 mouse plaque rupture model.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. This study does not involve testing of a therapeutic or diagnostic agent in animal models. Please see the Major Resources Table in the Data Supplement.
Animal Studies
To begin development of tissue protein extraction techniques for enrichment of ECM proteins, aortic segments were obtained postmortem from a chow-fed rabbit that was euthanized in the course of unrelated experiments.31 All other animal experiments were performed with mice. All mice were Apoe−/− and were progeny of at least 10 generations of C57BL/6 backcrosses. Mice with macrophage-specific overexpression of urokinase-type plasminogen activator (SR-uPA+/0 mice) were progeny of mice generated in our laboratory.27 These mice are hemizygous for a transgene that includes the murine Plau gene driven by the human scavenger receptor promoter. SR-uPA+/0 mice were bred with nontransgenic mice (SR-uPA0/0) to yield SR-uPA+/0 and SR-uPA0/0 littermate controls. We used aortas of four 20-week-old mice (both SR-uPA+/0 and SR-uPA0/0) to further develop protein extraction techniques; the remaining experimental mice (n=93, plus 18 BM donors) are described in detail below. We used only female experimental mice because the accelerated atherosclerosis and plaque rupture phenotypes that we investigated here were described in female mice.27,29 These phenotypes have never been investigated in males. All mice were housed in a specific-pathogen-free facility and genotyped by PCR for the SR-uPA allele.27
To generate a first set of samples for proteomic analyses, 5-week-old SR-uPA+/0 (n=12) and SR-uPA0/0 (n=13) littermates were fed a diet containing 21% fat and 0.15% cholesterol by weight (TD88137; Harlan-Teklad). One SR-uPA+/0 mouse died before its planned harvest. After 15 weeks on diet, proximal thoracic aortas (described in more detail below) were harvested and were either frozen immediately (6 SR-uPA+/0 and 6 SR-uPA0/0) or were explanted and cultured to generate conditioned medium (5 SR-uPA+/0 and 7 SR-uPA0/0). Two of the SR-uPA0/0 samples were later excluded for technical reasons (evidence of significant blood contamination in one sample and low peptide counts in a second sample), leaving 5 conditioned medium samples per group.
A separate cohort of SR-uPA0/0 mice (n=68) was maintained on a normal laboratory diet until 35 weeks of age, then lethally irradiated with 10.5 Gy (Cesium-137 gamma-ray source) and transplanted by tail vein injection of ≈1.2×107 BM cells from 12- to 19-week-old donors. Forty-five mice received BMT from SR-uPA+/0 donors (n=10 donors), and 23 mice received BMT from SR-uPA0/0 donors (n=8 donors). BMT recipients received neomycin-containing water (2 mg/mL) for 1 week before and 2 weeks after BMT and were maintained on a normal laboratory diet post-BMT. Mice were enrolled as BMT recipients only if they appeared in excellent health. After enrollment, mice were not randomized between the 2 BMT donor genotypes because of their genetic homogeneity and their uniformly healthy appearance at the time of BMT. Allocation concealment was also not performed because the experimenter was aware of which BM genotype the next enrollees were going to receive. However, this awareness came only at a late stage: as BMT recipients aged toward 35 weeks, their treatment allocation was determined by availability of donor mice of each BM genotype and whether enrollment in one or the other group had been completed. In addition, the genetic homogeneity of the BMT recipients, their uniformly healthy condition, and the severe phenotype resulting from BMT with the experimental (SR-uPA+/0) versus control (SR-uPA0/0) BM make it highly unlikely that lack of allocation concealment would alter the results. After BMT, mice were monitored for signs of distress such as inactivity and failure to groom. Twelve SR-uPA+/0 BMT recipients and 2 SR-uPA0/0 BMT recipients died or were euthanized (due to deteriorating health) before their planned harvests (8 weeks post-BMT). Aortas were harvested from 33 SR-uPA+/0 BMT recipients and 21 SR-uPA0/0 BMT recipients.
The individual responsible for enrolling mice, performing BMT, harvesting aortas, and collecting conditioned medium was not blinded to BM donor genotype. Lack of blinding was due to practical considerations (to avoid mixing up experimental and control mice) and to our assessment based on previous studies that the severe phenotype engendered by transplantation of SR-uPA+/0 BM was unlikely to be altered by minor variations in handling of the mice and their tissues. We did not perform formal sample size and power calculations before commencing the study. Instead, the number of mice enrolled was selected prospectively based on a goal of achieving n=6 in all experimental groups. Selection of n=6 was based on our extensive experience with tissue proteomics, as well as our expectation that pooled samples would be needed to generate a sufficient amount of protein for PROTOMAP. The number of human plaques selected for protein extraction and analysis was also set prospectively at 6. In both cases, members of the team with experience in proteomic analyses suggested that the use of 6 independent biological replicates would be adequate to detect biologically important differences in the vascular proteome. Mice were housed in a specific-pathogen-free facility in cages with absorbent bedding (Natural Absorbent ¼ inches Bed-o’Cobs; Andersons Lab Bedding). All animal protocols and procedures were approved by the University of Washington Office of Animal Welfare.
Studies on Human Carotid Plaques
Patients scheduled for carotid endarterectomy at the University of Washington Medical Center, Harborview Medical Center or the VA Puget Sound Health Care System provided informed consent for their tissues to be studied and for de-identified clinical information to be abstracted from their medical records. Inclusion criteria for the study were preoperative for carotid endarterectomy, medically stable, age 50 to 80, English speaking, willing to participate in study, and able to give informed consent before the day of surgery. Potential subjects at the VA Puget Sound Health Care System were excluded if they were already enrolled in a different (imaging-based) study to which their surgically removed carotid plaques were already committed. There were no other specific exclusion criteria. All protocols involving human subjects were approved by the Human Subjects Division of the University of Washington and the Institutional Review Board of the VA Puget Sound Health Care System.
Processing of Mouse Aortic Tissue and Generation of Aorta-Conditioned Medium
At the time of harvest (15 weeks after initiating the high-fat diet, for mice not receiving BMT; 8 weeks after BMT for the BMT recipients) mice were anesthetized with ketamine (140 mg/kg) and xylazine (40 mg/kg), saline perfused, and exsanguinated via cardiac puncture. For mice not receiving BMT, the proximal thoracic aorta was removed from the point at which it exits the heart to a point halfway between the left subclavian artery and the diaphragm. For BMT recipients, the same aortic segment was removed, with the innominate artery attached. Aortic segments destined for protein extraction were trimmed free of periadventitial fat in situ, excised, then snap-frozen in liquid nitrogen, and stored at −80°C. To obtain aortic segments for explant culture, mice were anesthetized and perfused in the same manner. Aortas were trimmed in situ, excised (from aortic root to midway between the left subclavian artery and the diaphragm, without attached innominate), washed 3× at 37°C in 2 mL of M199 (Gibco 11043-023; 20 minutes per wash), then placed in wells of a 96-well plate in 110 µL of M199 medium at 37°C. After 20 hours, the conditioned medium was collected and stored at −80°C.
Human Plaque Harvest and Processing
A total of 22 carotid plaques were removed in the operating room as part of a clinically indicated procedure. Freshly removed plaques were placed in a tube containing ice-cold PBS with 25 mmol/L EDTA, 1X Halt protease-inhibitor cocktail (Pierce Biotechnology, no. 87786) and 1X Halt phosphatase-inhibitor cocktail (Pierce Biotechnology, no. 78420) and transported to the laboratory. As part of the surgical procedure, some plaques were divided with a single axial incision that opened up the vessel lumen. Other plaques arrived intact and were divided axially via a single incision extending through the common carotid segment then through the internal carotid artery segment. The specimens were then rinsed 5× with 50 mL ice-cold PBS, placed in the inverted lid of a tissue culture dish with the axial incision facing upwards, and covered with ice-cold PBS with protease and phosphatase inhibitors/EDTA.
Using forceps for traction, we then exposed the lumen of the carotid plaque and examined the lumen surface for presence of an ulcerated lesion with thrombus/hemorrhage (a ruptured plaque). Such lesions were typically located at the bifurcation, opposite the external carotid artery takeoff. Plaques without gross evidence of rupture were not studied further. The area with the ulcerated lesion, including the full thickness of the plaque specimen below it, was trimmed with a scalpel to remove tissue adjacent to the ulcerated lesion. These ruptured plaque specimens were typically rectangular solids, with sides of ≈0.5 to 1.0 cm and a thickness of ≈1 mm. If the ulcerated area was large, we divided it into 2 pieces, to provide a backup sample in case we encountered technical difficulties extracting and analyzing the first sample. A scalpel was then used to cut one thin, full thickness slice from both the caudal and cranial ends of each sample. These slices of ruptured plaque tissue were placed in 10% formalin for 48 hours, then stored in 70% ethanol for later histological analysis. The remainder of the ruptured plaque specimens was snap-frozen in liquid nitrogen and stored at −80°C for later protein extraction. To select stable plaque tissue, we then examined the lumen of the remaining plaque tissue to locate areas that did not have surface ulceration, thrombosis, or hemorrhage and could be easily dissected free. These areas were typically at or near the caudal end of the specimen and often included tissue from the common carotid artery. Segments of this plaque area (stable plaque tissue)—approximately equal in surface area to the ruptured specimen, but thinner (<1 mm)—were dissected free. If possible, more than 1 stable segment was dissected free per sample. A scalpel was used to cut thin slices from the caudal and cranial ends of these stable plaque specimens, and the slices were placed in 10% formalin for 48 hours, then transferred to 70% ethanol. The remainder of the stable plaque specimens were snap-frozen in liquid nitrogen and stored at −80°C for later protein extraction.
We initially collected 16 carotid plaques. One plaque was delivered as a fragment and was excluded from further study. On gross examination, 8 plaques did not show any evidence of luminal thrombosis, one plaque had equivocal evidence of luminal thrombosis, and one plaque had only a small area of luminal thrombosis; all were excluded. Extracts of the remaining 6 ruptured plaques (and of adjacent stable plaque tissue) were analyzed using shotgun proteomics. We planned to analyze these same extracts using PROTOMAP; however, the extracts were lost in a laboratory accident. Therefore, we collected an additional 6 carotid plaques. Extracts of this second set of 6 plaques—plus new extracts of back-up ruptured and stable segments of one plaque from the first set of 6 plaques—were processed for PROTOMAP analyses. Because of technical issues (eg, disconnected tubing resulting in sample loss), only 5 of these 7 samples yielded PROTOMAP data. Extracts of the second set of 6 plaques were also analyzed with shotgun proteomics as a validation cohort for discoveries made with the first set of 6 plaques.
Protein Extraction From Tissues
To develop a method that would enrich the extracts in ECM proteins, we tested 2 tissue extraction protocols. According to the first protocol (P1; similar to a published protocol),32 frozen tissue was pulverized in liquid nitrogen and resuspended in 20 mmol/L phosphate buffer pH 7.0 with 10 mmol/L EDTA, 1X Halt protease-inhibitor cocktail, and 1X Halt phosphatase-inhibitor cocktail. The suspension was incubated at 37°C for 10 minutes then centrifuged at 12 000g for 15 minutes at 4°C. The supernatant (fraction 1; termed P1F1) was stored at −80°C. The pellet was resuspended in 4 mol/L guanidine HCl in 50 mmol/L acetate buffer (pH 5.8), incubated overnight on a rotator at 4°C, then spun at 16 000g for 15 minutes at 4°C. The supernatant (fraction 2; termed P1F2) was dialyzed against distilled water at 4°C overnight and either stored at −80°C (for shotgun proteomics) or lyophilized (for PROTOMAP).
According to the second protocol (P2), frozen tissue was pulverized in liquid nitrogen and resuspended in ice-cold 50 mmol/L Tris 10 mmol/L EDTA (pH 8.0) with 1X Halt protease-inhibitor cocktail and 1X Halt phosphatase-inhibitor cocktail. The suspension was homogenized through sonication on ice: 3 cycles of 10-second on/10-second off, 5 minutes on ice, then another 3 cycles of sonication (10-second on/10-second off), using a sonic dismembrator (Fisher Scientific, no. FB120) at an amplitude setting of 40%. The suspension was then centrifuged at 100 000g for 45 minutes at 4°C. The supernatant (fraction 1, termed P2F1) was stored at −80°C. The pellet was resuspended in ice-cold 50 mmol/L Tris 10 mmol/L EDTA (pH 8.0), and sonication was performed followed by centrifugation, as for fraction 1. The resulting supernatant (fraction 2, termed P2F2) was stored at −80°C.
Because pilot experiments testing the protocols described above showed that the P1F2 fraction contained the largest number of ECM proteins (Results), we used the P1 protocol to extract protein from experimental mouse aortas and from human carotid plaques and performed all proteomics analyses on fraction 2. For mice that did not receive BMT, individual aortas (n=12) were extracted and extracts analyzed individually. For mice that received BMT, individual aortas (n=54) were pooled into groups of 3 and extracted (33 aortas from SR-uPA+/0 recipients were pooled to generate 11 samples; 21 aortas from SR-uPA0/0 recipients were pooled to generate 7 samples). We pooled samples to have sufficient protein for both shotgun proteomics and PROTOMAP analyses. For both SR-uPA+/0 and SR-uPA0/0 BMT recipients, the 6 pooled samples with the highest amounts of protein were used for proteomics analyses; other samples were not analyzed.
Sample Processing and Shotgun Proteomics Analysis
The concentration of protein in aortic and carotid extracts was measured with the Bradford assay. For trypsin digestion, an aliquot of protein extract corresponding to 10 μg of protein was diluted first to 50 μL with 100 mmol/L ammonium bicarbonate and subsequently with 1% RapiGest (Waters 186001861) in 100 mmol/L ammonium bicarbonate to 100 μL (final concentration 0.5% RapiGest). Samples were then denatured and reduced with 5 mmol/L dithiothreitol by heating at 65°C for 1 hour, and alkylated with 15 mmol/L iodoacetamide (30 minutes at room temperature in the dark). Excess iodoacetamide was quenched with additional 5 mmol/L dithiothreitol, and the sample was digested with trypsin (Promega, V5111) at 1:20 w/w ratio overnight at 37°C with mixing. After digestion, RapiGest was hydrolyzed by addition of 1% trifluoroacetic acid, and the pellet was separated by centrifugation at 14 000g for 10 minutes. The samples were desalted by solid-phase extraction using Oasis HLB 96-well µElution Plate, dried down and stored at −80°C until LC-MS analysis. Before analysis, samples were reconstituted with 0.1% formic acid (FA) in 5% acetonitrile to a concentration of 0.2 μg/μL.
The digested peptides (0.3 μg for lysates) were injected on a trap column (in-house packed 40×0.1 mm, 5 µm XBridge BEH C18, Waters), desalted for 5 minutes at a flow of 4 μL/min and separated on a pulled tip analytical column (in-house packed 280×0.075 mm, XBridge BEH C18, 3.5 μm, Waters) heated to 50°C with a 3-segment linear gradient of acetonitrile, 0.1% FA (B) in water, 0.1% FA (A) as follows: 0 to 5 minutes 1% to 10% B, 5 to 155 minutes 10% to 25% B, 155 to 185 minutes 25% to 35% B, followed by column wash at 80% B and reequilibration at a flow rate 0.4 µL/min (Waters NanoACQUITY UPLC). MS/MS (tandem mass spectrometry) spectra were acquired on Orbitrap XL (Thermo Scientific) operated in data-dependent mode on charge states 2 to 4 with 8 MSMS scans with dynamic exclusion for 30 seconds, collision-induced dissociation (CID) fragmentation (NCE 35%) and MSMS acquisition in the linear ion trap. MS spectra were acquired at resolution 60 000 in the Orbitrap, and MSMS spectra (precursor selection window 2.0 Da) were acquired in the linear ion trap. Peptides and proteins were identified using the Comet search engine,33 with PeptideProphet and ProteinProphet validation34,35 (search criteria included 20 ppm tolerance window for precursors and nominal resolution for products, Cys alkylation, and Met oxidation as fixed and variable modifications, respectively). Except for analyses performed during method development, we applied stringent protein identification criteria: protein identifications were considered valid only if at least 3 unique peptide-spectrum matches were detected in at least 4 samples in an analyzed group of experimental or control samples. Proteins that failed this test were not included in later analyses. Even after applying this test, some of the identified proteins were not detected in all of the samples. For samples in which a protein was not detected, we used 0 for the number of peptide-spectrum matches for that protein.
Relative quantification of proteins was accomplished using spectral counting and the PepC statistical approach (PepC version 1.0.).36 We used PepC, a statistical procedure developed for proteomic data analysis and validated in multiple studies36–39 as a method for identifying proteins that are differentially abundant between groups of tissue extracts or between groups of tissue culture medium (CM). PepC combines dual statistical tests, t test, and G test, with random permutation testing to allow computation of false discovery rates (FDR). Before applying the PepC algorithm, the measured peptide-spectrum matches for each protein were normalized to the total peptide-spectrum matches across all proteins in that sample. Individuals responsible for performing the proteomics experiments and for the initial analyses of proteomics data were blinded both to mouse genotype and to whether individual plaque extracts were from ruptured or stable areas.
Sample Processing and Analysis by PROTOMAP Approach
A 100 µg aliquot of each protein sample was separated via a 10% SDS-PAGE gel for 850-volt hours. The gel was washed in water and manually excised into 0.5 cm bands. Bands that corresponded to the migration of molecular weight markers were noted, and this information was used to estimate the molecular weights of proteins migrating in each band. Bands were subjected to in-gel trypsin digestion as previously described.30 Briefly, bands were washed in 100 mmol/L ammonium bicarbonate, and proteins were reduced in 10 mmol/L tris(2-carboxyethyl) phosphine at 37°C for 0.5 hours and then alkylated with 55 mmol/L iodoacetamide in the dark for 0.5 hours. The bands were then dehydrated by washing in 1:1 acetonitrile:100 mmol/L ammonium bicarbonate. Gel bands were then dried and resuspended in 40 µL of trypsin at 10 ng/µL. Upon re-swelling of the gel bands, 25 mmol/L ammonium bicarbonate was added to a final volume of 200 µL, and the gel bands were placed at 37°C overnight. Supernatants containing peptides were removed, and the gel bands were further extracted with 5% FA and acetonitrile. The pooled supernatants were dried and stored at –80°C until analysis. Before analysis, samples were resuspended in 10 µL buffer A (95% H2O, 5% acetonitrile, 0.1% FA) and autosampler loaded onto a 100 µm (inner diameter) fused silica capillary column with a 5 µm tip containing 10 cm of C18 resin (Aqua 5 µm, Phenomenex).
LC-MS/MS (liquid chromatography-tandem mass spectrometry) analysis was performed on an LTQ ion trap mass spectrometer (ThermoFisher) coupled to an Agilent 1100 series high-performance liquid chromatography. Peptides were eluted from the column using a 2-hour gradient of 5% to 100% buffer B (buffer B: 20% H2O, 80% acetonitrile, 0.1% FA). The flow rate through the column was 0.25 µL/min, and the spray voltage was 2.5 kV. The LTQ spectrometer was operated in data-dependent scanning mode, with one full MS scan (400–1800 m/z) followed by an MS2 scan of the most abundant ion, followed by an MS2 scan of the second most abundant ion, sequentially through the seventh most abundant ion, with dynamic exclusion enabled (20-second duration). A subset of the human plaque samples was analyzed on an LTQ-Velos Elite Orbitrap mass spectrometer (ThermoFisher) coupled to an Agilent 1200 series high-performance liquid chromatography in similar fashion to the ones described above, but the full MS scan (120 000 resolution) was followed by twenty MS2 scans of the top 20 most abundant ions with dynamic exclusion enabled (20-second duration).
PROTOMAP Data Collection and Analysis
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE40 partner repository with the data set identifiers PXD020405 and PXD020406. Raw mass spectrometry data were converted to MS2 format using RawXtract (version 1.9.9.2).41 Peptide spectral matching was accomplished with ProLuCID.42 ProLuCID was configured to allow for differential oxidation of methionine (16 or 15.9949 amu for LTQ or Elite, respectively) and required cysteines to be carboxamidomethylated (+ 57, 57.0215 amu for LTQ or Elite, respectively). The data were searched using a human or mouse reverse-concatenated nonredundant (gene-centric) FASTA database that was assembled from the UniProt database (http://www.uniprot.org/) downloaded on 11/5/2012 or 11/9/2012, respectively. The resulting matched MS2 spectra were assembled into protein identifications and filtered using DTASelect (version 2.0.47) using default settings. The data were then processed using custom Perl scripts as previously described.30 For each resulting peptograph, spectral counts were averaged for each band and condition and displayed with error bars representing standard errors of the mean.
Individual proteins were identified by PROTOMAP if at least 20 total spectral counts were detected among all of the samples analyzed. To use the PROTOMAP M.W. COMPARE tool to identify proteins with a shift in average migration on SDS-PAGE between extracts of experimental and control samples, we used a spectral count minimum of 20 per protein and a minimum average migration shift of 3 bands between experimental and control samples. To use the PROTOSORT algorithm to identify proteins that display changes in abundance and/or migration, we used a fold-change of 3 between the experimental and control samples, a mimimum of 10 spectral counts for the fragment to be recognized, and a mimimum of 25 total spectral counts per protein.
Histology
Slices of fixed human plaque tissue (2 slices from each of the ruptured and stable specimens) were processed into paraffin. From each tissue block, five 5-µm-thick sections were cut at eight 50-µm steps (5 sections, then 25 µm discarded tissue, then repeat × 7) covering a total of 400 µm. Hematoxylin and eosin and Masson trichrome stains were performed on eight 50-µm step sections per tissue slice and were examined with the assistance of a vascular pathologist (R.F. Nicosia). An observer blinded to specimen identity graded the slides for the presence or absence of intimal hemorrhage/thrombosis (ie, extravascular red blood cells or fibrin clot, detected as a bright red intimal mass on trichrome-stained slides), cholesterol clefts, and calcification. Intimal hemorrhage/thrombosis was graded as definite (large amount of red blood cells/clot in intima), possible (small amount of free red blood cells in intima, either with minor cap disruption or no cap disruption; thought potentially due to processing artifact), or absent. Cholesterol clefts and calcification were graded as present or absent.
Immunohistochemistry
Sections of both stable and ruptured human plaque segments described above were used for immunohistochemistry. All immunostaining was performed using a Leica BOND RX Automated IHC Research Stainer, with reagents from Leica Biosystems except as indicated. Initial studies were performed only with sections of stable plaques, with the goal of localizing LAMA5 (laminin subunit alpha 5), HSPG2 (heparin sulfate proteoglycan 2), and COL18A1 (collagen type XVIII alpha 1 chain) and confirming that these proteins are present in caps of advanced yet stable atherosclerotic lesions. For all slides, antigen retrieval was performed with proteinase K for 15 minutes at 37°C. Primary antibodies and dilutions were rabbit anti-mouse laminin α5 serum (clone 504; a gift from Dr Lydia Sorokin, Munster University; 1:1000),43 mouse monoclonal anti-mouse endostatin (a fragment of COL18A1; Santa Cruz Biotechnology sc-32720; 1:100), and rat monoclonal anti-HSPG2 (Abcam ab2501; 1:100). Negative control primary antibodies and dilutions were rabbit IgG (R&D Systems AB-105-C; 1:1000), mouse IgG (Mouse Negative Control Clone MOPC-21; PA0996; 1.7 mg/L), and rat IgG2b K isotype Control (BD Biosciences 553986; 1:1000). Bound primary antibodies were detected with the anti-rabbit IgG Polymer Detection System, the anti-mouse IgG Polymer Detection System, and mouse-adsorbed unconjugated rabbit anti-rat IgG.
To test whether LAMA5 and HSPG2 were depleted in caps of ruptured human plaques, we performed additional immunohistochemical stains of sections of 3 ruptured human plaque segments and one stable human plaque segment, using antibodies validated by Rickelt and Hynes for use in immunohistochemistry.44 Primary antibodies (mouse monoclonal anti-LAMA5; AMAb91124; Atlas Antibodies and rabbit polyclonal anti-HSPG2; Boster Biological Technology; PB9277) were applied at 1:250 and 1:750 dilutions, respectively. According to data provided by the manufacturers (available on product sheets), both of these antibodies detect the corresponding full-length proteins (400 and 468 kDa, respectively). Negative control primary antibodies are listed above. Primary antibodies were applied after antigen retrieval with the Leica HIER 2 reagent (EDTA; for LAMA5) or HIER 1 reagent (citrate; for HSPG2), with both solutions applied for 20 minutes at 100°C. Bound LAMA5 antibody was detected with rabbit anti-mouse IgG, followed by incubation with HRP (horseradish peroxidase)-polymerized goat anti-rabbit IgG. Bound HSPG2 antibody was detected with HRP-polymerized goat anti-rabbit IgG. Peroxidase activity was detected with diaminobenzidine substrate, and slides were counterstained with hematoxylin.
Immunoblotting
We performed Western blot analysis of tissue extracts from the same stable and ruptured human plaque segments (7 each) used for shotgun proteomics. For analysis of HSPG2 and LAMA5, 30 µg of the extracts were loaded onto 3% to 8% Tris-acetate SDS-PAGE gels and transferred to nitrocellulose membranes. HSPG2 was visualized with 0.375 µg/mL of HSPG2 antibody (PB9277; Boster Biological Technology) in 5% milk/PBST (phosphate-buffered saline/Tween) overnight. LAMA5 was visualized with a 1:800 dilution of LAMA5 antibody (AMAb91124, Atlas Antibodies) in 5% milk/PBST overnight, after stripping the HSPG2 blot. For analysis of COL18A1, 30 µg of the same tissue extracts were run on 4% to 12% Tris-Glycine SDS-PAGE gel, transferred to a nitrocellulose membrane, and visualized with COL18A1 antibody (ab207162 Abcam) diluted 1:1000 in 5% milk/PBST overnight. In all cases, bound antibody was detected with an appropriate secondary antibody coupled with HRP and SuperSignal West Femto Maximum Sensitivity Substrate (no. 34096, ThermoFisher Scientific).
Statistics
We assessed the correlation of individual protein spectral counts among extracts of mouse aortas and extracts of human plaque segments by calculating pairwise Spearman correlation coefficients. We applied correspondence analysis, a form of multidimensional scaling45 as implemented in the TM4 software46 (https://sourceforge.net/projects/mev-tm4/), to determine whether global variation in the ensemble of extracted proteins distinguished the 2 experimental groups of mouse aortas and the 2 experimental groups of human plaque segments. Multiple hypothesis testing was addressed using a permutation-based FDR analysis within the PepC algorithm and with an FDR threshold of <0.05 to determine significant differential abundance. PepC is sensitive to the distribution and dispersion of spectral counts. In the second shotgun proteomics study of human plaques, differences in abundance of 10 prospectively identified proteins were analyzed with the nonparametric Wilcoxon rank-sum test. To facilitate comparison of data sets, we also applied the Wilcoxon rank-sum test retrospectively to analyze differences in human and mouse ECM proteins that we had already identified as statistically significant using PepC, a method that controls for multiple hypothesis testing. Two-dimensional hierarchical clustering of differentially abundant proteins was performed, and the primary branch point was used to segregate samples and proteins.46
For functional enrichment analyses, we used web-based programs (Webgestalt: http://www.webgestalt.org/ and Reactome: https://reactome.org/), along with proteins identified by PepC as differentially abundant. These programs use several databases: Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genome, Reactome, Disgenet, Mammalian Phenotype Ontology, Human Phenotype Ontology, and Online Mendelian Inheritance in Man. Functional enrichment was determined using a hypergeometric test, and over-represented categories were identified after adjusting enrichment P values for multiple hypothesis testing using a strict FDR cutoff <0.01 (more-complete lists using FDR <0.05 are provided in the Tables in the Data Supplement).
By leveraging available gene-product interaction resources (https://analysis.ingenuity.com/pa/, https://string-db.org/), a protein interaction network47,48 was constructed based on differentially abundant proteins extracted from mouse aortas. To increase confidence in biological relevance, the connectivity among network members was limited to relationships based on experimentally verified direct interactions.
Results
Development of a Protocol to Extract Aortic ECM Proteins
Because we hypothesized that plaque rupture is initiated by proteolysis of artery wall ECM, we developed a tissue extraction protocol that maximizes extraction of arterial ECM proteins. We compared 2 protocols for their ability to extract ECM proteins, using rabbit aortas because they are larger than mouse aortas and, therefore, more suitable for protocol development. Both protocols (termed P1 and P2; Methods) provide 2 protein fractions per sample (termed F1 and F2). Analysis of the 4 fractions (P1F1, P1F2, P2F1, and P2F2) using shotgun proteomics suggested that P1F2 was most enriched in ECM proteins (data not shown). To confirm this, we next compared P1 and P2 using thoracic aortas of Apoe−/− mice. We extracted 2 aortas with P1 and 2 aortas with P2, yielding 8 total samples (2 each of P1F1, P1F2, P2F1, and P2F2), and confirmed that the P1F2 fractions contained the largest number of unique proteins as well as the most unique proteins in the GO category ECM (Data Set I in the Data Supplement). Of the 73 unique ECM proteins detected in any of the 4 fractions, 68 (93%) were present in P1F2 (Figure I in the Data Supplement). We, therefore, used the P1F2 fractions for all remaining mouse and human tissue analyses. Importantly, although the P1F2 fraction extracts were optimized for enrichment with ECM proteins, these fractions also contained many cell-associated proteins, as described below.
Proteomics of Aortic Extracts From SR-uPA+/0 and SR-uPA0/0 Apoe−/− Mice
To begin to discover how elevated protease expression alters the atherosclerotic plaque proteome, we extracted proteins from thoracic aortas of 20-week-old SR-uPA+/0 and SR-uPA0/0 mice (both Apoe−/−; n=6 per group) and analyzed them by shotgun proteomics. This analysis identified 775 unique proteins (Data Set II in the Data Supplement). The relative abundance of individual proteins in the extracts was highly reproducible among the 12 samples with mean pairwise Spearman correlation coefficients of 0.86 (range 0.79–0.89) within each genotype and 0.78 (range 0.74–0.82) between the 2 genotypes.
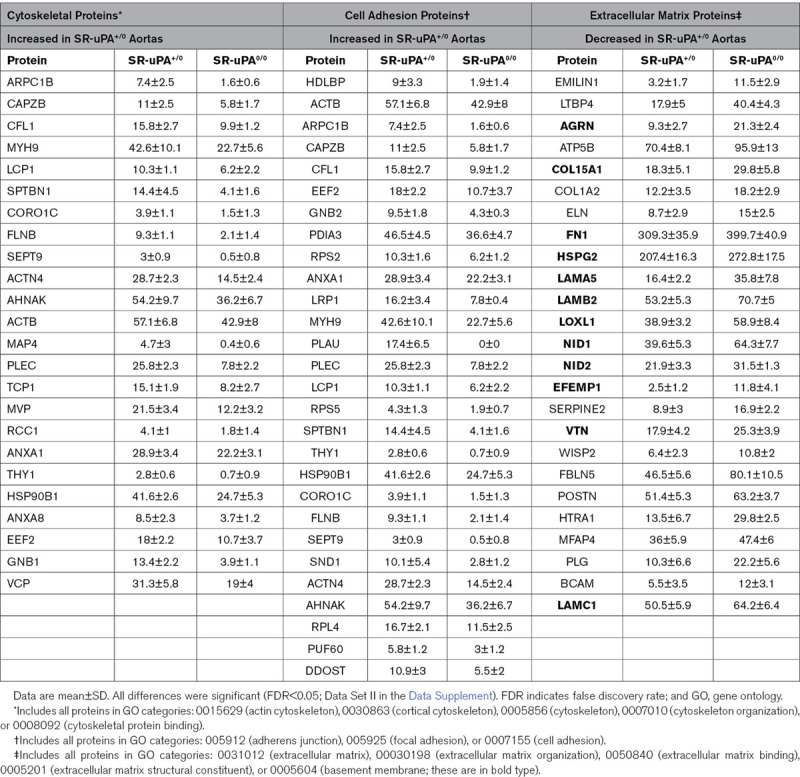
Correspondence analysis based on variability in abundance across all 775 unique proteins clearly segregated SR-uPA+/0 from SR-uPA0/0 aortas, confirming global differences between the aortic proteomes of the 2 genotypes (Figure 1A). Using a previously validated statistical procedure (PepC),36,37 we detected significant differences in individual protein abundance between the SR-uPA+/0 and SR-uPA0/0 aortas. One hundred proteins (13% of all identified proteins) were differentially abundant in SR-uPA+/0 versus SR-uPA0/0 aortas at FDR<0.05 (Data Set II in the Data Supplement).37 Proteins increased in SR-uPA+/0 aortas included PLAU as well as 2 other extracellular proteases that are involved in ECM remodeling (MMP-2 matrix [metalloproteinase 2] and MMP-3 matrix [metalloproteinase 3]). Surprisingly, the remaining 58 more abundant proteins included many intracellular proteins related to the cytoskeleton and cell adhesion (Table 1), suggesting that cytoskeletal structure and cell adhesion are altered in response to elevated vascular protease activity. Furthermore, elevated extracellular protease activity in SR-uPA+/0 aortas29 led to decreased abundance of numerous (25) aortic ECM proteins. Notably, 12 of the 25 (48%) were basement-membrane proteins (Table 1).
Figure 1.
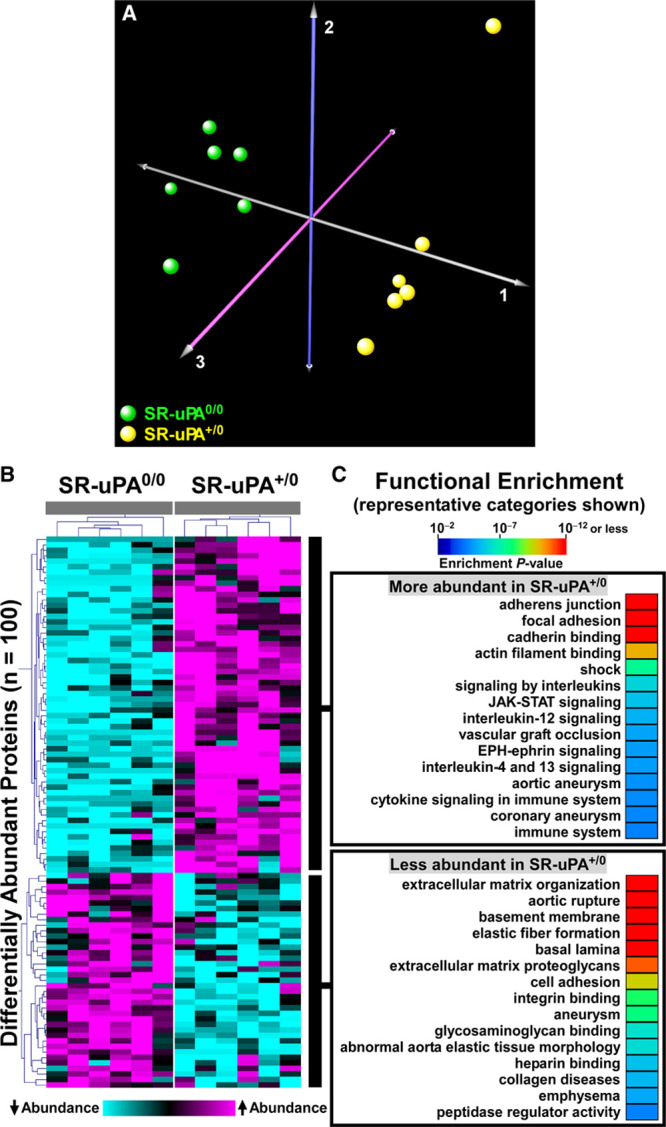
Proteomic analyses identify global differences in protein abundance and biological processes in aortas of SR-uPA+/0 and SR-uPA0/0 mice.
A, Correspondence analysis was applied to 775 proteins identified in extracts of aortas of SR-uPA+/0 and SR-uPA0/0 mice. Individual mice of the 2 genotypes are represented by colored spheres. B, Heatmap of 2-dimensional hierarchical cluster analysis for the 100 differentially abundant proteins (false discovery rate [FDR]<0.05). A complete list of these proteins is in Data Set II in the Data Supplement. C, Functional enrichment analysis of the differentially abundant proteins reveals over-represented biological processes. A complete list of these categories is in Data Sets III and IV in the Data Supplement.
Table 1.
Cytoskeletal, Cell Adhesion, and Extracellular Matrix Proteins With Significantly Different Abundance in Aortas of SR-uPA+/0 Mice
To obtain a more global view of the pathways that connect elevated vascular protease activity to plaque rupture, we performed unsupervised hierarchical cluster analysis on the 100 differentially abundant proteins (Figure 1B). We also interrogated multiple gene annotation, pathway, and mammalian disease databases, with a goal of applying functional enrichment analysis to the 2 distinct profiles that emerged from the cluster analysis. These analyses confirmed that the group of proteins significantly more abundant in SR-uPA+/0 aortas was highly enriched in functional categories related to cell adhesion (eg, adherens junction, focal adhesion, and cadherin binding; FDR<5×10−10 for all), cytoskeleton (eg, actin cytoskeleton, cytoskeletal protein binding, cytoskeleton organization; FDR<2×10−4 for all) as well as immuno-inflammatory programs (eg, signaling by interleukins, JAK-STAT (Janus kinase-signal transducer and activator of transcription) signaling, and cytokine signaling; FDR<1.5×10−3 for all; Figure 1C and Data Set III in the Data Supplement, in which proteins in each over-represented category are listed). A review of functional categories over-represented among proteins that were less abundant in SR-uPA+/0 aortas confirmed significant reductions in ECM and basement-membrane proteins (eg, ECM organization, basement membrane, elastic fiber formation, basal lamina, and ECM proteoglycans; FDR<4×10−10 for all; Figure 1C and Data Set IV in the Data Supplement, in which proteins in each over-represented category are listed).
A protein interaction network was built based on known direct interactions among the differentially abundant proteins (Figure 2 and Data Set V in the Data Supplement) and confirmed that the differentially abundant proteins that are associated with the extracellular space tended to have decreased abundance in SR-uPA+/0 aortas; whereas, the differentially abundant proteins that are located in the cell membrane, cytoplasm, and nucleus tended to have increased abundance in SR-uPA+/0 aortas. This observation was further corroborated by GO cellular component enrichment analysis (Data Sets III and IV in the Data Supplement). The protein interaction network analysis also identified several densely connected hubs, including FN1 (fibronectin 1), MYH9 (myosin heavy chain 9), and ACTB (beta actin), that may represent drivers of the network’s function.49,50 Although extracellular and structural proteins, such as FN1, MYH9, and ACTB, may not be thought of as likely network drivers, ample evidence from humans and mice suggests that alterations of extracellular and structural proteins can profoundly impact complex intracellular processes.51–53 Collectively, these data indicate that PLAU overexpression in mouse atherosclerotic tissue—which can cause plaque rupture29—activates proinflammatory signals, depletes ECM components including basement-membrane proteins, and upregulates intracellular pathways related to cell-cell adhesion, cell-matrix adhesion, and the cytoskeleton.
Figure 2.
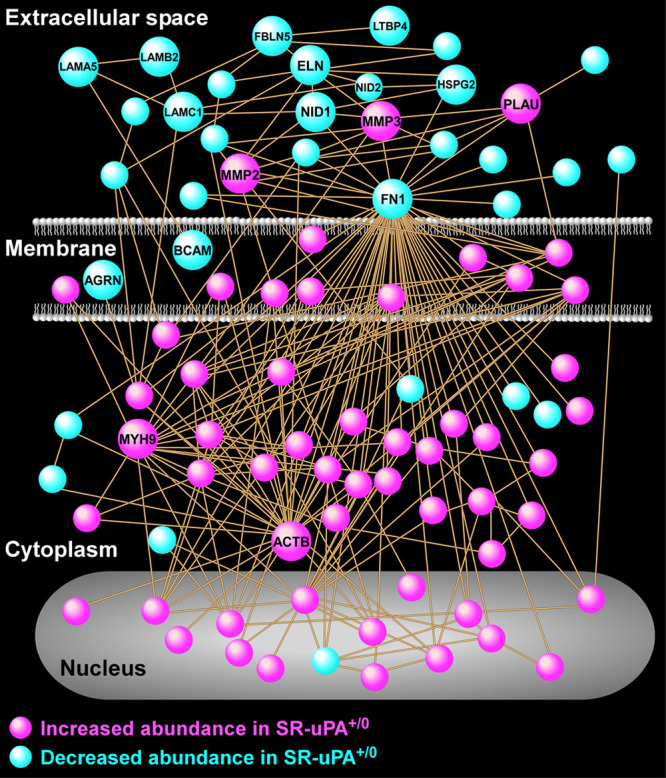
Protein interaction network analysis reveals numerous interactions of proteins that are differentially abundant in aortas of SR-uPA+/0 mice.
A protein-protein relational network was built based on experimentally validated direct interactions. The network is comprised of 87 proteins, each portrayed as a circular node (all nodes are identified in Data Set V in the Data Supplement). Key highly connected nodes (hubs) are labeled together with 2 members of the matrix metalloproteinase family of extracellular proteases and several extracellular matrix components. ACTB indicates beta actin; AGRN, agrin; BCAM, basal cell adhesion molecule; ELN, elastin; FBLN5, fibulin 5; FN1, fibronectin 1; HSPG2, heparan sulfate proteoglycan 2; LAMA5, laminin subunit alpha 5; LAMB2, laminin subunit beta 2; LAMC1, laminin subunit gamma 1; LTBP4, latent transforming growth factor binding protein 4; MMP2, matrix metalloproteinase 2; MMP3, matrix metalloproteinase 3; MYH9, myosin heavy chain 9; NID1, nidogen1; NID2, nidogen 2; and PLAU, urokinase-type plasminogen activator.
Proteomics of Medium Conditioned by SR-uPA+/0 and SR-uPA0/0 Aortas
Several groups have investigated the pathogenesis of plaque rupture via proteomic analyses of the plaque secretome (ie, proteins released in culture by explanted human plaques).54–57 Therefore, as a complementary approach to our aortic tissue extract analyses, we used the same shotgun proteomics methods to analyze the secretome of SR-uPA+/0 and SR-uPA0/0 aortas (n=5 per group). Proteomic analysis of aortic tissue CM detected 921 unique proteins (Data Set VI in the Data Supplement). This approach offered a distinct view of the aortic proteome because nearly half of these proteins (431; 47%) were not identified in tissue extracts.
Statistical analysis with PepC revealed that 45 of the 921 proteins (4.9%) were differentially abundant (FDR<0.05) in CM of SR-uPA+/0 versus SR-uPA0/0 explants. Functional enrichment analysis of the 45 differentially abundant CM proteins revealed—congruently with results obtained with aortic tissue extracts—that categories related to cell adhesion and cytoskeleton (eg, focal adhesion, cell-substrate junction, adherens junction, and cytoskeleton) were significantly over-represented among proteins that were increased in SR-uPA+/0 CM (FDR≤3×10−6 for all; Data Set VII in the Data Supplement, in which proteins in each over-represented category are listed). Functional categories related to apoptosis (eg, apoptotic execution phase, apoptotic cleavage of cellular proteins, and apoptosis) were also significantly over-represented among proteins that were increased in SR-uPA+/0 CM (FDR≤5×10−3 for all; Data Set VII in the Data Supplement). Also congruent with the aortic tissue extract data, ECM-related categories as well as other categories related to the extracellular space (eg, extracellular space, ECM organization, blood coagulation, fibrinolysis, complement, and coagulation cascades) were significantly over-represented among proteins that were less abundant in SR-uPA+/0 CM (FDR≤10−5 for all; Data Set VIII in the Data Supplement, in which proteins in each over-represented category are listed).
Proteomics of Aortic Extracts From SR-uPA+/0 and SR-uPA0/0 BMT Recipients
Older SR-uPA+/0 mice cannot be used to discover the impact of increased vascular protease activity on the proteome of more-advanced atherosclerotic lesions because SR-uPA+/0 mice die suddenly between 10 and 30 weeks of age.27 We, therefore, used a BMT approach and 35-week-old Apoe−/− recipients to determine how increased protease activity alters the proteome of advanced atherosclerotic lesions (which are present in innominate arteries of older Apoe−/− mice).28,58 Eight weeks after BMT from either SR-uPA+/0 or SR-uPA0/0 donors, we removed recipient innominate arteries (with proximal aortas attached), extracted proteins, and analyzed the extracts with shotgun proteomics. We included the proximal aorta because we were unable to extract sufficient protein from innominate arteries alone. Importantly, although lesions in these 43-week-old mice are more advanced than lesions in the 20-week-old SR-uPA+/0 mice described above, the SR-uPA+/0 BMT recipients are exposed to elevated levels of PLAU for a substantially shorter period of time (8 versus 20 postnatal weeks).
Analysis of aortic extracts of BMT recipients (n=6 samples per group, each pooled from 3 mice) revealed 1465 unique proteins. PepC analysis identified 56 proteins that were differentially abundant between SR-uPA+/0 versus SR-uPA0/0 BMT recipients (3.8% of the total proteins identified; FDR<0.05; Data Set IX in the Data Supplement). Pathway analyses revealed that extracellular space was the only functional category that was over-represented among the 32 more abundant proteins (FDR=8.5×10−5; Data Set X in the Data Supplement, in which proteins in each over-represented category are listed). Functional categories significantly over-represented among the 24 less-abundant proteins also included extracellular space (FDR=4×10−8; Data Set XI in the Data Supplement, in which proteins in each over-represented category are listed). Other categories over-represented among less-abundant proteins again included ECM as well as glycosaminoglycan binding, proteinaceous ECM, focal adhesion, and complement and coagulation cascades (FDR≤1×10−3 for all; Data Set XI in the Data Supplement).
Proteomics of Ruptured and Stable Areas of Human Carotid Artery Plaques
To determine whether the changes in protein abundance that we detected in the mouse models are also present in ruptured human plaques, we applied shotgun proteomics to extracts of ruptured and stable areas of carotid artery plaques removed from 6 individuals (12 total samples; all plaques were removed for clinical indications). Demographic and clinical characteristics of the 6 patients from whom these plaques were removed are in Table I in the Data Supplement (cohort 1). Histological sections of ruptured and stable areas were examined for evidence of advanced atherosclerosis, including intimal hemorrhage/thrombosis, cholesterol clefts, and calcification (Figure 3). As expected, intimal hemorrhage/thrombosis was far more common in ruptured areas: sections from all 6 ruptured areas had either definite (5) or possible (1) intimal hemorrhage/thrombosis whereas none of the sections from stable areas showed definite intimal hemorrhage/thrombosis and only one showed possible intimal hemorrhage. Cholesterol clefts and calcification were equally common in sections from both ruptured and stable areas (≈50% of sections in both groups showed cholesterol clefts, calcification, or both). Therefore, advanced atherosclerosis was present in all samples, but intimal hemorrhage/thrombosis was essentially confined to samples taken from areas judged by gross examination to be ruptured plaques.
Figure 3.
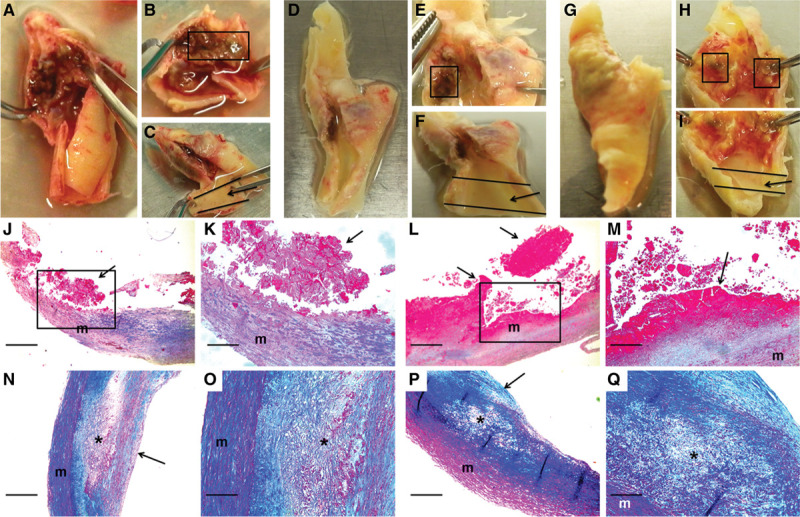
Macroscopic and histological images of ruptured and stable segments of human carotid plaques.
Human carotid plaques were removed for clinical indications. Images are of 3 freshly harvested plaques (A–C, D–F, and G–I) and stained sections of 2 ruptured (J–K and L–M) and 2 stable (N–O and P–Q) plaques. Ruptured segments (boxes in B, E, and H) and stable segments (arrows in C, F, and I) were dissected free. A scalpel was used to cut thin slices from the caudal and cranial edges of the dissected segments. These slices were embedded in paraffin, and the remainder of each segment was used for protein extraction. Sections of 2 ruptured plaque segments (J–M; boxes in J and L are expanded in K and M) show disrupted intima with adherent thrombus (arrows; thrombus fragmentation is likely sectioning artifact). Sections of 2 stable plaque segments (N–O and P–Q; O and Q are expanded from N and P, respectively) show intact fibrous caps (arrows in N and P) and lipid-rich necrotic cores (asterisks) containing cholesterol clefts and foam cells. J–Q, m=vascular media. Size bars are 50 µm (J, L, N, and P) and 20 µm (K, M, O, and Q). J–Q, Masson trichrome stain.
Shotgun proteomics of the 12 plaque extracts identified 1161 unique proteins (Data Set XII in the Data Supplement). Relative abundance of individual proteins in the extracts was highly reproducible among both the ruptured and stable samples, with mean pairwise Spearman correlation coefficients of 0.86 (range 0.79–0.90) among the stable samples and 0.80 (range 0.71–0.85) among the ruptured samples. In contrast, the relative protein abundance was much less correlated among the ruptured and stable samples: mean pairwise Spearman correlation coefficients of 0.60 (range 0.37–0.78). Correspondence analysis based on variability in abundance across all 1161 proteins clearly segregated the ruptured and stable samples (Figure 4A). Differential protein abundance analysis using PepC found that 489 of the 1161 proteins (42%) were differentially abundant between ruptured and stable areas. Among the 150 more abundant proteins, only one (0.7%) was also more abundant in aortic extracts of 20-week SR-uPA+/0 versus SR-uPA0/0 mice: HIST1H1B (histone cluster 1 H1 family member B). Among the 339 less-abundant proteins, only 13 (4%) were also less abundant in aortic extracts of 20-week SR-uPA+/0 versus SR-uPA0/0 mice. Strikingly, 7 of the 13 proteins that were significantly less abundant in both ruptured human plaques and SR-uPA+/0 aortas were basement-membrane proteins (AGRN [agrin], HSPG2, LAMA5, LAMB2 [laminin subunit beta 2], LAMC1 [laminin subunit gamma 1], NID1 [nidogen 1], and NID2 [nidogen 2]), and another 4 of the 13 proteins were also ECM proteins (ELN [elastin], FBLN5 [fibulin 5], LTBP4 [latent transforming growth factor binding protein 4], and MFAP4 [microfibril-associated protein 4]). The cell-surface laminin receptor (BCAM [basal cell adhesion molecule]) was also less abundant in both SR-uPA+/0 aortas and ruptured human plaques. Therefore, at the individual protein level, overlap between SR-uPA+/0 aortas and ruptured human plaques is highly focused on a small number of basement-membrane and other ECM proteins.
Figure 4.
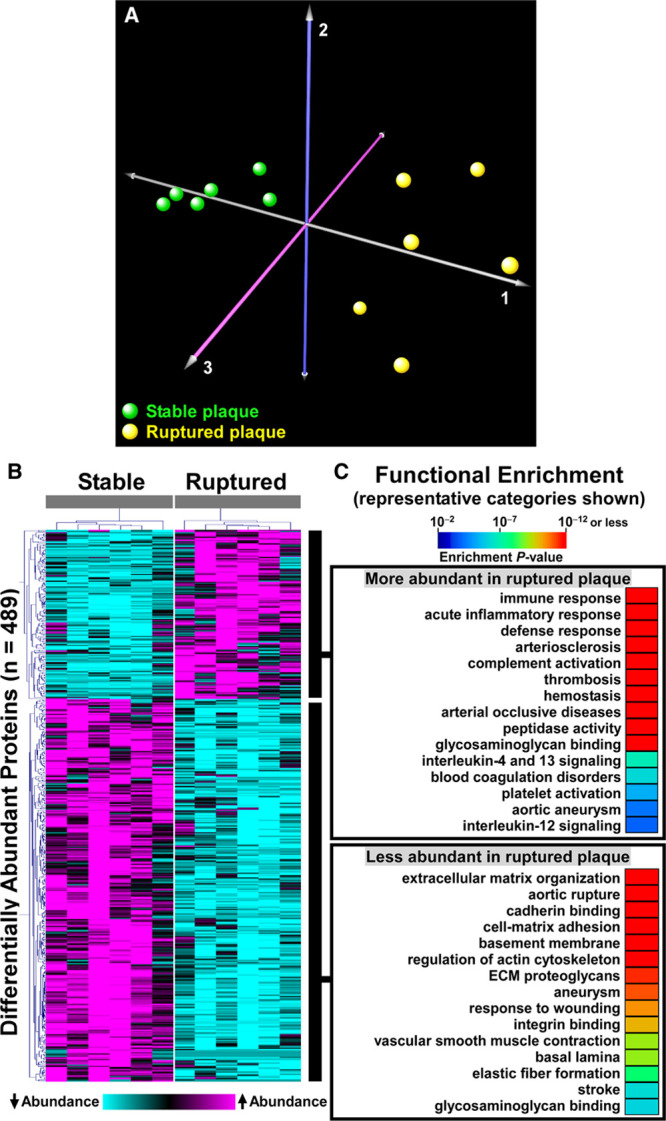
Proteomic analyses identify global differences in protein abundance and biological processes in paired samples of ruptured and stable human carotid artery plaque segments.
A, Correspondence analysis was applied to the 1161 identified proteins. Individual plaques from the 2 areas are represented by colored spheres. B, Heatmap of 2-dimensional hierarchical cluster analysis for the 489 proteins that are differentially abundant (false discovery rate [FDR]<0.05) between ruptured and stable plaque segments. A complete list of these proteins is in Data Set XII in the Data Supplement. C, Functional enrichment analysis of the differentially abundant proteins reveals over-represented biological processes. A complete list is in Data Sets XIII and XIV in the Data Supplement.
Unsupervised hierarchical cluster analysis of the 489 differentially abundant proteins revealed distinct protein abundance patterns in ruptured versus stable plaque areas (Figure 4B). Functional enrichment analysis further revealed that categories over-represented among proteins that were more abundant in ruptured areas (Figure 4C and Data Set XIII in the Data Supplement, in which proteins in each over-represented category are listed) included those related to inflammation (eg, activation of immune response, acute inflammatory response, defense response, and complement activation; FDR<1×10−15 for all), atherosclerosis (eg, arteriosclerosis and arterial occlusive disease; FDR<5×10−12 for both), blood coagulation (eg, thrombosis, hemostasis, blood coagulation, and platelet degranulation; FDR<5×10−12 for all) and apoptotic cell clearance (FDR<6×10−3). Similar to our results comparing aortas of SR-uPA+/0 versus SR-uPA0/0 mice, categories over-represented among proteins that were less abundant in the ruptured areas included several ECM and basement-membrane protein-related categories (eg, ECM, basement membrane, basal lamina, and laminin complex; FDR<8×10−5 for all; Figure 4C and Data Set XIV in the Data Supplement, in which proteins in each over-represented category are listed). Other categories over-represented among less-abundant proteins included cell adhesion (adherens junction, focal adhesion, cell-substrate junction, cadherin binding, and integrin binding; FDR<2×10−8 for all), and cytoskeleton (cytoskeleton, actin cytoskeleton, regulation of cytoskeleton organization FDR<3×10−10 for all).
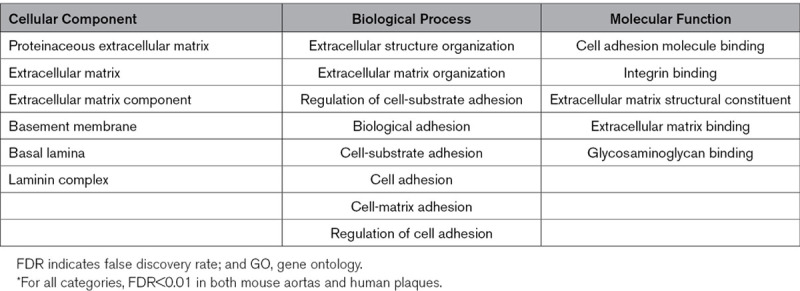
Despite little overlap at the individual protein level between the SR-uPA+/0 model (ie, 20-week-old transgenic mice) and ruptured human plaques, several of the functional categories that were over-represented among the differentially abundant proteins in ruptured human plaques overlap congruently with functional categories that were over-represented among the differentially abundant proteins in aortic extracts of SR-uPA+/0 mice (Figures 1C and 4C). This overlap between human and mouse results was particularly robust for functional categories related to the less-abundant proteins, including ECM and basement-membrane proteins (Table 2). Among proteins with increased abundance both in ruptured human plaques and in the SR-uPA+/0 model, categories related to the immune response (eg, interleukin-4 and -13 signaling) were highly over-represented. Among proteins with decreased abundance in both settings, categories related to the ECM and basement membrane were highly over-represented (eg, ECM, proteinaceous ECM, ECM organization, ECM component, ECM proteoglycans, ECM binding, ECM structural constituent, basement membrane, basal lamina, laminin complex, elastic fiber formation, basal lamina, integrin binding, and cell adhesion molecule binding; Figures 1C and 4C, Table 2, Data Sets III, IV, XIII, and XIV in the Data Supplement). However, enrichment patterns in some functional categories were discordant. For example, proteins in GO categories adherens junction, focal adhesion, and actin cytoskeleton were more abundant in SR-uPA+/0 aortas but less abundant in ruptured human plaques (FDR<5×10−6 for all).
Table 2.
GO Categories Significantly* Over-represented by Less-Abundant Proteins in Both SR-uPA+/0 Aortas and Ruptured Segments of Human Plaques
We also compared the human plaque proteomic data to results obtained with the mouse BMT model. Among the 150 more abundant proteins in ruptured plaque segments, only one protein (0.7%) was also more abundant in aortic extracts of 20-week SR-uPA+/0 versus SR-uPA0/0 BMT recipients (LTF [lactotransferrin]). Among the 339 less-abundant proteins in ruptured plaque segments, only 6 (2%) were also less abundant in aortic extracts of 20-week SR-uPA+/0 versus SR-uPA0/0 BMT recipients. Four of the 6 proteins are in the GO category ECM (ABI3BP [ABI family member 3 binding protein], LTBP2, LTBP4, and MYL6 [myosin light chain 6]), and one is an adhesion-plaque protein (ZYX [zyxin]); none were basement-membrane proteins. Compared with this small number of congruently abundant proteins, there was a more substantial overlap in functional category over-representation between the human plaques and aortic extracts of the BMT recipients. The only category over-represented among more abundant proteins in aortic extracts of SR-uPA+/0 BMT recipients (extracellular space; FDR=8.5×10−5) was also highly over-represented among the more abundant proteins in ruptured human plaques (FDR<1×10−15). Ten of the 18 (56%) functional categories over-represented among less-abundant proteins in aortic extracts of SR-uPA+/0 BMT recipients (Data Set XI in the Data Supplement; FDR<0.01 for all) were also over-represented among less-abundant proteins in ruptured human plaques (Data Set XIV in the Data Supplement; FDR<1×10−3 for all). These categories were again focused on ECM and included ECM, proteinaceous ECM, ECM organization, focal adhesion, collagen binding, glycosaminoglycan binding, and others.
PROTOMAP Analysis of Extracts of Human Carotid Plaques and Mouse Aortas
The reduced abundance of several structural ECM proteins—including basement-membrane proteins—in both SR-uPA+/0 aortas and ruptured human plaques suggested that these proteins might be targets of proteolysis. We, therefore, analyzed the human plaque extracts with PROTOMAP, a proteomic analysis methodology that identifies proteolytic events and peptide fragments in complex protein mixtures.30 For these analyses, we collected 6 additional carotid plaques (all removed for clinical indications; Table I in the Data Supplement, cohort 2) and extracted proteins from a ruptured area and a stable area of each plaque. We also extracted proteins from backup segments of one plaque from the first patient cohort (Table I in the Data Supplement, cohort 1). Technically satisfactory data were obtained for both ruptured and stable areas of 5 of the 7 plaques. Demographic and clinical characteristics of the 5 patients whose plaques yielded satisfactory PROTOMAP data are in Table I in the Data Supplement (cohort 3).
A total of 2059 proteins were identified by PROTOMAP. Primary data are accessible at http://www.scripps.edu/cgi-bin/cravatt/pmap_project_page.pl?pname=Dichek (data set 161027 BvA). To detect evidence of increased proteolysis in ruptured versus stable plaque segments—and to potentially identify proteolytic fragments that are markers of plaque rupture—we first used the PROTOMAP M.W. COMPARE tool to identify proteins with a shift in average migration on SDS-PAGE between extracts of ruptured and stable plaques. This tool identified 49 proteins with differential migration in ruptured versus stable areas. However, only 5 of the 49 proteins had a higher average migration distance in the ruptured plaque samples (ie, a shift to lower molecular weight, suggestive of increased proteolysis). The remaining 44 proteins had a lower average migration distance (shift to higher molecular weight) in the ruptured plaque samples.
We next used the PROTOSORT algorithm, which identifies proteins that display changes in abundance and/or migration. We hypothesized that increased proteolysis of structural ECM components (or other proteins) in ruptured plaques would yield more abundant small peptides, with increased migration. We examined the peptographs (graphs of peptide abundance versus gel-slice number; a surrogate for molecular weight)30 of the 593 candidate proteins identified by PROTOSORT. In most cases, the peptographs indicated that these proteins were identified by PROTOSORT based on differences in protein abundance. However, we also identified several proteins that appeared more fragmented in extracts of ruptured areas (eg, CP [ceruloplasmin], AGT [angiotensinogen], ITIH4 [inter-alpha-trypsin inhibitor heavy chain], GPLD1 [phosphatidylinositol-glycan-specific phospholipase D], FA5 [coagulation factor 5], and SERPIND1 [serpin family D member 1]) and a smaller number of proteins that appeared more fragmented in extracts of stable areas (eg, LAMB2 [laminin subunit beta 2], SVIL [supervillin]; Table II in the Data Supplement and Figure 5A). Notably, although most of the proteins that appeared more fragmented in ruptured areas are in the GO category extracellular space and at least 2 are listed in GO categories related to ECM (cell-matrix adhesion and ECM organization for AGT; ECM for GPLD1), none of these proteins are structural ECM components.
Figure 5.
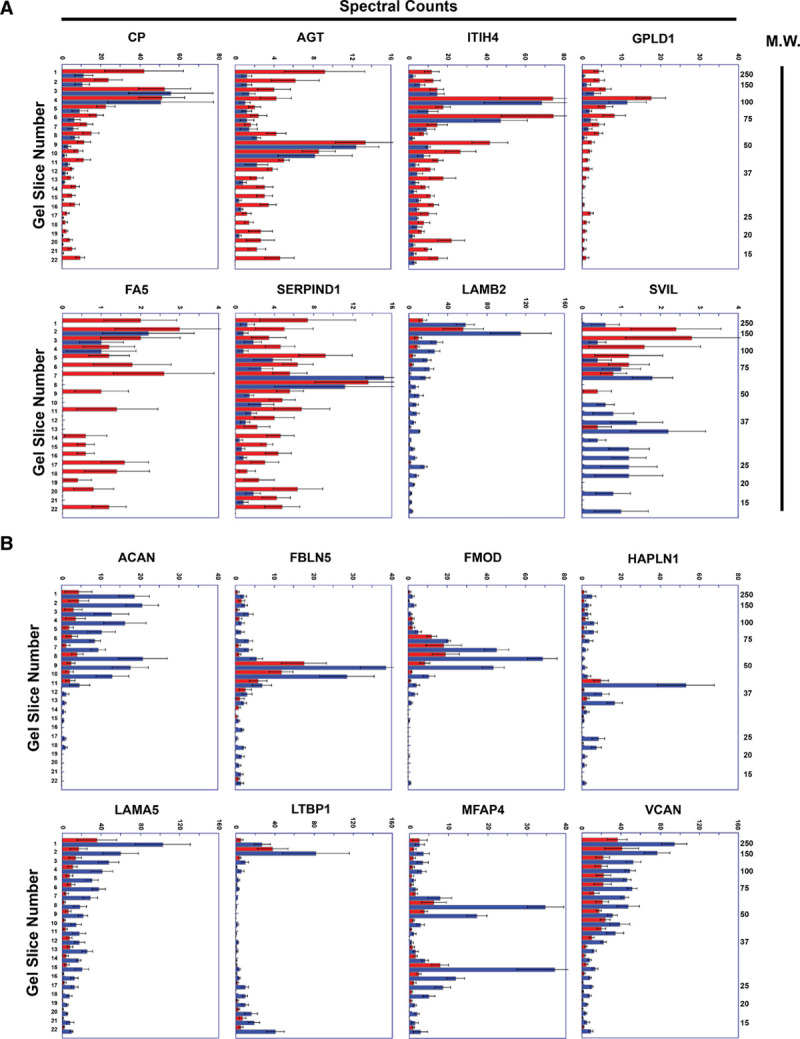
Representative peptographs of extracts of ruptured vs stable human plaque segments.
A and B, Extracts of ruptured (red) and adjacent stable (blue) segments of 5 human carotid plaques were analyzed using the PROTOMAP protocol. The extracts were subjected to SDS-PAGE, and the gels were cut into 22 slices, each corresponding to a molecular weight range. After in-gel trypsin digestion, peptides were extracted, identified by tandem mass spectrometry, and spectral counts were aggregated over all 22 slices. A, Proteins with differential abundance of lower-molecular weight peptides in extracts of ruptured vs. stable segments: ceruloplasmin (CP), angiotensinogen (AGT), ITIH4 (inter-alpha-trypsin inhibitor heavy chain), GPLD1 (phosphatidylinositol-glycan-specific phospholipase D), FA5 (coagulation factor V), SERPIND1 (serpin family D member 1), LAMB2 (laminin subunit beta-2), and SVIL (supervillin). B, ECM (Extracellular matrix) proteins that are significantly less abundant in extracts of ruptured vs stable human plaque segments: ACAN (aggrecan core protein), FBLN5 (fibulin 5), FMOD (fibromodulin), HAPLN (hyaluronan and proteoglycan link protein 1), LAMA5 (laminin subunit alpha 5), MFAP4 (microfibril-associated glycoprotein 4), LTBP1 (latent-transforming growth factor beta-binding protein 1), and VCAN (versican). A and B, Horizontal bars in each peptograph portray the total spectral counts for protein-specific peptides in each of the 22 gel slices (mean±SEM; n=5). Gel-slice number is on the leftward y-axis; molecular weight of the gel slices (in kilodaltons) is on the rightward y-axis.
To look more specifically for evidence of increased proteolysis of structural ECM proteins, we examined the peptographs of proteins extracted from the ruptured plaque samples. We identified—among the 339 proteins found by shotgun proteomics to be significantly less abundant in ruptured areas of the first set of human plaque extracts (Data Set XII in the Data Supplement)—69 proteins that are in the GO category ECM (Table III in the Data Supplement). All 69 proteins were also identified by PROTOMAP analysis of the second set of plaque extracts. Concordant with data from the shotgun analyses, PROTOMAP tools found that 21 (30%) of these ECM proteins were significantly less abundant (P<0.05) in ruptured versus stable areas, and an additional 14 (20%) tended to be less abundant (ie, 0.05≤P≤0.2) in ruptured areas (Table III in the Data Supplement). However, a review of all 69 peptographs (Figure 5B and data not shown) did not provide any additional evidence that these ECM components had undergone increased proteolysis in ruptured plaque tissue (ie, we did not find increased abundance of low-molecular weight peptides in ruptured plaque tissue).
We next repeated all of the PROTOMAP analyses described above on aortic extracts of SR-uPA+/0 and SR-uPA0/0 BMT recipients. As with the human plaque data, these analyses did not associate significant decreases in individual protein abundance (in aortas of SR-uPA+/0 BMT recipients) with concomitant increases in small fragments of the decreased abundance proteins. For example, of the 39 proteins found by shotgun proteomics to be significantly less abundant in SR-uPA+/0 aortas (Data Set II in the Data Supplement), 36 were also identified by PROTOMAP in aortic extracts of SR-uPA+/0 and SR-uPA0/0 BMT recipients. Peptographs of these 36 proteins revealed only one protein (NID2) with a modest trend towards increased abundance of lower-molecular weight fragments in aortic extracts of SR-uPA+/0 BMT recipients (Figure II in the Data Supplement).
Validation of Decreased Abundance of Basement-Membrane Proteins in Ruptured Human Plaques
In parallel with the PROTOMAP analyses of human plaque extracts, we performed shotgun proteomics (Data Set XV in the Data Supplement) on extracts of the 6 new plaques initially collected for the PROTOMAP experiment; Table I in the Data Supplement, cohort 2). Because 7 basement-membrane proteins were less abundant in both SR-uPA+/0 aortas and in ruptured human plaques in the initial discovery cohort, we focused our analysis on these 7 proteins. We also focused on the 3 collagens that we had identified as less-abundant in ruptured versus stable plaque segments of the discovery cohort (Data Set XII in the Data Supplement). Therefore, this second set of plaque extracts served as an independent validation cohort to test hypotheses generated from the first set of human plaque extracts. In agreement with the discovery cohort analysis, 5 of the 7 basement-membrane proteins were significantly less abundant in ruptured versus stable segments (P<0.005 for LAMC1, LAMA5, LAMB2, and HSPG2; P<0.05 for NID1; Figure 6A) while the other 2 showed a strong trend towards decreased abundance (P<0.1 for AGRN and NID2). In contrast, of the 3 collagens, only COL18A1—which is a basement-membrane protein59—was also significantly less abundant in the validation cohort (P<0.005). Reanalysis of the mouse data using the Wilcoxon rank-sum test (Figure 6B) confirmed that all 7 of these basement-membrane proteins (and only COL18A1 of the 3 collagens) were significantly less abundant in SR-uPA+/0 aortas (Figure 6B).
Figure 6.
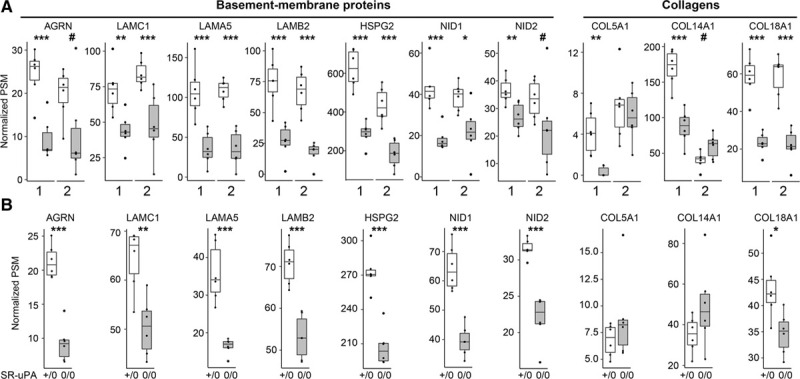
Depletion of basement-membrane proteins in ruptured human carotid plaques and in aortas of SR-uPA+/0 mice.
A, Data from 2 sets of human carotid plaques. Shotgun proteomics was performed on stable (open bars) and ruptured (shaded bars) segments of 2 independent sets of plaques (1 and 2). Analyses were performed independently, 1 y apart. Proteins were selected for this analysis based on their significantly decreased abundance in the first set of human plaques and—for basement-membrane proteins—congruently decreased abundance in SR-uPA+/0 mouse aortas. B, Data from aortas of 20-week-old SR-uPA+/0 and SR-uPA0/0 mice. A and B, Individual data points (n=6 per group) indicate individual plaque segments (A) or aortas (B); group medians and interquartile ranges are shown. PSM indicates peptide-spectrum match. P: #<0.1, *<0.05, **<0.01, and ***<0.005.
Immunoblotting of Human Plaque Extracts
We then used immunoblotting in an attempt to validate our mass-spectrometry-based detection of basement-membrane protein loss in ruptured human plaque segments. Loss of basement-membrane proteins was of particular interest because these proteins were decreased in both ruptured human plaques and in SR-uPA+/0 aortas. Western blots of 7 paired protein extracts of stable and ruptured segments (1 from cohort 1; 6 from cohort 2) were probed to detect LAMA5 and HSPG2. Due to depletion of first sample, a separate blot of 6 paired extracts was probed to detect COL18A1 (Figure III in the Data Supplement). The LAMA5 antibody detected a band of ≈400 kDa (consistent with intact LAMA5) in 6 of the 7 extracts of stable segments and in 0 of the 7 extracts of ruptured segments. Moreover, lower-molecular weight bands were more prominent in extracts of the ruptured plaque segments. When the blot was probed with the HSPG2 antibody, higher-molecular weight bands (≈200–300 kDa) were again more prominent in the extracts of stable plaques, and lower-molecular weight bands were more prominent in extracts of ruptured plaques. The immunoblot probed for COL18A1 revealed far more immunoreactive bands in extracts of ruptured versus stable plaques (consistent with increased proteolysis), although some of these fragments were of relatively high molecular weight. Bands corresponding to the molecular weight of the intact protein were not reliably detected on immunoblots for either HSPG2 or COL18A1.
Location of Basement-Membrane Proteins in Advanced Human Plaques
To verify the presence and begin to determine the location of basement-membrane proteins that were revealed by mass spectrometry to be depleted in ruptured plaques (and were also depleted in SR-uPA+/0 aortas), we performed immunohistochemistry on sections of stable segments of each of the 6 human plaques from cohort 1. We could not perform extensive or quantitative immunohistochemistry that compared stable and ruptured plaque tissue because nearly all of the ruptured plaque tissue was used for protein extraction, and only thin slices cut from the ends of the plaque segments were available for sectioning and staining. Moreover, most of the sections of ruptured plaques contained no plaque cap tissue. We stained the sections for LAMA5, HSPG2, and COL18A1 because these were the 3 most abundant basement-membrane proteins in the stable plaque segments, and all 3 were significantly and substantially decreased in ruptured versus stable areas of both cohorts 1 and 2 (Figure 6A). All 3 proteins were abundant in the vascular media but were also present in and around smooth muscle cell (SMC)-like cells in the plaque fibrous caps. LAMA5 and HSPG2 were also abundant in the endothelium/subendothelium; however, COL18A1 was detected in this location only rarely and at apparently low levels (Figures IV and V in the Data Supplement).
Last, we attempted to confirm the loss of HSPG2 and LAMA5 from ruptured plaque tissue and localize loss of these basement-membrane proteins to the plaque fibrous caps. We focused on HSPG2 and LAMA5 because well-validated antibodies are available for detecting these proteins in tissue sections.44 Importantly, both of these antibodies detect the corresponding full-length proteins on Western blots (Methods). Using sections stained with hematoxylin and eosin or Masson trichrome, a vascular pathologist (R.F. Nicosia) could locate small fragments of cap tissue in sections of only 3 of the ruptured plaques. Using adjacent slides and a validated antibody to HSPG2,44 we found only faint staining in the 3 ruptured plaque cap fragments. In all 3 samples, the plaque cap fragments stained less intensely than adjacent vascular media (Figure VI panels A through I in the Data Supplement). In contrast, sections from a stable plaque segment stained in parallel had strong staining for HSPG2 in the fibrous cap, with more faint staining in the vascular media (Figure VIJ through VIL in the Data Supplement). Parallel studies with a validated LAMA5 antibody44 did not yield consistent results.
Discussion
To identify the biological pathways through which increased vascular proteolysis leads to plaque rupture, to assess whether the SR-uPA+/0 mouse model replicates biochemical features of human plaque rupture, and to gain new insights into the mechanisms of human plaque rupture, we performed proteomic analyses of atherosclerotic aortas of mice with macrophage-specific overexpression of PLAU (SR-uPA+/0 mice) and of ruptured human carotid plaques. Our major findings are as follows: (1) Germline overexpression of PLAU in atherosclerotic aortas of 20-week-old mice reproducibly alters the abundance of subsets of extractable aortic proteins and of proteins in the aortic secretome: ECM proteins are decreased; MMPs, as well as proteins related to cell adhesion, cytoskeleton, inflammatory signaling, and apoptosis, are increased. (2) Introduction of PLAU-overexpressing macrophages into advanced atherosclerotic mouse lesions (for 8 weeks) primarily alters extracellular proteins, including decreases in cell adhesion and ECM proteins. (3) The proteome of a ruptured area of a human carotid plaque differs markedly from the proteome of adjacent stable plaque tissue: proteins related to inflammation, atherosclerosis, and blood coagulation are increased, whereas ECM, cell adhesion, and cytoskeletal proteins are decreased. (4) Proteomes of atherosclerotic aortas of SR-uPA+/0 mice and of ruptured human carotid plaques have limited overlap in the differential abundance of specific proteins but show strikingly similar decreases in protein abundance within several ECM-associated functional categories and have congruent decreases in several individual basement-membrane proteins.
We initially generated SR-uPA+/0 mice with a goal of developing a mouse model of protease-induced plaque rupture,27 and later found that transplantation of SR-uPA+/0 BM into 35-week-old nontransgenic Apoe−/− recipients28 reproduced critical histological features of human plaque rupture.29 We concluded that these histological findings—along with increased aortic PLAU and MMP activity in SR-uPA+/0 BMT recipients—validated the SR-uPA+/0 mouse as a model of protease-induced plaque rupture. However, this conclusion was based primarily on anatomic data. Other than documenting increased uPA and MMP activity in SR-uPA+/0 aortas, we did not generate any biochemical data that might explain how increased proteolytic activity caused plaque rupture. We, therefore, conceived the present study to search for these biochemical data, and thereby identify molecular and cellular mechanisms underlying plaque rupture.
We designed the present study based on 3 assumptions: (1) that analyses of arterial tissue of 20-week-old SR-uPA+/0 mice would reveal effects of increased protease activity on early mouse lesions; (2) that analyses of arterial tissue of SR-uPA+/0 BMT recipients (8 weeks after BMT) would reveal effects of increased protease activity on more-advanced mouse lesions (including ruptured plaques); and (3) that analyses of ruptured human plaques would reveal the effects of increased protease activity on advanced human lesions. However, our results showed that alterations in the arterial proteome are far more extensive in aortas of 20-week-old SR-uPA+/0 mice than in aorta/innominate arteries of the SR-uPA+/0 BMT recipients. This observation, likely due to the longer duration of uPA overexpression in 20-week-old SR-uPA+/0 mice, prompted us to analyze results from the 3 experiments according to the duration of exposure of plaque tissue to increased protease activity: SR-uPA+/0 BMT recipients (8 weeks), SR-uPA+/0 mice (20 weeks), then ruptured human plaques (>>20 weeks; Figure 7).
Figure 7.
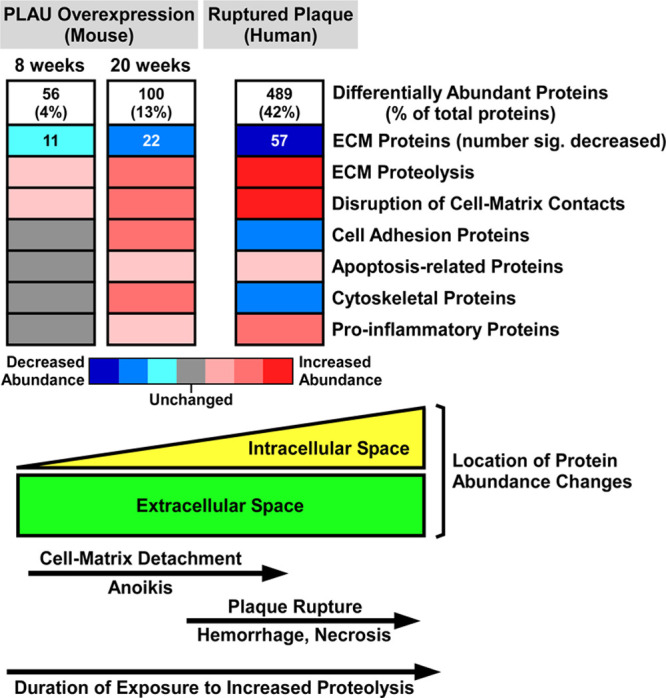
Shotgun proteomic analyses of mouse and human plaques suggest molecular and cellular mechanisms that connect extracellular proteolysis with plaque rupture.
Proteomic data from the mouse model of PLAU (urokinase-type plasminogen activator) overexpression (2 time points) and from human plaques show stepwise increases in the number of differentially abundant proteins (Data Sets II, IX, and XII in the Data Supplement) and corresponding decreases in ECM protein abundance (Data Sets IV, XI, and XIV in the Data Supplement). Decreased ECM (extracellular matrix) protein abundance is likely caused by increased proteolysis. Eight weeks exposure to elevated PLAU activity initiates disruption of cell-matrix contacts, which increases by 20 wk. Also by 20 wk, ECM protein loss stimulates homeostatic increases in cell adhesion and cytoskeletal proteins, inflammation, as well as programmed cell death (anoikis) triggered by loss of cell-matrix contacts. Advanced human plaques have more extensive proteolysis and disruption of cell-matrix contacts, causing anoikis, and cell necrosis with attendant loss of cell adhesion and cytoskeletal proteins. Plaque cell death weakens plaque structure, leading to rupture and an inflammatory response to tissue injury. The lower part of the figure illustrates the predominance of extracellular protein abundance changes at early stages of plaque proteolysis and the shift towards intracellular protein abundance changes at later stages.
When analyzed in this manner, our proteomic data—from both SR-uPA+/0 mice and ruptured human plaques—identify biochemical and cellular pathways that connect elevated vascular protease activity to plaque rupture. Aortas of mice exposed to elevated vascular protease activity for only 8 weeks (ie, SR-uPA+/0 BMT recipients) have relatively few differentially abundant proteins, predominantly located in the extracellular space. These aortas likely model the initiation of protease-mediated ECM degradation and cell-matrix detachment. In contrast, the aortas of mice exposed to elevated vascular protease activity for 20 weeks (ie, SR-uPA+/0 mice) have a far-larger number of differentially abundant proteins that include cell-associated as well as extracellular proteins. Our data suggest that this stage of plaque development likely models upregulation of cytoskeletal and cell adhesion proteins in response to progressive ECM degradation. The hypothesis that upregulation of cytoskeletal and cell adhesion proteins is a homeostatic cellular reaction to enhanced extracellular proteolysis is consistent with the observed global pattern of altered protein abundance in SR-uPA+/0 aortas (Figure 2): proteins with decreased abundance are overwhelmingly extracellular, whereas proteins with increased abundance are overwhelmingly cell-associated. Moreover, this hypothesis is consistent with substantial in vitro data that connect extracellular proteolysis with alterations in cell adhesion and cytoskeletal proteins.60–64 The aortic proteome of 20-week-old SR-uPA+/0 mice also shows elevated levels of apoptosis-related proteins, likely released from cells that are undergoing anoikis: programmed cell death that results from loss of cell-ECM contacts.65
The final biochemical steps associated with protease-mediated plaque rupture—that are not accessible in the germline transgenic SR-uPA+/0 mice due to their premature deaths27—are reflected in the proteome of ruptured human plaques (Figure 7). At this stage, matrix proteolysis has progressed, and plaque cell death has occurred along with loss of endothelial barrier function and appearance of intraplaque hemorrhage and thrombosis. Accordingly, the human ruptured plaque proteome is characterized by decreased levels of ECM proteins as well as cell adhesion and cytoskeletal proteins and increased levels of proteins associated with blood cells and clotting (eg, hemoglobin, fibrinogen, and prothrombin). Taken together, our proteomic data map a temporal series of biochemical and cellular events in SR-uPA+/0 aortas and human carotid plaques that lead from increased extracellular proteolysis to plaque rupture (Figure 7).
The present study was also designed to leverage proteomics to evaluate whether the SR-uPA+/0 mouse model recapitulates key biochemical features of human plaque rupture. With few exceptions,66,67 mouse models of plaque rupture have been evaluated, promoted, and criticized based solely on their histological—rather than their biochemical—resemblance to ruptured human plaques.68–74 We are not aware of any mouse plaque rupture studies in which biochemical data from the mouse model were compared directly with similar data generated from ruptured human plaques. Generation of data that enabled this comparison was challenging, because our SR-uPA+/0 mouse proteomic data were largely from stable plaques (as any ruptured innominate artery plaques in tissue from SR-uPA+/0 BMT recipients represent only a small fraction of the total extracted arterial tissue), whereas our human proteomic data were generated specifically from ruptured plaque tissue. Despite these limitations, our data strongly suggest that several key biochemical processes that are well described in unstable and ruptured human plaques—and are detected in our ruptured human plaques—are also present in atherosclerotic plaques of SR-uPA+/0 mice. These processes include elevated levels of MMPs and reduced levels of ECM proteins17,18; activation of inflammatory pathways2,13,75,76; and activation of apoptosis in association with ECM degradation.77–79
The overlap in biochemical processes between SR-uPA+/0 aortas and ruptured human plaques is striking because—as mentioned above—comparison of differentially abundant proteins in 20-week-old SR-uPA+/0 aortas with differentially abundant proteins in ruptured human plaques juxtaposes different disease stages and is, therefore, biased against finding common features. Indeed, there were few individual proteins that were commonly differentially abundant in both data sets: of the 100 differentially abundant proteins in the mouse data set and the 489 differentially abundant proteins in the human data set, only 27 proteins were differentially abundant in both. Nevertheless, these 27 proteins included several proteins in the GO category basement membrane that were significantly decreased in both data sets: AGRN, HSPG2, LAMA5, LAMB2, LAMC1, NID1, and NID2. These 7 basement-membrane proteins represent 18% of the 39 proteins that were significantly decreased in 20-week-old SR-uPA+/0 mouse aortas, suggesting that basement-membrane loss is a biochemical feature of human plaque rupture that is particularly well modeled in SR-uPA+/0 mouse aortas. We speculate that loss of basement-membrane proteins—most likely because of proteolysis—is an important and under-recognized contributor to plaque rupture.
We considered whether our results could shed light on whether human plaque rupture is driven by PLAU. If true, the SR-uPA+/0 mouse—in which basement-membrane protein loss and plaque rupture are definitely caused by elevated PLAU activity—would reproduce human disease even more closely. Others have hypothesized that human plaque PLAU activity precipitates rupture because PLAU is present in advanced human plaques, localizes at rupture-prone areas, and its abundance is correlated with plaque stability.80–83 However, because measuring PLAU in human plaques was not an objective of this study, we did not optimize tissue processing protocols for detecting PLAU protein or activity. Accordingly, PLAU was not detected in human plaques by shotgun proteomics and was detected in only one sample by PROTOMAP analysis. It is, nevertheless, possible to use our results to propose a causal relationship between loss of specific basement-membrane proteins in ruptured human plaques and elevated plaque PLAU activity. Laminin, nidogen, HSPG2, and collagen XVIII are cleaved by plasmin, MMP9, or MMP12. These 3 proteases are elevated in ruptured human plaques and are activated either directly or indirectly by PLAU.84,85 Therefore, our results are consistent with the hypothesis that elevated human plaque PLAU activity precipitates basement-membrane protein loss and plaque rupture, although our experimental methods cannot establish causality.
Basement-membrane protein loss due to pericellular matrix proteolysis could lead to plaque rupture via several mechanisms. The best-described mechanism involves SMC and EC apoptosis (anoikis) that is caused by loss of cell attachments to surrounding basement-membrane proteins.7,9,77,78,86 Death of EC and SMC would weaken cap structure while also triggering a local inflammatory response that includes entry of protease-secreting inflammatory cells (eg, macrophages, T cells, and mast cells) to the artery wall.7,9 Proteolysis of subendothelial basement-membrane proteins such as laminins increases artery wall permeability, facilitating inflammatory cell entry,87 and potentially increases lipoprotein entry as well. Migration and retention of inflammatory cells are further stimulated by basement-membrane proteolysis that releases chemotactic protein fragments and exposes adhesive protein domains (well described for laminins, agrin, other HSPGs, and nidogens).88–90 Proteolysis of basement-membrane proteins can also shift SMC away from a contractile phenotype, weakening the fibrous cap.91 A critical role for basement-membrane proteolysis that causes cap thinning and SMC death is supported by GWAS data that link coronary artery disease to a COL4A2 variant associated with lower COL4A1 and COL4A2 expression, increased SMC apoptosis, and fibrous cap thinning.92 Other GWAS data associate SMC phenotypic modulation (a consequence of basement-membrane proteolysis)91 with unstable coronary artery disease.93
Loss of plaque fibrillar collagen—not of basement-membrane components—has long been considered a hallmark of human plaque rupture.17,18 Consistent with this, several functional categories related to collagen were over-represented among less-abundant proteins in both SR-uPA+/0 aortas and ruptured human plaques (eg, collagen trimer in SR-uPA+/0 aortas; FDR<3×10−3; and collagen binding in ruptured human plaques; FDR<3×10−4). However, at the individual protein level, no collagen molecule was decreased in both murine and human settings, and the number of less-abundant individual collagen molecules was small in both settings and nonoverlapping: only COL14A1, COL18A1, and COL5A1 were lower in the discovery cohort of ruptured human plaques; only COL1A2, COL15A1, and COL6A3 were lower in SR-uPA+/0 aortas. Therefore, neither type I nor type III (ie, fibrillar) collagen—thought to be the major vascular collagens providing tensile strength and resistance to plaque rupture94–96—were reduced in ruptured human plaques. Indeed, few collagen molecules were less abundant in ruptured versus stable plaque extracts: in the first set of human plaques, only 3 of 15 identified collagens were significantly reduced. In the second set, only 2 of 13 identified collagens were significantly reduced in ruptured plaques extracts, and 2 were significantly increased. In contrast to these modest changes in collagen abundance, several basement-membrane proteins were decreased in both settings (9 individual basement-membrane proteins in SR-uPA+/0 aortas; 17 in the first set of ruptured human plaques; FDR<2×10−11 for basement-membrane protein loss in both settings). Moreover, decreases in specific human basement-membrane proteins were highly reproducible in the second (independent) set of ruptured human plaques. Our unbiased proteomic data thus suggest that basement-membrane protein loss may be more critical than fibrillar collagen loss in promoting plaque rupture.
We also hypothesized that if ECM components were proteolyzed either in the SR-uPA+/0 mouse or in ruptured human plaques, we would detect increased levels of proteolytic fragments in the corresponding tissue extracts. To address this question, we used PROTOMAP, a technology designed to identify protease substrates and that relies, in part, on detection of elevated levels of substrate fragments.30 Identification of these substrates might help both to identify the proteases that are responsible for plaque rupture and to identify peptide markers of plaque rupture.24,25 It is unclear why PROTOMAP did not identify ECM substrate fragments, especially because both SR-uPA+/0 aortas and ruptured human plaques have significant increases in protease abundance and significant decreases of several ECM proteins. It is possible that protein fragments released during ECM proteolysis are: (1) rapidly cleared from the interstitial space; (2) unstable in arterial tissue in vivo; (3) inefficiently extracted from arterial tissue; or (4) lost during processing of the extracts. It is also possible that low size-resolution of the gel-LC (liquid chromatography) approach and inherent variability of in-gel digestion interferes with detection of proteolytic fragments. Additional work optimizing the extraction and identification of ECM peptide fragments is likely required before PROTOMAP can be used to identify the in vivo substrates of atherosclerotic plaque proteases.
We focused our study on identifying mechanisms of plaque rupture because plaque rupture is the most common mechanism underlying coronary and carotid artery thrombosis.4,15 Plaque erosion can also precipitate coronary and carotid thrombosis, although it is a less common etiology of both.4,15 Because the design of our study precluded systematic histological analysis of thrombosed human plaque tissue, we cannot exclude that some of our ruptured plaques may have been eroded. However, for 2 principal reasons, we think it is unlikely that plaque erosion accounted for a majority of our thrombosed carotid plaques. First, in most clinical series examining thrombosed carotid plaques, plaque rupture is more common (60% in the largest study),97 with plaque erosion estimated to be responsible for <25% of carotid plaque thrombosis.4 Second, one of us (T. Kohler; unpublished data) recently completed a systematic histological study of 111 carotid plaques collected at the same institutions as in the present study. Ruptured fibrous caps were present in 54% of the plaques, luminal surface disruption with mural thrombus was present in 36% of the plaques, and some plaques had both features (as reported elsewhere).15 Accordingly, it is highly unlikely that the thrombosed carotid segments studied herein were not predominantly ruptured plaques. Moreover, key biochemical features—including relatively low levels of ECM and SMC-specific proteins (eg, ACTA2 [actin alpha 2, smooth muscle], MYH11, and CNN1 [calponin 1]) along with high levels of proteins associated with atherogenic lipoproteins (APOB [apolipoprotein B] and APOE)—were generally shared by all of the ruptured plaque segments. These biochemical features are more consistent with the substrate of plaque rupture versus plaque erosion.76 These observations notwithstanding, a compelling argument has been made that basement-membrane proteolysis contributes importantly to plaque erosion as well as rupture.9 According to this model, proteolysis of EC basement membrane would precipitate endothelial desquamation and plaque erosion, whereas proteolysis of the basement membrane surrounding SMC (especially fibrous cap SMC) would trigger plaque rupture.9,98
We were concerned whether the significant reductions in abundance of basement-membrane proteins in both the mouse and human studies were tightly linked to plaque rupture processes (such as proteolysis) or whether they were driven primarily by other compositional differences between SR-uPA and nontransgenic mouse aortas and between ruptured and stable segments of human plaques. These other compositional differences could include increased macrophages and lipid in SR-uPA aortas27 and increased plasma proteins in ruptured human plaque segments. Because these structural differences cannot be eliminated as experimental variables, this question may be impossible to answer. However, several observations argue that the lower abundance of basement-membrane proteins in ruptured human plaques is not driven by increased plasma protein abundance. First, as mentioned above, abundance of specific basement-membrane proteins is reproducibly lower in ruptured human plaques, but abundance of specific collagens is not. Second, if the decrease in basement-membrane proteins (as a percentage of total protein) in ruptured human plaque segments were due to accumulation of plasma proteins in ruptured plaque segments, immunoblots that detect basement-membrane proteins should show reproducible decreases in band intensities in extracts of ruptured versus stable plaque segments; yet they do not. Third, peptides derived from albumin (the most abundant plasma protein) are no more abundant in extracts of ruptured versus stable human plaque segments (mean 384 versus 337, P=0.54 in cohort 1; mean 806 versus 577, P=0.14 in cohort 2), discounting plasma protein abundance as a major uncontrolled variable.
Our study has several limitations. Unlike human plaques, mouse aortic plaques are too small to be specifically extracted, requiring that we extract proteins from larger segments of mouse aorta. These segments may contain areas without advanced atherosclerosis, diluting signals from the plaque-derived proteins. Similar considerations related to physical size prevented us from performing proteomic studies solely on innominate arteries of SR-uPA+/0 BMT recipients, in which plaque rupture is common.29 Interpretation of our human data must take into consideration that human plaques cannot be harvested immediately before they rupture and that ruptured/thrombosed plaques removed in the operating room have already entered a healing phase, with corresponding changes in the plaque proteome. This limitation might be addressed in future studies in which proteomic analyses are performed on carotid plaques that are removed based on noninvasive assessment as high-risk but have not yet ruptured.99,100 Our ability to use the proteomics data to identify novel pathways and relationships is limited because our functional enrichment and network analyses are based on known biological processes and gene-product interactions. However, the primary aim of our analyses was to compartmentalize the complex molecular events occurring during plaque rupture within the context of well-defined canonical annotations and experimentally verified interactions. Another limitation is that our extraction protocol (developed with a focus on enrichment of ECM proteins) may not capture some important proteins and peptides. Absence of these proteins would limit comparison of our data with proteomic data generated by others using different extraction methods.25,26 In addition, because the murine and human data exhibit only modest overlap in altered expression of individual proteins, our data suggest important differences between the biochemistry of SR-uPA+/0 aortas and human carotid plaques. For example, the proteases that are more abundant in SR-uPA+/0 aortas (MMP2 and MMP3) differ from the proteases that are more abundant in ruptured human plaques (MMP9, MMP12, MMP19, neutrophil elastase, Cathepsins B, D, G, and Z, chymase, carboxypeptidase B2, carboxypeptidase N, and mast cell carboxypeptidase). Accordingly, development of pharmacological strategies that target specific plaque proteases will need to rely on human rather than mouse data. Finally, proteolysis is the most likely cause of ECM loss from both SR-uPA+/0 aortas and ruptured human plaques, and our immunoblots provide some support that ECM proteolysis is increased in ruptured plaque segments. However, a mass spectrometry-based assay (PROTOMAP) did not detect increased numbers of proteolytic fragments of the less-abundant ECM proteins. This could be a result of technical limitations of PROTOMAP methodology.
Unbiased discovery studies such as ours, which test numerous hypotheses simultaneously, risk generating falsely positive results, due only to chance. Concerns regarding false positivity are most definitively resolved when study predictions are borne out (eg, through validation of our results by other groups using similar methodology or demonstration that interventions that alter basement-membrane proteolysis have predicted effects on plaque rupture). Nevertheless, we offer several reasons why our results are unlikely to be explained by chance: (1) we applied rigorous false discovery rate thresholds to both the peptide abundance data and the functional enrichment data; (2) in both murine and human studies, the statistical significance of over-representation of basement-membrane proteins among the decreased abundance proteins was far below the FDR threshold (P<2×10−11 for both); (3) 6 of the 7 basement-membrane proteins that were differentially abundant in both mouse samples and human cohort 1 were in the top 30% of differentially abundant proteins in cohort 1 (ie, these were not borderline differences); and (4) the key findings on basement-membrane protein abundance in plaques from cohort 1 were reproduced independently in a second set of plaques (cohort 2). All 8 basement-membrane proteins (including COL18A1) were also decreased in this second set, with P<0.005 for 5 of the 8 proteins.
In summary, shotgun proteomic studies performed with murine and human tissues identify biological pathways that link elevated vascular protease activity with plaque rupture and reveal that several biochemical features of human plaque rupture (eg, inflammatory signaling, MMP abundance, decreased ECM proteins) are replicated in SR-uPA+/0 mouse aortas. Our results also suggest that loss of basement-membrane proteins may play a central role in precipitating plaque rupture.
Acknowledgments
We are grateful to Kristi Pimentel for help with human subject recruitment and to the University of Washington Histology and Imaging Core for assistance with histochemical staining. We thank Ilona Babenko for assistance with proteomics sample preparation and Western blot analysis. This material is the result of work supported by resources from the VA Puget Sound Health Care System, Seattle, Washington.
Sources of Funding
This work was supported by the National Heart Lung and Blood Institute grant (R21HL113405, to D.A. Dichek), by the National Institute of Diabetes, Digestive, and Kidney Disease (P30DK017047, supporting the Quantitative and Functional Proteomics Core and the Cellular and Molecular Imaging Core of the University of Washington Diabetes Research Center), by the University of Washington Proteomics Resource (UWPR95794), and by the John L. Locke Jr. Charitable Trust. S.A. Gharib was supported by the National Institute of Allergy and Infectious Disease (R01AI137111). T. Vaisar and J. Heinecke were supported by P01HL092969 and P01HL128203. J. Heinecke was supported by R01HL149685.
Disclosures
None.
Supplemental Materials
Major Resources Table
Online Figures I–VI
Online Tables I–III
Online Data Sets I–XV
Supplementary Material
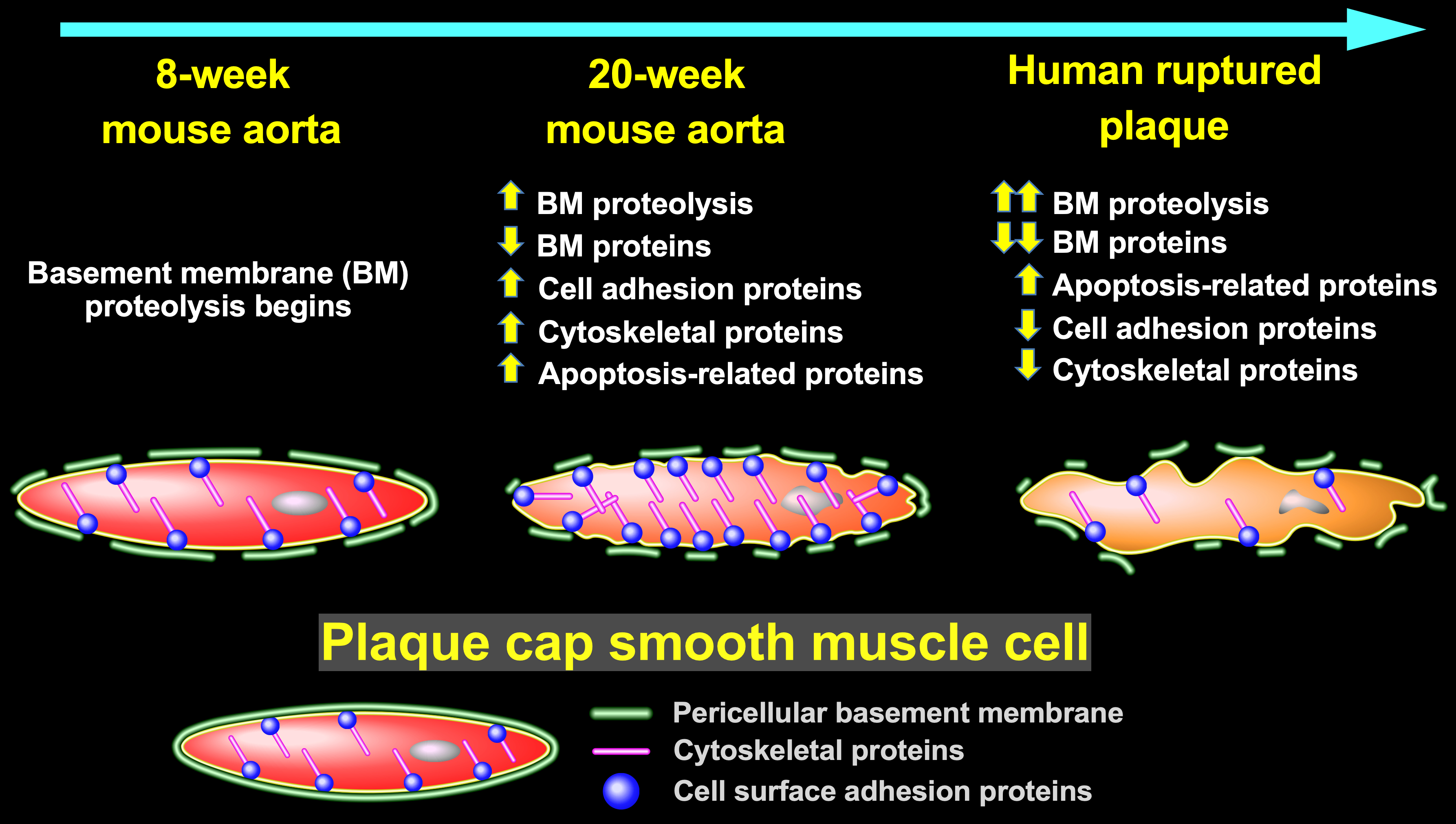{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- BM
- bone marrow
- BMT
- bone marrow transplantation
- CM
- culture medium
- ECM
- extracellular matrix
- FA
- formic acid
- FDR
- false discovery rate
- GO
- gene ontology
- LAMA
- laminin subunit alpha
- MMP
- matrix metalloproteinase
- PROTOMAP
- protein topography and migration analysis platform
- SR-uPA
- a transgene including the human scavenger receptor promoter and the mouse Plau gene
T.V., J.H.H., S.A.G., and D.A.D. contributed equally to this article.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.317295.
For Sources of Funding and Disclosures, see page 1019 and 1020.
Novelty and Significance
What Is Known?
Most myocardial infarctions and many strokes are caused by rupture of atherosclerotic arterial plaques.
Candidate-gene approaches have associated elevated artery wall protease activity and proteolysis of plaque structural proteins—especially collagens—with plaque rupture in humans.
Mouse models have established a causal relationship between elevated plaque protease activity and histological features of plaque rupture.
What New Information Does This Article Contribute?
Unbiased proteomic studies performed in a mouse model of protease-induced plaque rupture and in ruptured human plaques identify specific basement-membrane proteins that are depleted in both the mouse and human plaques.
Bioinformatics analyses of the proteomics data identify extracellular proteolysis, loss of cell-substrate adhesion, inflammation, and apoptosis as key vascular wall processes that precede plaque rupture.
Loss of plaque basement-membrane proteins predominates over loss of plaque collagens both in the mouse model and in ruptured human plaques.
The mouse model of plaque rupture replicates biochemical processes—particularly those associated with extracellular matrix loss—that are found in ruptured human plaques.
Plaque rupture is the proximate cause of most athero sclerosis-associated morbidity and mortality (including most heart attacks and many strokes); however, the underlying molecular mechanisms are unknown. Identification of these mechanisms would guide development of therapeutics and might identify diagnostics that predict plaque rupture. Identification of plaque rupture mechanisms would be facilitated by availability of an animal model that reproduced biochemical processes underlying plaque rupture and in which vascular tissue was available before plaque rupture. Instead of using a candidate-gene approach, we used unbiased proteomics and bioinformatics to identify specific proteins and biochemical processes associated with plaque rupture. Moreover, we performed these studies both in ruptured human plaques and in a mouse model of protease-induced plaque rupture. We confirmed key findings in a human validation cohort. Extracellular proteolysis, loss of basement-membrane proteins, loss of cell-matrix adhesion, and inflammation were common to both ruptured human plaques and the mouse model. Surprisingly, plaque collagens were minimally altered in both settings. Although specific proteolytic fragments were not identified, combined data from the mouse and human studies illuminated a series of biochemical processes through which elevated plaque protease activity leads to basement-membrane proteolysis, apoptosis, inflammation, and plaque rupture. These findings suggest novel and attractive targets for interventions aimed at preventing plaque rupture.
References
- 1.Tabas I. 2016 Russell ross memorial lecture in vascular biology: molecular-cellular mechanisms in the progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:183–189. doi: 10.1161/ATVBAHA.116.308036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the unstable plaque. Prog Cardiovasc Dis. 2002;44:349–356. doi: 10.1053/pcad.2002.122475 [DOI] [PubMed] [Google Scholar]
- 3.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. doi: 10.1161/ATVBAHA.108.179739 [DOI] [PubMed] [Google Scholar]
- 4.Kolodgie FD, Yahagi K, Mori H, Romero ME, Trout HH, Rd, Finn AV, Virmani R. High-risk carotid plaque: lessons learned from histopathology. Semin Vasc Surg. 2017;30:31–43. doi: 10.1053/j.semvascsurg.2017.04.008 [DOI] [PubMed] [Google Scholar]
- 5.Slager CJ, Wentzel JJ, Gijsen FJ, Thury A, van der Wal AC, Schaar JA, Serruys PW. The role of shear stress in the destabilization of vulnerable plaques and related therapeutic implications. Nat Clin Pract Cardiovasc Med. 2005;2:456–464. doi: 10.1038/ncpcardio0298 [DOI] [PubMed] [Google Scholar]
- 6.Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018 [DOI] [PubMed] [Google Scholar]
- 7.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459 [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M, Bennett M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–1919. doi: 10.1161/CIRCULATIONAHA.115.016457 [DOI] [PubMed] [Google Scholar]
- 9.Lindstedt KA, Leskinen MJ, Kovanen PT. Proteolysis of the pericellular matrix: a novel element determining cell survival and death in the pathogenesis of plaque erosion and rupture. Arterioscler Thromb Vasc Biol. 2004;24:1350–1358. doi: 10.1161/01.ATV.0000135322.78008.55 [DOI] [PubMed] [Google Scholar]
- 10.Dunmore BJ, McCarthy MJ, Naylor AR, Brindle NP. Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J Vasc Surg. 2007;45:155–159. doi: 10.1016/j.jvs.2006.08.072 [DOI] [PubMed] [Google Scholar]
- 11.Dickhout JG, Colgan SM, Lhoták S, Austin RC. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome: a balancing act between plaque stability and rupture. Circulation. 2007;116:1214–1216. doi: 10.1161/CIRCULATIONAHA.107.728378 [DOI] [PubMed] [Google Scholar]
- 12.Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303:H619–H628. doi: 10.1152/ajpheart.00036.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libby P. The molecular mechanisms of the thrombotic complications of atherosclerosis. J Intern Med. 2008;263:517–527. doi: 10.1111/j.1365-2796.2008.01965.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah PK. Molecular mechanisms of plaque instability. Curr Opin Lipidol. 2007;18:492–499. doi: 10.1097/MOL.0b013e3282efa326 [DOI] [PubMed] [Google Scholar]
- 15.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721 [DOI] [PubMed] [Google Scholar]
- 16.Newby AC. Proteinases and plaque rupture: unblocking the road to translation. Curr Opin Lipidol. 2014;25:358–366. doi: 10.1097/MOL.0000000000000111 [DOI] [PubMed] [Google Scholar]
- 17.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sukhova GK, Schönbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503 [DOI] [PubMed] [Google Scholar]
- 19.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041 [DOI] [PubMed] [Google Scholar]
- 20.Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. doi: 10.1161/CIRCULATIONAHA.107.707448 [DOI] [PubMed] [Google Scholar]
- 21.Quillard T, Tesmenitsky Y, Croce K, Travers R, Shvartz E, Koskinas KC, Sukhova GK, Aikawa E, Aikawa M, Libby P. Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2464–2472. doi: 10.1161/ATVBAHA.111.231563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YC, Huang AL, Kyaw TS, Bobik A, Peter K. Atherosclerotic plaque rupture: identifying the straw that breaks the camel’s back. Arterioscler Thromb Vasc Biol. 2016;36:e63–e72. doi: 10.1161/ATVBAHA.116.307993 [DOI] [PubMed] [Google Scholar]
- 24.Stegemann C, Didangelos A, Barallobre-Barreiro J, Langley SR, Mandal K, Jahangiri M, Mayr M. Proteomic identification of matrix metalloproteinase substrates in the human vasculature. Circ Cardiovasc Genet. 2013;6:106–117. doi: 10.1161/CIRCGENETICS.112.964452 [DOI] [PubMed] [Google Scholar]
- 25.Langley SR, Willeit K, Didangelos A, Matic LP, Skroblin P, Barallobre-Barreiro J, Lengquist M, Rungger G, Kapustin A, Kedenko L, et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J Clin Invest. 2017;127:1546–1560. doi: 10.1172/JCI86924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matic LP, Jesus Iglesias M, Vesterlund M, Lengquist M, Hong MG, Saieed S, Sanchez-Rivera L, Berg M, Razuvaev A, Kronqvist M, et al. Novel multiomics profiling of human carotid atherosclerotic plaques and plasma reveals biliverdin reductase B as a marker of intraplaque hemorrhage. JACC Basic Transl Sci. 2018;3:464–480. doi: 10.1016/j.jacbts.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cozen AE, Moriwaki H, Kremen M, DeYoung MB, Dichek HL, Slezicki KI, Young SG, Véniant M, Dichek DA. Macrophage-targeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death. Circulation. 2004;109:2129–2135. doi: 10.1161/01.CIR.0000127369.24127.03 [DOI] [PubMed] [Google Scholar]
- 28.Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587 [DOI] [PubMed] [Google Scholar]
- 29.Hu JH, Du L, Chu T, Otsuka G, Dronadula N, Jaffe M, Gill SE, Parks WC, Dichek DA. Overexpression of urokinase by plaque macrophages causes histological features of plaque rupture and increases vascular matrix metalloproteinase activity in aged apolipoprotein e-null mice. Circulation. 2010;121:1637–1644. doi: 10.1161/CIRCULATIONAHA.109.914945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dronadula N, Wacker BK, Van Der Kwast R, Zhang J, Dichek DA. Stable in vivo transgene expression in endothelial cells with helper-dependent adenovirus: roles of promoter and interleukin-10. Hum Gene Ther. 2017;28:255–270. doi: 10.1089/hum.2016.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eng JK, Hoopmann MR, Jahan TA, Egertson JD, Noble WS, MacCoss MJ. A deeper look into Comet–implementation and features. J Am Soc Mass Spectrom. 2015;26:1865–1874. doi: 10.1007/s13361-015-1179-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- 35.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- 36.Heinecke NL, Pratt BS, Vaisar T, Becker L. PepC: proteomics software for identifying differentially expressed proteins based on spectral counting. Bioinformatics. 2010;26:1574–1575. doi: 10.1093/bioinformatics/btq171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker L, Gharib SA, Irwin AD, Wijsman E, Vaisar T, Oram JF, Heinecke JW. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010;11:125–135. doi: 10.1016/j.cmet.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reardon CA, Lingaraju A, Schoenfelt KQ, Zhou G, Cui C, Jacobs-El H, Babenko I, Hoofnagle A, Czyz D, Shuman H, et al. Obesity and insulin resistance promote atherosclerosis through an IFNγ-regulated macrophage protein network. Cell Rep. 2018;23:3021–3030. doi: 10.1016/j.celrep.2018.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–625. doi: 10.1016/j.cmet.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR., 3rd MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun Mass Spectrom. 2004;18:2162–2168. doi: 10.1002/rcm.1603 [DOI] [PubMed] [Google Scholar]
- 42.Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. ProLuCID: an improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics. 2015;129:16–24. doi: 10.1016/j.jprot.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garbe JH, Göhring W, Mann K, Timpl R, Sasaki T. Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin alpha3B and alpha5 chains. Biochem J. 2002;362:213–221. doi: 10.1042/0264-6021:3620213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rickelt S, Hynes RO. Antibodies and methods for immunohistochemistry of extracellular matrix proteins. Matrix Biol. 2018;71-72:10–27. doi: 10.1016/j.matbio.2018.04.011 [DOI] [PubMed] [Google Scholar]
- 45.Fellenberg K, Hauser NC, Brors B, Neutzner A, Hoheisel JD, Vingron M. Correspondence analysis applied to microarray data. Proc Natl Acad Sci U S A. 2001;98:10781–10786. doi: 10.1073/pnas.181597298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01 [DOI] [PubMed] [Google Scholar]
- 47.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. ; Inflamm and Host Response to Injury Large Scale Collab. Res. Program A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985 [DOI] [PubMed] [Google Scholar]
- 48.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454 [DOI] [PubMed] [Google Scholar]
- 51.Milewicz DM, Østergaard JR, Ala-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, Bradley TJ, Olney AH, Adès L, Maher JF, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A:2437–2443. doi: 10.1002/ajmg.a.33657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papke CL, Cao J, Kwartler CS, Villamizar C, Byanova KL, Lim SM, Sreenivasappa H, Fischer G, Pham J, Rees M, et al. Smooth muscle hyperplasia due to loss of smooth muscle α-actin is driven by activation of focal adhesion kinase, altered p53 localization and increased levels of platelet-derived growth factor receptor-β. Hum Mol Genet. 2013;22:3123–3137. doi: 10.1093/hmg/ddt167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522 [DOI] [PubMed] [Google Scholar]
- 54.Durán MC, Martín-Ventura JL, Mohammed S, Barderas MG, Blanco-Colio LM, Mas S, Moral V, Ortega L, Tuñón J, Jensen ON, et al. Atorvastatin modulates the profile of proteins released by human atherosclerotic plaques. Eur J Pharmacol. 2007;562:119–129. doi: 10.1016/j.ejphar.2007.01.077 [DOI] [PubMed] [Google Scholar]
- 55.de la Cuesta F, Barderas MG, Calvo E, Zubiri I, Maroto AS, Darde VM, Martin-Rojas T, Gil-Dones F, Posada-Ayala M, Tejerina T, et al. Secretome analysis of atherosclerotic and non-atherosclerotic arteries reveals dynamic extracellular remodeling during pathogenesis. J Proteomics. 2012;75:2960–2971 [DOI] [PubMed] [Google Scholar]
- 56.Rocchiccioli S, Pelosi G, Rosini S, Marconi M, Viglione F, Citti L, Ferrari M, Trivella MG, Cecchettini A. Secreted proteins from carotid endarterectomy: an untargeted approach to disclose molecular clues of plaque progression. J Transl Med. 2013;11:260 doi: 10.1186/1479-5876-11-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aragonès G, Auguet T, Guiu-Jurado E, Berlanga A, Curriu M, Martinez S, Alibalic A, Aguilar C, Hernández E, Camara ML, et al. Proteomic profile of unstable atheroma plaque: increased neutrophil defensin 1, clusterin, and apolipoprotein e levels in carotid secretome. J Proteome Res. 2016;15:933–944. doi: 10.1021/acs.jproteome.5b00936 [DOI] [PubMed] [Google Scholar]
- 58.Johnson JL, Jackson CL. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis. 2001;154:399–406. doi: 10.1016/s0021-9150(00)00515-3 [DOI] [PubMed] [Google Scholar]
- 59.Saarela J, Rehn M, Oikarinen A, Autio-Harmainen H, Pihlajaniemi T. The short and long forms of type XVIII collagen show clear tissue specificities in their expression and location in basement membrane zones in humans. Am J Pathol. 1998;153:611–626. doi: 10.1016/S0002-9440(10)65603-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0 [DOI] [PubMed] [Google Scholar]
- 61.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogier C, Bernard A, Chollet AM, LE Diguardher T, Hanessian S, Charton G, Khrestchatisky M, Rivera S. Matrix metalloproteinase-2 (MMP-2) regulates astrocyte motility in connection with the actin cytoskeleton and integrins. Glia. 2006;54:272–284. doi: 10.1002/glia.20349 [DOI] [PubMed] [Google Scholar]
- 63.von Nandelstadh P, Gucciardo E, Lohi J, Li R, Sugiyama N, Carpen O, Lehti K. Actin-associated protein palladin promotes tumor cell invasion by linking extracellular matrix degradation to cell cytoskeleton. Mol Biol Cell. 2014;25:2556–2570. doi: 10.1091/mbc.E13-11-0667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mead TJ, Du Y, Nelson CM, Gueye NA, Drazba J, Dancevic CM, Vankemmelbeke M, Buttle DJ, Apte SS. ADAMTS9-regulated pericellular matrix dynamics governs focal adhesion-dependent smooth muscle differentiation. Cell Rep. 2018;23:485–498. doi: 10.1016/j.celrep.2018.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, Haviv I, Agrotis A, Htun NM, Jowett J, et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res. 2013;113:252–265. doi: 10.1161/CIRCRESAHA.113.301562 [DOI] [PubMed] [Google Scholar]
- 67.Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, Van Der Linden A, Bauters D, Lijnen HR, Sluimer JC, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. 2015;36:1049–1058. doi: 10.1093/eurheartj/ehu041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20 [DOI] [PubMed] [Google Scholar]
- 69.Jackson CL, Bennett MR, Biessen EA, Johnson JL, Krams R. Assessment of unstable atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:714–720. doi: 10.1161/01.ATV.0000261873.86623.e1 [DOI] [PubMed] [Google Scholar]
- 70.Falk E, Schwartz SM, Galis ZS, Rosenfeld ME. Putative murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:969–972. doi: 10.1161/01.ATV.0000261572.33474.e0 [DOI] [PubMed] [Google Scholar]
- 71.Jackson CL. Defining and defending murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:973–977. doi: 10.1161/01.ATV.0000261545.53586.f0 [DOI] [PubMed] [Google Scholar]
- 72.Schapira K, Heeneman S, Daemen MJ. Animal models to study plaque vulnerability. Curr Pharm Des. 2007;13:1013–1020. doi: 10.2174/138161207780487575 [DOI] [PubMed] [Google Scholar]
- 73.Matoba T, Sato K, Egashira K. Mouse models of plaque rupture. Curr Opin Lipidol. 2013;24:419–425. doi: 10.1097/MOL.0b013e3283646e4d [DOI] [PubMed] [Google Scholar]
- 74.Silvestre-Roig C, de Winther MP, Weber C, Daemen MJ, Lutgens E, Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114:214–226. doi: 10.1161/CIRCRESAHA.114.302355 [DOI] [PubMed] [Google Scholar]
- 75.Hellings WE, Peeters W, Moll FL, Pasterkamp G. From vulnerable plaque to vulnerable patient: the search for biomarkers of plaque destabilization. Trends Cardiovasc Med. 2007;17:162–171. doi: 10.1016/j.tcm.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 76.Crea F, Libby P. Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation. 2017;136:1155–1166. doi: 10.1161/CIRCULATIONAHA.117.029870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choy JC, Hung VH, Hunter AL, Cheung PK, Motyka B, Goping IS, Sawchuk T, Bleackley RC, Podor TJ, McManus BM, et al. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin: involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol. 2004;24:2245–2250. doi: 10.1161/01.ATV.0000147162.51930.b7 [DOI] [PubMed] [Google Scholar]
- 78.Lindstedt KA, Kovanen PT. Proteolysis of pericellular matrix: a process linking inflammation to plaque destabilization and rupture. Arterioscler Thromb Vasc Biol. 2004;24:2205–2206. doi: 10.1161/01.ATV.0000149753.74793.88 [DOI] [PubMed] [Google Scholar]
- 79.Michel JB. Anoikis in the cardiovascular system: known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol. 2003;23:2146–2154. doi: 10.1161/01.ATV.0000099882.52647.E4 [DOI] [PubMed] [Google Scholar]
- 80.Lupu F, Heim DA, Bachmann F, Hurni M, Kakkar VV, Kruithof EK. Plasminogen activator expression in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1995;15:1444–1455. doi: 10.1161/01.atv.15.9.1444 [DOI] [PubMed] [Google Scholar]
- 81.Kienast J, Padró T, Steins M, Li CX, Schmid KW, Hammel D, Scheld HH, van de Loo JC. Relation of urokinase-type plasminogen activator expression to presence and severity of atherosclerotic lesions in human coronary arteries. Thromb Haemost. 1998;79:579–586 [PubMed] [Google Scholar]
- 82.Sayed S, Cockerill GW, Torsney E, Poston R, Thompson MM, Loftus IM. Elevated tissue expression of thrombomodulatory factors correlates with acute symptomatic carotid plaque phenotype. Eur J Vasc Endovasc Surg. 2009;38:20–25. doi: 10.1016/j.ejvs.2009.02.020 [DOI] [PubMed] [Google Scholar]
- 83.Svensson PA, Olson FJ, Hägg DA, Ryndel M, Wiklund O, Karlström L, Hulthe J, Carlsson LM, Fagerberg B. Urokinase-type plasminogen activator receptor is associated with macrophages and plaque rupture in symptomatic carotid atherosclerosis. Int J Mol Med. 2008;22:459–464 [PubMed] [Google Scholar]
- 84.Gong Y, Fan Y, Hoover-Plow J. Plasminogen regulates stromal cell-derived factor-1/CXCR4-mediated hematopoietic stem cell mobilization by activation of matrix metalloproteinase-9. Arterioscler Thromb Vasc Biol. 2011;31:2035–2043. doi: 10.1161/ATVBAHA.111.229583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–333 [PubMed] [Google Scholar]
- 86.Meilhac O, Ho-Tin-Noé B, Houard X, Philippe M, Michel JB, Anglés-Cano E. Pericellular plasmin induces smooth muscle cell anoikis. FASEB J. 2003;17:1301–1303. doi: 10.1096/fj.02-0687fje [DOI] [PubMed] [Google Scholar]
- 87.Song J, Zhang X, Buscher K, Wang Y, Wang H, Di Russo J, Li L, Lütke-Enking S, Zarbock A, Stadtmann A, et al. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. 2017;18:1256–1269. doi: 10.1016/j.celrep.2016.12.092 [DOI] [PubMed] [Google Scholar]
- 88.Graf J, Iwamoto Y, Sasaki M, Martin GR, Kleinman HK, Robey FA, Yamada Y. Identification of an amino acid sequence in laminin mediating cell attachment, chemotaxis, and receptor binding. Cell. 1987;48:989–996. doi: 10.1016/0092-8674(87)90707-0 [DOI] [PubMed] [Google Scholar]
- 89.Celie JW, Rutjes NW, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T, Dräger AM, Zweegman S, Kessler FL, Beelen RH, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170:1865–1878. doi: 10.2353/ajpath.2007.070061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Senior RM, Gresham HD, Griffin GL, Brown EJ, Chung AE. Entactin stimulates neutrophil adhesion and chemotaxis through interactions between its Arg-Gly-Asp (RGD) domain and the leukocyte response integrin. J Clin Invest. 1992;90:2251–2257. doi: 10.1172/JCI116111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Steffensen LB, Rasmussen LM. A role for collagen type IV in cardiovascular disease? Am J Physiol Heart Circ Physiol. 2018;315:H610–H625. doi: 10.1152/ajpheart.00070.2018 [DOI] [PubMed] [Google Scholar]
- 92.Yang W, Ng FL, Chan K, Pu X, Poston RN, Ren M, An W, Zhang R, Wu J, Yan S, et al. Coronary-heart-disease-associated genetic variant at the COL4A1/COL4A2 locus affects COL4A1/COL4A2 expression, vascular cell survival, atherosclerotic plaque stability and risk of myocardial infarction. PLoS Genet. 2016;12:e1006127 doi: 10.1371/journal.pgen.1006127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iyer D, Zhao Q, Wirka R, Naravane A, Nguyen T, Liu B, Nagao M, Cheng P, Miller CL, Kim JB, et al. Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 2018;14:e1007681 doi: 10.1371/journal.pgen.1007681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burleigh MC, Briggs AD, Lendon CL, Davies MJ, Born GV, Richardson PD. Collagen types I and III, collagen content, GAGs and mechanical strength of human atherosclerotic plaque caps: span-wise variations. Atherosclerosis. 1992;96:71–81. doi: 10.1016/0021-9150(92)90039-j [DOI] [PubMed] [Google Scholar]
- 95.Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation. 1993;87:1179–1187. doi: 10.1161/01.cir.87.4.1179 [DOI] [PubMed] [Google Scholar]
- 96.Katsuda S, Okada Y, Minamoto T, Oda Y, Matsui Y, Nakanishi I. Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies. Arterioscler Thromb. 1992;12:494–502. doi: 10.1161/01.atv.12.4.494 [DOI] [PubMed] [Google Scholar]
- 97.Redgrave JN, Lovett JK, Gallagher PJ, Rothwell PM. Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation. 2006;113:2320–2328. doi: 10.1161/CIRCULATIONAHA.105.589044 [DOI] [PubMed] [Google Scholar]
- 98.Mäyränpää MI, Heikkilä HM, Lindstedt KA, Walls AF, Kovanen PT. Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis. 2006;17:611–621. doi: 10.1097/01.mca.0000224420.67304.4d [DOI] [PubMed] [Google Scholar]
- 99.Fleg JL, Stone GW, Fayad ZA, Granada JF, Hatsukami TS, Kolodgie FD, Ohayon J, Pettigrew R, Sabatine MS, Tearney GJ, et al. Detection of high-risk atherosclerotic plaque: report of the NHLBI Working Group on current status and future directions. JACC Cardiovasc Imaging. 2012;5:941–955. doi: 10.1016/j.jcmg.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khan AA, Sikdar S, Hatsukami T, Cebral J, Jones M, Huston J, Howard G, Lal BK. Noninvasive characterization of carotid plaque strain. J Vasc Surg. 2017;65:1653–1663. doi: 10.1016/j.jvs.2016.12.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.