

SUMMARY
The majority of mosquito-borne illness is spread by a few mosquito species that have evolved to specialize in biting humans, yet the precise causes of this behavioral shift are poorly understood. We address this gap in the arboviral vector Aedes aegypti. We first collect and characterize the behavior of mosquitoes from 27 sites scattered across the species’ ancestral range in sub-Saharan Africa, revealing previously unrecognized variation in preference for human versus animal odor. We then use modeling to show that over 80% of this variation can be predicted by two ecological factors – dry season intensity and human population density. Finally we integrate this information with whole genome sequence data from 375 individual mosquitoes to identify a single underlying ancestry component linked to human preference, with genetic changes concentrated in a few chromosomal regions. Our findings suggest that human-biting in this important disease vector originally evolved as a by-product of breeding in human-stored water in areas where doing so provided the only means to survive the long, hot dry season. Our model also predicts that the rapid urbanization currently taking place in Africa will drive further mosquito evolution, causing a shift towards human-biting in many large cities by 2050.
eTOC
Rose et al. demonstrate that the evolution of human biting in Aedes aegypti mosquitoes across Africa is associated with long, hot dry seasons and recent increases in human population density. This behavioral shift has a shared genomic basis inside and outside Africa, with genetic changes concentrated in key chromosomal regions.
INTRODUCTION
Mosquitoes spread pathogens that make approximately 100 million people sick every year [1]. There are roughly 3,500 mosquito species worldwide [2], and the vast majority are generalists – they bite a variety of vertebrate animals with which they come into contact. Most cases of human disease, however, are caused by the bites of just a few species that specifically target us [3,4]. Understanding where and why mosquitoes evolve to specialize in biting humans is therefore critical for controlling and predicting disease spread.
Why might mosquitoes specialize in biting humans? Most researchers speculate that human-biting would have posed no particular advantage to mosquitoes before the development of agriculture and dense, sedentary human societies approximately 10,000 years ago [5–7]. After this time, abundant humans living together may have provided an easy and reliable resource. Genomic data is consistent with the idea that key human specialist taxa evolved within this time frame [8–10]. Nevertheless, it is not clear why mosquitoes would strongly prefer humans over the domestic animals that almost always accompany us in domestic environments unless behavioral, physiological, or morphological trade-offs exist between traits required for biting humans and those that allow efficient use of non-human animals.
Human-specialist mosquitoes don’t just bite humans; they also tend to breed in human habitats. Mosquitoes lay their eggs in water, and humans are unique among animals in the ways that we manipulate water, channeling it into ditches for irrigation and bringing it into our homes for drinking, cooking, and washing. Many authors have thus speculated that dependence on human sources of water for breeding, particularly in arid regions, may also play a role in mosquito specialization [6,7,11].
Aedes aegypti provides an excellent opportunity to investigate these possibilities. The globally invasive subspecies, Ae. aegypti aegypti, thrives in urban habitats across the American and Asian tropics, where its proclivity for biting humans makes it the primary vector of dengue, Zika, chikungunya, and yellow fever [12]. Host-seeking females take up to 95% of their blood meals from humans in nature [3]. This human-biting specialist is thought to have evolved from generalist ancestors in Africa 5,000–10,000 years ago, possibly in northern Senegal or Angola [8,13]. However, in at least a few places in East Africa, the contemporary African subspecies Ae. aegypti formosus remains a generalist [14]. Little is known about the host-seeking behavior of Ae. aegypti in other parts of Africa, and no work to date has explicitly examined the ultimate drivers of human-biting in mosquitoes. Here we use a combination of field collection, laboratory behavior assays, ecological modelling, and genome sequencing to infer the historical and contemporary evolutionary forces that shape mosquito preference for humans in this important disease vector.
RESULTS
We first set out to assemble a set of Ae. aegypti colonies representing diverse populations across the species native range in sub-Saharan Africa. We used ovitraps to collect mosquito eggs from multiple outdoor sites in each of 27 locations (Figure 1A–C, Table S1). The collections spanned a wide range of human population densities, with some egg traps placed among assemblages of plastic and concrete in large cities with over 2,000 people per square kilometer and others placed among trees and undergrowth in wild areas where mosquitoes rarely encounter humans (Figure 1A,C). They also spanned a wide range of climates, from highly seasonal, semi-arid habitats in the northwest to forest ecosystems with year-round rain in Central Africa (Figure 1B–C). We used Ae. aegypti eggs from independent traps to establish two replicate laboratory colonies for each of 23 populations, and a single colony for the remaining 4 populations (n=50 colonies total, Table S2).
Figure 1. Preference for human odor varies widely in Aedes aegypti mosquitoes across Africa.
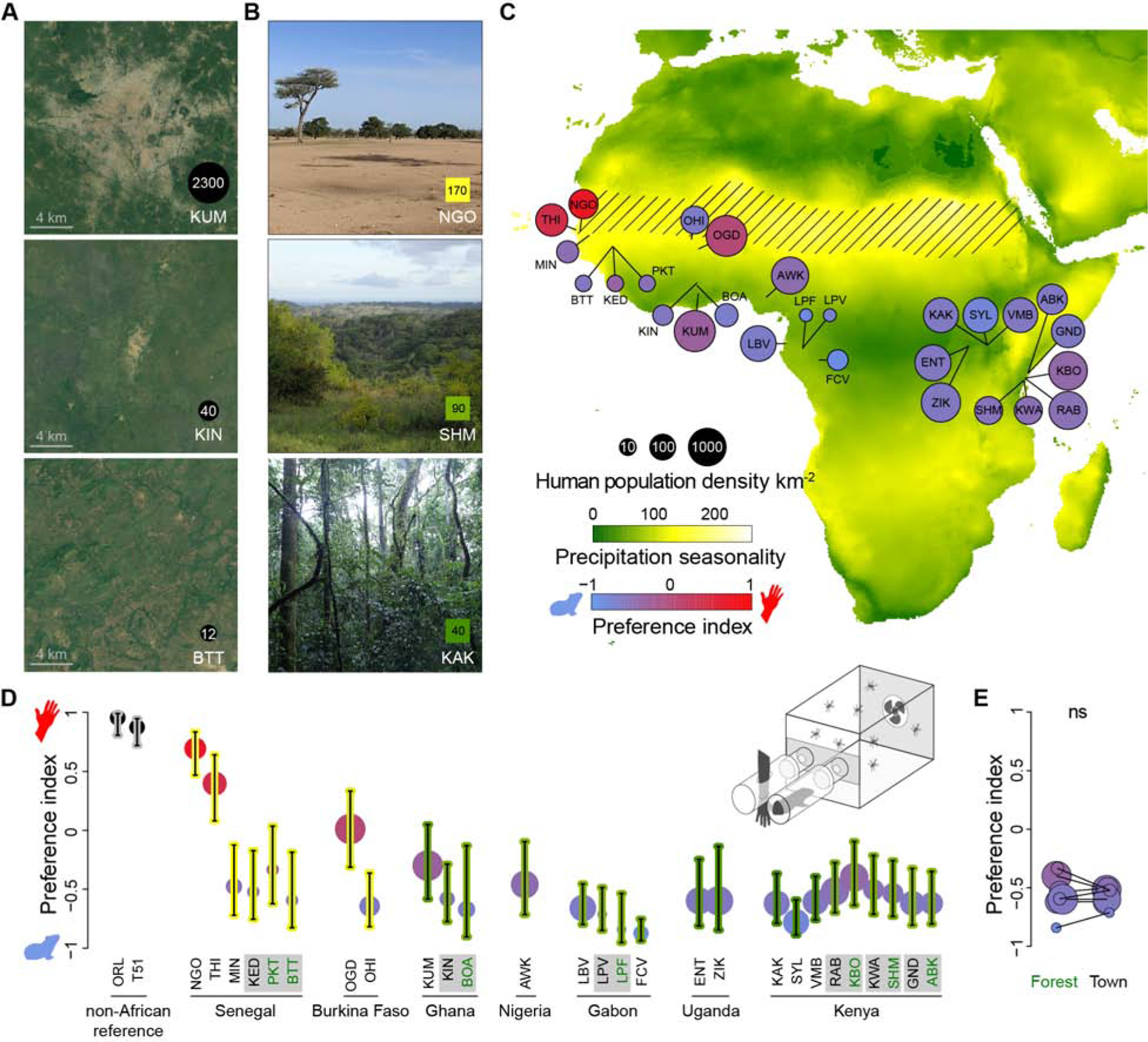
(A-B) Satellite images or photographs of mosquito collection localities with different human population densities (A) or levels of precipitation seasonality (B). Satellite images are from Google Earth, copyright Maxar Technologies and CNES/Airbus. (C) Map of collection localities. Diagonal hatched lines mark the Sahel ecoclimatic zone. Table S1 has full location names. (D) Host preference measured in a two-port olfactometer (inset) for all African localities plus a reference colony from Thailand (T51) and a lab colony most likely to have originated in the United States (ORL) (n=3–14 trials with 25–110 females per trial; details in Table S2). Bars indicate 95% confidence intervals. Circle sizes and bar colors show population density and precipitation seasonality, respectively, as in (C). Grey boxes around location names highlight adjacent forest-town pairs (forest in green text). (E) Females from adjacent forest and town habitats did not differ in preference (linear model P>0.05, ns=not significant). See also Figure S1A–F.
Preference for human odor varies widely across sub-Saharan Africa
Mosquitoes choose hosts based largely on body odor [4]. Ae. aegypti females from human-biting populations show a robust preference for human odor, while those from generalist populations tend to prefer the odor of non-human animals [15]. We tested the odor preference of colony females from each population in a two-port olfactometer (Figure 1D inset) and estimated preference using a beta-binomial mixed model that accounts for trial structure (Figure 1D, Figure S1A–B, Table S2, see STAR Methods). A single human and one of two guinea pigs provided the stimuli in most trials, but results were generalizable in follow-up tests with a different human and second animal species (Figure S1C).
Preference for humans varied significantly among sites (likelihood ratio test P<2.2×10−16), and the behavior of replicate colonies from the same site was strongly correlated (R2=0.60, linear model P=1.5×10−5, Figure S1D). Most populations preferred animals, but one population from Central Africa stood out as having an extreme animal preference (Franceville, Gabon; FCV), and three from West Africa showed either no preference (Ouagadougou, Burkina Faso; OGD) or clear human preference (Thies and Ngoye, Senegal; THI, NGO) (Figure 1D). As seen in previous work, overall response rates mirrored preference, with females from animal-preferring colonies being less likely to choose either host in the assay (Figure S1E–F) [15,16]. These females might respond more strongly to an untested animal species, but they are known to bite a wide variety of taxa in at least some locations [14]. Alternatively, animal-preferring mosquitoes may be less aggressive and/or motivated to seek hosts in enclosed spaces.
Preference variation is largely explained by two ecological factors
Variation in mosquito preference may be explained by local differences in human abundance. For example, we expected Ae. aegypti from towns to be more human-preferring than those from nearby forests. Such a pattern was previously documented in the Rabai region of Kenya where behaviorally divergent ‘domestic’ and ‘forest’ forms coexisted from at least the mid-1900s to early 2000s. However, the ‘domestic’ form from Rabai likely originated from a localized reintroduction of non-African Ae. aegypti aegypti mosquitoes rather than in situ evolution, and it could not be found when the field work for this study was carried out in 2017. We instead observed no effect of native habitat on behavior in a systematic comparison of forest and town mosquitoes across six paired sites, including one from Rabai (Figure 1E).
Gene flow might homogenize the behavior of mosquitoes in adjacent habitats despite divergent selection. We therefore took a broader perspective and asked whether human population density could explain preference variation on a regional scale. Linear modelling of behavior across all locations supported this prediction. The number of humans living within a 20–50km radius around collection localities was a strong predictor of preference (Figure 2A, likelihood ratio test all P≤0.002; compare grey and black lines in Figure S1G). This effect helped explain why mosquitoes from cities in Burkina Faso (Ouagadougou, OGD), Ghana (Kumasi, KUM), and Gabon (Libreville, LBV) were all more responsive to human odor, on average, than those from less populated areas of the same countries (Figure 1D).
Figure 2. Human population density and dry season intensity explain variation in preference.
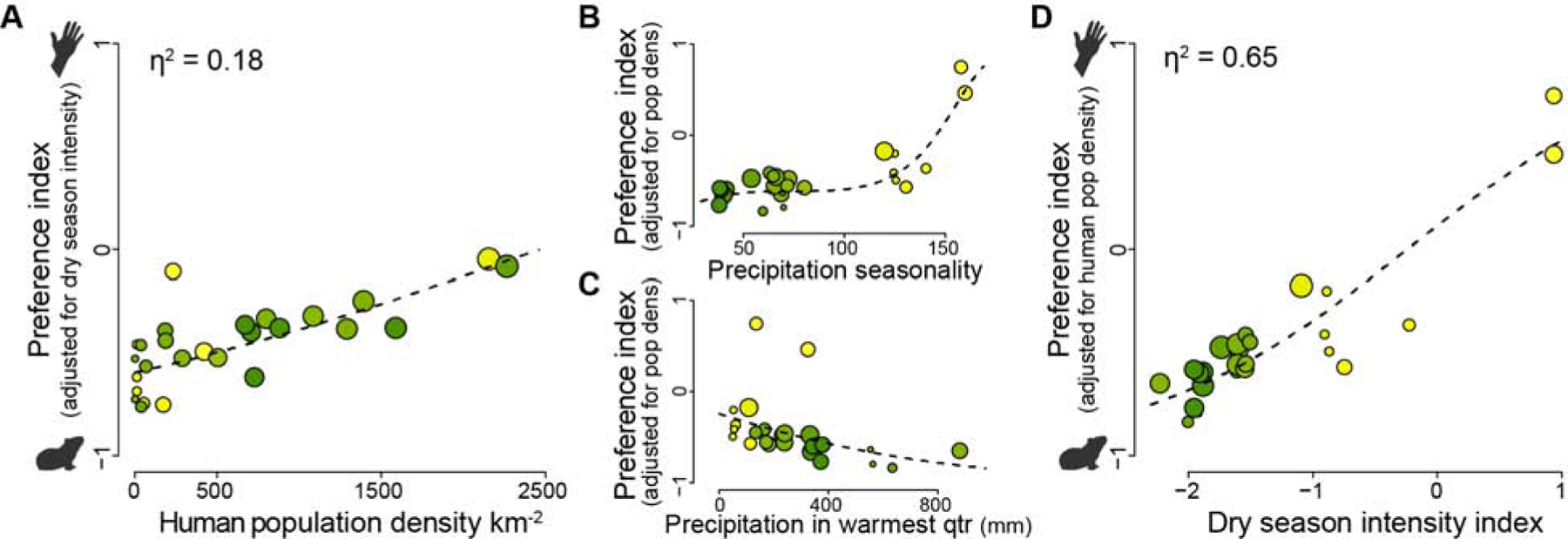
(A) Preference for humans increases significantly with human population density, which explains 18% of variation in the final model (linear fit, density calculated within 20km radius, likelihood ratio test [LRT] P=1.0×10−5). (B-D) Preference for humans increases in habitats with highly seasonal rainfall (B, cubic monotonic polynomial fit, LRT P=1.2×10−8) and decreases in habitats with more rain at the warmest time of year (C, linear fit, LRT P=0.016 for model that already includes seasonality). (D) Climate variables in (B-C) can be combined in an index of overall dry season intensity, which explains 65% of variation in mosquito preference in the final model (LRT P=3.0×10−9). All analyses were carried out with logit-transformed preference indices subsequently back-transformed for plotting. Point color and size correspond to precipitation seasonality (scale in Figure 1C), and human population density (proportional to scale in Figure 1C), respectively. See also Figure S1G–I.
The population density model included latitude and longitude as covariates to control for clear geographic trends in the data (higher preference for humans in the northwest, Figure 1C). We wondered whether climate might explain some of this additional variation. Remarkably, when we used stepwise model selection to replace latitude and longitude with ecologically relevant climate variables (Bio1-Bio19 from the WorldClim 2 dataset; Figure S1H–I; see STAR Methods) [17], the best climate variables explained much more behavioral variation than human population density itself. In the final model, human population density explained 18% of variation (Figure 2A, likelihood ratio test P=1.0×10−5, density measured within 20 km radius). The strongest climate predictor was precipitation seasonality (Figure S1H), a measure of how variable rainfall is from month to month. A third-degree polynomial provided the best fit (Figure 2B, likelihood ratio test P=1.2×10−8), helping to explain the abrupt emergence of preference for humans in the Sahel ecoclimatic zone of West Africa, where it is dry for up to 9 months of the year and all rainfall comes during a short, intense rainy season (Figure 1C). A second climate variable, level of precipitation during the warmest quarter of the year, also contributed significantly to our model (Figure 2C, Figure S1I, likelihood ratio test P=0.014) and helped explain behavior across populations in the animal-preferring range. Mosquitoes were more attracted to human odor in places with less rain at the hottest time of year.
Taken together, these two climate variables capture the challenges mosquitoes face during the dry season. Ae. aegypti lay their eggs on wet substrate just above the water line in tree holes, rock pools, or artificial containers [18]. If the eggs remain wet, they can hatch immediately. However, eggs laid in wild areas at the end of the rains must pause development and survive the duration of the dry season until rain returns – a particularly difficult challenge when the dry season is long (i.e. precipitation seasonality is high) and hot (i.e. precipitation is low at the warmest time of year) [18,19]. Human water storage helps Ae. aegypti in harsh environments by providing a year-round aquatic niche for larval development. We put the two climate variables together into a single index of dry season intensity that explains 65% of variation in host odor preference across Africa (Figure 2D, likelihood ratio test P=2.3×10−9). These findings point to long, hot dry seasons as the key selective factor driving Ae. aegypti specialization on human hosts, likely as a by-product of dependence on human-stored water for breeding [7,11,20].
Preference for humans within and outside Africa has a single genomic origin
Females of the globally invasive human specialist subspecies are characterized by light scaling on the back of the abdomen (first tergite, Figure 3A inset) [21,22], and previous work documented this trait in the Sahel of northern Senegal where we observed preference for humans [20,23]. We therefore wondered whether it might be linked to behavior in a continuous way across our full sample set. Indeed, abdominal scaling was strongly correlated with preference for humans across Africa (R2=0.78, linear model P=1.3×10−8, Figure 3A,D). The trend was driven not only by the most extreme phenotypic variation in Senegal but also by modest variation in other regions (R2=0.46, linear model P=0.002, Senegal excluded).
Figure 3. Specialization on humans has a single genomic origin.
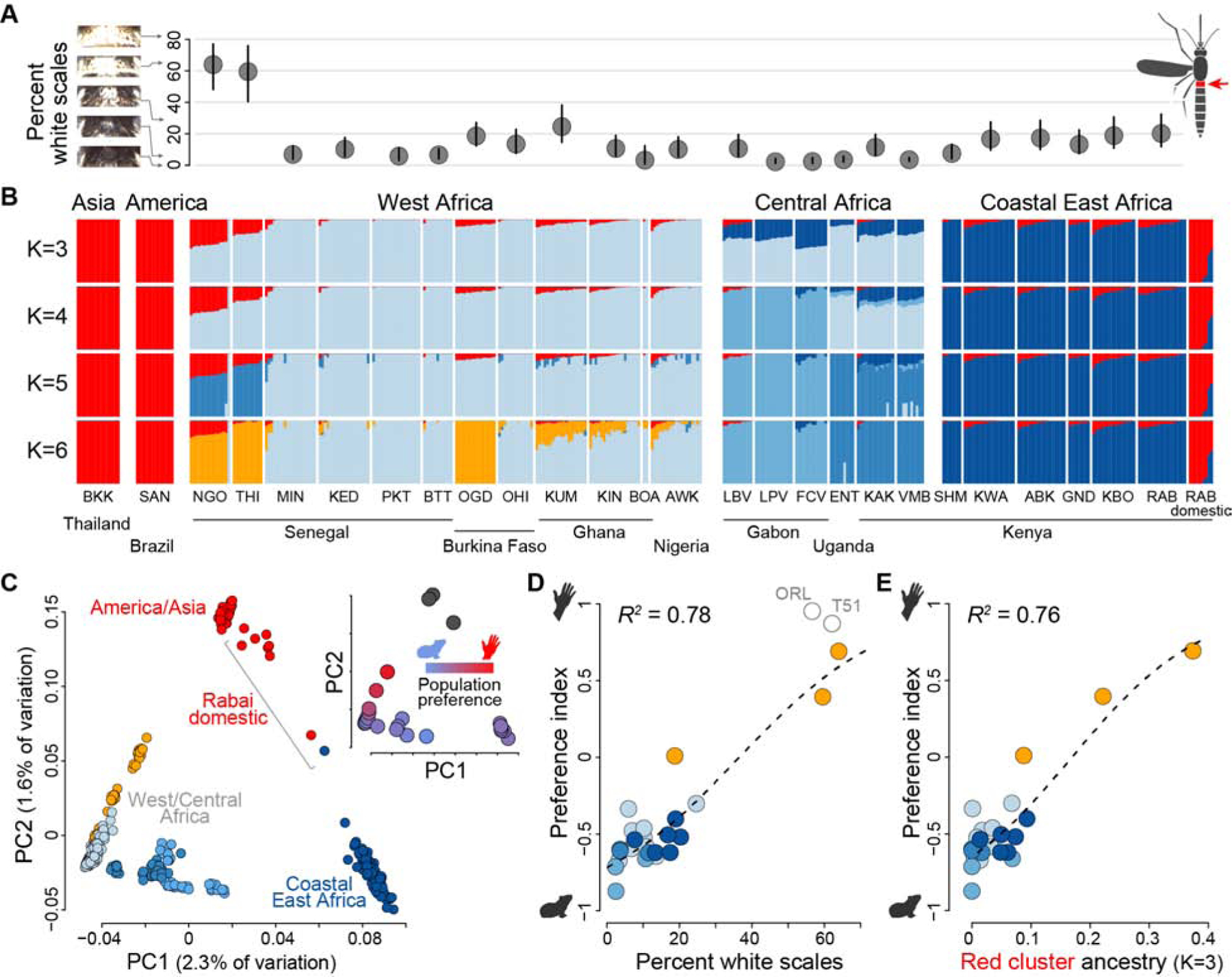
(A) Percentage of the first abdominal tergite of adult females (red in inset, right) covered in white scales. Dots and lines show mean and 95% confidence intervals, respectively, for n=7–29 females from the populations listed in panel (B). (B) ADMIXTURE analysis of population structure. Bar plots show ancestry proportions for 375 mosquitoes when specifying K=3–6 ancestry components. K=3 minimized cross-validation error. (C) Principal components analysis of population structure. Points in the main plot represent individuals and are colored according to primary ancestry assignment from the K=6 ADMIXTURE analysis in (B). Points in the inset represent population means and are colored according to host preference. (D-E) Preference for humans is tightly correlated with both population mean abdominal scaling (D, linear model P=1.3×10−8) and mean proportion ancestry assigned to the red component in the K=3 ADMIXTURE analysis (E, linear model P=2.7×10−8). Point colors are as in the main plot of (C), except they represent primary ancestry assignment for each population rather than individual. Open grey circles in (D) show non-African reference populations (not included in correlation analysis).
The morphological resemblance of human-preferring mosquitoes within and outside Africa suggests shared ancestry. To test this hypothesis, we sequenced the genomes of ~15 field-collected individuals (see STAR methods) from 24 sites in our current study plus one site in South America and one in Asia (n=366 genomes after exclusion of relatives, ~15x coverage). We also sequenced 9 previously collected individuals of the human-biting domestic form from Rabai [15]. Analyses of overall population structure were consistent with earlier work [13,24]. ADMIXTURE [25] and principal components analysis (PCA) revealed strong support for a model with three genomic clusters or ancestry components corresponding to coastal East Africa, West/Central Africa, and globally invasive human specialists (Figure 3B–C). The Rabai (RAB) domestic form was the only African population to group unambiguously with non-African human specialists, consistent with its putative origin as a localized reintroduction of the global subspecies [24]. However, many populations across sub-Saharan Africa showed some level of ancestry from the human specialist component (red in Figure 3B, PC2 in Figure 3C), and like abdominal scaling, this signal was strongly correlated with preference for humans (Figure 3E, R2=0.76, linear model P=2.7×10−8; Figure 3D inset).
The shared ancestry of human-preferring mosquitoes within and outside Africa has two potential explanations – contemporary admixture due to back-to-Africa gene flow or ancestral population structure present before the species left Africa. Admixture has almost certainly occurred in Kenya, where the reintroduced domestic form once thrived. However, a recent exome study suggested that a supposedly highly ‘admixed’ population from the West African Sahel may instead be ancestral to bottlenecked, non-African populations [8]. Consistent with this interpretation, the three most human-seeking Sahelian populations in our dataset (NGO, THI, OGD) formed a unified genomic cluster, distinct from both the globally invasive subspecies and nearby animal-preferring populations, in an ADMIXTURE analysis with six clusters (orange in Figure 3B, K=6).
Loci associated with specialization are clustered in the genome
Mosquito preference for human odor likely has a complex genetic basis. Moreover, its strong association with a single human specialist ancestry component suggests that the underlying causal variants covary across populations with those that regulate other traits important for survival and reproduction in human environments [7,22]. To identify candidate loci and genomic regions underlying adaptation to humans, we used the program PCAdapt to look for single nucleotide variants that were more strongly associated with human specialist ancestry than would be expected under genetic drift alone [26]. Significant variants were scattered across the entire genome (Figure 4A–B, n=16,782 SNPs at Bonferroni-adjusted P<0.05), but particularly concentrated in a few genomic regions (grey shading in Figure 4B, permutation false discovery rate<0.001). These included a large area at the distal end of the first chromosome containing an odorant receptor (Or4) previously linked to preference for humans [15]. This example reinforces the idea that outlier regions harbor loci important for specialization on humans. We emphasize, however, that the pattern almost certainly extends beyond a single locus or behavior. Each outlier region likely contains multiple variants that regulate diverse human-adaptive traits. The highest peak in the chromosome 1 outlier region, for example, falls not in Or4, but near an unannotated gene 10 Mb upstream (AAEL019513; see Table S3 for full list of genes containing outlier SNPs).
Figure 4. Specialization on humans is associated with changes concentrated in a few key chromosomal regions.

(A-B) Results from a PCAdapt scan for genomic variants associated with human specialist ancestry in African genomes. (A) Preference for humans was tightly correlated with the mean population value of the second principal component (PC2) from a principal components analysis, indicating that this component captures genomic variation associated with specialization (linear model P=6.1×10−11). See Figure 3C inset for a similar pattern in a PCA that also included non-African genomes. (B) Thousands of SNPs were more strongly associated with PC2 than expected under neutral evolution (n=16,782 SNPs in red, Bonferroni-adjusted P<0.05), suggesting positive selection. Blue line shows the percentage of SNPs that were outliers in a sliding 10 Mb window. Grey shading indicates >=10 Mb regions where significant SNPs were concentrated (false discovery rate<0.001). Points with unadjusted P>0.01 not plotted. Grey triangle marks the position of Or4, an odorant receptor previously linked to preference for human odor [15]. (C) Polarized genomic divergence (Population Branch Statistic, PBS, 10 Mb sliding window) along the lineages leading to three human-seeking populations (top; NGO, THI, OGD) and two animal-seeking populations (bottom; KED, OHI) from the Sahel region. (D) Proportion of derived variation (fD, 10 Mb sliding window) shared by Sahelian human-seeking populations (NGO, THI, OGD) with non-African human specialists from Bangkok, Thailand (BKK). See also Figure S2, Figure S3, and Table S3.
The outlier regions identified above come from a holistic analysis of genomic variation associated with human specialist ancestry across Africa. We can also look more narrowly at patterns of genomic variation in the most strongly human-seeking populations from the Sahel (Figure 1C). Here we expect selection to drive and maintain divergence at human-adaptive loci despite gene flow from nearby animal-seeking mosquitoes. Outlier regions should therefore show enhanced divergence from other African populations and increased sharing of derived alleles with non-African human specialists. Our data support both predictions. Using the Population Branch Statistic (PBS) [27], we confirmed that divergence along the branches leading to human-seeking mosquitoes from Ngoye (NGO), Thies (THI), and Ouagadougou (OGD) was greatest in outlier regions (Figure 4C, Fisher’s exact test, all P<2.2×10−16). In contrast, branches leading to two animal-seeking populations showed uniform divergence across the genome (Figure 4C, blue traces). Using the fD statistic [28], we also found that human-seeking populations were more likely to share derived alleles with non-African populations in outlier regions (Figure 4D; Fisher’s exact test, all P<2.2×10−16). Separate analyses of absolute differentiation between populations (dxy), diversity within populations (π), and the site frequency spectrum (Tajima’s D, and Fay and Wu’s H) were also consistent with the idea that PCAdapt outlier regions have been affected by strong selection during the establishment of human specialist ecology, followed by the accumulation of sequence differences via subsequent selection against gene flow (Figure S2) [29–31].
The conflict between selection and gene flow should be greatest at the transition zone where behaviorally divergent populations live in close proximity. Animal-preferring mosquitoes in Mindin, Senegal (MIN), for example, live just 100 km from human-preferring mosquitoes in Ngoye (NGO) (Figure 1C, Figure S3A). Interestingly, mosquitoes from Mindin exhibited a pattern of genomic variation in the prominent chromosome 1 outlier region that was discretely different from that seen in animal-preferring populations located farther from the transition zone. They showed elevated divergence in this region from both human-preferring mosquitoes (Figure S3B) and other animal-preferring mosquitoes (Figure S3C). They also showed reduced nucleotide diversity (Figure S3E), and decreased values of both Tajima’s D and Fay and Wu’s H (Figure S3F–G). These patterns suggest a recent sweep, possibly driven by selection for maintenance of animal preference in the face of gene flow from nearby human-biting populations. Moreover, the discrete plateau of altered population genetic statistics on a genomic backdrop that is otherwise typical of animal-preferring populations could potentially be explained by a chromosomal inversion specific to the ecological transition zone. Long-range sequencing and higher-resolution sampling across the Sahel are needed to test this hypothesis. Previous work provided evidence for inversions in Aedes aegypti [32–34], but not in exactly this position.
Rapid urbanization may drive a shift towards human biting by 2050
Both climate and human population density are changing rapidly in Africa [35,36]. We therefore incorporated publicly available climate and human population projections into our model to explore how the behavior of African Ae. aegypti mosquitoes might be expected to evolve over the next 30–50 years (see STAR Methods). Projected changes in relevant precipitation variables are modest and unlikely to drive substantial shifts in preference (Figure 5A, Figure S4). Rapid urbanization, in contrast, is expected to result in dramatic increases in human population density, and may trigger transitions to human biting in many cities across the continent by 2050 (Figure 5, Figure S4). As noted below, this prediction comes with the important caveat that the highest projected population densities are well above those used to fit our model.
Figure 5. Rapid urbanization may favor a shift towards human-biting in large African cities by 2050.
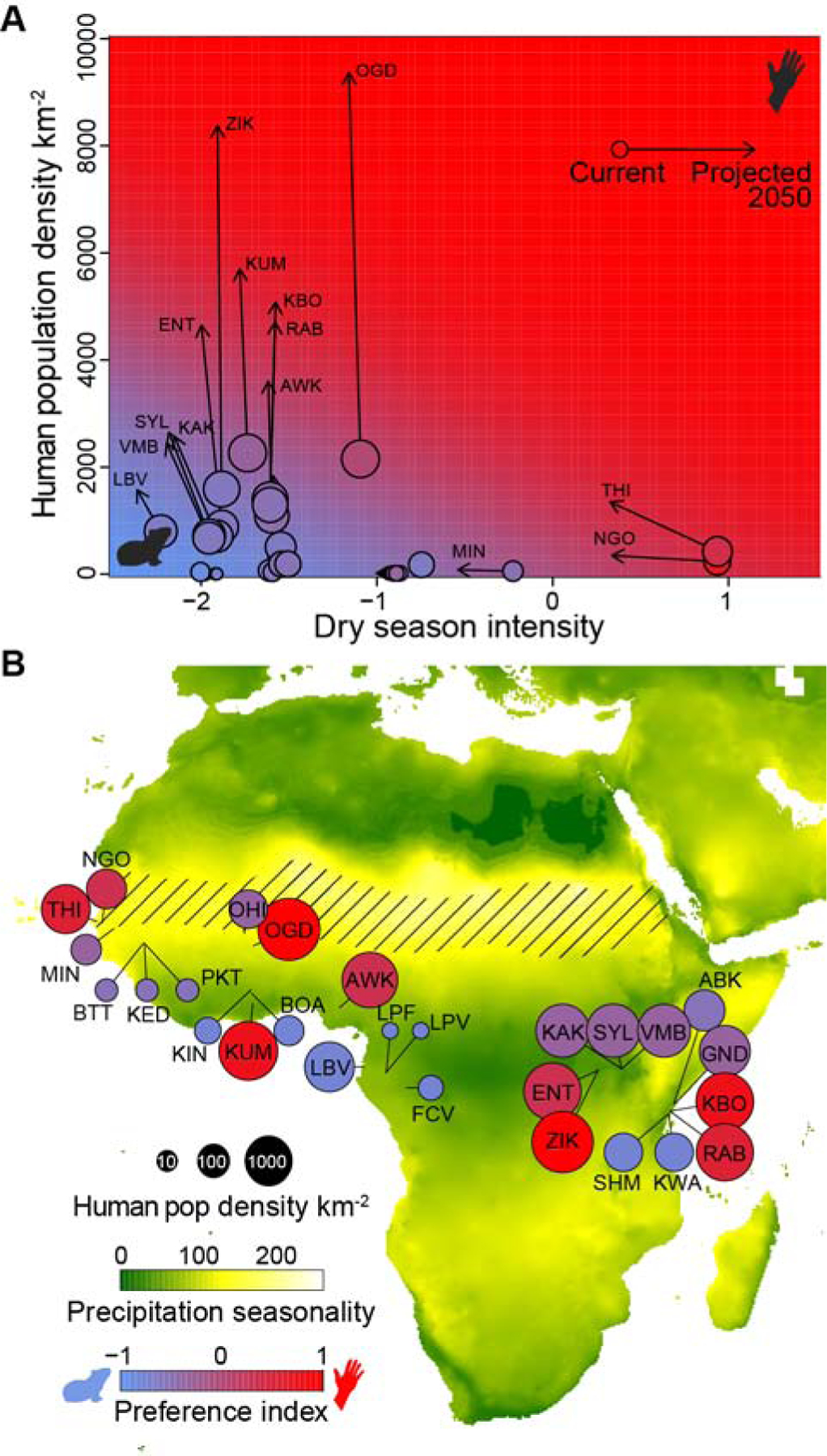
(A) Current (circles) and projected (arrows) dry season intensity and human population density for each site. Circle colors indicate current host preference. Plot background color indicates the host preference predicted by the model described in Figure 2 for the given combination of environmental parameters. (B) Map from Figure 1C showing projected (rather than current) human population densities (circle size), precipitation seasonality (map background color), and host odor preference (circle color) for the year 2050. See also Figure S4.
DISCUSSION
In this study, we used field collections, colony generation, and laboratory behavioral tests to show that preference for human odor varies widely in Ae. aegypti mosquitoes across sub-Saharan Africa (Figure 1). A remarkable 83% of this geographic variation can be explained by two ecological factors – dry season intensity and regional human population density (Figure 2). Genome sequencing further showed that preference variation is tightly correlated with levels of ancestry from a single human specialist ancestry component (Figure 3), involving shared derived alleles concentrated in a few key chromosomal regions (Figure 4). Application of our ecological model to projected future conditions suggested that near-term climate change will not drive selection for major changes in mosquitoes preference, but that rapid urbanization may drive a shift to human-biting in many cities across sub-Saharan Africa by 2050 (Figure 5).
The strong association between preference for humans and dry season intensity provides the first concrete support for speculation that human specialization in Ae. aegypti was driven by reliance on human-stored water for breeding [7,11]. Laying eggs in human-stored water may be the only way for Ae. aegypti to survive a long, hot dry season. Once dependent on humans for breeding sites, mosquitoes may evolve preference for humans as a form of habitat fidelity, or due to trade-offs between traits that promote effective use of locally abundant human targets and those necessary for finding and biting animals [37]. There are other indications that human biting in Ae. aegypti is intertwined with the ability to breed at dry times of year. A decades-old study found that globally invasive specialists from Asia and the Americas, as well as a single population from the African Sahel, were all more resistant to desiccation than strains from other parts of Africa [38]. Moreover, human-biting is accompanied by a genetically based preference for laying eggs in human water-storage vessels in at least some areas [7].
While the hypothesis outlined above is compelling, we cannot rule out other selective links between climate and the evolution of human-biting. For example, the sparsity of natural breeding sites (trees) and alternative hosts (wild animals) in highly seasonal environments may contribute to reliance on humans even during the rainy season. It would also be helpful to further refine our model of the underlying climate-preference association by characterizing additional Ae. aegypti populations. The link between behavior and precipitation variables was present across our entire sample (Figure 2C), but the abrupt emergence of strong human preference in the most extreme seasonal environments was driven largely by two populations in Senegal (Figure 2B). Sampling across the central and eastern Sahel would help test this feature of the model. We expect human-preferring populations to extend into these areas unless selective barriers associated with human cultural variation (e.g. nomadism or lack of consistent water storage) prevent establishment. Our model does not predict strong human preference in any non-urban area of southern Africa, but this would also be worth investigating.
Beyond climate, human population density explained an additional 18% of contemporary variation in mosquito preference for humans across Africa, presumably because selection favors use of locally abundant hosts. However, the effect involved population densities (thousands of people per square kilometer, see Figure 2A) that almost certainly did not exist during initial specialization on humans thousands of years ago [39]. Instead, human density effects on mosquito host preference are likely to be a more recent phenomenon associated with urbanization, a process that continues to accelerate across sub-Saharan Africa. Indeed, our model predicts that the rapid growth of many African cities will drive a further shift toward human-biting over the coming decades (Figure 5). There are several important caveats to this prediction. First, future population densities are expected to exceed those observed in this study, and thus used to generate our model (compare arrowheads to circles along the y-axis of Figure 5A). Extrapolation of the linear trend seen across contemporary sites (Figure 2A) suggests mosquitoes in these large cities will evolve strong preference for humans. Alternatively, the effect of human density could plateau at intermediate preference (willingness to bite humans) without driving the true specialization we see in highly seasonal environments. In addition, limits to gene flow (e.g. across the Congolian basin or the Great Rift Valley) could prevent the establishment of human specialists in some eastern cities where our model suggests they will be favored. Despite these ambiguities, the speed and scale of ongoing urbanization argue strongly for careful monitoring of potential shifts in Ae. aegypti behavior (or correlated morphology/genetics) across Africa.
Our genomic data are consistent with the hypothesis that human specialization is not only favored in the seasonal Sahel, but also first arose there before seeding globally invasive populations [7,8,11]. While climates have fluctuated substantially over the millenia, a zone similar to the Sahel, on the southern edge of a shifting Saharan Desert, has likely existed in some form for the past 5,000 years [40]. Further work is needed to determine exactly where and when human specialists arose, incorporating genome-wide data from a wider range of global populations. Ae. aegypti from Angola [13] and northern Argentina [24], for example, show similar patterns of ancestry to populations in the Sahel, and present day distributions may not reflect those at the time when human specialization first occurred.
Regardless of origins, specialization on humans in this mosquito clearly involves shifts in the frequency of a large number of variants, scattered throughout the genome but particularly concentrated in a few key regions. These regions contain large numbers of genes, many of which likely contribute to adaptation to human hosts and habitats. More broadly, the tight correlations between ancestry, behavior, and environment reveal a dynamic situation playing out across the continent as a whole, with selection and gene flow fine-tuning the frequency of human-adaptive alleles, and thus levels of attraction to human hosts, according to local climate and human population density. We urgently need to incorporate such environmentally-structured variation into epidemiological models and other efforts to predict and manage the transmission of Ae. aegypti-borne disease in Africa.
STAR METHODS:
RESOURCE AVAILABILITY
Lead Contact
Correspondence and requests for materials should be addressed to Lead Contact, Carolyn McBride (csm7@princeton.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Raw genomic data is available in the NCBI SRA under the accession code PRJNA602495 (https://www.ncbi.nlm.nih.gov/sra/PRJNA602495). Other raw data and scripts are available at github.com/noahrose.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics and regulatory information
Mosquito eggs were collected and exported with permission from local institutions and/or governments as required (Kenya SERU No. 3433; Uganda permit 2014-12-134; Gabon AR0013/16/MESRS/CENAREST/CG/CST/CSAR and AE16008/PR/ANPN/SE/CS/AEPN) and imported to the USA under USDA permit 129920. The use of non-human animals in olfactometer trials was approved and monitored by the Princeton University Institutional Animal Care and Use Committee (protocols 1998–17 and 2113–17). The participation of humans in olfactometer trials was approved and monitored by the Princeton University Institutional Review Board (protocol 8170). All human subjects gave their informed consent to participate in work carried out at Princeton University. Human-blood feeding conducted for colony maintenance did not meet the definition of human subjects research, as determined by the Princeton University IRB (Non Human-Subjects Research Determination 6870).
Field collections
We collected mosquito eggs in each African sampling location by distributing 20–60 ‘ovitraps’ at regular intervals across the landscape. Ovitraps consisted of 32 oz black plastic cups (The Executive Advertising), each lined with a 38 × 15 cm piece of 76 lb (34.5 kg) seed germination paper (Anchor Paper Co.) and filled with 3–8 cm of water. In all locations except coastal Kenya, the water was infused with a mixture of fresh or dry mango leaves collected from the leaf litter (n=~20 leaves per 10 liters of water) for 1–2 days before use. In coastal Kenya and Uganda, we used tap water or made a similar infusion with twigs, bark, and leaves from unidentified broad-leafed trees. Anecdotally, water source did not appear to affect egg numbers. Each ovitrap had a hole in the side at a height of ~3 inches to allow rainwater to drain from the trap. We attempted to spread ovitraps at approximately 100 meter intervals, but placed them at intervals as small as 10 meters in areas with limited access. We left ovitraps in the field for two nights before returning to collect egg-impregnated seed papers. We then dried the papers slowly on beds of paper towels over the course of 24 hours and stored them in airtight, whirl-pak bags during transport back to the laboratory. The only exception to this approach was at Bantata, Senegal (BTT), where we collected Saba senegalensis husks from the forest floor, flooded them with water, and collected hatchling larvae over the following few days. Most collections were carried out in 2017 and 2018, but Ugandan collections were carried out in 2015.
Generation and maintenance of laboratory colonies
We hatched egg-impregnated papers from each ovitrap in separate pans of hatch broth, made by dissolving finely ground Tetramin Tropical Tablets fish food (Spectrum Brands, Inc.) in deoxygenated water (¼ tablet/liter). We continued to feed larvae Tetramin ad libitum through to pupation, and transferred pupae from each seed paper to separate 32 oz HDPE plastic cages (VWR). Eclosing males and females were able to mate with each other in the cages and had access to 10% sucrose solution. Other mosquito species sometimes hatched from papers alongside Ae. aegypti and were eventually removed from cages without hindering our breeding efforts. However, Ae. albopictus males are known to satyrize Ae. aegypti females, rendering them infertile [41]. In areas where Ae. albopictus was present (Nigeria and Gabon), we therefore separated the male and female pupae reared from any given seed paper and let them eclose separately before identifying adults to species and recombining Ae. aegypti males and females only. We set aside 2–20 g0 (i.e. raised from field-collected eggs) adults from each population for genome sequencing, using only a single individual per ovitrap/cage where possible to reduce the probability of sequencing siblings (n=10–20 individuals for each of 20 locations; n=2–9 for each of 4 additional locations).
We used mosquitoes from independent ovitraps to establish two replicate laboratory colonies for 23 locations and a single colony for the remaining 4 locations (Table S2). Each colony was founded using eggs from 4–43 females, except the Lope Forest colony which was founded with eggs from a single female (Table S2). Founding females were fed on human volunteers (see ethics subsection) and allowed to lay eggs individually on wet filter paper cones (Whatman 55 mm Grade 1 filter paper) in small shell vials (Applied Scientific Drosophila Vials, 28.5 mm diameter, 95 mm height). We gave females multiple opportunities to feed in order to ensure high feeding rates (typically >90%) and thus reduce the potential for selection on host preference. However, it was sometimes difficult to coax recalcitrant females to lay eggs in the lab. Oviposition rates ranged from 35 to 100% in the first generation. In subsequent generations, we maintained population sizes of 300–600 individuals per colony, continued to ensure blood-feeding rates >90%, and tried to maximize oviposition by forcing females into contact with wet, potting soil-infused, seed germination paper cones in small 8.5 oz HDPE plastic cups (VWR) for 2 days (30 females/cup). Eggs were dried and stored at 16°C, 80%RH for up to 6 months between generations. The only exception to these breeding procedures applied to the first 2 generations of the colony from Zika, Uganda (ZIK), which was fed on a membrane and laid eggs on cups of water placed inside a large breeding cage.
We included two reference colonies of non-African origin in behavioral and morphological studies (Table S2). These were a colony from Thailand (T51) generated as described above and a laboratory colony originally maintained at the USDA labs in Orlando, Florida (ORL) that is of uncertain origin but was most likely supplemented for decades with local Floridian mosquitoes [42].
METHOD DETAILS
Behavior
We tested the host odor preference of 7–14 day old females that had been housed over night with access to water only (no sucrose). Different colonies were hatched on the same day, females mated freely with males after eclosion, and females were matched for age on testing days. We used a two-port olfactometer as previously described (Figure 1D inset) [15,16], with small modifications. Instead of using a large box fan to pull air through the device from the back of the olfactometer, we used a smaller fan to pull air through an 10.2 × 10.2 cm opening in the back panel. Instead of pulling air from the room, carbon-filtered, conditioned air was supplied to the two olfactometer ports from an independent building source. Inflow and outflow was balanced to achieve a rate of approximately 0.3m/s as measured at the traps. In each trial, 25–110 females were allowed to acclimate for 5 minutes in the large holding chamber before turning on the fan and opening a sliding door to expose them to streams of air coming from two alternative cylindrical traps and host chambers. One host chamber contained an awake guinea pig (Cavia porcellus, pigmented breed) or button quail (Coturnix coturnix). The other contained a section of the arm of a human volunteer (middle of forearm to middle of upper arm; silicone sheeting used to seal the holes through which the arm was inserted). The breath of the animal mixed with its odor in the animal odor stream. To add human breath on the human side, we asked the human subject to breathe gently through a nasal mask into the host chamber every 30 seconds. Trials lasted 10 minutes, and mosquitoes choosing to fly upwind towards either host odor stream during this time were trapped in small ports and counted at the end.
We carried out host preference trials in two main waves, with a 28-year old, European-American male serving as the human subject and one of two female guinea pigs serving as the animal subject. In the first wave, we tested second-generation colonies from Kenya and Gabon. In the second wave, we tested second generation colonies from Nigeria, Ghana, Burkina Faso, and Senegal, eighth- or ninth-generation colonies from Uganda, and two reference colonies of non-African origin. We also repeat tested a representative set of first wave Kenyan and Gabonese colonies (RAB, VMB, FCV, LBV; by then in their fourth generation) in the second wave to ensure that results were comparable between waves. At least one of two colonies from every population included in a given wave was tested on every experimental day in order to balance random day-to-day variation with the population effects we were trying to estimate (Figure S1A–B). Overall, we carried out 3–4 trials for each colony, except one of two replicate colonies from BTT, KED, and KUM, which were tested only twice. This resulted in a total of 7 trials for most populations, 3–6 trials for the six populations represented by only a single colony or for which one of two colonies had fewer trials, and 14 trials for the four populations tested in both waves. In total, we carried out 206 trials including 17,856 female mosquitoes, of which 7,385 responded to one or the other host odor.
After the two main waves, we carried out a smaller set of trials with one colony from each of four representative African populations (FCV, OGD, AWK, NGO) and a wider array of host comparisons. In one set of trials, we substituted a 22-year old Nigerian-American female for the original 28-year old European-American male, and in another set we substituted a button quail for the guinea pig (Figure S1C, n=3–5 trials per colony x host combination).
Whole genome resequencing and variant calling
As part of an ongoing 1200 Aedes aegypti genomes project, we extracted gDNA from 480 field-collected mosquitoes using the Chemagic DNA tissue protocol and sequenced them to 15x coverage with PE 151bp reads using the Illumina HiSeqX platform. The sequenced mosquitoes included 397 individuals from 24 sub-Saharan African populations collected for this study, 29 additional individuals from sites in Uganda, Kenya, and Burkina Faso that were not included in the main study, 12 individuals of the domestic form collected in 2009 or 2011 in Rabai, Kenya [15], 20 individuals from Bangkok, Thailand, 18 individuals from Santarem, Brazil, and 4 Ae. mascarensis mosquitoes for use as an outgroup.
We initially mapped all sequence data to the L5 reference genome [43]. We identified and removed close relatives from our sample as follows. First, we generated a matrix of relatedness coefficients using the --relatedness2 subprogram from VCFtools [44] with a set of randomly selected 109,267 biallelic SNPs (MAF>0.05, >1 read in 90% of individuals) preliminarily called with bcftools. Second, we hierarchically clustered the coefficients using the R function hclust (method=‘average’). Third, we grouped close relatives using the R function cutree, with a relatedness cutoff of 0.05 for African samples (corresponding to first cousin or closer relationships) and 0.2 for non-African populations (corresponding to siblings). The more permissive cutoff was used for non-African populations because they are more inbred/bottlenecked, with many individuals showing cousin-like relationships. Finally, we removed all but one randomly-chosen individual from each group of relatives. This left us with 345 sub-Saharan African Ae. aegypti genomes from our 24 focal study sites, 14 Ae. aegypti from other sites in sub-Saharan Africa, 30 Ae. aegypti genomes from outside continental Africa and 4 Ae. mascarensis genomes. Most relatives came from the same ovitrap (we sequenced more than one individual from a single ovitrap when ovitrap/egg limited). A smaller number came from nearby ovitraps in the same general location. The 14 Ae. aegypti genomes from non-focal sites were used for variant discovery and included in ADMIXTURE and PCA-based analyses (see below) in order to ensure we were sampling as much diversity as possible, but they are not plotted in figures.
We then used three iterative mapping steps to construct an updated African reference based on data from a geographically distributed (Africa only) set of 100 unrelated male mosquitoes (Figure S5). We chose to use males for the update because the L5 reference was constructed using data from males. In each of three iterative mapping steps, we (1) mapped sequence data from the mapping set to the reference using bwa mem (MAPQ cutoff of 10), (2) called consensus biallelic SNP genotypes using bcftools (“bcftools mpileup −BI | bcftools call −vmOu | bcftools view −v snps -q 0.5:alt1 | bcftools norm −Ou -m - | bcftools norm −Oz −d snps”), and (3) substituted the consensus base into our reference sequence using bcftools consensus [45,46]. We used PicardTools [47] to characterize read mapping quality after each iteration on a set of individuals not used for alternate reference construction (20 individuals; male-female pairs from 10 African populations) (Figure S5). We used a permissive MAPQ cutoff of 10 for the mapping steps because analyses suggested that high levels of sequence divergence from the L5 reference were disrupting initial alignments (Figure S5A–D). Finally, we remapped data from all 480 genomes to the updated third-iteration reference; this included non-African and outgroup samples, which also mapped well to the updated reference (Figure S5E–F). Males and females mapped similarly, except in the region around the sex-determining M-locus (Figure S5G–H). After remapping, we realigned reads near insertions and deletions to improve variant discovery in these regions using GATK IndelRealigner [48].
We took two different approaches to variant calling, both of which were confined to regions of the genome we inferred to be non-repetitive (repeat masked using the RepeatMasker intervals from the L5 genome) and single copy (mean coverage between 5–30X across individuals). We used the program ANGSD [49] to calculate population-level allele frequencies and genetic diversity, as well as to carry out genotype-likelihood based analyses (see below) for 161,713,099 biallelic single nucleotide polymorphisms (SNPs, P<10−6). We also called individual genotypes for a filtered set of 14,045,728 high quality biallelic SNPs using bcftools. These SNPs were filtered for coverage across the entire sample (covered by at least 1 read in 90% of individuals) and then called for any individual with sample depth > 8 reads and genotype quality score >30. After individual genotype calling we implemented a further filter for the fraction of individuals genotyped (>75% at any given SNP) and minor allele frequency (MAF>1%). We used the same permissive MAPQ cutoff of 10 for variant calling as used for generating the updated reference genome (see above) in order to minimize potential problems with aligning alternate haplotypes. Note that our additional MAF and genotyping filters help protect against SNP calls from false positive alignments. Hard genotype calls (or subsets thereof) were used for ADMIXTURE, principal components analyses (PCA), PCAdapt, and Dsuite analyses (see below).
QUANTIFICATION AND STATISTICAL ANALYSIS
Behavior
We used a beta-binomial mixed generalized linear model as implemented in the R [50] package glmmTMB [51] to model the probability of choosing a human versus animal host for each population. This model assumes independence of individual females within trials but accounts for trial structure and the fact that preference varies more from trial to trial than is expected for a binomial model (is overdispersed) due to random sources variation (e.g. exact starting position of females within the acclimation chamber at the start of a trial, small differences in airflow between right and left ports, uncontrollable trial-to-trial variation in live host stimuli etc.). Replicate colonies and trial day were included as random factors, while population was modelled as a fixed factor. We switched the guinea pig used and the side of the human versus animal host between days such that these effects would be subsumed under the trial day random factor. We used the R package emmeans [52] to extract from our glm the fitted probability of choosing a human host with 95% confidence intervals. For purposes of data visualization, we transformed each probability (p) into a preference index (PI) ranging from −1 to 1 using the formula PI=2p-1. An index of zero means the mosquitoes were equally likely to choose either host (no preference), while an index above or below zero means the mosquitoes were more likely to choose the human or animal, respectively. We used a likelihood ratio test to compare our glm to a null model accounting for day-to-day variation but not population of origin. The same beta-binomial mixed generalized linear model was used to model the probability of responding to either host (overall response rates, Figure S1F).
Ecological modeling
We first compared the behavior of mosquitoes from paired forest and town populations within 5–60km of each other. In one case, a single town population (KED) was paired with two forest sites (BTT and PKT). We estimated the effect of forest habitat using a linear model that estimated preference for each pair (or group for KED, BTT, and PKT) and a coefficient for forest or town habitat. This is conceptually very similar to carrying out a paired t-test, except it allowed us to take into account the two different forest sites near KED.
We next explored the ecological factors associated with preference for humans across all populations in the sample set, again using a linear modelling framework. In this set of analyses, each population was represented by a single logit-transformed preference probability (generated by the beta-binomial model described in the previous section) and a single estimate of each ecological descriptor extracted from public datasets using the mean latitude and longitude of the ovitraps that contributed to the corresponding colony (or the mean of the two independent colony means for populations with two colonies).
While immediate habitat had no effect on behavior, we hypothesized that human population density might be relevant when calculated across a larger spatial scale. We therefore used a 2.5-minute resolution population density raster from the United Nations World Population Prospects (UNWPP, 2015 population densities adjusted to country totals) [53] to compare the effect of density across buffers of the following radiuses: 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 200, and 300 km. In a simple human population density model that also takes regional variation into account (logit(prob) ~ human_pop_density + Latitude*Longitude), human population density had a significant effect across a wide range of spatial scales, but was strongest with ~20–50km buffers (black line in Figure S1G).
To better understand the regional drivers of variation in preference, we used the WorldClim 2 bioclimatic variables (Bio1–19) as a set of candidate predictors [17]. Because some of these variables are correlated, we also considered the predictive value of the first three principal components from a PCA analysis of Bio1–19 variation across our populations. In preliminary tests, Bio15 (precipitation seasonality) clearly showed the strongest single-variable association with preference. This was true both in a comparison of simple correlations between each variable and preference (Bio15 r=0.65) and when we included each variable in a linear model with human population density (20km buffer) (red circles in Figure S1H). However, the relationship with Bio15 appeared to be strongly nonlinear, if still monotonic (Figure 2B). We therefore used a two-step procedure to model the nonlinearity. We first used the R package MonoPoly [54] to fit monotonic polynomials of different degrees to logit-transformed preference probabilities, and then included the fitted values as an offset (i.e. removed their effects before fitting) in the linear model (logit(prob) ~ human_pop_density_20km + offset(fitted(monotonic polynomial Bio15))). We found that a third-order monotonic polynomial significantly improved model performance, minimizing the Akaike Information Criterion (AIC). Rechecking the performance of different human population density buffers in this new model context showed that 20km yielded a much lower AIC than other buffers (light blue line in Figure S1G). Note that this buffer most likely reflects the balance between selection and dispersal, and is not a direct reflection of adult dispersal patterns per se.
We were concerned that nonlinear relationships could have obscured another better predictor in our initial survey of single-variable correlations. However, after fitting monotonic polynomials of degree 1–4 for all 19 bioclim variables and the first three PC axes, a third-order monotonic polynomial fit for Bio15 still had the lowest AIC (Figure S1H).
To check whether additional climate variables could further improve our model, we regressed logit-transformed preference and the other bioclimate variables on our third-order fit for Bio15 and tested if residual variation in preference could be explained by the other variables. We again used a two-step procedure to model these effects. We used the R package MonoPoly to fit monotonic polynomials of different degrees to logit-transformed preference probability residuals, and then included the fitted values as an offset (i.e. removed their effects before fitting) in the linear model (logit(prob) ~ human_pop_density_20km + offset(fitted(monotonic polynomial Bio15)) + offset(fitted(monotonic polynomial BioX))). We found that including a linear Bio18 (precipitation in the warmest quarter) term further reduced AIC (Figure S1I). Because Bio18 and our fitted Bio15 polynomial relationship were modestly correlated (r=−.29) and we had selected the variables in a stepwise way, we wondered if including them in a single linear model would change our estimates of their effects. In this full, final model (logit(prob) ~ human_pop_density_20km + fitted(monotonic polynomial Bio15) + Bio18), the estimated coefficient for the fitted Bio15 component was close to 1 (1.04), indicating that fitting the effects sequentially or together didn’t make a major difference, but we used the model where both were fit together going forward. Using both Bio15 and Bio18 as covariates, we again found that using a buffer of 20km for calculating population density yielded a much lower AIC than other buffers (Figure S1G).
Bio15 and Bio18 both have clear connections to the length and temperature of the dry season - an important factor in survival of dormant Aedes aegypti eggs. We therefore combined them into a single Dry Season Intensity Index by adding together the fitted Bio15 and Bio18 terms for each location. This simple transformation yields a single linear climate term that predicts the host odor preference of mosquitoes from a given location.
Morphological analyses
We pinned 7–29 female mosquitoes from each location (field-collected [AWK, BOA, FCV, KED, KIN, KUM, LBV, LPV, MIN, NGO, OGD, OHI, PKT, THI] or lab colony [ABK, BTT, ENT, GND, KAK, KBO, KWA, LPF, ORL, RAB, SHM, T51, VMB, ZIK]) as previously described [15] and captured light microscope images of the dorsal abdomen under constant lighting and magnification (3X) on a Nikon SMZ1270 microscope. We then estimated the proportion of white scales on the first abdominal tergite by converting each image to 8-bit grayscale in ImageJ, selecting the region of interest, and calculating the area with brightness values above 128. Area estimates were only weakly sensitive to the precise cutoff since the white and black scales differ markedly in brightness; we therefore chose a value in the middle of the range. We used the R function lm to fit a linear model with logit-transformed scaling proportion as the response variable and population of origin as the predictor. We then used the R package emmeans to calculate 95% confidence intervals for each population.
Population structure analyses
We characterized population structure using two alternative approaches based on a set of 1,000,000 unlinked SNPs selected in PLINK (step size 100, cutoff 0.1, --thin-count 1000000) [55]. First, we used ADMIXTURE [25] to assign individuals to variable numbers of population clusters for K=2–10, with K=3 minimizing cross-validation error. Second, we used PLINK to carry out principal components analysis (PCA). One sample from Ngoye, Senegal (NGO) was a clear outlier in ancestry, showing strong affinity with West African generalist populations while all other individuals from this population showed consistent affiliation with human specialists (Figure 3B); we excluded this putative recent migrant from subsequent FST, PBS, ABBA-BABA, π, and dxy analyses involving Ngoye (see below).
Gene flow and divergence analyses
We used PCAdapt [25] to test whether specific SNPs were associated with specialist ancestry (represented by PC2, see Figure 4A) across the subset of high-quality, biallelic SNPs from our hard-called set that had minor allele frequency >0.05 (n=5,369,564 SNPs) (PCAdapt parameters: K=3, method “componentwise”, and LD clumping with a size of 200 and a cutoff of 0.1). We used a permutation testing approach to identify outlier regions with a significantly increased concentration of outlier SNPs as follows: First, we counted the number of outlier and non-outlier SNPs in each 100kb window across the chromosomal scaffolds. We then calculated the proportion of outlier SNPs in a 10Mb sliding window with a 100kb step across the genome. This represents our observed distribution. To generate a null distribution, we shuffled the genomic position of 100kb windows (thus preserving local linkage patterns) across the genome 100 times. We only permuted windows among coordinates matched for the same decile of nucleotide diversity (calculated across all populations), since variation in diversity levels across the genome could affect the propensity of windows to harbor outliers. We then used these permuted windows to calculate an empirical cumulative distribution function for the proportion of outlier SNPs in a 10Mb window using the R function ecdf. Comparison of the observed distribution to this null distribution allowed us to identify ≥10Mb regions with an elevated proportion of outlier SNPs at a two-tailed false discovery rate of 0.001. The choice of a 10Mb window restricts us to larger regions that are either relatively new (giving recombination limited time to break up divergent chromosomal regions) or contain several tightly linked loci. However, using smaller regions (e.g. 1Mb or 100kb) gave similar results.
We used ANGSD (subprogram realSFS) to calculate pairwise FST between populations and a custom script to turn these FST values into the Population Branch Statistic (PBS, essentially polarized FST) for NGO, THI, OGD, KED, and OHI, using BTT as a nearby generalist reference population and FCV as an outgroup [27]. Using alternative reference and outgroup populations yielded similar results.
We used ABBA-BABA-related statistics to further explore patterns of divergence between populations. These statistics test for an excess of shared derived variation between lineages in order to distinguish gene flow or ancestral population structure from the incomplete lineage sorting (ILS) that can occur during a simple tree-like branching process. For more on expected genome-wide and locus-specific patterns of derived allele sharing under ILS and gene flow, see [28]. First, we used Dsuite [56] to confirm that the populations in our dataset did not conform to the strict tree-like model, which is expected since all populations belong to the same species and almost certainly exchange genes. Indeed, we strongly rejected the null tree-like hypothesis (block jacknife P<10−7) for all three-population trees with Ae. mascarensis as an outgroup.
We then explored potential heterogeneity in gene flow across the genome using the fD statistic (calculated in 10Mb windows with a 100kb step). The fD statistic uses shared derived genetic variation to estimate the fraction of ancestry at a specific locus derived from gene flow between branches in a specified tree [28]. We calculated fD from ANGSD population allele frequencies using a custom python script for the tree (BTT, X; BKK, mascarensis) to identify regions of the genome showing elevated levels of shared derived variation between the focal population (X=NGO, THI, or OGD) and non-African human specialists (BKK). We do not think such shared derived variation is necessarily derived from introgression back to Africa from non-African populations. Instead, it may reflect relationships present in ancestral populations, before Ae. aegypti left Africa. Differentiating between these two hypotheses is out-of-scope for this study but will be addressed in a future study incorporating much more genomic data from outside Africa. Regardless, we expect shared derived variation between human-preferring populations within and outside Africa to be present in regions that code for human-adaptive traits and thus experience reduced gene flow between human- and animal-preferring populations within Africa. We tested whether high PBS and high fD windows (defined as 100kb windows in the top 10% of genome-wide values for each statistic in turn) were significantly concentrated in PCAdapt outlier regions using Fisher’s exact test.
To help interpret measures of between-population divergence (i.e. FST and PBS), we used ANGSD to estimate levels of genetic diversity (π), Tajima’s D, and Fay and Wu’s H [30,31] across the genome for focal populations, and we used the perl script getDxy.pl (modified to skip variant sites not covered in one population) from ngsTools [57] to calculate dxy (Figure S2A–F). We also calculated normalized dxy (Figure S2G) by dividing dxy for a given population pair by mean dxy for all pairs of NGO, THI, OGD, BTT, and FCV. We calculated normalized π (Figure S2G) by dividing population π by mean π across all populations.
Climate and population projections
We predicted future changes in host odor preference at each sampling location by plugging climate and human population density change projections for 2050 into our final, fitted, ecological model – including human population density (calculated within 20km radius), precipitation seasonality (Bio15, third degree monotonic polynomial) and precipitation in the warmest quarter (Bio18, linear).
Climate change projection data came from the Coupled Model Intercomparison Project Phase 5 (CMIP5) based on Representative Concentration Pathway 8.5 (RCP8.5) scenarios [36]. RCP8.5 is considered the business-as-usual scenario for future greenhouse gas concentrations, reflecting minimal mitigation efforts. The CMIP5 effort contributed to the International Panel on Climate Change (IPCC) Fifth Assessment Report. A new modeling effort, CMIP6, is currently underway but complete data are not yet available. Projected climate data from a global climate model (GCM) cannot be directly compared to present-day observational climate data due to model biases and measurement error. Failing to account for these biases can result in misinterpreting structural differences between the two datasets as potential climate change effects. Instead, the projection data must first be bias-corrected by calculating the relative or absolute change between current and future climates for the variable of interest, using solely the GCM output. This relative or absolute change can then be applied to observational data. Depending on the resolution of the observational climate data, projections may also be downscaled – i.e. the resolution of the model output improved. The Worldclim projection data has undergone both downscaling and bias-correction processes such that it can be compared with the observational data used in our present-day analysis. Absolute changes were used for temperature and relative changes were used for precipitation. Further details of these processes are available at https://www.worldclim.org/downscaling. There is relatively high agreement across models in terms of the spatial distribution of projected Bio15 and Bio18 changes (Figure S4).
Population projection data came from the United Nations medium-variant scenario [53]. This scenario assumes existing high fertility populations will experience a fertility decline over the coming century, as economic development increases. Despite falling fertility, sub-Saharan Africa is expected to see an increase in the total number of births over the next several decades relative to the recent past. High birth numbers coupled with increasing life expectancy will lead to 1.05 billion increase in population in sub-Saharan African countries by 2050, 52% of the additional global population in this timeline [58]. We used urban and rural, medium-variant projections for each country and calculated growth rates by comparing 2050 numbers with those from 2015. Note, our field collections were conducted between 2015 and 2018 (mostly 2017–2018) making 2015 numbers more applicable than any other available estimates. We then applied these growth rates to the baseline population data for each location. Urban and rural locations were considered separately because urban populations are expected to grow at a faster rate than rural populations over this time period. Urban populations were defined as those with current population density > 400 humans/km2 calculated with a 20km buffer (Table S1). These populations were all from areas that we observed to be dominated by human structures and activities (Table S1). A few intermediate density locations fell below this cutoff and were classified as rural. More specifically, KWA, OHI and NGO are rural towns, while ABK and SHM are wild areas on the far outskirts of what most would consider urban areas (densities 173–288 humans/km2; Table S1). The other sites classified as rural had much lower densities (1–67 humans/km2; Table S1).
Supplementary Material
Table S3. List of genes containing PCAdapt outlier SNPs. Related to Figure 4. Genes come from the AaegL5 v2 gene set and are given in bed interval format. SNP coordinates use one-based indexing. The last column indicates (0 or 1) if a gene also falls within a PCAdapt outlier region (where outlier SNPs were significantly clustered, FDR<0.001).
Key Resource Table
Highlights.
African populations of Ae. aegypti vary in preference for human vs. animal odor
Preference for humans is associated with intense dry seasons and urbanization
Preference for humans has a single, shared genomic basis inside and outside Africa
Rapid urbanization may further increase human biting in many African cities by 2050
ACKNOWLEDGEMENTS:
The authors thank Siyang Xia, Christophe Paupy, and Bryan Grenfell for valuable feedback on early results, Boy Ponlawat for generously providing mosquito eggs used to establish a laboratory reference colony from Thailand, Francis Mulwa, Gilbert Rotich, Gilbert Bianquinche, Marc F. Ngangué for field assistance, the National Park Services and rangers of Kenya, Gabon, and Ghana for providing access to forest areas, and a large number of local residents who gave advice and assistance at all field sites. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of CDC.
Funding: This work was funded by Pew Scholars, Searle Scholars, Klingenstein-Simons, and Rosalind Franklin/Gruber Foundation awards (to C.S.M.), the National Institutes of Health (R00DC012069 to C.S.M.; R01AI101112 and U01AI115595 to J.R.P.), a Helen Hay Whitney Postdoctoral Fellowship (to N.H.R.) and undergraduate thesis funding from the Princeton University Department of Ecology and Evolutionary Biology and African Studies Program (to N.I and E.G.E.); Verily Life Sciences funded all genome sequencing. The research was also supported by the New York Stem Cell Foundation. C.S.M. is a New York Stem Cell Foundation – Robertson Investigator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS: The authors declare no competing interests.
REFERENCES
- 1.WHO (2020). WHO | Vector-borne diseases. WHO. Available at: http://www.who.int/mediacentre/factsheets/fs387/en/ [Accessed April 3, 2017].
- 2.Clements AN (1999). The Biology of Mosquitoes Volume 2: Sensory Reception and Behavior. Wallingford: CABI Publishing. [Google Scholar]
- 3.McBride CS (2016). Genes and Odors Underlying the Recent Evolution of Mosquito Preference for Humans. Current Biology 26, R41–R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takken W, and Verhulst NO (2013). Host Preferences of Blood-Feeding Mosquitoes. Annual Review of Entomology 58, 433–453. [DOI] [PubMed] [Google Scholar]
- 5.Besansky NJ, Powell JR, Caccone A, Hamm DM, Scott JA, and Collins FH (1994). Molecular phylogeny of the Anopheles gambiae complex suggests genetic introgression between principal malaria vectors. PNAS 91, 6885–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayala FJ, and Coluzzi M (2005). Chromosome speciation: Humans, Drosophila, and mosquitoes. PNAS 102, 6535–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen J (1977). Behavioral Differences in Two Sub-species of Aedes aegypti (L.)(diptera, Culicidae) in East Africa. PhD Thesis, University of Notre Dame. [Google Scholar]
- 8.Crawford JE, Alves JM, Palmer WJ, Day JP, Sylla M, Ramasamy R, Surendran SN, Black WC, Pain A, and Jiggins FM (2017). Population genomics reveals that an anthropophilic population of Aedes aegypti mosquitoes in West Africa recently gave rise to American and Asian populations of this major disease vector. BMC Biology 15, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, Jiang X, Hall AB, Catteruccia F, Kakani E, et al. (2015). Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonseca DM, Keyghobadi N, Malcolm CA, Mehmet C, Schaffner F, Mogi M, Fleischer RC, and Wilkerson RC (2004). Emerging Vectors in the Culex pipiens Complex. Science 303, 1535–1538. [DOI] [PubMed] [Google Scholar]
- 11.Powell JR, Gloria-Soria A, and Kotsakiozi P (2018). Recent history of Aedes aegypti: vector Genomics and Epidemiology records. BioScience 68, 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christophers SR (1960). Aedes aegypti: The Yellow Fever Mosquito (Cambridge: Cambridge University Press; ). [Google Scholar]
- 13.Kotsakiozi P, Evans BR, Gloria-Soria A, Kamgang B, Mayanja M, Lutwama J, Le Goff G, Ayala D, Paupy C, and Badolo A (2018). Population structure of a vector of human diseases: Aedes aegypti in its ancestral range, Africa. Ecology and Evolution 8, 7835–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McClelland GAH, and Weitz B (1963). Serological identification of the natural hosts of Aedes aegypti (L.) and some other mosquitoes (Diptera, Culicidae) caught resting in vegetation in Kenya and Uganda. Annals of Tropical Medicine & Parasitology 57, 214–224. [DOI] [PubMed] [Google Scholar]
- 15.McBride CS, Baier F, Omondi AB, Spitzer SA, Lutomiah J, Sang R, Ignell R, and Vosshall LB (2014). Evolution of mosquito preference for humans linked to an odorant receptor. Nature 515, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gouck HK (1972). Host preferences of various strains of Aedes aegypti and A. simpsoni as determined by an olfactometer. Bulletin of the World Health Organization 47, 680. [PMC free article] [PubMed] [Google Scholar]
- 17.Fick SE, and Hijmans RJ (2017). WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. International Journal of Climatology 37, 4302–4315. [Google Scholar]
- 18.Silver JB (2007). Mosquito ecology: field sampling methods (springer science & business media).
- 19.Trpiš M (1972). Dry season survival of Aedes aegypti eggs in various breeding sites in the Dar es Salaam area, Tanzania. Bulletin of the World Health Organization 47, 433. [PMC free article] [PubMed] [Google Scholar]
- 20.Paupy C, Brengues C, Ndiath O, Toty C, Hervé J-P, and Simard F (2010). Morphological and genetic variability within Aedes aegypti in Niakhar, Senegal. Infection, Genetics and Evolution 10, 473–480. [DOI] [PubMed] [Google Scholar]
- 21.Mattingly PF (1957). Genetical Aspects of the Aedes aegypti Problem: I. Taxonomy and Bionomics. Annals of Tropical Medicine & Parasitology 51, 392–408. [PubMed] [Google Scholar]
- 22.McClelland GAH (1974). A worldwide survey of variation in scale pattern of the abdominal tergum of Aedes aegypti (L.)(Diptera: Culicidae). Transactions of the Royal Entomological Society of London 126, 239–259. [Google Scholar]
- 23.Sylla M, Bosio C, Urdaneta-Marquez L, Ndiaye M, and Iv WCB (2009). Gene Flow, Subspecies Composition, and Dengue Virus-2 Susceptibility among Aedes aegypti Collections in Senegal. PLOS Negl. Trop. Dis 3, e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gloria-Soria A, Ayala D, Bheecarry A, Calderon-Arguedas O, Chadee DD, Chiappero M, Coetzee M, Elahee KB, Fernandez-Salas I, Kamal HA, et al. (2016). Global genetic diversity of Aedes aegypti. Molecular Ecology 25, 5377–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexander DH, Novembre J, and Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Research 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luu K, Bazin E, and Blum MG (2017). pcadapt: an R package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources 17, 67–77. [DOI] [PubMed] [Google Scholar]
- 27.Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, and Korneliussen TS (2010). Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin SH, Davey JW, and Jiggins CD (2014). Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Molecular Biology and Evolution 32, 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruickshank TE, and Hahn MW (2014). Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Molecular Ecology 23, 3133–3157. [DOI] [PubMed] [Google Scholar]
- 30.Tajima F (1989). Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fay JC, and Wu C-I (2000). Hitchhiking Under Positive Darwinian Selection. Genetics 155, 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernhardt SA, Blair C, Sylla M, Bosio C, and Black IV WC (2009). Evidence of multiple chromosomal inversions in Aedes aegypti formosus from Senegal. Insect Molecular Biology 18, 557–569. [DOI] [PubMed] [Google Scholar]
- 33.Dickson LB, Sharakhova MV, Timoshevskiy VA, Fleming KL, Caspary A, Sylla M, and Black WC (2016). Reproductive Incompatibility Involving Senegalese Aedes aegypti (L) Is Associated with Chromosome Rearrangements. PLoS Negl. Trop. Dis 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redmond SN, Sharma A, Sharakhov I, Tu Z, Sharakhova M, and Neafsey DE (2020). Linked-read sequencing identifies abundant microinversions and introgression in the arboviral vector Aedes aegypti. BMC Biology 18, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerland P, Raftery AE, Ševčíková H, Li N, Gu D, Spoorenberg T, Alkema L, Fosdick BK, Chunn J, Lalic N, et al. (2014). World population stabilization unlikely this century. Science 346, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor KE, Stouffer RJ, and Meehl GA (2011). An Overview of CMIP5 and the Experiment Design. Bull. Amer. Meteor. Soc 93, 485–498. [Google Scholar]
- 37.Futuyma DJ, and Moreno G (1988). The Evolution of Ecological Specialization. Annual Review of Ecology and Systematics 19, 207–233. [Google Scholar]
- 38.Machado-Allison CE, and Craig GB (1972). Geographic Variation in Resistance to Desiccation in Aedes aegypti and A. atropalpus (Diptera: Culicidae). Ann. Entomol. Soc. Am 65, 542–547. [Google Scholar]
- 39.Ehret C (2002). The Civilizations of Africa: A History to 1800 (Charlottesville: University Press of Virginia; ). [Google Scholar]
- 40.Kröpelin S, Verschuren D, Lézine A-M, Eggermont H, Cocquyt C, Francus P, Cazet J-P, Fagot M, Rumes B, Russell JM, et al. (2008). Climate-Driven Ecosystem Succession in the Sahara: The Past 6000 Years. Science 320, 765–768. [DOI] [PubMed] [Google Scholar]
- 41.Tripet F, Lounibos LP, Robbins D, Moran J, Nishimura N, and Blosser EM (2011). Competitive Reduction by Satyrization? Evidence for Interspecific Mating in Nature and Asymmetric Reproductive Competition between Invasive Mosquito Vectors. Am. J. Trop. Med. Hyg 85, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gloria-Soria A, Soghigian J, Kellner D, and Powell JR (2019). Genetic diversity of laboratory strains and implications for research: The case of Aedes aegypti. PLOS Negl. Trop. Dis 13, e0007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthews BJ, Dudchenko O, Kingan SB, Koren S, Antoshechkin I, Crawford JE, Glassford WJ, Herre M, Redmond SN, Rose NH, et al. (2018). Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 563, 501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, and Sherry ST (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWAMEM. arXiv preprint arXiv:1303.3997. [Google Scholar]
- 46.Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. http://broadinstitute.github.io/picard.
- 48.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, and Daly M (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korneliussen TS, Albrechtsen A, and Nielsen R (2014). ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.R Core Team (2013). R: A language and environment for statistical computing.
- 51.Brooks ME, Kristensen K, van Benthem KJ, Magnusson A, Berg CW, Nielsen A, Skaug HJ, Machler M, and Bolker BM (2017). glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. The R Journal 9, 378–400. [Google Scholar]
- 52.Lenth R, Singmann H, and Love J (2018). Emmeans: Estimated marginal means, aka least-squares means. R Package. [Google Scholar]
- 53.United Nations, Department of Economic and Social Affairs, Population Division (2019). World Population Prospects 2019, Online Edition.
- 54.Turlach BA, Murray K, and Turlach MBA (2019). MonoPoly. R Package.
- 55.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, and Daly MJ (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malinsky M (2019). Dsuite-fast D-statistics and related admixture evidence from VCF files. BioRxiv, 10.1101/634477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fumagalli M, Vieira FG, Linderoth T, and Nielsen R (2014). ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30, 1486–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.United Nations, Department of Economic and Social Affairs, Population Division (2019). World Population Prospects 2019: Highlights.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. List of genes containing PCAdapt outlier SNPs. Related to Figure 4. Genes come from the AaegL5 v2 gene set and are given in bed interval format. SNP coordinates use one-based indexing. The last column indicates (0 or 1) if a gene also falls within a PCAdapt outlier region (where outlier SNPs were significantly clustered, FDR<0.001).
Data Availability Statement
Raw genomic data is available in the NCBI SRA under the accession code PRJNA602495 (https://www.ncbi.nlm.nih.gov/sra/PRJNA602495). Other raw data and scripts are available at github.com/noahrose.