

Abstract
Objective
Diagnostic criteria from 2002 classify transverse myelitis (TM) as idiopathic or disease associated but predate the discovery of aquaporin-4 (AQP4)–immunoglobulin G (IgG) and myelin oligodendrocyte glycoprotein (MOG)-IgG, which associate with TM. Prior incidence estimates of idiopathic TM (ITM) range from 1 to 6.2 per 1 million. We sought to determine whether the population-based incidence and prevalence of ITM were reduced by testing patients with ITM for AQP4/MOG-IgG and reclassifying seropositive cases as having disease-associated TM.
Methods
For this observational study, we retrospectively identified all cases of incident (January 1, 2003–December 31, 2016) and prevalent (December 31, 2016) ITM in Olmsted County (85% white) by using the Rochester Epidemiology Project medical records linkage system. ITM was defined by the 2002 Transverse MyelitisConsortium Working Group diagnostic criteria. Available sera were tested for AQP4-IgG and MOG-IgG.
Results
Twenty-four patients (incident 22, prevalent 17) initially met 2002 ITM criteria (longitudinally extensive TM [LETM] 6). Sera were tested for AQP4-IgG in 22 of 24 (92%) and MOG-IgG in 21 of 24 (88%). Three seropositive cases (AQP4-IgG 2, MOG-IgG 1) were identified and reclassified as having disease-associated TM, accounting for 14% of total incident and 12% of total prevalent cases. AQP4-IgG and MOG-IgG seropositive cases represented 50% (3 of 6) of idiopathic LETM. After reclassification of seropositive patients, the final ITM incidence was 8.6 per 1,000,000 and prevalence was 7.9 per 100,000. Three cases of ITM (14%) subsequently fulfilled multiple sclerosis criteria within the study period.
Conclusions
The availability of AQP4-IgG and MOG-IgG modestly reduced ITM incidence and prevalence, which remained higher than previously reported in this predominantly white population. Incorporation of these biomarkers into future revisions of TM diagnostic criteria should be considered.
Idiopathic transverse myelitis (ITM) is a subgroup of inflammatory myelopathies of unclear etiology. In 2002, diagnostic criteria were published to differentiate ITM from disease-associated myelopathies secondary to irradiation, vascular disorders (e.g., spinal cord infarction), connective tissue/granulomatous disorders (e.g., sarcoidosis), infections (e.g., cytomegalovirus), multiple sclerosis (MS), and other CNS demyelinating diseases (based on brain MRI suggestive of MS or prior optic neuritis).1 After the exclusion of disease-associated myelopathies, ITM diagnosis requires bilateral (although not necessarily symmetric) symptoms/signs of spinal cord dysfunction (sensory, motor, or autonomic) evolving over 4 hours to 21 days, a sensory level on the trunk, and evidence of inflammation (MRI gadolinium enhancement or CSF pleocytosis/elevated immunoglobulin G [IgG] index). When evidence of inflammation is lacking, a possible ITM diagnosis is allowed. The goal of these criteria was to identify a homogeneous group of patients for clinical studies.1 During the last 15 years, the identification of aquaporin-4 (AQP4)–IgG and myelin oligodendrocyte glycoprotein (MOG)–IgG as biomarkers of transverse myelitis (TM) has provided a definable etiology to some myelopathies previously thought to be idiopathic.2 Recent findings suggest that a definite etiology can be identified in up to 70% of myelopathies labeled as ITM, prompting a need for contemporary reassessment of ITM epidemiology and characteristics.2 We conducted a population-based study in Olmsted County to determine ITM incidence, prevalence, and characteristics with AQP4-IgG and MOG-IgG testing availability.
Methods
Standard protocol approvals, registrations, and patient consents
The study was approved by the institutional review boards of the Mayo Clinic and Olmsted Medical Center. All patients consented to the use of their medical records for research purposes.
Setting
The Olmsted County region of Minnesota is situated in the Upper Midwest of the United States with a population of 153,183 (December 31, 2016) and white predominance (85.3%). The Rochester Epidemiology Project medical records linkage system includes all medical practitioners in Olmsted County and associated medical records since 1966.
Identification of ITM cases
By using the Rochester Epidemiology Project medical records linkage system, we initially identified all cases of CNS inflammatory demyelinating diseases who resided in Olmsted County during the study period (January 1, 2003–December 31, 2016), similar to prior descriptions.3 Three neurology investigators (E.S., E.S., E.P.F.) reviewed electronic and paper medical records to include isolated myelitis cases fulfilling the 2002 criteria for definite/possible ITM1; consensus was reached after discussion in cases of disagreement. We excluded patients with prior myelitis episodes (including 2 AQP4-IgG–positive cases), concomitant/preceding extraspinal CNS manifestations (including 1 pediatric case with MOG-IgG–positive acute disseminated encephalomyelitis), and myelopathies not meeting the 2002 criteria (figure 1).
Figure 1. Flowchart for identification of patients with ITM.

Flowchart shows the search strategy used to identify patients with isolated myelitis fulfilling the 2002 idiopathic transverse myelitis (ITM) criteria in Olmsted County during the study period. Twenty-one patients did not fulfill the 2002 criteria because of 1 or more of the following: onset to nadir >21 days (48%), unilateral symptoms/signs (48%), and lack of sensory level (57%). These patients also had the following characteristics: age at myelitis onset of 42 (17–56) years; female sex in 15 of 21 (71%); white in 21 of 21 (100%); longitudinally extensive transverse myelitis (LETM) in 1 of 21 (5%); CSF pleocytosis in 7 of 14 (50%); oligoclonal bands in 4 of 12 (33%); elevated immunoglobulin G (IgG) index in 5 of 13 (38%); Expanded Disability Status Scale score at nadir of 2 (0–3); and Expanded Disability Status Scoring at last follow-up of 1 (0–2.5). Five patients subsequently fulfilled multiple sclerosis (MS) criteria during the study period (24%). Seventeen (81%) and 15 (71%) of 21 patients were tested for aquaporin-4 (AQP4)–IgG and myelin oligodendrocyte glycoprotein (MOG)–IgG, respectively, and all were negative. *Myelitis episode with concomitant detection of brain MRI abnormalities consistent with MS. **One patient had a low positivity for antinuclear antibodies. ADEM = acute disseminated encephalomyelitis; CIS = clinically isolated syndrome; HTLV-1 = human T-cell leukemia virus-1; NMOSD = neuromyelitis optica spectrum disorders; ON = optic neuritis; RIS = radiologically isolated syndrome; SLE = systemic lupus erythematosus.
Autoantibody testing
Cases of ITM were tested for AQP4-IgG and/or MOG-IgG by live cell–based assay (fluorescence-activated cell sorting) at the Mayo Clinic Neuroimmunology Laboratory, Rochester, MN, with previously described methodologies.4 Archived samples drawn before AQP4/MOG-IgG testing availability were retested.
Definitions
Longitudinally extensive TM (LETM) was defined as sagittal T2 hyperintensity spanning ≥3 vertebral segments on spine MRI with axial images confirming a single lesion.
Follow-up
Patients were followed up until last contact/MS diagnosis (2017 McDonald criteria).5
Statistical methodology
Prevalence was calculated on December 31, 2016, as the number of cases of ITM per 100,000. Incidence rate was the number of patients with incident ITM (from myelitis onset) divided by the number of person-years at risk during the study period, reported per 1 million person-years. Age standardization to the world population was performed for comparison with other studies.6 Details on the incidence/prevalence calculation and source populations used are outlined in the table. Continuous and categorical variables were reported as median (range) and percentages.
Table.
Incidence and prevalence estimates for Olmsted County and after age standardization to the world population


Data availability
Anonymized data used during this study are available from the corresponding author.
Results
Patients reclassified from ITM to disease-associated TM on the basis of AQP4/MOG-IgG seropositivity
Twenty-four patients initially met definite (n = 22) or possible (n = 2) 2002 ITM criteria (LETM = 6). Serum was tested for AQP4-IgG in 22 of 24 (92%) and MOG-IgG in 21 of 24 (88%). Samples were obtained at the time of myelitis (AQP4-IgG 82%, MOG-IgG 68%) or during remission. Three of 22 cases of incident (14%; AQP4-IgG 2, MOG-IgG 1) and 2 of 17 cases of prevalent (12%; AQP4-IgG 2) ITM were reclassified as having disease-associated TM: all had LETM (figure 2) and CSF pleocytosis (white cells >5/μL); 1 AQP4-IgG–positive case had abnormal CSF oligoclonal bands; and the other had positive antinuclear antibodies but negative SSA/SSB. No patients had sicca symptoms. The AQP4-IgG–positive patients received maintenance rituximab, while the MOG-IgG–positive case received no maintenance treatment. No relapses had occurred by the last follow-up at a median of 8 (8–20) months. AQP4/MOG-IgG seropositivity accounted for 50% (3 of 6) of idiopathic LETM in the population.
Figure 2. Representative examples of spinal cord MRI of patients with AQP4-IgG associated myelitis, and MOG-IgG associated myelitis.
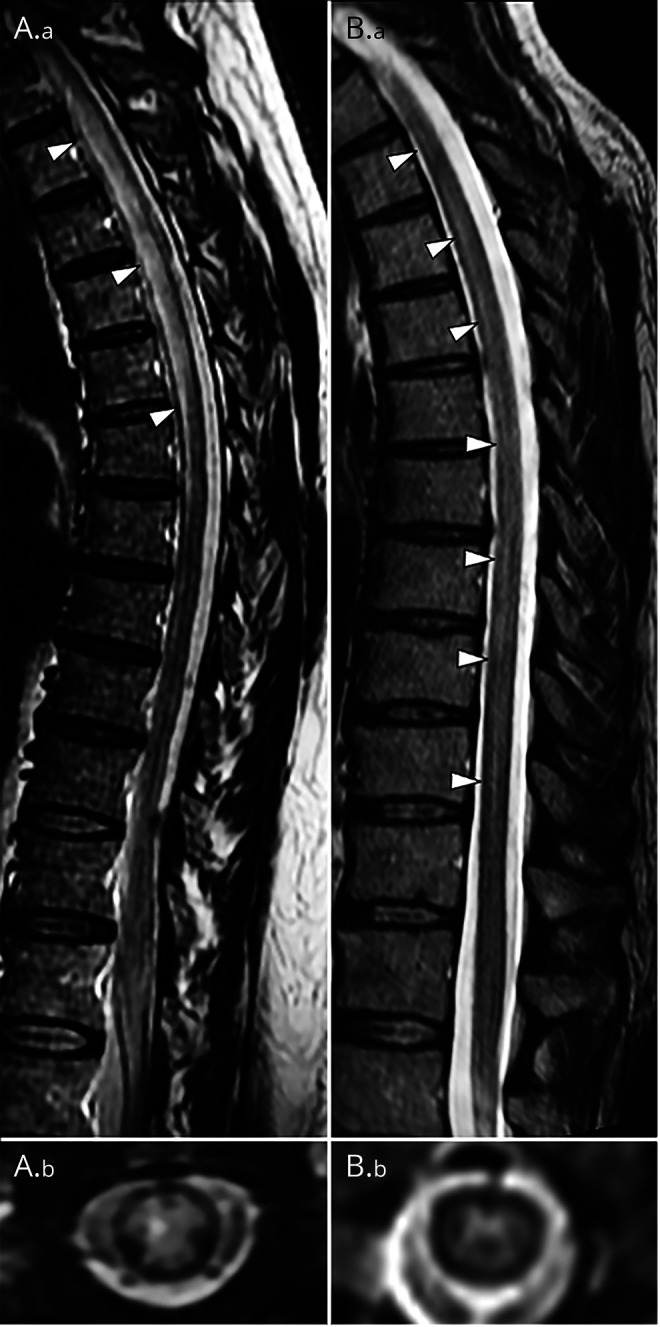
Sagittal T2-weighted images reveal longitudinally extensive lesions along the thoracic spine of one of the two patients with AQP4-IgG (A.a) and the patient with MOG-IgG (B.a) associated myelitis. Axial T2-weighted images reveal AQP4-IgG involving grey and white matter (A.b) and grey matter alone in a H-pattern with MOG-IgG (B.b).
ITM epidemiology
Incidence and prevalence estimates are shown in the table. The temporal distribution of identified cases is illustrated in figure 3.
Figure 3. Temporal distribution of the 21 cases with incident and prevalent ITM who resided in Olmsted County between 2003 and 2016.

Every patients with idiopathic transverse myelitis (ITM) is represented in the graph by a horizontal line that begins at ITM onset. In the first 2 lines from the bottom, onset of ITM precedes January 1, 2003. Only cases with ITM who resided in Olmsted County at myelitis onset were considered for incidence calculation (blue dots). Only the blue nondotted lines that reach the prevalence day (December 31, 2016) contribute to prevalence calculation (green dots). MS = multiple sclerosis
ITM clinical characteristics
Median age at myelitis onset was 41 (22–69) years; 14 of 21 (67%) were female; and 18 of 19 (95%) were white. CSF findings included pleocytosis in 7 of 18 (39%), oligoclonal bands in 6 of 16 (38%), and IgG index >0.85 in 5 of 16 (31%). The median Expanded Disability Status Scale score at nadir was 2.5 (0–7.5) and at last neurology follow-up was 1.5 (0–4.5) at a median of 26 (0.5–213) months from onset. Four patients were lost to follow-up before the prevalence day. Three patients (14%) subsequently fulfilled MS criteria within the study period (figure 3) by developing additional MRI lesions (n = 1) or clinical relapse (n = 2).
Discussion
The discovery of AQP4-IgG and MOG-IgG since the publication of ITM criteria in 2002 results in a modest reduction in ITM incidence and prevalence, which remained higher than previously reported. Incorporation of AQP4-IgG and MOG-IgG testing into TM diagnostic criteria is needed to allow reclassification of seropositive patients as having disease-associated TM.
Few population-based studies have investigated ITM epidemiology in the MRI era, and they predate AQP4/MOG-IgG testing availability. A US study (Albuquerque, NM) reported an ITM incidence of 1 per 1 million using different ITM requirements (e.g., concomitant presence of sensory, motor, and autonomic symptoms/signs).7 Another study (North Canterbury, New Zealand) using the 2002 criteria reported an ITM incidence of 6.2 per 1 million, which is closer to our findings (8.6 per 1 million).8 In both studies, however, patient identification was based on hospital registries, which may fail to detect less severe cases not hospitalized or residents seen outside the expected referral centers. In our study, 14% of cases of ITM subsequently fulfilled MS criteria, which is similar to the 13% reported in a prior hospital-based series with longer follow-up.9 The absence of pediatric cases of ITM in our study may reflect the relatively small study population, the lower proportion of children compared to the world population (table), or the exclusion of pediatric TM cases who met criteria for other diseases (e.g., acute disseminated encephalomyelitis).
Although not incorporated into the 2002 criteria, lesion length on spine MRI has been shown to be very useful in discriminating TM accompanied by AQP4/MOG-IgG from MS, and these antibodies accounted for 50% of idiopathic LETM in this study.10 A prior hospital-based study on ITM before MOG-IgG availability reported an LETM frequency of 30%, but after the exclusion of both AQP4-IgG and MOG-IgG disease-associated TM, we found it to be 14%.9 Notably, short-TM cases often had peripheral/incomplete MS-like spinal cord lesions but still fulfilled the 2002 criteria clinical requirement of bilateral symptoms/signs (e.g., Brown-Sequard syndrome). Stratification by lesion length on spinal cord MRI (LETM or short-TM) may help identify more homogeneous subgroups of myelitis.
Our findings are generalizable to the Upper Midwest of the United States, but the proportion of AQP4-IgG disease-associated TM could be higher in populations with a higher prevalence of nonwhite ethnicities.3 It is also possible that some cases of ITM without sera available or with sera drawn during remission (particularly relevant for MOG-IgG in which transient seropositivity is common) could have led to underestimation of AQP4/MOG-IgG seropositivity.4 However, the vast majority had sera available for testing, and most were attack samples.
Acknowledgment
The authors thank Dr. Jennifer St. Sauver for her suggestions on how to report epidemiologic estimates.
Glossary
- AQP4
aquaporin-4
- IgG
immunoglobulin G
- ITM
idiopathic transverse myelitis
- LETM
longitudinally extensive transverse myelitis
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- TM
transverse myelitis
Appendix. Authors
Study funding
This study was supported by a research fellowship funded by the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology. This study was made possible with the resources of the Rochester Epidemiology Project, which is supported by the National Institute on Aging of the NIH under award R01AG034676. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosure
E. Sechi, E. Shosha, and J. Williams report no disclosures relevant to the manuscript. S. Pittock has financial interest in patents (12/678,350 filed in 2010 and 12/573,942 filed in 2008) that relate to functional AQP4/neuromyelitis optica (NMO)–IgG assays and NMO-IgG as a cancer marker. He has served as a consultant to Alexion Pharmaceuticals and MedImmune. He has received research funding from Alexion, MedImmune, and Grifols. B. Weinshenker receives royalties from RSR Ltd, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr. Volkmann und Kollegen GbR for a patent of NMO-IgG as a diagnostic test for NMO and related disorders. He serves as a member of an adjudication committee for clinical trials in NMO being conducted by Viela Bio and Alexion Pharmaceutical. He is a consultant for Caladrius Biosciences, Brainstorm Therapeutics, Roivant Sciences, and Chugai Pharma regarding potential clinical trials for NMO. B. Keegan was funded by Biogen and receives publishing royalties for Common Pitfalls in Multiple Sclerosis and CNS Demyelinating Diseases. He is an Editorial Board member of Multiple Sclerosis and Related Disorders. N. Zalewski, A. Lopez-Chiriboga, and J. Jitprapaikulsan report no disclosures relevant to the manuscript. E. Flanagan receives research support as a site principal investigator in a randomized placebo-controlled clinical trial of inebilizumab (A CD19 inhibitor) in NMO spectrum disorders funded by MedImmune/Viela Bio. Go to Neurology.org/N for full disclosures.
References
- 1.Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology 2002;59:499–505. [DOI] [PubMed] [Google Scholar]
- 2.Zalewski NL, Flanagan EP, Keegan BM. Evaluation of idiopathic transverse myelitis revealing specific myelopathy diagnoses. Neurology 2018;90:e96–e102. [DOI] [PubMed] [Google Scholar]
- 3.Flanagan EP, Cabre P, Weinshenker BG, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol 2016;79:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol 2018;75:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 6.Ahmad OB, Boschi-Pinto C, Lopez AD, Murray CJL, Lozano R, Inoue M. Age Standardization of Rates: A New Who Standard. Geneva: World Health Organization; 2001. [Google Scholar]
- 7.Jeffery DR, Mandler RN, Davis LE. Transverse myelitis: retrospective analysis of 33 cases, with differentiation of cases associated with multiple sclerosis and parainfectious events. Arch Neurol 1993;50:532–535. [DOI] [PubMed] [Google Scholar]
- 8.Young J, Quinn S, Hurrell M, Taylor B. Clinically isolated acute transverse myelitis: prognostic features and incidence. Mult Scler 2009;15:1295–1302. [DOI] [PubMed] [Google Scholar]
- 9.Cobo Calvo A, Mañé Martínez MA, Alentorn-Palau A, Bruna Escuer J, Romero Pinel L, Martínez-Yélamos S. Idiopathic acute transverse myelitis: outcome and conversion to multiple sclerosis in a large series. BMC Neurol 2013;13:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zalewski NL, Flanagan EP. Autoimmune and paraneoplastic myelopathies. Semin Neurol 2018;38:278–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data used during this study are available from the corresponding author.