

Abstract
Hypoxia-inducible factor 1α (HIF-1α) is recognized as a prime molecular target for metastatic cancer. However, no specific HIF-1α inhibitor has been approved for clinical use. Here, we demonstrated that in vivo efficacy of echinomycin in solid tumors with HIF-1α overexpression is formulation-dependent. Compared to previously-used Cremophor-formulated echinomycin, which was toxic and ineffective in clinical trials, liposomal-echinomycin provides significantly more inhibition of primary tumor growth and only liposome-formulated echinomycin can eliminated established triple-negative breast cancer (TNBC) metastases, which is the leading cause of death from breast cancer as available therapies remain minimally effective at this stage. Pharmacodynamic analyses reveal liposomal-echinomycin more potently inhibits HIF-1α transcriptional activity in primary and metastasized TNBC cells in vivo, the latter of which are HIF-1α enriched. The data suggests that nanoliposomal-echinomycin can provide safe and effective therapeutic HIF-1α inhibition and could represent the most potent HIF-1α inhibitor in prospective trials for metastatic cancer.
Keywords: HIF-1α, Echinomycin, Nanomedicine, Liposome, Breast Cancer
Graphical Abstract:
In solid tumors with HIF-1α overexpression, the in vivo efficacy of HIF-1α inhibitor echinomycin is formulation-dependent. Formulated in Cremophor, echinomycin performed poorly in clinical trials of solid tumors and, here, is shown to be similarly toxic and ineffective in mouse models of solid tumors. In contrast, reformulation of echinomycin in nanoliposomes widened therapeutic index, more potently inhibited HIF-1α transcription in tumor cells, and enabled direct elimination of established metastasis in vivo. Our data indicate that, with proper formulation, echinomycin may fulfil the unmet need for safe and effective therapeutic HIF inhibition in metastatic cancer.
Background
Metastatic breast cancer is the most frequent cause of cancer-related death in women1. Hypoxia-inducible factor 1α (HIF-1α) is a master transcription factor that is highly expressed in cancer tissues, including breast cancer, and is particularly enriched in triple-negative breast cancer (TNBC)2. Generally, HIF-1α is degraded under normoxia, but it can be stabilized by oxygen-independent mechanisms, especially in cancer cells3. HIF-1α activates transcription by binding to hypoxia response elements (HRE) in the promoters of its target genes. In breast cancer, HIF-1α acts as a master regulator for the expression of genes that promote tumor growth, vascularization, and metastasis4. HIF-1α has been identified as one of five predictive signature markers to predict disease outcome among node-negative breast cancer patients5. HIF-1α inhibition has shown success as a therapeutic strategy in mouse models of metastatic breast cancer6, 7. However, no specific HIF inhibitor has been approved for clinical use8.
Echinomycin is a potent small-molecule inhibitor of HIF-1α, which functions through sequence-specific binding to HRE to competitively inhibit HIF-1α binding to its target genes9. Before HIF-1α was identified as molecular target of echinomycin, the drug was tested in multiple phase I and II trials for treatment of solid tumors10-21. However, clinical development was discontinued because echinomycin was not found to be effective for patients with solid tumors that were refractory to all available treatments.
Several factors could have contributed to failure of the trials. First, preclinical studies had not identified HIF-1α as molecular target of echinomycin, making it impossible to judge the on-target activity of echinomycin in vivo. Second, no methods have been developed to evaluate the drug availability in vivo. Echinomycin is extremely insoluble in water, which complicates its formulation into a suitable dosage form. In earlier clinical trials, echinomycin was formulated with Cremophor EL® (CrEL). CrEL has been under scrutiny for its allergenic activity and, in clinical trials with CrEL-echinomycin, hypersensitivity reactions were observed12, 18, 19, 21-23 CrEL-echinomycin also caused severe nausea and vomiting, constituting its dose-limiting toxicity11, 12, 15-21.
We have demonstrated that echinomycin, dissolved in dimethyl sulfoxide (DMSO) is active against leukemia in mice24, 25. However, this formulation has shown little efficacy in treatment of solid tumors in mice (our unpublished observations). Since liposomes can improve drug delivery to solid tumors, we explored their potential as an enabling technology for echinomycin in solid tumors26. We report here that reformulating echinomycin in liposomes reduced toxicity and provided strong therapeutic effects with effective in vivo targeting of the HIF-1α pathway. Our data demonstrate that, with proper formulation, echinomycin provides a safe and effective approach for in vivo targeting of HIF-1α activity to inhibit growth and metastasis of solid tumors.
Methods
Materials
All cell lines were purchased from American Type Culture Collection (Manassas, VA) except for SUM-159 (gift from Dr. Max Wicha’s laboratory, University of Michigan) and TUBO (gift from Dr. Yangxin Fu’s laboratory, University of Texas). All experiments were performed with mycoplasma-free cells. Anti-HIF-1α (GTX127309) was purchased from GeneTex (Irvine, CA), anti-Vimentin (NBP2-12472) from Novus Biologicals (Centennial, CO), anti-cleaved Capase3 from Cell Signaling (Danvers, MA), and anti-Ki67 (sc-101861) from Santa Cruz Biotech (Santa Cruz, CA). Hydrogenated soy L-α-phosphatidylcholine (HSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (18:0 PEG2000 PE), and cholesterol were from Avanti Polar Lipids, Inc. (Alabaster, AL). Echinomycin was provided by Oncoimmune (Rockville, MD). LenticrisprV2 vector plasmid was from Addgene (Cambridge, MA).
Liposomal-echinomycin preparation and characterization
Preparation.
HSPC, cholesterol, and 18:0 PEG2000 PE were dissolved in 2:1 chloroform:methanol (v/v) at 56.7:5.3:38 (mol:mol) ratio. Echinomycin was added at a 1:20 (drug:lipid, wt:wt) ratio. Solvent was evaporated with nitrogen to produce a lipid film, which was hydrated in 300 mM sucrose for 7.1 mg/mL lipid. Liposomes were extruded 21x across 50 nm polycarbonate with a MiniExtruder (Avanti Polar Lipids, Alabaster, AL), then sterile-filtered through 0.22 μm polyethersulfone (MilliporeSigma, Burlington, MA). The filtrate was sampled for high-performance liquid chromatography (HPLC) and dynamic light scattering (DLS)/zeta potential and stored at 4°C. Drying, hydration, and extrusion steps were carried out at 65°C; all other steps were at room temperature. Hydrodynamic diameter, polydispersity index (PdI), and zeta potential of liposomal-echinomycin was determined using a Malvern Zetasizer Nano ZS. TEM measurement conditions: 3.5 μL undiluted sample plunge frozen using FEI Vitrobot; Humidity 100%, Blot Time 2.5 seconds, Blot Force 5.
Echinomycin Release.
Echinomycin release from liposomal-echinomycin was determined by dialysis. Liposomal-echinomycin was diluted 1:4 in release medium (DI water), and transferred into individual dialysis tubes as aliquots (GE Healthcare, Chicago, IL. Then, the dialysis tubes were placed in a reservoir containing 4 L of release medium and dialyzed at room temperature with stirring. At each time point, a dialysis tube was removed and the contents were filtered by a fresh 0.22 μm polyethersulfone (PES) filter. The filtrate was diluted in acetonitrile to match HPLC mobile phase, and run on HPLC to determine echinomycin concentration. Percent release was calculated as follows: %release = [1–(echinomycin concentrationtimepoint)/(echinomycin concentrationinitial)]x100.
Stability Assay.
Liposomal-echinomycin was stored at 4°C and sampled for DLS/zeta potential and echinomycin content analysis at indicated time points. Echinomycin content was determined by sub-micron filtration27, 28, with 0.22 μm polyethersulfone.
Mice
All procedures involving experimental animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Children’s Research Institute or University of Maryland School of Medicine. NOD.Cg-PrkdcscidIl2rγtm1Wjl/SZJ (NSG) mice were from Jackson Laboratory (Bar Harbor, ME) and maintained under specified pathogen-free conditions at the Children’s Research Institute or University of Maryland School of Medicine.
Breast cancer xenograft model.
Tumor cells (0.5x106-1x106) were suspended in RPMI 1640 and injected into the 2nd mammary fat pad of 6-8 weeks-aged female NSG mice. Tumor volume was calculated as follows: Volume = (AxB2)/2, where A is the shorter diameter. Mice were euthanized by CO2 asphyxiation when tumors reached 20mm. For full surgical resection (FSR) experiments, primary tumors were resected once 5x5mm as previously described29. All agents were administered by intravenous (i.v.) injection. CrEL-echinomycin (dissolved in 1:1 volume of Cremophor:ethanol), or liposomal-echinomycin were diluted using normal saline.
Histology, Immunofluorescence
Formalin-fixed tissues were paraffin-embedded and sectioned for hematoxylin and eosin (H&E) or immunofluorescence staining. Toxicological analyses were performed double-blind by a certified veterinary pathologist. Metastasis was quantified in liver or lung tissues by counting human vimentin-positive nodules/view at 10x magnification. For immunofluorescence, 5μm-thick sections were treated by sequential deparaffinization, rehydration, and antigen-retrieval with 0.5 M Tris-HCl plus 5% urea buffer, and microwave-boiled for 20 min. Antibodies were used to stain 0.3% Triton-permeabilized sections in 2% bovine serum albumin blocking buffer.
Statistics
Statistical analyses were performed using GraphPad Prism 7. To determine statistical significance P-values were calculated using Student’s t-test (two tailed) for pair-wise comparisons, or one-way ANOVA with Bonferonni’s posttest for experiments involving multiple groups, and two-way ANOVA for tumor kinetics (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns, not significant). Sample sizes were determined by power calculation or prior experience. All experiments have been performed at least three times, producing similar results. Representative data is shown.
Results
Echinomycin Effectively Targets HIF-1α-dependent Breast Cancer
Although HIF-1α is normally degraded under normoxia, non-canonical mechanisms exist to promote its stability regardless of oxygen tension in cancer cells, including breast cancer3. We evaluated HIF-1α protein in breast cancer cell lines under normoxia, with or without the hypoxia mimetic CoCl2. Although detectable in HER2+ cell lines, HIF-1α protein was particularly higher in TNBC cell lines SUM-159 and MDA-MB-231 (Figure 1A). HIF-1α protein levels under normoxia corresponded to the breast cancer cells’ susceptibility to echinomycin after 24 hours (Figure 1B) or 48 hours (Figure S1) treatment, suggesting oncogenic addiction. We analyzed in vitro activity of echinomycin on expression of HIF-1α targets known to be critical for tumor growth and metastasis30-35 in SUM-159. mRNA levels of CITED2, VEGFA, SLC2A3, and SDF1 were reduced in a dose-dependent manner (Figure 1C). Primer sequences are listed in Table S1. To test the specificity of echinomycin on HIF-1α inhibition in breast cancer, we generated HIF1A knockout SUM-159 cells by Crispr/Cas9 guided gene editing method (Figure 1D). HIF1A(−/−) SUM-159 cells were less sensitive to echinomycin compared to empty vector (WT). The apparent HIF-1α-dependence provides strong genetic evidence that HIF-1α is the major target of echinomycin (Figure 1E). Double-knockdown of HIF1A and HIF2A in MDA-MB-231 cells was reported to impair primary tumor growth and lung metastasis in vivo6. Indeed, our analysis using SUM-159 cells with Crispr/Cas9 knockout of HIF1A confirmed that HIF1A is required for TNBC growth and metastatic potential, even under normoxia. Knockout of HIF1A alone severely inhibited SUM-159 growth by colony forming assay (Figure S2A) and metastatic potential determined by transwell migration and wound healing assays (Figure S2B). Accordingly, genetic depletion of the HIF1A isoform was sufficient to abrogate tumor growth in xenogeneic recipient mice (Figure S2C).
Figure 1.

Echinomycin Effectively Targets HIF-1α-dependent Breast Cancer.
A) Immunoblot shows relative amounts of HIF-1α protein accumulation in TNBC and HER2+ breast cancer cells cultured under normoxia with/without 6-hour 250 μM CoCl2 treatment. B) Sensitivity of MCF-7, SUM-159, or MDA-MB-231 to echinomycin. Cells were incubated in triplicate with echinomycin (0.05, 0.15, 0.45, 1.35, or 4.05 nM) for 24 hours, and viability was calculated by MTT assay. Mean optical density (OD) ± SEM is shown as % of DMSO control. P-values shown for MCF-7 vs SUM-159 and MDA-MB-231. C) mRNA expression of HIF-1α target genes in SUM-159 cells with/without echinomycin treatment. Triplicate wells were incubated with DMSO or echinomycin (0.45 or 1.35 nM) for 24 hours, and mRNA expression was determined by qPCR, normalized to L37. D) Immunoblot of HIF-1α protein in WT or HIF1A(−/−) (KO) SUM-159 cells. KO was performed by Crispr/Cas9 gRNA strategy and clones 3 to 6 are mutually exclusive KOs. All cells were treated by 250 μM CoCl2 for 6 hours. E) Sensitivity of WT or HIF1A(−/−) SUM-159 to echinomycin. Triplicate wells were incubated for 24 hours with DMSO or echinomycin and viability determined by MTT assay. Mean OD ± SEM is shown as % of DMSO control. P-values shown for WT vs HIF1A-KO-3 and HIF1A-KO-5. F) Efficacy of CrEL-echinomycin in SUM-159 mouse model. Mice received SUM-159 cells on day 0. Mice were randomized (n=5 mice/group) to receive CrEL (Vehicle) or CrEL-echinomycin, 0.1 mgkg−1, once every other day for 15 doses, starting day 15. Mean tumor volumes ± SEM are shown.
Formulation and Characterization of Liposomal-echinomycin
Having confirmed HIF1A-dependent breast cancer growth and inhibition by echinomycin in vitro, we set out to test if the CrEL-echinomycin formulation used for earlier clinical trials can target HIF-1α in vivo. We transplanted SUM-159 cells into NSG mice and treated them with CrEL-echinomycin at a maximally-tolerable regimen once 3x3 mm tumors formed. However, no tumor growth inhibition was observed (Figure 1 F). These data demonstrate that, even for HIF1A-dependent tumor cells that can be effectively targeted by echinomycin in vitro, the clinical formulation was largely ineffective. The data suggest that lack of clinical efficacy may be due to inadequate delivery to tumor cells in vivo.
Since liposomes can improve the therapeutic index of incorporated drugs, we tested if a liposomal platform could achieve the same for echinomycin. Liposomal-echinomycin displayed an average hydrodynamic diameter of 98 nm and high uniformity (Figure 2A&B, Table S2), with narrow polydispersity index (PdI) of ~0.05 by DLS (Figure 2C, Table S2). Capillary electrophoresis revealed an average zeta potential of −30 mV (Figure 2C). These physiochemical characteristics were highly reproducible between batches (Table S2). We studied the drug release profile by dialysis followed by HPLC and found that echinomycin was released over ~168 hours under this condition (Figure 2D, Figure S3). We next examined stability of liposomal-echinomycin at 4°C. Hydrodynamic diameter, PdI, and zeta potential remained stable for at least 18 months (Figure 2E&F). To study drug content loss, we filtered the product at various time points with 0.22 μm polyethersulfone for the removal of any echinomycin precipitates, and analyzed the filtrates by HPLC. No evidence of drug content loss was observed throughout the study (Figure 2G). To verify that the potency of echinomycin drug substance remained unchanged, we treated SUM-159 cells with liposomal-echinomycin kept in storage for various periods of time. Liposomal-echinomycin kept for 3 or 18 months in storage was indistinguishable in potency from of a fresh batch of liposomal-echinomycin or free echinomycin (Figure 2H). Therefore, the potency of echinomycin is unaltered by its entrapment into the liposomes, and the potency of liposomal-echinomycin remains stable in storage for at least 18 months.
Figure 2.

Formulation and Characterization of Liposomal-echinomycin.
A) Size distribution of liposomal-echinomycin measured by DLS. B) Representative TEM depicting liposomal-echinomycin. C) Hydrodynamic diameter, zeta potential (ζ), and polydispersity index (PdI) of liposomal-echinomycin. Data shown as mean ± SD for 6 batches (detailed in Table S2). D) Release echinomycin from liposomal-echinomycin by dialysis. Percent release over time summarized as mean ± SD for triplicate runs on HPLC (Left), corresponding HPLC chromatograms are depicted (Right). E – H) Storage/Stability of liposomal-echinomycin. Liposomal-echinomycin was stored for 18 months at 4°C and analyzed at various time points for DLS size distribution (E), PdI (D, annotated), hydrodynamic diameter and ζ (F), echinomycin content loss (G), and potency against SUM-159 cells in vitro (24 hour incubation, performed in triplicate wells), determined by MTT assay (H). Data for D-G shown as mean ± SEM. EM, echinomycin.
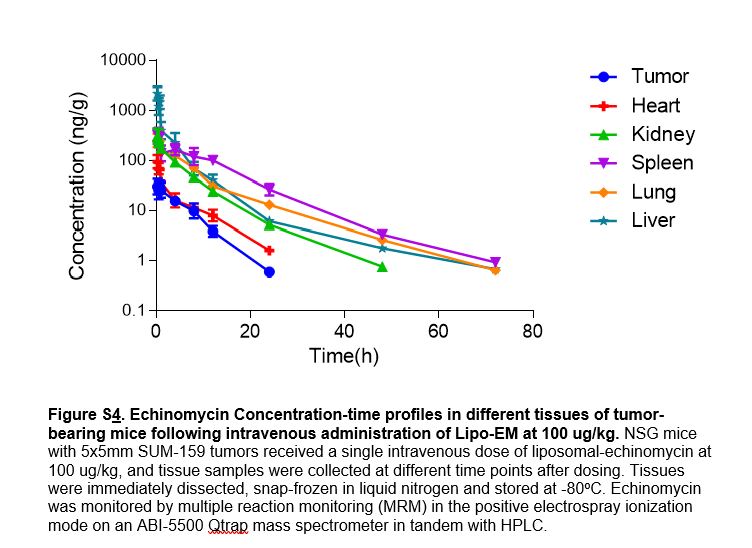
We have recently reported that liposomal formulation increased the systemic exposure of Echinomycin in the blood circulation36. We studied the tissue biodistribution of echinomycin in tumor-bearing mice following i.v. administration of Lipo-EM at 0.1 mgkg−1. Figure S4 and Table S3 show echinomycin concentration-time profiles and the pharmacokinetic parameters of echinomycin in different tissues, respectively. After i.v. administration, echinomycin reached most tissues with Tmax between 0.25-1 hour (Table S3). The highest amount of echinomycin was found in liver, followed by kidney, spleen, lung, heart, and tumor (Figure S4). Terminal half-life (T1/2) and mean residence time (MRT) ranged between 3.37-13.43 hours and 4.42-10.86 hours, respectively, and nearly all of the drug was eliminated by 72 hours post-dosing (Figure S4, Table S3).
Liposomal-echinomycin Is Less Toxic than Conventionally-formulated Echinomycin
Since we encountered a toxicity ceiling for CrEL-echinomycin in the SUM-159 model, we tested if reformulation resulted in reduced toxicity by comparing acute toxicity of CrEL-echinomycin and liposomal-echinomycin in mice. First, we evaluated single-dose acute toxicity at the equivalent dose of 1 mgkg−1. While 1 mgkg−1 of CrEL-echinomycin was 100% lethal by day 7 post-administration, the same dose of liposomal-echinomycin did not reach LD50 (Figure 3A). CrEL-echinomycin also caused 100% mortality in repeat dosing at 0.35 mgkg−1, (Figure 3B). While these mice experienced rapid, progressive weight loss, mice receiving the same dose/schedule of liposomal-echinomycin were visually indistinguishable from control mice (Figure 3C). Thus, liposomal-echinomycin is less toxic than CrEL-echinomycin. In terms of body weight loss, 0.35 mgkg−1 liposomal-echinomycin was equitoxic to 0.25 mgkg−1 of CrEL-echinomycin (Figure 3B).
Figure 3.

Liposomal-echinomycin Is Less Toxic than CrEL-Echinomycin.
A) Survival of mice receiving single dose of CrEL-echinomycin or liposomal-echinomycin at 1 mgkg−1, or equivalent excipient dose (n=7 mice/group), administered day 0. B – F) Acute toxicity of mice receiving CrEL-echinomycin or liposomal-echinomycin at 0.15 mgkg−, 0.25 mgkg−1 or 0.35 mgkg−1, or corresponding vehicle controls (n=5 mice/group). Agents were administered days 0, 3, and 6. B) Body weight loss of mice throughout therapeutic cycle. P-values shown for CrEL-echinomycin 0.25 mgkg−1 vs liposomal-echinomycin 0.25 mgkg−1. C) Representative photograph of mice taken on day 7. D) Longitudinal analysis of cumulative food consumption throughout therapeutic cycle. P-values indicate CrEL-echinomycin 0.25 mgkg−1 vs liposomal-echinomycin 0.25 mgkg−1. Comparison of average daily food consumption 3 days prior to (before) and 15 days following (after) dose one, shown as mean ± SEM. F) Representative H&E staining of liver, kidney, and lung tissues for mice on day 7. Panel i: CrEL-vehicle mouse liver showing normal glycogen levels; panel ii: liposomal-echinomycin 0.35 mgkg−1 mouse liver showing mild glycogen depletion; panel iii: p CrEL-echinomycin 0.25 mgkg−1 mouse liver showing glycogen depletion with mild atrophy, reduced hepatocyte size; panels iv, v: CrEL-echinomycin 0.25 mgkg−1 mice livers showing hepatitis (panel iv) and steatosis (panel v); panel vi: normal kidney, CrEL-vehicle mouse; panel vii: normal kidney, liposomal-echinomycin 0.35 mgkg−1 mouse; panels viii, ix: abnormal kidneys from CrEL-echinomycin mice showing massive necrosis of proximal tubule epithelium (panel viii) and pyelonephritis (panel ix); panel x: normal lung, CrEL-vehicle mouse; panel xi: normal lung, liposomal-echinomycin 0.35 mgkg−1 mouse; panel xii: abnormal lung from CrEL-echinomycin 0.25 mgkg−1 mouse showing multi-focal pleuritis with mineralization, hemorrhage, and necrosis.
We monitored food consumption as a surrogate marker for safety of echinomycin formulations. Mice that received 0.25 mgkg−1 of CrEL-echinomycin, or 0.35 mgkg−1 of liposomal-echinomycin consumed significantly less throughout the observation period compared to mice that received empty liposomes, CrEL vehicle, liposomal-echinomycin 0.25 mgkg−1, or either formulation at 0.15 mgkg−1 (Figure 3D). As shown in Figure 3E, compared to each group’s respective pre-treatment values, mice that received CrEL-echinomycin, but not liposomal-echinomycin, demonstrated a significant decrease in mean daily food consumption at doses as low as 0.15 mgkg−1. Starting at 0.25 mgkg−1, there was significant decrease in mean daily food consumption for all mice receiving echinomycin, regardless of the formulation. However, at 0.25 mgkg−1, the decrease was more pronounced in mice that received CrEL-echinomycin vs liposomal-echinomycin, and post-treatment averages in daily food intake were also significantly different between the two groups. Compared to pre-treatment values, the reduction in average daily food intake for mice that received CrEL-echinomycin 0.25 mgkg−1 was 81.4% of the initial, compared to 79.5% of the initial for liposomal-echinomycin 0.35 mgkg−1, and the post-treatment averages in daily food intake were not significantly different between the two groups. This analysis demonstrated that liposomal-echinomycin was approximately 30% less toxic than CrEL-echinomycin in terms of body weight loss and food consumption.
We performed post-mortem histological analysis of H&E stained liver, kidney and adrenal gland, lung, heart, small intestine, pancreas, and muscle tissues of the mice euthanized on day 7. In the liver, all echinomycin-treated groups showed evidence of glycogen depletion, consistent with the reduction in food intake that we observed at these doses (Figure 3F, panels ii, iii). In addition, hepatitis and steatosis were observed in recipients of CrEL-echinomycin (Figure 3F, panels iv and v, respectively). The remainder of the analysis revealed lesions only in CrEL-echinomycin recipients. In the kidneys, lesions included massive necrosis of proximal tubular epithelium (Figure 3F, panel viii) and pyelonephritis with atrophy, degeneration and dilation of the tubules (Figure 3F, panel ix). Lungs showed evidence of multi-focal pleuritis with mineralization, hemorrhage, and necrosis (Figure 3F, panel xii). We found no lesions in the heart, adrenal gland, small intestine, pancreas, or muscle tissues for any mice. Altogether, histology indicated less toxicity for liposomal-echinomycin vs CrEL-echinomycin, even at an “equitoxic” dose in terms of body weight loss and food consumption (i.e. liposomal-echinomycin 0.35 mgkg−1 vs. CrEL-echinomycin 0.25 mgkg−1).
We completed our toxicology analyses by examining complete blood count and clinical biochemistry parameters of mice treated with different echinomycin formulations. Apart from slightly decreased red blood cell counts in CrEL-echinomycin-treated mice, the blood indices revealed no significant differences between formulations, and all values fell within the normal ranges (Table S4). On average, ALT and AST liver enzymes were slightly higher in CrEL-echinomycin- vs liposomal-echinomycin-treated mice, although the trend was not statistically significant (Table S4).
Liposomal-echinomycin Is Effective Against HIF-1α-expressing Breast Cancer in Vivo
To test the therapeutic effect of liposomal-echinomycin, we used MDA-MB-231 and SUM-159 xenograft models and treated the mice with liposomal-echinomycin or CrEL-echinomycin at equitoxic, or equivalent doses, or CrEL-vehicle control. At 0.25 mgkg−1, its maximum-tolerated dose (MTD), CrEL-echinomycin demonstrated slight to modest therapeutic activity against SUM-159 and MDA-MB-231 primary tumor growth, respectively (Figure 4A&B), which can be attributed to the increase in dose strength compared to our preliminary studies (Figure 1F). Liposomal-echinomycin more potently inhibited primary tumor growth in both models at its MTD (0.35 mgkg−1), and even at the equivalent dose of 0.25 mgkg−1 (Figure 4A&B). The enhanced efficacy of liposomal-echinomycin at the equivalent dose to CrEL-echinomycin suggested that liposomes improved in vivo delivery of echinomycin to tumor cells. To gain insight to the effects of liposomal-echinomycin on tumor cells in vivo, we performed histological analyses of the fixed tumor tissues. H&E staining revealed tumor necrosis corresponding to the dose and formulation of echinomycin (Figure 4C). Immunofluorescence analysis of Ki67 and cleaved-caspase 3 in the primary tumors indicated that liposomal-echinomycin more potently inhibited the proliferation of tumor cells and induced their apoptosis, respectively, vs CrEL-echinomycin (Figure 4D). Reductions in HIF-1α levels also followed the same dose- and formulation-dependent trend in the tumor sections (Figure 4E). We further analyzed tumor tissues of the xenograft mice for mRNA levels of HIF targets involved in tumor growth/metastasis30-35. In a dose- and formulation-dependent manner mirroring the efficacy data, echinomycin decreased expression of HIF target genes CITED2, VEGFA, SLC2A3, and PGK1 in primary tumor tissues of MDA-MB-231 and SUM-159 xenografts (Figure 4F&G).
Figure 4.


Liposomal-echinomycin Is More Effective than CrEL-Echinomycin. A&B) Female NSG mice were injected with 1.0x106 SUM-159 (A) or MDA-MB-231 (B) cells. Treatment was initiated when tumors measured 3x3 mm, on day 10 (SUM-159) or day 14 (MDA-MB-231). Mice were randomized (n=7 mice/group) to receive vehicle, CrEL-echinomycin or liposomal-echinomycin, once every three days for 5 doses. Mean tumor volumes shown; P-values indicate CrEL-echinomycin 0.25 mgkg−1 vs liposomal-echinomycin (0.25 mgkg−1 and 0.35 mgkg−1) groups. SUM-159 or MDA-MB-231 xenograft mice were euthanized on days 25 or 40, respectively, and tumor, liver and lung tissues were formalin-fixed. C) Tumor tissues were H&E stained to detect necrotic regions in SUM-159 and MDA-MB-231 tumor tissues. D) Representative immunofluorescence staining of Ki67 and cleaved-caspase 3 protein and 4,6-diamidino-2-phenylindole (DAPI) in fixed primary tumor tissues from SUM-159 or MDA-MB-231 xenograft mice. E) Representative immunofluorescence staining of HIF-1α protein and DAPI in fixed primary tumor tissues from SUM-159 xenograft mice. F&G) mRNA expression of HIF-1α target genes in primary tumor cells from SUM-159 (F) and MDA-MB-231 (G) xenograft mice treated with different dose/formulation of echinomycin or vehicle. mRNA was isolated from tumors of 5 mice/group and cDNA was pooled for each group prior to analysis by qPCR. Mean expression ± SEM is shown, normalized to L37. P-values annotated beside CrEL-echinomycin indicate comparison with vehicle; those beside liposomal-echinomycin indicate comparison with CrEL-echinomycin.
Metastasis is the leading cause of death from breast cancer1, 37-39. We next investigated if reformulation of echinomycin had any impact on metastasis in the mouse models of TNBC. H&E analysis paired with immunofluorescence staining of human vimentin revealed extensive lung metastasis in vehicle-treated SUM-159 (Figure 5A&B) or MDA-MB-231 (Figure 5E&F) recipients, and extensive liver metastasis from MDA-MB-231 (Figure 5C&D). In comparison, metastasis was reduced in echinomycin recipients, regardless of dose or formulation, for both cell lines. However, the data revealed particularly profound inhibition of metastasis for liposomal-echinomycin compared to CrEL-echinomycin, which was significant between the two formulations, even at the equivalent dose (Figure 5A-F). At 0.35 mgkg−1, liposomal-echinomycin apparently eradicated liver metastasis in the MDA-MB-231 model (Figure 5C&D), and nearly eradicated lung metastasis in both models (Figure 5A&B, Figure 5E&F).
Figure 5.

Liposomal-echinomycin Reduces Metastasis of MDA-MB-231 and SUM-159 in Xenograft Mice. A-F) NSG mice were transplanted with 1.0x106 MDA-MB-231 (A-D) or SUM-159 (E-F) and treated with CrEL-echinomycin, liposomal-echinomycin or vehicle and euthanized as described in Figure 3A&B. Fixed lung and liver tissues were stained with H&E or anti-human Vimentin antibody. Representative H&E staining (B, D, F, top panels) and immunofluorescence staining for human Vimentin (B, D, F, bottom panels) are shown for fixed lung (B&F) and liver (D) tissues for mice xenografted with MDA-MB-231 (B&D) or SUM-159 (F) tumors. Summarized data corresponding to B, D and F are shown in A, C, and E, respectively, displaying mean number of metastatic nodules ± SEM found in lung and liver tissues.
It is known that HIF-1α expression in primary tumor breast cancer cells promotes metastasis, although from a clinical perspective this concept has limited value since prognosis weighs heavily on the stage of progression at time of surgical resection39. To improve survival rates for breast cancer, it is essential to understand how potential treatments impact progression of established metastases. To distinguish between effects of echinomycin on the process of metastasis and on the metastasized cells themselves, we performed full-surgical resection (FSR) of primary SUM-159 or MDA-MB-231 tumors in xenografted mice prior to initiating treatment. H&E paired with immunofluorescence staining of human vimentin in lung and liver revealed that, as in non-FSR studies, FSR mice that received liposomal-echinomycin were nearly free of lung metastasis in MDA-MB-231 (Figure 6A&B) and SUM-159 (Figure 6E&F) models, and free of liver metastasis in the MDA-MB-231 model (Figure 6C&D). These results demonstrated that echinomycin directly eliminates metastasized breast cancer cells in the lung and liver and further suggested an essential role for HIF-1α in the established secondary tumors. To test if this is the case, we isolated SUM-159 cells from the local tumor or lungs of xenograft mice and used FACS to readout HIF-1α activity via lentiviral EGFP-reporter25. Compared to cultured primary cells, lung-metastatic SUM-159 cells had higher HIF-1α transcriptional activity (Figure 6G). We performed RT-PCR of HIF-1α target genes in the metastasized cells in lung tissue of vehicle- and liposomal-echinomycin-treated FSR mice. Consistent with high HIF-1α transcriptional activity in lung-metastatic cells (Figure 6G), we found that the expression of HIF-1α target genes was significantly decreased in the metastasized cells, indicating the targeted effect of liposomal-echinomycin on the HIF-1α pathway in established metastasis (Figure 6H).
Figure 6.

Liposomal-echinomycin Eliminates Established Breast Cancer Metastases A-G) NSG mice were injected with 1.0x106 MDA-MB-231 or SUM-159 cells. Tumors measuring 5x5 mm were surgically resected, and the mice were treated with empty liposomes or liposomal-echinomycin 0.35 mgkg−1 (n=7/group), once every three days for 5 doses starting day 7 post-surgery. Mice were euthanized 25 days after surgery, and fixed lung and liver tissues were H&E or anti-human Vimentin stained to quantify metastasis. Representative H&E staining (B, D, F, top panels) and immunofluorescence staining (B, D, F, bottom panels) are shown for lung (B, F) and liver (D) tissues for MDA-MB-231 (B, D) or SUM-159 (D) xenograft mice. Summarized data corresponding to B, D, and F, are shown in A, C, and E, respectively, as mean number of human vimentin positive nodules ± SEM in the tissues. G) Higher HIF1α activity in lung-metastasized SUM-159 cells compared to primary SUM-159 cells. The primary tumor (Local) or lung-metastasis (LM) derived SUM159 cells were introduced with lentiviral HRE-EGFP reporter or mutated control (HRE-Mt) and cultured for 48 hrs. 2μg/ml puromycin was added and cells were cultured another 48 hrs prior to detection of EGFP expression by FACS. EGFP intensity histogram is shown for one representative mouse (Upper) and the summarized data for EGFP+ cells is shown (Lower). H) qPCR of HIF1α targets in lung-metastasized SUM-159 or MDA-MB-231 cells from liposomal-echinomycin or vehicle treated xenograft mice. Tumors were established according to the FSR method detailed in Figure 6A-G and 7 days after FSR the mice received liposomal-echinomycin 0.35 mgkg−1 or vehicle once every three days for three doses. On day 25 post-FSR, mice were sacrificed and primary or metastasized tumor cells were isolated and analyzed by qPCR.
Discussion
HIF-1α is overexpressed in 70% of human cancers40. HIF-1α drives tumor progression because it orchestrates fundamental processes such as angiogenesis, glycolytic switch, and metastasis8. It is therefore of interest to consider whether poor outcomes in previous trials were due to improper formulation, or poor choice of targets/targeting agents. The termination of echinomycin in clinical trials immediately preceded the first clinical report identifying HIF-1α as a potential target for cancer8, 10, 40. Nearly a decade later, echinomycin was first identified as a HIF-1α inhibitor9. Therefore, while echinomycin was not effective as a putative cytotoxic therapy, it was never examined clinically as a HIF-1α inhibitor. Here, through the use of HIF1A-addicted breast cancer cell lines, we provide the first evidence indicating that the outcomes in early clinical trials with echinomycin were likely skewed by formulation-related issues that were unknown at the time. First, dose-limiting toxicities of CrEL-echinomycin may have prevented therapeutically effective doses from being reached. Our data showed that MTD of CrEL-echinomycin provided minimal, or even no tumor growth inhibition, depending on the treatment schedule. The effect of MTD CrEL-echinomycin on metastasis was suboptimal, and only liposomal-echinomycin could eliminate liver metastasis altogether in the MDA-MB-231 model. Second, the efficiency in which CrEL delivers the drug to its target is suboptimal; although CrEL-echinomycin and liposomal-echinomycin are equally effective in vitro, liposomal-echinomycin is significantly more effective in vivo. Therefore, liposomes delivered echinomycin to relevant cells more efficiently in vivo, and could potentially provide the same advantage in humans.
Clinical studies have provided considerable safety data for echinomycin10-12, 14-18, 41-45 Echinomycin has an advantage over untested HIF-1α inhibitors because safe doses have already been established in over a dozen clinical trials. Liposomal-echinomycin also achieved superior in vivo targeting, evidenced by more potent reduction in HIF-1α targets. Therefore, liposomal-echinomycin is safer and more effective than CrEL-echinomycin, and should be tolerated better in clinical trials.
Our results showed that echinomycin remained stably associated with the liposomes in long-term storage but was released within days under dialysis conditions. The difference in release profiles under the two conditions can be attributed to bilayer exchange, which has been reported for a variety of lipophilic, poorly water soluble drugs that partition into the liposomal bilayer, as reviewed by Fahr and colleagues46. As such drugs continuously partition/repartition between bilayers of neighboring vesicles according to an equilibrium, echinomycin remains solubilized by the liposome bilayer in storage because the equilibrium between bilayer-partitioning is already established and remains unchanged over time.
Altogether, our data provides insight about the performance of echinomycin documented in early clinical trials involving Cremophor. More importantly, it indicates that reformulation of echinomycin via a nanoliposomal platform enables successful targeting of HIF-1α in metastatic breast cancer. Since metastasis is the leading cause of breast cancer-related mortality, our work provides a new therapeutic approach to improve survival in patients with metastatic breast cancer. Given the extensive data showing the critical function of HIF-1α in human cancer, it is of great interest to evaluate this new formulation in clinical trials.
Supplementary Material
{kind=link}
Acknowledgement:
The formulation described herein was characterized by the Nanotechnology Characterization Laboratory as part of its free Assay Cascade characterization service for cancer nanomedicines, which is supported by the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E.
Funding: This study was supported by the grants from the National Institutes of Health National Cancer Institute (CA171972, CA183030 (Yang Liu), CA164469 (Yin Wang)) and a grant from OncoImmune, Inc. Christopher M. Bailey received stipend support from the Institute of Biomedical Sciences at The George Washington University during part of the studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest: Yang Liu and Pan Zheng are co-founders of OncoImmune, Inc., which has licensed the technology from the Children’s National Medical Center. No potential conflicts of interest were disclosed by other authors.
References:
- 1.Weigelt B, Peterse JL and van 't Veer LJ. Breast cancer metastasis: markers and models. Nature reviews Cancer 2005; 5: 591–602. DOI: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70. DOI: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iommarini L, Porcelli AM, Gasparre G, et al. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front Oncol 2017; 7: 286 2017/December/13 DOI: 10.3389/fonc.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilkes DM and Semenza GL. Role of hypoxia-inducible factors in breast cancer metastasis. Future oncology 2013; 9: 1623–1636. DOI: 10.2217/fon.13.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charpin C, Tavassoli F, Secq V, et al. Validation of an immunohistochemical signature predictive of 8-year outcome for patients with breast carcinoma. International journal of cancer 2012; 131: E236–243. DOI: 10.1002/ijc.27371. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Wong CC, Wei H, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012; 31: 1757–1770. 2011/August/24 DOI: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Schito L, Rey S, Tafani M, et al. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2012; 109: E2707–2716. DOI: 10.1073/pnas.1214019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soni S and Padwad YS. HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta oncologica 2017; 56: 503–515. DOI: 10.1080/0284186X.2017.1301680. [DOI] [PubMed] [Google Scholar]
- 9.Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res 2005; 65: 9047–9055. 2005/October/06 DOI: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 10.Chang AY, Kim K, Boucher H, et al. A randomized phase II trial of echinomycin, trimetrexate, and cisplatin plus etoposide in patients with metastatic nonsmall cell lung carcinoma: an Eastern Cooperative Oncology Group Study (E1587). Cancer 1998; 82: 292–300. 1998/January/28 DOI: . [DOI] [PubMed] [Google Scholar]
- 11.Gradishar WJ, Vogelzang NJ, Kilton LJ, et al. A phase II clinical trial of echinomycin in metastatic soft tissue sarcoma. An Illinois Cancer Center Study Investigational new drugs 1995; 13: 171–174. Clinical Trial Clinical Trial, Phase II Research Support, U.S. Gov't, P.H.S; 1995/January/01. [DOI] [PubMed] [Google Scholar]
- 12.Wadler S, Tenteromano L, Cazenave L, et al. Phase II trial of echinomycin in patients with advanced or recurrent colorectal cancer. Cancer chemotherapy and pharmacology 1994; 34: 266–269. [DOI] [PubMed] [Google Scholar]
- 13.Shevrin DH, Lad TE, Guinan P, et al. Phase II trial of echinomycin in advanced hormone-resistant prostate cancer. An Illinois Cancer Council study. Invest New Drugs 1994; 12: 65–66. 1994/January/01. [DOI] [PubMed] [Google Scholar]
- 14.Chang AY, Tu ZN, Bryan GT, et al. Phase II study of echinomycin in the treatment of renal cell carcinoma ECOG study E2885. Invest New Drugs 1994; 12: 151–153. 1994/January/01. [DOI] [PubMed] [Google Scholar]
- 15.Muss HB, Blessing JA and DuBeshter B. Echinomycin in recurrent and metastatic endometrial carcinoma. A phase II trial of the Gynecologic Oncology Group. American journal of clinical oncology 1993; 16: 492–493. [DOI] [PubMed] [Google Scholar]
- 16.Marshall ME, Wolf MK, Crawford ED, et al. Phase II trial of echinomycin for the treatment of advanced renal cell carcinoma. A Southwest Oncology Group study. Investigational new drugs 1993; 11: 207–209. [DOI] [PubMed] [Google Scholar]
- 17.Taylor SA, Crowley J, Townsend J, et al. Phase II evaluation of echinomycin (NSC-526417) in patients with central nervous system malignancies. A Southwest Oncology Group study. J Neurooncol 1993; 15: 181–184. 1993/February/01. [DOI] [PubMed] [Google Scholar]
- 18.Schilsky RL, Faraggi D, Korzun A, et al. Phase II study of echinomycin in patients with advanced breast cancer: a report of Cancer and Leukemia Group B protocol 8641. Invest New Drugs 1991; 9: 269–272. 1991/August/01. [DOI] [PubMed] [Google Scholar]
- 19.Harvey JH, McFadden M, Andrews WG, et al. Phase I study of echinomycin administered on an intermittent bolus schedule Cancer treatment reports 1985; 69: 1365–1368. Research Support, U.S. Gov't, P.H.S; 1985/December/01. [PubMed] [Google Scholar]
- 20.Kuhn JG, Von Hoff DD, Hersh M, et al. Phase I trial of echinomycin (NSC 526417), a bifunctional intercalating agent, administered by 24-hour continuous infusion European journal of cancer & clinical oncology 1989; 25: 797–803. Research Support, U.S. Gov't, P.H.S; 1989/May/01. [DOI] [PubMed] [Google Scholar]
- 21.Pazdur R, Haas CD, Baker LH, et al. Phase I study of echinomycin Cancer treatment reports 1987; 71: 1217–1219. Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S; 1987/December/01. [PubMed] [Google Scholar]
- 22.Bernabeu E, Cagel M, Lagomarsino E, et al. Paclitaxel: What has been done and the challenges remain ahead. International journal of pharmaceutics 2017; 526: 474–495. DOI: 10.1016/j.ijpharm.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Gelderblom H, Verweij J, Nooter K, et al. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer 2001; 37: 1590–1598. Review 2001/August/31. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Liu Y, Tang F, et al. Echinomycin protects mice against relapsed acute myeloid leukemia without adverse effect on hematopoietic stem cells. Blood 2014; 124: 1127–1135. 2014/July/06 DOI: 10.1182/blood-2013-12-544221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Liu Y, Malek SN, et al. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell stem cell 2011; 8: 399–411. DOI: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pattni BS, Chupin VV and Torchilin VP. New Developments in Liposomal Drug Delivery. Chem Rev 2015; 115: 10938–10966. 2015/May/27 DOI: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- 27.Hong SS, Choi JY, Kim JO, et al. Development of paclitaxel-loaded liposomal nanocarrier stabilized by triglyceride incorporation. International journal of nanomedicine 2016; 11: 4465–4477. 2016/September/24 DOI: 10.2147/IJN.S113723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kan P, Tsao CW, Wang AJ, et al. A liposomal formulation able to incorporate a high content of Paclitaxel and exert promising anticancer effect. Journal of drug delivery 2011; 2011: 629234 2011/April/15 DOI: 10.1155/2011/629234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munoz R, Man S, Shaked Y, et al. Highly efficacious nontoxic preclinical treatment for advanced metastatic breast cancer using combination oral UFT-cyclophosphamide metronomic chemotherapy Cancer research 2006; 66: 3386–3391. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't; 2006/April/06 DOI: 10.1158/0008-5472.CAN-05-4411. [DOI] [PubMed] [Google Scholar]
- 30.Jayaraman S, Doucet M and Kominsky SL. CITED2 attenuates macrophage recruitment concordant with the downregulation of CCL20 in breast cancer cells. Oncol Lett 2018; 15: 871–878. 2018/February/06 DOI: 10.3892/ol.2017.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minemura H, Takagi K, Sato A, et al. CITED2 in breast carcinoma as a potent prognostic predictor associated with proliferation, migration and chemoresistance. Cancer Sci 2016; 107: 1898–1908. 2016/September/15 DOI: 10.1111/cas.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krzeslak A, Wojcik-Krowiranda K, Forma E, et al. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res 2012; 18: 721–728. 2012/January/25 DOI: 10.1007/s12253-012-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arjaans M, Schroder CP, Oosting SF, et al. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: from bench to bedside. Oncotarget 2016; 7: 21247–21258. 2016/January/21 DOI: 10.18632/oncotarget.6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee D and Zhao J. The Role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res 2013; 3: 46–57. 2013/January/30. [PMC free article] [PubMed] [Google Scholar]
- 35.Sun S, Liang X, Zhang X, et al. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. British journal of cancer 2015; 112: 1332–1339. 2015/April/14 DOI: 10.1038/bjc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Liu Y, Bailey C, et al. Therapeutic targeting of TP53-mutated acute myeloid leukemia by inhibiting HIF-1alpha with echinomycin. Oncogene 2020. 2020/February/16 DOI: 10.1038/s41388-020-1201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu G, Dustin D and Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Current opinion in pharmacology 2016; 31: 97–103. Review 2016/November/25 DOI: 10.1016/j.coph.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Bianchini G, Balko JM, Mayer IA, et al. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nature reviews Clinical oncology 2016; 13: 674–690. DOI: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Mahmood S, Sapiezynski J, Garbuzenko OB, et al. Metastatic and triple-negative breast cancer: challenges and treatment options. Drug Deliv Transl Res 2018; 8: 1483–1507. 2018/July/07 DOI: 10.1007/s13346-018-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999; 59: 5830–5835. 1999/December/03. [PubMed] [Google Scholar]
- 41.Muss HB, Blessing JA and Malfetano J. Echinomycin (NSC 526417) in squamous-cell carcinoma of the cervix. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 1990; 13: 191–193. 1990/June/01. [DOI] [PubMed] [Google Scholar]
- 42.Muss HB, Blessing JA, Baker VV, et al. Echinomycin (NSC 526417) in advanced ovarian cancer. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 1990; 13: 299–301. 1990/August/01. [DOI] [PubMed] [Google Scholar]
- 43.Taylor SA, Metch B, Balcerzak SP, et al. Phase II trial of echinomycin in advanced soft tissue sarcomas. A Southwest Oncology Group study. Invest New Drugs 1990; 8: 381–383. 1990/November/01. [DOI] [PubMed] [Google Scholar]
- 44.Muss HB, Blessing JA, Eddy GL, et al. Echinomycin (NSC 526417) in recurrent and metastatic squamous cell carcinoma of the cervix. A phase II trial of the Gynecologic Oncology Group (GOG). Invest New Drugs 1992; 10: 25–26. 1992/April/01. [DOI] [PubMed] [Google Scholar]
- 45.Muss HB, Blessing JA, Hanjani P, et al. Echinomycin (NSC 526417) in recurrent and metastatic nonsquamous cell carcinoma of the cervix. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 1992; 15: 363–364. 1992/August/01. [DOI] [PubMed] [Google Scholar]
- 46.Fahr A, van Hoogevest P, May S, et al. Transfer of lipophilic drugs between liposomal membranes and biological interfaces: consequences for drug delivery. Eur J Pharm Sci 2005; 26: 251–265. 2005/August/23 DOI: 10.1016/j.ejps.2005.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.