

Abstract
Protists provide insights into the diversity and function of RNA viruses in marine systems. Among them, marine macroalgae are good targets for RNA virome analyses because they have a sufficient biomass in nature. However, RNA viruses in macroalgae have not yet been examined in detail, and only partial genome sequences have been reported for the majority of RNA viruses. Therefore, to obtain further insights into the distribution and diversity of RNA viruses associated with marine protists, we herein examined RNA viruses in macroalgae and a diatom. We report the putative complete genome sequences of six novel RNA viruses from two marine macroalgae and one diatom holobiont. Four viruses were not classified into established viral genera or families. Furthermore, a virus classified into Totiviridae showed a genome structure that has not yet been reported in this family. These results suggest that a number of distinct RNA viruses are widespread in a broad range of protists.
Keywords: RNA virus, algae, dsRNA
Marine macroalgae are classified into multiple eukaryotic lineages that are phylogenetically distinct from each other. For example, green algae and red algae belong to Chloroplastida and Rhodophyceae in Archaeplastida, respectively, while brown algae belongs to Stramenopiles in the SAR supergroup (i.e., Stramenopiles, Alveolata, and Rhizaria) (Adl et al., 2012). Although diatoms, which belong to the Stramenopiles lineage, are recognized as microalgae, they sometimes form macrocolonies. These marine macroalgae are considered to be a type of holobiont, a functional ecosystem, because of the relationship between macroalgae and diverse microorganisms. Thus, to elucidate the functional relationship between macroalgae and associated microbes, metagenomic approaches have been applied to macroalgal holobionts (Egan et al., 2013; Lachnit et al., 2015).
Recent studies suggested that viruses, in addition to cellular microorganisms, are involved in the homeostasis and evolution of holobiont systems (Marhaver et al., 2008; Barr et al., 2013; Bruwer et al., 2017; Thurber et al., 2017). However, viruses in macroalgal holobionts have not yet been examined in detail. To date, two DNA viruses, Ectocarpus siliculosus virus 1 (Lanka et al., 1993) and Feldmannia species virus (Henry and Meints, 1992), have been isolated from marine macroalgae. Only two virome analyses have been conducted on RNA viruses (Lachnit et al., 2015; Waldron et al., 2018). One analysis involved the identification of partial viral RNA sequences in the Tombus-Noda, Bunya-Arena, and Narna-Levi clades from a metatranscriptome in a mixture of brown algae (Fucus serratus) (Waldron et al., 2018). The other consisted of a combination of virus particle purification and RNA sequencing from two individuals of red algae (Delisea pulchra): the sequences of members of Totiviridae, Partitiviridae, and Picornavirales were commonly detected (Lachnit et al., 2015). In addition, unassigned mitochondria-associated dsRNA and chloroplast-associated dsRNA have been reported from the green alga Bryopsis cinicola (Koga et al., 1998, 2003).
Complete genome sequences are essential for understanding the diversity and evolution of RNA viruses (Shi et al., 2016; Dolja and Koonin, 2017; Shi et al., 2018; Wolf et al., 2018). However, conventional RNA sequencing methods are technically challenging. Thus, several novel molecular techniques have been developed to obtain complete genome information on RNA viruses (Lambden et al., 1992; Vreede et al., 1998; Maan et al., 2007; Potgieter et al., 2009; Darissa et al., 2010; Inoue et al., 2010). Fragmented and primer ligated dsRNA sequencing (FLDS) is a novel method that provides full-length viral RNA genome segments and reconstructs putative complete genomes (Urayama et al., 2016, 2018, 2020). Using this method, a sequencing library is constructed from cellular dsRNA molecules consisting of dsRNA virus genomes and replicative intermediates of ssRNA viruses (Morris and Dodds, 1979), and the entire sequences of dsRNA molecules are reconstructed in silico.
In the present study, to investigate the distribution and diversity of RNA viruses associated with marine macroalgae holobionts, FLDS was applied to three marine macroalgae holobionts that contained sufficient amounts of dsRNA to be observed by agarose gel electrophoresis. We successfully obtained the putative complete genome sequences of six novel RNA viruses and identified a highly novel RNA virus with a new genome structure that was not recognized based on partial sequence information.
Materials and Methods
Sample collection
Based on morphology, we collected marine macroalgae in front of the National Research Institute of Fisheries and Environment of Inland Sea, Japan Fisheries Research and Education Agency (34°16'29.2"N 132°15'57.7"E) on February 18, 2016. After carefully washing the samples with autoclaved seawater (121°C, 15 min) that was passed through polycarbonate membrane filters with a pore size of 0.2 μm (GE Healthcare Life Sciences), excess water was removed using paper towels. Treated samples were stored at –80°C until analyzed.
RNA extraction
Macroalgae samples 1–6 (0.40, 0.46, 0.37, 0.30, 0.81, and 0.96 g wet weight, respectively) were disrupted in liquid nitrogen with a mortar. Regarding dsRNA purification, samples were suspended in 2× STE (0.2 M Tris–HCl, 0.2 M NaCl, and 2 mM EDTA, pH 6.8) containing 0.1% (v/v) β-mercaptoethanol, and total nucleic acids were manually extracted with SDS-phenol. dsRNA was purified twice through a Poly-Prep Empty Chromatography Column (Bio-Rad) and microspin column (empty Bio-spin column; Bio-Rad) containing cellulose powder (Cellulose D; ADVANTEC). To remove the remaining DNA and ssRNA, eluted dsRNA was further treated with amplification grade DNase I (Invitrogen) and S1 nuclease (Invitrogen) as described previously (Urayama et al., 2018).
To purify ssRNA, part of the pulverized sample was treated with a TRIzol Plus RNA Purification Kit (Invitrogen) according to the manufacturer’s protocol. The total ssRNA fraction was treated with amplification grade DNase I (Invitrogen) and purified using RNA Clean & Concentrator-5 (Zymo Research).
Library construction and sequencing
cDNA libraries were constructed from purified dsRNA and ssRNA as described previously (Urayama et al., 2018). In brief, dsRNA obtained from each sample was converted into a cDNA library using the FLDS method. The U2 primer was ligated to the 3′ end of fragmented dsRNA, and cDNA was synthesized using the SMARTer RACE 5′/3′ Kit (Takara Bio) with the U2-comp primer. Regarding total RNA-seq, ssRNA was converted into a cDNA library using the SMARTer Universal Low Input RNA Kit according to the manufacturer’s protocol (Takara Bio). After PCR amplification, cDNA was fragmented by an ultrasonicator (Covaris S220). Illumina sequencing libraries were then constructed using KAPA Hyper Prep Kit Illumina platforms (Kapa Biosystems) and evaluated using the KAPA Library Quantification Kit (Kapa Biosystems). The libraries were sequenced using the Illumina MiSeq v3 Reagent Kit (600 cycles) with 300-bp paired-end reads on the Illumina MiSeq platform.
Data processing
The raw sequence reads obtained from FLDS libraries were processed to remove low-quality, adaptor, rRNA, and low-complexity sequences as described previously (Urayama et al., 2018). The remaining reads were subjected to de novo assembly using CLC GENOMICS WORKBENCH version 11.0 (CLC Bio) with the following parameters: a minimum contig length of 500, word value set to auto, and bubble size set to auto. To obtain putative complete viral RNA genomes from FLDS data, the ends of the contigs with at least 250× average coverage were extended using CLC GENOMICS WORKBENCH version 11.0, Genetyx version 14 (Genetyx), and Tablet viewer version 1.19.09.03 (Milne et al., 2010). In our previous study, contigs, for which both ends were termini, were identified as full-length genome segments and viral genomes were reconstructed (Urayama et al., 2018). To analyze the total RNA virome of three marine macroalgae, total FLDS data were used for de novo assembly, and the assembled contigs with at least 3× average coverage and 500 nt in length were clustered at 90% identity using VSEARCH (Rognes et al., 2016). The cluster centroid sequences were selected as representative sequences.
The raw sequence reads obtained from total RNA-seq libraries were trimmed as described previously (Urayama et al., 2016). Small subunit (SSU) rRNA sequences were reconstructed from total RNA-seq reads with EMIRGE, which is an iterative template-guided assembler that relies on a database of 16S rRNA genes (Miller et al., 2011). As the reference database, the SILVA SSU version 132 database was downloaded by running emirge_makedb.py (option: -i 0.99), and we added the mitochondrial 16S rRNA gene sequences of morphologically identified macroalgae species (Scytosiphon lomentaria and Ectocarpus siliculosus) to the SILVA database because these sequences were absent in the original SILVA database. In the surveillance of RNA-dependent RNA polymerase (RdRp)-coding sequences from total RNA-seq data, trimmed reads were assembled as described above.
RNA viral genes were identified using the BlastX program against the NCBI non-redundant (nr) database with an e-value ≤1×10–5. To identify the taxonomic status, the taxonomic information of the top hit virus sequences was used.
Phylogenetic analysis
The phylogenetic positions of the RNA viruses identified were analyzed based on the deduced amino acid sequences of RdRp genes using the maximum-likelihood method, specifically RAxML (Stamatakis, 2014). Related RdRp sequences were collected and aligned with identified RdRp sequences using MUSCLE (Edgar, 2004) in MEGA6 (Tamura et al., 2013). Ambiguous positions in the alignment were removed using trimAl with the option gt=1 (Capella-Gutiérrez et al., 2009). The best-fitting model of amino acid substitutions was tested in Aminosan (Tanabe, 2011) and judged by the corrected Akaike information criterion (Sugiura, 1978). Bootstrap tests were conducted with 1,000 samplings. FigTree (Rambaut, 2014) was used to illustrate the resulting phylogenies.
Data accessibility
Datasets supporting the results of the present study are available in the GenBank database repository (Accession Nos. DDBJ: LC521321–LC521329) and Short Read Archive database (Accession No. DDBJ: DRA009245).
Results
Major RNA viruses and cellular rRNAs
In the surveillance of RNA viruses associated with the marine macroalgae samples, one or two dsRNA band(s) that suggested the presence of RNA viruses were detected (Fig. 1 and Table 1). We then performed FLDS on these dsRNAs and reconstructed viral RNA genomes in silico (see Materials and Methods). We found twelve viral segments and eight viral RNA genomes were reconstructed (Table 1 and S1). Among them, two RNA viruses occupied approximately 0.4 and 7% of the reads in each FLDS library. The other six RNA viruses dominated approximately 19–74% of reads in each FLDS library (Table S2). Since the percentage of reads mapped to the genomes of these six RNA viruses was particularly high in each library, we did not expect the presence of other segments of their genomes. Therefore, we conducted a more detailed analysis of the six dominant RNA viruses individually and summarized the entire RNA viromes of these three marine macroalgae, including the minor populations.
Fig. 1.

dsRNA-positive marine macroalgae used in the present study. (A) They were identified by morphology and SSU rRNA sequences reconstructed from total RNA-seq data. (B) Agarose gel electrophoresis of the total nucleic acids (T) and dsRNA (ds) of marine macroalgae samples 1–3. Arrowheads indicate dsRNA bands. Nucleic acids were stained with ethidium bromide.
Table 1.
Summary of dsRNA detection and sequence analysis.
| Sample name |
Category | dsRNA band(s) |
Virus name | Family | Length | Average coverage |
Accession no. |
|---|---|---|---|---|---|---|---|
| 1 | Brown algae | 13 kbp | Brown algae endornavirus 1 | Endornaviridae | 13,603 | 18,432.2 | LC521321 |
| Brown algae endornavirus 2 | Endornaviridae | 12,572 | 8,181.6 | LC521322 | |||
| 9 kbp | Brown algae RNA virus 1 | unclassified | 8,290 | 15,180.5 | LC521323 | ||
| 2 | Diatom | 7 kbp | Diatom RNA virus 1 | unclassified | 6,743 | 18,090.4 | LC521324 |
| 4 kbp | Diatom totivirus 1 | Totiviridae | 3,671 | 13,489.8 | LC521325 | ||
| 3,564 | 3,961.4 | LC521326 | |||||
| 3 | Red algae | — | Red algae totivirus 1 | Totiviridae | 5,031 | 318.8 | LC521327 |
| 3 kbp | 2,627 | 1,156.3 | LC521328 | ||||
| 2,623 | 531.3 | LC521329 |
To clarify the active holobiont population of samples, SSU rRNA sequences were reconstructed from the reads obtained from total RNA-seq. Their compositions were estimated based on mapped read numbers on the SSU rRNA sequences (Miller et al., 2011). Brown algae, diatoms, and red algae occupied more than 80% of all SSU rRNA reads in the total RNA-seq libraries from samples 1, 2, and 3, respectively (Fig. S1). This result indicated that sample 2 was a diatom holobiont, but not a macroalgae holobiont. Therefore, we designated samples 1, 2, and 3 as a brown algae holobiont, diatom holobiont, and red algae holobiont, respectively (Fig. S1). For example, the SSU rRNA reads of the red algae holobiont included Pyropia suborbiculata (red algae, 69.6%), Pyropia yezoensis chloroplast (red algae, 24.0%), Pyropia tenera (red algae, 5.2%), Halochlorococcum dilatatum (diatom, 1.2%), P. yezoensis mitochondria (red algae, 0.1%).
RNA viruses in the brown algae holobiont
Three putative complete genomes (13,603 nt, 12,572 nt, and 8,290 nt) were obtained in the brown algae holobiont (Fig. 2A). Based on a homology search with the predicted amino acid sequence, particularly the open reading frame (ORF) encoding RdRp, the sequences with 13,603 nt and 12,572 nt were named brown algae endornavirus 1 (BraEV1) and brown algae endornavirus 2 (BraEV2), respectively. The 8,290-nt sequence was named brown algae RNA virus 1 (BraRV1). BraEV1 and BraEV2 corresponded to a band of approximately 13 kbp, and BraRV1 to a 9-kbp band (Fig. 1, sample 1). A phylogenetic analysis with the RdRp amino acid sequence revealed that BraEV1 and BraEV2 belonged to the established family Endornaviridae, and BraRV1 formed a single unclassified deep branch with St97 virga-like virus 1 (Accession: BDQD01000142) (Urayama et al., 2018) identified from surface seawater from the north Pacific Ocean (Fig. 2B and C). Based on the results of a BlastP analysis, the closest isolate of BraRV1 was the plant ssRNA virus, raspberry bushy dwarf virus (RBDV). A stretch of cytosine is present at the 3′ end of many viruses in the family Endornaviridae (Okada et al., 2018a). In the present study, the stretch of cytosine was found in BraEV1, but not in BraEV2. The polyproteins encoded by BraEV1 and BraEV2 contained a cysteine-rich region with conserved CXCC motifs shared among several viruses in the family Endornaviridae (Okada et al., 2011).
Fig. 2.

Genome organization and phylogenetic position of RNA viruses identified from a brown algae sample. (A) Predicted ORFs and identified domains: Met, viral methyltransferase superfamily; Hel, viral helicase 1 superfamily; RdRp, RdRP_2 superfamily. (B) Maximum-likelihood tree of RdRp amino acid sequences from representative members of the Hepe-Virga clade (Shi et al., 2016) and three RNA viral sequences obtained in the present study. Open and closed circles represent bootstrap values of 50–90% and ≥90%, respectively. The best-fitting substitution model was [LG+I+G+F]. A virus identified from surface seawater (Urayama et al., 2018) is marked in blue. Brown circles indicate previously established RNA virus families or order. The scale bar represents the number of amino acid substitutions per site. (C) Enlarged view of the phylogenetic tree of Endornaviridae in Fig. 2B. Viruses identified from plants are marked in green. Orange and gray indicate oomycete endornavirus and fungal endornavirus, respectively.
RNA viruses in the red algae holobiont and diatom holobiont
Three viral RNA genomes were obtained from the diatom and red algae holobiont: diatom RNA virus 1 (DiRV1) and diatom totivirus 1 (DiTV1) from the diatom holobiont and red algae totivirus 1 (RaTV1) from the red algae holobiont (Fig. 3A and B). DiRV1 with 6,743 nt appeared to correspond to the 7-kbp band observed in the electrophoresis results (Fig. 1, sample 2). A BlastX search revealed that DiRV1 only had significant similarity with diatom colony associated dsRNA virus 16, an unclassified RNA virus identified from a diatom colony in a tidal pool (Urayama et al., 2016).
Fig. 3.

Genome organization and phylogenetic position of RNA viruses identified from diatom and red algae samples. (A, B) Predicted ORFs and identified domains: Coat, Totivirus_coat superfamily; RdRp, RdRP_4 superfamily. (C) Maximum-likelihood tree of RdRp amino acid sequences from representative members of the family Totiviridae, two RNA viral sequences obtained in the present study, and their relatives. Open and closed circles represent bootstrap values of 50–90% and ≥90%, respectively. The best-fitting substitution model was [LG+I+G+F]. Viruses identified from red algae (Lachnit et al., 2015) are marked in blue. Brown circles indicate previously established RNA virus genera. The scale bar represents the number of amino acid substitutions per site.
However, RaTV1 and DiTV1 were related to non-segmented dsRNA viruses in Totiviridae (Fig. 3C and see below). DiTV1 consisted of 3,671-nt and 3,564-nt segments and were named DiTV1 RNA1 and RNA2, respectively. These segments corresponded to the 4-kbp dsRNA band (Fig. 1, sample 2). The terminal sequences of DiTV1 RNA1 and RNA2 were shared with each other (Fig. S2), which is one of the hallmarks of the segmented genome of an RNA virus (Hutchinson et al., 2010). RNA1 and RNA2 were predicted to encode an capsid protein (CP) and RdRp, respectively. In the case of RaTV1, although RNA1 was not detected in gel electrophoresis, RNA2 and RNA3 with 2,627 and 2,623 nt, respectively, appeared to correspond to the 3-kbp band (Fig. 1, sample 3). The terminal sequences were also highly conserved among the three segments (Fig. S2). RaTV1 RNA1 showed the typical ORF structure of a virus in Totiviridae. Similar to many viruses in the genus Victorivirus (family Totiviridae) (Jamal et al., 2019), an overlap region was observed between the CP start and RdRp stop codons (AUGA; nt 2,520 to 2,523). The predicted amino acid sequences of RNA2 and RNA3 did not show significant similarities to known proteins in the NCBI nr database or to conserved motifs in the Pfam database. Since some viruses in Totiviridae contain a satellite RNA called M (Tipper and Schmitt, 1991), these RNAs were expected to be satellite RNAs of RaTV1. The results of a phylogenetic analysis showed that DiTV1 and RaTV1 were not classified in any previously established genera in Totiviridae (Fig. 3C). To date, viruses classified into Totiviridae (DpTV, AMB17466–17469 in Fig. 3C) in D. pulchra (red algae) have been reported (Lachnit et al., 2015); however, their phylogenetic position is distinct from DiTV1 and RaTV1.
Other viral sequences
To analyze the total RNA virome, we focused on contigs encoding RdRp. Ninety-eight representative contigs showed significant similarity with RdRp genes in public databases. Based on the Blast top hit sequences, more than 50% of these contigs were designated as unclassified lineages, and the other contigs were identified as members of Totiviridae, Endornaviridae, endorna-like, or Narnaviridae (Fig. 4). In addition, 67% of the RdRp contigs appeared to represent new viral species with less than 50% amino acid identity to the known viral sequence (Fig. 5).
Fig. 4.
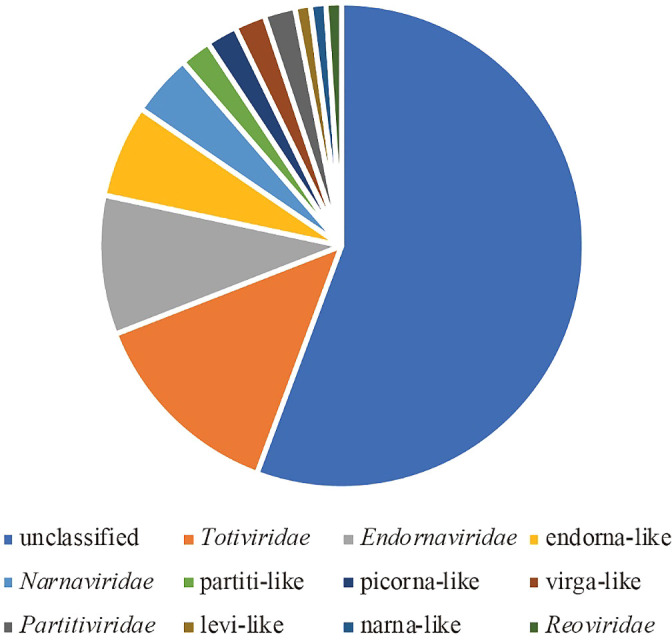
Classification of 98 representative contigs based on taxonomic information on their top hit virus.
Fig. 5.
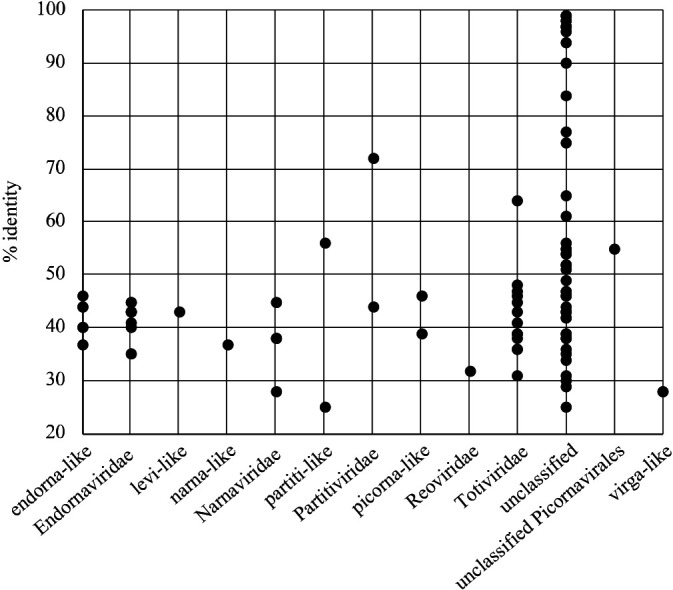
Plot of amino acid % identity for each centroid encoding RdRp to a known viral sequence.
Previous studies reported that the degree of concentrations for viral RNA sequences in dsRNA-seq data differed from that in virus species (Urayama et al., 2016; Decker et al., 2019). We cannot rule out the possibility that some RNA viruses are easier to detect in total RNA-seq than in dsRNA-seq. However, RNA viruses specific for total RNA-seq data were not identified in the present study (data not shown).
Discussion
In the present study, we identified three dsRNA-positive samples among 6 samples (Fig. S3). The frequency of detectable dsRNA in marine macroalgae holobionts reflected the high infection rate of RNA viruses in fungi and plants and the broad distribution of viral dsRNA in eukaryotes (Koga et al., 1998). The present results suggest that marine macroalgae holobionts harbor diverse RNA viruses and include novel RNA viruses, such as fungi (Kotta-Loizou, 2019), plants (Roossinck et al., 2010; Kamitani et al., 2019), and animals (Shi et al., 2016; 2018; Urayama et al., 2020). Although data are limited, the present results support the hypothesis that protists harbor diverse RNA viruses. To prove this hypothesis, further information is needed on the diversity and distribution of RNA viruses in taxonomically diverse hosts.
Based on phylogenetic analyses of the conserved polymerase, helicase, and methyltransferase motifs (Koonin et al., 1993), RBDV belongs to the ancestral lineage of the family Bromoviridae (Isogai et al., 2019). The genome size of BraRV1 is similar to that of viruses in Bromoviridae (8.0 kb) and RBDV (7.7 kb), but differs from those of Bromoviridae viruses that have the three-segmented genome. These results suggest that BraRV1 is distantly related to an established RNA virus family and forms a novel RNA virus group with St97 virga-like virus 1. All known viruses in Totiviridae harbor undivided dsRNA genomes with two large ORFs (King et al., 2012). The 5′-proximal ORF encodes a CP, and the downstream 3′-proximal ORF encodes an RdRp. Therefore, DiTV1 appears to be a novel member of the family Totiviridae with a unique genome structure.
Although the effects of these RNA virus infections are not predictable, some RNA viruses impact the phenotype of host organisms, such as viral toxin production (Magliani et al., 1997) and host toxin production (Okada et al., 2018b), cytological alterations in cellular organelles (Newhouse et al., 1983), and stress tolerance (Marquez et al., 2007). To understand the influence of these widely distributed RNA viruses in marine macroalgae holobionts, we need to isolate and cultivate possible macroalgae hosts and compare virus-infected and virus-free strains. For example, algal strains for culturing laver (nori farming) may be good targets because cultivation systems under laboratory conditions have already been established; however, it currently remains unclear whether these strains harbor RNA viruses. Since difficulties are associated with both the cultivation of host organisms and isolation of viruses from diverse hosts, metagenomics is also required to elucidate the host-RNA virus relationship, as in the case of DNA viruses in diverse marine ecosystems (Silveira et al., 2020). The present results will promote further research on the functions and distribution of RNA viruses in protists, particularly marine macroalgae.
Citation
Chiba, Y., Tomaru, Y., Shimabukuro, H., Kimura, K., Hirai, M., Takaki, Y., et al. (2020) Viral RNA Genomes Identified from Marine Macroalgae and a Diatom. Microbes Environ 35: ME20016.
https://doi.org/10.1264/jsme2.ME20016
Supplementary Material
Acknowledgements
This research was supported in part by Grants-in-Aid for Scientific Research (26892031, 18K19235, and 18H05368) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and Grants-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT) of Japan (No. 16H06429, 16K21723, and 16H06437).
References
- Adl S.M., Simpson A.G.B., Lane C.E., Lukes J., Bass D., Bowser S.S., et al. (2012) The revised classification of eukaryotes. J Eukaryot Microbiol 59: 429–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr J.J., Auro R., Furlan M., Whiteson K.L., Erb M.L., Pogliano J., et al. (2013) Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 110: 10771–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruwer J.D., Agrawal S., Liew Y.J., Aranda M., and Voolstra C.R. (2017) Association of coral algal symbionts with a diverse viral community responsive to heat shock. BMC Microbiol 17: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., and Gabaldón T. (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darissa O., Willingmann P., and Adam G. (2010) Optimized approaches for the sequence determination of double-stranded RNA templates. J Virol Methods 169: 397–403. [DOI] [PubMed] [Google Scholar]
- Decker C.J., Steiner H.R., Hoon-Hanks L.L., Morrison J.H., Haist K.C., Stabell A.C., et al. (2019) dsRNA-Seq: identification of Viral Infection by Purifying and Sequencing dsRNA. Viruses 11: 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolja V.V., and Koonin E.V. (2017) Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res 244: 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan S., Harder T., Burke C., Steinberg P., Kjelleberg S., and Thomas T. (2013) The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol Rev 37: 462–476. [DOI] [PubMed] [Google Scholar]
- Henry E.C., and Meints R.H. (1992) A persistent virus infection in Feldmannia (Phaeophyceae) J Phycol 28: 517–526. [Google Scholar]
- Hutchinson E.C., von Kirchbach J.C., Gog J.R., and Digard P. (2010) Genome packaging in influenza A virus. J Gen Virol 91: 313–328. [DOI] [PubMed] [Google Scholar]
- Inoue E., Wang X., Osawa Y., and Okazaki K. (2010) Full genomic amplification and subtyping of influenza A virus using a single set of universal primers. Microbiol Immunol 54: 129–134. [DOI] [PubMed] [Google Scholar]
- Isogai M., Matsudaira T., Ito M., and Yoshikawa N. (2019) The 1b gene of raspberry bushy dwarf virus is a virulence component that facilitates systemic virus infection in plants. Virology 526: 222–230. [DOI] [PubMed] [Google Scholar]
- Jamal A., Sato Y., Shahi S., Shamsi W., Kondo H., and Suzuki N. (2019) Novel victorivirus from a Pakistani isolate of Alternaria alternata lacking a typical translational stop/restart sequence signature. Viruses 11: 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani M., Nagano A.J., Honjo M.N., and Kudoh H. (2019) A survey on plant viruses in natural Brassicaceae communities using RNA-seq. Microb Ecol 78: 113–121. [DOI] [PubMed] [Google Scholar]
- King, A.M.Q., Adams, M.J., Carstens, E.B., and Lefkowitz, E.J. (2012) Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. London: Elsevier. [Google Scholar]
- Koga R., Fukuhara T., and Nitta T. (1998) Molecular characterization of a single mitochondria-associated double-stranded RNA in the green alga Bryopsis. Plant Mol Biol 36: 717–724. [DOI] [PubMed] [Google Scholar]
- Koga R., Horiuchi H., and Fukuhara T. (2003) Double-stranded RNA replicons associated with chloroplasts of a green alga, Bryopsis cinicola. Plant Mol Biol 51: 991–999. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Dolja V.V., and Morris T.J. (1993) Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit Rev Biochem Mol Biol 28: 375–430. [DOI] [PubMed] [Google Scholar]
- Kotta-Loizou, I. (2019) Mycoviruses. Basel: MDPI AG (Multidisciplinary Digital Publishing Institute). [Google Scholar]
- Lachnit T., Thomas T., and Steinberg P. (2015) Expanding our understanding of the seaweed holobiont: RNA viruses of the red alga delisea pulchra. Front Microbiol 6: 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambden P., Cooke S., Caul E., and Clarke I. (1992) Cloning of noncultivatable human rotavirus by single primer amplification. J Virol 66: 1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanka S.T., Klein M., Ramsperger U., Müller D.G., and Knippers R. (1993) Genome structure of a virus infecting the marine brown alga Ectocarpus siliculosus. Virology 193: 802–811. [DOI] [PubMed] [Google Scholar]
- Maan S., Rao S., Maan N.S., Anthony S.J., Attoui H., Samuel A.R., and Mertens P.P.C. (2007) Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J Virol Methods 143: 132–139. [DOI] [PubMed] [Google Scholar]
- Magliani W., Conti S., Gerloni M., Bertolotti D., and Polonelli L. (1997) Yeast killer systems. Clin Microbiol Rev 10: 369–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhaver K.L., Edwards R.A., and Rohwer F. (2008) Viral communities associated with healthy and bleaching corals. Environ Microbiol 10: 2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez L.M., Redman R.S., Rodriguez R.J., and Roossinck M.J. (2007) A virus in a fungus in a plant: three-way symbiosis required for thermal tolerance. Science 315: 513–515. [DOI] [PubMed] [Google Scholar]
- Miller C.S., Baker B.J., Thomas B.C., Singer S.W., and Banfield J.F. (2011) EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol 12: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I., Bayer M., Cardle L., Shaw P., Stephen G., Wright F., and Marshall D. (2010) Tablet—next generation sequence assembly visualization. Bioinformatics 26: 401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T.J., and Dodds J.A. (1979) Isolation and analysis of double-stranded-Rna from virus-infected plant and fungal tissue. Phytopathology 69: 854–858. [Google Scholar]
- Newhouse J.R., Hoch H.C., and MacDonald W.L. (1983) The ultrastructure of Endothia parasitica. Comparison of a virulent with a hypovirulent isolate. Can J Bot 61: 389–399. [Google Scholar]
- Okada R., Kiyota E., Sabanadzovic S., Moriyama H., Fukuhara T., Saha P., et al. (2011) Bell pepper endornavirus: molecular and biological properties, and occurrence in the genus Capsicum. J Gen Virol 92: 2664–2673. [DOI] [PubMed] [Google Scholar]
- Okada R., Alcalá-Briseño R.I., Escalante C., Sabanadzovic S., and Valverde R.A. (2018a) Genomic sequence of a novel endornavirus from Phaseolus vulgaris and occurrence in mixed infections with two other endornaviruses. Virus Res 257: 63–67. [DOI] [PubMed] [Google Scholar]
- Okada R., Ichinose S., Takeshita K., Urayama S.-I., Fukuhara T., Komatsu K., et al. (2018b) Molecular characterization of a novel mycovirus in Alternaria alternata manifesting two-sided effects: Down-regulation of host growth and up-regulation of host plant pathogenicity. Virology 519: 23–32. [DOI] [PubMed] [Google Scholar]
- Potgieter A., Page N., Liebenberg J., Wright I., Landt O., and Van Dijk A. (2009) Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J Gen Virol 90: 1423–1432. [DOI] [PubMed] [Google Scholar]
- Rambaut, A. (2014) FigTree 1.4. 2 software. Institute of Evolutionary Biology, University of Edinburgh. URL: http://tree.bio.ed.ac.uk/software/figtree/.
- Rognes T., Flouri T., Nichols B., Quince C., and Mahé F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck M.J., Saha P., Wiley G.B., Quan J., White J.D., Lai H., et al. (2010) Ecogenomics: using massively parallel pyrosequencing to understand virus ecology. Mol Ecol 19 Suppl 1: 81–88. [DOI] [PubMed] [Google Scholar]
- Shi M., Lin X.D., Tian J.H., Chen L.J., Chen X., Li C.X., et al. (2016) Redefining the invertebrate RNA virosphere. Nature 540: 539–543. [DOI] [PubMed] [Google Scholar]
- Shi M., Lin X.D., Chen X., Tian J.H., Chen L.J., Li K., et al. (2018) The evolutionary history of vertebrate RNA viruses. Nature 556: 197–202. [DOI] [PubMed] [Google Scholar]
- Silveira C.B., Coutinho F.H., Cavalcanti G.S., Benler S., Doane M.P., Dinsdale E.A., et al. (2020) Genomic and ecological attributes of marine bacteriophages encoding bacterial virulence genes. BMC Genomics 21: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura N. (1978) Further analysts of the data by akaike’s information criterion and the finite corrections. Commun Stat Theory Methods 7: 13–26. [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe A.S. (2011) Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour 11: 914–921. [DOI] [PubMed] [Google Scholar]
- Thurber R.V., Payet J.P., Thurber A.R., and Correa A.M. (2017) Virus-host interactions and their roles in coral reef health and disease. Nat Rev Microbiol 15: 205–216. [DOI] [PubMed] [Google Scholar]
- Tipper D.J., and Schmitt M.J. (1991) Yeast dsRNA viruses: replication and killer phenotypes. Mol Microbiol 5: 2331–2338. [DOI] [PubMed] [Google Scholar]
- Urayama S., Takaki Y., and Nunoura T. (2016) FLDS: A comprehensive dsRNA sequencing method for intracellular RNA virus surveillance. Microbes Environ 31: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama S., Takaki Y., Nishi S., Yoshida-Takashima Y., Deguchi S., Takai K., and Nunoura T. (2018) Unveiling the RNA virosphere associated with marine microorganisms. Mol Ecol Resour 18: 1444–1455. [DOI] [PubMed] [Google Scholar]
- Urayama S., Takaki Y., Hagiwara D., and Nunoura T. (2020) dsRNA-seq reveals novel RNA virus and virus-like putative complete genome sequences from hymeniacidon sp. sponge. Microbes Environ 35: ME19132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreede F., Cloete M., Napier G., Van Dijk A., and Viljoen G. (1998) Sequence-independent amplification and cloning of large dsRNA virus genome segments by poly (dA)-oligonucleotide ligation. J Virol Methods 72: 243–247. [DOI] [PubMed] [Google Scholar]
- Waldron F.M., Stone G.N., and Obbard D.J. (2018) Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet 14: e1007533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y.I., Kazlauskas D., Iranzo J., Lucia-Sanz A., Kuhn J.H., Krupovic M., et al. (2018) Origins and evolution of the global RNA virome. mBio 9: e02329-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets supporting the results of the present study are available in the GenBank database repository (Accession Nos. DDBJ: LC521321–LC521329) and Short Read Archive database (Accession No. DDBJ: DRA009245).