

Abstract
Cell-penetrating peptides (CPPs) are capable of delivering membrane-impermeable cargoes (including small molecules, peptides, proteins, nucleic acids, and nanoparticles) into the cytosol of mammalian cells and have the potential to revolutionize biomedical research and drug discovery. However, the mechanism of action of CPPs has remained poorly understood, especially how they escape from the endosome into the cytosol following endocytic uptake. We show herein that CPPs exit the endosome by inducing budding and collapse of CPP-enriched vesicles from the endosomal membrane. This mechanism provides a theoretical basis for designing CPPs and other delivery vehicles of improved efficiencies.
Keywords: Cell-penetrating peptide, cyclic peptide, drug delivery, endosome escape, mechanism
Graphical Abstract
INTRODUCTION
Cell-penetrating peptides (CPPs) are capable of delivering a variety of membrane-impermeable cargos (e.g., peptides, proteins, nucleic acids, and nanoparticles) into the cytosol of mammalian cells and therefore have enormous potential in biomedical research and drug discovery.1–3 It is generally established that at low concentrations, CPPs enter the cell primarily through one or more endocytosis mechanisms followed by endosomal escape, whereas at high concentrations some of the CPPs can also directly translocate across the plasma membrane in an energy-independent fashion.4–6 Regardless of the entry mechanism (endocytosis or direct translocation), the CPPs must topologically cross a lipid bilayer at some point in order to reach the cytosolic space. The mechanism by which CPPs translocate across the lipid bilayer, especially when conjugated to a macromolecular cargo, remains poorly understood.
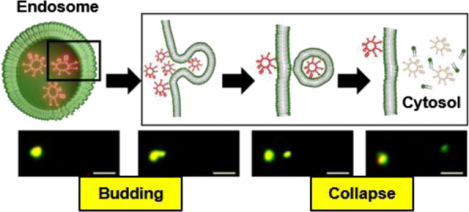
We recently discovered a family of highly active cyclic CPPs which enter the cell by endocytosis.7–9 Unlike most of the previous CPPs, the cyclic CPPs efficiently escape from the early endosome into the cytosol. Based on experimental observations with model membranes, we hypothesized that the cyclic CPPs exit the endosome by a vesicle budding and collapse mechanism (Fig. 1A).9 In brief, cyclic CPPs bind to the endosomal membrane to form CPP-enriched lipid domains, which bud off the endosome as small CPP-loaded vesicles. The budded vesicles disintegrate into amorphous peptide/lipid aggregates, either as or shortly after they bud off the endosomal membrane, thereby releasing their luminal contents into the cytosol. The acidic pH inside the endosome facilitates this process by increasing the binding affinity of the CPPs for the endosomal membrane.9 Since the CPPs do not physically traverse any cell membrane, this mechanism readily explains how CPPs deliver macromolecular cargoes (e.g., proteins and nanoparticles) into the cytosol of mammalian cells. In this study, we demonstrate for the first time that CPPs and a nonpeptidic cell-penetrating molecule exit the endosome by the vesicle budding and collapse mechanism in live mammalian cells.
Figure 1.
Vesicle budding and collapse in live cells induced by cyclic CPP12. (A) The proposed mechanism for the endocytic uptake of cyclic CPPs and subsequent escape from the early/late endosome by inducing budding and collapse of vesicles. (B) The structure of CPP12pHAb. (C) Time-lapse confocal microscopy images of a single endosome inside a HeLa cell. Cells were incubated with PSTopFluor (1.5 μM, green channel) for 15 min on ice, washed with DPBS, treated with CPP12pHAb (2.5 μM, red channel) for 10 min, and imaged every 6 s. Left panel, merged image of a HeLa cell (DIC, green, and red channels). The area boxed in red is enlarged and shown in the right panel. Right panel, images of a single endosome at 119–143 s after initiation of imaging. The endosome split into two vesicles at 137 s and at 143 s, the smaller vesicle traveled to the upright corner and collapsed while the larger one remained intact. Scale bars = 2 and 20 μm.
RESULTS AND DISCUSSION
Cyclic CPP12 induces vesicle budding and collapse from endosomal membrane.
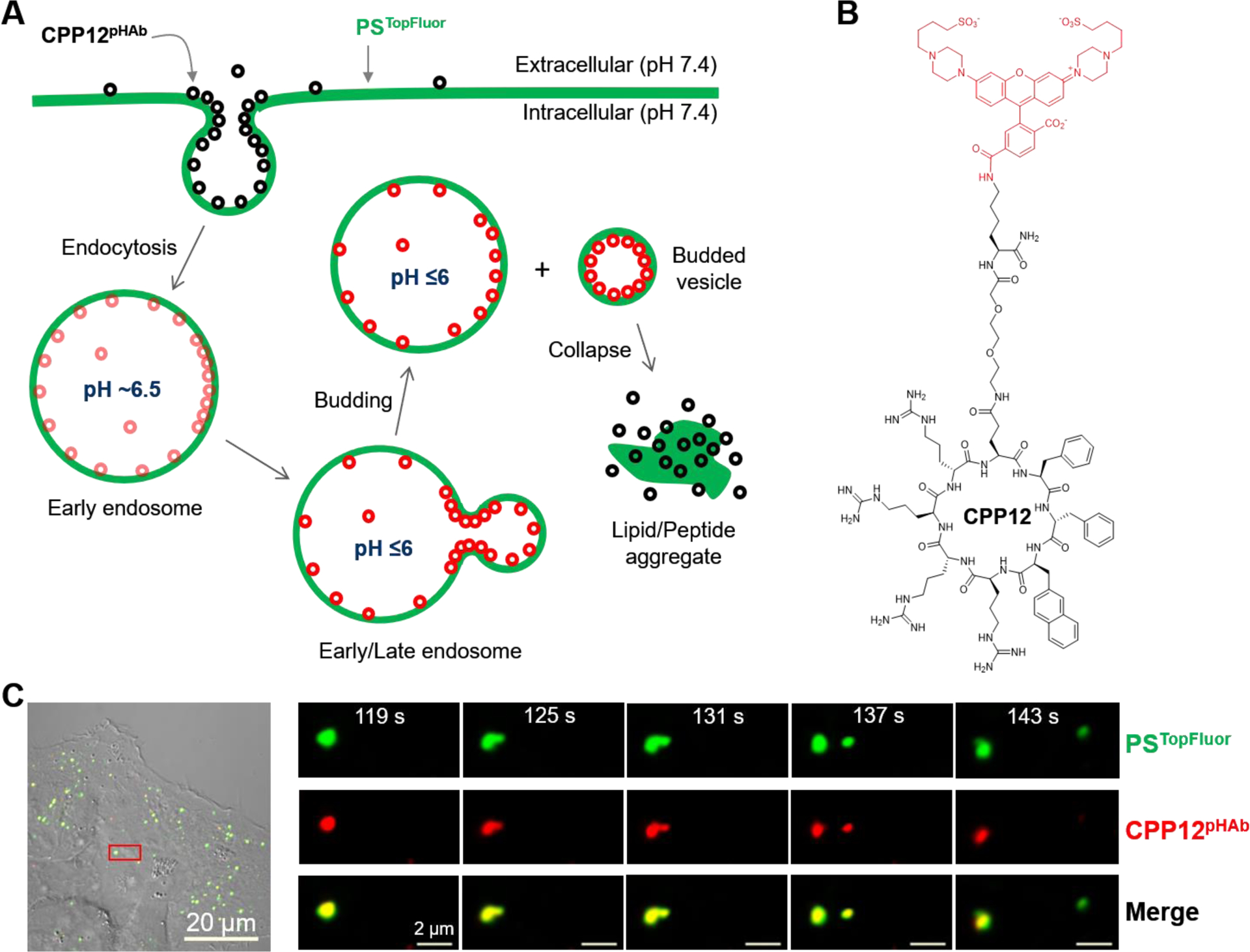
To test the vesicle budding and collapse hypothesis in a cellular context, we labeled CPP12,9 one of the most active cyclic CPPs, with a pH-sensitive fluorescent dye, 3′,6′-bis(4-(4-sulfobutyl)piperazin-1-yl) rhodamine (5′,6′) carboxylic acid (pHAb),10 through a flexible linker (Fig. 1B). With an apparent pKa value of 6.2, pHAb emits strong fluorescence (λEM 560 nm) when inside the acidic environments of the endosome (pH 5.5–6.5) and lysosome (pH 4.5–5.5), but is only weakly fluorescent in the cytosolic/nuclear or extracellular space (pH 7.4). Prior to treatment with the pHAb-labeled CPP12 (CPP12pHAb), HeLa (human cervical cancer) cells were pulse labeled with phospholipid 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phospho-L-serine (PSTopFluor) to visualize the plasma and intracellular membranes (green channel). The cells were then imaged by time-lapse, live-cell confocal microscopy at 6-s intervals. According to the proposed mechanism (Fig. 1A), endosomes and lysosomes inside the treated cells should display both red (CPP12pHAb) and green fluorescence (PSTopFluor). Immediately after a small vesicle buds off an endosome, the budded vesicle as well as the remaining endosome should also have both red and green fluorescence. However, once the budded vesicle collapses and loses membrane integrity, exposing the resulting lipid/peptide aggregate to the cytosol (pH 7.4), CPP12pHAb fluorescence (red) should immediately disappear whereas PSTopFluor fluorescence (green) should stay. As shown in Fig. 1C, Fig. S1, and Movie S1, this sequence of events was indeed captured by confocal microscopy in live HeLa cells upon treatment with PSTopFluor and CPP12pHAb. All together, we observed six unambiguous CPP12-induced endosomal budding and collapse events, which typically spanned ≤60 s. The lingering fluorescence of PSTopFluor demonstrates that phospholipids are a component(s) of the vesicle remnants, which gradually dissolve into the cytosolic milieu. It is worth noting that, due to technical limitations of confocal microscopy, we were only able to observe the rare events during which both the budded vesicle and the remaining endosome stayed within the same focal plane over the entire budding and collapse process. More frequently, we observed budding of vesicles from endosomes but not the subsequent collapse before the budded vesicles moved out of focus. Some endosomes proceeded to various vesicle budding intermediates but eventually failed to bud. We did not observe any collapse event without a prior or simultaneous budding event.
We also treated HeLa cells simultaneously with equal concentrations of CPP12pHAb and CPP12 labeled with fluorescein isothiocyanate (CPP12FITC). CPP12pHAb and CPP12FITC should bind to cell membranes (and potentially other cellular components) and enter the cell similarly. However, CPP12FITC should remain fluorescent (green) throughout the entire cellular entry process (i.e., inside the endosome/lysosome as well as the cytosol/nucleus). We again observed budding and collapse of small vesicles from intracellular membranous structures (total 5 events), as evidenced by the selective disappearance of CPP12pHAb fluorescence (Fig. 2, Fig. S2 and Movie S2). Retention of CPP12FITC fluorescence indicates that CPP12 is also a component of the lipid/peptide aggregate. The cells contained more green (than red) fluorescence puncta; in fact, for every red fluorescence punctum, there was usually a corresponding green signal, but the reverse was not true (Fig. 2 and Fig. S3). Some of the green fluorescence puncta were quite large (up to 2 μm in length). These green-only puncta are likely the aggregates of disrupted vesicles but could also be other intracellular organelles bound by cytosolic CPP12FITC.
Figure 2.
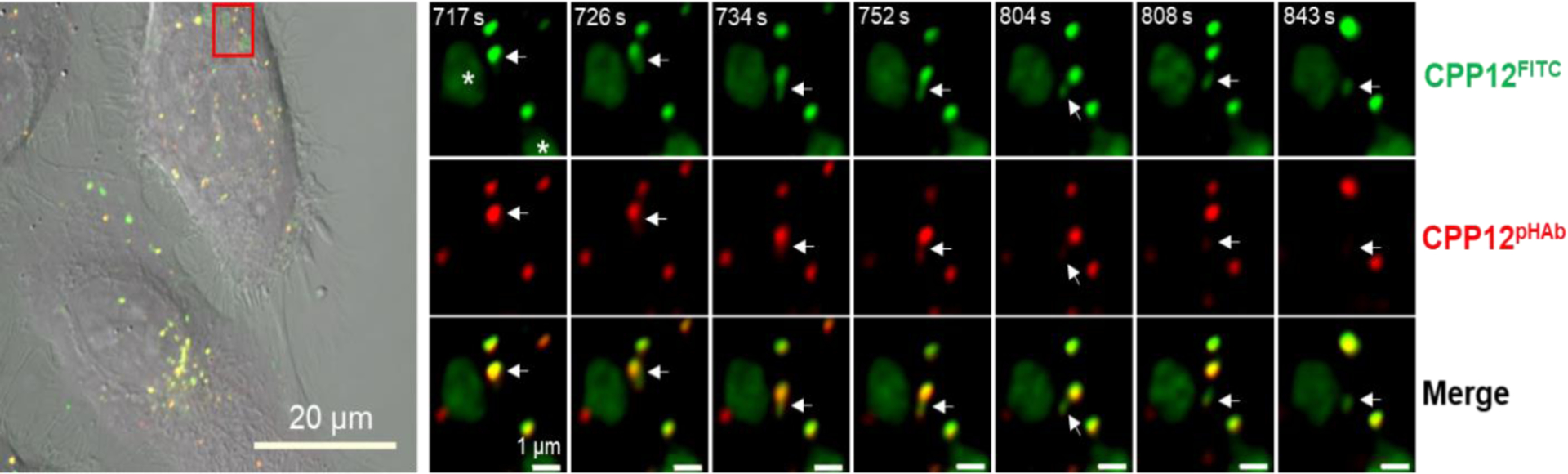
A vesicle budding and collapse event visualized by dual labeled CPP12. HeLa cells were treated simultaneously with CPP12FITC (2 μM, green channel) and CPP12pHAb (2 μM, red channel) for 30 min, washed, and imaged every 4.5 s. Left panel, merged image of a HeLa cell (DIC, green, and red channels). The area boxed in red is enlarged and shown in the panel to the right. Right panel, the endosome undergoing vesicle budding and collapse is indicated by a white arrow. Green fluorescent structures marked by “*” are likely lipid/peptide aggregates derived from collapsed vesicles or intracellular organelles bound with cytosolic CPP12FITC. Scale bars = 1 and 20 μm.
Visualization of vesicle budding and collapse intermediates in enlarged endosomes.
To gain insight into how CPPs induce vesicle budding and collapse from the endosomal membrane, we enlarged the endosomes by treating cells with a phosphatidylinositol-3-phosphate kinase-FYVE inhibitor, YM201636,11 prior to the addition of tetramethylrhodamine (TMR)-labeled CPP12 (CPP12TMR) and AlexaFluor488-labeled 10-kDa dextran (DextranAlexa). Treatment of HeLa cells with 800 nM YM201636 for 2 h increased the diameters of their endosomes from ~0.5 to ~2 μm, allowing us to visualize the various intermediates along the vesicle budding/collapse pathway by live-cell confocal microscopy (Fig. 3). At the earliest stage (stage I), CPP12TMR bound to and was relatively evenly distributed over the endosomal membrane, while DextranAlexa (an endosomal marker) was uniformly distributed throughout the endosomal volume (Fig. 3A). At stage II, CPP12TMR clustered into one or several highly fluorescent foci on the endosomal membrane (Fig. 3B). At stage III, endosomes in the midst of vesicle budding and collapse were readily visible (Fig. 3C, D). In some cases, the budded small vesicle showed both red (CPP12TMR) and green fluorescence (DextranAlexa), indicating that budding was ongoing or had just taken place but before the budded vesicle collapsed. In other cases, the budded vesicle/aggregate, which was usually in the vicinity of an intact endosome(s), showed intensively red but little green fluorescence. This is consistent with a recently collapsed vesicle, as the fluorescence of CPP12TMR is insensitive to pH and remains constant before and after endosomal escape, whereas the water-soluble DextranAlexa rapidly diffuses away from the collapsed vesicle. In still other cases, instead of budding of small vesicles, tubular structures extended from endosomes and collapsed. Finally, at stage IV, irregularly shaped red fluorescence puncta, which were not matched by any DextranAlexa signal, were often (although not always) found in the vicinity of intact endosomes (Fig. 3D). These likely represent the remnants of recently collapsed vesicles/tubules. A representative budding and collapse event encompassing all above stages is shown in Fig. 3E and Movie S3.
Figure 3.
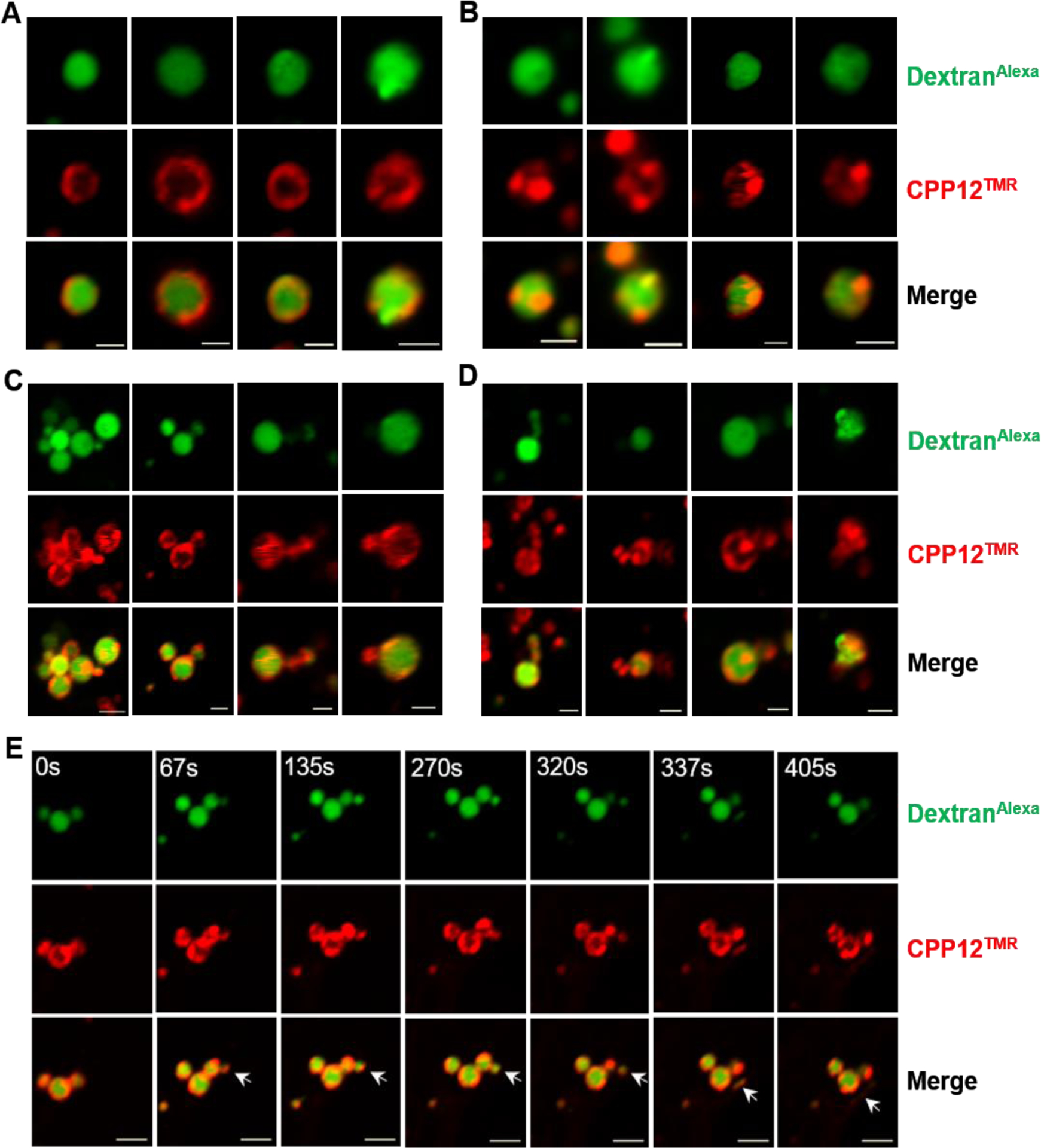
CPP12-induced vesicle budding and collapse from enlarged endosomes. HeLa cells were pretreated with YM201636 (800 nM) for 2 h, then CPP12TMR (2 μM, red channel) and DextranAlexa (50 μg/mL, green channel) were added. After incubation for 30–40 min, the cells were washed and imaged by live-cell confocal microscopy. (A-D) Images of different endosomes at different stages of vesicle budding and collapse. (E) Time-lapse images showing a vesicle budding and collapse event from a single endosome (indicated by an arrow). Scale bars = 2 μm.
Cyclic CPP12 induces endosomal escape of unassociated cargo.
Each vesicle budding and collapse event is accompanied by the release of a small endosomal volume into the cytosol (Fig. 1A), suggesting that CPPs should enhance the endosomal escape of molecules that are neither covalently attached to nor physically associated with the CPPs. We tested this notion by examining the effect of CPP12 and Tat (YGRKKRRQRRR), a prototypical linear CPP,12 on the cellular entry of TMR-labeled (DextranTMR) and naphthofluorescein (NF)-labeled 10-kDa dextran (DextranNF) by flow cytometry. Neither CPP12 nor Tat (at 5 μM) had significant effect on the mean fluorescence intensity (MFI) of HeLa cells treated with DextranTMR, which represents the total cellular uptake of dextran by endocytic mechanisms (Fig. 4A,B). On the other hand, CPP12 (but not Tat) increased the MFI value of HeLa cells incubated with DextranNF by 2.2-fold (Fig. 4C,D). Since NF (pKa = 7.8) is fluorescent in the cytosol/nucleus but non-fluorescent inside the acidic endosome/lysosome, the MFI of DextranNF treated cells reflects the amount of dextran that successfully reached the cytosol.13 These results demonstrate that CPP12 (but not Tat) significantly enhanced the endosomal release of dextran. Other CPPs have also been reported to enhance the endosomal release of non-associated cargoes.14–16
Figure 4.
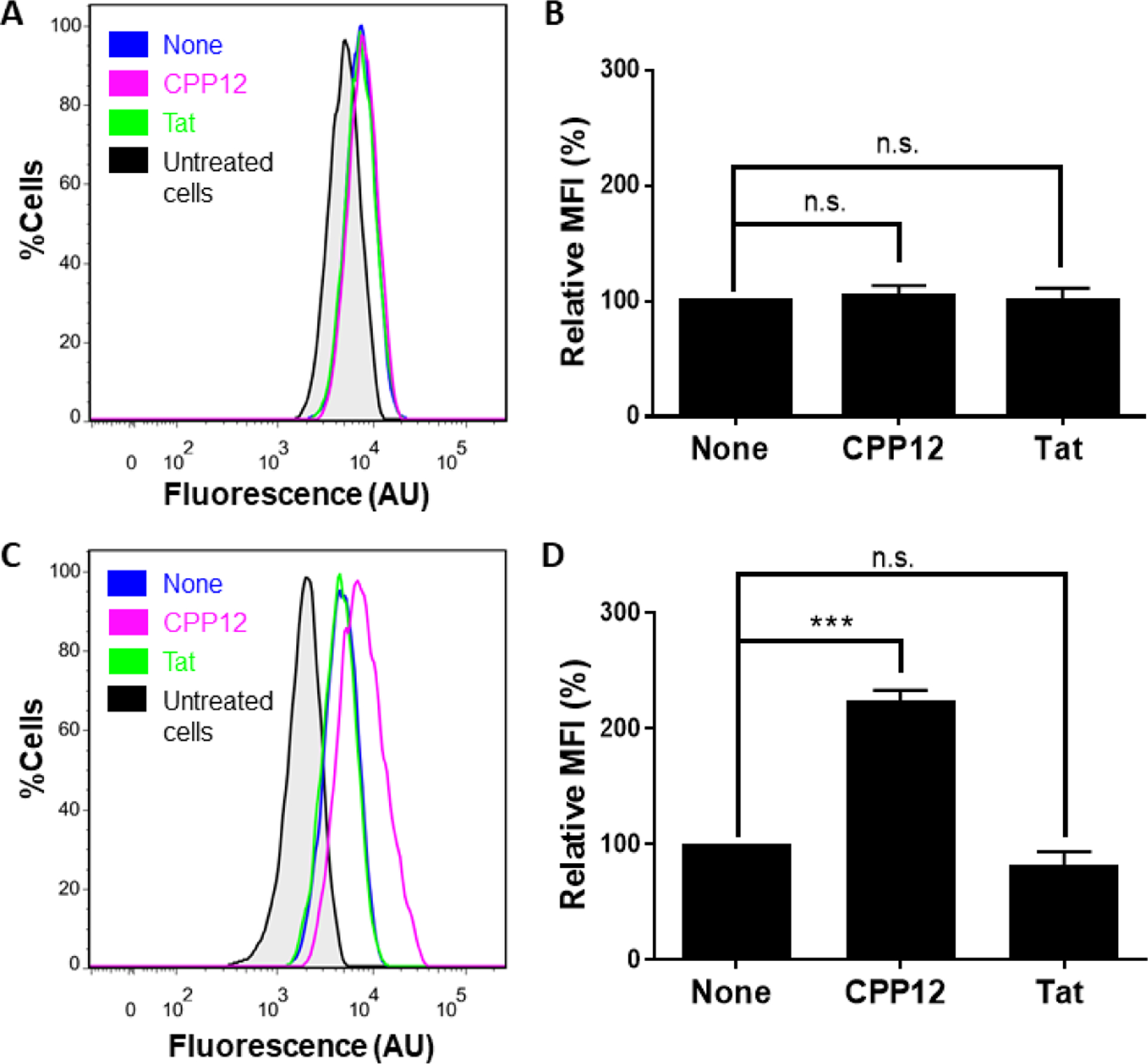
Effect of CPPs on the total cellular uptake (A,B) and endosomal release of dextran (C,D). HeLa cells were incubated for 2 h with 200 μg/mL DextranTMR (A,B) or DextranNF (C,D) in the absence or presence of 5 μM CPP12 or Tat and analyzed by flow cytometry (A and C). (B,D) Comparison of the MFI values of different cell cultures from A and C, respectively. The MFI values are relative to that in the absence of CPP (none; 100%) and represent the mean ± SD for ≥3 sets of independent experiments. n.s., p ≥ 0.05; ***, p < 0.001.
Induction of vesicle budding and collapse by other cell-penetrating molecules.
To test whether the vesicle budding and collapse mechanism is unique to CPP12, we repeated the live-cell imaging experiments with two other cell-penetrating molecules, Tat and CPM3 (Figure 5A). As a prototypical first-generation CPP, Tat efficiently enters mammalian cells by endocytic mechanisms17–20 but has poor endosomal escape efficiency [~18-fold lower than that of CPP12].9 CPM3, on the other hand, is a member of the recently discovered nonpeptidic cell-penetrating motifs (CPM), which exit the endosome almost completely and are subsequently localized into the mitochondrial matrix.21 As expected, CPM3 behaved similarly to CPP12 and induced robust vesicle budding and collapse in HeLa cells (Fig. 5B,C and Fig. S4). In contrast, vesicle budding and especially collapse events were infrequent in Tat-treated cells. Despite many attempts, we have only observed a single Tat-mediated vesicle budding and collapse event (Fig. 6).
Figure 5.
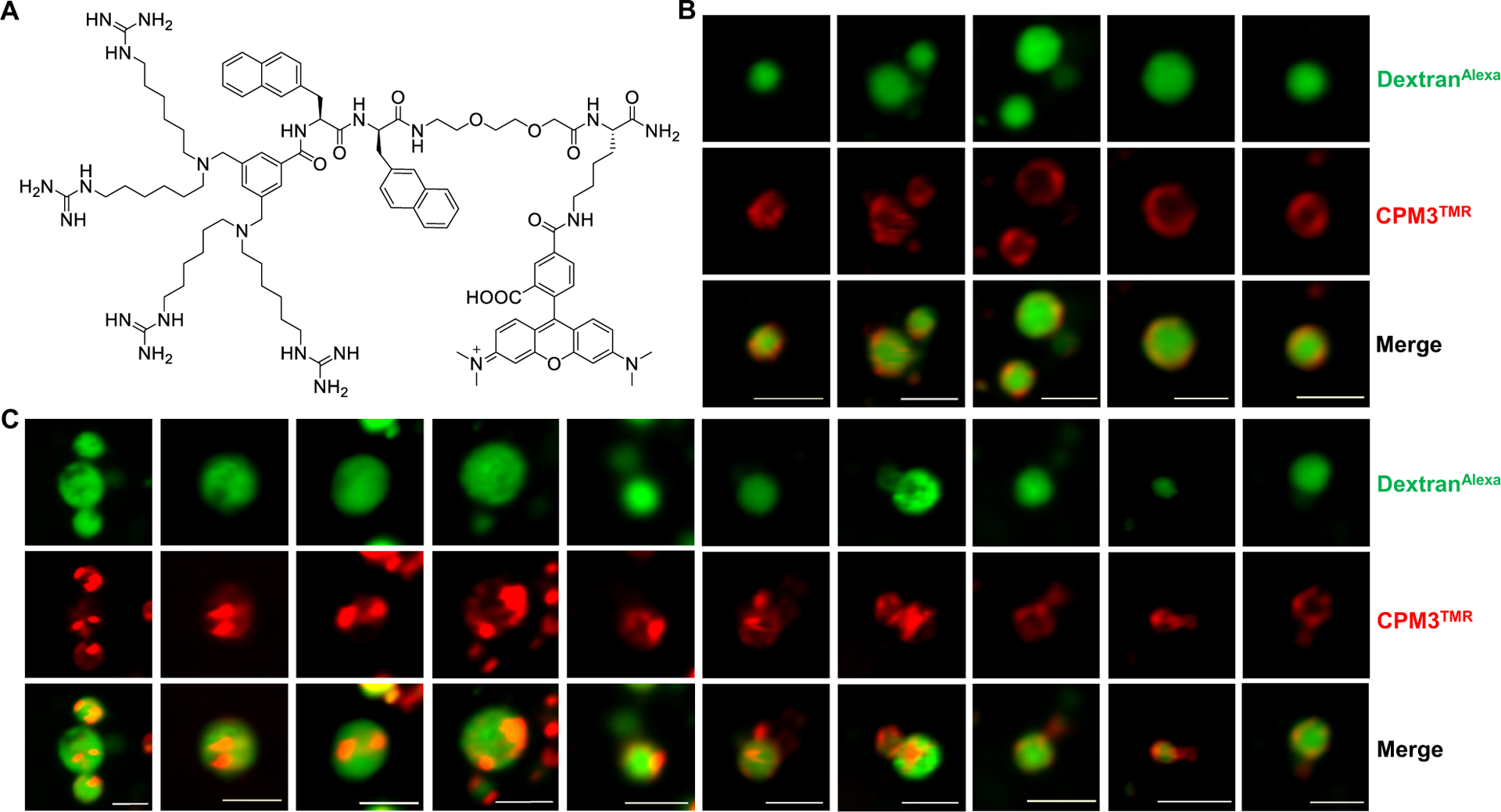
CPM3-induced vesicle budding and collapse from enlarged endosomes. (A) Structure of CPM3TMR. (B,C) Live-cell confocal microscopic images of endosomes at different stages of vesicle budding and collapse process. HeLa cells were pretreated with YM201636 (800 nM) for 2 h, and CPM3TMR (2 μM, red channel) and DextranAlexa (50 μg/mL, green channel) were added. After incubation for another 30–40 min, the cells were washed and imaged by live-cell confocal microscopy. The images shown are snapshots of different endosomes from multiple cells. Scale bars = 2 μm.
Figure 6.
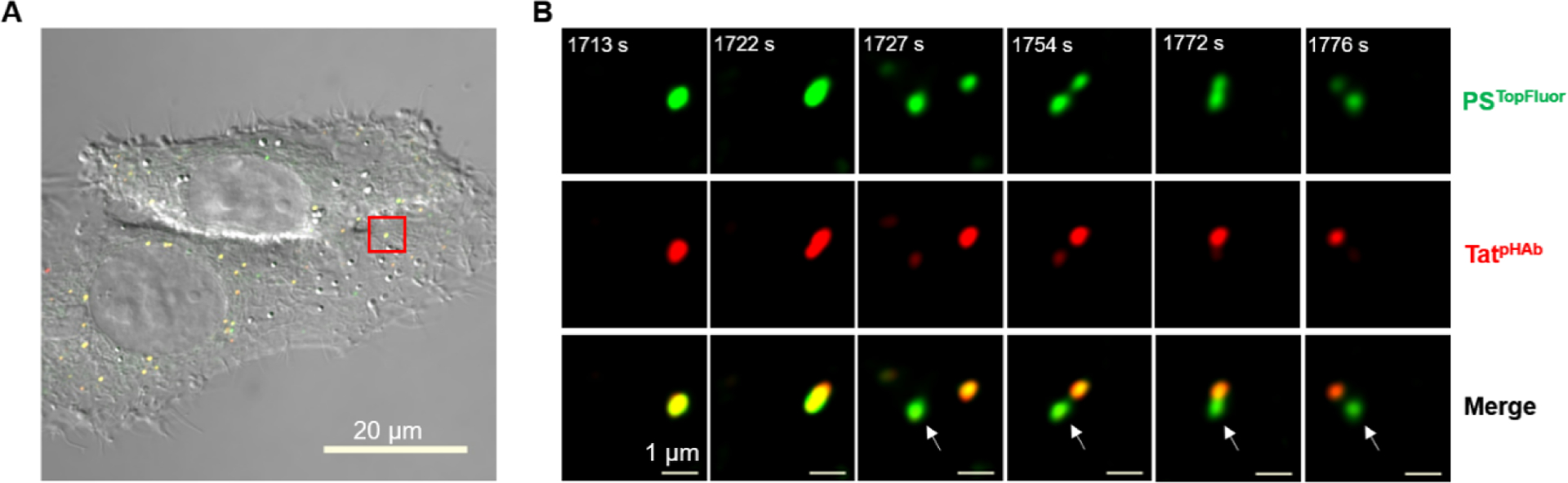
Vesicle budding and collapse in live cells induced by Tat. Cells were incubated with psTopFluor (1.5 μM, green channel) for 15 min on ice, washed with DPBS, treated with TatpHAb (2.5 μM, red channel) for 5 min, and imaged every 4.5 s. (A) Merged image of two HeLa cells (DIC, green, and red channels). (B) Time-lapse confocal microscopic images of the area boxed in red from (A) at 1717–1731 s after initiation of imaging. A single endosome split into two vesicles at 1727 s and one of the vesicles collapsed (marked by a white arrow) while the other remained intact. Scale bars = 1 and 20 μm.
Discussion.
In this work, we provide direct evidence that CPPs and CPMs escape the endosome by inducing vesicle budding and collapse from the endosomal membrane. This mechanism reconciles many of the previously confusing and/or conflicting observations in the CPP field, e.g., how CPPs deliver macromolecular cargos across a lipid bilayer,8,14,22 or how they enhance the endosomal release of cargos that are not conjugated or physically associated with the CPPs.14–16 Most importantly, the mechanism offers critical insights into what structural features facilitate endosomal escape and how to design intracellular delivery vehicles of improved endosomal escape and cytosolic entry efficiencies. In principle, the endosomal escape efficiency is governed by the probability of each endosome to undergo vesicle budding and collapse, how fast each budding and collapse event occurs, and the percentage of endosomal cargo released by each budding and collapse event. Our results show that each budding and collapse event occurs quickly, typically spanning <60 s (although the process appears to be slower in enlarged endosomes). Although we have not yet precisely determined the percentage of CPP/CPM molecules released by each budding and collapse event, the relative fluorescence intensities of the budded vesicles and the remaining endosomes suggest that this value is also relatively uniform and typically from a few percent to 50%. The endosomal escape efficiency for a given CPP/CPM is therefore primarily determined by the frequency of the CPP/CPM-induced budding and collapse events. Indeed, CPP12 and CPM3 induced robust vesicle budding and collapse in HeLa cells and exhibit high endosomal escape efficiencies.9,21 In contrast, Tat was much less effective in promoting vesicle budding/collapse and remains largely entrapped inside the endosome. During a vesicle budding event, the budding neck features acute Gaussian curvatures (i.e., simultaneous positive and negative membrane curvatures in orthogonal directions) and high potential energy (Fig. 1A). To promote vesicle budding, a CPP/CPM needs to bind selectively to the budding neck and stabilize the membrane curvatures at the neck.23 Among the recently discovered CPPs and CPMs of high endosomal escape efficien-cies,9,15,21,24 most are structurally constrained (e.g., by cyclization or α-helix formation) and amphipathic (i.e., containing both guanidinium and hydrophobic groups). It has been hypothesized that structural constraints enhance the membrane binding affinity, while the hydrophobic and guanidinium groups induce positive and negative membrane curvatures, respectively.23 It is not yet clear what causes the budded vesicles to collapse, although their small size and high membrane curvature likely render them intrinsically unstable.
Several other mechanistic hypotheses have previously been put forth to explain the endosomal release of CPPs and other cell-permeable modalities, including “proton sponge effect” and osmotic lysis of the endosome,25 vesicle fusion with the endosomal membrane,26 pore formation on the endosomal membrane,27,28 and local disruption of the endosomal membrane.29,30 Among them, pore formation and local membrane disruption have been invoked for CPP-mediated endosomal escape.31,32 While the current study does not disprove the previous hypotheses, the availability of an alternative mechanism (budding and collapse) calls for a reevaluation of the experimental observations used to support the former. Because pore formation and local membrane disruption mechanisms require a cargo molecule to physically cross a lipid bilayer, they are inherently incompatible with macromolecular cargos. It remains to be determined whether they contribute to the endosomal release of low-molecular-weight entities (e.g., CPPs and CPP-cargo conjugates), along with vesicle budding and collapse.
CONCLUSION
In conclusion, our current as well as previous results7–9 demonstrate that CPPs and CPMs enter mammalian cells by endocytosis and exit the endosome by inducing vesicle budding and collapse from the endosomal membrane. This mechanistic understanding should facilitate the design of CPPs and other drug delivery vehicles of improved endosomal escape efficiencies.
EXPERIMENTAL SECTION
Materials.
Reagents for peptide synthesis were purchased from Chem-Impex (Wood Dale, IL), NovaBiochem (La Jolla, CA), or Anaspec (San Jose, CA). Rink amide resin (100–200 mesh, 0.43 mmol/g) and fluorescein isothiocyanate isomer I were purchased from Chem-Impex (Wood Dale, IL). Tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] was purchased from Sigma-Aldrich (St. Louis, MO) and phenylsilane was purchased from TCI America. O-Benzotriazole-N,N,N,N-tetramethyl-uronium hexafluorophosphate (HATU) and (benzotriazol-1-yloxy)-tripyrrolidinophosphonium hexafluorophosphate (PyBOP) were from Matrix Scientific (Columbia, SC). HeLa cells were obtained from ATC (Manassas, VA). All solvents and other chemical reagents were obtained from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and were used without further purification. Cell culture media, fetal bovine serum (FBS), penicillin-streptomycin, 0.25% trypsin-EDTA, DPBS (2.7 mM potassium chloride, 1.5 mM monopotassium phosphate, 8.9 mM disodium hydrogen phosphate, and 137 mM sodium chloride) were purchased from Sigma-Aldrich. 5(6)-Carbox-ynaphthofluorescein succinimidyl ester (NF-NHS) was purchased from Setareh Biotech (Eugene, OR). 5(6)-Carboxytet-ramethylrhodamine succinimidyl ester (TMR-NHS ester) was from ThermoFisher Scientific (Waltham, MA). Fluorescent pHAb dyes (amine reactive and thiol reactive) were purchased from Promega (Madison, WI). PSTopFluor (ammonium salt) was purchased from Avanti Polar Lipids (Alabaster, AL). Endosome maturation inhibitor YM201636 was purchased from Cayman Chemical Company (Ann Arbor, MI). Dextran AlexaFluor488 (10,000 MW, anionic), dextran tetramethylrhodamine (10,000 MW, neutral), and dextran amino (10,000 MW, 2.5 moles amine/mole) were purchased from Invitrogen (Carlsbad, CA).
Peptide synthesis.
Peptides were manually synthesized on Rink amide resin LS (0.43 mmol/g) using standard Fmoc chemistry. The typical coupling reaction contained 5 equiv. of Fmoc-amino acid, 5 equiv. of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluoro-phosphate (HATU), 10 equiv. of diisopropylethylamine (DIPEA), and was allowed to proceed with mixing for 40 min at room temperature (RT). For CPP12, after the addition of the last (N-terminal) residue, the allyl group on the C-terminal Glu residue was removed by treatment with Pd(PPh3)4 and phenylsilane (0.3 and 10 equiv., respectively) in anhydrous dichloromethane (3 × 15 min). The N-terminal Fmoc group was removed by treatment with 20% piperidine in DMF and the peptide was cyclized by treatment with PyBOP/HOBt/DIPEA (5, 5, and 10 equiv.) in DMF for 3 h. N-Terminal acetylation (for linear peptides, e.g., Tat) was performed by treating the resin-bound peptide with 10 equiv. of acetic anhydride and DIPEA in dichloromethane for 15 min (twice and with mixing). CPM3 was synthesized as previously described.21 After synthesis, the peptides were deprotected and released from the resin by treatment with 92.5:2.5:2.5:2.5 (v/v) TFA/water/1,3-dimethoxybenzene/triisopropylsilane for 3 h at RT. For cysteine-containing peptides, 1,2-ethanedithiol (2.5% v/v) was included in the cleavage cocktail. The solvents were removed by flowing a stream of N2 over the crude peptide solution, following which the peptide residue was triturated with cold diethyl ether (3 times). The crude peptides were purified by reversed-phase HPLC equipped with a semi-preparative Waters XBridge C18 column, eluting with linear gradients of acetonitrile in ddH2O (each containing 0.05% TFA). The purity of the peptides (>95%) was assessed by reversed-phase HPLC equipped with an analytical Waters XBridge C18 column. The authenticity of each peptide was confirmed by high-resolution mass spectrometry (HR-MS) using a custom Bruker 15-Tesla MALDI-FT-ICR instrument. The structures and analytical data of peptides used in this study are provided in Fig. S5.
Fluorescent labeling of peptides.
Peptide labeling with pHAb (amine reactive or thiol reactive), FITC, or TMR-NHS was carried out in the solution phase. For FITC labeling, 2 mg of CPP12-miniPEG-Lys (lyophilized solid) was dissolved in 20 μL of DMF and the pH was adjusted to 8.5 by the addition of a 0.2 M sodium bicarbonate solution. FITC (1.2 equiv.) dissolved in DMF (5 μL) was added and the mixture was incubated at RT for 2 h on a shaker. Peptide labeling with TMR-NHS or NF-NHS followed a similar protocol, except that the reaction was carried out in at pH 8.0 by using a 0.2 M sodium bicarbonate solution. Labeling with pHAb-maleimide also followed a similar protocol but in PBS (pH 7.4). The labeled peptides were purified by HPLC and analyzed by MALDI-FT-ICR mass spectrometry as previously described.
Cell culture.
HeLa cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (hereafter referred to as Abs). Cells were cultured in a humidified incubator at 37 °C in the presence of 5% CO2.
Confocal Microscopy.
All microscopy experiments were performed on live cells. Three hundred μL of HeLa cell suspension (5 × 104 cells/mL) was seeded per compartment in a 35/10 mm glass-bottomed microwell dish with four compartments (Grenier Bio-One) and cultured overnight in DMEM containing 10% FBS and 1% Abs. During incubation and imaging, phenol red-free DMEM (1% FBS, 1% Abs) was used unless specified otherwise. Cells were imaged on a Nikon A1R live-cell confocal laser scanning microscope (ECLIPSE Ti-E automated, inverted) equipped with a 100X oil objective (1.45 N.A.) and a heated (37 °C) chamber supplied with 5% CO2. The galvano scanner was used. Nikon’s Perfect Focus System (PFS) was used for all time-lapse image acquisitions. Differential interference contrast (DIC) images were also obtained to ensure that cells were healthy throughout the imaging process. Data was processed and analyzed on NIS-Elements AR. Typically, images were denoised using Nikon’s “Denoise.ai” feature, and scale bars and time stamps were added where needed. Images were exported to the 8-bit TIFF format. Detailed protocols for cell treatment and imaging parameters are described below.
PSTopFluor and CPP12pHAb.
Before the experiment, cells were incubated on ice for 10 min and washed twice with chilled DPBS. TopFluor-labeled PS (1.5 μM) in chilled media (700 μL per well) was added to the cells and the cells were incubated on ice for 15 min. The cells were gently washed with cold DPBS (4 × 300 μL) and incubated with warm phenol red-free DMEM media containing 1% FBS and 1% Abs (300 μL per well) containing 2.5 μM CPP12pHAb. After 10–15 min, time-lapse imaging was commenced, with images acquired every 6 s. For the red channel (CPP12pHAb), the laser line with λEx 561 nm was set at 0.6% laser power, while for the green channel (PSTopFluor), the laser line with λEx 487 nm was set at 1.0 % laser power. Experiments with TatpHAb were similarly carried out.
CPP12FITC and CPP12pHAb.
Cells were washed twice with warm DPBS and then incubated with 2 μM CPP12FITC and 2 μM CPP12pHAb in phenol red-free DMEM containing 1% FBS and 1% Abs (total volume = 300 μL per well) for 30 min at 37 °C. The cells were gently washed with warm DPBS (twice) and incubated with 300 μL phenol red-free DMEM media. After ~10 min, time-lapse imaging was commenced, with images acquired every 4.5 s. For the red channel (CPP12pHAb), the laser line with λEx 561 nm was set at 1.0% laser power. For the green channel (CPP12FITC), the laser line with λEx 487 nm was set to 0.6% laser power.
Enlarged Endosomes with YM201636.
Cells were washed twice with warm DPBS and then incubated with 800 nM YM201636 in phenol red-free DMEM containing 1% FBS and 1% Abs (300 μL per well) for 2 h. CPP12TMR (2 μM) and DextranAlexa (50 μg/mL) were added to the cells and the cells were incubated for another 30–40 min. The cells were gently washed with warm DPBS (twice) to remove the compounds and incubated in phenol red-free DMEM (300 μL). Cells with swollen intracellular vesicles were isolated and subjected to time-lapse imaging at 10- to 30-s intervals. For the green channel (DextranAlexa488), the laser line with λEx 487 nm was set at 0.9% laser power. For the red channel (CPP12TMR), the laser line with λEx 561 nm was set to 0.3% laser power. Experiments with CPM3tmr, or with 2 μM CPP12FITC and 2 μM CPP12pHAb, were similarly carried out.
Dextran labeling with NF.
For flow cytometry experiments, dextran amine (10,000 MW, 2.5 moles of amine/mole) was labeled with 5(6)-carboxynaphthofluorescein (NF) dye. Dextran amine was dissolved in PBS buffer (50 mg/mL final concentration), and the pH was adjusted to 8.0 using a sodium bicarbonate solution. NF succinimidyl ester (1.5 equiv; dissolved in DMSO) was added to the dextran solution and the mixture was incubated on a shaker for 1.5 h at room temperature. The solution was dialyzed into doubly distilled water overnight to remove any excess dye using a Slide-A-Lyzer Dialysis cassette (7,000 MWCO, ThermoFisher Scientific) and lyophilized to a powder. The stoichiometry of labeling was estimated to be ~0.14 dye/dextran by measuring the absorbance at 595 nm and the dry weight after lyophilization. For DextranTMR, the degree of labeling was estimated to be ≤3 dye molecules/dextran by the manufacturer, using a similar method.
Flow Cytometry.
HeLa cells were cultured in 12-well plates (1.5 × 105 cells per well) overnight. The cells were washed twice with DPBS and incubated in DMEM containing 1% FBS, 1% Abs, 200 μg/mL DextranTMR (or DextranNF), and 5 μM CPP12 (or Tat) at 37 °C for 2 h. The total treatment volume per well was 1 mL. Cells treated with DextranTMR (or DextranNF) but no CPP were used as the control. Cells treated with media only (i.e. no CPP or dextran) were used as the “blank”. After incubation, the cells were washed with cold DPBS twice, detached from the plate with 0.25% trypsin, diluted into cold DPBS and pelleted at 300 g for 5 min at 4 °C. The supernatant was discarded and the cells were washed twice with cold DPBS in a similar manner. Finally, the cells were resuspended in 200 μL of cold DPBS and analyzed on a BD FACS LSR II flow cytometer. For DextranTMR, a 561-nm laser was used for excitation and the fluorescence was analyzed in the PE channel. For DextranNF, a 633-nm laser was used for excitation and the fluorescence emission was analyzed in the APC channel. Data were analyzed using FlowJo. The mean fluorescence intensity (MFI) value for the “blank” was subtracted from that of other samples. The MFI values reported are relative to that in the absence of CPP (100%) and represent the mean ± SD of three (DextranNF) or four independent experiments (DextranTMR). Excel was used to calculate the P values for the Student’s t test using the formula for a two-tailed distribution (two-sample assuming equal variances).
Supplementary Material
ACKNOWLEDGMENT
We thank S. Cole for technical assistance and advice for confocal imaging experiments.
Funding Sources
This work was supported by the National Institutes of Health (GM122459). Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. This facility is supported in part by grant P30 CA016058, National Cancer Institute, Bethesda, MD.
ABBREVIATIONS
- CPP
cell penetrating peptide
- CPM
cell penetrating motif
Footnotes
Supporting Information. Associated data provided:
Figures S1 to S5 (PDF)
Captions for Movies S1 to S3:
Movie S1. A CPP12-induced vesicle budding and collapse event in a HeLa cell.
Movie S2. A vesicle budding and collapse event visualized with dual labeled CPP12.
Movie S3. A CPP12-induced vesicle budding and collapse event from an enlarged endosome.
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): D.P. is a co-founder and shareholder of Entrada Therapeutics.
REFERENCES
- (1).Futaki S Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Delivery Rev 2005, 57, 547–558. [DOI] [PubMed] [Google Scholar]
- (2).Stanzl EG; Trantow BM; Vargas JR; Wender PA Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc. Chem. Res 2013, 46, 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bechara C; Sagan S Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- (4).Madani F; Lindberg S; Langel U; Futaki S; Gräslund A Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys [Online] 2011, 2011, Article 414729. 10.1155/2011/414729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Palm-Apergi C; Lönn P; Dowdy SF Do cell-penetrating peptides actually “penetrate” cellular membranes? Mol. Ther 2012, 20, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Takeuchi T; Futaki S Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull 2016, 64, 1431–1437. [DOI] [PubMed] [Google Scholar]
- (7).Qian Z; Liu T; Liu Y-Y; Briesewitz R; Barrios AM; Jhiang SM; Pei D Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Qian Z; LaRochelle JR; Jiang B; Lian W; Hard RL; Sel-ner NG; Luechapanichkul R; Barrios AM; Pei D Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Robers MB; Binkowski BF; Cong M; Zimprich C; Corona C; McDougall M; Otto G; Eggers CT; Hartnett J; Machleidt T; Fan F; Wood KV A luminescent assay for real-time measurements of receptor endocytosis in living cells. Anal. Biochem 2015, 489, 1–8. [DOI] [PubMed] [Google Scholar]
- (11).Jefferies HBJ; Cooke FT; Jat P; Boucheron C; Koizumi T; Hayakawa M; Kaizawa H; Ohishi T; Workman P; Waterfield MD; Parker PJ A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts en-domembrane transport and retroviral budding. EMBO Rep. 2008, 9, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vives E; Brodin P; Lebleu B A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem 1997, 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- (13).Qian Z; Dougherty PG; Pei D Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluor-ophore. Chem. Commun 2015, 51, 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wadia JS; Stan RV; Dowdy SF Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med 2004, 10, 310–315. [DOI] [PubMed] [Google Scholar]
- (15).Erazo-Oliveras A; Najjar K; La Dayani L; Wang TY; Johnson GA; Pellois JP Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014. 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu H-H; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem 2017, 9, 751–761. [DOI] [PubMed] [Google Scholar]
- (17).Ferrari A; Pellegrini V; Arcangeli C; Fittipaldi A; Giacca M; Beltram F Caveolae-mediated internalization of extracellular HIV-1 Tat fusion proteins visualized in real time. Mol. Ther 2003, 8, 284–294. [DOI] [PubMed] [Google Scholar]
- (18).Fittipaldi A; Ferrari A; Zoppe M; Arcangeli C; Pellegrini V; Beltram F; Giacca MJ Biol. Chem 2003, 278, 34141–34149. [DOI] [PubMed] [Google Scholar]
- (19).Richard JP; Melikov K; Brooks H; Prevot P; Lebleu B; Chernomordik LV Cellular uptake of unconjugated Tat peptide involves clathrin dependent endocytosis and heparan sulfate receptors. J. Biol. Chem 2005, 280, 15300–15306. [DOI] [PubMed] [Google Scholar]
- (20).Kaplan IM; Wadia JS; Dowdy SF Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Controlled Release 2005, 102, 247–253. [DOI] [PubMed] [Google Scholar]
- (21).Appiah Kubi G; Qian Z; Amiar S; Sahni A; Stahelin RV; Pei D Non-peptidic cell-penetrating motifs for mitochondrion-specific cargo delivery. Angew. Chem. Int. Ed. Engl 2018, 57, 17183–17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Morris MC; Vidal P; Chaloin L; Heitz F; Divita G A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dougherty PG; Sahni A; Pei D. Understanding cell penetration of cyclic peptides. Chem Rev. 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Appelbaum JS; LaRochelle JR; Smith BA; Balkin DM; Holub JM; Schepartz A Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem. Biol 2012, 19, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Behr J-P The proton sponge: a trick to enter cells the viruses did not exploit. CHIMIA Int. J. Chem 1997, 51, 34–36. [Google Scholar]
- (26).White JM; Whittaker GR Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shai Y Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [DOI] [PubMed] [Google Scholar]
- (28).Jenssen H; Hamill P; Hancock RE Peptide antimicrobial agents. Clin. Microbiol. Rev 2006, 19, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Varkouhi AK; Scholte M; Storm G; Haisma HJ Endosomal escape pathways for delivery of biologicals. J. Controlled Release 2011, 151, 220–228. [DOI] [PubMed] [Google Scholar]
- (30).Bus T; Traeger A; Schubert US The great escape: how cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [DOI] [PubMed] [Google Scholar]
- (31).Herce HD; Garcia AE; Litt J; Kane RS; Martin P; Enrique N; Rebolledo A; Milesi V Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J 2009, 97, 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brock DJ; Kondow-McConaghy HM; Hager EC; Pellois JP Endosomal escape and cytosolic penetration of macromolecules mediated by synthetic delivery agents. Bio-conjug. Chem 2019, 30, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.