

ABSTRACT
The E2F1 transcription factor and RB tumor suppressor are best known for their roles in regulating the expression of genes important for cell cycle progression but, they also have transcription-independent functions that facilitate DNA repair at sites of damage. Depending on the type of DNA damage, E2F1 can recruit either the GCN5 or p300/CBP histone acetyltransferases to deposit different histone acetylation marks in flanking chromatin. At DNA double-strand breaks, E2F1 also recruits RB and the BRG1 ATPase to remodel chromatin and promote loading of the MRE11-RAD50-NBS1 complex. Knock-in mouse models demonstrate important roles for E2F1 post-translational modifications in regulating DNA repair and physiological responses to DNA damage. This review highlights how E2F1 moonlights in DNA repair, thus revealing E2F1 as a versatile protein that recruits many of the same chromatin-modifying enzymes to sites of DNA damage to promote repair that it recruits to gene promoters to regulate transcription.
KEYWORDS: E2F1, ATM/ATR, TopBP1, GCN5, p300/CBP, BRG1
Introduction
Agents that cause DNA damage, including certain chemicals and radiation, can induce the genetic mutations that underlie cancer but also provide a strategy for treating cancer. Thus, understanding how cells respond to DNA damage is important for understanding both the causes of cancer and the biological responses to many cancer therapeutics. The ATM, ATR, and DNA-PK kinases recognize DNA damage and relay this information throughout the cell to regulate transcription, cell cycle progression and DNA repair [1]. The phosphorylation of target proteins by these kinases, along with signals from other DNA damage sensors, like poly-(ADP-ribose) polymerase 1 (PARP1), results in the efficient recruitment of appropriate DNA repair enzymes to repair different types of DNA damage. An important component of the response to DNA damage is the dynamic modification of chromatin structure at sites of damage that must be coordinated with DNA repair [2–5].
Dozens of sequence-specific, DNA-binding transcription factors have been reported to localize to laser-induced DNA damage tracks [6]. Although recruitment of these transcription factors to sites of DNA damage is likely sequence-independent, the DNA-binding domains of many of these transcription factors are still required for their enrichment at sites of DNA damage [6]. Exceptions include E2F1, ATF2, NR4A, and SP1, which localize to sites of DNA damage independently of their DNA binding domains but may require phosphorylation by ATM, ATR or DNA-PK [7–13]. Loss-of-function experiments demonstrate that E2F1, ATF2, NR4A and SP1 each play important roles in DNA damage response signaling and/or in enhancing DNA repair efficiency, independent of transcription [7,8,12,14]. How these transcription factors directly regulate DNA damage response signaling and repair is largely unknown.
E2F1 has a well-established role in regulating the periodic expression of genes important for cell proliferation [15]. An important regulator of E2F1 is the RB tumor suppressor, which directly binds E2F1 and a subset of other E2F family members. The interaction with RB switches these E2F family members from transcriptional activators to transcriptional repressors. E2F factors can bind RB both in its unphosphorylated form and following RB mono-phosphorylation by cyclin D-CDK4/6 complexes [16]. When RB is hyperphosphorylated by CDK2 during the late G1 and S phases of the cell cycle, E2F factors are released to stimulate the transcription of target genes. Thus, E2F transcriptional activity is tightly controlled by RB during the cell cycle and in non-proliferating cells. Mutations that disrupt this regulation are a hallmark of cancer [17].
In this review, we discuss how E2F1 is converted from a regulator of transcription into a regulator of DNA repair through post-translational modifications induced in response to DNA damage. We also discuss how the RB tumor suppressor protein cooperates with E2F1 at DNA double-strand breaks (DSBs) to promote DNA end-resection and homologous recombination (HR) repair. This expands the known genome maintenance functions of RB and reveals a potential vulnerability of cancer cells lacking RB. E2F1 emerges as a paradigm for factors that regulate both transcription and DNA repair by recruiting the same chromatin-modifying enzymes and directing the same changes to chromatin structure at both gene promoters and at sites of DNA damage.
Regulation of E2F1 in response to DNA damage
In response to DNA damage, both ATM and ATR can phosphorylate E2F1 at a site (serine 31 in humans) that is not conserved in other E2F family members [18]. When phosphorylated at this site, the 14-3-3τ protein binds to and stabilizes E2F1 by inhibiting its ubiquitination and degradation [19]. In response to agents that cause DSBs, E2F1 is phosphorylated by ATM and this is associated with the induction of apoptosis through the transcriptional activation of target genes such as TP73 [18–20]. In contrast, in response to ultraviolet (UV) radiation, E2F1 is phosphorylated by ATR, which is associated with neither the induction of apoptosis nor the transcriptional activation of TP73 [21,22]. Phosphorylation of E2F1 also leads to the formation of a complex containing TopBP1 that suppresses E2F1 transcriptional activity [23].
TopBP1 contains nine BRCT domains and binds to E2F1 through a phospho-specific interaction between its sixth BRCT domain (BRCT6) and the E2F1 serine 31 motif [11]. This interaction was one of the first examples of a BRCT domain binding to a phosphorylated protein motif, now recognized as a general feature of BRCT domains [24,25]. Other TopBP1 BRCT domains bind to phosphorylated motifs on other DNA damage response factors, including RAD9, 53BP1 and MDC1, which helps to recruit TopBP1 to damaged DNA [26,27]. In turn, TopBP1 oligomerization at sites of DNA damage allows the BRCT6 domain to bind and recruit phosphorylated E2F1 [11,28]. Notably, E2F1 localization to damaged DNA requires the ATM/ATR phosphorylation site but not the DNA binding or transcriptional activation domains of E2F1 [9,11].
Localization of E2F1 at sites of UV-induced DNA damage is associated with the efficient recruitment of nucleotide excision repair (NER) factors like XPA and XPC [9,29]. Likewise, localization of E2F1 at DSBs is associated with the recruitment and/or retention of DNA repair factors such as NBS1, MRE11, RPA and RAD51 [14,30,31]. In both cases, the absence of E2F1 results in inefficient DNA repair [14,32]. A knock-in mutation (S29A in mice) that prevents phosphorylation of E2F1 by ATM/ATR, also prevents its accumulation at sites of DNA damage, impairs the recruitment of DNA repair factors, and reduces the efficiency of repair of both UV photoproducts and DSBs [21,33].
RB forms one or more complexes with E2F1 in response to DNA damage. These complexes are associated with not only the transcriptional repression of cell cycle-related genes but also, and unexpectedly, the transcriptional activation of pro-apoptotic genes like TP73 and CASP7 [20,34,35]. RB stabilizes phosphorylated E2F1 and the E2F1-TopBP1 interaction that occurs in response to ionizing radiation (IR) [33]. This is consistent with earlier reports that the interaction between RB and E2F1 protects E2F1 from ubiquitination and proteasomal degradation [36,37]. Moreover, the binding of RB to E2F1 recruits RB to DSBs, whereas the loss of RB prevents E2F1 accumulation at DSBs [33]. Thus, E2F1 and RB are mutually dependent on each other for their localization to DSBs. Further, loss of RB, like loss of E2F1, results in defective DSB repair, particularly by the HR pathway [33,38–40].
In sharp contrast to DSB repair, the loss of RB enhances the kinetics of repair for UV-induced DNA photoproducts, possibly through the upregulation of XPC and other NER-related genes due to the deregulation of E2F activity [41–43]. In agreement with these findings, we were unable to detect recruitment of RB to sites of UV-induced DNA damage under the same conditions in which we observed E2F1 co-localization with DNA photoproducts and NER factors [9]. This suggests that in response to UV there is an RB-independent mechanism that stabilizes both phosphorylated E2F1 and the TopBP1-E2F1 interaction. The differential impact of RB loss on HR repair and NER has important implications for therapeutic sensitivities and vulnerabilities of cancers lacking RB [33,39,40].
E2F1 recruits GCN5 and induces H3K9 acetylation at UV-induced DNA damage
E2F1 stimulates transcription by recruiting a variety of histone acetyltransferases to gene promoters, leading to a more open chromatin conformation that allows the transcriptional machinery access to DNA [44–52]. As with transcription, DNA repair also requires remodeling of chromatin structure to provide the DNA repair machinery access to DNA. Pioneering studies by Smerdon and others demonstrated that chromatin structure becomes relaxed in response to UV radiation, that this process is important for efficient NER, and that chromatin structure is generally restored following repair [53,54]. This Access-Repair-Restore model has been refined by Almouzni and coworkers to describe the dynamic and active role of chromatin in DNA repair [4]. Not surprisingly, many of the histone modifying enzymes and nucleosome remodeling complexes involved in regulating gene transcription are also important for DNA repair; however, the mechanisms that target these proteins to sites of DNA damage are poorly understood.
We hypothesized that E2F1 might promote DNA repair through a mechanism similar to that used to regulate transcription, i.e. by recruiting proteins with chromatin-modifying activities to sites of DNA damage. Indeed, we found that the GCN5 acetyltransferase, which is known to cooperate with E2F1 in transcription [44,47], associates with E2F1 in response to UV irradiation and is recruited to sites of UV-induced DNA damage in an E2F1-dependent manner [32]. Moreover, both E2F1 and GCN5 are required for the induction of histone H3 lysine 9 acetylation (H3K9ac) at sites of UV-induced DNA damage, which is associated with chromatin relaxation and the efficient re-localization of NER factors XPC and XPA to damaged DNA [32].
As mentioned above, we made a knock-in mutant mouse allele, E2f1S29A, that prevents E2F1 from being phosphorylated by ATM/ATR in response to DNA damage [21]. E2f1S29A/S29A knock-in cells have normal cell cycle checkpoint and apoptotic responses to UV and express E2F target genes, including those involved in DNA repair, at levels comparable to wild-type cells [21]. However, the E2F1 S29A mutation prevents both E2F1 and GCN5 from associating with UV-irradiated DNA and impairs H3K9 acetylation in UV damaged chromatin [21]. Preventing E2F1 and GCN5 enrichment at sites of UV-induced DNA damage correlates with reduced NER efficiency and increased numbers of epidermal cells with p53 mutations [21]. Moreover, E2f1S29A/S29A mice are significantly more sensitive to UV-induced skin carcinogenesis, thus linking E2F1’s ability to directly stimulate DNA repair with tumor suppression.
E2F1 and RB recruit p300 and CBP to DNA double-strand breaks to induce H3K18 and H3K56 acetylation
Like UV-induced DNA damage, other types of DNA damage also lead to dynamic changes in chromatin structure that facilitate DNA repair. At DSBs, chromatin structure is transiently compacted and then relaxed, a process that is coordinated with DNA repair [55,56]. Given that GCN5 is enriched at DSBs, where it is associated with increased histone acetylation [57–60], we predicted that E2F1 would also recruit GCN5 to DSBs. In contrast to our expectations, we observed that loss of E2F1 had no effect on either the enrichment of GCN5 at a DSB or on the levels of H3K9ac in chromatin flanking the break.
Instead, we found that two other acetyltransferases, p300 and CBP, directly associate with E2F1 in response to IR and that E2F1 is required for their recruitment to DSBs [31]. Just as RB is required for stabilizing the TopBP1-E2F1 complex at DSBs, RB is also required for the recruitment of p300 and CBP to DSBs (Figure 1). E2F1- and RB-dependent recruitment of p300 and CBP to DSBs is associated with increased acetylation of histone H3 lysine 18 (H3K18ac) and histone H3 lysine 56 (H3K56ac) in flanking chromatin, rather than H3K9ac, as observed in response to UV damage [31,32]. The E2F1 S29A knock-in mutation, which blocks E2F1 phosphorylation and thus its interaction with TopBP1, also prevents p300/CBP recruitment and induction of H3K18ac and H3K56ac at DSBs [31].
Figure 1.
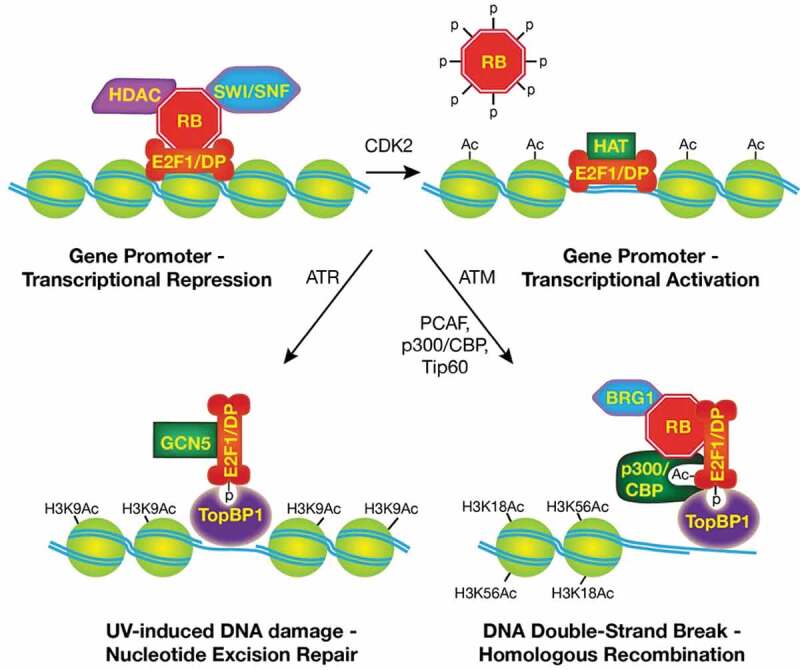
Regulation of E2F1 function during the cell cycle and in response to DNA damage. E2F1/DP dimers bind DNA sequences present in target gene promoters. When associated with RB, E2F1/DP can recruit chromatin-modifying enzymes to repress transcription (top left). RB is hyperphosphorylated and inactivated by CDK2 during cell cycle progression, which allows E2F1 to recruit histone acetyltransferases to activate transcription (top right). E2F1 is phosphorylated by either ATM or ATR in response to DNA damage, resulting in E2F1 localization to damaged DNA through an interaction with TopBP1. In turn, E2F1 recruits GCN5 to sites of UV-induced DNA damage to promote NER (bottom left). At DSBs, E2F1 is also acetylated and recruits p300/CBP, RB, and BRG1 to modify chromatin to facilitate HR repair (bottom right).
These findings raise the question of how E2F1 is regulated in response to different types of DNA damage to ensure it recruits the proper acetyltransferase to correctly mark the chromatin flanking different forms of DNA damage. One clue comes from the finding that E2F1 is acetylated in response to agents that cause DSBs but not in response to UV radiation [22]. E2F1 is acetylated by one or more acetyltransferases, including PCAF, p300/CBP and Tip60, on three lysine residues located near its DNA binding domain [52,61,62]. Acetylation of E2F1 enhances its stability and in some contexts promotes the transcriptional activation of the pro-apoptotic gene TP73 [22]. The importance of E2F1 acetylation for DNA repair was discovered only recently.
Protein acetylation can create binding motifs for bromodomains, which are frequently present in proteins that associate with chromatin to regulate transcription and/or DNA damage responses [63,64]. To determine whether acetylated E2F1 might interact with a bromodomain-containing protein, we screened a protein domain microarray. We identified the bromodomain of p300 as specifically interacting with an acetylated E2F1 peptide but not an unacetylated E2F1 peptide [31]. Follow-up studies demonstrated that the closely related bromodomain of CBP could also bind to acetylated E2F1, but not unacetylated E2F1.
The importance of the p300 and CBP bromodomains for recruiting these acetyltransferases to DSBs was demonstrated using a small molecule bromodomain inhibitor specific for p300/CBP [65]. Treatment of cells with this inhibitor did not affect the enrichment of E2F1 or RB at DSBs, but it did prevent both the recruitment of p300/CBP and the acetylation of H3K18 and H3K56 at sites of damage [31]. Further, knock-in mutations that block E2F1 acetylation caused similar effects; E2F1 and RB were recruited to sites of damage but p300 and CBP were not, resulting in a lack of H3K18 and H3K56 acetylation [31]. Thus, E2F1 acetylation, which occurs in response to DSB formation but not UV damage, helps to specify p300/CBP, rather than GCN5, as the acetyltransferases recruited to DSBs by E2F1 (Figure 1).
E2F1 post-translational modifications direct chromatin remodeling at DNA double-strand breaks to facilitate repair
BRG1, a core ATPase subunit of SWI/SNF (BAF/PBAF) nucleosome remodeling complexes, is also recruited to DSBs dependent on E2F1 and RB [33]. Like p300/CBP, BRG1 associates with phosphorylated E2F1 and RB in response to IR. Although the mechanism of this interaction is not established, mutation of the E2F1 acetylation sites prevents BRG1 from associating with the TopBP1-E2F1-RB complex [31]. E2F1 recruits both p300/CBP and BRG1 to E2F1 target gene promoters, and p300/CBP-mediated histone acetylation is thought to cooperate with BRG1-containing complexes to reduce nucleosome density at these promoters to stimulate transcription [48,66,67]. In the context of DNA repair, E2F1-dependent recruitment of p300/CBP and BRG1 is also associated with a reduction in nucleosome density, specifically in chromatin flanking DSBs [31,33].
The Tip60 acetyltransferase is also recruited to DSBs and is important for increasing histone H4 acetylation and destabilizing nucleosomes in chromatin flanking DSBs [68,69]. Unexpectedly, depletion of E2F1 or RB, or mutation of E2F1 at sites of phosphorylation or acetylation, also impairs recruitment of Tip60 and histone H4 acetylation at DSBs [31]. However, unlike p300/CBP and BRG1, Tip60 does not appear to physically associate with phosphorylated E2F1 in response to DNA damage. This suggests that E2F1- and RB-dependent chromatin remodeling is indirectly involved in the recruitment and/or retention of Tip60 at DSBs.
Knock-in mutations that block either the phosphorylation or acetylation of E2F1 also inhibit enrichment of the MRE11-RAD50-NBS1 (MRN) complex at an induced DSB, suggesting that efficient loading of MRN onto chromatin requires E2F1-mediated histone acetylation and nucleosome remodeling [31]. A direct interaction between NBS1 and E2F1 could also contribute to the recruitment and/or retention of MRN at DSBs [14,70]. Defective loading of MRN onto chromatin flanking a DSB may explain, in part, why depletion or mutation of E2F1 impairs DNA end-resection and HR repair [14,33]. The physiological relevance of E2F1-mediated chromatin remodeling at DSBs is illustrated by the finding that mice harboring knock-in mutations that block E2F1 phosphorylation or acetylation are hypersensitive to IR, a hallmark of defective HR repair [31,33].
Are there universal mechanisms for E2F1-mediated regulation of transcription and DNA repair?
As discussed above, E2F1 recruits p300/CBP and BRG1 to DSBs and to some gene promoters to induce histone acetylation and reduce nucleosome density. Depending on the specific locus where E2F1 recruits these factors, the resulting modifications to chromatin structure can facilitate either DNA repair or transcription. In some cases, such as during osteoblast differentiation, RB also participates in the recruitment of BRG1 and cooperates with p300 in the transcriptional activation of tissue-specific E2F1 target genes [71,72]. How RB participates in E2F1-dependent transcriptional activation is unclear. RB binding is thought to mask the C-terminal transcriptional activation domain of E2F1, thus preventing its interaction with histone acetyltransferases and other co-activators [45,73–75]. However, because the bromodomains of p300 and CBP interact with the N-terminal acetylation sites of E2F1, this could allow E2F1 to simultaneously associate with RB and either p300 or CBP. While this appears to be the mechanism by which E2F1 recruits both RB and p300/CBP to DSBs, it remains to be determined whether this mechanism is also used by E2F1-RB complexes to activate transcription.
In other contexts, E2F1 can regulate both transcription and DNA repair by recruiting GCN5 to induce H3K9 acetylation, relax chromatin structure, and increase access to the transcription or NER machinery, respectively [21,32,44,47]. The mechanism by which E2F1 associates with GCN5 in response to UV radiation is unclear. Mutational analysis of E2F1 indicates that neither the DNA binding domain nor the transcriptional activation domain are necessary for E2F1 to promote NER [9]. Instead, the Marked box domain of E2F1, which is located between the DNA binding and transcriptional activation domains, is required for E2F1 to enhance NER factor recruitment to sites of UV damage [9]. The Marked box domain is used by E2F1 to bind a number of partner proteins, including prohibitin and Jab1 [76,77]. Whether this domain also binds GCN5 in response to UV radiation remains to be determined. Regardless of the mechanism, taken together, these studies reveal E2F1 to be a versatile protein that performs multiple jobs as a transcription factor while also moonlighting as a regulator of DNA repair by recruiting the same chromatin-modifying enzymes to either target genes or different types of DNA damage.
The distinction between transcription factors and DNA repair factors is becoming blurry
The histone code model posits key roles for writers, erasers, and readers of histone post-translational modifications in regulating transcription and this model has been extended to include DNA repair and other process that require access to DNA in the context of chromatin [3,78,79]. However, mechanisms must exist for targeting the writers, erasers and readers of the histone code to specific sites in the genome, such as gene promoters and sites of DNA damage. The role of sequence-specific DNA binding proteins like E2F1 in recruiting components of the histone code machinery to gene regulatory elements is well known but how the histone code machinery is targeted to sites of DNA damage is less well understood. Some DNA damage recognition factors may recruit chromatin-modifying enzymes to sites of DNA damage through direct interactions. For example, damaged DNA binding protein 2 (DDB2/XPE) interacts with and recruits the acetyltransferase HBO1 to sites of UV-induced DNA damage, resulting in increased acetylation of histone H3K14 and H4 [80]. This DDB2-HBO1 pathway appears to cooperate with the E2F1-GCN5 pathway in relaxing chromatin structure to allow the NER machinery access to damaged DNA for efficient repair.
Interestingly, DDB2 moonlights as a transcriptional regulator by associating with gene promoters and recruiting different components of the histone code machinery [81–84]. In fact, DDB2 can bind to and cooperate with E2F1 to activate transcription [85,86]. Another DNA damage recognition factor, XPC, also moonlights as a regulator of transcription, including E2F1-dependent transcription [87–90]. XPC interacts with both E2F1 and GCN5 and co-localizes with them at a subset of active gene promoters [87]. Depletion of XPC reduces the expression of these target genes, concomitant with a decrease in H3K9ac at their promoters [87].
If E2F1 and XPC cooperate to regulate both transcription and DNA repair [29,32,87], then the distinction between a transcription factor and a DNA repair factor begins to break down. There is no term for factors like E2F1 and XPC that regulate both transcription and DNA repair through similar mechanisms involving the recruitment of chromatin modifying enzymes, like GCN5, to specific loci in the genome. We and others have used the term “chromatin accessibility factors” to describe the functions of E2F1 and p53 in relaxing chromatin structure for NER [91,92]. However, this term does not imply a uniform mechanism as E2F1 co-localizes with sites of UV-induced DNA damage, whereas p53 does not, indicating that E2F1 plays a more direct role in modifying chromatin structure at sites of UV damage [9,93]. Moreover, E2F1-mediated chromatin remodeling may be important for more than simply increasing chromatin accessibility to cellular machinery.
Perhaps a good analogy for factors like E2F1, DDB2 and XPC is that they act like fishing lures (Figure 2). Like a lure, E2F1, DDB2 and XPC can attract and catch various chromatin-modifying enzymes using multiple protein-protein interaction domains as hooks. They can then reel in these chromatin modifiers to specific genomic loci. Just as different lures can be selected to target different fish, E2F1 can be post-translationally modified to capture different prey to differentially modulate chromatin structure depending on the context. It is quite possible that other transcription factors that localize to DNA damage also lure the same set of chromatin-modifying proteins to both promoters and sites of DNA damage to regulate gene transcription and DNA repair, respectively. It will be interesting to determine whether other DNA repair factors that moonlight as transcriptional regulators, like BRCA1 [94–99], also recruit the same chromatin-modifying enzymes and direct the same chromatin modifications for both transcription and DNA repair.
Figure 2.
The fishing lure analogy. Some factors, like E2F1, lure various components of the histone code machinery to specific genomic loci to regulate multiple DNA-dependent cellular processes in the context of chromatin.
Acknowledgments
We wish to thank Rebecca Deen and Briana Dennehey for help with manuscript preparation and editing, Joi Holcomb for figure graphics, and Jen Orona for her many years of technical and animal care support for our research program. Work described in this review was supported in part by grants from the Cancer Prevention and Research Institute of Texas (RP140222 to D.G.J.), the National Institutes of Health (CA079648 to D.G.J., CA214723 to D.G.J., and Cancer Core Support Grant CA016672), and institutional support from the Center for Cancer Epigenetics and the Center for Genetics and Genomics.
Funding Statement
This work was supported by the Division of Cancer Epidemiology and Genetics, National Cancer Institute [CA214723].
Disclosure statement
The authors declare no competing interests.
References
- [1].Blackford AN, Jackson SP.. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817. [DOI] [PubMed] [Google Scholar]
- [2].Chen H, Symington LS.. Overcoming the chromatin barrier to end resection. Cell Res. 2013;23:317–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Loizou JI, Murr R, Finkbeiner MG, et al. Epigenetic information in chromatin: the code of entry for DNA repair. Cell Cycle. 2006;5:696–701. [DOI] [PubMed] [Google Scholar]
- [4].Polo SE, Almouzni G. Chromatin dynamics after DNA damage: the legacy of the access-repair-restore model. DNA Repair (Amst). 2015;36:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Izhar L, Adamson B, Ciccia A, et al. A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep. 2015;11:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beishline K, Kelly CM, Olofsson BA, et al. Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol Cell Biol. 2012;32:3790–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bhoumik A, Takahashi S, Breitweiser W, et al. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol Cell. 2005;18:577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guo R, Chen J, Zhu F, et al. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem. 2010;285:19308–19315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Iwahori S, Yasui Y, Kudoh A, et al. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell Signal. 2008;20:1795–1803. [DOI] [PubMed] [Google Scholar]
- [11].Liu K, Lin FT, Ruppert JM, et al. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol. 2003;23:3287–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Malewicz M, Kadkhodaei B, Kee N, et al. Essential role for DNA-PK-mediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev. 2011;25:2031–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Munnur D, Somers J, Skalka G, et al. NR4A nuclear receptors target poly-ADP-ribosylated DNA-PKcs protein to promote DNA repair. Cell Rep. 2019;26:2028–2036 e2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen J, Zhu F, Weaks RL, et al. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle. 2011;10:1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer. 2019;19:326–338. [DOI] [PubMed] [Google Scholar]
- [16].Narasimha AM, Kaulich M, Shapiro GS, et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife. 2014;3:e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- [18].Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM- dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- [19].Wang B, Liu K, Lin FT, et al. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J Biol Chem. 2004;279:54140–54152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Carnevale J, Palander O, Seifried LA, et al. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol Cell Biol. 2012;32:900–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Biswas AK, Mitchell DL, Johnson DG. E2F1 responds to ultraviolet radiation by directly stimulating DNA repair and suppressing carcinogenesis. Cancer Res. 2014;74:3369–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pediconi N, Ianari A, Costanzo A, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5:552–558. [DOI] [PubMed] [Google Scholar]
- [23].Liu K, Luo Y, Lin FT, et al. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004;18:673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Manke IA, Lowery DM, Nguyen A, et al. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. [DOI] [PubMed] [Google Scholar]
- [25].Yu X, Chini CC, He M, et al. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. [DOI] [PubMed] [Google Scholar]
- [26].Bigot N, Day M, Baldock RA, et al. Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage checkpoint. Elife. 2019;8:e44353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wardlaw CP, Carr AM, Oliver AW. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst). 2014;22:165–174. [DOI] [PubMed] [Google Scholar]
- [28].Liu K, Paik JC, Wang B, et al. Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. Embo J. 2006;25:4795–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh RK, Dagnino L. E2F1 interactions with hHR23A inhibit its degradation and promote DNA repair. Oncotarget. 2016;7:26275–26292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Choi EH, Kim KP. E2F1 facilitates DNA break repair by localizing to break sites and enhancing the expression of homologous recombination factors. Exp Mol Med. 2019;51:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Manickavinayaham S, Velez-Cruz R, Biswas AK, et al. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat Commun. 2019;10:4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guo R, Chen J, Mitchell DL, et al. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011;39:1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Velez-Cruz R, Manickavinayaham S, Biswas AK, et al. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016;30:2500–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ianari A, Natale T, Calo E, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. Embo J. 2007;26:2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:2221–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hofmann F, Martelli F, Livingston DM, et al. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes Dev. 1996;10:2949–2959. [DOI] [PubMed] [Google Scholar]
- [38].Cook R, Zoumpoulidou G, Luczynski MT, et al. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015;10:2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Marshall AE, Roes MV, Passos DT, et al. RB1 deletion in retinoblastoma protein pathway-disrupted cells results in DNA damage and cancer progression. Mol Cell Biol. 2019;39:e00105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xiao H, Goodrich DW. The retinoblastoma tumor suppressor protein is required for efficient processing and repair of trapped topoisomerase II-DNA-cleavable complexes. Oncogene. 2005;24:8105–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bosco EE, Knudsen ES. Differential role of RB in response to UV and IR damage. Nucleic Acids Res. 2005;33:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lin PS, McPherson LA, Chen AY, et al. The role of the retinoblastoma/E2F1 tumor suppressor pathway in the lesion recognition step of nucleotide excision repair. DNA Repair (Amst). 2009;8:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Prost S, Lu P, Caldwell H, et al. E2F regulates DDB2: consequences for DNA repair in Rb-deficient cells. Oncogene. 2007;26:3572–3581. [DOI] [PubMed] [Google Scholar]
- [44].Chen L, Wei T, Si X, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem. 2013;288:14510–14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fry CJ, Pearson A, Malinowski E, et al. Activation of the murine dihydrofolate reductase promoter by E2F1. A requirement for CBP recruitment. J Biol Chem. 1999;274:15883–15891. [DOI] [PubMed] [Google Scholar]
- [46].Korah J, Falah N, Lacerte A, et al. A transcriptionally active pRb-E2F1-P/CAF signaling pathway is central to TGFbeta-mediated apoptosis. Cell Death Dis. 2012;3:e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lang SE, McMahon SB, Cole MD, et al. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J Biol Chem. 2001;276:32627–32634. [DOI] [PubMed] [Google Scholar]
- [48].Ogiwara H, Kohno T. CBP and p300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes. PLoS ONE. 2012;7:e52810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pediconi N, Guerrieri F, Vossio S, et al. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol Cell Biol. 2009;29:1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pillai S, Kovacs M, Chellappan S. Regulation of vascular endothelial growth factor receptors by Rb and E2F1: role of acetylation. Cancer Res. 2010;70:4931–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Taubert S, Gorrini C, Frank SR, et al. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol. 2004;24:4546–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Van Den Broeck A, Nissou D, Brambilla E, et al. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis. 2012;33:320–325. [DOI] [PubMed] [Google Scholar]
- [53].Bodell WJ, Cleaver JE. Transient conformation changes in chromatin during excision repair of ultraviolet damage to DNA. Nucleic Acids Res. 1981;9:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Smerdon MJ, Lieberman MW. Nucleosome rearrangement in human chromatin during UV-induced DNA- repair synthesis. Proc Natl Acad Sci U S A. 1978;75:4238–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bakkenist CJ, Kastan MB. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst). 2015;36:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gursoy-Yuzugullu O, House N, Price BD. Patching broken DNA: nucleosome dynamics and the repair of DNA breaks. J Mol Biol. 2016;428:1846–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bennett G, Peterson CL. SWI/SNF recruitment to a DNA double-strand break by the NuA4 and Gcn5 histone acetyltransferases. DNA Repair (Amst). 2015;30:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fu X, Zhang C, Meng H, et al. Oncoprotein Tudor-SN is a key determinant providing survival advantage under DNA damaging stress. Cell Death Differ. 2018;25:1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee HS, Park JH, Kim SJ, et al. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. Embo J. 2010;29:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005;25:4903–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Martinez-Balbas MA, Bauer UM, Nielsen SJ, et al. Regulation of E2F1 activity by acetylation. Embo J. 2000;19:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Marzio G, Wagener C, Gutierrez MI, et al. E2F family members are differentially regulated by reversible acetylation. J Biol Chem. 2000;275:10887–10892. [DOI] [PubMed] [Google Scholar]
- [63].Chiu LY, Gong F, Miller KM. Bromodomain proteins: repairing DNA damage within chromatin. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zaware N, Zhou MM. Bromodomain biology and drug discovery. Nat Struct Mol Biol. 2019;26:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Picaud S, Fedorov O, Thanasopoulou A, et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015;75:5106–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pietrzak J, Ploszaj T, Pulaski L, et al. EP300-HDAC1-SWI/SNF functional unit defines transcription of some DNA repair enzymes during differentiation of human macrophages. Biochim Biophys Acta Gene Regul Mech. 2019;1862:198–208. [DOI] [PubMed] [Google Scholar]
- [67].Sobczak M, Pitt AR, Spickett CM, et al. PARP1 co-regulates EP300-BRG1-dependent transcription of genes involved in breast cancer cell proliferation and DNA repair. Cancers (Basel). 2019;11:1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–99. [DOI] [PubMed] [Google Scholar]
- [69].Xu Y, Sun Y, Jiang X, et al. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol. 2010;191:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Maser RS, Mirzoeva OK, Wells J, et al. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol. 2001;21:6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Flowers S, Beck GR Jr., Moran E. Transcriptional activation by pRB and its coordination with SWI/SNF recruitment. Cancer Res. 2010;70:8282–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Flowers S, Xu F, Moran E. Cooperative activation of tissue-specific genes by pRB and E2F1. Cancer Res. 2013;73:2150–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Flemington EK, Speck SH, Kaelin WG Jr.. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci U S A. 1993;90:6914–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Trouche D, Cook A, Kouzarides T. The CBP co-activator stimulates E2F1/DP1 activity. Nucleic Acids Res. 1996;24:4139–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hallstrom TC, Nevins JR. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wang S, Nath N, Fusaro G, et al. Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol Cell Biol. 1999;19:7447–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Clouaire T, Legube G. A snapshot on the cis chromatin response to DNA double-strand breaks. Trends Genet. 2019;35:330–345. [DOI] [PubMed] [Google Scholar]
- [79].Kim JJ, Lee SY, Miller KM. Preserving genome integrity and function: the DNA damage response and histone modifications. Crit Rev Biochem Mol Biol. 2019;54:208–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Niida H, Matsunuma R, Horiguchi R, et al. Phosphorylated HBO1 at UV irradiated sites is essential for nucleotide excision repair. Nat Commun. 2017;8:16102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bommi PV, Chand V, Mukhopadhyay NK, et al. NER-factor DDB2 regulates HIF1alpha and hypoxia-response genes in HNSCC. Oncogene. 2020;39:1784–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Huang S, Fantini D, Merrill BJ, et al. DDB2 Is a Novel Regulator of Wnt Signaling in Colon Cancer. Cancer Res. 2017;77:6562–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhao R, Cui T, Han C, et al. DDB2 modulates TGF-beta signal transduction in human ovarian cancer cells by downregulating NEDD4L. Nucleic Acids Res. 2015;43:7838–7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhao R, Han C, Eisenhauer E, et al. DNA damage-binding complex recruits HDAC1 to repress Bcl-2 transcription in human ovarian cancer cells. Mol Cancer Res. 2014;12:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hayes S, Shiyanov P, Chen X, et al. DDB, a putative DNA repair protein, can function as a transcriptional partner of E2F1. Mol Cell Biol. 1998;18:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shiyanov P, Hayes SA, Donepudi M, et al. The naturally occurring mutants of DDB are impaired in stimulating nuclear import of the p125 subunit and E2F1-activated transcription. Mol Cell Biol. 1999;19:4935–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bidon B, Iltis I, Semer M, et al. XPC is an RNA polymerase II cofactor recruiting ATAC to promoters by interacting with E2F1. Nat Commun. 2018;9:2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Fong YW, Inouye C, Yamaguchi T, et al. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell. 2011;147:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Le May N, Mota-Fernandes D, Velez-Cruz R, et al. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38:54–66. [DOI] [PubMed] [Google Scholar]
- [90].Semer M, Bidon B, Larnicol A, et al. DNA repair complex licenses acetylation of H2A.Z.1 by KAT2A during transcription. Nat Chem Biol. 2019;15:992–1000. [DOI] [PubMed] [Google Scholar]
- [91].Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. Embo J. 2003;22:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Velez-Cruz R, Johnson DG. E2F1 and p53 transcription factors as accessory factors for nucleotide excision repair. Int J Mol Sci. 2012;13:13554–13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fitch ME, Cross IV, Turner SJ, et al. The DDB2 nucleotide excision repair gene product p48 enhances global genomic repair in p53 deficient human fibroblasts. DNA Repair (Amst). 2003;2:819–826. [DOI] [PubMed] [Google Scholar]
- [94].Bochar DA, Wang L, Beniya H, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. [DOI] [PubMed] [Google Scholar]
- [95].De Siervi A, De Luca P, Byun JS, et al. Transcriptional autoregulation by BRCA1. Cancer Res. 2010;70:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lee YH, Bedford MT, Stallcup MR. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 2011;25:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mullan PB, Quinn JE, Harkin DP. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 2006;25:5854–5863. [DOI] [PubMed] [Google Scholar]
- [98].Pao GM, Janknecht R, Ruffner H, et al. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc Natl Acad Sci U S A. 2000;97:1020–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci U S A. 1999;96:4983–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]