

Clostridium difficile is an anaerobic, multidrug-resistant, toxin-producing pathogen with major health impacts worldwide. It is the only widespread pathogen harboring a complete set of Wood-Ljungdahl pathway (WLP) genes; however, the role of the WLP in C. difficile is poorly understood. In other anaerobic bacteria and archaea, the WLP can operate in one direction to convert CO2 to acetic acid for biosynthesis or in either direction for energy conservation. Here, conditions are defined in which WLP levels in C. difficile increase markedly, functioning to support metabolism of carbohydrates. Amino acid nutritional requirements were better defined, with new insight into how the WLP and butyrate pathways act in concert, contributing significantly to energy metabolism by a mechanism that may have broad physiological significance within the group of nonclassical acetogens.
KEYWORDS: CO dehydrogenase, CODH, Clostridium difficile, Stickland reaction, Wood-Ljungdahl pathway, acetate, acetyl-CoA synthase, acsB, butyrate, carbon monoxide dehydrogenase
ABSTRACT
Clostridium difficile is the leading cause of hospital-acquired antibiotic-associated diarrhea and is the only widespread human pathogen that contains a complete set of genes encoding the Wood-Ljungdahl pathway (WLP). In acetogenic bacteria, synthesis of acetate from 2 CO2 molecules by the WLP functions as a terminal electron accepting pathway; however, C. difficile contains various other reductive pathways, including a heavy reliance on Stickland reactions, which questions the role of the WLP in this bacterium. In rich medium containing high levels of electron acceptor substrates, only trace levels of key WLP enzymes were found; therefore, conditions were developed to adapt C. difficile to grow in the absence of amino acid Stickland acceptors. Growth conditions were identified that produce the highest levels of WLP activity, determined by Western blot analyses of the central component acetyl coenzyme A synthase (AcsB) and assays of other WLP enzymes. Fermentation substrate and product analyses, enzyme assays of cell extracts, and characterization of a ΔacsB mutant demonstrated that the WLP functions to dispose of metabolically generated reducing equivalents. While WLP activity in C. difficile does not reach the levels seen in classical acetogens, coupling of the WLP to butyrate formation provides a highly efficient system for regeneration of NAD+ “acetobutyrogenesis,” requiring only low flux through the pathways to support efficient ATP production from glucose oxidation. Additional insights redefine the amino acid requirements in C. difficile, explore the relationship of the WLP to toxin production, and provide a rationale for colocalization of genes involved in glycine synthesis and cleavage within the WLP operon.
IMPORTANCE Clostridium difficile is an anaerobic, multidrug-resistant, toxin-producing pathogen with major health impacts worldwide. It is the only widespread pathogen harboring a complete set of Wood-Ljungdahl pathway (WLP) genes; however, the role of the WLP in C. difficile is poorly understood. In other anaerobic bacteria and archaea, the WLP can operate in one direction to convert CO2 to acetic acid for biosynthesis or in either direction for energy conservation. Here, conditions are defined in which WLP levels in C. difficile increase markedly, functioning to support metabolism of carbohydrates. Amino acid nutritional requirements were better defined, with new insight into how the WLP and butyrate pathways act in concert, contributing significantly to energy metabolism by a mechanism that may have broad physiological significance within the group of nonclassical acetogens.
INTRODUCTION
Clostridium difficile (also known as Clostridioides difficile) is a Gram-positive, rod-shaped, spore-forming anaerobic bacterium that is the leading cause of nosocomial antibiotic-associated diarrhea worldwide. The severity of C. difficile infection ranges from mild diarrhea to pseudomembranous colitis that can lead to toxic megacolon and death. Pathogenesis involves germination of spores, multiplication of vegetative cells and colonization, and subsequent production of two high-molecular-weight toxins, TcdA and TcdB (and in some strains, a third binary toxin, CDT), that elicit an intense inflammatory response (1–5). Throughout the infectious process, from the onset of antibiotic-induced dysbiosis to the final production of new endospores, nutrient availability varies in the gut, and evidence indicates that C. difficile undergoes substantial shifts in its carbon and energy metabolism in response to changes in the nutrient landscape (6–9).
As its major pathway of energy metabolism, C. difficile relies heavily on the Stickland reaction/process in which rapid oxidation of one amino acid (electron donor) is coupled to the reduction of the same or another amino acid (electron acceptor), with energy captured by substrate-level phosphorylation (10, 11). Proline, leucine, and glycine are the most important Stickland acceptors for C. difficile, acting to regenerate NAD+ as needed for the continued oxidation of various Stickland donors such as leucine, isoleucine, valine, and alanine (12–14).
The importance of Stickland acceptors leucine and proline became evident in early studies to define minimal growth requirements of C. difficile, which identified 6 amino acids considered to be essential, namely, proline, leucine, isoleucine, valine, tryptophan, and cysteine, such that omission of any one of these resulted in no observable growth (15–17). Later it was shown in studies on the effect of cysteine repression of toxin production (18), and in the present work, that cysteine can be eliminated from minimal medium simply by the addition of inorganic sulfide and that the requirement for tryptophan can be rationalized by the fact that no homologs for the key biosynthetic genes for that amino acid exist in the genome (19, 20).
In addition to amino acids, C. difficile is able to ferment some carbohydrate sources, including glucose, fructose, and, to a lesser extent, mannose (21, 22). Evidence indicates that sialic acid is also a fermentable substrate (23, 24), and although C. difficile lacks sialidase, in vivo studies in mice indicate that sialic acid liberated from host mucins by other bacteria in the gut supports growth of C. difficile (25). Nevertheless, despite the ability to metabolize sugars, to date, there have been no reports of culture conditions that allow for growth of C. difficile on carbohydrates as the principal source of carbon and energy in the absence of amino acid Stickland acceptors.
Given the high activity of Stickland acceptor reactions for rapid disposal of electrons, it is surprising that C. difficile harbors a complete set of genes encoding enzymes of the reductive acetyl coenzyme A (acetyl-CoA) pathway, also known as the Wood-Ljungdahl pathway (WLP), which is used by the strictly anaerobic acetogens as an electron sink to dispose of reducing equivalents by reduction of CO2 to acetate (Fig. 1A). A 15-gene, 18.4-kb operon (Fig. 1B) encoding the WLP pathway is found widely distributed in the sequenced strains, and WLP genes are represented in all major phylogenetic groups of C. difficile (26). Autotrophic acetogens take up H2 plus CO2, generating acetyl-CoA for biosynthetic purposes, whereas in heterotrophs, the pathway serves to accept electrons derived from oxidation of complex organic substrates (27). An incomplete WLP, discovered in an organohalide-respiring anaerobe, functions to cleave acetate and thereby generate methyl groups needed for methionine biosynthesis (28). In some anaerobic species able to use high-potential electron acceptors, the WLP serves yet another role, for the cleavage of acetate followed by complete oxidation to CO2 (29). Remarkably, C. difficile is the only widespread human pathogen known to contain the WLP, although it is also present in the less-well-known pathogen Clostridium sordellii (Paeniclostridium sordellii).
FIG 1.

(A) The Wood-Ljungdahl pathway and the conversion of its product acetyl-CoA to ethanol, acetate, and butyrate. Enzyme abbreviations are given in blue and gene names are in black with locus tags (19) in red. H4F stands for tetrahydrofolate. Separate methyl and carbonyl branches converge in the formation of acetyl-CoA. In the methyl branch, a reversible hydrogen-dependent CO2 reductase complex (HDCR), which contains formate dehydrogenase (fdh) and [Fe-Fe] hydrogenase components, reduces CO2 to formate using H2 as its direct electron donor. Formate is activated to produce N10-formyl-H4F (HCO-H4F) in an ATP-requiring reaction by N10-formyl-H4F synthetase (FTS). N5,N10-methenyl-H4F (CH≡H4F) is then generated via cyclization and dehydration by N5,N10-methenyl-tetrahydrofolate 5-hydrolase (formyl-H4F cyclohydrolase; fchA) (MTC). Bifunctional FolD protein, which includes N5,N10-methylene-H4F dehydrogenase (MTD) and additional formyl-H4F cyclohydrolase activity, converts CH≡H4F to N5,N10-methylene-H4F (CH2=H4F), which is then further reduced to N5-methyl-H4F (CH3-H4F) by N5,N10-methylene-H4F reductase (MTR) with catalytic subunit MetF and an accessory Fe/S- and Zn-containing subunit MetV. CH3-H4F then transfers its methyl group to a corrinoid iron-sulfur protein to form Co3+-methyl corrinoid iron-sulfur protein (CH3-CFeSP) catalyzed by CH3-H4F:corrinoid methyltransferase (MeTr). In the carbonyl branch, carbon monoxide dehydrogenase (CODH) produces CO contained within a protein channel, depicted as [CO], by reduction of CO2 using low-potential reduced ferredoxin (). Acetyl-CoA, shown at the center in the shaded oval, is then produced by condensation of the corrinoid methyl and channel-bound CO groups by acetyl-CoA synthase (ACS) acsB. In the pathway leading to ethanol, acetyl-CoA is reduced in two steps by bifunctional acetaldehyde-CoA/alcohol dehydrogenase (AdhE). An alternative pathway would be to convert acetate to ethanol by way of aldehyde dehydrogenase (Aldh). Acetate formation from acetyl-CoA is via phosphotransacetylase (Pta/Ptb) that produces acetyl phosphate (CD2683 is annotated in the KEGG database as a putative phosphotransacetylase ortholog, and other candidates potentially active with acetate could include phosphotransbutyrylase CD0112 and the two other annotated ptb homologs CD0715 and CD2425). Acetyl phosphate is then used to generate ATP by acetate kinase or butyrate kinase active with acetate (Ack/Buk). Formation of butyrate follows the typical pathway in which thiolase (acetyl-CoA acetyltransferase) (THL) generates acetoacetyl-CoA that is acted on by 3-hydroxybutyryl-CoA dehydrogenase (HBD) followed by 3-hydroxybutyryl-CoA dehydratase (crotonase) (CRT) to form crotonyl-CoA. The electron-bifurcating butyryl-CoA dehydrogenase (Bcd) CD1054 in complex with electron-transferring flavoprotein subunits EtfB and EtfA (CD1055 and CD1056), complex (Bcd-EtfAB)4, then generates butyryl-CoA (75–77). Thereafter, butyrate is produced by butyryl-CoA:acetate CoA-transferase activity (CoA transfer) using free acetate to reform one molecule of acetyl-CoA. (B) The Wood-Ljungdahl pathway operon in C. difficile. A 15-gene, 18.4-kb region (CD0716 to CD0730) is shown according to data in references 19 and 20. Gene designations and corresponding products are indicated.
Homoacetogenic fermentation of carbohydrates produces three molecules of acetate from glucose by the combined actions of glycolysis, pyruvate decarboxylation, and the WLP. The overall redox stoichiometry required to convert glucose to 2 acetates plus 2 CO2 molecules is balanced by the reduction of the CO2 to form a third molecule of acetate via the WLP. Three pairs of electrons are used to reduce one CO2 to the level of a methyl group via a series of tetrahydrofolate intermediates (Fig. 1A, methyl branch), and one pair of electrons (represented as ) reduces the other CO2 to carbon monoxide, which is sequestered in a channel in the enzyme CO dehydrogenase (depicted as [CO] in Fig. 1A, carbonyl branch) (30–32). CO and CH3 then condense in an unusual organometallic carbonylation reaction that takes place at a redox-active Ni- and Fe-containing metal center at the active site of the enzyme acetyl-CoA synthase (AcsB) (33–35). The combination of CO and CH3 groups on AcsB generates a high-energy acetyl intermediate which is then captured by CoA to form acetyl-CoA (36–39).
The question of what advantage C. difficile would gain from disposal of reducing equivalents through the WLP arises especially because, in addition to the critical amino acid Stickland acceptors proline, leucine, and glycine, C. difficile is also able to employ a variety of other substrates or processes to regenerate NAD+. These include the reduction of acetyl-CoA to both ethanol and butyrate, reduction of pyruvate to lactate, conversion of ethanolamine to ethanol (40), reduction of succinate, which is available in vivo as a product of fermentation by other microorganisms in the gut (41), and evolution of hydrogen. Moreover, in brain heart infusion medium supplemented with yeast extract and cysteine (BHIS) that is replete with Stickland acceptors, neither extracts of C. difficile nor extracts of the genetically and metabolically related bacterium Clostridium sticklandii grown on similar rich medium exhibit significant levels of CO dehydrogenase activity, as measured by sensitive viologen-based reactions down to the limits of detection of the assays (D. A. Grahame and T. C. Stadtman, unpublished results).
In this work, the role of the WLP in C. difficile was examined by developing a method for stepwise adaptation of cells to growth on glucose in the absence of Stickland acceptors. The results of fermentation analyses and enzymatic activity levels under the newly developed growth conditions and the characterization of a mutant deficient in acsB indicate that C. difficile uses the WLP as an efficient electron sink that is able to confer a substantial growth advantage even at relatively low levels of activity. Coupling of the WLP to butyrate formation is proposed as a means by which relatively low flux through both pathways is sufficient to regenerate substantial amounts of NAD+ to support the fermentation of carbohydrates. Overall, the findings agree with accumulating evidence that C. difficile exhibits a wider metabolic potential and is more adaptable to changing nutrient conditions than has been previously appreciated.
RESULTS
Minimal expression of the WLP in the presence of abundant Stickland acceptors.
To assess WLP enzyme expression during growth of C. difficile, Western blot analyses were carried out on acetyl-CoA synthase (AcsB), the central condensing enzyme in the WLP pathway. As shown in Fig. 2, almost no AcsB was produced during exponential growth and during the transition to stationary phase in cells grown in BHIS, a rich medium in which Stickland acceptors are abundant. Increased production of AcsB was observed only late in the stationary phase and corresponded to less than 0.01% of total protein, a level which is very low in comparison to that from a typical acetogenic organism such as Clostridium ljungdahlii grown in complex medium. Most of the increase in AcsB occurred near the time when an abrupt decrease in the optical density at 600 nm (OD600) was observed. This decline in OD600, which occurred between 16 and 24 h, suggests that substantial autolysis may be taking place. As a process connected with cellular stress, autolysis is under the control of a number of factors, among which is the activation of cell wall-associated murine hydrolases as a consequence of the loss of an energized membrane, which occurs upon final exhaustion of all utilizable energy-generating substrates (42–44). A switch to obtain energy via the WLP could help to delay the lytic phase somewhat. However, despite the increased expression, final AcsB levels were still very low, and the activity of the WLP was inadequate to provide sufficient energy to prevent autolysis.
FIG 2.

Trace levels of acetyl-CoA synthase (AcsB) detected during growth of C. difficile in BHIS. Growth was measured on four separate BHIS cultures prepared as described in Materials and Methods. (Inset) Western blot of a 12% acrylamide SDS-PAGE gel with samples taken from cultures at the times indicated (hours). Ten nanograms of purified recombinant AcsB was included as a reference on the outside lanes.
Adaptation to growth in the absence of proline and leucine.
Because only minimal levels of AcsB were detectable in BHIS, we attempted to grow C. difficile on glucose in the absence of favored amino acid Stickland acceptors to determine whether higher levels of WLP enzymes would be produced. Guided by the chemically defined medium developed as described in reference 17, we confirmed that C. difficile 630 fails to grow when either proline or leucine is omitted. Inoculation at high cell densities (initial OD600 in the range of 0.1 to 0.2) was explored to determine whether adaptation would occur over time; instead, the initial OD600 values decreased markedly within the first 12 h after inoculation, indicative of cell lysis in the absence of a usable carbon and energy source. Similarly, initial lysis and subsequent failure to adapt and grow were observed in ATCC PETC 1754 medium lacking yeast extract and other organic carbon substrates under an H2/CO2 atmosphere, regardless of whether the initial inoculum had been grown in BHIS or in reinforced clostridial medium (RCM; Difco) as used in reference 45.
Attempts were then made to determine whether stepwise downgrowth on lower levels of proline and leucine would decrease the dependence on these acceptors. For this purpose, adaptation medium (ADM; see Materials and Methods) was developed, containing low concentrations of proline and leucine (0.5 and 1.5 mM, respectively) in the presence of 5 mM glucose and 10 mM glycine. Following a short lag phase, cells obtained from BHIS day cultures grew well in ADM. As shown in Fig. 3B, within the first 5 h after inoculation, proline (solid brown triangles) was completely consumed, being reduced to 5-aminovalerate (open brown triangles). During this time, the rates of leucine, isoleucine, and valine consumption increased such that after 12 h all branched-chain amino acids had become exhausted. At that time, however, most of the glucose (Fig. 3A, + symbols) and glycine (Fig. 3A, green diamonds) still remained available, and the cultures continued to grow until glucose was finally consumed after approximately 25 h. Thereafter, extensive lysis was observed, which began at around 30 h, coinciding with the final depletion of glycine.
FIG 3.

Adaptation of C. difficile to growth on glucose in the absence of proline and leucine. To adapt cells to grow without proline and leucine, cells harvested from a BHIS day culture were used to inoculate adaptation medium (ADM) which contained 5 mM glucose, 10 mM glycine, and low concentrations of leucine, isoleucine, valine, proline, methionine, and tryptophan in a basal salt mixture supplemented with trace elements and vitamins, as described in Materials and Methods. Growths were monitored in triplicate by OD600 (*, □, and ×), aliquots were removed over time and centrifuged, and the supernatants were assayed for amino acids and glucose. (A) Concentrations are given for glucose (Glc), glycine (Gly), alanine (Ala), and ammonia (NH3). (B) The same cultures as in panel A, assayed for leucine (Leu), isoleucine (Ile), valine (Val), proline (Pro), and 5-aminovalerate (5-AV).
It was notable that substantial growth in ADM continued even after proline and leucine had been consumed. Therefore, cells grown in ADM were then transferred to media without proline and leucine but containing glucose plus 10 mM glycine, either without (Glc+Gly) or with (Gly/Ala) added alanine, as described in Materials and Methods. ADM-grown cells grew well in both media, and repeated transfers in these media resulted in continued good growth with similarly high maximum OD600 levels. Omission of methionine resulted in little or no growth, and the addition of 0.2 or 2 mM homocysteine was unable to substitute. Poor growth was observed when valine and isoleucine were omitted; however, in one trial, a Glc+Gly culture that lacked both valine and isoleucine grew to an OD600 of 0.18 after an extensive lag. This suggests that C. difficile might be able to grow also in the absence of all three branched-chain amino acids; however, further studies are needed for confirmation. No growth was found when tryptophan was withheld, which is consistent with the absence of genes for the part of the aromatic amino acid biosynthetic pathway that leads to tryptophan (19). These results indicate that C. difficile indeed is able to adapt to grow in the absence of proline and leucine and that it is possible to develop growth conditions that are devoid of these preferred Stickland acceptors for analyzing the effects on activation of alternative electron accepting pathways such as the WLP.
Growth in Gly/Ala medium.
C. difficile is readily able either to consume or to produce alanine, depending on the growth conditions (46). Alanine has long been recognized as a very effective Stickland donor (47). Therefore, alanine addition was used to augment the amount of oxidizable carbon as a strategy to promote the expression of the WLP, possibly acting to dispose of the higher load of reducing equivalents. Growth was most rapid in Glc+Gly medium in the absence of alanine, and addition of up to 10 mM alanine caused the maximum growth rate to decrease somewhat but had little effect on the final OD600 (data not shown). Cells transferred from Gly/Ala cultures quickly resumed growth in Glc+Gly medium; however, growth was not as rapid when Glc+Gly cells were used to inoculate Gly/Ala medium. One possibility is that the activity of electron acceptor pathways supporting glucose oxidation in Glc+Gly cells was insufficient to maintain redox homeostasis when the cells were shifted to more-reducing conditions imposed by 10 mM alanine.
Typical growth characteristics in Gly/Ala medium are shown in Fig. 4. As in ADM cultures, the OD600 continued to increase until the point at which glucose (+ symbols) was finally depleted. Furthermore, the major lytic phase was delayed until around 55 h, at roughly the same time that glycine-alanine fermentation had ceased, upon final depletion of glycine (green diamonds). The glycine-to-alanine uptake stoichiometry was very close to 1:1 throughout most of the fermentation. This suggests that pyruvate produced from the oxidation of alanine was being converted to acetyl-CoA without further production of NADH or reduced ferredoxin. This would be expected for C. difficile using pyruvate-formate lyase (PFL) rather than either pyruvate dehydrogenase (PDH) or pyruvate:ferredoxin oxidoreductase (PFOR). In contrast, if pyruvate were being further oxidized, then a 2:1 stoichiometry would be expected, requiring the reduction of 2 mol glycine to balance the overall process, as found in Clostridium sporogenes (47, 48).
FIG 4.

Glycine-alanine fermentation of C. difficile growing in the absence of proline and leucine. Cells obtained from ADM cultures grown to an OD600 of ca. 0.6 were used to inoculate glycine-alanine (Gly/Ala) medium lacking proline and leucine and in which glycine was the only remaining Stickland acceptor (see Materials and Methods). The growth substrates available in Gly/Ala medium were 5 mM glucose, 10 mM glycine, 10 mM alanine, and low concentrations of isoleucine, valine, methionine, and tryptophan. In addition, 2 mM sodium acetate and 3.9 mM NH4Cl were present. Triplicate growths were monitored, and amino acids and glucose were assayed as indicated for Fig. 3. OD600 (*, □, and ×) and concentrations of glucose (Glc), glycine (Gly), alanine (Ala), and ammonia (NH3) are shown.
Growth on glucose in the absence of Stickland acceptors.
For growth in the absence of all Stickland acceptors, Gly/Ala-grown cells were used to inoculate glucose-only medium (see Materials and Methods) containing 5 mM glucose and 0.21 mM valine, 0.1 mM isoleucine, 0.22 mM methionine, and 0.12 mM tryptophan as the only amino acids provided. Reproducible growth was observed and rates were lower than when glycine was added and typically yielded lower maximum OD600 (Fig. 5).
FIG 5.

Growth of C. difficile on glucose in the absence of Stickland acceptors. A glucose-containing medium in which both glycine and alanine were omitted (Glc only) was used to grow C. difficile in the absence of all amino acid Stickland acceptors. Glc-only medium was similar to Gly/Ala medium without glycine and alanine. OD600 (* and □) and concentrations (mM) of glucose (Glc), glycine (Gly), alanine (Ala), and ammonia (NH3) are shown. Gly/Ala cultures were used to inoculate the Glc-only tubes, accounting for the small amounts of glycine and alanine present initially.
Induction of WLP expression under the newly developed growth conditions.
The growth conditions described above were examined to assess the amount of WLP pathway expression based on the amounts of acetyl-CoA synthase (AcsB) protein detected on Western blots. For this, buffer-soluble supernatant fractions were prepared from extracts of C. difficile cells obtained from large-scale growths. The cell yields, final OD600, and other data from these growths are given in Materials and Methods. As shown in Fig. 6, comparing equivalent amounts of extract protein loaded, the largest amounts of AcsB were produced in the Gly/Ala growths (Gly/Ala 0.5 was harvested later than Gly/Ala 2.2, referring to 0.5 and 2.2 mM glucose remaining at harvest, respectively). By comparison to the band intensity produced by purified AcsB loaded as a standard, AcsB accounted for around 0.20% of total protein in the Gly/Ala 0.5 cell extract, whereas values of 0.03%, 0.04%, and 0.08% were estimated for Glc+Gly, Glc only, and Gly/Ala 2.2 cells. At the same amount of total protein loaded, AcsB was hardly detectable in the BHIS extracts. Addition of alanine resulted in markedly increased expression, such that AcsB was 3.0 and 7.7 times higher in Gly/Ala 2.2 and Gly/Ala 0.5 cells, respectively, than in Glc+Gly cells. Even though cells grew considerably faster in the Glc+Gly medium, omission of glycine resulted in noticeably increased production of AcsB in the Glc-only growth. This result is consistent with glycine reduction acting to support glucose utilization in Glc+Gly cells. Thus, in the absence of glycine, a greater need exists for the WLP acting to dispose of electrons. Similarly, BHIS growths that are rich in electron acceptor substrates produced the smallest amounts of AcsB, indicating little or no utilization of the WLP when Stickland acceptors are plentiful. Interestingly, the largest amounts of AcsB were not produced in the slower growing Glc-only cultures in the absence of glycine but rather were found upon addition of alanine to challenge faster growing cultures with an excess of oxidizable substrate under the Gly/Ala conditions.
FIG 6.

Western blot analysis of AcsB levels in different growth media. Cell extracts prepared from the large-scale growths indicated were analyzed by SDS-PAGE. Gels containing 12.5 μg total protein per lane were stained with Coomassie blue for protein (bottom, lanes 1 to 6), and 37.5 μg per lane of the same samples was applied for Western blot assays of AcsB (top). Gly/Ala 2.2 and Gly/Ala 0.5 indicate Gly/Ala growths with 2.2 and 0.5 mM glucose remaining at the time of harvest. Purified AcsB, 10 ng, was applied on the outer lanes of the Western blot, and molecular weight markers (M) were included on the stained gel. The estimation of equivalent amounts of protein loaded was verified by densitometric analysis of the stained gel (center).
Assays of key enzymes as indicators of functional energy-generating pathways.
(i) CODH, MTR, and glycine reductase. To help identify which energy-generating pathways or combinations thereof are most active under the different growth conditions, cell extracts were prepared and assayed for a number of critical enzymes. As shown in Fig. 7, the activity of carbon monoxide dehydrogenase (CODH) and N5,N10-methylenetetrahydrofolate reductase (MTR), both essential enzymes in the WLP, paralleled the AcsB expression pattern seen on Western blots. As with AcsB, the highest levels of these enzymes were found in Gly/Ala cells, followed by the Glc-only growth. Addition of glycine suppressed the amounts of both CODH and MTR relative to the Glc-only condition, and almost no activity was observed in BHIS. Glycine reductase (GR) was most active in the Glc+Gly cells and Gly/Ala cells, where glycine remains as the only Stickland acceptor available. Indeed, addition of glycine in complex growth media was previously shown to stimulate the production of glycine reductase (14, 49). GR is a selenoenzyme complex that reductively deaminates glycine to yield acetyl phosphate, a process which provides energetic benefits both for substrate level phosphorylation (SLP) to generate ATP directly and by acting as an efficient Stickland acceptor (reviewed in reference 50). Genes encoding the selenocysteine-containing subunits GrdA and GrdB are present in an 8-gene cluster (within the region CD2348 to CD2357) in an identical arrangement to that found in C. sticklandii (51). Near the end of growth in Gly/Ala medium, as glucose availability diminished, GR activity also declined (cf. Gly/Ala 2.2 versus Gly/Ala 0.5), while the WLP activities of CODH and MTR and AcsB protein levels all increased. These results suggest that as electron flux through GR declines, C. difficile compensates by switching to increase its reliance on the WLP pathway.
FIG 7.

Enzyme activities in C. difficile that reflect metabolic pathways active under different growth conditions. Enzymes involved in a number of key pathways were assayed in anaerobic cell extracts prepared from large-scale growths of C. difficile in BHIS and defined media indicated at the top. Gly/Ala 2.2 and Gly/Ala 0.5 indicate Gly/Ala growths with 2.2 and 0.5 mM glucose remaining at the time of harvest. The substrates and products in bold font were measured directly in the assays described in Materials and Methods. Enzyme specific activities are given in units per milligram extract protein. MV, methyl viologen; BV, benzyl viologen; H4F, tetrahydrofolate.
(ii) Formate dehydrogenase and hydrogenase. Formate dehydrogenase (FDH) assays showed almost no activity in cells grown on BHIS; however, substantial activity was observed in cells fermenting glucose in all of the defined media (Glc+Gly, Glc only, and Gly/Ala). FDH would reasonably function in C. difficile to support glucose utilization by disposing of formate produced from pyruvate by the pyruvate-formate lyase reaction, and C. difficile 630 harbors a gene cluster CD3313 to CD3317 encoding the components of a reversible hydrogen-dependent CO2 reductase complex, HDCR, reviewed in reference 52, that includes a molybdenum/tungsten- and selenocysteine-dependent formate dehydrogenase (FdhF) and an [Fe-Fe] hydrogenase (HydA) likely involved in converting formate to CO2 plus H2. The intrinsic hydrogenase activity of the HydA component in HDCR may account for a portion of the separately measured hydrogenase activities under all of the defined medium conditions. However, the hydrogenase activity observed in cells grown in BHIS is probably due to a different enzyme, because almost no FDH activity was produced in BHIS.
A different hydA gene in C. difficile is part of a three-gene cluster, CD3405 to CD3407, encoding homologs of the HydABC proteins with high sequence similarity to the subunits in the trimeric [Fe-Fe] electron-bifurcating hydrogenases that have been best characterized in species such as Thermotoga maritima, Acetobacterium woodii, Moorella thermoacetica, and Ruminococcus albus (53–56). Reversible electron-bifurcating hydrogenases (operating in the direction of confurcation to form H2) allow for reoxidation of NADH by coupling the otherwise endergonic process of proton reduction by NADH to the exergonic oxidation of reduced ferredoxin. Evolution of H2 by C. difficile growing in BHIS (see Table S1 in the supplemental material) would allow for greater net substrate oxidation and generate more ATP per mole of substrate consumed than would be possible if NAD+ regeneration were to proceed by other means that would require a portion of the available carbon substrates to be used as electron acceptors. Three other genes encoding [Fe-Fe] hydrogenase homologs, with cysteine ligands at appropriate positions to potentially form an active site H cluster, are also present in the genome, namely, CD0893, CD0894, and CD3258; however, it is uncertain to what extent these other proteins contribute to the measured hydrogenase activity or what their functions may be.
(iii) Serine deaminase/dehydratase. Growth on BHIS is characterized by extensive amino acid catabolism, with leucine, serine, threonine, cysteine, and isoleucine being consumed in the largest amounts (see Table S2). Deamination reactions produce a total of around 20 mM ammonia, and a substantial amount of nitrogen is also disposed of by conversion of pyruvate to alanine, with as much as 9 to 10 mM alanine being secreted during growth. Glycolysis and the deamination of serine and cysteine are potentially the largest sources of pyruvate and, in comparison with defined media that contain neither serine nor cysteine, serine deaminase activity was found to be especially high in BHIS-grown cells (Fig. 7). As a likely candidate, C. difficile sdaB CD3222 encodes a serine deaminase (also termed serine dehydratase) that is a member of the pyridoxal-5′-phosphate (PLP)-independent, iron sulfur cluster-containing, single polypeptide chain, type 2 l-serine dehydratases that have been best studied in Legionella pneumophila and are widely distributed in prokaryotes (57, 58).
(iv) Transaminases supporting leucine and alanine metabolism. Extensive catabolism of leucine requires as a first step the conversion to its keto acid form. The usual pathway involves transamination with α-ketoglutarate (α-KG) to produce glutamate plus α-ketoisocaproic acid, with regeneration of α-KG by oxidative deamination of glutamate by glutamate dehydrogenase (GDH), producing NH3 and NADH. The gluD gene CD0179 encodes GDH, and recombinant C. difficile NAD-dependent GDH has been purified and characterized (59). As a secreted enzyme measured clinically in stool samples as one tool to diagnose C. difficile infection (CDI) (60), GDH confers resistance to H2O2 (61) and is important for colonization in a hamster model (62). In contrast, it is unclear what gene(s) encodes transaminase activities for branched-chain amino acid (BCAA) utilization. In particular, no orthologs have yet been identified for α-KG- or alanine-synthesizing BCAA aminotransferases for C. difficile in the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway database. However, as shown in Fig. 7, high levels of leucine:α-KG aminotransferase activity are indeed observed in the extracts. Substantial amounts of glutamate:pyruvate aminotransferase activity are present as well. In BHIS-grown cells, the Leu→Glu activity was higher than in cells grown in defined media; by contrast, Glu→Ala activity was usually lower in BHIS. Moreover, the ratio of the activities (Leu→Glu versus Glu→Ala) was always greater than 1 in BHIS and substantially less than 1 in the defined media. These results indicate that a selective branched-chain or leucine-selective transaminase likely exists for degradation of leucine that is yet to be identified. Furthermore, by excretion of alanine generated from readily available sources of pyruvate (e.g., glucose, serine, and cysteine), the combination of aminotransferase activities affords a means for C. difficile to convert leucine to its keto form by a redox-independent process to regenerate α-KG, without involving GDH and the need to dispose of NADH.
(v) Proline reductase produced in the absence of proline. Considering that proline is entirely absent in the defined media, it seemed at first counterintuitive to find a large increase in proline reductase (PR) activity versus that in BHIS (Fig. 7). PR serves an important Stickland acceptor function, as reducing equivalents are consumed and NAD+ is regenerated in the conversion of proline to 5-aminovalerate. PR is a selenoenzyme complex encoded by the prd operon CD3237 to CD3247 that is upregulated in a proline-responsive manner by PrdR (49). Acting as a σ54-dependent activator protein, PrdR is a member of the bacterial enhancer binding proteins (bEBPs), many of which respond to environmental signals through ligand-binding regulatory domains (63, 64). Our finding of high PR activity in the absence of proline indicates that transcription of the prd operon is unlikely to be activated solely by a PrdR-dependent mechanism and suggests the additional involvement of σ70 or a different alternative sigma factor. Moreover, by analysis of the catabolite control protein CcpA regulon in C. difficile, Antunes et al. (65) showed that expression of prd genes is induced by glucose and positively controlled by CcpA, and they confirmed by electrophoretic mobility shift assays (EMSAs) that the promoter of prdA contains a catabolite responsive cre site to which CcpA binds. Thus, our data showing the increased expression of PR in the absence of proline can be reasonably interpreted as a result of induction by glucose under the control of CcpA.
(vi) CoA transferase activity in the production of butyrate. Butyryl-CoA:acetate CoA transferase catalyzes the formation of butyrate by reaction of acetate with butyryl-CoA. Instead of converting butyryl-CoA to butyryl phosphate and generating ATP via butyrate kinase, this enzyme allows the cell to directly maintain the pool of acetyl-CoA by using acetate from the medium to recapture the thioester bond of butyryl-CoA as butyrate is released. Therefore, assays were performed to determine the activity under the different growth conditions. As shown in Fig. 7, activity was readily detected in cell extracts of both BHIS and the defined media. Activity was highest in BHIS and the Glc-only growths and notably lowest in Glc+Gly cells. Two candidate CoA transferase genes (cat1 CD2343 and cat2 CD2339) are present within a cluster of genes assumed to be involved in succinate and succinate semialdehyde conversion to butyrate, and another more distantly related succinyl-CoA:acetoacetate transferase (two subunits encoded by CD2677 and CD2678) is also annotated, presumably involved in a different pathway. An additional CoA transferase (hadA CD0395) involved in the reduction of leucine can be excluded, because it has high substrate specificity and does not act on acetyl-CoA or butyryl-CoA (66).
As a substrate for the transferase, acetate is known to stimulate butyrate production and increase the growth rates of a number of Clostridia and other anaerobic butyrate-producing species relevant to the gut microbiome, e.g., in Clostridium thermobutyricum (67), and in intestinal species such as Roseburia and Faecalibacterium (68). Acetate also stimulates and helps to stabilize solvent production during repeated subculture of Clostridium beijerinckii (69) and is absolutely required in the medium for cultivation of the predominantly butyrate-forming Clostridium kluyveri (70). By comparison with C. kluyveri, the most likely candidate for butyryl-CoA:acetate CoA transferase in C. difficile is the cat2 gene CD2339 product whose sequence is 55% identical to authentic C. kluyveri cat3 protein active in the pathway of acetyl-CoA reduction to butyrate (71).
The activity of butyryl-CoA:acetate CoA transferase in BHIS growths was lower at 5 h but increased substantially by 12 and 17 h (Fig. 7). Our data indicate that formation of butyrate occurs late during growth in media containing leucine (e.g., BHIS and others) and that little production of butyrate takes place as long as more-preferred electron acceptors such as leucine are available. This agrees with findings of others (46) who showed late production of butyrate in C. difficile minimal medium (CDMM) in which leucine was consumed entirely and showed that no butyrate was formed at all in a minimal defined medium (MDM) (72) containing higher initial levels of proline and in which substantial leucine still remained at the end of the growth. During the early phase of growth in BHIS, the low rate of butyrate production that occurs while better electron acceptor substrates are still available would explain why butyryl-CoA transferase activity is initially lower and then subsequently increases over time as the more favorable substrates become depleted. Because the Glc+Gly cells contain highly active glycine reductase, there is no need to dispose of electrons by reducing acetyl-CoA to butyrate, which explains why very low levels of butyryl-CoA:acetate CoA transferase are produced. In contrast, high levels of activity are observed in the Glc-only growth, which would be consistent with the pathway of acetyl-CoA reduction to butyrate functioning to support glucose utilization by these cells.
Effect of acsB deletion on growth, glucose uptake, and acetate production.
To better delineate the role of the WLP in C. difficile, a mutant containing a defined in-frame deletion of acsB was produced by allele exchange using Escherichia coli codA as a heterologous counterselection marker according to the procedure developed by the Minton laboratory (73), as described in Materials and Methods. In comparison with the wild type, the mutant C. difficile 630 ΔacsB exhibited little or no difference in the OD600 versus time growth curves obtained in BHIS. Among the various newly defined media, a small but reproducible decrease in growth rate was seen under the Glc-only condition, but no noticeable differences were found under other conditions, including Gly/Ala medium, which yields especially precise and reproducible growth curves for comparisons.
In the course of exploring different compositions of defined media, it was found that some of the conditions lacking acetate produced a pronounced lag phase that could be shortened or eliminated altogether by addition of relatively low levels of sodium acetate in the range of 2 to 5 mM. As shown in Fig. 8, acetate limitation had a substantially greater effect on the acsB mutant (Fig. 8, open symbols) than on the wild type (Fig. 8, closed symbols). As the amount of acetate added to the medium was decreased below 2 mM, both the growth rate and the maximum OD600 were decreased in the mutant to a much greater extent than in the wild type. In addition, the glucose uptake profiles (Fig. 8, red circles) showed that as acetate became increasingly scarce, the time course for substrate utilization by the mutant became increasingly longer, as the maximum rate at which the cells were able to use glucose declined. Measurements of the amount of acetate formed over time (Fig. 8, right, blue symbols) showed a marked decrease in the ability of the mutant to produce acetate, regardless of the growth rate or the final extent of growth or the amount of acetate initially added. Complementation of the mutant by introduction of a plasmid containing acsB under the control of the native WLP promoter (see Materials and Methods) provided an increase in both the growth and acetate production compared with that in the acsB mutant. However, neither growth nor acetate production was completely restored to the wild-type levels (see Fig. S1, green × and + symbols and blue + symbols). Notably, the maximum rates of glucose utilization achieved by the complemented strain were much higher than those in the mutant, particularly at low acetate levels, and nearly equal to those of the wild type (Fig. S1, red + symbols).
FIG 8.

C. difficile wild-type and ΔacsB mutant growth, glucose consumption, and acetate production in glucose-only (Glc-only) medium under acetate-limiting conditions. Glc-only medium containing decreasing amounts of sodium acetate was used to compare growth characteristics of the wild type (closed symbols) and the ΔacsB mutant (open symbols). Graphs on the left show growth (OD600, black symbols) and glucose consumption (red symbols), whereas those on the right show acetate production (blue symbols) from the same growths and the corresponding glucose consumption curves for comparison.
Attempts to interpret the effects of inactivating the WLP on acetate production under other growth conditions, such as in Gly/Ala medium, are complicated by the fact that large amounts of acetate are generated from other substrates, e.g., reduction of glycine and oxidation of alanine, both of which are present in high concentrations in Gly/Ala medium and extensively metabolized to acetate. However, the data from growth conditions that contain only glucose and CO2 as carbon sources, shown in Fig. 8, provide convincing evidence that the WLP acts both to increase the production of acetate and to support the metabolism of glucose.
Fermentation product analyses of the wild type and acsB mutant—evidence for WLP coupling to butyrate formation.
To obtain insight into how the WLP promotes the utilization of glucose in C. difficile, fermentation products were quantified by gas chromatography (GC) and high-performance liquid chromatography (HPLC) methods as described in Materials and Methods. The major products present at the end of the Glc-only fermentation were ethanol, formate, acetate, butyrate, and small amounts of lactate. In addition to these, other end products that were readily identified in growths on BHIS and under other conditions were not detectable in the Glc-only growths at the sensitivity limits of our assay methods. These included 1-butanol, isobutanol, isobutyric acid, n-valeric and isovaleric acids, 2-methylbutyric acid, n-caproic and isocaproic acids, and the α-keto acids (pyruvic, α-ketoisovaleric, and α-ketoisocaproic acids). As shown in Fig. 9, the mutant (orange) produced notably increased amounts of reduced products ethanol and lactate regardless of the initial acetate concentration in the culture, whereas in the wild type, production of acetate was substantially higher (Fig. 9, dark plum). These data indicate that pathways of ethanol and lactate formation were being used to a greater extent for the disposal of reducing equivalents in the cells lacking a functional WLP. As already suggested by the AcsB levels and enzyme activities under different growth conditions (Fig. 6 and 7), the increased formation of reduced products in the acsB mutant provides further evidence for a role of the WLP in C. difficile as a terminal electron sink to support growth on glucose.
FIG 9.

Fermentation product comparisons of C. difficile wild type and ΔacsB mutant. Samples removed from cultures of C. difficile wild type and ΔacsB mutant grown in Glc-only medium at different levels of added acetate (0 to 2 mM), presented in Fig. 8, were taken over time and analyzed for glucose and volatile organic compounds by GC, HPLC, and enzymatic assays, as described in Materials and Methods. The maximum rates of glucose consumption during the growths are compared, along with the final concentrations of products acetate, butyrate, lactate, ethanol, and formate present at the end of the fermentations. The averages from duplicate cultures are plotted, error bars indicate the differences between them.
The amount of formate generated was largely independent of the initial acetate concentration or other parameters such as growth rate or the maximal extent of growth. This likely reflects an equilibrium concentration of formate, which is calculated to be in the range of 10 to 20 mM on the basis of the E0′ values for the CO2/formate and H+/H2 couples (74) at the final pH and the concentrations of H2 and CO2 available in the atmosphere. However, accumulation of formate was notable even in the absence of added H2, for example, in BHIS and in the Gly/Ala growths, which produce around 3 to 4 mM formate.
In addition, Fig. 9 shows that in contrast to that of ethanol and lactate, the production of butyrate was substantially lower in the acsB mutant, and the amount of butyrate formed dropped sharply as acetate availability decreased. Why butyrate, which is also a reduced product, was markedly diminished whereas ethanol and lactate were increased can be rationalized along two lines. First, acetate is required by the butyryl-CoA:acetate CoA transferase reaction to generate butyrate. Therefore, the production of butyrate would be expected to decline as the level of acetate decreased. Indeed, regardless of the amount of acetate initially added, much more acetate was ultimately produced by the wild type containing a functional WLP than by the acsB mutant. This indicates that the WLP acts to supply acetate that can be used to support the formation of butyrate.
The second consideration is direct redox coupling of the WLP to the pathway of acetyl-CoA conversion to butyrate. As indicated in Fig. 1A, butyryl-CoA is generated by the electron bifurcating butyryl-CoA dehydrogenase-electron transferring flavoprotein complex (Bcd-EtfAB)4 which consumes 2 NADH to reduce 1 mol of crotonyl-CoA and transfers two low-potential electrons to ferredoxin (75–77). Because the WLP provides a means to reoxidize the reduced ferredoxin formed by the electron-bifurcating enzyme, knockout of the WLP is also expected to decrease the formation of butyrate by impairing the ability to return ferredoxin to its oxidized state. Other means to reoxidize ferredoxin, such as by H2-evolving hydrogenase, are made less favorable under the 1.4-atm H2 partial pressure added to the Glc-only tubes; however, hydrogen is not inhibitory to the WLP. Thus, the data suggest that coupling of the WLP and butyrate pathways provides an energetically efficient means to regenerate NAD+, with a theoretical value of 5 NADH oxidized for each acetyl-CoA converted to butyrate (see Discussion).
Toxin production under the different growth conditions.
Toxin expression by C. difficile is affected by a number of environmental signals, among which nutritional factors, such as carbon source and the availability of certain amino acids, play a critical role. Regulatory systems involving carbon catabolite repression mediated by the regulatory protein CcpA, and the response to the level of GTP and branched-chain amino acids acting through the global regulator CodY, control the expression of TcdA and TdcB toxins by binding upstream of the tcdR gene, which encodes a sigma factor specifically involved in transcription of the toxin genes (65, 78–82). To assess the relative levels of toxin expression in cell extracts from the various growth conditions, Western blots were made using anti-TcdA antibodies. The analysis shown in Fig. 10, which used equivalent loading amounts of the same samples as for AcsB in Fig. 6, indicates that the amount of TcdA was much greater in cells grown on BHIS than in any of the defined media, up to 13 times higher on the basis of TcdA band signal intensity. The toxin levels in BHIS cells were relatively constant throughout the time course of growth, from exponential through stationary phase (see Fig. S2). Amino acid starvation is associated with toxin induction, and cysteine and proline are the most potent amino acids that suppress toxin formation (72, 83). However, neither of these amino acids is present in the defined media developed here. In addition, although both Cys and Pro are rapidly consumed early in BHIS growths, which could result in a lack of toxin suppression in that medium, their absence in the defined media would not explain the low level of toxin production there. Moderate repression of toxin production by other amino acids, including alanine and glycine, has also been reported (83), which at first glance might explain the low levels of toxin in the Gly/Ala and Glc+Gly media. However, low levels of toxin are also found in the Glc-only medium that contains only low concentrations of a minimal set of amino acids (methionine, tryptophan, isoleucine, and valine). These results point to factors other than amino acid availability for suppression of toxin under the defined medium conditions. Here, the mechanism of glucose control over toxin expression appears to predominate, and CcpA-mediated regulation in response to glucose seems likely.
FIG 10.
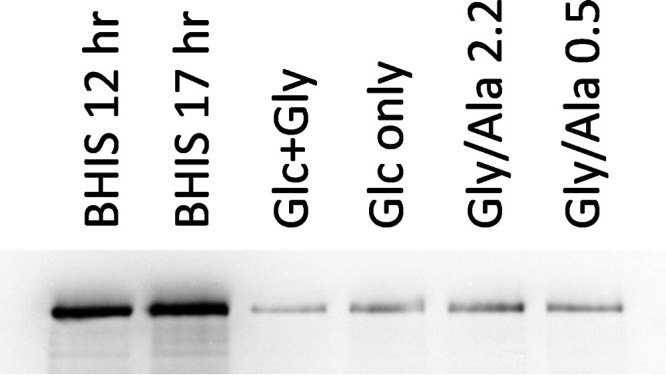
Relative TcdA toxin levels in cells grown in BHIS and in defined media. Cell extracts prepared from the large-scale growths indicated were analyzed by SDS-PAGE on an 8% acrylamide gel with equivalent amounts of protein, 37.5 μg, applied per lane. The loading pattern was the same as for Fig. 6. Western blot analysis employed mouse monoclonal anti-TcdA as the primary antibody and was developed and imaged as indicated in Materials and Methods. Relative TcdA band intensities were 10.8, 13.0, 1.00, 1.66, 2.11, and 1.55 for the BHIS 12 h, BHIS 17 h, Glc+Gly, Glc-only, Gly/Ala 2.2, and Gly/Ala 0.5 extracts, respectively.
DISCUSSION
In this study, the role of the Wood-Ljungdahl pathway in C. difficile was investigated by developing novel conditions for growth in the absence of favored amino acid Stickland acceptors. Only minimal levels of key WLP enzymes CO dehydrogenase (CODH) and acetyl-CoA synthase (AcsB) were expressed in rich media containing large amounts of electron acceptor substrates. Under these conditions, CODH activity was at the limit of detection in sensitive enzymatic assays, and AcsB was estimated to be at or below 0.01% of total protein on Western blots. Similarly, CODH was virtually undetectable in extracts of C. sticklandii, a genetically and metabolically related bacterium that also harbors a complete set of WLP genes (51). Given the wide variety of electron-accepting pathways available to C. difficile, we hypothesized that if the WLP acts in a reductive role, then higher levels of WLP proteins might be observed under conditions in which the more-preferred acceptors were absent. Our attempts to simply transfer cells obtained from various growing or stationary-phase cultures to nonpermissive conditions, under which they are metabolically unprepared to maintain energy requirements, did not result in a lag phase with eventual growth but instead caused extensive autolysis and thereafter no subsequent increase in OD600 over time. In contrast to an earlier report (45), we were unable to demonstrate autotrophic growth of C. difficile under a pressurized gas phase of H2 plus CO2. Since our results in defined media verified that tryptophan is absolutely required for growth, as expected by the lack of genes needed for its biosynthesis (19), we also attempted autotrophic growths with added tryptophan, which were also unsuccessful. Although we were not able to obtain autotrophic growth, it was possible to shift cells to more restrictive conditions and by adaptation to overcome the problem of autolysis.
Adaptation to decrease the dependence on Stickland acceptors redefines C. difficile essential amino acid requirements.
Adaptation to growth in the absence of proline and leucine was achieved during growth in ADM, which contains low concentrations of these preferred Stickland acceptors, consumed early in the fermentation, while most of the glucose still remains available to support continued growth (Fig. 3). Following adaptation, C. difficile was able to grow in the absence of added proline and leucine, conditions that would be less conducive to regeneration of NAD+, especially considering how rapidly these amino acids are reduced in complex media. In vivo metabolomic studies by others revealed an utmost importance of proline (by inference from its product 5-aminovalerate), which undergoes large changes in concentration soon after infection (8), and it has been reported that a C. difficile mutant in which the proline reductase prdB gene was disrupted exhibited a decreased ability to colonize germfree mice transplanted with human fecal samples from either dysbiotic or healthy individuals (84). The same prdB mutant was originally described and shown in reference 49 to grow almost as well as the wild type in complex media. C. difficile strains, including C. difficile 630, have been reported to be auxotrophic for proline, and our findings agree with others (17, 84) that growth does not occur when proline is simply omitted from defined media. However, our data show that C. difficile 630 indeed is able to grow in minimal media without proline provided that prior adaptation has been established. Thus, proline can no longer be considered an essential amino acid for C. difficile.
Biosynthesis of proline is required under the minimal nutritional conditions described here; however, it is unlikely to occur by the standard pathway involving glutamate 5-kinase and glutamate-5-semialdehyde dehydrogenase in the first two steps, because C. difficile lacks proB and proA homologs encoding these enzymes. Instead, the less common pathway, in which proline is derived directly from ornithine, is indicated, because C. difficile appears to contain a complete set of ornithine biosynthetic genes and a putative ornithine cyclodeaminase (OCD) gene CD0544 is also present. Furthermore, CD0544 is highly similar to OCD genes found in C. sporogenes and Clostridium botulinum, species whose extracts actively convert ornithine to proline (85). Therefore, it seems unlikely that authentic proline auxotrophy would be a general trait in C. difficile.
Leucine is another amino acid that our data indicate is not essential for C. difficile. When available to the cells, leucine is a highly preferred Stickland acceptor. In the leucine-rich BHIS, it is entirely consumed and almost exclusively reduced to isocaproic acid, with less than 2% undergoing oxidation, as judged by the relative amounts of isocaproic and isovaleric acids found (see Table S2 in the supplemental material). This is consistent with the likely function of leucine as a positive activator of the reductive catabolic pathway, which is thought to be regulated by a leucine-responsive bacterial enhancer binding protein LeuR required for σ54-dependent transcription of the hadAIBC-acdB-etfBA operon (64). The reductive pathway relies on a “radicals by reduction” (86) strategy that involves ATP-dependent low-potential activation of the key enzyme 2-hydroxyisocaproyl-CoA dehydratase, whose catalytic role is to generate a remarkable ketyl radical intermediate from the substrate 2-hydroxyisocaproyl-CoA, which can then undergo dehydration and subsequent conversion to the product isocaprenoyl-CoA (87, 88). Thereafter, reduction of isocaprenoyl-CoA and conversion to isocaproic acid proceeds by way of flavin-based electron bifurcation to regenerate 2 NAD+ and capture two low-potential equivalents in reduced ferredoxin (89, 90). Growth in the absence of leucine deprives C. difficile of this very efficient reductive pathway and, at the same time, also requires a biosynthetic route to supply leucine for anabolic processes. Homologs of all the genes in the ilv and leu pathways needed for de novo synthesis of the keto forms of Ile, Val, and Leu are present in the genome. Although a transaminase candidate then needed in the final step to complete the formation of the BCAAs has yet to be identified in the KEGG database, our enzyme assays (Fig. 7) indicate that a selective branched-chain or leucine-selective transaminase may be involved.
In defined media lacking leucine, the amounts of its precursor keto acid required to support protein synthesis potentially could be produced from valine supplied in the medium after conversion to the common intermediate α-ketoisovalerate. This intermediate is also needed for biosynthesis of pantothenate and coenzyme A, and in Gram-positive bacteria, branched-chain keto acids serve as precursors for branched-chain fatty acids that make up major components of membrane lipids. Upregulation of ilv and leu pathways in BCAA-limited media, involving the global regulator CodY as sensor of the availability of GTP and BCAAs, is indicated by the strong induction of ilv and leu genes in a C. difficile codY null mutant (80). The need to provide low levels of isoleucine and valine in the defined media devoid of leucine described here may reflect a high demand for BCAAs and their keto acid precursors for multiple anabolic pathways that could be difficult to satisfy completely by de novo biosynthesis acting alone.
Reductive function of the WLP in C. difficile.
Our results indicate that C. difficile uses the WLP to dispose of metabolically generated reducing equivalents, and hence the WLP is included in this bacterium’s repertoire of multiple diverse reductive pathways. This is supported by the following lines of evidence. First, WLP expression increases progressively as Stickland acceptors are increasingly withheld from the various media, as indicated by Western blot analysis of AcsB (Fig. 6). The levels of AcsB rise markedly when proceeding from BHIS, a complex medium that contains large amounts of efficient electron acceptors such as proline and leucine, to the Glc+Gly medium, in which glycine is the only Stickland acceptor provided. A further increase in AcsB is seen when comparing the Glc+Gly medium to the Glc-only condition, in which glycine has been removed, leaving no remaining amino acids able to act as electron acceptors. The data indicate that C. difficile readily takes up and excretes alanine from the medium as a means of balancing redox status, and AcsB is maximally elevated when 10 mM alanine is added to the Glc+Gly medium. Thus, the presence of large amounts of alanine in the Gly/Ala medium creates a demand for additional activity and flux through reductive pathways due to the added load of oxidizable substrate, consequently leading to the upregulation of the WLP. A second line of evidence comes from the comparison of enzyme activities in cell extracts. As expected for key enzymes in the pathway, the activities of CO dehydrogenase (CODH) and methylenetetrahydrofolate reductase (MTR) follow the pattern seen for AcsB expression in the various extracts (Fig. 7). Moreover, enzymes in other reductive pathways, such as proline reductase (PR) and glycine reductase (GR), most often also show increased activity in the defined media compared with that in BHIS, which again points to a greater demand for removal of reducing equivalents. Compared with the Glc-only medium, GR activity in the glycine-containing Glc+Gly medium exhibits an inverse relationship with respect to the WLP, as indicated by the activities of CODH and MTR (Fig. 7) and the levels of AcsB (Fig. 6). A similar inverse pattern is seen with GR and the WLP when the two Gly/Ala conditions are compared with one another. Taken as a whole, this suggests that at low GR activity the WLP steps up to take on its reductive function. In addition, in the defined media in the absence of glycine, the high activity of butyryl-CoA:acetate CoA transferase suggests an increased reliance on the reductive pathway converting acetyl-CoA to butyrate. A third line of evidence that the WLP functions in a reductive capacity in C. difficile comes from the comparison of fermentation products generated by wild-type cells and by the acsB mutant (Fig. 9), in which loss of a functional WLP results in a marked decrease in the amount of acetate produced accompanied by the excretion of greater amounts of other reduced products ethanol and lactate.
The effects of limiting the availability of electron acceptors, increasing the load of oxidizable donors, and disabling the WLP by deletion of acsB all point to an increased requirement for regeneration of NAD+. In C. difficile and many other Gram-positive bacteria, global regulation of various reductive pathways involves the redox-sensing transcriptional repressor Rex, which directly responds to the NADH/NAD+ ratio (91–94). Rex binds to DNA under low NADH/NAD+ conditions, repressing its target genes; as the NADH/NAD+ ratio increases, binding of NADH to Rex results in a conformational change, dissociation of Rex from its operator sites, and derepression of genes in NAD+-regenerating pathways. In C. difficile, various reductive pathways in which Rex binding to target genes has been demonstrated by EMSA analyses include butyrate formation from both succinate and acetyl-CoA, ethanol production, and glycine reductase (95). We note that putative Rex-binding sites exist in the C. difficile WLP promoter, and a Rex-binding motif was also identified previously in the WLP promoter in C. ljungdahlii and Clostridium carboxidivorans and confirmed by EMSA (96). Therefore, in our defined media from which favored acceptors are withheld, as well as when cells are challenged with large amounts of alanine, the expected increased NADH/NAD+ ratio may upregulate the expression of the WLP potentially by means of Rex-mediated derepression.
Finally, additional regulatory controls of the WLP are not unexpected. Several potential CodY-binding sites can be found in the C. difficile WLP promoter, and transcriptome sequencing (RNA-Seq) studies on cells in exponential growth, involving a panel of CodY point mutants with different levels of residual activity, show substantial increased transcription of genes throughout the WLP operon, with the largest amount (4- to 5-fold) observed in the CodY null mutant (97). Regulation in response to glucose, possibly involving CcpA or other means (65), may also be integrated with Rex and CodY in a more complex network.
Connection of the WLP to glucose utilization and butyrate production.
Enzyme activity data and quantitative Western blot assays of AcsB taken as a whole indicate that the WLP pathway is on the order of 50- to 100-fold more active in cells growing in the defined medium Gly/Ala than in BHIS in which activity is barely above the limits of detection. However, even the highest levels found here are still relatively low compared to that of typical acetogenic bacteria, even when grown heterotrophically. The specific activity of CO dehydrogenase in glucose-grown Clostridium thermoaceticum (Moorella thermoacetica) (98) is 18 times higher than the maximum activity in C. difficile reported here (Fig. 7). In addition, our assays of extracts from fructose-grown C. ljungdahlii indicate a 43-fold higher CODH specific activity, and Western blots show much higher intensities of C. ljungdahlii AcsB, even considering that C. difficile antibodies were used (data not shown). As shown in Fig. 9, wild-type C. difficile with an active WLP produces substantially greater amounts of acetate than the acsB mutant. However, well-characterized acetogens can produce concentrations of acetate on the order of ∼50 mM, which is roughly 10- to 12-fold higher than wild-type C. difficile growing under comparable glucose- or fructose-fermenting conditions under an H2/CO2 atmosphere.
It is clear that nature has chosen to maintain a functional WLP operon in C. difficile; however, if it was never intended to be expressed at very high levels, then what advantage does only a modest level of activity confer? One possibility is that low flux through the pathway operates during glucose fermentation to increase the efficiency of ATP production in connection with butyrate metabolism. This is supported by the finding that butyrate formation is greatly diminished in the acsB mutant, whereas other reduced products such as ethanol and lactate are elevated (Fig. 9). As depicted in Fig. 11, coupling of the WLP to butyrate production increases the efficiency of ATP formation to 3.6 ATPs/glucose, compared with values of 2.0 and 3.0 for lactate and ethanol fermentations, respectively. Homoacetogenic fermentation, which employs the WLP and produces 3 molecules of acetate from glucose, would give a maximum of 4.0 ATPs/glucose, assuming no involvement of electron-bifurcating hydrogenase and Rnf-type chemiosmotic mechanisms to capture additional energy from reduced ferredoxin. In Acetobacterium woodii, that additional capture is estimated to be 0.3 ATP/glucose, which is included in the 4.3 ATPs/glucose total calculated for the homoacetogenic fermentation of glucose (99). Autotrophic acetogenesis, which produces 0.25 to 0.3 ATP per acetate formed from 4 H2 plus 2 CO2 (100) demands a much higher flux through the WLP. The required flux is related to the amount of acetate that must be formed by the WLP to support the overall ATP yield from the fermentation. Thus, in autotrophic growth, 3 to 4 acetates must be produced via the WLP for each ATP generated, which can be compared with the much lower flux of 0.23 acetate/ATP in the homoacetogenic case (where only one acetate formed in the WLP is all that is needed to balance the redox requirements to produce 4.3 ATPs). As shown in Fig. 11, when coupled to butyrate formation, the WLP needs only to generate 0.4 acetate to support a total of 3.6 ATPs, giving rise to the lowest flux level through the WLP of 0.11 acetate/ATP. When glucose is available in limited amounts or at low concentrations, it would be advantageous for C. difficile to maintain a high efficiency of ATP formation. Furthermore, by acting in concert, much lower levels of expression of the WLP and butyrate pathway enzymes would be sufficient to satisfy the redox requirements, which can be achieved by diverting much less acetyl-CoA to form butyrate. Because of the high efficiency of NAD+ regeneration by this type of “acetobutyrogenesis,” high rates of ATP generation are possible even with relatively low levels of expression of the WLP. A similar strategy may be more widely distributed in other organisms such as C. sticklandii that contain the WLP but are not considered to be typical acetogens.
FIG 11.

Approximate ATP yields from glucose in various anaerobic fermentation pathways and using the Wood-Ljungdahl pathway coupled to butyrate formation. The efficiencies of ATP production from glucose are compared. For simplicity, contributions from chemiosmotic mechanisms and membrane complexes, such as Rnf, are excluded. In general, the efficiency of ATP production is highest when organic products of the pathway are not required to serve as electron acceptors, which increases the amount of acetyl-CoA available to produce ATP via SLP. A maximal level of 4.0 ATPs/glucose is expected, provided that another pathway is used to fully regenerate NAD+ and assuming that pyruvate is converted to acetyl-CoA without producing additional redox equivalents either as NADH or reduced ferredoxin. This assumption is reasonable for C. difficile given that formate is produced even in the absence of added H2 and that glycine and alanine are consumed in a 1:1 stoichiometry in the Gly/Ala fermentation (see Results and Fig. 4). By comparison, the lactic acid fermentation yields the lowest ATP/glucose ratio of 2.0, because no SLP from acetyl-CoA is possible. Diversion of one-half of the acetyl-CoA to ethanol regenerates the necessary amount of NAD+ and improves the ratio to 3.0. Connection of the WLP pathway to the pathway for butyrate formation from acetyl-CoA results in a further increase to 3.6, closer to the theoretical maximum. A total of 5 NADHs are consumed per mol butyrate produced by this arrangement: 2 NADHs are used by the WLP to produce each acetate, which enters into the butyryl-CoA:acetate CoA transferase reaction to form butyrate and regenerate acetyl-CoA, and 3 NADHs are taken up for each acetoacetyl-CoA converted to butyryl-CoA (1 NADH from the 3-hydroxybutyryl-CoA dehydrogenase reaction, and 2 NADHs from the reduction of crotonyl-CoA involving electron bifurcation by the Bcd-EtfAB complex that also conserves energy by producing 1 , which is then used to reduce CO2 to CO in the WLP). However, only 2 NAD+s instead of 5 are needed per glucose oxidized so that formation of 0.4 mol butyrate is sufficient to balance the fermentation using butyrate/WLP coupling.
In the gut, this system would be less likely to operate in the presence of other preferred electron acceptors. However, in vivo transcriptomic and in vivo metabolomic studies demonstrate that C. difficile undergoes substantial changes in its metabolic profile in response to decreasing nutrient availability that occurs during growth over time postinfection (6–8, 101). In one study using mice pretreated with cefoperazone and inoculated with C. difficile spores, a large increase in the differential expression of all WLP genes, on average 50-fold, was found between 12 and 24 h postinfection (8). The 24-h time point is particularly noteworthy, because in studies on the dynamics of infection, extensive proliferation occurs by that time at which maximum levels of C. difficile CFU per milliliter in the cecum and colon are detectable (102). Our results that identify new growth environments in vitro provide further indications that the metabolic diversity and adaptability of C. difficile to changes in the nutrient environment are greater than may have been previously appreciated.
Glycine cleavage/synthase complex subunits encoded within the WLP operon.
Colocalization of the genes gcvL CD0723 and gcvH CD0729 involved in the reversible glycine cleavage system (GCS) within the WLP operon (Fig. 1B) is now recognized to be considerably more widespread than originally indicated in reference 51. In addition to eight of the acetogens compared in reference 103, there are at least 8 other acetogenic and related bacteria that contain both gcvL and gcvH located within the WLP operon (Fig. 1B). Note that species that contain colocalized WLP and GCS genes include C. sticklandii, C. sordellii, Clostridium bartlettii, Clostridium glycolicum, Clostridium bifermentans, Tepidanaerobacter acetatoxydans, Alkaliphilus metalliredigens, and Peptostreptococcaceae bacterium VA2, in addition to the species listed in reference 103: Clostridium aceticum, C. difficile, Clostridium drakei, Clostridium scatologenes, C. carboxidivorans, Clostridium autoethanogenum, Clostridium ragsdalei, and C. ljungdahlii. The reason for this colocalization is not well understood; however, our finding of the inverse expression pattern of the WLP and the glycine reductase (GR) system in C. difficile may provide a clue. The GCS comprises four enzymes that reversibly oxidize glycine in reaction with tetrahydrofolate to produce N5,N10-methylenetetrahydrofolate, CO2, and NH3. Besides gcvL (encoding a flavoprotein dihydrolipoyl dehydrogenase) and gcvH (encoding a heat-stable protein with covalently bound lipoamide), C. difficile possesses genes for the other two GCS proteins located elsewhere in the chromosome gcvP CD1658 (encoding the P protein, glycine decarboxylase-containing pyridoxal phosphate) and gcvT CD1657 (gene for the T protein, which catalyzes release of NH3 from the dihydrolipoamide-bound aminomethyl group and the transfer of the methylene moiety to tetrahydrofolate).
In various species of aerobic and anaerobic bacteria, as well as in plants and animals, GCS acts in the physiological direction of glycine decomposition. Recently, a biosynthetic role for the system acting in the opposite direction of glycine synthesis in cooperation with the WLP was proposed in the acetogen C. drakei under autotrophic conditions (104). A clear demonstration that the GCS functions in the direction of glycine synthesis for biomass assimilation from C1 substrates was provided from metabolic engineering studies (105) in which unequivocal 13C isotopic labeling experiments proved that the GCS functions as a central module in an engineered strain of E. coli capable of using CO2 and either formate or methanol as the sole source of carbon and energy for growth. We propose that the rationale for colocalization of components of the GCS within the WLP operon could be related to the metabolic changes that are required as cells adjust between abundant versus nutrient-deprived conditions. Glycine, which is generally available in nutrient-rich environments, can both participate in the highly favorable reductive pathway involving glycine reductase and enter directly into biosynthetic pathways in which it acts as a direct precursor to supply critical portions of the overall biomass. With glycine and other Stickland acceptors in abundance, WLP activity is minimal, as its reductive function is superseded by GR and other reductive processes and glycine biosynthesis is not required. However, as glycine and other electron acceptors become increasingly scarce during nutrient depletion, the upregulation of the WLP would then serve not only to dispose of reducing equivalents no longer possible by way of GR but also to initiate carbon flow to synthesize glycine, by virtue of coexpression of the required previously repressed GCS genes integrated within the operon. Thus, during starvation, in the absence of glycine, induction of the WLP operon would fulfill the biosynthetic requirement for glycine at the same time that it steps in to function as a terminal electron sink. Glycine formed in this way could then be converted to serine by serine hydroxymethyltransferase (SHMT) and thereafter form pyruvate by a PFOR- and ferredoxin-independent pathway, which would help to conserve reduced ferredoxin. Indeed, SHMT activity is barely detectable in extracts of cells grown on BHIS but is easily assayed at much higher levels in cells grown on the newly defined media (data not shown). Marked increases are found as well for a number of other amino acid biosynthetic enzymes (preliminary results from proteomic studies, not shown). Our data are consistent with such a colocalization/coinduction mechanism to provide control over carbon assimilation through the biosynthesis of glycine; however, further studies are needed to elucidate the precise modes of interaction between the WLP and GCS systems.
Toxin formation in relation to the WLP.
The expression of C. difficile toxins TcdA and TcdB is governed by several global regulators and is a complex process that incorporates controls responsive to nutrient availability that regulate the activities of key metabolic pathways (82). Studies in vitro indicate that toxin expression increases as cells enter the stationary phase (106), and in peptone-yeast extract medium, C. difficile toxin synthesis in the stationary phase was correlated with increased expression of alternative energy-conserving pathways, including butyrate formation and the WLP (107). Our results agree with an increased level of WLP expression in the stationary phase (Fig. 2), although toxin levels in cells grown on BHIS appear to remain relatively constant (Fig. S2). It should be noted that the data presented here represent intracellular levels of TcdA, as extracellular amounts were not measured. However, it is known that even among individual cells, the intracellular content can vary, such that a bimodal distribution of toxin in the population was observed by fluorescence reporter assays (108). In contrast with the situation where toxin and WLP expression both tend to increase in the stationary phase, the WLP activity increases as much as 50- to 100-fold in the Gly/Ala medium relative to that in BHIS, but toxin levels show the opposite pattern, such that TcdA is up to 13-fold higher in BHIS than in defined media (cf. Fig. 6 and 10). Thus, depending on nutrient availability and metabolic state, toxin expression and the WLP may be either coordinately or divergently regulated. The results are consistent with repression of toxin synthesis in response to glucose as the overriding mechanism in the defined media, while at the same time, the lack of adequate electron acceptors results in derepression of the WLP.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
(i) BHIS. C. difficile 630 (ATCC BAA-1382) was routinely cultivated in crimp-sealed stoppered 18-mm by 150-mm anaerobic culture (Balch) tubes at 37°C in BHIS, which is brain heart infusion (BHI) medium supplemented with yeast extract and cysteine (109) and modified to contain resazurin, CO2, and bicarbonate. Modified BHIS contained per liter 37 g BD Bacto brain heart infusion, 5 g Bacto yeast extract, 1.68 g NaHCO3, 1 μg/ml resazurin, and 0.5 g cysteine hydrochloride. All solid components except cysteine were dissolved in anaerobic H2O inside a Coy-type anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) and 10-ml aliquots were added to the tubes and sealed. CO2 gas, 5.0 ml, was then injected into the tube headspace to produce an atmosphere after equilibration of ca. 18% CO2 in a balance of N2. After autoclaving, the tubes were stored in the anaerobic chamber, and prior to inoculation, 200 μl of a sterile anaerobic 2.5% cysteine-HCl solution was injected. Reproducible growth of BHIS day cultures was obtained by inoculation with 0.12 ml from BHIS starter cultures grown to an OD600 of 0.6 to 0.8.
BHIS for plates contained per liter 37 g Bacto brain heart infusion, 5 g Bacto yeast extract, 15 g Bacto agar, and 1 μg/ml resazurin. Before use, plates were incubated in the anaerobic chamber for several hours, and 100 μl of a 10% cysteine solution was spread out per ∼25 ml agar. When appropriate, antibiotics were used at the following concentrations: 250 μg/ml d-cycloserine, 8 μg/ml cefoxitin, and 15 μg/ml thiamphenicol. Plates were incubated at 37°C inside anaerobic jars under positive pressure with an atmosphere of 87% N2, 9% CO2, and 4% H2. C. difficile minimal medium (CDMM) (110) containing 50 μg/ml 5-fluorocytosine was used for selection. Difco reinforced clostridial medium (RCM) was from Becton Dickinson. E. coli strains were grown at 37°C in LB medium supplemented with 100 μg/ml ampicillin or 25 μg/ml chloramphenicol or 50 μg/ml kanamycin as needed. Clostridium ljungdahlii was grown in ATCC 1754 PETC medium.
(ii) Adaptation medium. Growth on adaptation medium (ADM) was used to eliminate the dependence on proline and leucine. ADM contains 5 mM glucose, 10 mM glycine, 1.5 mM leucine, 0.75 mM isoleucine, 0.25 mM valine, 0.5 mM proline, 0.22 mM methionine, and 0.16 mM tryptophan in a basal salt mixture composed of 15.4 mM NaCl, 5 mM NaH2PO4, 5 mM K2HPO4, and 130 mM NaHCO3, equilibrated with 100% CO2 to give a final pH 6.9. In addition, 0.136 mM CaCl2, 0.45 mM MgCl2, 1.5 mM sodium sulfide, 20 μM Fe(NH4)2(SO4)2, and 1 μM Na2SeO3 were present along with modified Wolfe’s vitamin stock solution (10 ml of 100× stock per liter) and trace element stock solution (10 ml of 100× stock per liter).
Vitamin solution (100× modified Wolfe's stock) contained per liter 2 mg biotin, 2 mg folic acid, 10 mg pyridoxine·HCl, 5 mg thiamine·HCl, 5 mg riboflavin, 5 mg nicotinamide, 5 mg calcium d-pantothenate, 5 mg p-aminobenzoic acid, 5 mg thioctic acid, and 0.05 ml of a 2 mg/ml vitamin B12 solution and was stored frozen in aliquots. Before addition to the medium, thawed samples were made anaerobic by bubbling with N2 and filter sterilized.
Trace element solution (100× stock) contained per liter 2.46 g nitrilotriacetic acid disodium salt, 1.0 g MnCl2·4H2O, 0.2 g CoCl2·6H2O, 0.1 g NiCl2·6H2O, 1 ml of a 10 mM ZnSO4 solution, 1 ml of a 10 mM CuSO4 solution, 20 mg Na2MoO4·2H2O, and 12 mg Na2WO4·2H2O and was prepared, filter sterilized, and stored dark under anaerobic conditions.
To prepare ADM, 6 ml of basal salt mixture was added to Balch tubes, bubbled with 100% CO2, crimp sealed, autoclaved, and stored inside the anaerobic chamber. On the day of inoculation, the tubes were injected with the following sterile anaerobic components in order: (i) 90 μl of 0.1 M sodium sulfide, (ii) a mixture of glucose, amino acids, CaCl2 and MgCl2 prepared from concentrated stock solutions to which was added 60 μl of vitamin solution, and (iii) 66 μl of a mixture made from 60 μl of trace element solution supplemented with 0.1 mM Na2SeO3 and 6 μl of 20 mM Fe(NH4)2(SO4)2. For inoculation, a BHIS day culture was grown for 5 h to an OD600 of around 0.6, and 1.5-ml aliquots were centrifuged for 5 min at 3,000 × g inside the anaerobic chamber. The pellets were resuspended in 1.2 ml of ADM, and 0.22 ml of cell suspension was used to inoculate each tube of ADM. Growth at 37°C was monitored by OD600.
(iii) Glycine/alanine medium. Gly/Ala medium was used for growth in the absence of proline and leucine and contained glycine as the only Stickland acceptor. The medium contains 5 mM glucose, 10 mM glycine, 10 mM alanine, 0.1 mM isoleucine, 0.21 mM valine, 0.22 mM methionine, and 0.12 mM tryptophan with the same basal salt, vitamin, and trace element composition as ADM. The preparation was identical to that for ADM tubes except that in step ii the mixture also contained 6 μl of 2 M sodium acetate and 5 μl of 4.67 M NH4Cl. Gly/Ala tubes were inoculated with 0.4 ml of an ADM culture which had been grown for 20 to 22 h to an OD600 around 0.6.
(iv) Glucose plus glycine medium. Glc+Gly medium was identical to Gly/Ala medium except that alanine was omitted. Preparation of the tubes and inoculation with cells grown on ADM was the same as for Gly/Ala growths.
(v) Glucose-only medium. Glc-only medium was similar to Glc+Gly medium except that glycine was omitted, such that the only amino acids present were valine, isoleucine, methionine, and tryptophan. Cells were grown under 2.3 atm pressure containing 60% H2 and 40% CO2. Tubes were inoculated with 0.6 ml of a Gly/Ala culture which had been grown for 22 to 24 h to an OD600 of around 0.45.
Cell extracts from large-scale growths.
The same medium compositions were used in the scale-up to larger volume growths for harvest and preparation of soluble cell extracts. Large-scale cultures of 1.2 liters total were obtained by growth in two 1,000-ml serum stoppered anaerobic bottles (Chemglass Life Sciences, LLC) each containing 600 ml of medium. Forty milliliters of an ADM culture with an OD600 of ∼0.5 was used to inoculate each bottle of 600 ml Gly/Ala medium. For glucose-only growths, three 1,000-ml bottles were used, containing 400 ml Glc-only medium each and pressurized to 2.3 atm with 60% H2 and 40% CO2, as with tubes. Each bottle of 400 ml Glc-only medium was inoculated with 27 ml of a Gly/Ala culture at an OD600 of ∼0.42. After reaching the desired amount of growth, cultures were prechilled and harvested anaerobically by centrifugation at 6,000 × g for 10 min at 4°C. The pellets were combined, and the final cell paste (collected after recentrifugation) was resuspended in 50 mM morpholinepropanesulfonic acid (MOPS; pH 7.2), 100 mM NaCl using approximately 2.5 ml of buffer solution per g cell paste and then frozen by dripping into liquid N2. Cell yields, OD600 values measured at harvest, and other parameters for the large-scale growths are given in Table 1. To prepare soluble extracts, 1 g of drip-frozen pellets was ground with a mortar and pestle chilled with liquid N2 inside the anaerobic chamber. The powder was transferred to an O-ring screw-cap microcentrifuge tube and allowed to thaw in the presence of 1 μg DNase and 0.5 mM MgCl2. The supernatant obtained after anaerobic centrifugation at 11,000 × g for 10 min at 4°C was then drip frozen in liquid N2 and stored in the vapor phase of a liquid N2 storage unit.
TABLE 1.
Large-scale growth metrics
| Growth condition | Vol (liters) | OD600 at harvest | Growth phase at harvesta | Amount |
||
|---|---|---|---|---|---|---|
| Cell mass (g) | g/liter | g/liter/OD | ||||
| BHIS 5 h | 0.8 | 1.073 | exp | 1.00 | 1.25 | 1.17 |
| BHIS 12 h | 0.5 | 1.815 | stat | 1.96 | 3.91 | 2.15 |
| BHIS 17 h | 0.5 | 1.720 | stat | 1.81 | 3.62 | 2.10 |
| Glc+Gly | 1.2 | 0.492 | lexp | 0.85 | 0.71 | 1.44 |
| Glc only | 1.2 | 0.354 | lexp | 0.93 | 0.78 | 2.19 |
| Gly/Ala 2.2b | 1.2 | 0.356 | exp | 0.58 | 0.48 | 1.36 |
| Gly/Ala 0.5c | 1.2 | 0.462 | lexp | 1.00 | 0.83 | 1.80 |
exp, exponential phase; lexp, late exponential phase; stat, stationary phase.
Harvested with 2.2 mM glucose remaining.
Harvested with 0.5 mM glucose remaining.
Enzyme assays.
All enzyme assays were carried out under strictly anaerobic conditions. One unit of activity is the amount of enzyme required to catalyze the formation/disappearance of 1 μmol of product/substrate per min. Specific activities were obtained on the basis of protein concentration in the extracts, assayed by the dye-binding method of Bradford (111).
CO dehydrogenase.
CO dehydrogenase activity was measured in 100% CO gas saturated 0.1 M Tris-HCl (pH 8.0) containing 10 mM methyl viologen (MV) at 25°C. Reactions were initiated by the addition of extract and followed spectrophotometrically using an absorptivity value of reduced MV of 13.1 mM−1 cm−1 at 600 nm (112). Units are reported based on moles of CO oxidized, which was taken as one-half the rate of MV reduction.
Formate dehydrogenase.
Formate dehydrogenase assays were modified from the methods used in reference 113 and contained 25 mM sodium formate, 50 mM MOPS sodium salt (pH 6.5), and 5 mM benzyl viologen (BV) at 25°C under a 100% N2 atmosphere. The rate of formate oxidation was taken as one-half the rate of BV reduction calculated using 11.0 mM−1 cm−1 as the absorptivity of reduced BV at 600 nm.
Hydrogenase.
Hydrogenase was measured in reaction mixtures containing 50 mM Tris-HCl (pH 7.5) and 5 mM benzyl viologen equilibrated and maintained under an atmosphere of 100% H2 at 25°C. Rates were determined as described for formate dehydrogenase and are reported as units of H2 oxidized.
N5,N10-Methylenetetrahydrofolate reductase.
The assay for methylenetetrahydrofolate reductase was developed on the basis of previously reported methods (114–116). The principle of the assay was HPLC quantification of both the disappearance of N5,N10-methylene-H4F and the formation of N5-methyl-H4F in aliquots removed over time from reaction mixtures containing dithionite-reduced benzyl viologen as reducing agent. Fresh substrate solution N5,N10-methylene-H4F was prepared in advance by separate nonenzymatic reaction of 5 mM formaldehyde with 2 mM tetrahydrofolate (H4F) in 20 mM potassium phosphate (pH 7.5). For this, H4F was added from a 23 mM anaerobic stock solution obtained by dissolving 50 mg of (6S)-tetrahydrofolic acid, purity >95% (Cayman Chemical Co., Ann Arbor, MI), in 5 ml of 0.1 M HEPES sodium salt (pH 7.5), 1 mM dithiothreitol. MTR reaction mixtures (200 μl final volume) contained 1 mM methylene-H4F (100 μl of substrate solution) and 1.8 mM benzyl viologen in 50 mM HEPES buffer (pH 7.5) with sufficient sodium dithionite added from a standardized stock solution calibrated to bring about approximately 85% reduction of the benzyl viologen. Reactions at 37°C were initiated by adding 10 μl of extract to obtain the final volume, and at timed intervals, 10-μl aliquots were removed, mixed with 230 μl 0.1 M sodium phosphate (pH 6.3), and immediately frozen in liquid N2.
Quantitative HPLC analysis of H4 folates was carried out on a C18 reversed-phase column (Grace Genesis 4 μm AQ, 150 by 4.6 mm, Hichrom Ltd., UK) equilibrated in 2% acetonitrile, 25 mM sodium phosphate (pH 6.3) (solvent A) at a flow rate of 0.60 ml/min. Gradient elution was performed by increasing the amount of acetonitrile (solvent B, composed of 40% acetonitrile in H2O). Solvents were continuously degassed with helium to help maintain anaerobic conditions, and samples were thawed under N2 and immediately withdrawn into an N2-purged gas-tight syringe for injection. Absorbance at 300 nm was followed, and H4F, methylene-H4F, and methyl-H4F were quantified on the basis of their integrated peak areas using relative absorptivity values determined empirically.
Serine deaminase/dehydratase.
Serine deamination catalyzed by the extracts was measured by quantification of serine and ammonia by amino acid analysis carried out using the precolumn derivatization method of Cohen (117). Reaction mixtures contained 25 mM serine in 0.1 M MOPS sodium salt (pH 7.7) and extract protein in a total volume of 80 μl. The reactions were initiated at 37°C by addition of 4 μl of extract, and thereafter, aliquots of 4 μl were removed at timed intervals, mixed with 156 μl of borate derivatization buffer (117), and frozen in liquid N2. Immediately upon thawing, samples were brought to room temperature, and 40 μl of a solution containing 3 mg/ml of 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC; Gold Biotechnology, Inc., St. Louis, MO) in acetonitrile was added with rapid mixing to generate stable unsymmetrical urea derivatives. The derivatives were then analyzed by reversed-phase HPLC using a Grace Apex ODS column (250 by 4.6 mm) eluted at 0.65 ml/min using an acetonitrile-25 mM sodium phosphate (pH 6.0) solvent system with UV detection at 248 nm.
Glycine reductase.
The assay for glycine reductase was developed on the basis of previously described methods (118–120). Reaction mixtures contained 10 mM glycine, 20 mM dithiothreitol (DTT), 4 mM Tris(2-carboxyethyl)phosphine-HCl (TCEP), 0.1 M potassium phosphate (pH 7.5), and the extract sample to be analyzed in a total volume of 80 μl. In addition, 0.75 mM α-aminoadipic acid was included as an internal standard (supplied from a fresh stock solution). Reactions at 37°C were initiated by the addition of 16 μl of extract, and aliquots (4 μl) were removed at timed intervals, mixed with borate derivatization buffer, frozen, and analyzed for amino acid content exactly as described above for serine deaminase. The time course of glycine disappearance matched well with that for the production of NH3, even at relatively low reaction rates. The use of an internal standard was not essential but produced more precise measurements of glycine consumption, particularly at low rates of glycine conversion. The classic ferric acetohydroxamate colorimetric method for direct detection of acetyl phosphate (121) in a scaled-down format was also used to assay glycine reductase but was not applied to all of the extracts because it required considerably larger amounts of protein due to lower sensitivity and larger reaction volumes.
Proline reductase.
Amino acid analysis by HPLC was used to measure the enzymatic conversion of d-proline to 5-aminovaleric acid (5AV) under reaction conditions similar to those described previously (122). The substrates 5 mM d-proline and 20 mM DTT were incubated at 37°C in 0.1 M potassium phosphate buffer (pH 7.5) in a final volume, including extract sample, of 80 μl. Reactions were initiated by the addition of 4 μl extract, and aliquots of 4 μl were removed over time, processed, and analyzed by HPLC exactly as described above for serine deaminase.
Transaminases.
Transaminase reactions were carried out at 37°C in 0.1 M MOPS sodium salt (pH 7.7) in a total volume of 80 μl, including extract. Leucine:α-ketoglutarate aminotransferase reactions contained 5 mM leucine and 20 mM α-ketoglutarate. Glutamate:pyruvate aminotransferase reactions contained 5 mM glutamate and 20 mM pyruvate. Solutions of α-ketoglutaric acid and pyruvic acid were neutralized by titration with 1 M NaOH to pH 6.8, adjusted to give 0.1 M stock concentration, and stored frozen before use in the respective assays. Reactions were initiated by the addition of 4 μl extract, and aliquots of 4 μl were removed over time, processed, and analyzed for substrate and product amino acids by HPLC exactly as described above for serine deaminase. Initial rates were obtained from fits of the time course data as an apparent first-order approach to equilibrium.
Butyryl-CoA:acetate CoA transferase.
The acetate-dependent formation of acetyl-CoA from butyryl-CoA was followed by HPLC analyses of CoA substrate and product derivatives over time. Butyryl-CoA:acetate CoA transferase reaction mixtures contained 20 mM sodium acetate, 0.5 mM butyryl-CoA, 50 mM HEPES (pH 7.5), and extract protein in a final volume of 200 μl. Reactions at 37°C were initiated by the addition of 4 μl of diluted extract (one-third to one-sixth of original concentration in 50 mM HEPES [pH 7.5]). Aliquots of 20 μl were removed over time, mixed with 180 μl of 0.1 M sodium citrate (pH 4.0), and frozen in liquid N2 prior to HPLC analysis using a previously described procedure (123). Reaction progress curves were linear up to at least 40% conversion of butyryl-CoA.
Expression of C. difficile AcsB in E. coli, purification, and antibody preparation.
The C. difficile acsB gene sequence was codon optimized for expression in E. coli. The synthetic acsB gene was cloned as an untagged construct into the NcoI- and BamHI-digested pETDuet-1 plasmid (Novagen) and used for transformation of E. coli DH5α. Following sequence verification, anaerobic expression was carried out in E. coli NM522(λDE3). Anaerobic growth of E. coli and induction with isopropyl-β-d-thiogalactopyranoside (IPTG) was carried out as described previously (124). Soluble cell extracts were prepared, and the protein was purified under anaerobic conditions by anion-exchange chromatography on Q Sepharose fast flow (124). Ni reconstitution, which is required to form active acetyl-CoA synthase produced in E. coli, was modified to limit the amount of excess NiCl2 to 2:1 (Ni/protein) and carried out at a lower pH of 7.2. Although reconstitution of other heterologously expressed AcsB proteins from archaea (Methanosarcina thermophila) and bacteria (Carboxydothermus hydrogenoformans) was effective at higher ratios of Ni/protein and higher pH, the activity of the C. difficile protein was suboptimal under those protocols. The turnover number for C. difficile AcsB in the synthesis of acetyl-CoA from CO, CoA, and methylcobinamide was 20.6 min−1, assayed as described in reference 123, which compares favorably with the other AcsB proteins studied.
For immunization, the protein was further purified by extraction from SDS slab gel strips. Bands were revealed as cloudy zones of potassium dodecyl sulfate precipitate by briefly soaking the gel in buffer containing 0.25 M KCl, and strips were then cut, macerated, and extracted overnight in an excess volume of 20 mM Tris-HCl (pH 7.8) containing 0.1% SDS and 1 mM DTT. After filtration through a 0.22-μm filter, the filtrate was concentrated by ultrafiltration, diafiltered with 20 mM Tris-HCl (pH 7.8), and samples of the frozen concentrate were used for production of rabbit antisera (Covance Immunology Services, Denver, PA). Protein A-based purification of the antisera was used to produce the final anti-AcsB antibody preparation.
Western blotting.
Aliquots (1 ml) taken from growing cultures were centrifuged, and the pellets were frozen in liquid nitrogen. The pellets were resuspended in 100 μl 1× Tris-glycine SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 10% glycerol, 2% [wt/vol] SDS, 0.005% bromophenol blue). After thorough vortex mixing, the samples were incubated at 95°C for 5 min and centrifuged for 3 min. Samples, 8 to 16 μl, were run on 12% acrylamide SDS-PAGE (or 8% gels for TcdA) 1-mm-thick gels (125) and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked in TBS-T (20 mM Tris-HCl [pH 7.6], 137 mM NaCl, 0.1% Tween 20) with 1% (wt/vol) nonfat dried milk. After incubation with the primary antibody (protein A-purified anti-C. difficile AcsB polyclonal rabbit IgG at 1:2,400 or C. difficile toxin A mouse monoclonal antibody PCG4.1 from Novus Bio at 1:1,000) for 90 min, the membranes were washed and incubated with secondary antibody (blotting-grade goat anti-rabbit IgG–horseradish peroxidase conjugate, Bio-Rad, 1:3,000, or goat anti-mouse IgG–horseradish peroxidase conjugate, Bio-Rad, 1:1,000) for 60 min. Blots were developed using Amersham ECL enhanced chemiluminescence reagent (GE Healthcare). The components were combined according to the manufacturer’s instructions, and 2% H2O2 was added to the final mixture. Images for quantification were obtained with a cooled charge-coupled-device (CCD) camera using a FluorChem HD2 imaging system (Alpha Innotech/Protein Simple, Inc.). Integrated band intensities were linear versus camera exposure time but were nonlinear with respect to the amount of AcsB protein loaded. Therefore, AcsB estimations were made by use of a standard curve.
Fermentation substrate/product quantifications.
The amino acid contents in samples from culture supernatants were determined by reversed-phase HPLC using the precolumn AQC derivatization method, as described above for the assay for serine deaminase. Organic acids and alcohols were quantified by GC and HPLC methods. HPLC employed mixed-mode chromatography on a Bio-Rad fermentation monitoring column (150 mm by 7.6 mm) at 65°C with a mobile phase of 5 mM H2SO4 and flow rate of 0.70 ml/min. An Agilent 1100 series model G1362A instrument was used for refractive index (RI) detection. In addition to the HPLC analyses, glucose was also determined by spectrophotometric assays of reduced pyridine nucleotide formation in coupled reaction mixtures containing hexokinase plus glucose 6-phosphate dehydrogenase (glucose assay reagent, G3293; Sigma). In our experience, with the culture supernatants analyzed here, the enzymatic method was largely free from interference by other substances, and values agreed well with the HPLC method. GC analyses were performed on a 30-m Agilent J&W Ultra Inert DB-624 column with He carrier gas at 1.4 ml/min attached to an Agilent HP 6890 GC system with an HP 5793 mass-selective detector. Samples from culture supernatants were acidified using HCl, and thereafter, methanol was added to a final concentration of 80% immediately prior to injection of 1 μl using a 10.8:1 split ratio. Oven temperature was held at 45°C for 2 min and then raised to 155°C in 10 min. Solvent delay was set to 2.2 min. Formic acid was difficult to quantify by this method, because the recovery was highly sensitive to the degree of acidification and the peak area was variable. Therefore, HPLC was used for all formate determinations.
C. difficile 630 ΔacsB mutant: construction of the codA allele exchange plasmid and allele exchange cassette.
The cytosine deaminase codA gene from E. coli was used as a heterologous counterselection marker for C. difficile 630, as developed by the laboratory of N. Minton (73). The codA gene from E. coli was placed under the control of the Clostridium pasteurianum fdx promoter. A 231-bp fragment was synthetized that contained the C. pasteurianum fdx promoter (126) modified such that after fusion to the codA gene, the gene possessed a suboptimal spacer (12 nt) between the Shine-Dalgarno sequence and the GTG start codon and a competing open reading frame (ORF) encoding a 39-amino-acid peptide with a favorable spacer of 10 nt and an ATG start codon (73). The 5′ end of the construct with the modified promoter was PCR amplified with primers fdx-codA-BamHI-A-f and fdx-codA-B-r and gel purified. The 3′ end, i.e., the codA gene, was PCR amplified from E. coli XL1-Blue strain using primers fdx-codA-C-f and fdx-codA-NcoI-D-r and gel purified. The purified fragments (synthetic fragment and the codA gene) were then used as the templates for splicing by overhang extension (SOE) PCR (127) using the outer primers fdx-codA-BamHI-A-f and fdx-codA-NcoI-D-r. The PCR fdx-codA fragment was cloned into the pCR-Blunt II-TOPO vector and sequenced. Thereafter, the fdx-codA fragment was excised with BamHI and NcoI and ligated into BamHI/NcoI-digested pMTL83151 (128) (a generous gift from the laboratory of N. Minton) to produce pMTL83151::codA.
For in-frame deletion of the C. difficile acsB gene via homologous recombination, an acsB allele exchange cassette was constructed comprising upstream and downstream regions flanking the acsB gene, whose coding sequence carried a deletion between nucleotides 61 to 2049. For the allele exchange cassette, the 5′ PCR fragment was amplified with primers acsE-PmeI-A-f and acsB-5-pdel-B-r and the 3′ PCR fragment with acsB-3 pdel-C-f and gcvH-PmeI-D-r. Both fragments were gel purified and used in the SOE PCR with the flanking primers acsE-PmeI-A-f and gcvH-PmeI-D-r. The allele exchange construct was cloned into the PmeI site of pMTL83151::codA giving rise to pSG1217.
The plasmid pSG1217 was transferred via conjugation from the S17-1 E. coli donor strain SG-Ec2270 to C. difficile 630 (129, 130), spotting the mixed cell suspension onto BHIS agar. After overnight incubation, the growth was harvested and spread onto BHIS agar plates supplemented with 250 μg/ml d-cycloserine, 8 μg/ml cefoxitin, and 15 μg/ml thiamphenicol. Transconjugants were restreaked onto the same medium to select for single-crossover integrants, which were verified by PCR using primer pairs acsB-AE-1f and PL-AE-PmeI-r and PL-AE-PmeI-f and acsB-AE-1r. The single-crossover clones were subcultured without selection on BHIS to allow sufficient time for the rare second recombination event to occur. After harvesting the growth and dilution of the cell suspension, the cells were plated onto CDMM plates (110) supplemented with 50 μg/ml 5-fluorocytosine to select for fluorocytosine-resistant (FCr) colonies. The FCr colonies were then patch plated onto BHIS and BHIS plus thiamphenicol to screen for the loss of plasmid (FCr Tms clones). The length of fragments obtained with acsB-AE-1f and acsB-AE-1r primers was used to identify double-crossover recombinants, versus WT revertants, which were then sequenced to verify the accuracy of the in-frame deletion. A listing of the strains and plasmids is given in Table S3 in the supplemental material, and oligonucleotide primer sequences are listed in Table S4.
Complementation plasmid.
To confirm that the phenotypic changes observed in the mutant were directly attributable to the deletion of acsB, complementation experiments were performed. Two PCR fragments were amplified. The first encompassed the WLP promoter region upstream of the acsA gene and was amplified using primers pro-BamHI-A-f and pro-acsB-B-r. The second included the acsB gene amplified using primers pro-acsB-C-f and acsB-AatII-D-r. SOE PCR using the outer primer pair produced a fragment that was cloned into the pMTL84151 (128) vector between the BamHI and AatII sites to give plasmid pMTL84151::acsB containing the native promoter region of the WLP operon upstream of the acsB gene. The complementation plasmid was transferred via conjugation from the E. coli donor SG-Ec2280 into the C. difficile 630 ΔacsB mutant strain.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nigel Minton, University of Nottingham, UK, for the generous gift of plasmids pMTL83151 and pMTL84151.
This work was supported by NIH, NIAID grant 1R03AI137615.
We declare no conflicts of interest with the contents of this article. Any opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the view of the Department of Defense or the Uniformed Services University of the Health Sciences.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Abt MC, McKenney PT, Pamer EG. 2016. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol 14:609–620. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Farrell RJ, LaMont JT. 2000. Pathogenesis and clinical manifestations of Clostridium difficile diarrhea and colitis. Curr Top Microbiol Immunol 250:109–125. doi: 10.1007/978-3-662-06272-2_6. [DOI] [PubMed] [Google Scholar]
- 4.Aktories K, Schwan C, Jank T. 2017. Clostridium difficile toxin biology. Annu Rev Microbiol 71:281–307. doi: 10.1146/annurev-micro-090816-093458. [DOI] [PubMed] [Google Scholar]
- 5.Aktories K, Papatheodorou P, Schwan C. 2018. Binary Clostridium difficile toxin (CDT) - a virulence factor disturbing the cytoskeleton. Anaerobe 53:21–29. doi: 10.1016/j.anaerobe.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. 2013. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun 81:3757–3769. doi: 10.1128/IAI.00515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenior ML, Leslie JL, Young VB, Schloss PD. 2017. Clostridium difficile colonizes alternative nutrient niches during infection across distinct murine gut microbiomes. mSystems 2:e00063-17. doi: 10.1128/mSystems.00063-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fletcher JR, Erwin S, Lanzas C, Theriot CM. 2018. Shifts in the gut metabolome and Clostridium difficile transcriptome throughout colonization and infection in a mouse model. mSphere 3:e00089-18. doi: 10.1128/mSphere.00089-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hryckowian AJ, Pruss KM, Sonnenburg JL. 2017. The emerging metabolic view of Clostridium difficile pathogenesis. Curr Opin Microbiol 35:42–47. doi: 10.1016/j.mib.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stickland LH. 1934. Studies in the metabolism of the strict anaerobes (genus Clostridium): the chemical reactions by which Cl. sporogenes obtains its energy. Biochem J 28:1746–1759. doi: 10.1042/bj0281746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker HA. 1981. Amino acid degradation by anaerobic bacteria. Annu Rev Biochem 50:23–40. doi: 10.1146/annurev.bi.50.070181.000323. [DOI] [PubMed] [Google Scholar]
- 12.Britz ML, Wilkinson RG. 1982. Leucine dissimilation to isovaleric and isocaproic acids by cell suspensions of amino acid fermenting anaerobes: the Stickland reaction revisited. Can J Microbiol 28:291–300. doi: 10.1139/m82-043. [DOI] [PubMed] [Google Scholar]
- 13.Elsden SR, Hilton MG. 1978. Volatile acid production from threonine, valine, leucine and isoleucine by clostridia. Arch Microbiol 117:165–172. doi: 10.1007/BF00402304. [DOI] [PubMed] [Google Scholar]
- 14.Jackson S, Calos M, Myers A, Self WT. 2006. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol 188:8487–8495. doi: 10.1128/JB.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haslam SC, Ketley JM, Mitchell TJ, Stephen J, Burdon DW, Candy DC. 1986. Growth of Clostridium difficile and production of toxins A and B in complex and defined media. J Med Microbiol 21:293–297. doi: 10.1099/00222615-21-4-293. [DOI] [PubMed] [Google Scholar]
- 16.Yamakawa K, Kamiya S, Meng XQ, Karasawa T, Nakamura S. 1994. Toxin production by Clostridium difficile in a defined medium with limited amino acids. J Med Microbiol 41:319–323. doi: 10.1099/00222615-41-5-319. [DOI] [PubMed] [Google Scholar]
- 17.Karasawa T, Ikoma S, Yamakawa K, Nakamura S. 1995. A defined growth medium for Clostridium difficile. Microbiology 141:371–375. doi: 10.1099/13500872-141-2-371. [DOI] [PubMed] [Google Scholar]
- 18.Dubois T, Dancer-Thibonnier M, Monot M, Hamiot A, Bouillaut L, Soutourina O, Martin-Verstraete I, Dupuy B. 2016. Control of Clostridium difficile physiopathology in response to cysteine availability. Infect Immun 84:2389–2405. doi: 10.1128/IAI.00121-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 20.Dannheim H, Riedel T, Neumann-Schaal M, Bunk B, Schober I, Spröer C, Chibani CM, Gronow S, Liesegang H, Overmann J, Schomburg D. 2017. Manual curation and reannotation of the genomes of Clostridium difficile 630Δerm and C. difficile 630. J Med Microbiol 66:286–293. doi: 10.1099/jmm.0.000427. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura S, Nakashio S, Yamakawa K, Tanabe N, Nishida S. 1982. Carbohydrate fermentation by Clostridium difficile. Microbiol Immunol 26:107–111. doi: 10.1111/j.1348-0421.1982.tb00159.x. [DOI] [PubMed] [Google Scholar]
- 22.Seddon SV, Borriello SP. 1989. A chemically defined and minimal medium for Clostridium difficile. Lett Appl Microbiol 9:237–239. doi: 10.1111/j.1472-765X.1989.tb00334.x. [DOI] [Google Scholar]
- 23.Scaria J, Chen JW, Useh N, He H, McDonough SP, Mao C, Sobral B, Chang YF. 2014. Comparative nutritional and chemical phenome of Clostridium difficile isolates determined using phenotype microarrays. Int J Infect Dis 27:20–25. doi: 10.1016/j.ijid.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson KH, Perini F. 1988. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun 56:2610–2614. doi: 10.1128/IAI.56.10.2610-2614.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar N, Browne HP, Viciani E, Forster SC, Clare S, Harcourt K, Stares MD, Dougan G, Fairley DJ, Roberts P, Pirmohamed M, Clokie MRJ, Jensen MBF, Hargreaves KR, Ip M, Wieler LH, Seyboldt C, Noren T, Riley TV, Kuijper EJ, Wren BW, Lawley TD. 2019. Adaptation of host transmission cycle during Clostridium difficile speciation. Nat Genet 51:1315–1320. doi: 10.1038/s41588-019-0478-8. [DOI] [PubMed] [Google Scholar]
- 27.Drake HL, Gössner AS, Daniel SL. 2008. Old acetogens, new light. Ann N Y Acad Sci 1125:100–128. doi: 10.1196/annals.1419.016. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang WQ, Yi S, Bill M, Brisson VL, Feng X, Men Y, Conrad ME, Tang YJ, Alvarez-Cohen L. 2014. Incomplete Wood-Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc Natl Acad Sci U S A 111:6419–6424. doi: 10.1073/pnas.1321542111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thauer RK. 1988. Citric-acid cycle, 50 years on. Modifications and an alternative pathway in anaerobic bacteria. Eur J Biochem 176:497–508. doi: 10.1111/j.1432-1033.1988.tb14307.x. [DOI] [PubMed] [Google Scholar]
- 30.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. 2002. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science 298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 31.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. 2003. Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol 10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 32.Doukov TI, Blasiak LC, Seravalli J, Ragsdale SW, Drennan CL. 2008. Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry 47:3474–3483. doi: 10.1021/bi702386t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindahl PA. 2004. Acetyl-coenzyme A synthase: the case for a Ni(p)(0)-based mechanism of catalysis. J Biol Inorg Chem 9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 34.Ragsdale SW, Pierce E. 2008. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta 1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gencic S, Kelly K, Ghebreamlak S, Duin EC, Grahame DA. 2013. Different modes of carbon monoxide binding to acetyl-CoA synthase and the role of a conserved phenylalanine in the coordination environment of nickel. Biochemistry 52:1705–1716. doi: 10.1021/bi3016718. [DOI] [PubMed] [Google Scholar]
- 36.Gencic S, Grahame DA. 2008. Two separate one-electron steps in the reductive activation of the A cluster in subunit beta of the ACDS complex in Methanosarcina thermophila. Biochemistry 47:5544–5555. doi: 10.1021/bi7024035. [DOI] [PubMed] [Google Scholar]
- 37.Tan X, Surovtsev IV, Lindahl PA. 2006. Kinetics of CO insertion and acetyl group transfer steps, and a model of the acetyl-CoA synthase catalytic mechanism. J Am Chem Soc 128:12331–12338. doi: 10.1021/ja0627702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhaskar B, DeMoll E, Grahame DA. 1998. Redox-dependent acetyl transfer partial reaction of the acetyl-CoA decarbonylase/synthase complex: kinetics and mechanism. Biochemistry 37:14491–14499. doi: 10.1021/bi9812423. [DOI] [PubMed] [Google Scholar]
- 39.Lu WP, Ragsdale SW. 1991. Reductive activation of the coenzyme A/acetyl-CoA isotopic exchange reaction catalyzed by carbon monoxide dehydrogenase from Clostridium thermoaceticum and its inhibition by nitrous oxide and carbon monoxide. J Biol Chem 266:3554–3564. [PubMed] [Google Scholar]
- 40.Nawrocki KL, Wetzel D, Jones JB, Woods EC, McBride SM. 2018. Ethanolamine is a valuable nutrient source that impacts Clostridium difficile pathogenesis. Environ Microbiol 20:1419–1435. doi: 10.1111/1462-2920.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. 2014. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16:770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice KC, Bayles KW. 2008. Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev 72:85–109. doi: 10.1128/MMBR.00030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calamita HG, Doyle RJ. 2002. Regulation of autolysins in teichuronic acid-containing Bacillus subtilis cells. Mol Microbiol 44:601–606. doi: 10.1046/j.1365-2958.2002.02872.x. [DOI] [PubMed] [Google Scholar]
- 44.Jolliffe LK, Doyle RJ, Streips UN. 1981. The energized membrane and cellular autolysis in Bacillus subtilis. Cell 25:753–763. doi: 10.1016/0092-8674(81)90183-5. [DOI] [PubMed] [Google Scholar]
- 45.Kopke M, Straub M, Durre P. 2013. Clostridium difficile is an autotrophic bacterial pathogen. PLoS One 8:e62157. doi: 10.1371/journal.pone.0062157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neumann-Schaal M, Hofmann JD, Will SE, Schomburg D. 2015. Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiol 15:281. doi: 10.1186/s12866-015-0614-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stickland LH. 1935. Studies in the metabolism of the strict anaerobes (genus Clostridium): III. The oxidation of alanine by Cl. sporogenes. IV. The reduction of glycine by Cl. sporogenes. Biochem J 29:889–898. doi: 10.1042/bj0290889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter J, Schindler F, Wildenauer FX. 1987. Fermentation of alanine and glycine by pure and syntrophic cultures of Clostridium sporogenes. FEMS Microbiology Lett 45:153–161. doi: 10.1111/j.1574-6968.1987.tb02351.x. [DOI] [Google Scholar]
- 49.Bouillaut L, Self WT, Sonenshein AL. 2013. Proline-dependent regulation of Clostridium difficile Stickland metabolism. J Bacteriol 195:844–854. doi: 10.1128/JB.01492-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreesen JR. 2004. Glycine reductase mechanism. Curr Opin Chem Biol 8:454–461. doi: 10.1016/j.cbpa.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Fonknechten N, Chaussonnerie S, Tricot S, Lajus A, Andreesen JR, Perchat N, Pelletier E, Gouyvenoux M, Barbe V, Salanoubat M, Le Paslier D, Weissenbach J, Cohen GN, Kreimeyer A. 2010. Clostridium sticklandii, a specialist in amino acid degradation: revisiting its metabolism through its genome sequence. BMC Genomics 11:555. doi: 10.1186/1471-2164-11-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuchmann K, Chowdhury NP, Muller V. 2018. Complex multimeric [FeFe] hydrogenases: biochemistry, physiology and new opportunities for the hydrogen economy. Front Microbiol 9:2911. doi: 10.3389/fmicb.2018.02911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schut GJ, Adams MW. 2009. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol 191:4451–4457. doi: 10.1128/JB.01582-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuchmann K, Muller V. 2012. A bacterial electron-bifurcating hydrogenase. J Biol Chem 287:31165–31171. doi: 10.1074/jbc.M112.395038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, Huang H, Kahnt J, Thauer RK. 2013. A reversible electron-bifurcating ferredoxin- and NAD-dependent [FeFe]-hydrogenase (HydABC) in Moorella thermoacetica. J Bacteriol 195:1267–1275. doi: 10.1128/JB.02158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng Y, Kahnt J, Kwon IH, Mackie RI, Thauer RK. 2014. Hydrogen formation and its regulation in Ruminococcus albus: involvement of an electron-bifurcating [FeFe]-hydrogenase, of a non-electron-bifurcating [FeFe]-hydrogenase, and of a putative hydrogen-sensing [FeFe]-hydrogenase. J Bacteriol 196:3840–3852. doi: 10.1128/JB.02070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu XL, Grant GA. 2013. Identification and characterization of two new types of bacterial l-serine dehydratases and assessment of the function of the ACT domain. Arch Biochem Biophys 540:62–69. doi: 10.1016/j.abb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Xu XL, Grant GA. 2016. Mutagenic and chemical analyses provide new insight into enzyme activation and mechanism of the type 2 iron-sulfur l-serine dehydratase from Legionella pneumophila. Arch Biochem Biophys 596:108–117. doi: 10.1016/j.abb.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Anderson BM, Anderson CD, Van Tassell RL, Lyerly DM, Wilkins TD. 1993. Purification and characterization of Clostridium difficile glutamate dehydrogenase. Arch Biochem Biophys 300:483–488. doi: 10.1006/abbi.1993.1065. [DOI] [PubMed] [Google Scholar]
- 60.Planche T, Wilcox MH. 2015. Diagnostic pitfalls in Clostridium difficile infection. Infect Dis Clin North Am 29:63–82. doi: 10.1016/j.idc.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 61.Girinathan BP, Braun SE, Govind R. 2014. Clostridium difficile glutamate dehydrogenase is a secreted enzyme that confers resistance to H2O2. Microbiology 160:47–55. doi: 10.1099/mic.0.071365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Girinathan BP, Braun S, Sirigireddy AR, Lopez JE, Govind R. 2016. Importance of glutamate dehydrogenase (GDH) in Clostridium difficile colonization in vivo. PLoS One 11:e0160107. doi: 10.1371/journal.pone.0160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Danson AE, Jovanovic M, Buck M, Zhang X. 2019. Mechanisms of sigma54-dependent transcription initiation and regulation. J Mol Biol 431:3960–3974. doi: 10.1016/j.jmb.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nie X, Dong W, Yang C. 2019. Genomic reconstruction of sigma54 regulons in Clostridiales. BMC Genomics 20:565. doi: 10.1186/s12864-019-5918-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, Rodionov DA, Martin-Verstraete I, Dupuy B. 2012. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res 40:10701–10718. doi: 10.1093/nar/gks864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Darley D, Selmer T, Buckel W. 2006. Characterization of (R)-2-hydroxyisocaproate dehydrogenase and a family III coenzyme A transferase involved in reduction of l-leucine to isocaproate by Clostridium difficile. Appl Environ Microbiol 72:6062–6069. doi: 10.1128/AEM.00772-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Canganella F, Kuk SU, Morgan H, Wiegel J. 2002. Clostridium thermobutyricum: growth studies and stimulation of butyrate formation by acetate supplementation. Microbiol Res 157:149–156. doi: 10.1078/0944-5013-00140. [DOI] [PubMed] [Google Scholar]
- 68.Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. 2002. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol 68:5186–5190. doi: 10.1128/aem.68.10.5186-5190.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen CK, Blaschek HP. 1999. Effect of acetate on molecular and physiological aspects of Clostridium beijerinckii NCIMB 8052 solvent production and strain degeneration. Appl Environ Microbiol 65:499–505. doi: 10.1128/AEM.65.2.499-505.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bornstein BT, Barker HA. 1948. The nutrition of Clostridium kluyveri. J Bacteriol 55:223–230. doi: 10.1128/JB.55.2.223-230.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seedorf H, Fricke WF, Veith B, Bruggemann H, Liesegang H, Strittmatter A, Miethke M, Buckel W, Hinderberger J, Li F, Hagemeier C, Thauer RK, Gottschalk G. 2008. The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc Natl Acad Sci U S A 105:2128–2133. doi: 10.1073/pnas.0711093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karlsson S, Burman LG, Akerlund T. 1999. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 145:1683–1693. doi: 10.1099/13500872-145-7-1683. [DOI] [PubMed] [Google Scholar]
- 73.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180. doi: 10.1128/MMBR.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demmer JK, Pal Chowdhury N, Selmer T, Ermler U, Buckel W. 2017. The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-CoA dehydrogenase complex from Clostridium difficile. Nat Commun 8:1577. doi: 10.1038/s41467-017-01746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 77.Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol 190:843–850. doi: 10.1128/JB.01417-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 79.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 80.Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bouillaut L, Dubois T, Sonenshein AL, Dupuy B. 2015. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol 166:375–383. doi: 10.1016/j.resmic.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin-Verstraete I, Peltier J, Dupuy B. 2016. The regulatory networks that control Clostridium difficile toxin synthesis. Toxins (Basel) 8:153. doi: 10.3390/toxins8050153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. 2000. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68:5881–5888. doi: 10.1128/iai.68.10.5881-5888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Battaglioli EJ, Hale VL, Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, Rekdal VM, Till LM, Huq L, Smits SA, Moor WJ, Jones-Hall Y, Smyrk T, Khanna S, Pardi DS, Grover M, Patel R, Chia N, Nelson H, Sonnenburg JL, Farrugia G, Kashyap PC. 2018. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci Transl Med 10:eaam7019. doi: 10.1126/scitranslmed.aam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Costilow RN, Laycock L. 1971. Ornithine cyclase (deaminating). Purification of a protein that converts ornithine to proline and definition of the optimal assay conditions. J Biol Chem 246:6655–6660. [PubMed] [Google Scholar]
- 86.Jarrett JT. 2008. Biochemistry: radicals by reduction. Nature 452:163–164. doi: 10.1038/452163a. [DOI] [PubMed] [Google Scholar]
- 87.Kim J, Darley DJ, Buckel W, Pierik AJ. 2008. An allylic ketyl radical intermediate in clostridial amino-acid fermentation. Nature 452:239–242. doi: 10.1038/nature06637. [DOI] [PubMed] [Google Scholar]
- 88.Knauer SH, Buckel W, Dobbek H. 2012. On the ATP-dependent activation of the radical enzyme (R)-2-hydroxyisocaproyl-CoA dehydratase. Biochemistry 51:6609–6622. doi: 10.1021/bi300571z. [DOI] [PubMed] [Google Scholar]
- 89.Buckel W. 2019. Enzymatic reactions involving ketyls: from a chemical curiosity to a general biochemical mechanism. Biochemistry 58:5221–5233. doi: 10.1021/acs.biochem.9b00171. [DOI] [PubMed] [Google Scholar]
- 90.Buckel W, Thauer RK. 2018. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem Rev 118:3862–3886. doi: 10.1021/acs.chemrev.7b00707. [DOI] [PubMed] [Google Scholar]
- 91.Nakamura A, Sosa A, Komori H, Kita A, Miki K. 2007. Crystal structure of TTHA1657 (AT-rich DNA-binding protein; p25) from Thermus thermophilus HB8 at 2.16 Å resolution. Proteins 66:755–759. doi: 10.1002/prot.21222. [DOI] [PubMed] [Google Scholar]
- 92.McLaughlin KJ, Strain-Damerell CM, Xie K, Brekasis D, Soares AS, Paget MS, Kielkopf CL. 2010. Structural basis for NADH/NAD+ redox sensing by a Rex family repressor. Mol Cell 38:563–575. doi: 10.1016/j.molcel.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 93.Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL, Rodionov DA. 2012. Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol 194:1145–1157. doi: 10.1128/JB.06412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, von Wachenfeldt C. 2008. Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol Microbiol 69:466–478. doi: 10.1111/j.1365-2958.2008.06295.x. [DOI] [PubMed] [Google Scholar]
- 95.Bouillaut L, Dubois T, Francis MB, Daou N, Monot M, Sorg JA, Sonenshein AL, Dupuy B. 2019. Role of the global regulator Rex in control of NAD+ -regeneration in Clostridioides (Clostridium) difficile. Mol Microbiol 111:1671–1688. doi: 10.1111/mmi.14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang L, Nie X, Ravcheev DA, Rodionov DA, Sheng J, Gu Y, Yang S, Jiang W, Yang C. 2014. Redox-responsive repressor Rex modulates alcohol production and oxidative stress tolerance in Clostridium acetobutylicum. J Bacteriol 196:3949–3963. doi: 10.1128/JB.02037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Daou N, Wang Y, Levdikov VM, Nandakumar M, Livny J, Bouillaut L, Blagova E, Zhang K, Belitsky BR, Rhee K, Wilkinson AJ, Sun X, Sonenshein AL. 2019. Impact of CodY protein on metabolism, sporulation and virulence in Clostridioides difficile ribotype 027. PLoS One 14:e0206896. doi: 10.1371/journal.pone.0206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Daniel SL, Hsu T, Dean SI, Drake HL. 1990. Characterization of the H2- and CO-dependent chemolithotrophic potentials of the acetogens Clostridium thermoaceticum and Acetogenium kivui. J Bacteriol 172:4464–4471. doi: 10.1128/jb.172.8.4464-4471.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schuchmann K, Müller V. 2016. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microbiol 82:4056–4069. doi: 10.1128/AEM.00882-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schuchmann K, Müller V. 2014. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol 12:809–821. doi: 10.1038/nrmicro3365. [DOI] [PubMed] [Google Scholar]
- 101.Kansau I, Barketi-Klai A, Monot M, Hoys S, Dupuy B, Janoir C, Collignon A. 2016. Deciphering adaptation strategies of the epidemic Clostridium difficile 027 strain during infection through in vivo transcriptional analysis. PLoS One 11:e0158204. doi: 10.1371/journal.pone.0158204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, Young VB. 2015. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 83:934–941. doi: 10.1128/IAI.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poehlein A, Cebulla M, Ilg MM, Bengelsdorf FR, Schiel-Bengelsdorf B, Whited G, Andreesen JR, Gottschalk G, Daniel R, Dürre P. 2015. The complete genome sequence of Clostridium aceticum: a missing link between Rnf- and cytochrome-containing autotrophic acetogens. mBio 6:e01168-15. doi: 10.1128/mBio.01168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Song Y, Lee JS, Shin J, Lee GM, Jin S, Kang S, Lee JK, Kim DR, Lee EY, Kim SC, Cho S, Kim D, Cho BK. 2020. Functional cooperation of the glycine synthase-reductase and Wood-Ljungdahl pathways for autotrophic growth of Clostridium drakei. Proc Natl Acad Sci U S A 117:7516–7523. doi: 10.1073/pnas.1912289117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim S, Lindner SN, Aslan S, Yishai O, Wenk S, Schann K, Bar-Even A. 2020. Growth of E. coli on formate and methanol via the reductive glycine pathway. Nat Chem Biol 16:538–545. doi: 10.1038/s41589-020-0473-5. [DOI] [PubMed] [Google Scholar]
- 106.Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, von Eichel-Streiber C. 1997. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur J Biochem 244:735–742. doi: 10.1111/j.1432-1033.1997.t01-1-00735.x. [DOI] [PubMed] [Google Scholar]
- 107.Karlsson S, Burman LG, Akerlund T. 2008. Induction of toxins in Clostridium difficile is associated with dramatic changes of its metabolism. Microbiology 154:3430–3436. doi: 10.1099/mic.0.2008/019778-0. [DOI] [PubMed] [Google Scholar]
- 108.Ransom EM, Kaus GM, Tran PM, Ellermeier CD, Weiss DS. 2018. Multiple factors contribute to bimodal toxin gene expression in Clostridioides (Clostridium) difficile. Mol Microbiol 110:533–549. doi: 10.1111/mmi.14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith CJ, Markowitz SM, Macrina FL. 1981. Transferable tetracycline resistance in Clostridium difficile. Antimicrob Agents Chemother 19:997–1003. doi: 10.1128/aac.19.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cartman ST, Minton NP. 2010. A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl Environ Microbiol 76:1103–1109. doi: 10.1128/AEM.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 112.Graentzdoerffer A, Rauh D, Pich A, Andreesen JR. 2003. Molecular and biochemical characterization of two tungsten- and selenium-containing formate dehydrogenases from Eubacterium acidaminophilum that are associated with components of an iron-only hydrogenase. Arch Microbiol 179:116–130. doi: 10.1007/s00203-002-0508-1. [DOI] [PubMed] [Google Scholar]
- 113.Axley MJ, Grahame DA, Stadtman TC. 1990. Escherichia coli formate-hydrogen lyase. Purification and properties of the selenium-dependent formate dehydrogenase component. J Biol Chem 265:18213–18218. [PubMed] [Google Scholar]
- 114.Mock J, Wang S, Huang H, Kahnt J, Thauer RK. 2014. Evidence for a hexaheteromeric methylenetetrahydrofolate reductase in Moorella thermoacetica. J Bacteriol 196:3303–3314. doi: 10.1128/JB.01839-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mock J, Zheng Y, Mueller AP, Ly S, Tran L, Segovia S, Nagaraju S, Kopke M, Durre P, Thauer RK. 2015. Energy conservation associated with ethanol formation from H2 and CO2 in Clostridium autoethanogenum involving electron bifurcation. J Bacteriol 197:2965–2980. doi: 10.1128/JB.00399-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bertsch J, Oppinger C, Hess V, Langer JD, Müller V. 2015. Heterotrimeric NADH-oxidizing methylenetetrahydrofolate reductase from the acetogenic bacterium Acetobacterium woodii. J Bacteriol 197:1681–1689. doi: 10.1128/JB.00048-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cohen SA. 2003. Amino acid analysis using pre-column derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Analysis of hydrolyzed proteins and electroblotted samples. Methods Mol Biol 211:143–154. [PubMed] [Google Scholar]
- 118.Stadtman TC, Davis JN. 1991. Glycine reductase protein C. Properties and characterization of its role in the reductive cleavage of Se-carboxymethyl-selenoprotein A. J Biol Chem 266:22147–22153. [PubMed] [Google Scholar]
- 119.Schrader T, Andreesen JR. 1992. Purification and characterization of protein PC, a component of glycine reductase from Eubacterium acidaminophilum. Eur J Biochem 206:79–85. doi: 10.1111/j.1432-1033.1992.tb16903.x. [DOI] [PubMed] [Google Scholar]
- 120.Wagner M, Sonntag D, Grimm R, Pich A, Eckerskorn C, Sohling B, Andreesen JR. 1999. Substrate-specific selenoprotein B of glycine reductase from Eubacterium acidaminophilum. Biochemical and molecular analysis. Eur J Biochem 260:38–49. doi: 10.1046/j.1432-1327.1999.00107.x. [DOI] [PubMed] [Google Scholar]
- 121.Lipmann F, Tuttle LC. 1945. A specific micromethod for the determination of acyl phosphates. J Biol Chem 159:21–28. [Google Scholar]
- 122.Seto B. 1979. Proline reductase: a sensitive fluorometric assay with O-phthalaldehyde. Anal Biochem 95:44–47. doi: 10.1016/0003-2697(79)90183-0. [DOI] [PubMed] [Google Scholar]
- 123.Grahame DA. 2011. Methods for analysis of acetyl-CoA synthase applications to bacterial and archaeal systems. Methods Enzymol 494:189–217. doi: 10.1016/B978-0-12-385112-3.00010-X. [DOI] [PubMed] [Google Scholar]
- 124.Gencic S, Grahame DA. 2003. Nickel in subunit beta of the acetyl-CoA decarbonylase/synthase multienzyme complex in methanogens. Catalytic properties and evidence for a binuclear Ni-Ni site. J Biol Chem 278:6101–6110. doi: 10.1074/jbc.M210484200. [DOI] [PubMed] [Google Scholar]
- 125.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 126.Graves MC, Mullenbach GT, Rabinowitz JC. 1985. Cloning and nucleotide sequence determination of the Clostridium pasteurianum ferredoxin gene. Proc Natl Acad Sci U S A 82:1653–1657. doi: 10.1073/pnas.82.6.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Horton RM. 1997. In vitro recombination and mutagenesis of DNA. SOEing together tailor-made genes. Methods Mol Biol 67:141–149. doi: 10.1385/0-89603-483-6:141. [DOI] [PubMed] [Google Scholar]
- 128.Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 129.Purdy D, O'Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP. 2002. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol 46:439–452. doi: 10.1046/j.1365-2958.2002.03134.x. [DOI] [PubMed] [Google Scholar]
- 130.Kirk JA, Fagan RP. 2016. Heat shock increases conjugation efficiency in Clostridium difficile. Anaerobe 42:1–5. doi: 10.1016/j.anaerobe.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.