

Abstract
Hematopoiesis has long served as a paradigm of stem cell biology and tissue homeostasis. In the past decade, the genomics revolution has ushered in powerful new methods for investigating the hematopoietic system that have provided transformative insights into its biology. As part of the genomics revolution, increasingly accurate deep sequencing and novel methods of cell tracking have revealed hematopoiesis to be more of a continuous and less of a discrete and punctuated process than originally envisioned. In part, this continuous nature of hematopoiesis is made possible by the emergent outcomes of vast, interconnected regulatory networks that influence cell fates and lineage commitment. It is also becoming clear how these mechanisms are modulated by genetic variation present throughout the population. This review describes how these recently uncovered complexities are reshaping our concept of tissue development and homeostasis, while opening up a more comprehensive future understanding of hematopoiesis.
Sankaran eTOC blurb
Sankaran et al.. review our evolving understanding of the mechanisms of hematopoiesis, emphasizing how insights from genomics technologies are driving a deeper understanding of lineage commitment, gene regulatory mechanisms, and the impact of human genetic variation.
INTRODUCTION
The hematopoietic system is a remarkable tissue that participates in nutrient transport, immune function, hemostasis, and wound healing, and in humans generates a staggering 500 billion cells each day (Fliedner et al., 2002). The tissue is responsible for the production of the adaptive and innate immune systems, the red blood cells that transport oxygen, and the megakaryocytes that are responsible for producing platelets necessary for blood clotting. As a relatively accessible tissue, the hematopoietic system has served a pivotal role in advancing our understanding of fundamental aspects of stem cell biology. The hierarchical organization of the hematopoietic system has also been illustrative of how tissues can rely on a small pool of stem and progenitor cells for decades, while still effectively and continuously generating large numbers of mature functional cells. While numerous studies over the last century have illuminated how hematopoiesis is regulated, the genomics revolution has offered new opportunities to obtain a more comprehensive understanding of this complex process. It is important to note that dramatic differences in hematopoiesis exist across various organisms and in the course of development, and while these differences are fascinating in their own right and have been extensively reviewed elsewhere (Banerjee et al., 2019; Dzierzak and Bigas, 2018; Stachura and Traver, 2011), in this review we focus largely on observations made through studies of adult hematopoiesis in mice and humans.
It has been a continuous challenge to understand the behavior of tissues at the cellular level, in a way that encompasses the relationships between cells and their dynamics during differentiation. The genomics revolution has enabled entire tissues such as the hematopoietic system to be comprehensively interrogated at single-cell resolution, revealing a previously unappreciated degree of functional and phenotypic variation within previously presumed homogeneous cell compartments. This heterogeneity is revealing how cells make fate decisions and differ in their contributions to the hematopoietic system. Combined with the power of human genetics, we are gaining a deeper appreciation for how genetic variation can modulate the process of hematopoiesis in surprising and consequential ways. In the words of Bram Stoker in his novel Dracula, “There are mysteries which [people] can only guess at, which age by age they may solve only in part” (Stoker, 1897). In this review, we illustrate how this concept echoes in the field of hematopoiesis and discuss how the last decade has led to ongoing work that leverages the enumerable advances in genomics to refine our understanding of this process.
OVERVIEW OF HEMATOPOIESIS
Hematopoietic tissue hierarchy
The hematopoietic system is classically divided into two major branches, the myeloid and lymphoid lineages. The myeloid lineage is responsible for three major functions: oxygen transport, hemostasis, and innate immunity. Erythrocytes are produced through myeloid progenitors and enable effective oxygen transport throughout the body. Megakaryocytes are the cellular precursors that give rise to platelets that are necessary for blood clotting and are produced through the myeloid lineage, although recent findings suggest that these cells may arise directly from stem cells (Carrelha et al., 2018; Rodriguez-Fraticelli et al., 2018; Sanjuan-Pla et al., 2013). Innate immune cells are produced by the myeloid lineage and include monocytes, neutrophils, eosinophils, basophils, and dendritic cells which provide a broad, general defense against foreign pathogens, as well as facilitate the initiation of adaptive immune responses. The lymphoid lineage produces B, T, and NK cells that are responsible for adaptive immunity. While these two lineages have been thought of as distinct, recent studies suggest some complexity in this classic bifurcation, with innate lymphoid cells that have key roles in innate immunity arising from lymphoid progenitors and some contribution of lymphoid-biased progenitors to lineages typically thought to be of myeloid origin (Pucella et al., 2020; Vivier et al., 2018).
An improved understanding of the stem and progenitor cells mediating production of myeloid and lymphoid cells has been enabled by the use of cell surface markers and flow cytometry to separate out distinct states that could be ordered by their degree of differentiation. This organization led to a model in which mammalian hematopoiesis was initiated within a homogeneous pool of multipotential long-term reconstituting hematopoietic stem cells (LTHSCs) that produced a cascade of increasingly more lineage-restricted cells in order to produce all mature blood cells (Figure 1A) (Orkin and Zon, 2008). Such a model often proposed that LTHSCs remain relatively quiescent in order to minimize cell-cycle associated DNA damage, relying instead on short-term hematopoietic stem cells (ST-HSCs) and downstream progenitors to carry most of the replicative burden necessary for steady state hematopoiesis. In order to maintain its population over an organism’s lifespan, this LT-HSC population can self-renew, while more differentiated pools of cells progressively lose this ability (Morrison and Weissman, 1994; Morrison et al., 1997).
Figure 1. Evolving Models of Hematopoiesis.
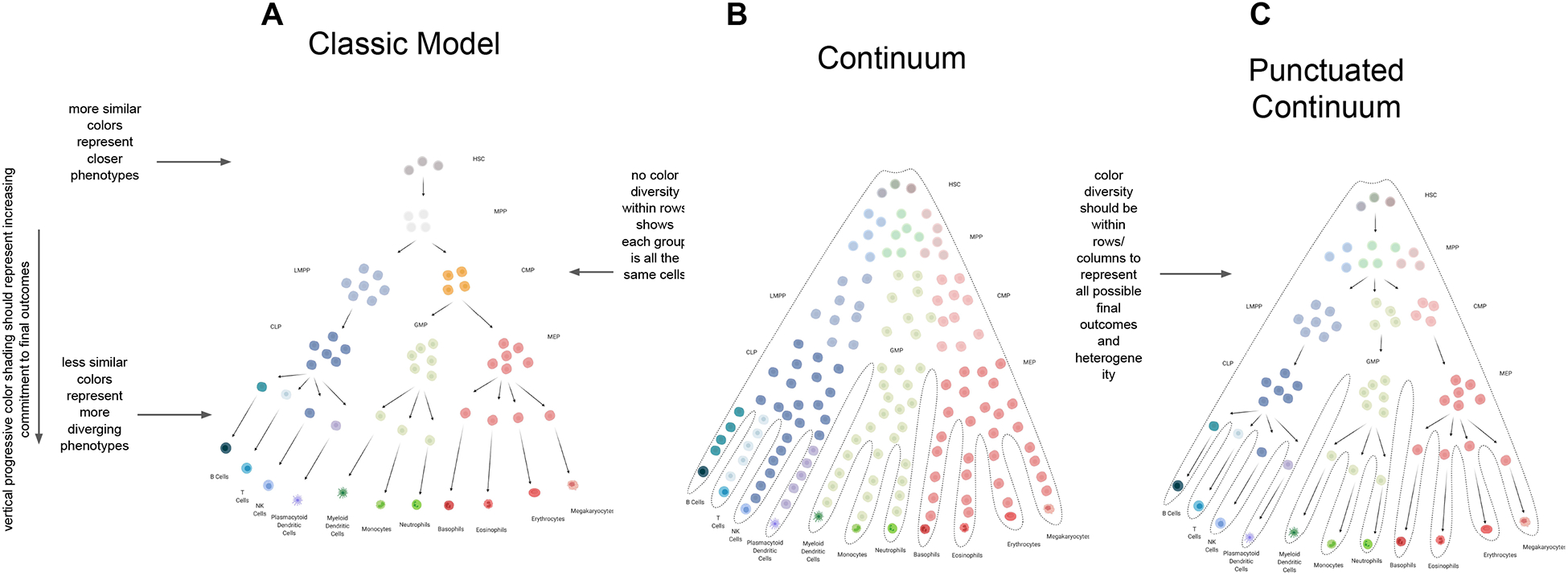
(A) The “classic model” of hematopoiesis involves discrete states that contain cells of uniform potential, as illustrated by cell color. Cell fate choices are made at each of these states, and are depicted as punctuated transformations of one phenotype to another (Orkin and Zon, 2008). (B) In the “continuum” model, cells are variably lineage biased as indicated by coloration. However, punctuated transitions are absent and a truly continuous process of differentiation emerges (Laurenti and Göttgens, 2018; Velten et al., 2017). Abbreviations: hematopoietic stem cell (HSC), multipotent progenitor (MPP), common myeloid progenitor (CMP), megakaryocyte/erythroid progenitor (MEP), granulocyte/macrophage progenitors (GMP), common lymphoid progenitor (CLP), lymphoid-primed multi-potential progenitor (LMPP). (C) The “punctuated continuum” model shares some of the discrete phenotypic transformations of the classic model, but is distinct in its representation of cell states as heterogeneous pools of cells (Laurenti and Göttgens, 2018; Pietras et al., 2015). Within each state, cells are variably primed to give rise to different hematopoietic lineages, as indicated by the variation in color. The magnitude of this lineage commitment increases with color intensity as cells proceed along the differentiation hierarchy.
A fundamental aspect of the classic model of hematopoiesis is that at each stage of differentiation, cells are uniformly lineage committed, and only further commit by undergoing binary fate choices at these discrete cell states (Figure 1A). A characteristic example of such a binary decision is the split from multipotent progenitors into lineage-restricted common myeloid progenitors (CMP) and common lymphoid progenitors (CLP) (Akashi et al., 2000). This punctuated model of differentiation has served as the basis for studies in other tissues (Barker et al., 2007; Snippert et al., 2010), and has shaped our understanding of pathological changes, such as those observed in aging and cancer (Derényi and Szöllősi, 2017; Rozhok et al., 2015; Tomasetti and Vogelstein, 2015).
Hematopoietic stem cells at the apex of this hierarchy
While today the concept of a stem cell may seem unremarkable, it was first formally proposed at the beginning of the twentieth century, but proved notoriously difficult to experimentally identify (Maximow, 1909). Direct evidence for the existence of a blood-forming stem cell came nearly half a century later. In search of primitive blood-forming cells, the circulatory systems of lethally irradiated mice and healthy mice were connected to demonstrate that the healthy hematopoietic tissue could rescue the lost function of the irradiated tissue (Brecher and Cronkite, 1951). While successful rescue in these experiments relied on the unrecognized behavior of HSCs to periodically egress from the bone marrow (BM) (Bhattacharya et al., 2009), direct injection of BM cells also rescued irradiated mice (Ford et al., 1956). Fortuitously, it was noted that injected BM formed colonies consisting of various hematopoietic cells within the spleens of the recipient mice (Becker et al., 1963; Till and McCULLOCH, 1961). When reinjected into irradiated mice, some of these colonies themselves could rescue hematopoiesis (Siminovitch et al., 1963). A few decades later, surface-marker staining enabled, for the first time, the prospective purification of a BM-reconstituting population of hematopoietic stem cells (HSCs) (Spangrude et al., 1988). Shortly after this discovery, competitive BM transplants were used to estimate that about 10,000 HSCs existed in the mouse hematopoietic system (Szilvassy et al., 1990).
Improved cell labelling later revealed the existence of subpopulations of hematopoietic cells, and resulted in the separation of the stem cell compartment into cells from least differentiated to most differentiated LT-HSCs, ST-HSCs, and multipotent progenitors (MPPs) (Orkin and Zon, 2008). Through early studies of these populations, their lineage relationships were modeled as a uniform progression from LT-HSCs, which could provide long term multilineage reconstitution after transplantation, through ST-HSCs that could only provide short term reconstitution after transplantation, to MPPs that gave limited reconstitution, after which cells became increasingly lineage restricted (Morrison et al., 1997). This hierarchy has now been observed not only in mice, but also in humans and other organisms (Doulatov et al., 2010; Majeti et al., 2007; Notta et al., 2011), and has continued to expand in complexity, incorporating further subpopulations and subdivision of downstream progenitors.
DECIPHERING HEMATOPOIETIC CELL STATES AND FATES FROM SINGLE CELL GENOMICS
Revisions to the lineage tree model of hematopoiesis
The now classic arrangement of hematopoietic hierarchy presents an enticingly concise model of punctuated lineage decisions being made within pools of functionally homogeneous cells (Figure 1A). In retrospect, the limited diversity of available surface markers likely resulted in the oversimplification of a more complex process (Doulatov et al., 2012). Indeed, as additional cell-surface markers were employed (Nolan, 2011), models of hematopoiesis grew in complexity, incorporating revisions to the timing of lineage commitment decisions (Adolfsson et al., 2005; Doulatov et al., 2010; Goardon et al., 2011; Sanjuan-Pla et al., 2013; Yamamoto et al., 2013) and the subdivision of cell states into increasing numbers of subgroups (Cabezas-Wallscheid et al., 2014; Pietras et al., 2015). Of course, as models grew in complexity, inconsistencies in their organization emerged, especially in how cell states were defined (Adolfsson et al., 2005; Görgens et al., 2013; Haas et al., 2015; Perié et al., 2015; Yamamoto et al., 2013). These inconsistencies have made it attractive to turn to single-cell genomic methods that can more comprehensively identify various cell populations and states, and better reveal the timing of cell decision-making during differentiation.
The long and rich history of research in the field of hematopoiesis has helped lay the foundation for the use and value of single-cell genomics. Early work provided methods of cell purification and transplantation of single HSCs into mice in order to reveal functional differences, such as lineage output biases (Dykstra et al., 2007; Muller-Sieburg et al., 2004; Osawa et al., 1996), and these approaches were complemented by single-cell progenitor functional assays (Purton and Scadden, 2007). Preliminary HSC fate tracking was performed through retroviral integration (Lemischka et al., 1986). Even before the first draft of the human genome, it was possible to perform single cell transcriptomics on hematopoietic cells (Brady et al., 1990). With time, these methods grew in throughput, allowing thousands of hematopoietic cells to be simultaneously profiled, and consequently revealed a great deal of heterogeneity within the mammalian hematopoietic system (Adolfsson et al., 2005; Goardon et al., 2011; Kohn et al., 2012; Miyawaki et al., 2017; Notta et al., 2016; Pronk et al., 2007; Psaila et al., 2016; Sanada et al., 2016).
From homogeneous to heterogeneous cell states in hematopoiesis
Early studies of hematopoiesis relied on the ability to group cells into populations by surface marker staining. As available staining panels were limited, these populations were functionally and phenotypically distinct, representing dramatically different points throughout the process of hematopoiesis. As a result, initial models of hematopoiesis reflected the discrete nature of these surface staining methods (Figure 1A). As single-cell transcriptomics became possible, there was a unique opportunity to simultaneously assay a significant proportion of the hematopoietic system and hierarchically organize cells by their similarity in gene expression (Gulati et al., 2020; Shin et al., 2015; Trapnell et al., 2014).
When computationally reconstructed, single-cell RNA sequencing (scRNA-seq) profiling of the mammalian hematopoietic system typically produces a largely continuous distribution of cells from the earliest HSCs through the characteristic hematopoietic lineages (Nestorowa et al., 2016; Olsson et al., 2016; Paul et al., 2015; Pellin et al., 2019; Tusi et al., 2018; Weinreb et al., 2020). Such observations were made both for analyses involving the entire hematopoietic system and for assessments of populations of cells that previously were thought to be homogeneous. Similar differentiation continuity can also be seen in complementary but distinct methods such as RT-PCR (Guo et al., 2013). This transcriptional continuity has contributed to a revised model of the hematopoietic lineage tree, often termed a continuum model of hematopoiesis (Figure 1B). In this continuum model, the process of differentiation from HSCs to progenitors to mature functional cells is envisioned as occurring constantly and without the distinct punctuated phenotypic changes that exist within the classic model of hematopoiesis (Laurenti and Göttgens, 2018; Velten et al., 2017). This continuity represents an important divergence from the classical model of hematopoiesis in which cells only make a handful of fate decisions and are functionally identical to other cells that have made those same decisions. The original states themselves would also largely reflect the arbitrary borders created by limited samplings of hematopoiesis rather than biologically genuine cell states. It is still unclear how continuous lineage decisions could be made, but there is support for cells expressing distinct modules of transcriptional programs that cooperate to subtly and continuously prime cells to differentiate along target lineages (Giladi et al., 2018; Velten et al., 2017). Of course, gene expression can vary widely across phenotypically similar cells (Kaufmann and van Oudenaarden, 2007; Marinov et al., 2014), which may cause enough transcriptome diversity in scRNA-seq that distinct populations of cells blend together and become indistinguishable from each other. Thus, while the continuum model clearly addresses some of the oversimplifications of the classic model of hematopoiesis, gene expression alone may be insufficient to distinguish discrete cell populations and may miss other relevant aspects of biology (Olsson et al., 2016).
Functional interrogation of hematopoietic cell states can complement single cell transcriptomics, and may reveal underlying biology that may be missed by scRNA-seq alone. As mentioned earlier, the use of increased numbers of surface markers have expanded the number of possible hematopoietic subgroups, especially within the stem and progenitor cell compartments. Coupling gene expression variation with assessment of different subpopulations has enabled distinct functional groups to be defined (Pietras et al., 2015). Furthermore, with improved abilities to simultaneously interrogate gene expression, while perturbing key master regulators of this process, such as hematopoietic transcription factors (TFs), it has become clear that there are distinct transition points that occur over this continuous transcriptomic landscape, suggesting the presence of punctuated transitions (Giladi et al., 2018). It must therefore be considered that while hematopoiesis may be far more of a continuous process than originally modeled, punctuated transitions across this continuous gene expression landscape may still exist, and represent functionally distinct groups of cells (Figure 1C) (Laurenti and Göttgens, 2018). In the future, it will be important to consider the limitations of scRNA-seq and couple these types of assays with functional perturbations and other multi-omic measurements to comprehensively dissect the process of hematopoiesis.
We expect that future work will combine multiple technologies together with more sophisticated computational models that are more capable of unravelling the underlying complex mechanisms of hematopoiesis (Gulati et al., 2020). Additionally, while the focus of this review is on steady state hematopoiesis, the generation of the tissue during development involves unique biology (Copley and Eaves, 2013; Dzierzak and Bigas, 2018; Ivanovs et al., 2017) and may therefore require uncharacteristic and intriguing cell behaviors. Indeed, single-cell studies are already uncovering distinct states involved in the generation and early development of HSCs (Pijuan-Sala et al., 2019; Popescu et al., 2019; Vink et al., 2020; Zeng et al., 2019a, 2019b; Zhou et al., 2016). Moreover, while our focus has been on studies involving the intrinsic regulation of hematopoiesis, it is clear that microenvironmental factors and niche cells play a key role in this process, which has been covered in detail in other reviews (Crane et al., 2017; Pinho and Frenette, 2019). Recent studies have started to dissect the nature of these cells through scRNA-seq (Baccin et al., 2019; Baryawno et al., 2019; Tikhonova et al., 2019). With progressive improvements in single-cell methods to dissect functional interactions (Giladi et al., 2020), further insights into the hematopoietic microenvironment will undoubtedly be made in the coming years. Finally, while single-cell genomic approaches have produced compelling evidence that observed cellular heterogeneity reflects downstream lineage decisions (Herring et al., 2018; La Manno et al., 2018; Rodriguez-Fraticelli et al., 2018), in the absence of fate tracking, all inferred biases in native hematopoiesis are speculative (Pina et al., 2012; Velten et al., 2017).
Lineage commitment and fate heterogeneity
Both scRNA-seq and single-cell transplant experiments suggest significant heterogeneity in the lineage commitment of HSCs and other progenitors that otherwise appear to be functionally similar (Dong et al., 2020). Within the last decade, it has become possible through the combination of cell barcoding and scRNA-seq to directly trace cell fates and understand the extent of lineage commitment present within a given cell compartment and how deterministic these are (Figure 2).
Figure 2. Lineage Tracing and Fate Mapping in Hematopoiesis.
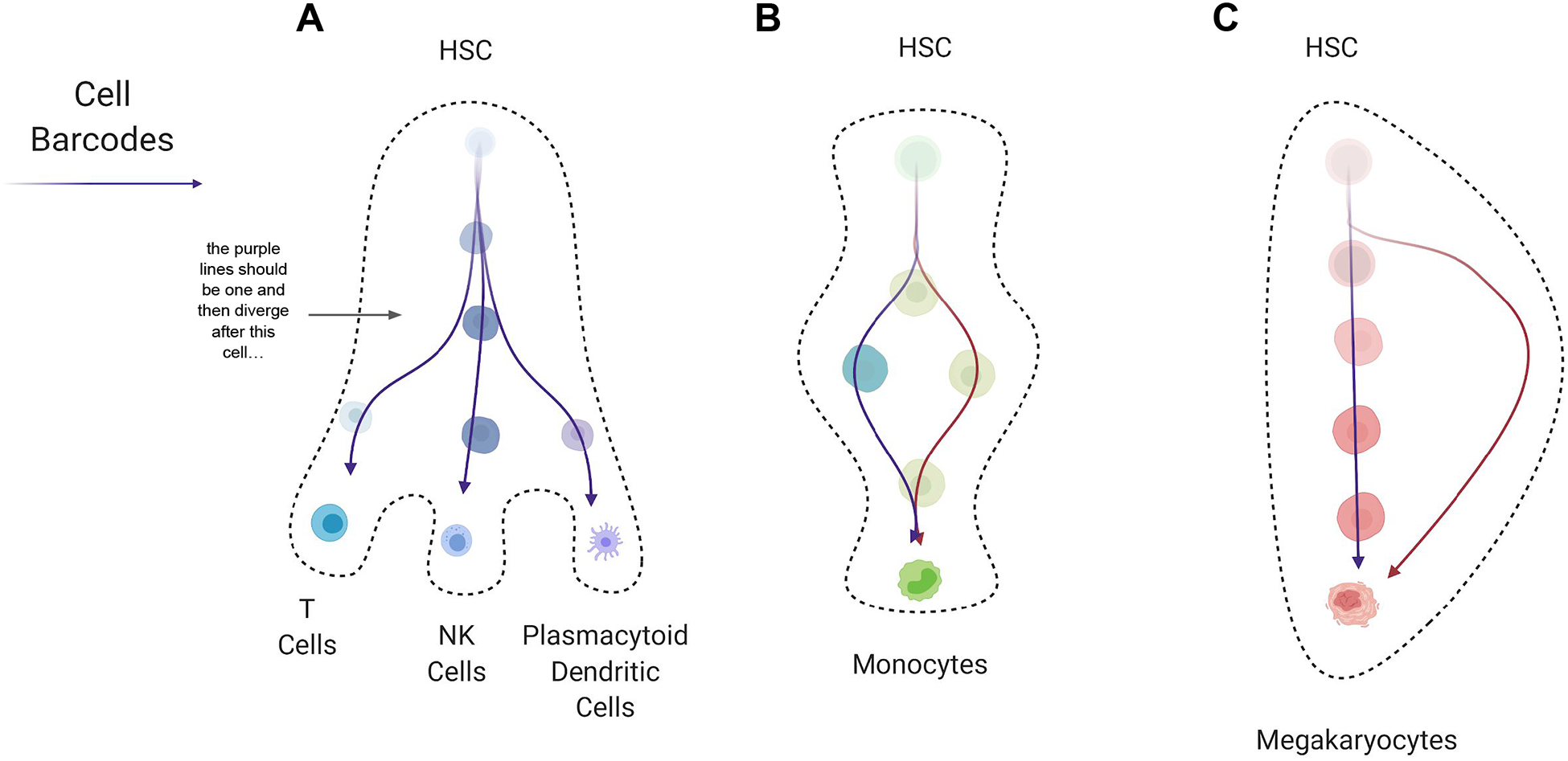
Hematopoietic differentiation pathways as observed by cell barcoding and indicated by colored arrows. (A) Barcoding of an HSPC results in the barcode being carried through three lineages (T cells, NK cells and plasmacytoid dendritic cells) to which the clone contributes, showing the multipotential behavior of such cells. (B) Two different barcodes originate in the same compartment in early hematopoiesis, but are found within distinct progenitor lineages before giving rise to monocytes (Weinreb et al., 2020). This illustrates how a single differentiation state can be produced through multiple distinct paths. (C) A barcode originating within early HSPCs can be found along the classic path of differentiation, through progenitor states on the way to producing mature megakaryocytes. Alternatively, HSCs can also directly generate megakaryocytes and no barcodes will be found within the classic differentiation path in this case (Carrelha et al., 2018). This is another example of how a differentiated cell type can be produced via multiple paths. Abbreviations: hematopoietic stem cell (HSC).
While cell-sorting based methods often show a multipotent HSC and progenitor compartment that gives rise to unipotent lineage-committed cells (Notta et al., 2016), the combination of scRNA-seq and cell barcoding has now directly demonstrated that the early fate biases in hematopoietic stem and progenitor cells (HSPCs) significantly impact eventual fate (Weinreb et al., 2020). Moreover, cell barcoding approaches suggest that CMPs are a heterogeneous population of cells that have already made deterministic fate choices of lineage output (Perié et al., 2015). While barcoding efficiencies may complicate the interpretation of how different progenitors contribute to hematopoiesis, these observations appear to directly indicate that cells within the same compartment do not equally contribute to a particular lineage or to overall hematopoietic output. In the future, it will be helpful to understand how committed HSPCs remain to generating a certain lineage, how variable this is when comparing ex vivo manipulation of cells with steady-state labeling approaches (Sun et al., 2014b; Weinreb et al., 2020), and how this may change in response to injury or disease (Giladi et al., 2018; Khajuria et al., 2018). Further complicating the picture of hematopoiesis, it appears that there are multiple differentiation paths that cells can follow to produce the same lineage, such as is observed for monocyte differentiation (Figure 2) (Weinreb et al., 2020). Signatures of this differentiation route decision are even maintained within mature cells. It is still uncertain, however, if these signatures impact the final behavior of the mature cells or if the various differentiation routes are a way of responding to the ever-changing needs of the hematopoietic system.
While the HSC and immediate downstream MPP compartments give rise to all the lineages of the hematopoietic system, the mechanism by which this occurs still remains uncertain (Grün et al., 2016; Notta et al., 2016; Paul et al., 2015). Future studies are necessary to understand when lineage commitment decisions are made, in order to understand the output of individual HSCs during steady state hematopoiesis and how flexible this commitment is in homeostatic and disease states. Furthermore, the dynamics of hematopoiesis in early stem and progenitor cell pools is still unclear. Classic models put most replicative burden on progenitor cells, and expect HSCs to contribute to hematopoiesis only sporadically. Emerging cell barcoding and fate tracing methods may help to clarify this process as well.
HSC contributions to hematopoiesis
In classic models of hematopoiesis, replication burden exponentially increased with level of differentiation (Morrison et al., 1997), allowing HSCs to remain quiescent and unscathed from DNA damage in order to ensure their longevity (Cheshier et al., 1999; Passegué et al., 2005; Wilson et al., 2008). While there have been attempts to understand the relative contributions of HSPCs to steady state hematopoiesis, it has been challenging to develop a clear understanding of the direct contributions typically made by HSCs (Dykstra et al., 2007; Eaves, 2015; Muller-Sieburg et al., 2004; Shizuru et al., 2005).
Recent attempts to understand the typical quiescence of HSCs in mice have leveraged new genomic tools in order to better illuminate their direct contribution during steady state hematopoiesis. One innovative approach employed a doxycycline-inducible transposase system in mice to barcode cells by transposon integration site (Sun et al., 2014b). During steady state hematopoiesis, barcodes found within mature cells appeared to originate primarily from MPPs, suggesting that progenitors, rather than HSCs, carry most of the tissue-generation burden. Direct genetic barcoding in HSCs has also resulted in similar findings (Rodriguez-Fraticelli et al., 2018). However, barcodes produced through Cre-lox recombination in different hematopoietic cells confirmed a major replicative role of MPPs, while also demonstrating a consistent contribution of HSCs (Busch et al., 2015). Similarly, direct fluorescent labeling of mouse HSCs under steady state conditions illustrated a significant and continued contribution of HSCs to nearly all steady state hematopoiesis (Chapple et al., 2018; Sawai et al., 2016; Säwen et al., 2018).
It remains unclear why different methods demonstrate such variable contributions of HSCs to murine steady state hematopoiesis. It is possible that labeling inefficiencies overlook the contributions of rare HSCs or low frequency differentiation, just as labeling saturation may overrepresent the contribution of HSCs that share barcodes with progenitors. It must also be considered that underlying assumptions in such varied models may be artificially muddling our view of the process. At the current time, there is substantial evidence that HSCs make regular and consistent contributions to hematopoiesis, although further studies are needed to delineate the precise nature of these contributions, while accounting for limitations that exist for these different experimental approaches (Pucella et al., 2020).
Clonal hematopoiesis
As HSC behavior during steady state hematopoiesis is more precisely defined, it will become clearer how the process can be disrupted. There are many ways to impair tissue functionality, as is observed in non-malignant and malignant blood disorders. A recently uncovered phenomenon called clonal hematopoiesis (CH) provides an illustrative example of how increasingly sensitive genomic approaches can reveal key perturbations to hematopoiesis and provide new insights into this process (Figure 3) (Jaiswal and Ebert, 2019; Liggett et al., 2019).
Figure 3. Clonal Hematopoiesis.
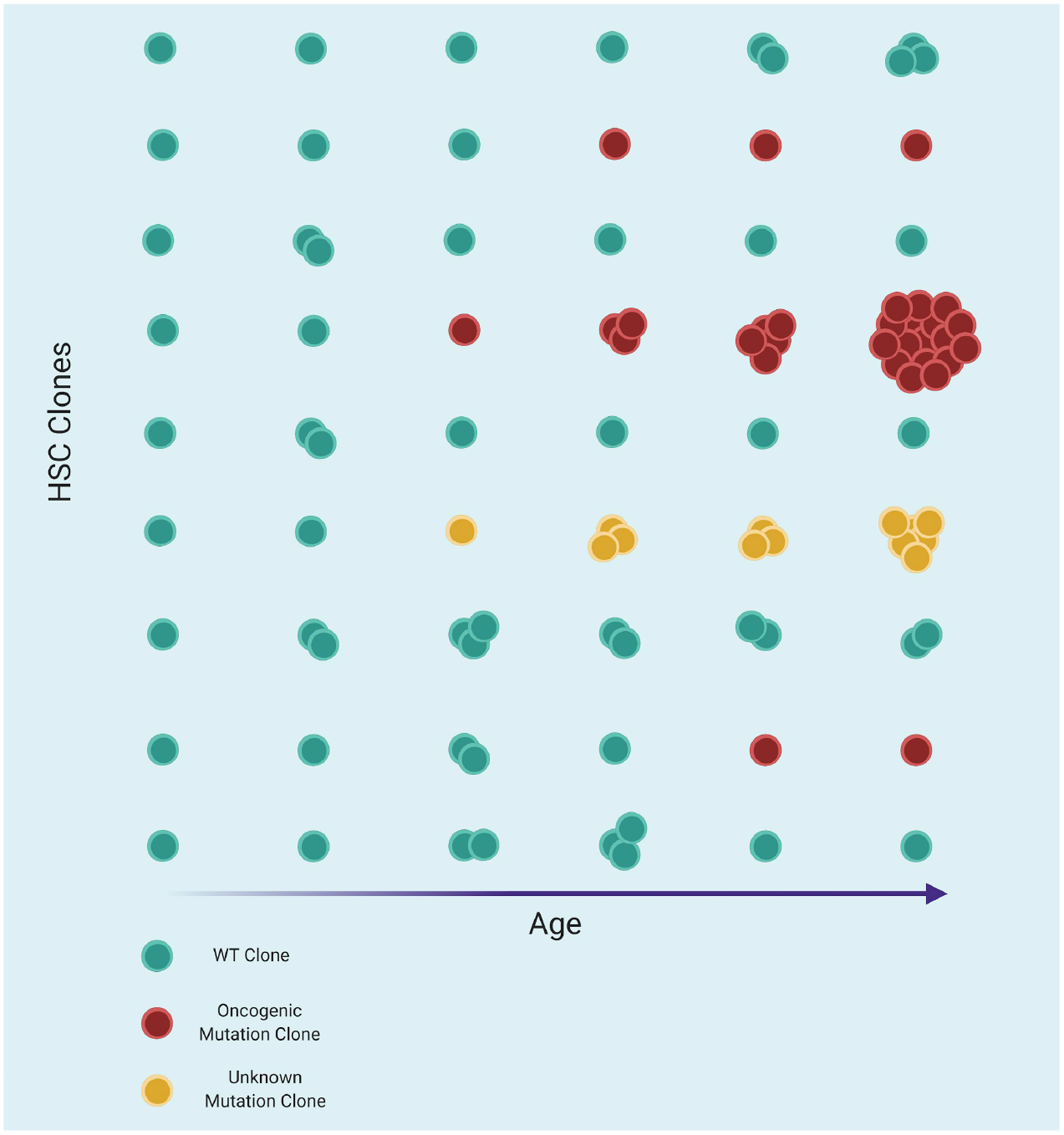
The HSC compartment is depicted here as columns, with each cell being a representative clone from the population. The leftmost column depicts early human life, in which HSC clones are largely of similar sizes and typically free of red, oncogenic mutant clones. With age, HSCs sporadically acquire mutations, most of which will be of little functional consequence; cells will remain blue, and continue to fluctuate in clone size over time as they compete against their peers without any acquired genetic advantage. Some cells will acquire putatively oncogenic mutations, as indicated in red. These mutant cells might clonally expand and represent a significant proportion of hematopoiesis, as occurs in clonal hematopoiesis of indeterminate potential (CHIP) and as depicted in the fourth row from the top. Yellow cells represent those that have unknown genetic alterations and increase in clonal size, thereby also representing CH. With time, clones can also sporadically be lost or otherwise outcompeted, thereby eliminating them. By old age in humans, the HSC compartment will often have become clonal, as depicted in the final column, and will contain more observable oncogenic mutant clones.
As humans age, they typically experience an increase in total phenotypic HSC numbers as the cell pool grows with time (de Haan and Van Zant, 1999; Pang et al., 2011; Sudo et al., 2000). However, in some elderly individuals, HSCs acquire oncogenic mutations that allow them to outcompete their neighbors and clonally expand. As these HSCs clonally expand, they can account for a substantial fraction of steady state hematopoiesis, termed clonal hematopoiesis of indeterminate potential (CHIP) (Busque et al., 1996; Champion et al., 1997; Vas et al., 2012). While the likelihood of CHIP increases with age, it appears in only a fraction of the population, and it increases the risk of cancer, cardiovascular disease, and other age-related morbidities (Bick et al., 2019a; Genovese et al., 2014; Jacobs et al., 2012; Jaiswal et al., 2014, 2017; Laurie et al., 2012; Xie et al., 2014). Though ostensibly the direct result of oncogenic mutation acquisition, it is unclear why increased HSC clonality itself would increase the risk of non-cancer morbidities, such as cardiovascular disease. The phenomenon also appears to be counterintuitively well tolerated, where HSCs containing oncogenic mutations can overtake most of the hematopoietic system without transforming into leukemia (Jaiswal et al., 2014).
With an ever growing ability to more precisely track individual clones at single cell resolution (Nam et al., 2019), it is likely to become more apparent why CHIP occurs in some individuals and not others, and why it predominantly appears to be limited to the elderly population. Moreover, recent work has identified several genetic loci that confer risk of developing CHIP and myeloproliferative disorders with age, suggesting that alterations in fundamental cellular processes may promote clonal expansion (Bao et al., 2019a; Bick et al., 2019b). New genomic tools may help to interrogate the functional consequences of these genetic risk factors and define the impact of disease in disrupting the HSC population in a manner that promotes the development of CH.
Future work to further decipher hematopoietic cell state and fate
In the future, new genomic methods enabling interrogation of cell heterogeneity and dynamics will help to understand the diversity of cell states that exist, and choices that cells make in order to stably maintain the hematopoietic tissue. One promising avenue is the development of barcoding strategies that allow computational reconstruction of the entire heritage of a cell, while avoiding many of the non-native conditions created by transplantation (Höfer et al., 2016). In one recent development, a barcoding system was created that employs CRISPR–Cas9-directed mutagenesis and scRNA-seq in order to label cells with continuously evolving barcodes as a way of overcoming the constraint of a limited barcode pool (Chan et al., 2019). Such approaches may help to untangle the behavior of entire tissues and organisms with cellular resolution (McKenna et al., 2016). Of course, improvements in lineage tracing will also necessitate advances in computational approaches to enable ever more sophisticated lineage inferences to be made using the more in depth genomic data generated from such assays (Jones et al., 2020).
Another important consideration when deciphering hematopoiesis is that the process differs widely across organisms, and the ability to lineage trace in humans will be necessary to gain comprehensive insights into human hematopoiesis. Unfortunately, genetic barcoding or inducible labeling systems, while powerful, cannot be used to study in vivo human hematopoiesis. Instead, some studies have leveraged endogenous genomic DNA mutations to track hematopoietic clonal dynamics (Lee-Six et al., 2018; Osorio et al., 2018; Wang et al., 2012). While effective, human genomic mutation rates tend to be low, which limits the resolution of clone tracking that can be achieved. As an increasing number of gene therapy and editing trials are being performed in humans, the unique genomic changes in each clone have been used as an alternative to effectively track transplanted human hematopoietic cells (Biasco et al., 2016; Six et al., 2020), although the resulting events might not entirely mimic steady state hematopoiesis. In order to truly reflect cell behavior during steady state hematopoiesis, one promising alternative in humans involves leveraging the high rate of mitochondrial mutations to track clonal dynamics within an unperturbed hematopoietic system (Lareau et al., 2019; Ludwig et al., 2019a; Xu et al., 2019). The scaling of mitochondrial mutation-based lineage tracing approaches should enable broader insights into human hematopoiesis. Additionally, inference methods using variation in telomere length or in other evolving biomarkers can be valuable for dissecting this process in humans (Werner et al., 2015). While lineage tracing will help to understand the outcomes of cell decisions during hematopoiesis, they are not sufficient to explain the mechanisms underlying these decisions.
EXPLORING HEMATOPOIETIC GENE REGULATION WITH GENOMICS
Improved fate and state-mapping methods have provided increasingly more comprehensive insights into the heterogeneity that exists within the hematopoietic system and the continuum of differentiation states. However, the underlying regulatory mechanisms that govern these differentiation decisions often remain mysterious. Recent advances in genomic approaches are providing insights into the mechanisms that underlie the key fate decisions in hematopoiesis. Early studies of hematopoiesis focused on uncovering the specific roles of master regulator TFs in this process (Orkin and Zon, 2008). These studies often relied on genetically ablating the function of key master TFs or epigenetic regulators, such as GATA1, SPI1 (PU.1), CEBPA, GFI1, BMI1, IKZF1 (Ikaros), and ETV6 in mice to assess their physiological roles in hematopoiesis (Doulatov et al., 2012; Fujiwara et al., 1996; Hock et al., 2004a, 2004b; Lessard and Sauvageau, 2003; Nichogiannopoulou et al., 1999; Park et al., 2003; Scott et al., 1994; Zeng et al., 2004; Zhang et al., 2004). More recently, genomic approaches have helped to unravel the mechanisms by which these factors alter hematopoietic cell differentiation (Göttgens, 2015). As we discuss in this section, such approaches include methods to identify TF binding events, chromatin modifications in specific genomic regions, and long-range chromatin interactions. The resulting highly detailed and rich data has enabled large networks of interacting elements to be generated that begin to explain the phenotypic diversity that seemingly exists within different hematopoietic cell compartments. Such analyses have also revealed the limitations of prior perturbation approaches. For instance, in most cases, the contribution of specific transcriptional regulatory elements to hematopoiesis remains unclear. Uncoupling the pleiotropic roles of any TF requires in depth study, beyond the complete loss-of-function techniques that have commonly been employed in this field.
Transcription factor profiling and interactions
To better understand the complex mechanisms involved in hematopoietic differentiation, we need to more accurately understand TF binding events. Historically, chromatin immunoprecipitation with sequencing (ChIP-seq) has been a valuable tool for accurately identifying TF binding events (Rhee and Pugh, 2011). While ChIP is not equally adept at measuring all interactions (Baranello et al., 2016; Teytelman et al., 2013), recent methods have overcome some of these limitations (Janssens et al., 2018; Skene and Henikoff, 2017; Skene et al., 2018; Zhu et al., 2019). Additionally, the assay for transposase-accessible chromatin using sequencing (ATAC-seq) and DNase hypersensitivity analyses have also enabled important insights into alterations in regulatory regions with differentiation (Klemm et al., 2019). Such genomic advances have enabled researchers to more thoroughly understand the mechanisms that underlie hematopoietic cell fate decisions.
As genomic tools improve in their throughput and accuracy, overlooked TF interactions are now being identified. For instance, these methods have helped to identify previously unappreciated roles for the TF BCL11A in silencing the expression of the fetal γ-globin genes via the direct occupancy of the γ-globin-encoding HBG1/2 gene promoters (Liu et al., 2018). These improved methods have also revealed that many TFs can work together in connected regulatory networks to produce a unique phenotype and set of lineage fate choices. For instance, the combinatorial occupancy of chromatin by multiple master TFs has revealed regulatory networks that are important for hematopoiesis (Beck et al., 2013; Schütte et al., 2016), as well as key regulatory elements necessary for HSC self-renewal (Bahr et al., 2018). These methods also enable epigenomic changes in primary hematopoietic cells to be deciphered in altered settings, such as is observed with aging (Sun et al., 2014a).
Single-cell measurements of transcriptional states, chromatin accessibility, and DNA methylation, have also provided a better understanding of the heterogeneity that exists in such regulatory mechanisms during hematopoiesis (Buenrostro et al., 2018; Cao et al., 2018; Ludwig et al., 2019b; Ulirsch et al., 2019). Such single-cell profiling has illustrated how transcriptional dynamics change with time and state. Furthermore, it has become possible to observe the shifts in the epigenome and consequently the transcriptome that occur during the continuous process of hematopoietic differentiation as separate lineages are favored. In the future, as costs decrease and throughput improves, it will become possible to profile the hematopoietic system more comprehensively, particularly in the setting of a range of perturbations. As this happens, fundamental questions about hematopoiesis can be addressed, such as which regulatory mechanisms underlie lineage bias in specific clones, and how specific subpopulations respond to stress and disease. It will additionally be important to understand how TFs associate with their target genes to regulate their activity.
3D genomic interactions
Early methods used to identify key hematopoietic regulatory elements relied on regulatory elements being genetically nearby to the genes they regulated. Within the last decade, however, hematopoietic regulatory elements have been identified that are quite distant from their associated genes, often separated by hundreds of kilobases (Amano et al., 2009; Mifsud et al., 2015; Sanyal et al., 2012; Schoenfelder et al., 2015). We are beginning to understand how chromatin conformation changes can bring such distant regulatory elements into close proximity with the genes they control (Figure 4). In the last decade, proximity ligation methods have played an important role in identifying these key interactions, such as promoter and enhancer associations (Kempfer and Pombo, 2020; Oudelaar et al., 2018; Quinodoz et al., 2018). By identifying such long-range associations, the complexity of the gene regulatory networks that coordinate hematopoietic differentiation can be more fully appreciated.
Figure 4. Long Range Transcriptional Regulation.
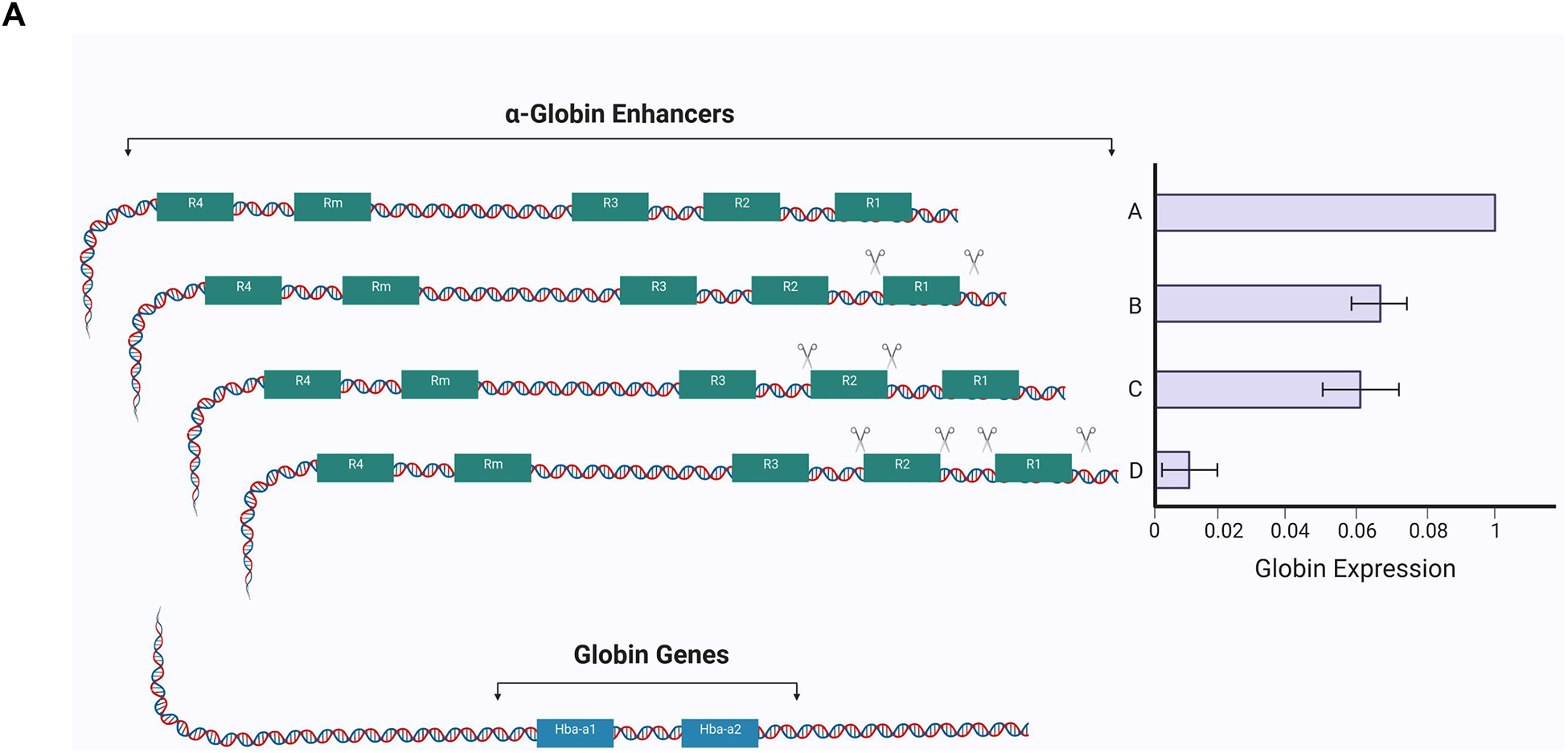
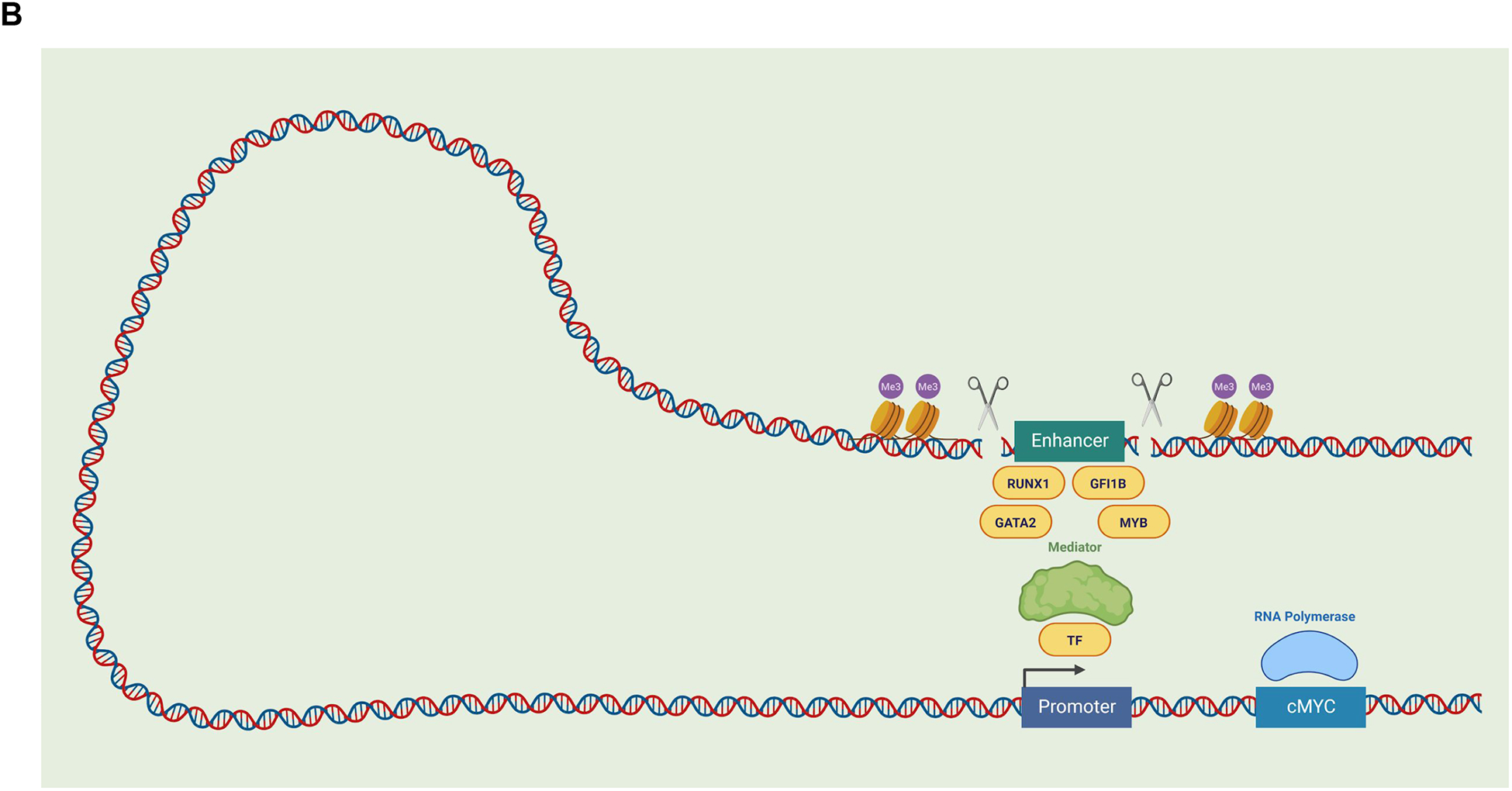
(B) The α-globin locus is regulated by the upstream enhancer elements, R1–R4. Targeted perturbtation of individual and combination regulatory elements within this α-globin enhancer, demonstrating additive regulation by each element in mice. As shown in scenarios B and C, disruption of individual enhancer elements reduces α-globin expression compared with the wildtype scenario A. The combined disruption of multiple enhancer elements appears to further reduce globin expression in an additive rather than synergistic manner, suggesting the α-globin enhancer is a cluster of conventional enhancers (Hay et al., 2016). (B) The long-range regulation of MYC expression involves distal enhancer elements that are bound by the hematopoietic master TFs GATA2, RUNX1, GFI1B, and MYB. As illustrated, chromatin looping can bring these enhancer elements and the promoter region into close proximity in order to regulate MYC gene expression. The disruption of such enhancer elements (through perturbation approaches, as illustrated with scissors) can help decipher the role of these elements in particular phenotypes, as has been performed for a hematopoietic-specific enhancer of MYC (Bahr et al., 2018). Abbreviations: transcription factor (TF), cellular myelocytomatosis (cMYC), runt-related transcription factor 1 (RUNX1), growth factor independent 1B (GFI1B), GATA binding protein 2 (GATA2), MYB proto-oncogene (MYB), histone methylation (Me3).
These proximity ligation approaches have contributed new insights into already well-studied genomic loci involved in hematopoiesis. One illustrative example is the α-globin gene cluster, which is carefully regulated to control the transcription of the α-globin genes during erythroid maturation (Sankaran and Weiss, 2015). Despite decades of study, many of the distant regulatory elements remained poorly characterized. With the use of proximity ligation, regulatory elements that control α-globin expression were discovered over 70 kb away. Incorporating specific chromatin modifications and boundary elements at these distant regulatory sites has created a more complete picture of the underlying regulation at this locus (Brown et al., 2018; Hanssen et al., 2017; Oudelaar et al., 2017). Furthermore, targeting disruption of specific enhancer elements has demonstrated that many of these long range regulatory elements additively enhance transcription (Hay et al., 2016) (Figure 4A).
Future explorations of hematopoietic regulatory mechanisms
The past decade has seen a dramatic increase in the use of genomic tools to decipher regulatory mechanisms, which generate a wealth of rich genomic data. Unique opportunities now arise with this rich genomic data and with the development of scalable perturbation approaches to decipher the regulatory mechanisms that underlie key hematopoietic differentiation decisions. Even studies at a single locus can be illuminating, as exemplified by the identification of a hematopoietic-specific enhancer of MYC that is required by HSCs (Bahr et al., 2018) (Figure 4B). Scalable approaches using genome editing tools, such as CRISPR/Cas9, enable cis-regulatory elements to be dissected in cell lines and even in primary cell contexts (Fulco et al., 2016; Simeonov et al., 2017). Such approaches have contributed to our understanding of the relationship between enhancer elements and the genes they regulate (Giladi et al., 2018). Future use of these tools in primary hematopoietic cells will dramatically improve our understanding of the cooperating mechanisms that underlie cellular decisions in hematopoiesis. Furthermore, with ongoing advances in human genetics, we can begin to understand how allelic differences in the human population underlie subtle differences in gene and regulatory element function in order to modify hematopoiesis.
EXPLORING HUMAN GENETIC VARIATION IN HEMATOPOIESIS
The focus of this review has, until now, has been on understanding how hematopoiesis occurs as an invariant process, particularly through studies in model organisms (Rowe et al., 2016). However, it is clear from studies of human variation that there is considerable inter-individual heterogeneity in this process. While environmental factors undoubtedly explain some of this variation, genetic factors appear to have a considerable role, both in healthy individuals and in those affected by disease. Advances in genomics over the past decade have enabled the genotyping of large cohorts and the sequencing of individuals using massively parallel methods, such as exome or genome sequencing, and have uncovered common variation that affects hematopoiesis and also rare mutations that alter this process (Bao et al., 2019b).
Human genetic variants that affect hematopoiesis
Population-based genetic variation has largely been studied for blood cell traits, and over ten thousand loci have been defined across the allelic spectrum that explain inter-individual variation in such traits (Astle et al., 2016; Chen et al., 2020; Vuckovic et al., 2020). While this catalog of variants is impressive and likely among the largest known for any complex trait or disease (Claussnitzer et al., 2020), these loci have also revealed some fascinating biology, both in the case of individual variants and through more holistic analyses. For instance, prior to the first genome-wide association studies (GWAS) into variation that impacts fetal hemoglobin (HbF) levels - an important ameliorating factor for sickle cell disease and β-thalassemia - the regulators of HbF were poorly understood (Basak et al., 2020). The follow up of these GWAS for HbF revealed BCL11A to be a key regulator of HbF silencing (Basak et al., 2015; Sankaran et al., 2008, 2009), a finding that has now led to further biological and therapeutic insights (Liu et al., 2018). In other cases, the interrogation of specific loci identified through studies of population-based variation has uncovered key regulatory mechanisms, such as the role of specific GFI1B isoforms in megakaryopoiesis or the identification of a distinct CEBPA enhancer involved in the production of basophils (Guo et al., 2017; Polfus et al., 2016).
With hundreds to thousands of loci now identified for each trait, such blood cell-associated variants also harbor considerable potential for gaining insights into the global mechanisms that underlie the genetic variation that alters hematopoiesis. For instance, functional assessments of red blood cell trait-associated variants have revealed common mechanisms by which the activity of the master TF GATA1 might be affected by genetic variants (Ulirsch et al., 2016). In addition, the ability to fine-map variants associated with blood cell traits and overlap these variants with accessible chromatin in hematopoietic progenitors has provided unprecedented insight into how genetic variation can influence a range of cell states in hematopoiesis (Ulirsch et al., 2019; Vuckovic et al., 2020). For instance, variants that influence multiple blood traits tend to impact chromatin that is accessible in early HSPCs. Variants that modify function only within single hematopoietic lineages instead tend to map to more mature, lineage committed cells. These associations suggest that human variation may modulate regulatory mechanisms across the full hematopoietic hierarchy (Figure 1) (Ulirsch et al., 2019).
While scalable genotyping and sequencing technologies have empowered numerous population-based studies, these advances have also enabled the discovery of rare genetic variation that underlie blood diseases. The rate of new causal gene identification for such disorders, as with many other rare diseases, has been incredible in the past decade as a result of advances in massively parallel sequencing (Claussnitzer et al., 2020). Beyond the ability to decipher the genetic basis for a range of previously enigmatic disorders that affect hematopoiesis, key biological insights have also emerged as a result. For instance, Diamond-Blackfan anemia (DBA) is a rare disorder that is characterized by impaired erythropoiesis; most patients have loss-of-function mutations in one of twenty-six distinct ribosome protein genes (Ulirsch et al., 2018). Why mutations that affect ubiquitously expressed ribosomes cause a selective defect in erythropoiesis, and not defects in other hematopoietic lineages, was unknown. Through exome sequencing, rare individuals with DBA containing loss-of-function mutations in the hematopoietic master TF GATA1 were identified (Sankaran et al., 2012). Inspired by this discovery and through functional follow up, it was shown that the more common ribosomal gene mutations impair the translation of GATA1 mRNA and thus result in impaired erythropoiesis (Khajuria et al., 2018; Ludwig et al., 2014). Similar discoveries have uncovered mutations in other hematopoietic TFs that alter homeostatic hematopoiesis and that in some cases may increase the risk of developing blood cancers (Churpek and Bresnick, 2019; Kennedy and Shimamura, 2019). Advances in sequencing capability are uncovering an even greater number of genetic mutations that can cause rare blood disorders; these findings are likely to provide key insights into both normal and pathologic hematopoiesis in the coming years (Turro et al., 2020). The coupling of these genetic discoveries with the single-cell genomic tools we have discussed in this review greatly increases our potential to further refine our understanding of hematopoiesis, as has been done elegantly in recent studies of congenital neutropenia (Muench et al., 2020).
Future opportunities in human genetics
The genomic advances that have supported studies of human genetic variation have also constrained this field. Studies in this field have generally focused on identifying single variants that are significantly associated with a trait or disease of interest through GWAS or have focused on disease-causal variants found in particular genes through sequencing-based approaches (Figure 5). These approaches have led to numerous important discoveries, but have also painted a dichotomized picture of the human genetic variation that affects hematopoiesis, with rare variants having a big impact on disease and common genetic variants subtly tuning this process.
Figure 5. The Genetic Architecture of Blood Diseases.
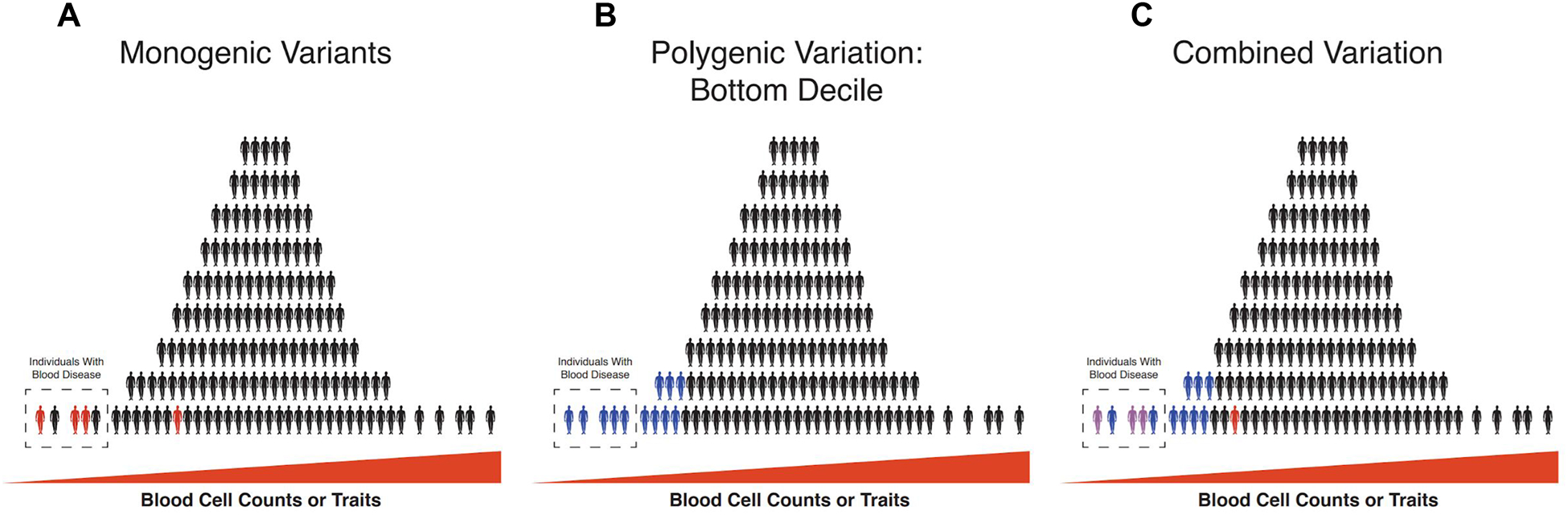
Blood cell trait/ count variation is depicted in each panel, with each human shape representing an individual. The distribution represents a general distribution of a trait that might be expected within a population, where few people are at the extremes. (A) The effect of monogenic variants of high effect size (red person) can be used to explain some fraction of the phenotypically extreme individuals that might be diagnosed as having a blood disease, but this effect often cannot explain all disease cases and these variants might have variable penetrance. (B) With the advent of genome-wide association studies (GWAS), it has become possible to identify polygenic common variation that underlies those at the bottom decile of a trait of interest. Blue colored individuals represent those that express an extreme phenotype that is caused by polygenic common variants rather than monogenic variants with high effect size. (C) In a combined approach, monogenic variation is assessed within the context of a polygenic background in an individual (purple color represents a combined impact of monogenic red variants and blue polygenic variation), to better explain the phenotypic trait variation that can result in blood diseases at the extremes. This emerging picture is modified as a result of improvements in genomic approaches enabling the full spectrum of genetic variation to be studied, instead of dichotomizing such variation between common and rare alleles.
With larger-scale, population-based studies, important opportunities have arisen and key observations are now being made (Vuckovic et al., 2020). First, alleles associated with monogenic blood disorders have begun to emerge from these larger GWASs. Second, it is clear that a spectrum of allelic variation collectively affects the risk of having a blood disorder. The importance of polygenic variation on large-effect, monogenic variants has been clearly demonstrated for rare alleles, such as for the BRCA1 and BRCA2 variants that predispose to breast cancer (Fahed et al., 2019; Gallagher et al., 2020). The emerging picture is therefore that a spectrum of polygenic variation and large-effect monogenic variants collectively influence hematopoiesis to shape one’s risk of having a blood disorder or altered hematopoiesis (Vuckovic et al., 2020). In the coming years, further studies of this sort and in cohorts of patients with blood disorders will likely reveal the complex genetic architecture that underlies normal hematopoiesis and how it is altered in disease (Figure 5). Importantly, there are likely to be key mechanistic studies performed in the coming years to unravel how such genetic variation has an impact on the fundamental biological mechanisms involved in hematopoiesis (Nandakumar et al., 2020).
In addition, beyond studies of blood cell traits, there are likely to be important opportunities to study cryptic phenotypes that might occur because of altered hematopoiesis. For instance, while clonal blood disorders or cancers are thought to primarily arise due to the acquisition of somatic mutations in HSCs or in other hematopoietic cells, it is clear that inherited genetic variation can alter this risk significantly (Bao et al., 2019a; Bick et al., 2019b; Vijayakrishnan et al., 2018; Went et al., 2018). Understanding the mechanisms that underlie such germline genetic predisposition, both for common and rare genetic variants, will undoubtedly advance our knowledge of how genetic variation can influence hematopoiesis in different ways.
OUTSTANDING QUESTIONS AT THE INTERSECTION OF HEMATOPOIESIS AND GENOMICS
The hematopoietic system has served as fertile ground for important biological discoveries. Aided by the genomics revolution, we are gaining a finer appreciation of the diverse and heterogeneous cell behaviors that are necessary to drive hematopoiesis. While the last decade has seen more comprehensive genomics, accurate lineage tracing, and genetic variation studies that have all been important for understanding hematopoiesis, further insights are yet to come. We envision that future genomic advances will help to illuminate many aspects of hematopoiesis that have been challenging to study in the past and below we outline what we see as important opportunities in the coming years.
In many ways, we have yet to understand some fundamental aspects of hematopoietic biology. An important example is HSC compartment size, for which estimates vary substantially. Such estimates of HSC numbers have existed since the first observations of hematopoietic colony-forming cells and more sophisticated genomics methods have now contributed insights (Lee-Six et al., 2018). However, methods to directly quantify HSC compartment size have remained elusive. As such, it is unclear how large and diverse HSC compartments are, how they might vary within the human population, and what the impact of such variation might be. For example, if stem cell compartment sizes differ across individuals, would an expanded pool of stem cells increase the probability of an HSC acquiring oncogenic mutations and thereby increase an individual’s risk of cancer? Or would a larger HSC pool reduce the risk of the hematopoietic system becoming more clonal with time and therefore mitigate the risk of developing leukemia? The size of the HSC compartment also changes dramatically during development, and considerably increases in the transition into adulthood (Ganuza et al., 2017; Werner et al., 2015). It is unclear if this effect results from fewer available niches in early life or if there is hematopoietic cell-intrinsic control of this.
Typically, many currently employed barcoding approaches are only efficient enough to label a fraction of hematopoietic stem and progenitor cells. This limitation might significantly underrepresent the contributions of rare cell populations and the variation in differentiation dynamics. More comprehensive lineage tracing methods are needed to help us to understand if bipotential or multipotential cells exist within the stem and progenitor compartment, or if individual cells commit to just a single lineage of output (Olsson et al., 2016; Paul et al., 2015). It is also unclear how long progenitor cells contribute to mature cell output, and how often they are replaced by stem cell divisions. It will be important to develop strategies that minimize manipulations to better understand the dynamics of steady state hematopoiesis. Current approaches that take advantage of endogenous somatic mutations and the expansion of these approaches are likely to provide new insights into hematopoiesis, particularly in humans (Behjati et al., 2014; Biezuner et al., 2016; Lee-Six et al., 2018; Ludwig et al., 2019a; Osorio et al., 2018). We must also expect that new technologies will come along and take advantage of other aspects of cell biology that may enable higher resolution clonal tracking.
As the dynamics of steady state hematopoiesis become clearer, we can begin understand to how the system can be perturbed. For example, if HSCs are lineage biased in a heritable manner, then it will be fascinating to understand how CH, in which hematopoietic cells arise from only a small number of HSCs, impacts the system. Older individuals can develop HSPC compartments that are highly clonal and yet still continually regenerate functional tissue and have ostensibly normal hematopoiesis. This possibility might indicate a significant degree of flexibility in the lineage output of HSPCs. Such flexibility might depend on the ability of the HSPC microenvironment to direct the differentiation trajectories and it is still unclear how much of this cell fate choice is intrinsic to the cells themselves.
Conclusions
We have highlighted only a few of the key questions in this field and realize that much more will be learned in the coming years. In many ways, the hematopoietic system has led researchers through some of the most fundamental puzzles of human biology. We now understand a great deal of how the tissue behaves as a whole, but are just gaining the ability to understand how the combined functions of all cells in the tissue produce these emergent behaviors. As the scale of single cell genomics continues to grow in the coming years, we expect a much more mechanistic understanding of how individual genes are regulated in every cell in the hematopoietic system to consistently provide the blood cells an organism needs. Furthermore, we expect that as cohorts of human genetic data continue to grow, that we will begin to understand how inter-individual genetic variation can modulate the process of hematopoiesis in ways that may increase disease risk. Despite hematopoiesis being a well-studied process that has uncovered numerous insights over past decades, there is undoubtedly much more that is waiting to be uncovered. In the spirit of Stoker’s writing in Dracula, we hope that in time we may solve additional mysteries in hematopoiesis, at least in part.
ACKNOWLEDGMENTS
We apologize for the inability to cite many key papers and topics in the field of hematopoiesis due to space constraints. We are grateful to members of the Sankaran Laboratory for valuable comments and critiques of this work. L.A.L. was supported by a National Institutes of Health training grant T32 HL066987. The Sankaran Laboratory is supported by National Institutes of Health grants R01 DK103794 and R01 HL146500, the New York Stem Cell Foundation, a gift from the Lodish Family to Boston Children’s Hospital, the MPN Research Foundation, and the Leukemia & Lymphoma Society. V.G.S. is a New York Stem Cell Foundation-Robertson Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
REFERENCES
- Adolfsson J, Månsson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge O-J, Thoren LAM, et al. (2005). Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 121, 295–306. [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. [DOI] [PubMed] [Google Scholar]
- Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, and Shiroishi T (2009). Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev. Cell 16, 47–57. [DOI] [PubMed] [Google Scholar]
- Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. (2016). The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167, 1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, Nombela-Arrieta C, Steinmetz LM, Trumpp A, and Haas S (2019). Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. (2018). A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. [DOI] [PubMed] [Google Scholar]
- Banerjee U, Girard JR, Goins LM, and Spratford CM (2019). Drosophila as a Genetic Model for Hematopoiesis. Genetics 211, 367–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao EL, Nandakumar SK, Liao X, Bick A, Karjalainen J, Tabaka M, Gan OI, Havulinna A, Kiiskinen T, Lareau CA, et al. (2019a). Genetic predisposition to myeloproliferative neoplasms implicates hematopoietic stem cell biology.
- Bao EL, Cheng AN, and Sankaran VG (2019b). The genetics of human hematopoiesis and its disruption in disease. EMBO Mol. Med 11, e10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Kouzine F, Sanford S, and Levens D (2016). ChIP bias as a function of cross-linking time. Chromosome Res. 24, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, et al. (2019). A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 177, 1915–1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M, Vlckova M, Muzikova K, Cermak J, Trka J, et al. (2015). BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J. Clin. Invest 125, 2363–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A, Munschauer M, Lareau CA, Montbleau KE, Ulirsch JC, Hartigan CR, Schenone M, Lian J, Wang Y, Huang Y, et al. (2020). Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat. Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D, Thoms JAI, Perera D, Schütte J, Unnikrishnan A, Knezevic K, Kinston SJ, Wilson NK, O’Brien TA, Göttgens B, et al. (2013). Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood 122, e12–e22. [DOI] [PubMed] [Google Scholar]
- Becker AJ, McCULLOCH EA, and Till JE (1963). Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 197, 452–454. [DOI] [PubMed] [Google Scholar]
- Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, et al. (2014). Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Czechowicz A, Ooi AGL, Rossi DJ, Bryder D, and Weissman IL (2009). Niche recycling through division-independent egress of hematopoietic stem cells. J. Exp. Med 206, 2837–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, Scaramuzza S, Baricordi C, Ferrua F, Cicalese MP, et al. (2016). In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell 19, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, and Natarajan P (2019a). Genetic IL-6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Leventhal MJ, Bao EL, Nasser J, Zekavat SM, Szeto MD, Laurie C, et al. (2019b). Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes.
- Biezuner T, Spiro A, Raz O, Amir S, Milo L, Adar R, Chapal-Ilani N, Berman V, Fried Y, Ainbinder E, et al. (2016). A generic, cost-effective, and scalable cell lineage analysis platform. Genome Res. 26, 1588–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady G, Barbara M, and Iscove NN (1990). Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol. Cell. Biol 2, 17–25. [Google Scholar]
- Brecher G, and Cronkite EP (1951). Post-radiation parabiosis and survival in rats. Proc. Soc. Exp. Biol. Med 77, 292–294. [DOI] [PubMed] [Google Scholar]
- Brown JM, Roberts NA, Graham B, Waithe D, Lagerholm C, Telenius JM, De Ornellas S, Oudelaar AM, Scott C, Szczerbal I, et al. (2018). A tissue-specific self-interacting chromatin domain forms independently of enhancer-promoter interactions. Nat. Commun 9, 3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, and Rodewald H-R (2015). Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546. [DOI] [PubMed] [Google Scholar]
- Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, Maragh M, and Gilliland DG (1996). Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood 88, 59–65. [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrelha J, Meng Y, Kettyle LM, Luis TC, Norfo R, Alcolea V, Boukarabila H, Grasso F, Gambardella A, Grover A, et al. (2018). Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 554, 106–111. [DOI] [PubMed] [Google Scholar]
- Champion KM, Gilbert JG, Asimakopoulos FA, Hinshelwood S, and Green AR (1997). Clonal haemopoiesis in normal elderly women: implications for the myeloproliferative disorders and myelodysplastic syndromes. Br. J. Haematol 97, 920–926. [DOI] [PubMed] [Google Scholar]
- Chan MM, Smith ZD, Grosswendt S, Kretzmer H, Norman TM, Adamson B, Jost M, Quinn JJ, Yang D, Jones MG, et al. (2019). Molecular recording of mammalian embryogenesis. Nature 570, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple RH, Tseng Y-J, Hu T, Kitano A, Takeichi M, Hoegenauer KA, and Nakada D (2018). Lineage tracing of murine adult hematopoietic stem cells reveals active contribution to steady-state hematopoiesis. Blood Adv 2, 1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M-H, Raffield LM, Mousas A, Sakaue S, Huffman JE, Jiang T, Akbari P, Vuckovic D, Bao EL, Moscati A, et al. (2020). Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. [DOI] [PMC free article] [PubMed]
- Cheshier SH, Morrison SJ, Liao X, and Weissman IL (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. U. S. A 96, 3120–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churpek JE, and Bresnick EH (2019). Transcription factor mutations as a cause of familial myeloid neoplasms. J. Clin. Invest 129, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, Kathiresan S, Kenny EE, Lindgren CM, MacArthur DG, et al. (2020). A brief history of human disease genetics. Nature 577, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, and Eaves CJ (2013). Developmental changes in hematopoietic stem cell properties. Exp. Mol. Med 45, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GM, Jeffery E, and Morrison SJ (2017). Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590. [DOI] [PubMed] [Google Scholar]
- Derényi I, and Szöllősi GJ (2017). Hierarchical tissue organization as a general mechanism to limit the accumulation of somatic mutations. Nat. Commun 8, 14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Hao S, Zhang S, Zhu C, Cheng H, Yang Z, Hamey FK, Wang X, Gao A, Wang F, et al. (2020). Differentiation of transplanted haematopoietic stem cells tracked by single-cell transcriptomic analysis. Nat. Cell Biol 22, 630–639. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, and Dick JE (2010). Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat. Immunol 11, 585–593. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, and Dick JE (2012). Hematopoiesis: a human perspective. Cell Stem Cell 10, 120–136. [DOI] [PubMed] [Google Scholar]
- Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee S-J, Brinkman R, and Eaves C (2007). Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1, 218–229. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, and Bigas A (2018). Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 22, 639–651. [DOI] [PubMed] [Google Scholar]
- Eaves CJ (2015). Hematopoietic stem cells: concepts, definitions, and the new reality. Blood 125, 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, Lai C, Brockman D, Philippakis A, Ellinor PT, et al. (2019). Polygenic background modifies penetrance of monogenic variants conferring risk for coronary artery disease, breast cancer, or colorectal cancer (medRxiv). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliedner TM, Graessle D, Paulsen C, and Reimers K (2002). Structure and function of bone marrow hemopoiesis: mechanisms of response to ionizing radiation exposure. Cancer Biother. Radiopharm 17, 405–426. [DOI] [PubMed] [Google Scholar]
- Ford CE, Hamerton JL, Barnes DW, and Loutit JF (1956). Cytological identification of radiation-chimaeras. Nature 177, 452–454. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Browne CP, Cunniff K, Goff SC, and Orkin SH (1996). Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. U. S. A 93, 12355–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher S, Hughes E, Wagner S, Tshiaba P, Rosenthal E, Roa BB, Kurian AW, Domchek SM, Garber J, Lancaster J, et al. (2020). Association of a Polygenic Risk Score With Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw Open 3, e208501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganuza M, Hall T, Finkelstein D, Chabot A, Kang G, and McKinney-Freeman S (2017). Lifelong haematopoiesis is established by hundreds of precursors throughout mammalian ontogeny. Nat. Cell Biol 19, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, Jaitin D, Cabezas-Wallscheid N, Dress R, Ginhoux F, et al. (2018). Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol 20, 836–846. [DOI] [PubMed] [Google Scholar]
- Giladi A, Cohen M, Medaglia C, Baran Y, Li B, Zada M, Bost P, Blecher-Gonen R, Salame T-M, Mayer JU, et al. (2020). Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol 38, 629–637. [DOI] [PubMed] [Google Scholar]
- Goardon N, Marchi E, Atzberger A, Quek L, and Schuh A (2011). Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. [DOI] [PubMed] [Google Scholar]
- Görgens A, Radtke S, Möllmann M, Cross M, Dürig J, Horn PA, and Giebel B (2013). Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 3, 1539–1552. [DOI] [PubMed] [Google Scholar]
- Göttgens B (2015). Regulatory network control of blood stem cells. Blood 125, 2614–2620. [DOI] [PubMed] [Google Scholar]
- Grün D, Muraro MJ, Boisset J-C, Wiebrands K, Lyubimova A, Dharmadhikari G, van den Born M, van Es J, Jansen E, Clevers H, et al. (2016). De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell 19, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Luc S, Marco E, Lin T-W, Peng C, Kerenyi MA, Beyaz S, Kim W, Xu J, Das PP, et al. (2013). Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell 13, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo MH, Nandakumar SK, Ulirsch JC, Zekavat SM, Buenrostro JD, Natarajan P, Salem RM, Chiarle R, Mitt M, Kals M, et al. (2017). Comprehensive population-based genome sequencing provides insight into hematopoietic regulatory mechanisms. Proc. Natl. Acad. Sci. U. S. A 114, E327–E336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan G, and Van Zant G (1999). Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood 93, 3294–3301. [PubMed] [Google Scholar]
- Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, Wurzer S, Prendergast ÁM, Schnell A, Hexel K, et al. (2015). Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 17, 422–434. [DOI] [PubMed] [Google Scholar]
- Hanssen LLP, Kassouf MT, Marieke Oudelaar A, Biggs D, Preece C, Downes DJ, Gosden M, Sharpe JA, Sloane-Stanley JA, Hughes JR, et al. (2017). Tissue-specific CTCF–cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol 19, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen L, Kassouf MT, Marieke Oudelaar AM, Sharpe JA, Suciu MC, et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet 48, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CA, Chen B, McKinley ET, and Lau KS (2018). Single-Cell Computational Strategies for Lineage Reconstruction in Tissue Systems. Cell Mol Gastroenterol Hepatol 5, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, and Orkin SH (2004a). Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431, 1002–1007. [DOI] [PubMed] [Google Scholar]
- Hock H, Meade E, Medeiros S, Schindler JW, Valk PJM, Fujiwara Y, and Orkin SH (2004b). Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 18, 2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höfer T, Busch K, Klapproth K, and Rodewald H-R (2016). Fate Mapping and Quantitation of Hematopoiesis In Vivo. Annu. Rev. Immunol 34, 449–478. [DOI] [PubMed] [Google Scholar]
- Ivanovs A, Rybtsov S, Ng ES, Stanley EG, Elefanty AG, and Medvinsky A (2017). Human haematopoietic stem cell development: from the embryo to the dish. Development 144, 2323–2337. [DOI] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner M-J, et al. (2012). Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet 44, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, and Ebert BL (2019). Clonal hematopoiesis in human aging and disease. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens DH, Wu SJ, Sarthy JF, Meers MP, Myers CH, Olson JM, Ahmad K, and Henikoff S (2018). Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MG, Khodaverdian A, Quinn JJ, Chan MM, Hussmann JA, Wang R, Xu C, Weissman JS, and Yosef N (2020). Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann BB, and van Oudenaarden A (2007). Stochastic gene expression: from single molecules to the proteome. Curr. Opin. Genet. Dev 17, 107–112. [DOI] [PubMed] [Google Scholar]
- Kempfer R, and Pombo A (2020). Methods for mapping 3D chromosome architecture. Nat. Rev. Genet 21, 207–226. [DOI] [PubMed] [Google Scholar]
- Kennedy AL, and Shimamura A (2019). Genetic predisposition to MDS: clinical features and clonal evolution. Blood 133, 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR, et al. (2018). Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 173, 90–103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- Kohn LA, Hao Q-L, Sasidharan R, Parekh C, Ge S, Zhu Y, Mikkola HKA, and Crooks GM (2012). Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat. Immunol 13, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau CA, Ludwig LS, and Sankaran VG (2019). Longitudinal assessment of clonal mosaicism in human hematopoiesis via mitochondrial mutation tracking. Blood Adv 3, 4161–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E, and Göttgens B (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, et al. (2012). Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet 44, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, Øbro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly BJP, Martincorena I, Anderson E, et al. (2018). Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemischka IR, Raulet DH, and Mulligan RC (1986). Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 45, 917–927. [DOI] [PubMed] [Google Scholar]
- Lessard J, and Sauvageau G (2003). Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423, 255–260. [DOI] [PubMed] [Google Scholar]
- Liggett LA, Sharma A, De S, and DeGregori J (2019). FERMI: A Novel Method for Sensitive Detection of Rare Mutations in Somatic Tissue. G3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, et al. (2014). Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med 20, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, Pelka K, Ge W, Oren Y, Brack A, et al. (2019a). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Lareau CA, Bao EL, Nandakumar SK, Muus C, Ulirsch JC, Chowdhary K, Buenrostro JD, Mohandas N, An X, et al. (2019b). Transcriptional States and Chromatin Accessibility Underlying Human Erythropoiesis. Cell Rep. 27, 3228–3240.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R, Park CY, and Weissman IL (2007). Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 1, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, and Wold BJ (2014). From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 24, 496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximow A (1909). Untersuchungen über Blut und Bindegewebe. II. über die Histogenese der Thymus bei Säugetieren. Arch. F. Mikr. Anat 74, 525. [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, and Shendure J (2016). Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al. (2015). Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet 47, 598–606. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Iwasaki H, Jiromaru T, Kusumoto H, Yurino A, Sugio T, Uehara Y, Odawara J, Daitoku S, Kunisaki Y, et al. (2017). Identification of unipotent megakaryocyte progenitors in human hematopoiesis. Blood 129, 3332–3343. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, and Weissman IL (1994). The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1, 661–673. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, and Weissman IL (1997). Identification of a lineage of multipotent hematopoietic progenitors. Development 124, 1929–1939. [DOI] [PubMed] [Google Scholar]
- Muench DE, Olsson A, Ferchen K, Pham G, Serafin RA, Chutipongtanate S, Dwivedi P, Song B, Hay S, Chetal K, et al. (2020). Mouse models of neutropenia reveal progenitor-stage-specific defects. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Sieburg CE, Cho RH, Karlsson L, Huang J-F, and Sieburg HB (2004). Myeloid-biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood 103, 4111–4118. [DOI] [PubMed] [Google Scholar]
- Nam AS, Kim K-T, Chaligne R, Izzo F, Ang C, Taylor J, Myers RM, Abu-Zeinah G, Brand R, Omans ND, et al. (2019). Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 571, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar SK, Liao X, and Sankaran VG (2020). In The Blood: Connecting Variant to Function In Human Hematopoiesis. Trends Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Göttgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C, and Georgopoulos K (1999). Defects in hemopoietic stem cell activity in Ikaros mutant mice. J. Exp. Med 190, 1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan GP (2011). Flow cytometry in the post fluorescence era. Best Pract. Res. Clin. Haematol 24, 505–508. [DOI] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, and Dick JE (2011). Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333, 218–221. [DOI] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Hanada K, Hamada H, and Nakauchi H (1996). Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273, 242–245. [DOI] [PubMed] [Google Scholar]
- Osorio FG, Rosendahl Huber A, Oka R, Verheul M, Patel SH, Hasaart K, de la Fonteijne L, Varela I, Camargo FD, and van Boxtel R (2018). Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 25, 2308–2316.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudelaar AM, Hanssen LLP, Hardison RC, Kassouf MT, Hughes JR, and Higgs DR (2017). Between form and function: the complexity of genome folding. Hum. Mol. Genet 26, R208–R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudelaar AM, Davies JOJ, Hanssen LLP, Telenius JM, Schwessinger R, Liu Y, Brown JM, Downes DJ, Chiariello AM, Bianco S, et al. (2018). Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat. Genet 50, 1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, and Weissman IL (2011). Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. U. S. A 108, 20012–20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I-K, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, and Clarke MF (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagers AJ, Giuriato S, Anderson WC, and Weissman IL (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med 202, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y.‘ara, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Pellin D, Loperfido M, Baricordi C, Wolock SL, Montepeloso A, Weinberg OK, Biffi A, Klein AM, and Biasco L (2019). A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun 10, 2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perié L, Duffy KR, Kok L, de Boer RJ, and Schumacher TN (2015). The Branching Point in Erythro-Myeloid Differentiation. Cell 163, 1655–1662. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang Y-A, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Göttgens B, and Passegué E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser RCV, Ho DLL, et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C, Fugazza C, Tipping AJ, Brown J, Soneji S, Teles J, Peterson C, and Enver T (2012). Inferring rules of lineage commitment in haematopoiesis. Nat. Cell Biol 14, 287–294. [DOI] [PubMed] [Google Scholar]
- Pinho S, and Frenette PS (2019). Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol 20, 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polfus LM, Khajuria RK, Schick UM, Pankratz N, Pazoki R, Brody JA, Chen M-H, Auer PL, Floyd JS, Huang J, et al. (2016). Whole-Exome Sequencing Identifies Loci Associated with Blood Cell Traits and Reveals a Role for Alternative GFI1B Splice Variants in Human Hematopoiesis. Am. J. Hum. Genet 99, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu D-M, Botting RA, Stephenson E, Green K, Webb S, Jardine L, Calderbank EF, Polanski K, Goh I, Efremova M, et al. (2019). Decoding human fetal liver haematopoiesis. Nature 574, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJH, Rossi DJ, Månsson R, Attema JL, Norddahl GL, Chan CKF, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Psaila B, Barkas N, Iskander D, Roy A, Anderson S, Ashley N, Caputo VS, Lichtenberg J, Loaiza S, Bodine DM, et al. (2016). Single-cell profiling of human megakaryocyte-erythroid progenitors identifies distinct megakaryocyte and erythroid differentiation pathways. Genome Biol. 17, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucella JN, Upadhaya S, and Reizis B (2020). The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol [DOI] [PubMed] [Google Scholar]
- Purton LE, and Scadden DT (2007). Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell 1, 263–270. [DOI] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, and Pugh BF (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RG, Mandelbaum J, Zon LI, and Daley GQ (2016). Engineering Hematopoietic Stem Cells: Lessons from Development. Cell Stem Cell 18, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhok AI, Wahl GM, and DeGregori J (2015). A Critical Examination of the “Bad Luck” Explanation of Cancer Risk . Cancer Prev. Res 8, 762–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada C, Xavier-Ferrucio J, Lu Y-C, Min E, Zhang P-X, Zou S, Kang E, Zhang M, Zerafati G, Gallagher PG, et al. (2016). Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood 128, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, and Weiss MJ (2015). Anemia: progress in molecular mechanisms and therapies. Nat. Med 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HKA, Hirschhorn JN, Cantor AB, and Orkin SH (2008). Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, et al. (2009). Developmental and species-divergent globin switching are driven by BCL11A. Nature 460, 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio J-A, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, et al. (2012). Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Invest 122, 2439–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, and Dekker J (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, Goloborodko A, Feng J, Fujisaki J, Ding L, et al. (2016). Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 45, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säwen P, Eldeeb M, Erlandsson E, Kristiansen TA, Laterza C, Kokaia Z, Karlsson G, Yuan J, Soneji S, Mandal PK, et al. (2018). Murine HSCs contribute actively to native hematopoiesis but with reduced differentiation capacity upon aging. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre B-M, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, et al. (2015). The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütte J, Wang H, Antoniou S, Jarratt A, Wilson NK, Riepsaame J, Calero-Nieto FJ, Moignard V, Basilico S, Kinston SJ, et al. (2016). An experimentally validated network of nine haematopoietic transcription factors reveals mechanisms of cell state stability. Elife 5, e11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, and Singh H (1994). Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming G-L, et al. (2015). Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell 17, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizuru JA, Negrin RS, and Weissman IL (2005). Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med 56, 509–538. [DOI] [PubMed] [Google Scholar]
- Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, Satpathy AT, Lee Y, Bray NL, Chan AY, et al. (2017). Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siminovitch L, McCulloch EA, and Till JE (1963). The distribution of colony-forming cells among spleen colonies. J. Cell. Comp. Physiol 62, 327–336. [DOI] [PubMed] [Google Scholar]
- Six E, Guilloux A, Denis A, Lecoules A, Magnani A, Vilette R, Male F, Cagnard N, Delville M, Magrin E, et al. (2020). Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood 135, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, and Weissman IL (1988). Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62. [DOI] [PubMed] [Google Scholar]
- Stachura DL, and Traver D (2011). Cellular dissection of zebrafish hematopoiesis. Methods Cell Biol. 101, 75–110. [DOI] [PubMed] [Google Scholar]
- Stoker B (1897). Dracula: A Mystery Story (W. R. Caldwell).
- Sudo K, Ema H, Morita Y, and Nakauchi H (2000). Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med 192, 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. (2014a). Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho Y-J, Klein A, Hofmann O, and Camargo FD (2014b). Clonal dynamics of native haematopoiesis. Nature 514, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, and Eaves CJ (1990). Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proceedings of the National Academy of Sciences 87, 8736–8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Thurtle DM, Rine J, and van Oudenaarden A (2013). Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U. S. A 110, 18602–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till JE, and McCULLOCH EA (1961). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res 14, 213–222. [PubMed] [Google Scholar]
- Tomasetti C, and Vogelstein B (2015). Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, Allen HL, Sanchis-Juan A, Frontini M, Thys C, et al. (2020). Whole-genome sequencing of patients with rare diseases in a national health system. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, Waisman A, Huh JR, Klein AM, and Socolovsky M (2018). Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, et al. (2016). Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, Handsaker RE, Abdulhay NJ, Fiorini C, Genovese G, et al. (2018). The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet 103, 930–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, et al. (2019). Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet 51, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vas V, Senger K, Dörr K, Niebel A, and Geiger H (2012). Aging of the microenvironment influences clonality in hematopoiesis. PLoS One 7, e42080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, Hirche C, Lutz C, Buss EC, Nowak D, et al. (2017). Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol 19, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakrishnan J, Studd J, Broderick P, Kinnersley B, Holroyd A, Law PJ, Kumar R, Allan JM, Harrison CJ, Moorman AV, et al. (2018). Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat. Commun 9, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink CS, Calero-Nieto FJ, Wang X, Maglitto A, Mariani SA, Jawaid W, Göttgens B, and Dzierzak E (2020). Iterative Single-Cell Analyses Define the Transcriptome of the First Functional Hematopoietic Stem Cells. Cell Rep. 31, 107627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, Chen M-H, Raffield LM, Tardaguila M, Huffman JE, et al. (2020). The Polygenic and Monogenic Basis of Blood Traits and Diseases (medRxiv). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fan HC, Behr B, and Quake SR (2012). Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Rodriguez-Fraticelli A, Camargo FD, and Klein AM (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Went M, Sud A, Försti A, Halvarsson B-M, Weinhold N, Kimber S, van Duin M, Thorleifsson G, Holroyd A, Johnson DC, et al. (2018). Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun 9, 3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner B, Beier F, Hummel S, Balabanov S, Lassay L, Orlikowsky T, Dingli D, Brümmendorf TH, and Traulsen A (2015). Reconstructing the in vivo dynamics of hematopoietic stem cells from telomere length distributions. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, and Chang HY (2019). Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H (2013). Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- Zeng H, Yücel R, Kosan C, Klein-Hitpass L, and Möröy T (2004). Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 23, 4116–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, He J, Bai Z, Li Z, Gong Y, Liu C, Ni Y, Du J, Ma C, Bian L, et al. (2019a). Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 29, 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, Bian Z, Li Z, Ni Y, Yan J, et al. (2019b). Single-Cell RNA Sequencing Resolves Spatiotemporal Development of Pre-thymic Lymphoid Progenitors and Thymus Organogenesis in Human Embryos. Immunity 51, 930–948.e6. [DOI] [PubMed] [Google Scholar]
- Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, et al. (2004). Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 21, 853–863. [DOI] [PubMed] [Google Scholar]
- Zhou F, Li X, Wang W, Zhu P, Zhou J, He W, Ding M, Xiong F, Zheng X, Li Z, et al. (2016). Tracing haematopoietic stem cell formation at single-cell resolution. Nature 533, 487–492. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Liu N, Orkin SH, and Yuan G-C (2019). CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol. 20, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]